
Abstract
Peroxisomes are remarkably plastic and dynamic organelles, which fulfil important functions in hydrogen peroxide and lipid metabolism rendering them essential for human health and development. Despite great advances in the identification and characterization of essential components and molecular mechanisms associated with the biogenesis and function of peroxisomes, our understanding of how peroxisomes are incorporated into metabolic pathways and cellular communication networks is just beginning to emerge. Here we address the interaction of peroxisomes with other subcellular compartments including the relationship with the endoplasmic reticulum, the peroxisome-mitochondria connection and the association with lipid droplets. We highlight metabolic cooperations and potential cross-talk and summarize recent findings on peroxisome-peroxisome interactions and the interaction of peroxisomes with microtubules in mammalian cells.
Similar content being viewed by others

Keywords
- Peroxisomes
- Mitochondria
- Endoplasmic reticulum
- Lipid droplet
- Cytoskeleton
- Organelle interplay
- Organelle cross-talk
1.1 Introduction
Peroxisomes are highly dynamic, multifunctional organelles in eukaryotic cells. They contribute to several anabolic and catabolic cellular pathways, which renders them essential for human health and development (Islinger and Schrader 2011). A remarkable feature of peroxisomes is their ability to respond to cellular and/or environmental changes and stimuli with alterations in their biogenesis, morphology, number, protein composition and metabolic activity. The metabolism of H2O2 and the oxidation of fatty acids are their most common functions in animals, plants and fungi. However, the spectrum of peroxisomal tasks is far from being complete, and due to extensive research in the last years, is constantly widening. Several specialized and novel peroxisomal functions have been discovered, including a new biological role in anti-viral defence (see Chap. 4), H2O2 signalling in hypothalamic neurons (Diano et al. 2011), the synthesis of hormonal signal molecules in plants (see Chaps. 14 and 16), Ca++ signalling (see Chap. 7), or the synthesis of secondary metabolites in fungi (see Chaps. 8 and 9). It is obvious that peroxisomes do not represent isolated entities within the cell, but are functionally integrated into a complex network of communicating subcellular compartments. In this respect, a physical and metabolic interaction with the endoplasmic reticulum (ER) has long been known (see Sect. 1.3; Fig. 1.1a–c), and it became obvious in the last years that the ER can also contribute to peroxisome formation, although the degree of this contribution is debated and may vary among species (see Sect. 1.3). In addition, a closer connection between peroxisomes and mitochondria was revealed (see Sect. 1.4) (Schrader and Yoon 2007; Camoes et al. 2009; Schrader et al. 2012). Despite great advances in the identification and characterization of essential proteins (e.g., peroxins) and molecular mechanisms associated with the biogenesis and function of peroxisomes, our understanding of how peroxisomes are incorporated into metabolic pathways and cellular communication networks is just beginning to emerge. Here we address the interaction of peroxisomes with other subcellular compartments and highlight metabolic cooperations and potential cross-talk. We particularly focus on peroxisome interactions in animals, but where appropriate also refer to recent discoveries in plants and fungi.

(a–b) Physical interaction of peroxisomes (P) with the endoplasmic reticulum (ER). (a) Cytochemical localization of catalase in rat hepatic peroxisomes stained with the alkaline diamino-benzidine technique. Note the close association of the limiting membrane of peroxisomes with the ER (arrows). Magnification, ×33,600 (from Fahimi and Yokota 1981). (b) Electron micrograph from the liver of a rat treated with the peroxisome proliferator clofibrate for 7 days illustrating two peroxisomes with their limiting membranes touching each other, giving rise to a junction-like appearance. Note the segments of ER which surround the peroxisomes, forming close contacts with their limiting membranes (arrows). Magnification, ×38,000 (from Fahimi et al. 1980). (c) The association of ER and peroxisomes is well demonstrated in freeze-etch preparations from rat liver, revealing that peroxisomes are almost entirely engulfed by fenestrated ER. Note also the two fracture faces of the peroxisomal membrane, with the P-face (PF) containing more membrane associated particles then the E-face (EF). Magnification, ×40,000 (from Fahimi et al. 1980). (d) Contact of a peroxisome with two mitochondria in mouse myocardium (from Herzog and Fahimi 1976). (e, f) Intimate physical contacts of elongated peroxisomes with lipid droplets in primate (Macaca java) myocardium (from Hicks and Fahimi 1977). Peroxisomes were stained with the alkaline diamino-benzidine technique for catalase
1.2 Peroxisomes and the Nucleus
Unlike mitochondria, peroxisomes do not contain DNA. Thus, all peroxisomal genes are encoded in the nucleus, and their transcription and translation as well as protein import requires coordinated regulation. It is well known that the number and size of peroxisomes as well as their protein/enzyme concentration and composition can be modulated by nutritional factors and environmental stimuli. This so called peroxisome proliferation is mediated by transcription factors which are activated by endogenous ligands such as fatty acids or synthethic peroxisome proliferators (e.g., hypolipidemic compounds). In mammals, particularly in rodents, peroxisome proliferation is mediated by PPARα, the peroxisome proliferator-activated receptor α, a member of the nuclear receptor superfamily. Upon ligand activation, PPARα forms heterodimers with the retinoid X receptor, which enables binding to PPAR responsive elements on the DNA, thereby activating peroxisomal genes involved in fatty acid β-oxidation and peroxisome proliferation. Similar signalling pathways involving transcription factors such as Oaf1, Pip2, or FarA and FarB have been evolved in yeast and fungi (reviewed in Schrader et al. 2012). Upon withdrawal of the proliferative stimulus or altered nutritional conditions, excess peroxisomes are degraded by autophagic processes (Till et al. 2012). Degradation requires an interaction of peroxisomes with autophagic membranes and/or the endosomal-lysosomal compartment, which involves certain peroxins such as Pex14 and autophagy-related proteins. A specific “eat me” signal on peroxisomes prone for degradation has been predicted, but not yet been identified.
