
Abstract
Since the construction of the first generation of general cloning vectors in the early 1970s, a large number of cloning vectors have been developed. Despite the bewildering choice of commercial and other available vectors, the selection of cloning vector to be used can be decided by applying a small number of criteria: insert size, copy number, incompatibility, selectable marker cloning sites, and specialized vector functions. Several of these criteria are dependent on each other. Most general cloning plasmids can carry a DNA insert up to around 15 kb in size. Several types of vectors are available for cloning large fragments of DNA too. This chapter presents a consolidated account of some new generation of high-capacity vectors such as cosmid, yeast artificial chromosome (YAC) , bacterial artificial chromosome (BAC), P1 phage artificial chromosome (PAC), and human artificial chromosome (HAC).
You have full access to this open access chapter, Download chapter PDF
1.1 Introduction
A prime requisite for a gene cloning experiment is the selection of a suitable cloning vector, i.e., a DNA molecule that acts as a vehicle for carrying a foreign DNA fragment when inserted into it and transports it into a host cell, which is usually a bacterium, though other types of living cells can also be used. A wide variety of natural replicons exhibit the properties that allow them to act as cloning vectors, however, vectors may also be designed to possess certain minimum qualification to function as an efficient agent for transfer, maintenance, and amplification of target DNA.
An ideal cloning vehicle would have the following four properties:
-
Low-molecular weight
-
Ability to confer readily selectable phenotypic traits on host cells
-
Single sites for a large number of restriction endonucleases, preferably in genes with a scorable phenotype
-
Ability to replicate within the host cell, so that numerous copies of the recombinant DNA molecule can be produced and passed to daughter cells.
In 1970s, when recombinant DNA technology was being first developed, only a limited number of vectors were available based on either high-copy number plasmids or phage λ. Later phage M13 was developed as a specialist vector to facilitate DNA sequencing; over a time a series of specialist vectors were constructed for specific purpose. The examples of naturally occurring or artificially constructed vectors include vectors based on Escherichia coli plasmids, bacteriophages (e.g., λ, M13, P1), viruses (e.g., animal viruses—retrovirus, adenovirus, adeno-associated virus, Herpes Simplex virus, Vaccinia virus, etc.; insect viruses—baculo virus; plant viruses—cauliflower mosaic virus, potato virus X, Gemini virus, etc.), Agrobacterium tumefaciens based vectors, chimeric plasmids (e.g., cosmid, phagemid, phasmid, and fosmid), artificial chromosomes [e.g., YAC, BAC, PAC, MAC and HAC], and non-E. coli vectors (e.g., Bacillus and Pseudomonas vectors etc.). Table 1.1 gives an idea about the size of the insert possible with different types of vectors.
In order to determine the choice of vector for a particular cloning experiment, various factors need to be considered such as:
-
1.
Insert size: The insert size may vary for different types of vectors ranging from 5 to 25 kb for plasmid vectors to >2,000 kb for HACs.
-
2.
Vector size: The vector size range varies from 5 kb plasmid vectors to 6–10 megabases HAC high-capacity vectors.
-
3.
Restriction sites: The number of restriction sites found in vectors is highly variable. There may be a few restriction sites in small plasmid vectors but they may be increased by the insertion of multiple cloning sites in vectors.
-
4.
Copy number: Different cloning vectors are maintained at different copy numbers, dependent on the replicon of the plasmid. However, a high-copy number vector is desirable. The origin of replication determines the vector copy number, which could be in the range of 25–50 copies/cell if the expression vector is derived from the low-copy number plasmid pBR322, or between 150 and 200 copies/cell, if derived from the high-copy number plasmid pUC.
-
5.
Cloning efficiency: The ability to clone a DNA fragment inserted into a vector is known as the cloning efficiency of the vector.
-
6.
Ability to screen for inserts: For selection of recombinants, certain selectable markers should be present in vectors in order to distinguish them from non-recombinants.
-
7.
Types of downstream experiments required.
