
Abstract
In this chapter, the synthesis of carbamides and their analogues such as carbamoyl azides through N atoms incorporation strategy with C–H and/or C–C bond cleavage is summarized. Azides considered as nitrogen sources with various oxidants are efficiently employed in these transformations. Aldehydes, ketones, alcohols, and alkynes were converted into the corresponding carbamoyl azides and carbamides efficiently through this strategy.
Similar content being viewed by others

Keywords
5.1 Introduction
Carbamides (also called ureas) and their analogues such as carbamates are commonly encountered in the structures of biologically active compounds. Carbamides are widely used as agrochemicals, dyes, antioxidants and HIV inhibitors [1–3]. Carbamates are pivotal precursors for the synthesis of pesticides, fungicides, herbicides and drugs [4, 5]. In addition, carbamides, and their analogues such as carbamates, carbodiimides, and carbamoyl azides are also widely applied as key intermediates in organic synthesis [6–10]. Furthermore, by serving as hydrogen-bond donors, carbamides can also be used as efficient and air-stable organocatalysts [11] or ligands for transition metals [12]. Therefore, many methods have been developed to synthesize carbamides and their analogues [13, 14]. Generally, efficient approaches to carbamides and carbamates are achieved via isocyanate intermediates (Scheme 5.1) [15], which are commonly generated by coupling of amines with phosgene and its derivatives [16–22], reductive carbonylation of nitroaromatics [23–26], or Curtius rearrangement [27, 28]. Unfortunately, in most cases, either the precursors of these methods lack environmental friendliness, or the substrate scope is limited. Therefore, intense effort has been focused on the development of non-phosgene processes for the synthesis of carbamides and carbamates [29–34]. In this chapter, we will focus on the recent progresses about carbamides and their analogues synthesis through nitrogenation strategy with N atoms incorporation. In addition, some relative N group incorporation protocols by using azides as N-partners are also discussed in this chapter.

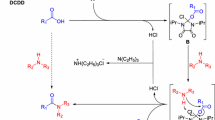
General synthesis of carbamide and carbamate
5.2 Carbamide Synthesis Through N Atom Incorporation
The thermal decomposition of acyl azides into isocyanate intermediates is known as Curtius rearrangement reaction (Scheme 5.2) [27, 28]. Conventionally used general methods to synthesize aroyl azides are limited to diazotization of hydrazides and reactions of NaN3 with acid chlorides, mixed anhydrides, and N-acyl benzotriazoles [35–38]. However, these procedures involve highly reactive chemicals which put significant limitations on functionalities of the substrate. The development of methodologically new, highly functional-group tolerant, catalytic routes to aroyl azides is particularly desirable.

Curtius rearrangement
In 2000, Chen et al. developed a method for the synthesis of aroyl azides from the corresponding aryl aldehydes with the aid of PhI(OAc)2 and sodium azide in high yields [39]. Simple stirring of a mixture of an aryl aldehyde, PhI(OAc)2 and NaN3 in CH2Cl2 under a N2 atmosphere at room temperature gave the desired aroyl azide free of Curtius rearrangement product (Scheme 5.3). This method is limited to the synthesis of thermally stable and isolable aroyl azides. As depicted in Scheme 5.3, the mechanism of this conversion may involve [bis(azido)iodo]benzene, which is formed by ligand exchange followed by homolytic decomposition to generate an azido radical. The starting aldehyde is azidonated via a usual H-abstraction and coupling process.