1.3 Relationship of Peroxisomes with the Endoplasmic Reticulum
In ultrastructural studies, peroxisomes have been found in close proximity to the ER, frequently virtually wrapped in ER cisternae implying a close relationship between both organelles (Fig. 1.1a–c) (Novikoff and Novikoff 1972; Zaar et al. 1987; Grabenbauer et al. 2000). Already in the early years of peroxisome research this striking spatial coincidence led to the hypothesis that peroxisomes are formed from terminal ER cisternae (Novikoff and Shin 1964), representing a specialized compartment of the cellular endomembrane system. This view was seriously challenged by the discovery that peroxisomal enzymes are synthesized on free polyribosomes and directly imported from the cytosol supporting the view of an autonomous, self-replicating organelle (Lazarow and Fujiki 1985). During recent years, however, the ER came back into focus as the place of peroxisome origin: deletion of the peroxins Pex3, Pex19 or Pex16 results in total loss of peroxisomal structures as these proteins are required for the maintenance of the peroxisomal membrane (South and Gould 1999; Hettema et al. 2000). Their reintroduction into correspondent deletion mutants leads to the de novo generation of peroxisomes, indicating that peroxisomes emerge from another cellular compartment. During this process, Pex3 and Pex19 have been observed to initially localize to the ER before maturing into import-competent peroxisomes (e.g., Hoepfner et al. 2005). That peroxisomes are not exceptionally formed from the ER under the somewhat artificial conditions of a peroxisome-free cell was subsequently suggested (Geuze et al. 2003; Kim et al. 2006; Karnik and Trelease 2007), and ER proteins involved in the secretory pathway such as Sec16B, Sec20, Sec29, Sec61 and Dsl1 have been proposed to contribute to this process (Perry et al. 2009; Yonekawa et al. 2011; Thoms et al. 2012). Recent in vitro studies reported the formation of vesicular structures from the ER carrying a specialized set of peroxins (Lam et al. 2010; Agrawal et al. 2011). As initially proposed (Titorenko and Rachubinski 2001), in yeast two biochemically different vesicle pools bearing individual sets of peroxisomal membrane proteins were described using GFP-split (BiFC) assays and are supposed to bud from specialized ER regions (van der Zand et al. 2012). They are reported to subsequently fuse with each other forming import-competent pre-peroxisomes. Those then mature into functional peroxisomes by importing further matrix components but do not fuse with pre-existing organelles. The authors consequently conclude that peroxisomes are exclusively formed by de novo synthesis from the ER. A vesicular ER-to-peroxisome protein transport would likely request a reciprocal sorting system in order to retarget misrouted ER-resident proteins and/or components of the sorting machinery to their original location. A study in plants described the routing of the tomato bushy stunt virus replication protein p33 from peroxisomes to the ER in concert with endogenous peroxisomal proteins pointing to a retrograde sorting system (McCartney et al. 2005). Under non-pathogenic conditions, however, the existence of peroxisome-to-ER sorting has yet not been convincingly proven. Other studies contradict the “ER-only” hypothesis for peroxisome biogenesis: expression of a mitochondria-targeted Pex3 variant in a correspondent ΔPex3 Saccharomyces cerevisiae mutant was recently reported to induce peroxisome reformation, thus questioning an obligate involvement of the ER (Rucktäschel et al. 2010). Morphologically documented by sequential steps of membrane elongation, constriction and final fission, peroxisomes are also observed to form by growth and division from pre-existing organelles using a division machinery shared with mitochondria (Schrader et al. 2012) (see Sect. 1.4). Furthermore, the dominating pathway for peroxisome formation in S. cerevisiae appears to be fission from pre-existing organelles as de novo formation is a significantly more time consuming process (Motley and Hettema 2007). Similar conclusions were drawn from studies using Hansenula polymorpha (Nagotu et al. 2008) and mammalian cells (Delille et al. 2010). Therefore, both pathways may contribute in parallel to the maintenance and proliferation of peroxisomes (Saraya et al. 2011). Their proportional contribution to total peroxisome numbers has still to be determined and may vary among species. Remarkably, in both processes peroxisomes are not formed as per se functional entities but involve a maturation pathway, where peroxisomal pre-compartments are subsequently supplied with proteins performing metabolic functions (Islinger et al. 2012). In either case, phospholipids are required to promote growth of pre-peroxisomal membrane structures and have thus to be delivered from the ER where they are generated. The peroxin-bearing vesicles described above, provided that they contribute to both de novo formation and growth and division, could deliver phospholipids as well as proteins to the nascent organelles, which would spare a specialized transport system for phospholipids. However, a non-vesicular transfer of ER-derived phospholipids to peroxisomes has been described (Raychaudhuri and Prinz 2008) suggesting that peroxisomal membrane growth can be independent from the vesicular transfer of peroxisomal membrane proteins from the ER when peroxisomes multiply by growth and division. As the transfer of lipids between the ER and peroxisomes is suggested to occur bidirectionally, an important function may also be to alter the lipid composition of the peroxisome membrane. Such alterations may modify the organelle’s mechanical properties thereby supporting membrane bending or elongation during peroxisome proliferation.
Disregarding the ongoing discussion about the role of the ER in peroxisome biogenesis, it is important to note that the relationship between both organelles includes other aspects of cooperation, in particular to facilitate various metabolic pathways shared by both compartments. This metabolic interplay is inter alia documented by the organelle alterations observed in conditional hepatic Pex5 knockout mice which cannot form functional peroxisomes (Dirkx et al. 2005). The knockout mice display severe morphological alterations of mitochondria, an accumulation of lipid droplets and a significant proliferation of the smooth ER. The most familiar pathway of the peroxisome-ER connection may be the biosynthesis of ether-phospholipids, which is initiated in peroxisomes and completed in the ER (Braverman and Moser 2012). Ether glycerolipids constitute about 15–20% of total cellular membranes and are especially enriched in brain, heart and immune cells of the blood. Briefly, their synthesis requires acylation of dihydroxyacetone phosphate by dihydroxyacetone phosphate acyltransferase (DHAPAT). The acyl group is then substituted with an alkyl group by alkyldihydroxyacetone phosphate synthase (alkyl-DHAP synthase or ADHAPS). Both enzymes form a heterotrimeric complex at the inner site of the peroxisomal membrane. Additional reactions associated with the cytosolic site of the peroxisomal membrane include the generation of a long chain alcohol for the substitution reaction described above performed by acyl-CoA reductase, and the reduction of alkyl-DHAP to alkylglycerol-3-phosphate by alkyl-DHAP reductase. All further reactions (acylation in position 2 of the glycerol, dephosphorylation and addition of ethanolamine/cholin in position 3) are carried out in the ER. The function of ether lipids is still unknown, but they are supposed to contribute to a reduction in membrane fluidity (e.g., in lipid rafts) and to act as scavengers for reactive oxygen species to prevent the oxidation of other vulnerable membrane lipids. A defect in ether lipid synthesis represented by the genetic disorder Rhizomelic Chondrodysplasia Punctata (RCDP) results in skeletal dysplasia, severe abnormalities in the central nervous system and cortical cataracts usually leading to death before adulthood (Braverman and Moser 2012). On the cellular level ether lipid deficiency is characterized by impaired membrane traffic and cholesterol distribution affecting the integrity of the plasma membrane (Thai et al. 2001; Gorgas et al. 2006). Recently, the peroxisomal part in ether lipid synthesis has been shown to be crucial for the formation of glycosyl phosphatidyl inositol (GPI)-anchored proteins in the ER, since in mammals, GPI-anchored proteins usually possess mainly 1-alkyl-2-acyl phosphatidyl inositol (Kanzawa et al. 2012). As GPI-anchored proteins fulfil important functions in cell–cell and cell–environment interactions and are prominent constituents of lipid microdomains, the disruption of their synthesis may significantly contribute to the severity of RCDP.
The production of polyunsaturated fatty acids is another biosynthetic process requiring cooperation between ER and peroxisomes. Usually the ER performs the synthesis of such metabolites by desaturation of correspondent saturated fatty acids but lacks an acyl-CoA-dependent 4 desaturase (Voss et al. 1991). To introduce a double bond at this position, 24 carbon n-6 and n-3 fatty acids synthesized in the ER are transferred to peroxisomes where they are partially degraded by β-oxidation until a double bond at position 4 of the carbonate chain is reached (Sprecher and Chen 1999; Su et al. 2001). Instead of further degradation, such fatty acids are exported and re-imported into the ER to be used for membrane lipid biosynthesis (Sprecher and Chen 1999). This process is of major significance for the production of docosahexaenoic acid, which is supposed to play an important role in neuronal migration during development, maintenance of synaptic plasticity in hippocampal neurons and the attenuation of neuroinflammation. Interestingly, the incorporation of docosahexaenoic acid into the peroxisomal membrane was found to stimulate peroxisome proliferation representing a positive feedback-loop for the coordination of peroxisome function and organelle abundance (Itoyama et al. 2012).
The synthesis of cholesterol and isoprenoids from acetyl-CoA was originally associated with the ER and the cytoplasm until a peroxisomal localization of 3-hydroxymethyl glutaryl-CoA reductase (HMG-CoA reductase) – the key regulatory enzyme in cholesterol biosynthesis – was reported (Keller et al. 1985). Gradually, the whole pre-squalene segment of the pathway up to the step of farnesylpyrophosphate (FPP) synthesis was partially or exclusively associated with peroxisomes whereas the incorporation of FPP into squalene was supposed to occur in the ER (Aboushadi et al. 1999). A peroxisomal contribution to further steps in cholesterol biosynthesis has been proposed (Appelkvist et al. 1990), but the peroxisomal localisation of the correspondent proteins has not been confirmed yet. The contribution of peroxisomes to isoprenoid/cholesterol biosynthesis in mammals remains under debate and is not ultimately clarified (Wanders and Waterham 2006; Kovacs et al. 2007). In plants, by contrast, new studies provide evidence that the final four steps of the pre-squalene segment (up to the generation of FPP) are localized to peroxisomes (Clastre et al. 2011; Simkin et al. 2011). As peroxisomal targeting sequences have also been described for the mammalian homologues (Kovacs et al. 2007), from an evolutionary standpoint this pathway was shared by both compartments in the eukaryotic ancestor and different localizations for some of the enzymes may have been evolved during evolution.