1.2 Vectors for Cloning Large Fragments of DNA
1.2.1 Cosmid Vectors
Cosmids are hybrids between a phage DNA molecule and a bacterial plasmid or are basically a plasmid that carries a cos site, the substrate for enzymes that package λ DNA molecule into phage coat proteins. The in vitro packaging reaction works not only with one genome but also with any DNA molecule that carries cos site separated by 37–52 kb of DNA. It also needs a selectable marker, such as ampicillin resistance gene, and a plasmid origin of replication, as cosmids lack all the λ genes, therefore do not produce plaques. Instead colonies are formed on selective media, just as with a plasmid vector. The loading capacity of cosmids varies depending on the size of the vector itself but usually lies around 40–45 kb—much more than a phage λ vector can accommodate. After packaging in vitro, the particle is used to infect suitable host. The recombinant cosmid DNA is injected into the cell where it circularizes like phage DNA but replicates as a normal plasmid without the expression of any phage functions. Transformed cells are selected on the basis of a vector drug resistance marker. The construct of a typical cosmid vector is shown in Fig. 1.1.

Construct of a cosmid vector
Cosmids provide an efficient means of cloning large pieces of DNA. Because of their capacity to carry large fragments of DNA, cosmids are particularly attractive for constructing libraries of eukaryotic genome fragments. Partial digestion with a restriction endonuclease provided suitably large fragments. However, there is potential problem associated with use of partial digests in this way. This is due to the possibility of two or more genome fragments joining together in the ligation reaction, hence creating a clone containing fragments that were not initially adjacent in the genome. The problem can be overcome by the size fractionation and dephosphorylation of the foreign DNA fragments so as to prevent their ligation together. But this method is very sensitive to the exact ratio of target-to-vector DNAs because vector-to-vector ligation can occur. Such difficulties have been overcome in a cosmid-cloning procedure devised by Ish-Horowicz and Burke (1981). By appropriate treatment of the cosmid vector pJB8, left-hand and right-hand vector ends are purified which are incapable of self-ligation but which accept dephosphorylated foreign DNA. Thus, the method eliminates the need to size the foreign DNA fragments and prevents formation of clones containing short foreign DNA or multiple vector sequences. Figure 1.2 describes the cosmid-cloning procedure devised by Ish-Horowicz and Burke (1981).

Cosmid-cloning procedure (Ish-Horowicz and Burke 1981)
Problems associated with lambda and cosmid cloning:
-
1.
Since repeats occur in eukaryotic DNA, rearrangements can occur via recombination of the repeats present on the DNA inserted into lambda or cosmid.
-
2.
Cosmids are difficult to maintain in a bacterial cell because they are somewhat unstable.
-
3.
Not easy to handle due to its very large size of approximately 50 kb.
1.2.2 Yeast Artificial Chromosomes
A YAC is a vector used to clone DNA fragments larger than 100 kb and up to 3,000 kb. YACs are useful for the physical mapping of complex genomes and for the cloning of large genes. First described in 1983 by Murray and Szostak, a YAC is an artificially constructed chromosome that contains a centromere (CEN), telomeres (TEL), and an autonomous replicating sequence (ARS) element which are required for replication and preservation of YAC in yeast cells. ARS elements are thought to act as replication origins. A YAC is built using an initial circular plasmid, which is typically broken into two linear molecules using restriction enzymes. DNA ligase is then used to ligate a sequence or gene of interest between the two linear molecules, forming a single large linear piece of DNA.
A plasmid-derived origin of replication (ori) and an antibiotic resistance gene allow the YAC vector to be amplified and selected for in E. coli. TRP1 and URA3 genes are included in the YAC vector to provide a selection system for identifying transformed yeast cells that include YAC by complementing recessive alleles trp1 and ura3 in yeast host cell. YAC vector cloning site for foreign DNA is located within the SUP4 gene. This gene compensates for a mutation in the yeast host cell that causes the accumulation of red pigment. The host cells are normally red, and those transformed with YAC only, will form colorless colonies. Cloning of a foreign DNA fragment into the YAC causes insertional inactivation, restoring the red color. Therefore, the colonies that contain the foreign DNA fragment are red.
1.2.2.1 Essential Components of YAC Vectors
-
1.
Large DNA (>100 kb) is ligated between two arms. Each arm ends with a yeast telomere so that the product can be stabilized in the yeast cell. Interestingly, larger YACs are more stable than shorter ones, which favors cloning of large stretches of DNA (Fig. 1.3a, b).