Transformation of aryl aldehydes to aroyl azides
Inspired by Chen’s work, Bols and co-workers found that TMSN3 and PhI(OAc)2 also promote high-yield azide substitution of aldehydes at zero to ambient temperature in acetonitrile to afford the primary acyl azide products, which were heated to 83 °C to give Curtius rearrangement to isocyanate intermediates that, under these conditions, reacts with azide ions to give carbamoyl azides [40]. For the azidonation an excess of equimolar amounts of PhI(OAc)2 and TMSN3 was necessary, but an extra equivalent of TMSN3 was added to form carbamoyl azide. A series of aliphatic and aromatic aldehydes were converted to carbamoyl azides in good yield with this procedure (Scheme 5.4). The preferred solvent was MeCN, while dichloromethane, benzene, EtOH and THF gave lower yields. The reaction is inhibited by the radical trap N-tert-butyl-a-phenylnitrone. A radical process was proposed for this transformation. The mixture of TMSN3 and PhI(OAc)2 lead to formation of PhI(N3)2 that decomposes giving azide radicals. The chain propagating steps are abstracting the aldehyde hydrogen atom by azide radical. The resulting carbon-centered radical reacts with iodine azide to produce an acyl azide and an iodine radical. The formed acyl azides undergo Curtius rearrangement to isocyanate intermediates that reacts with azide ions to give carbamoyl azides (Scheme 5.5).

Direct preparation of carbamoyl azides from aldehydes

The proposed mechanism
Moreover, Bols et al. developed another methodology for the synthesis of carbamoyl azides from aldehydes by treatment with iodine azide at reflux in acetonitrile [41]. The carbamoyl azides are obtained in 70–97 % yield from the aliphatic and aromatic aldehydes (Scheme 5.4). When the reaction of phenylpropanal with IN3 at 25 °C was performed in the presence of the radical trap, no acyl azide was observed, which was taken as support for a radical reaction mechanism. The mechanism shown in Scheme 5.6 is proposed for the reaction. Iodine radicals are formed by homolysis of the weak iodine-azide bond, abstracting the aldehyde hydrogen atom. The resulting carbon-centered radical reacts with iodine azide to produce an acyl azide. The following Curtius rearrangement provides carbamoyl azides.

The proposed mechanism
Futhermore, Bols et al. provides an alternative safe and convenient synthetic strategy for the synthesis of carbamoyl azides from aldehydes by treatment with polymer supported iodine azide in MeCN at 83 °C (Scheme 5.4) [42]. Considering the drawback of its potentially explosive nature, IN3 as a reagent cannot be widespread used. Therefore, a stable electrophilic polymer-bound reagent that synthetically behaves like iodine azide was employed in N atom incorporation strategy. The polymer supported iodine azide can be readily prepared by reacting the polystyrene bound iodide with phenyliodonium diacetate and subsequently with trimethysilyl azide, or by direct azido transfer after treatment of the polystyrene bound iodide with (diazido)benzene.
In 2010, Studer et al. reported N-heterocyclic carbene catalyzed oxidative amidations of various aldehydes to the corresponding acyl azides by using the readily available organic oxidant [43]. Acyl azides can readily be converted via thermal Curtius rearrangement to carbamoyl azides as shown for the transformation of benzaldehyde to phenyl carbamoylazide (Scheme 5.7), when t-BuOH was further added to the crude reaction mixture, which was then heated for 1 h to reflux. Therefore, a mild NHC-catalyzed oxidative azidation of aromatic aldehydes to form the corresponding acyl azides which can be rearranged to carbomoylazides in the same pot.

NHC-catalyzed oxidative azidation of aromatic aldehydes to carbomoylazides in one pot
In 2014, Jiao and coworkers group reported a simple ceric ammonium nitrate (CAN) catalyzed synthesis of carbamoyl azide from ketones and TMSN3 through C–C double bond cleavage [44]. The optimized conditions are using 20 mol% CAN, 1.0 equivalent of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) in EtOAc under O2 at 60 °C for 36 h (Scheme 5.8). This chemistry offers a simple approach leading to carbamoyl azides which are of versatile reactivity and synthetic value. Carbamoyl azide shows diverse transformations that it can be easily converted into carbamide and carbamate (Scheme 5.9). CAN is a single electron oxidant for this transformation and it also plays as a Lewis acid during the reaction. The reaction of TEMPO with the methylene group of ketones to form intermediate A is the key step for this C–C bond cleavage process (Scheme 5.10). Then an azide radical which is produced through this oxidative system reacts with intermediate A to produce the unstable intermediate B. Then it undergoes a N–O bond cleavage to generate intermediate C. Subsequently, intermediate C was attacked by the azido nucleophile to produces intermediate D with its resonance structure E. Then C–C bond cleavage occurred and benzoyl azide F and imidic acid G are formed. Finally, intermediate F undergoes Curtius rearrangement to produce carbamoyl azide and intermediate G undergoes tautomerization to produce amide.