By contrast, the peroxisomal role in bile acid synthesis/cholesterol degradation is well established. The conversion of cholesterol into the bile acid precursor trihydroxycholestanoic acid (THCA) is an extraperoxisomal process and enzymes from the ER, mitochondria and the cytosol participate in two alternative pathways (Russell 2003). THCA is then imported into peroxisomes as THCA-CoA and its side chain is shortened by peroxisomal β-oxidation to form cholic acyl-CoA. Finally, the CoA group of cholic acyl-CoA is substituted with taurine/glycine for excretion by the peroxisomal enzyme bile acid-CoA: amino acid N-acyltransferase (Ferdinandusse et al. 2009). For the conjugation of THCA with CoA, a pre-requisite for peroxisome import, two enzymes exist – bile acid-CoA synthetase and very long-chain acyl-CoA synthetase (VLACS) (Mihalik et al. 2002). Both are residents of the ER-membrane, however, VLACS has also been localized to peroxisomes. Nevertheless, peroxisomes show no THCA conjugating activity (Schepers et al. 1989), which is in line with the observation that VLACS localizes only at the inner site of the peroxisomal membrane (Smith et al. 2000). Thus, import of THCA into peroxisomes seems to rely on efficient metabolite activation at the ER and subsequent transport to peroxisomes.
As exemplified above, peroxisomal and ER metabolism are significantly intermingled requesting sophisticated regulation systems to link corresponding enzyme amounts and enzymatic activities. However, our current knowledge about those processes remains still fragmentary. An interesting example of a peroxisomal impact on ER physiology has been described recently: disruption of peroxisome functions in mice by Pex2 knockout reduced hepatic cholesterol levels to significant amounts which was paralleled by a pronounced up-regulation of sterol regulatory element binding proteins (SREBPs) (Kovacs et al. 2004) – transcription factors controlling cholesterol metabolism. Furthermore, the authors reported that ER stress pathways are activated in the Pex2 knockout mice, which in turn disturb the expression of SREBP-2, SREBP-1c and Insig-2a, leading to further deregulation of endogenous sterol response pathways (Kovacs et al. 2009). Even if not yet profoundly understood, metabolic connections between peroxisomes and the ER seem to give rise to reaction chains leading to a disruption of cholesterol metabolism in a peroxisome-deficient animal model. Likewise, further regulatory networks involved in interorganellar communication may be involved in the development of the complex phenotypes of peroxisomal disorders. With regard to the metabolic cooperations connecting peroxisomes and the ER, the intimate contacts observed in ultrastructural studies (Fig. 1.1a–c) may facilitate the efficient exchange of metabolites. How this organelle proximity is achieved and if it involves physical protein attachment sites is currently unclear. For mitochondria-ER contact sites, however, an involvement of the mitofusin Mfn2 has been reported (de Brito and Scorrano 2008).
Extending our view on the peroxisome-ER connection, new non-metabolism-related cooperative phenomena have been recently reported. MAVS are membrane-associated proteins involved in antiviral signalling and localize at the mitochondrial outer membrane, peroxisomes, and the MAM (mitochondrial-associated membrane) of the ER network (see Chap. 4). In this signalling network, the MAM was suggested to act as a synapse regulating innate immune signalling by MAVS from MAM, peroxisomes, and mitochondria, extending our view on organellar interactions to a dynamic, multiorganellar perspective.
1.4 The Peroxisome-Mitochondria Connection
It became evident in the last years that peroxisomes and mitochondria maintain a much closer interrelationship than previously anticipated (Schrader and Yoon 2007; Camoes et al. 2009; Islinger et al. 2012). The “peroxisome-mitochondria connection” (Fig. 1.2) includes metabolic cooperation in fatty acid β-oxidation to maintain lipid homeostasis (see Sect. 1.5), coordinated biogenesis by sharing key proteins of the organelle division machinery (Schrader et al. 2012), potential exchange by a novel vesicular trafficking pathway from mitochondria to peroxisomes (Neuspiel et al. 2008), a redox-sensitive relationship (Fransen et al. 2012) (see Chap. 3), and cooperation in anti-viral signalling and defence (see Chap. 4).

Schematic view of the peroxisome-mitochondria connection. Peroxisomes (left) and mitochondria (right) in mammals show metabolic cooperation in fatty acid β-oxidation (β-Ox), contribute to heat production, and have a redox-sensitive relationship. They share key components of their division machinery (e.g., DLP1, Mff, Fis1) and contribute to antiviral signalling via MAVS (mitochondrial antiviral-signalling protein). Moreover, novel trafficking pathways from mitochondria to peroxisomes (and lysosomes) involving mitochondria-derived vesicles (MDVs) have been reported (see Sect. 1.4 for details). Cat, peroxisomal catalase; VLCFA, LCFA, MCFA, very long-chain, long-chain and medium-chain fatty acids; RC respiratory chain (from Islinger et al. 2012)
In animals, peroxisomes and mitochondria cooperate in the degradation of fatty acids. This is different in many yeast species and plants, where fatty acid β-oxidation is solely peroxisomal (Poirier et al. 2006). Although the biochemical/enzymatic steps of fatty acid β-oxidation in both organelles are similar, each organelle harbours its specific set of enzymes. Peroxisomal fatty acid β-oxidation usually requires an acyl-CoA oxidase (dehydrogenation step), a bifunctional enzyme (hydration and dehydrogenation steps), and a thiolase (for thiolytic cleavage). These enzymes show substrate specificity; VLCFA for example, can only be degraded in peroxisomes. In addition, peroxisomes also perform α-oxidation of e.g., dietary fatty acids such as phytanic acid from dairy products (Wanders and Waterham 2006). As, in contrast to mitochondria, peroxisomes do not possess the proteins of the respiratory chain as electron acceptors, peroxisomal fatty acid β-oxidation results in the generation of H2O2 through the transfer of electrons to O2 by acyl-CoA oxidase. Hydrogen peroxide, which can be toxic to the cells, but also serves as a signalling molecule, is degraded by catalase, a prominent peroxisomal matrix enzyme (Schrader and Fahimi 2006b; Bonekamp et al. 2009; Fransen et al. 2012).
Furthermore, peroxisomal β-oxidation generates only chain-shortened fatty acids, and unlike in mitochondria, does not result in complete degradation of fatty acids. The medium chain fatty acids obtained in peroxisomes are routed to mitochondria for further oxidation and ATP production. Export of fatty acids from peroxisomes requires a carnitine shuttle system and the involvement of membrane pores, e.g., formed by PMP22 (Antonenkov and Hiltunen 2011). In addition to shuttle systems and proteinaceous pores, a vesicular trafficking pathway from mitochondria to peroxisomes has been suggested (Neuspiel et al. 2008). Mitochondria were observed to generate so called mitochondria-derived vesicles (MDVs). One population of MDVs carried the mitochondrial anchored protein ligase MAPL and was supposed to target to peroxisomes, but the physiological function of this trafficking pathway is still unclear.
More examples for a metabolic cooperation between peroxisomes and mitochondria (and even chloroplasts) are found in fungi and plants. It has recently been reported that mitochondria and peroxisomes cooperate in the synthesis of biotin in fungi (Tanabe et al. 2011).
Evidence for a coordinated biogenesis of peroxisomes and mitochondria under certain environmental conditions may arise from the discovery that both organelles share key proteins of their division machinery (reviewed in Schrader et al. 2012). Sharing division components appears to be an evolutionary conserved strategy among organisms. In mammals, these components include the dynamin-like GTPase DLP1/Drp1, which forms ring-like oligomeric structures around membrane constrictions and severs the peroxisomal and mitochondrial membranes in a GTP-dependent manner. The tail-anchored membrane proteins Fis1 and Mff are supposed to act as receptors for DLP1 at the organelle membranes. Whereas mitochondrial dynamics are regulated by balanced fusion and fission events, there is growing evidence that peroxisomes generally do not fuse to exchange matrix or membrane proteins (see Sect. 1.6) (Motley and Hettema 2007; Huybrechts et al. 2009; Bonekamp et al. 2012).