Fig. 1.3 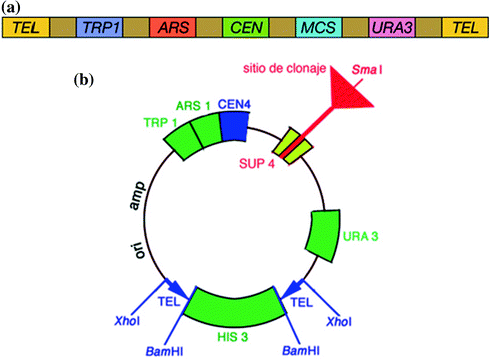
a Linear form of yeast artificial chromosome. b Circular form of yeast artificial chromosome
-
2.
One arm contains an autonomous replication sequence (ARS), a CEN, and TEL.
-
3.
amp r for selective amplification and markers such as TRP1 and URA3 for identifying cells containing the YAC vector.
-
4.
Recognition sites of restriction enzymes (e.g., EcoRI and BamHI).
-
5.
Insertion of DNA into the cloning site inactivates a mutant expressed in the vector DNA and red yeast colonies appear.
-
6.
Transformants are identified as those red colonies which grow in a yeast cell that is mutant for trp1 and ura3. This ensures that the cell has received an artificial chromosome with both TEL (because of complementation of the two mutants) and the artificial chromosome contains insert DNA (because the cell is red).

The procedure for cloning in YAC is as given below:
-
1.
The target DNA is partially digested by EcoRI and the YAC vector is cleaved by EcoRI and BamHI.
-
2.
The cleaved vector segment is ligated with a digested DNA fragment to form an artificial chromosome.
-
3.
Yeast cells are transformed to make a large number of copies.
1.2.2.2 Advantages and Disadvantages
Yeast expression vectors, such as YACs, yeast integrating plasmids (YIps), and yeast episomal plasmids (YEps) have an advantage over bacterial vectors (BACs) in that they can be used to express eukaryotic proteins that require post-translational modification. However, YACs are significantly less stable than BACs, producing “chimeric effects”: artifacts where the sequence of the cloned DNA actually corresponds not to a single genomic region but to multiple regions. Chimerism may be due to either coligation of multiple genomic segments into a single YAC, or recombination of two or more YACs transformed in the same host yeast cell. The incidence of chimerism may be as high as 50 %. Other artifacts are deletion of segments from a cloned region, and rearrangement of genomic segments (such as inversion). In all these cases, the sequence as determined from the YAC clone is different from the original, natural sequence, leading to inconsistent results, and errors in interpretation if the clone’s information is relied upon. Due to these issues, the Human Genome Project ultimately abandoned the use of YACs and switched to BACs, where the incidence of these artifacts is very low.
1.2.3 Bacterial Artificial Chromosome
As the Human Genome Project was underway in the early 1990s, there was a need to create high-resolution physical map of each human chromosome, which would permit the isolation of short DNA fragments for direct sequencing and other manipulations. In response to this, the YAC system was developed. Although yeast can carry the DNA as large as one Mb, subsequent studies indicated that yeast system presented several difficulties in the creation of a human genome map. Additionally, yeast cells were not as familiar to molecular biologist as E. coli. To circumvent these difficulties, a bacterial cloning system based on the well-characterized E. coli F factor, a low-copy plasmid that exist in a supercoiled form was developed by Hiroaki Shizuya in 1992.
A BAC is a DNA construct, based on a functional fertility plasmid (or F-plasmid), used for transforming and cloning in bacteria, usually E. coli. F-plasmids play a crucial role because they contain partition genes that promote the even distribution of plasmids after bacterial cell division. The BAC’s usual insert size is 150–350 kb. The replication of F factor is strictly controlled by the regulatory functions of E. coli; as a result F factor is maintained as a low-copy number (i.e., one or two copies per cell). This allows stable maintenance of large DNA inserts and reduces the potential for recombination between DNA fragments carried by the vector, which was a limitation observed with cosmid-cloning system. In addition to stable maintenance, the structural stability of F-factors allows complex genomic DNA inserts to be maintained with a great degree of structural stability in the E. coli host. The structure of a typical BAC is given in Fig. 1.4.