Synthesis of carbamoyl azides from ketones

Transformations of carbamoyl azide into carbamide and carbamate

The proposed mechanism
Recently, our group developed an efficient KI/TBHP catalyzed nitrogenation of aldehydes or alcohols for the direct synthesis of carbamoyl azides and ureas via a radical process [45]. A variety of substituted aryl aldehydes, even benzylamine, phenylacetaldehyde and benzyl alcohol performed well in this nitrogenation reaction to provide carbamoyl azides (Scheme 5.11). When NaOAc was added in the optimal conditions, symmetrical ureas could be directly preparation from the reaction of aryl aldehyde with azide (Scheme 5.12). The simple operating procedures, the readily available starting materials including aldehyde, alcohol and amines, as well as the utility of the products all make this strategy very attractive. A proposed mechanism is shown in Scheme 5.13. At first, tert-butoxyl radical is generated from TBHP through the catalyticcycle, where I2 play a role as the catalyst. Meanwhile, the substrate is attacked by the azide to produce the intermediate A. Secondly, tert-butoxyl radical abstractstwo hydrogen atoms from the intermediate to generate acyl azide. The acyl azide will undergo a Curtius rearrangement to generate aryl isocyanate, which reacts with another azide to produce carbamoyl azide. In the presence of base, carbamoyl azide is converted into urea easily.

Synthesis of carbamoyl azides from aldehydes and alcohols

Synthesis of carbamides from aldehydes

The proposed mechanism
Moreover, a gold catalyzed nitrogenation of alkynes for the synthesis of carbamides using TMSN3 as nitrogen source was realized in Jiao group recently [46]. The alkyne was split into three parts through C–C single bond and C≡C triple bond cleavage. The best reaction conditions are with 10 mol% PPh3AuCl/AgF, four equivalents TMSN3, four equivalents methanesulfonic acid (MSA) or Trifluoromethanesulfonic acid (TfOH), two equivalents H2O in trichloroethylene at room temperature for 24 h. Several internal and terminal alkynes bearing electron-donating and halo-substitutents were converted to the corresponding carbamides in moderate yields (Scheme 5.14). A plausible mechanism was shown in Scheme 5.15. Initially, the alkyne is activated by cationic Au(I) and attacked by TMSN3 to produce alkenyl azide A. Further protonation of A followed by an acid-catalyzed rearrangement process to generate intermediate C. Next, intermediate C reacts with TMSN3 once again to generate imino azide D. The subsequent protonation of D and rearrangement gives intermediate F. Finally, nucleophilic attack by H3O+ lead to the carbamide via a tautomerization process.

Synthesis of carbamides from alkynes

The proposed mechanism
5.3 Carbamide and Its Analogues Synthesis Through N Group Incorporation
N group incorporation is considered as alternative nitrogenation strategy for the synthesis of carbamides and their analogues such as carbamates, carbodiimides. Among them, Pd-catalyzed carbonylation is well established as one of the most important ways to synthesize compounds bearing carbonyl functionality. Guan and co-workers reported in 2012 an access to carbamates by palladium-catalyzed carbonylation of aromatic amines under an atmosphere of carbon monoxide with alcohol [47]. A variety of N-phenylcarbamates was easily synthesized in good to excellent yields from readily available aromatic amines under mild conditions (Scheme 5.16). Pd-catalyzed coupling of amines in the presence of CO and oxidant provided an alternative strategy to accessing ureas without alcohol. However, symmetrical urea was usually the dominate product, which is also the major byproduct in other urea formation strategies via transition-metal-catalyzed carbonylation in the presence of amines.