Loss of function of the key division proteins blocks peroxisomal and mitochondrial fission and leads to the accumulation of elongated organelles (Schrader et al. 2012). Elongated peroxisomes and mitochondria have been observed in fibroblasts of a patient who turned out to suffer from DLP1 deficiency based on a point mutation in the middle domain of DLP1 (Waterham et al. 2007) (see Sect. 1.7). The heterozygous, dominant-negative missense mutation A395D was shown to inhibit oligomerization of DLP1 (Chang et al. 2010). The resulting disorder combines peroxisomal and mitochondrial defects causing developmental abnormalities (e.g., in the brain) and early death. Similar observations have been made in DLP1 knockout mice (Ishihara et al. 2009; Wakabayashi et al. 2009). Interestingly, mitochondrial alterations have also been observed under conditions of peroxisomal dysfunction, e.g., in Zellweger syndrome, a peroxisome biogenesis disorder and in related knockout mouse models (see Sect. 1.3) (Baumgart et al. 2001; Dirkx et al. 2005). The lack of peroxisomal activity in Pex5 knockout mice compromised mitochondrial ATP production and resulted in the concomitant activation of AMP-activated kinase leading to elevated carbohydrate combustion (Peeters et al. 2011). Consequently, the Pex5 knockout mice loose body weight despite increased food intake.
If the mitochondrial alterations are based on secondary, indirect effects or are directly related to a loss of the peroxisome-mitochondria interplay, is currently unclear (Camoes et al. 2009). In this respect, recent evidence for a redox-sensitive relationship between peroxisomes and mitochondria (Ivashchenko et al. 2011) might point to the latter. The induction of peroxisomal reactive oxygen species (ROS) production by peroxisome-targeted KillerRed caused mitochondrial fragmentation and an altered mitochondrial redox potential. The mitochondrial redox balance is also disturbed in cells lacking functional peroxisomes or catalase. Correspondingly, catalase deficiency was reported to increase mitochondrial ROS in response to fatty acids in a diabetes mouse model (Hwang et al. 2012). Like mitochondria, peroxisomes contribute to cellular ROS metabolism and contain several enzymes which generate but also decompose ROS, among them acyl-CoA oxidases and catalase (Schrader and Fahimi 2006b; Antonenkov et al. 2010; Fransen et al. 2012; Bonekamp et al. 2011). Thus, peroxisomes are supposed to contribute to cellular ROS homeostasis, oxidative stress and ageing, but also to ROS-mediated cellular signalling. These topics are addressed in detail in Chaps. 3, 9, 13, and 15.
Evidence for a physical interaction of peroxisomes and mitochondria in mammalian cells is still scarce. Intimate contacts can be observed in ultrastructural studies (Fig. 1.1d), but their physiological relevance awaits experimental proof. Peroxisomes and mitochondria have been reported to cluster, e.g., after the expression of membrane proteins (Koch et al. 2005), and can be co-isolated from a specific density after gradient centrifugation and subsequently separated into individual fractions by repeating this centrifugation step (Islinger et al. 2006). As both organelles are in intimate contact with the ER (see Sect. 1.2), potential peroxisome-mitochondria contacts might as well be indirect and mediated by ER membranes. However, in the fission yeast Schizosaccharomyces pombe peroxisome movement in association with mitochondria has been observed (Jourdain et al. 2008). Furthermore, in the red algae Cyanidioschyzon merolae only one peroxisome and one mitochondrion exist. Their division is coordinated, and the peroxisome interacts with the mitochondrion to partition into the daughter cell (Miyagishima et al. 1999).
1.5 Peroxisomes and Lipid Droplets
Eukaryotic cells have the ability to accumulate neutral lipids such as triacylglycerol and cholesterol ester and to store them into lipid droplets (LD). The view that LDs represent simple lipid storage containers has gradually changed, and evidence for their complex nature and contribution to multiple cellular functions has been provided (Beller et al. 2010). LDs are dynamic and highly motile organelles. They grow through a fusion process mediated by SNARE proteins and move bi-directionally on microtubules. It is suggested that LD movement supports their interaction with other organelles, among them peroxisomes, endosomes, mitochondria and the ER, to distribute neutral lipids and phospholipids. Compelling evidence for an interaction of peroxisomes with LDs has been presented in early ultrastructural studies (Novikoff et al. 1980) (Fig. 1.1e, f). Using live-cell imaging the dynamic nature of the interaction between peroxisomes and LDs has been confirmed (Schrader 2001). In S. cerevisiae, intimate physical contacts between peroxisomes and LDs were observed (Binns et al. 2006). In some cases, peroxisomes formed processes that extended into the core of the LDs. This lipid droplet-peroxisome interaction may serve to link lipolysis mediated by LDs to fatty acid β-oxidation within the peroxisomes. Furthermore, lipids generated by peroxisomes might move into LDs. Defects in peroxisomal fatty acid β-oxidation have been linked to enlarged LDs in the nematode Caenorhabditis elegans (Zhang et al. 2010). Changes in the number and size of peroxisomes, mitochondria, and LDs have also been reported in morphometric studies after starvation and clofibrate treatment in mouse hepatocytes (Meijer and Afzelius 1989) and in peroxisome-deficient knockout mice (Dirkx et al. 2005). However, the underlying molecular mechanisms of the peroxisome-lipid droplet interaction remain to be elucidated. An interactome map of protein-protein contacts of LDs with mitochondria and peroxisomes in S. cerevisiae has recently been generated using a bimolecular fluorescence complementation assay (Pu et al. 2011). The LD proteins Erg6 and Pet10 were found to be involved in 75% of the interactions detected, including most of the peroxins tested. The physiological significance of these interactions awaits further experimental proof. Some of the interactions of LDs and other organelles (e.g., endosomes) may be regulated by Rab GTPases, which have been found on LDs (reviewed in Murphy et al. 2009).
1.6 Peroxisome-Peroxisome Interactions
As peroxisomes – like mitochondria – multiply by fission and even share key fission proteins (e.g., DLP1, Fis1, Mff) (see Sect. 1.4), it was long debated if peroxisomes can fuse to exchange matrix and membrane components and if they as well share key fusion proteins with mitochondria (e.g., Mfn1, Mfn2, Opa1). We recently addressed this question systematically by applying an in vitro fusion assay based on the co-cultivation of mammalian CHO cells stably expressing either red or green fluorescent peroxisomal matrix or membrane proteins. The interaction of red and green peroxisomes and a potential exchange of marker proteins were analyzed in hybridoma cells which were formed through cell fusion (Bonekamp et al. 2012). By combining epifluorescence microscopy, spinning disk confocal microscopy and live cell imaging, we demonstrated that mature peroxisomes in mammalian cells do not fuse and exchange matrix or membrane marker proteins in a mechanism analogous to mitochondria. Moreover, mitochondrial fusion proteins (e.g., Mfn1, Mfn2, Opa1) do not localize or target to peroxisomes and do not contribute to peroxisome dynamics (Bonekamp et al. 2012). These findings are in agreement with previous observations in yeast, plant and mammalian cells (Arimura et al. 2004; Motley and Hettema 2007; Huybrechts et al. 2009). However, our life cell studies revealed that peroxisomes in mammalian cells are engaged in several transient, but vivid and long term contacts (Bonekamp et al. 2012). Similar interactions were reported by real time imaging of GFP-labelled peroxisomes (Schrader et al. 2000). In many cases, peroxisomes moving along microtubules were observed to interact with other peroxisomes. Mathematical analysis showed that the transient interactions display so-called power law behaviour. Power law distributions in biological processes point to intricate dynamics which originate from diverse and yet specific mechanisms (Clauset et al. 2009). Thus, peroxisome interactions are more complex than previously assumed and represent a new dynamic behaviour of peroxisomes. By applying a simple computational model, we demonstrated that when combined with ATP-driven peroxisome movement along microtubules (see Sect. 1.7), the subsequent formation of inter-peroxisomal contacts can potentially contribute to the equilibration of cellular peroxisome pools. Peroxisomes are very heterogeneous in terms of density, protein composition and import competence (Heinemann and Just 1992; Luers et al. 1993; Islinger et al. 2010), thus an exchange of metabolic information might occur. However, an increase in heterogeneity among different peroxisome populations by manipulating ROS and fatty acid levels did not promote peroxisome interactions (Bonekamp et al. 2012), and questions the exchange of metabolites by inter-peroxisomal contacts. Although the physiological role of the transient complex peroxisomal interactions is unclear, they might contribute to a “signalling system” monitoring the state and/or distribution of peroxisome populations within the cell. Interestingly, the formation of small peroxisome groups with close apposition has been documented in ultrastructural studies (Stier et al. 1998; Zaar et al. 1984) (Fig. 1.3a). Peroxisomes which are attached to each other can also be isolated by gradient centrifugation from rat liver after treatment with peroxisome proliferators (Fig. 1.3b). These groups may represent functional units of peroxisomes which interact and cooperate via close but transient contacts. In addition, interactions with other organelles might occur.