Bacterial artificial chromosome
BACs have several advantages over YACs. It was observed that a large percentage of YACs carried chimeric inserts, making mapping efforts confusing and difficult. BACs in contrast are virtually free from chimerism. Another problem with YAC is that multiple YAC chromosomes may coexist in a single yeast cell, whereas in the BAC system the F factor encoded parA and parB gene are involved in exclusion of multiple F-factors, as a result multiple F-factors cannot coexist in a single cell.
1.2.3.1 BAC Vector Cloning Site
The cloning segment of BAC vector includes (1) two bacteriophage markers lambda cosN and P1 loxP, (2) three restriction enzyme sites (EcoRI, HindIII, and BamHI) for cloning, and (3) a GC- rich NotI restriction enzyme site for potential excision of inserts. The cosN site provides a fixed position for cleavage by bacteriophage lambda enzyme terminase, which allows the convenient generation of a linear form of the BAC DNA. The cosN site is also used to package approximately 50 kb DNA into bacteriophage lambda head as a particle. The method known as Fosmid for F-based cosmid system is extremely efficient, thus very useful when DNA is precious or available in small amounts. The P1 loxP site allows the retrofitting of additional components to BAC vector at a later stage. The loxP site is also utilized to linearize BACs through the P1 phage protein Cre, which catalyses strand exchange between two DNA strands at the loxP sites.
1.2.3.2 Uses
1.2.3.2.1 Inherited Disease
BACs are now being utilized to a greater extent in modeling genetic diseases, often alongside transgenic mice. BACs have been useful in this field as complex genes may have several regulatory sequences upstream of the encoding sequence, including various promoter sequences that will govern a gene’s expression level. BACs have been used to some degree of success with mice while studying neurological diseases such as Alzheimer’s disease or as in the case of aneuploidy associated with Down syndrome. There have also been instances when they have been used to study specific oncogenes associated with cancers. They are transferred over to these genetic disease models by electroporation/transformation, transfection with a suitable virus or microinjection. BACs can also be utilized to detect genes or large sequences of interest and then used to map them onto the human chromosome using BAC arrays. BACs are preferred for these kinds of genetic studies because they accommodate much larger sequences without the risk of rearrangement, therefore more stable than other types of cloning vectors.
1.2.3.2.2 Infectious Diseases
The genomes of several large DNA and RNA viruses have been cloned as BACs. These constructs are referred to as “infectious clones,” as transfection of the BAC construct into host cells is sufficient to initiate viral infection. The infectious property of these BACs has made the study of many viruses such as the herpes viruses, poxviruses, and coronaviruses more accessible. Molecular studies of these viruses can now be achieved using genetic approaches to mutate the BAC while it resides in bacteria. Such genetic approaches rely on either linear or circular targeting vectors to carry out homologous recombination.
1.2.3.2.3 Genome Sequencing
BACs are often used to sequence the genome of organisms in genome projects, for example the Human Genome Project. A short piece of the organism’s DNA is amplified as an insert in BACs, and then sequenced. Finally, the sequenced parts are rearranged in silico, resulting in the genomic sequence of the organism.
1.2.4 P1 Phage Derived Artificial Chromosome
The P1-derived artificial chromosomes are DNA constructs derived from the DNA of P1 bacteriophage and BAC. They can carry large amounts (about 100–300 kb) of other sequences for a variety of bioengineering purposes. It is one type of vector used to clone DNA fragments (100- to 300-kb insert size; average, 150 kb) in E. coli cells. PACs have a low-copy number origin of replication based on P1 bacteriophage, which is used for propagation. Similar to BACs, PACs allow replication of the clones at one copy per cell and replicate clones across 60–100 generations. In contrast to BACs, PACs have a negative selection against non-recombinants. PACs also have an IPTG- inducible high-copy number origin of replication that can be utilized for DNA production. These can accommodate larger inserts of DNA than a plasmid or many other types of vectors. Sometimes, the number of inserts can be as high as 300 kb (Fig. 1.5).

Phage artificial chromosome
1.2.4.1 Uniqueness of P1-bacteriophage
A P1 phage can exist in both lysogenic and lytic forms in the host cell, but its unique feature lies in its existence as an independent entity within the cell, rather than incorporating itself with the host chromosomes during the phase of ‘lysogeny’. Thus, it acts like a plasmid during its existence and can replace the function of a plasmid during processes, which entails this feature. However, the scientists consider P1 derived chromosome to contain features of both plasmids and ‘F’ factor, which is a unique plasmid like DNA sequence used in creating BAC.