Pd-catalyzed carbonylation of aromatic amines to synthesize carbamides and carbamates
Jiao and co-workers reported in 2014 a simple and practical catalytic methodology for the direct formation and application of isocyanates for the synthesis of carbamates [48]. This chemistry provides an efficient and practical approach to synthesize carbamates from simple organic azides, CO atmosphere and alcohols. The broad scope, mild and neutral conditions, and only N2 as the byproduct make this transformation very useful. Moreover, simple examples of modification of bioactive molecules and construction of macrocycles were achieved through this protocol. By utilizing this protocol, the marketed drugs Chlorzoxazone (a centrally acting muscle relaxant) and Chlorpropham (an important herbicide and sprout suppressant) were easily prepared from simple and readily available aromatic azides, CO, and alcohols in high yields (Scheme 5.17). The mechanism is proposed to start with the formation of a palladium nitrene species A from azides with the release of N2. The subsequent insertion of CO into A affords intermediate B. Then isocyanate C is afforded by the reductive elimination process of intermediate B with the regeneration of the Pd-catalyst. Finally, the nucleophilic attack of alcohols on isocyanate C promoted by the Pd-catalyst as a Lewis acid occurs and produces the desired product carbamate.

Pd-catalyzed assembly of organic azides, CO, and alcohols to synthesize carbamates
Zhang and co-workers reported latter an efficient palladium-catalyzed cross-coupling reaction of azides with isocyanides, providing a general synthetic route to unsymmetric carbodiimides with excellent yields [49]. This method shows a broad substrate scope, including not only aryl azides, but also unactivated benzyl and alkyl azides. Furthermore, from readily available substrates, Pd-catalyzed coupling with a tandem amine insertion cascade to obtain unsymmetric trisubstituted guanidines has been achieved in a one-pot fashion (Scheme 5.18). The mechanism is proposed to start with the formation of the probable palladium nitrene species A from azides simultaneously with the release of N2. Subsequently, insertion of isocyanide into Pd-nitrene species A occurred to give intermediate C. Finally, reductive elimination of intermediate C affords the product carbodiimide.

Pd-catalyzed cross-coupling reaction of azides with isocyanides to synthesize carbodiimides
Recently, studies in urea synthesis have focused on transition-metal-catalyzed reactions. Buchwald et al. reported a Pd-catalyzed cross-coupling of aryl chlorides with sodium cyanate, which represented a practical way to synthesize unsymmetrical ureas [50]. The protocol allows for the synthesis of unsymmetrical N,N′-di- and N,N,N′-trisubstituted ureas in one pot and is tolerant of a wide range of functional groups (Scheme 5.19). Insight into the mechanism of aryl isocyanate formation was gleaned through studies of the transmetalation and reductive elimination steps of the reaction, including the first demonstration of reductive elimination from an arylpalladium isocyanate complex to produce an aryl isocyanate. Mechanistic studies conducted on this system suggest that transmetalation is the rate-limiting step. Finally, the first example of reductive elimination from an arylpalladium isocyanate complex has also been demonstrated. Nonetheless, the application of this method is limited to aromatic ureas.

Pd-catalyzed cross-coupling of ArCl with NaOCN
Zhang and co-workers reported a novel product-derived bimetallic palladium complex catalyzes a sulfonylazide-transfer reaction with the σ-donor/π-acceptor ligand CO, and is advantageous given its broad substrate scope, high efficiency, and mild reaction conditions (atmospheric pressure of CO at room temperature) [51]. This methodology provides a new approach to sulfonylureas, which are present in both pharmaceuticals and agrochemicals. The synthesis of Glibenclamide on a gram scale further revealed the practical utility of this procedure (Scheme 5.20). Mechanistically, the generation of a bridged bimetallic palladium species derived from the product sulfonylurea is disclosed as the crucial step for this catalytic cycle. HRMS studies of [Pd2L2(MeCN)2] with sulfonylureas (H2L) indicates that disulfonylurea bridged bimetallic palladium was the real active palladium species in the mixture.