(a, b) Peroxisome-peroxisome interactions. (a) Electron micrograph of ordered stacks of peroxisomes in canine kidney. Magnification, ×120,000 (from Zaar et al. 1984). (b) Electron micrograph of closely associated peroxisomes in a purified peroxisome fraction isolated from bezafibrate-treated rat liver by gradient centrifugation. (c, d) Association of purified rat liver peroxisomes (Po) with microtubules (MT) in an in vitro assay. (c) Video-enhanced contrast microscopy (VECM) of microtubules and peroxisomes using a Zeiss Axiovert microscope equipped with differential interference contrast (DIC) optics and a Hamamatsu video camera (from Schrader et al. 2003). (d) Negative staining electron microscopy (EM) of isolated peroxisomes bound to microtubules (from Thiemann et al. 2000). Bars, 0.2 μm (c), 1 μm (d)
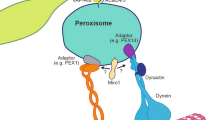
1.7 Peroxisomes and the Cytoskeleton
An association of mammalian peroxisomes with cytoskeletal elements was predicted from early ultrastructural observations (Gorgas 1985; Yamamoto and Fahimi 1987), and their direct interaction with and movement along microtubules has been well documented in vivo and in vitro (reviewed in Schrader et al. 2003; Schrader and Fahimi 2006a) (Fig. 1.3c, d). The peroxisome-microtubule interaction is crucial for long-range, (bi)-directional motility of peroxisomes and for the positioning of the organelle within the cell (e.g., to maintain a uniform intracellular distribution). Microtubule-based peroxisome movement involves the motor proteins dynein, kinesin and the dynein activator complex dynactin (Schrader et al. 2000; Kural et al. 2005). Whereas dynein can interact with organelle membranes via the dynactin complex, the recruitment of the microtubule plus-end kinesin motors often requires adaptor proteins. However, the role of different kinesins in peroxisome motility is poorly studied. Recently, a role for the peroxin Pex14 in microtubule-binding and peroxisome motility has been suggested (Bharti et al. 2011), but information on potential linker proteins and peroxisomal receptors for motor proteins is generally scarce. This is in part due to the fact that peroxisome motility and distribution in classical genetically accessible model organisms such as bakers’ yeast or plant cells depend on the actin cytoskeleton and type-V myosin motors, but not (or only partially) on microtubules. In plants, peroxisome motility can be modulated by ROS and Ca++ and may serve to protect the cell against oxidative stress (Rodríguez-Serrano et al. 2009). Peroxisome motility and positioning in S. cerevisiae is crucial for partitioning and inheritance of peroxisomes from the mother to the daughter cell (the forming bud), but also to retain peroxisomes in the mother. These processes are highly regulated and depend on actin dynamics, the myosin motor Myo2, and the peroxisomal inheritance proteins Inp1 and Inp2. Inp1 is crucial for retaining peroxisomes in the mother, whereas Inp2 recruits Myo2 to peroxisomes (Fagarasanu et al. 2010). Interestingly, S. cerevisiae harbours such a complex and tightly regulated inheritance machinery to guarantee proper distribution of peroxisomes to the bud although peroxisomes in the daughter can form de novo from the ER in case they are lost. An explanation might be that de novo formation is more energy-consuming, and thus represents a back-up system. Homologues of Inp1 and Inp2 have not been identified in mammals. In contrast to yeast cells which usually harbour 3–5 peroxisomes under non-proliferative conditions, mammalian cells contain hundred or more peroxisomes. Thus, complete loss of peroxisomes in the daughter cell is unlikely, and distribution during mitosis is supposed to be at random (Wiemer et al. 1997).
The analysis of peroxisome motility in cultured mammalian cells revealed, that only 10–15% of the peroxisome population is performing long-range, fast movements in a microtubule-dependent manner (Koch et al. 2003). We recently obtained evidence for a relationship between the percentage of fast moving peroxisomes, energy consumption and the mixing time of different peroxisome populations within a cell (Bonekamp et al. 2012). Our computational model indicates that approx. 15% of ATP-driven peroxisome movement represents an optimum allowing proper mixing/homogenisation of the peroxisomal compartment at minimal energy costs in a physiological time frame (see Sect. 1.6).
Microtubules are not required for matrix protein import into peroxisomes (Brocard et al. 2005), but may facilitate efficient and regulated sorting of proteins to peroxisomes (Chuong et al. 2005). Microtubules and dynein may as well contribute to the early stages of peroxisome biogenesis, as Pex16-mutant cells failed to restore peroxisome formation after microinjection complementation, when microtubules were depolymerised prior to microinjection, or when a dominant-negative CC1 subunit of the dynein/dynactin motor complex was co-expressed (Brocard et al. 2005). However, the membrane-deforming protein Pex11β was properly targeted to peroxisomes and induced membrane elongation in the absence of microtubules (our unpublished results).
In yeast, the actin cytoskeleton and the Myo2 motor have been suggested to exert pulling forces on the peroxisome membrane thus assisting in peroxisome fission. Indeed, Myo2 and Pex11 are required for the formation of tubular membrane extensions prior to peroxisome fission (Nagotu et al. 2008; Fagarasanu et al. 2010). In mammalian cells microtubules are not essential for membrane elongation of peroxisomes or for organelle division (Schrader et al. 2000). Mammalian peroxisomes can elongate in the complete absence of microtubules (Schrader et al. 1998), and remarkably, membrane elongation is promoted by microtubule-depolymerizing (but not stabilizing) agents (Schrader et al. 1996). Furthermore, the constriction and final fission of peroxisomes precedes independent of microtubules. However, microtubules are crucial for the proper intracellular distribution of peroxisomes after division, for fast and directed peroxisome motility, for their positioning and the maintenance of their uniform distribution within the cell (Schrader et al. 2003). In this respect, a mechanistic link between peroxisome multiplication (proliferation) and trafficking along microtubules has been proposed (Nguyen et al. 2006), suggesting that peroxisome division may trigger binding and transport of newly formed peroxisomes along microtubules.
It can be assumed that a loss of peroxisome motility and defects in the proper trafficking and distribution of peroxisomes result in a regional loss of essential peroxisomal functions and subsequently to cell damage and degeneration, especially in neurons. Correspondingly, loss of DLP1, which is essential for the division of peroxisomes and mitochondria (Koch et al. 2003, 2005; Schrader et al. 2012), has been linked to neurodegeneration and developmental defects in brain (Waterham et al. 2007; Ishihara et al. 2009; Wakabayashi et al. 2009). It is suggested that the block in peroxisomal and mitochondrial division, which results in elongated and enlarged organelles, inhibits their proper distribution within neurons and in the axon thus contributing to cell damage.