Bimetallic palladium complex catalyzed direct carbonylation of sulfonylazides
Zhang and co-workers subsequently reported a facile and efficient Pd/C-catalyzed carbonylation of both aliphatic and aromatic azides in the presence of amines [52]. Serving as the widely existed fragments in an array of biological pharmaceuticals, functionalized unsymmetrical ureas were straightforwardly synthesized by using readily available and cheap azides with amines under CO atmosphere, with the extrusion of N2 as the only byproduct. It was found that not only aryl azides but also benzyl and alkyl azides were suited for this methodology. Another feature of this procedure was the employment of a highly efficient palladium charcoal catalytic system (Scheme 5.21). Mechanistically, the probable palladium nitrene species A is formed from organic azide simultaneously with the release of N2. Subsequently, the insertion of CO into palladium nitrene species A occurred to give Pd-coordinated isocyanate B. Finally, nucleophilic attack of amine at isocyanate C, which is promoted by palladium/phosphine complexes, affords desired unsymmetrical ureas as the eventual product along with the regeneration of Pd catalyst.

Pd/C-catalyzed carbonylation of azides for synthesis of carbamide
5.4 Conclusion and Outlook
In this chapter, the recent progresses about carbamides and their analogues such as carbamoyl azides synthesis through N atom incorporation strategy with C–H and/or C–C bond cleavage are discussed in detail. Azides considered as nitrogen sources with various oxidants are efficiently employed in these transformations. Aldehydes, ketones, alcohols, and alkynes are converted into the corresponding carbamoyl azides and carbamides efficiently through this strategy. Alternatively, N group incorporation strategy with azides reagents for the synthesis of carbamides and their analogues such as carbamates, carbodiimides are mentioned as well. However, it is still challenging in this area although great progresses have been achieved over the past years. For instance, the synthesis of carbamides and their analogues from readily available amides and esters through N atom incorporation strategy with C–C bond activation are still highly desired. The development of sustainable catalytic systems using green oxidants such as O2 under mild conditions is still urgent.
References
Gallou IO (2007) Unsymmetrical ureas. Synthetic methodologies and application in drug design. Org Prep Proced Int 39(43):355–383
Zhang J, Zhou J, Ren X, Diao Y, Li H, Jiang H, Ding K, Pei D (2012) A new diaryl urea compound, D181, induces cell cycle arrest in the G1 and M phases by targeting receptor tyrosine kinases and the microtubule skeleton. Invest New Drugs 30(2):490–507
Anandan S-K, Webb HK, Do ZN, Gless RD (2009) Unsymmetrical non-adamantyl N,N′-diaryl urea and amide inhibitors of soluble expoxide hydrolase. Bioorg Med Chem Lett 19(15):4259–4263
Tai-The W, Huang J, Arrington ND, Dill GM (1987) Synthesis and herbicidal activity of.alpha.-heterocyclic carbinol carbamates. J Agric Food Chem 35(5):817–823
Chaturvedi D, Mishra N, Mishra V (2007) Various approaches for the synthesis of organic carbamates. Curr Org Synth 4:308–320
Clayden J, Hennecke U (2008) α-Pyridylation of chiral amines via urea coupling, lithiation and rearrangement. Org Lett 10(16):3567–3570
Lefranc J, Tetlow DJ, Donnard M, Minassi A, Galvez E, Clayden J (2011) Geometry-Selective synthesis of E or Z N-vinyl ureas (N-carbamoyl enamines). Org Lett 13(2):296–299
Yu S, Haight A, Kotecki B, Wang L, Lukin K, Hill DR (2009) Synthesis of a TRPV1 receptor antagonist. J Org Chem 74(24):9539–9542