1.8 Concluding Remarks
There is compelling evidence that peroxisomes are significantly more dynamic and interactive than previously expected. They do not function as isolated entities, but are integrated into a complex network of communicating endomembranes that is only beginning to emerge. Peroxisomes interact and cooperate with other organelles such as the ER, mitochondria, and lipid droplets to facilitate metabolic processes. Remarkably, they also act as signalling platforms, and contribute to the fine-tuning of cellular processes. The interaction of mammalian peroxisomes with microtubules likely serves to direct peroxisomal movement and to facilitate interactions with other endomembranes. However, how physical contacts between peroxisomes and other organelles are mediated, what components are transferred or exchanged, and how this transfer is mediated, is largely unknown. It is a great challenge for future investigations to identify and characterize the underlying molecular mechanisms and the physiological relevance of these interactions, and to elucidate their importance for health and disease.
References
Aboushadi N, Engfelt WH, Paton VG, Krisans SK (1999) Role of peroxisomes in isoprenoid biosynthesis. J Histochem Cytochem 47:1127–1132
Agrawal G, Joshi S, Subramani S (2011) Cell-free sorting of peroxisomal membrane proteins from the endoplasmic reticulum. Proc Natl Acad Sci USA 108:9113–9118
Antonenkov VD, Hiltunen JK (2011) Transfer of metabolites across the peroxisomal membrane. Biochim Biophys Acta 1822:1374–1386
Antonenkov VD, Grunau S, Ohlmeier S, Hiltunen JK (2010) Peroxisomes are oxidative organelles. Antioxid Redox Signal 13:525–537
Appelkvist EL, Reinhart M, Fischer R, Billheimer J, Dallner G (1990) Presence of individual enzymes of cholesterol biosynthesis in rat liver peroxisomes. Arch Biochem Biophys 282:318–325
Arimura S, Yamamoto J, Aida GP, Nakazono M, Tsutsumi N (2004) Frequent fusion and fission of plant mitochondria with unequal nucleoid distribution. Proc Natl Acad Sci USA 101:7805–7808
Baumgart E, Vanhorebeek I, Grabenbauer M, Borgers M, Declercq PE, Fahimi HD, Baes M (2001) Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am J Pathol 159:1477–1494
Beller M, Thiel K, Thul PJ, Jackle H (2010) Lipid droplets: a dynamic organelle moves into focus. FEBS Lett 584:2176–2182
Bharti P, Schliebs W, Schievelbusch T, Neuhaus A, David C, Kock K, Herrmann C et al (2011) PEX14 is required for microtubule-based peroxisome motility in human cells. J Cell Sci 124:1759–1768
Binns D, Januszewski T, Chen Y, Hill J, Markin VS, Zhao Y, Gilpin C et al (2006) An intimate collaboration between peroxisomes and lipid bodies. J Cell Biol 173:719–731
Bonekamp NA, Volkl A, Fahimi HD, Schrader M (2009) Reactive oxygen species and peroxisomes: struggling for balance. Biofactors 35:346–355
Bonekamp NA, Fahimi HD, Schrader M (2011) Oxidative stress in peroxisomes. In: Pantopoulos K, Shipper H (eds) Principles of free radical biomedicine. Nova Science Publishers, Hauppage, pp 333–358
Bonekamp NA, Sampaio P, de Abreu FV, Luers GH, Schrader M (2012) Transient complex interactions of mammalian peroxisomes without exchange of matrix or membrane marker proteins. Traffic 13:960–978
Braverman NE, Moser AB (2012) Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 1822:1442–1452
Brocard CB, Boucher KK, Jedeszko C, Kim PK, Walton PA (2005) Requirement for microtubules and dynein motors in the earliest stages of peroxisome biogenesis. Traffic 6:386–395
Camoes F, Bonekamp NA, Delille HK, Schrader M (2009) Organelle dynamics and dysfunction: a closer link between peroxisomes and mitochondria. J Inherit Metab Dis 32:163–180
Chang CR, Manlandro CM, Arnoult D, Stadler J, Posey AE, Hill RB, Blackstone C (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem 285:32494–32503
Chuong SD, Park NI, Freeman MC, Mullen RT, Muench DG (2005) The peroxisomal multifunctional protein interacts with cortical microtubules in plant cells. BMC Cell Biol 6:40
Clastre M, Papon N, Courdavault V, Giglioli-Guivarc’h N, St-Pierre B, Simkin AJ (2011) Subcellular evidence for the involvement of peroxisomes in plant isoprenoid biosynthesis. Plant Signal Behav 6:2044–2046
Clauset A, Shalizi CR, Newman MEJ (2009) Power-law distributions in empirical data. SIAM Rev 51:661–703
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
Delille HK, Agricola B, Guimaraes SC, Borta H, Luers GH, Fransen M, Schrader M (2010) Pex11pbeta-mediated growth and division of mammalian peroxisomes follows a maturation pathway. J Cell Sci 123:2750–2762
Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S et al (2011) Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med 17:1121–1127
Dirkx R, Vanhorebeek I, Martens K, Schad A, Grabenbauer M, Fahimi D, Declercq P et al (2005) Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology 41:868–878
Fagarasanu A, Mast FD, Knoblach B, Rachubinski RA (2010) Molecular mechanisms of organelle inheritance: lessons from peroxisomes in yeast. Nat Rev Mol Cell Biol 11:644–654
Fahimi HD, Yokota S (1981) Ultrastructural and cytochemical aspects of animal peroxisomes – some recent observations. In: Schweiger HG (ed) Internat Cell Biol, Springer Berlin-Heidelberg, pp 640–650
Fahimi HD, Kalmbach P, Stegmeier K, Stork H (1980) Comparison between the effects of clofibrate and bezafibrate upon the ultrastructure of rat heart and liver. In: Greten H, Lang PD, Schettler G (eds) Lipoprot & Coronary Disease, G Witzstrock Publishing House, New York/Baden-Baden/Cologne, pp 64–75
Ferdinandusse S, Denis S, Faust PL, Wanders RJ (2009) Bile acids: the role of peroxisomes. J Lipid Res 50:2139–2147
Fransen M, Nordgren M, Wang B, Apanasets O (2012) Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta 1822:1363–1373
Geuze HJ, Murk JL, Stroobants AK, Griffith JM, Kleijmeer MJ, Koster AJ, Verkleij AJ et al (2003) Involvement of the endoplasmic reticulum in peroxisome formation. Mol Biol Cell 14:2900–2907
Gorgas K (1985) Serial section analysis of mouse hepatic peroxisomes. Anat Embryol (Berl) 172:21–32
Gorgas K, Teigler A, Komljenovic D, Just WW (2006) The ether lipid-deficient mouse: tracking down plasmalogen functions. Biochim Biophys Acta 1763:1511–1526
Grabenbauer M, Satzler K, Baumgart E, Fahimi HD (2000) Three-dimensional ultrastructural analysis of peroxisomes in HepG2 cells. Absence of peroxisomal reticulum but evidence of close spatial association with the endoplasmic reticulum. Cell Biochem Biophys 32:37–49
Heinemann P, Just WW (1992) Peroxisomal protein import. In vivo evidence for a novel translocation competent compartment. FEBS Lett 300:179–182
Herzog V, Fahimi HD (1976) Identification of peroxisomes (microbodies) in mouse myocardium. J Mol Cell Cardiol 8:271–281
Hettema EH, Girzalsky W, van Den Berg M, Erdmann R, Distel B (2000) Saccharomyces cerevisiae pex3p and pex19p are required for proper localization and stability of peroxisomal membrane proteins. EMBO J 19:223–233
Hicks L, Fahimi HD (1977) Peroxisomes (microbodies) in the myocardium of rodents and primates. A comparative ultrastructural cytochemical study. Cell Tissue Res 175:467–481
Hoepfner D, Schildknegt D, Braakman I, Philippsen P, Tabak HF (2005) Contribution of the endoplasmic reticulum to peroxisome formation. Cell 122:85–95
Huybrechts SJ, Van Veldhoven PP, Brees C, Mannaerts GP, Los GV, Fransen M (2009) Peroxisome dynamics in cultured mammalian cells. Traffic 10:1722–1733
Hwang I, Lee J, Huh JY, Park J, Lee HB, Ho YS, Ha H (2012) Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 61:728–738
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H et al (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11:958–966
Islinger M, Schrader M (2011) Peroxisomes. Curr Biol 21:R800–R801
Islinger M, Luers GH, Zischka H, Ueffing M, Volkl A (2006) Insights into the membrane proteome of rat liver peroxisomes: microsomal glutathione-S-transferase is shared by both subcellular compartments. Proteomics 6:804–816
Islinger M, Li KW, Loos M, Liebler S, Angermuller S, Eckerskorn C, Weber G et al (2010) Peroxisomes from the heavy mitochondrial fraction: isolation by zonal free flow electrophoresis and quantitative mass spectrometrical characterization. J Proteome Res 9:113–124
Islinger M, Grille S, Fahimi HD, Schrader M (2012) The peroxisome: an update on mysteries. Histochem Cell Biol 137:547–574
Itoyama A, Honsho M, Abe Y, Moser A, Yoshida Y, Fujiki Y (2012) Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J Cell Sci 125:589–602
Ivashchenko O, Van Veldhoven PP, Brees C, Ho YS, Terlecky SR, Fransen M (2011) Intraperoxisomal redox balance in mammalian cells: oxidative stress and interorganellar cross-talk. Mol Biol Cell 22:1440–1451
Jourdain I, Sontam D, Johnson C, Dillies C, Hyams JS (2008) Dynamin-dependent biogenesis, cell cycle regulation and mitochondrial association of peroxisomes in fission yeast. Traffic 9:353–365
Kanzawa N, Shimozawa N, Wanders RJ, Ikeda K, Murakami Y, Waterham HR, Mukai S et al (2012) Defective lipid remodeling of GPI anchors in peroxisomal disorders, Zellweger syndrome and rhizomelic chondrodysplasia punctata. J Lipid Res 53:653–663
Karnik SK, Trelease RN (2007) Arabidopsis peroxin 16 trafficks through the ER and an intermediate compartment to pre-existing peroxisomes via overlapping molecular targeting signals. J Exp Bot 58:1677–1693
Keller GA, Barton MC, Shapiro DJ, Singer SJ (1985) 3-Hydroxy-3-methylglutaryl-coenzyme a reductase is present in peroxisomes in normal rat liver cells. Proc Natl Acad Sci USA 82:770–774
Kim PK, Mullen RT, Schumann U, Lippincott-Schwartz J (2006) The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J Cell Biol 173:521–532
Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M (2003) Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem 278:8597–8605
Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M (2005) A role for fis1 in both mitochondrial and peroxisomal fission in Mammalian cells. Mol Biol Cell 16:5077–5086
Kovacs WJ, Shackelford JE, Tape KN, Richards MJ, Faust PL, Fliesler SJ, Krisans SK (2004) Disturbed cholesterol homeostasis in a peroxisome-deficient PEX2 knockout mouse model. Mol Cell Biol 24:1–13
Kovacs WJ, Tape KN, Shackelford JE, Duan X, Kasumov T, Kelleher JK, Brunengraber H, Krisans SK (2007) Localization of the pre-squalene segment of the isoprenoid biosynthetic pathway in mammalian peroxisomes. Histochem Cell Biol 127:273–290
Kovacs WJ, Tape KN, Shackelford JE, Wikander TM, Richards MJ, Fliesler SJ, Krisans SK, Faust PL (2009) Peroxisome deficiency causes a complex phenotype because of hepatic SREBP/Insig dysregulation associated with endoplasmic reticulum stress. J Biol Chem 284:7232–7245
Kural C, Kim H, Syed S, Goshima G, Gelfand VI, Selvin PR (2005) Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science 308:1469–1472
Lam SK, Yoda N, Schekman R (2010) A vesicle carrier that mediates peroxisome protein traffic from the endoplasmic reticulum. Proc Natl Acad Sci USA 107:21523–21528
Lazarow PB, Fujiki Y (1985) Biogenesis of peroxisomes. Annu Rev Cell Biol 1:489–530
Luers G, Hashimoto T, Fahimi HD, Volkl A (1993) Biogenesis of peroxisomes: isolation and characterization of two distinct peroxisomal populations from normal and regenerating rat liver. J Cell Biol 121:1271–1280
McCartney AW, Greenwood JS, Fabian MR, White KA, Mullen RT (2005) Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 17:3513–3531
Meijer J, Afzelius B (1989) Effects of clofibrate treatment and of starvation on peroxisomes, mitochondria, and lipid droplets in mouse hepatocytes: a morphometric study. J Ultrastruct Mol Struct Res 102:87–94
Mihalik SJ, Steinberg SJ, Pei Z, Park J, Kim DG, Heinzer AK, Dacremont G et al (2002) Participation of two members of the very long-chain acyl-CoA synthetase family in bile acid synthesis and recycling. J Biol Chem 277:24771–24779
Miyagishima S, Nishimura M, Itoh R, Toda K, Kuroiwa H, Kuroiwa T (1999) Microbody proliferation and segregation cycle in the single microbody-alga Cyanidioschyzon merolae. Planta 208:326–336
Motley AM, Hettema EH (2007) Yeast peroxisomes multiply by growth and division. J Cell Biol 178:399–410
Murphy S, Martin S, Parton RG (2009) Lipid droplet-organelle interactions; sharing the fats. Biochim Biophys Acta 1791:441–447
Nagotu S, Saraya R, Otzen M, Veenhuis M, van der Klei IJ (2008) Peroxisome proliferation in Hansenula polymorpha requires Dnm1p which mediates fission but not de novo formation. Biochim Biophys Acta 1783:760–769
Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM (2008) Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18:102–108
Nguyen T, Bjorkman J, Paton BC, Crane DI (2006) Failure of microtubule-mediated peroxisome division and trafficking in disorders with reduced peroxisome abundance. J Cell Sci 119:636–645
Novikoff PM, Novikoff AB (1972) Peroxisomes in absorptive cells of mammalian small intestine. J Cell Biol 53:532–560
Novikoff AB, Shin WY (1964) The endoplasmic reticulum in the Golgi zone and its relations to microbodies, Golgi apparatus, and autophagic vacuoles in rat liver cells. J Microscopy 3:187–206
Novikoff AB, Novikoff PM, Rosen OM, Rubin CS (1980) Organelle relationships in cultured 3T3-L1 preadipocytes. J Cell Biol 87:180–196
Peeters A, Fraisl P, van den Berg S, Loren V, van Themaat E, Van Kampen A, Rider MH, Takemori H et al (2011) Carbohydrate metabolism is perturbed in peroxisome-deficient hepatocytes due to mitochondrial dysfunction, AMP-activated protein kinase (AMPK) activation, and peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) suppression. J Biol Chem 286:42162–42179
Perry RJ, Mast FD, Rachubinski RA (2009) Endoplasmic reticulum-associated secretory proteins Sec20p, Sec39p, and Dsl1p are involved in peroxisome biogenesis. Eukaryot Cell 8:830–843
Poirier Y, Antonenkov VD, Glumoff T, Hiltunen JK (2006) Peroxisomal beta-oxidation–a metabolic pathway with multiple functions. Biochim Biophys Acta 1763:1413–1426
Pu J, Ha CW, Zhang S, Jung JP, Huh WK, Liu P (2011) Interactomic study on interaction between lipid droplets and mitochondria. Protein Cell 2:487–496
Raychaudhuri S, Prinz WA (2008) Nonvesicular phospholipid transfer between peroxisomes and the endoplasmic reticulum. Proc Natl Acad Sci USA 105:15785–15790