Ali A, Reddy GSKK, Nalam MNL, Anjum SG, Cao H, Schiffer CA, Rana TM (2010) Structure-based design, synthesis, and structure–activity relationship studies of HIV-1 protease inhibitors incorporating phenyloxazolidinones. J Med Chem 53(21):7699–7708
Lieber E, Minnis RLJ, Rao CNR (1965) Carbamoyl azides. Chem Rev 65(3):377–384
Doyle AG, Jacobsen EN (2007) Small-molecule H-bond donors in asymmetric catalysis. Chem Rev 107(12):5713–5743
Yang T, Ferrali A, Sladojevich F, Campbell L, Dixon DJ (2009) Brønsted base/lewis acid cooperative catalysis in the enantioselective conia-ene reaction. J Am Chem Soc 131(26):9140–9141
Adams P, Baron FA (1965) Esters of carbamic acid. Chem Rev 65(5):567–602
Chaturvedi D (2011) Recent developments on the carbamation of amines. Curr Org Chem 15(10):1593–1624
Ulrich H (1997) Chemistry and technology of isocyanates. Wiley, Chichester
Slocombe RJ, Hardy EE, Saunders JH, Jenkins RL (1950) Phosgene derivatives. the preparation of isocyanates, carbamyl chlorides and cyanuric acid. J Am Chem Soc 72(5):1888–1891
Eckert H, Forster B (1987) Triphosgene, a crystalline phosgene substitute. Angew Chem Int Ed 26(9):894–895
Batey RA, Santhakumar V, Yoshina-Ishii C, Taylor SD (1998) An efficient new protocol for the formation of unsymmetrical tri- and tetrasubstituted ureas. Tetrahedron Lett 39(35):6267–6270
Babad H, Zeiler AG (1973) Chemistry of phosgene. Chem Rev 73(1):75–91
Norwick JS, Powell NA, Nguyen TM, Noronha G (1992) An improved method for the synthesis of enantiomerically pure amino acid ester isocyanates. J Org Chem 57(26):7364–7366
Shimizu M, Sodeoka M (2007) Convenient method for the preparation of carbamates, carbonates, and thiocarbonates. Org Lett 9(25):5231–5234
Kim JG, Jang DO (2009) Indium-catalyzed reaction for the synthesis of carbamates and carbonates: selective protection of amino groups. Tetrahedron Lett 50(22):2688–2692
Paul F (2000) Catalytic synthesis of isocyanates or carbamates from nitroaromatics using group viii transition metal catalysts. Coord Chem Rev 203(1):269–323
Dieck HA, Laine RM, Heck RF (1975) Low-pressure, palladium-catalyzed N,N′-diarylurea synthesis from nitro compounds, amines, and carbon monoxide. J Org Chem 40(19):2819–2822
Kim KD, Lee SM, Cho NS, Oh JS, Lee CW, Lee JS (1992) Palladium-catalyzed N,N′-diphenylurea synthesis from nitrobenzene, aniline, and carbon monoxide. Part 3. Evidence of carbamoyl intermediate. J Mol Catal 75(1):L1–L6
Gasperini M, Ragaini F, Remondini C, Caselli A, Cenini S (2005) The palladium–phenanthroline catalyzed carbonylation of nitroarenes to diarylureas: effect of chloride and diphenylphosphinic acid. J Organomet Chem 690(20):4517–4529
Banthorpe DV (1971) Rearrangements involving azido groups. In: Patai S (ed) The chemistry of the azido group. Wiley, New York, pp 397–400
Yagodkin A, Löschcke K, Weisell J, Azhayev A (2010) Straightforward carbamoylation of nucleophilic compounds employing organic azides, phosphines, and aqueous trialkylammonium hydrogen carbonate. Tetrahedron 66(12):2210–2221
Breitler S, Oldenhuis NJ, Fors BP, Buchwald SL (2011) Synthesis of unsymmetrical diarylureas via Pd-catalyzed C–N cross-coupling reactions. Org Lett 13(12):3262–3265
Hooker JM, Reibel AT, Hill SM, Schueller MJ, Fowler JS (2009) One-Pot, Direct Incorporation of [11C]CO2 into Carbamates. Angew Chem Int Ed 48(19):3482–3485
Peterson SL, Stucka SM, Dinsmore CJ (2010) Parallel synthesis of ureas and carbamates from amines and CO2 under mild conditions. Org Lett 12(6):1340–1343
Wei Y, Liu J, Lin S, Ding H, Liang F, Zhao B (2010) Acetoacetanilides as masked isocyanates: facile and efficient synthesis of unsymmetrically substituted ureas. Org Lett 12(19):4220–4223