Rodríguez-Serrano M, Romero-Puertas MC, Sparkes I, Hawes C, del Río LA, Sandalio LM (2009) Peroxisome dynamics in Arabidopsis plants under oxidative stress induced by cadmium. Free Radic Biol Med 47:1632–1639
Rucktäschel R, Halbach A, Girzalsky W, Rottensteiner H, Erdmann R (2010) De novo synthesis of peroxisomes upon mitochondrial targeting of Pex3p. Eur J Cell Biol 89:947–954
Russell DW (2003) The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174
Saraya R, Krikken AM, Veenhuis M, van der Klei IJ (2011) Peroxisome reintroduction in Hansenula polymorpha requires Pex25 and Rho1. J Cell Biol 193:885–900
Schepers L, Casteels M, Verheyden K, Parmentier G, Asselberghs S, Eyssen HJ, Mannaerts GP (1989) Subcellular distribution and characteristics of trihydroxycoprostanoyl-CoA synthetase in rat liver. Biochem J 257:221–229
Schrader M (2001) Tubulo-reticular clusters of peroxisomes in living COS-7 cells: dynamic behavior and association with lipid droplets. J Histochem Cytochem 49:1421–1429
Schrader M, Fahimi HD (2006a) Growth and division of peroxisomes. Int Rev Cytol 255:237–290
Schrader M, Fahimi HD (2006b) Peroxisomes and oxidative stress. Biochim Biophys Acta 1763:1755–1766
Schrader M, Yoon Y (2007) Mitochondria and peroxisomes: are the ‘Big Brother’ and the ‘Little Sister’ closer than assumed? Bioessays 29:1105–1114
Schrader M, Burkhardt JK, Baumgart E, Luers G, Spring H, Volkl A, Fahimi HD (1996) Interaction of microtubules with peroxisomes. Tubular and spherical peroxisomes in HepG2 cells and their alterations induced by microtubule-active drugs. Eur J Cell Biol 69:24–35
Schrader M, Reuber BE, Morrell JC, Jimenez-Sanchez G, Obie C, Stroh TA, Valle D et al (1998) Expression of PEX11beta mediates peroxisome proliferation in the absence of extracellular stimuli. J Biol Chem 273:29607–29614
Schrader M, King SJ, Stroh TA, Schroer TA (2000) Real time imaging reveals a peroxisomal reticulum in living cells. J Cell Sci 113:3663–3671
Schrader M, Thiemann M, Fahimi HD (2003) Peroxisomal motility and interaction with microtubules. Microsc Res Tech 61:171–178
Schrader M, Bonekamp NA, Islinger M (2012) Fission and proliferation of peroxisomes. Biochim Biophys Acta 822:1343–1357
Simkin AJ, Guirimand G, Papon N, Courdavault V, Thabet I, Ginis O, Bouzid S et al (2011) Peroxisomal localisation of the final steps of the mevalonic acid pathway in planta. Planta 234:903–914
Smith BT, Sengupta TK, Singh I (2000) Intraperoxisomal localization of very-long-chain fatty acyl-CoA synthetase: implication in X-adrenoleukodystrophy. Exp Cell Res 254:309–320
South ST, Gould SJ (1999) Peroxisome synthesis in the absence of preexisting peroxisomes. J Cell Biol 144:255–266
Sprecher H, Chen Q (1999) Polyunsaturated fatty acid biosynthesis: a microsomal-peroxisomal process. Prostaglandins Leukot Essent Fatty Acids 60:317–321
Stier H, Fahimi HD, Van Veldhoven PP, Mannaerts GP, Volkl A, Baumgart E (1998) Maturation of peroxisomes in differentiating human hepatoblastoma cells (HepG2): possible involvement of the peroxisome proliferator-activated receptor alpha (PPAR alpha). Differentiation 64:55–66
Su HM, Moser AB, Moser HW, Watkins PA (2001) Peroxisomal straight-chain Acyl-CoA oxidase and D-bifunctional protein are essential for the retroconversion step in docosahexaenoic acid synthesis. J Biol Chem 276:38115–38120
Tanabe Y, Maruyama J, Yamaoka S, Yahagi D, Matsuo I, Tsutsumi N, Kitamoto K (2011) Peroxisomes are involved in biotin biosynthesis in Aspergillus and Arabidopsis. J Biol Chem 286:30455–30461
Thai TP, Rodemer C, Jauch A, Hunziker A, Moser A, Gorgas K, Just WW (2001) Impaired membrane traffic in defective ether lipid biosynthesis. Hum Mol Genet 10:127–136
Thiemann M, Schrader M, Völkl A, Baumgart E, Fahimi HD (2000) Interaction of peroxisomes with microtubules. In vitro studies using a novel peroxisome-microtubule binding assay. Eur J Biochem 267:6264–6275
Thoms S, Harms I, Kalies KU, Gartner J (2012) Peroxisome formation requires the endoplasmic reticulum channel protein Sec61. Traffic 13:599–609
Till A, Lakhani R, Burnett SF, Subramani S (2012) Pexophagy: the selective degradation of peroxisomes. Int J Cell Biol 2012:512721
Titorenko VI, Rachubinski RA (2001) Dynamics of peroxisome assembly and function. Trends Cell Biol 11:22–29
van der Zand A, Gent J, Braakman I, Tabak HF (2012) Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell 149:397–409
Voss A, Reinhart M, Sankarappa S, Sprecher H (1991) The metabolism of 7,10,13,16,19-docosapentaenoic acid to 4,7,10,13,16,19-docosahexaenoic acid in rat liver is independent of a 4-desaturase. J Biol Chem 266:19995–20000
Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H (2009) The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186:805–816
Wanders RJ, Waterham HR (2006) Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem 75:295–332
Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV (2007) A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356:1736–1741
Wiemer EA, Wenzel T, Deerinck TJ, Ellisman MH, Subramani S (1997) Visualization of the peroxisomal compartment in living mammalian cells: dynamic behavior and association with microtubules. J Cell Biol 136:71–80
Yamamoto K, Fahimi HD (1987) Three-dimensional reconstruction of a peroxisomal reticulum in regenerating rat liver: evidence of interconnections between heterogeneous segments. J Cell Biol 105:713–722
Yonekawa S, Furuno A, Baba T, Fujiki Y, Ogasawara Y, Yamamoto A, Tagaya M, Tani K (2011) Sec16B is involved in the endoplasmic reticulum export of the peroxisomal membrane biogenesis factor peroxin 16 (Pex16) in mammalian cells. Proc Natl Acad Sci USA 108:12746–12751
Zaar K, Hartig F, Fahimi HD, Gorgas K (1984) Peroxisomal aggregates forming large stacks in the lipid segment of the canine kidney. Acta Histochem Suppl 29:165–168
Zaar K, Volkl A, Fahimi HD (1987) Association of isolated bovine kidney cortex peroxisomes with endoplasmic reticulum. Biochim Biophys Acta 897:135–142
Zhang SO, Trimble R, Guo F, Mak HY (2010) Lipid droplets as ubiquitous fat storage organelles in C. elegans. BMC Cell Biol 11:96
Acknowledgments
We would like to thank all laboratory members for stimulating discussions and we apologize to those whose work has not been cited owing to space limitations. This work was supported by the Portuguese Foundation for Science and Technology (FCT) and FEDER (PTDC/SAU-OSM/103647/2008; PTDC/BIA-BCM/099613/2008; PTDC/BIA-BCM/118605/2010; SFRH/BPD/74428/2010 to M. I.) and BBSRC (BB/K006231/1).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Schrader, M., Grille, S., Fahimi, H.D., Islinger, M. (2013). Peroxisome Interactions and Cross-Talk with Other Subcellular Compartments in Animal Cells. In: del Río, L. (eds) Peroxisomes and their Key Role in Cellular Signaling and Metabolism. Subcellular Biochemistry, vol 69. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6889-5_1
Download citation
DOI: https://doi.org/10.1007/978-94-007-6889-5_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6888-8
Online ISBN: 978-94-007-6889-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)