Dube P, Nathel NFF, Vetelino M, Couturier M, Aboussafy CL, Pichette S, Jorgensen ML, Hardink M (2009) Carbonyldiimidazole-mediated lossen rearrangement. Org Lett 11(24):5622–5625
Hutchby M, Houlden CE, Ford JG, Tyler SNG, Gagne MR, Lloyd-Jones GC, Booker-Milburn KI (2009) Hindered ureas as masked isocyanates: facile carbamoylation of nucleophiles under neutral conditions. Angew Chem Int Ed 48(46):8721–8724
Scriven EFV, Turnbull K (1988) Azides: their preparation and synthetic uses. Chem Rev 88(2):297–368
Brase S, Gil C, Knepper K, Zimmermann V (2005) Organic azides: an exploding diversity of a unique class of compounds. Angew Chem Int Ed 44(33):5188–5240
Brase S, Banert K (2010) Organic azides: syntheses and applications. Wiley, Chichester
Katritzky AR, Widyan K, Kirichenko K (2007) Preparation of polyfunctional acyl azides. J Org Chem 72(15):5802–5804
Chen DJ, Chen ZC (2000) Hypervalent iodine in synthesis. Part 54: one-step conversion of aryl aldehydes to aroyl azides using a combined reagent of (diacetoxyiodo)benzene with sodium azide. Tetrahedron Lett 41(38):7361–7363
Pedersen MC, Marinescu LG, Bols M (2005) Radical substitution with azide: TMSN3–PhI(OAc)2 as a substitute of IN3. Org Biomol Chem 3(5):816–822
Marinescu L, Thinggaard J, Thomsen IB, Bols M (2003) Radical azidonation of aldehydes. J Org Chem 68(24):9453–9455
Marinescu LG, Pedersen MC, Bols M (2005) Safe radical azidonation using polystyrene supported diazidoiodate(I). Tetrahedron 61(1):123–127
Sarkar SD, Studer A (2010) Oxidative amidation and azidation of aldehydes by NHC catalysis. Org Lett 12(9):1992–1995
Feng P, Sun X, Su Y, Li X, Zhang LH, Shi X, Jiao N (2014) Ceric ammonium nitrate (CAN) catalyzed modification of ketones via two C–C bond cleavages with the retention of the oxo-group. Org Lett 16(12):3388–3391
Feng P, Zou M, Jiao N, KI/TBHP catalyzed nitrogenation of aldehydes or alcohols: the direct synthesis of carbamoyl azides and ureas. Unpublished work
Qin C, Su Y, Shen T, Shi X, Jiao N (2016) Splitting a substrate into three parts: gold-catalyzed nitrogenation of alkynes by C–C and C≡C bond cleavage. Angew Chem Int Ed 55(1):350–354
Guan Z-H, Lei H, Chen M, Ren Z-H, Bai Y, Wang Y-Y (2012) Palladium-catalyzed carbonylation of amines: switchable approaches to carbamates and N,N′-disubstituted ureas. Adv Synth Catal 354(2–3):489–496
Ren L, Jiao N (2014) PdCl2 catalyzed efficient assembly of organic azides, CO, and alcohols under mild conditions: a direct approach to synthesize carbamates. Chem Commun 50(28):3706–3709
Zhang Z, Li Z, Fu B, Zhang Z (2015) Palladium-catalyzed cross-coupling reaction of azides with isocyanides. Chem Commun 51(91):16312–16315
Vinogradova EV, Fors BP, Buchwald SL (2012) Palladium-catalyzed cross-coupling of aryl chlorides and triflates with sodium cyanate: a practical synthesis of unsymmetrical ureas. J Am Chem Soc 134(27):11132–11135
Zhao J, Li Z, Song S, Wang M-A, Fu B, Zhang Z (2016) Product-derived bimetallic palladium complex catalyzes direct carbonylation of sulfonylazides. Angew Chem Int Ed 55(18):5545–5549
Zhao J, Li Z, Yan S, Xu S, Wang M-A, Fu B, Zhang Z (2016) Pd/C catalyzed carbonylation of azides in the presence of amines. Org Lett 18(8):1736–1739
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Li, X., Jiao, N. (2017). Nitrogenation Strategy for the Synthesis of Carbamides. In: Jiao, N. (eds) Nitrogenation Strategy for the Synthesis of N-containing Compounds. Springer, Singapore. https://doi.org/10.1007/978-981-10-2813-7_5
Download citation
DOI: https://doi.org/10.1007/978-981-10-2813-7_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-2811-3
Online ISBN: 978-981-10-2813-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)