In comparison with YACs, PACs offer certain advantages: (1) these are bacterial systems that are easy to manipulate, (2) libraries are generated using bacterial hosts with well defined properties, (3) transformation efficiency is higher than that obtained by YACs, (4) PACs are nonchimeric, and (5) PACs have very stable inserts and do not delete sequences.
1.2.4.2 Construction of PACs Through Electroporation
During the construction of PACs, P1 phage containing cells will undergo a process known as ‘electroporation’, which will increase the permeability of the cell membrane and allow DNA material to enter the cell and couple with the existing DNA. This process will give rise to PACs and from there onwards, the PACs can replicate within the cell through ‘lysogeny’, without destructing the cell or incorporating into rest of the chromosomes.
1.2.4.3 Uses of PACs
PACs are in high demand for cloning important biomedical sequences, which are essential for many scientific functions. One of its main uses is the genome analysis and map-based cloning of complex plants and animals, which requires isolation of large pieces of DNA rather than smaller segments. Furthermore, PAC-based cloning is useful in the study of ‘phage therapy’ and in scientific studies focusing on how antibiotics act on a particular bacteria.
Although there are other forms of artificial chromosomes which can accommodate more base pairs than PACs, relative user friendliness of these vectors makes them a popular choice among many biomedical researchers.
1.2.5 Human Artificial Chromosomes
The idea of using artificial chromosomes as potential vectors for gene therapy applications came as a consequence of the first studies involving chromosome manipulation, designed to understand human chromosome structure, and to identify elements necessary for their correct functioning. There are two approaches that can be used for the development of artificial chromosomes: top-down approach in which natural chromosomes are truncated by radiation or telomere-associated fragmentation; and the bottom-up approach in which a de novo chromosome is formed from the basic elements of CENs, TEL, and origins of replication.
The construction of YAC showed CENs, TEL, and origins of replication as the elements necessary for extrachromosomal retention and led to the development of mammalian chromosomes which are similar to yeast chromosomes. Many experiments designed to find mammalian origins of replication could not identify specific sequences responsible for mammalian genome replication. However, the structure of human TEL was soon described as an array. The confirmation came from the observation of newly formed TEL in the α globin gene, caused by insertion of (TTAGGG) n sequence. The discovery of the telomeric sequence as a tandemly repeated (TTAGGG) n sequence orientated 5′-3′ toward the end of the chromosome and its role in telomere formation led to the development of telomere-mediated chromosome fragmentation (top-down approach), which allowed the isolation of minichromosomes in somatic cell hybrid. The first “top down” approach or TACF (telomere-associated chromosome fragmentation) involved modifying natural chromosome into smaller defined minichromosomes in cultured cells. Following recombination and subsequent breakage between homologous sequences on the endogenous host chromosome and an incoming telomere containing the targeting vector, engineered minichromosomes as small as 450 kb in size in avian cells have been generated. The approach has been important for studying the structure, sequence organization, and size requirements of the human X and Y chromosomes.
Second approach, the “bottom up” or assembly approach involved generating HACs in human cells by introducing defined chromosomal sequences as naked DNA including human TEL, alpha satellite (alphoid) DNA and genomic fragmentation containing replication origins. The de novo HACs are generated following recombination and some amplification of the input DNA within the host cell. Together, the generation of minichromosomes and de novo HACs has identified alphoid DNA as the major sequence element of the CEN and determined the minimum size (~700–100 kb) required for CEN function and stability.
Second approach includes the generation of SATACs (satellite DNA-based artificial chromosomes) following integration of repetitive DNA into preexisting centromeric regions of host chromosomes and modifying small human marker chromosomes (minichromosomes derived from naturally occurring chromosomes). The two approaches are shown in Fig. 1.6.

Structural map of ‘top-down’ engineered mammalian artificial chromosome systems
The de novo HACs when introduced into the cell, undergo a process of recombination and amplification forming large (1–10 Mb) circular molecules (usually at one or two copies per cell) which are mitotically stable in the absence of any selection for 9 months in some cells. The efficiency of de novo HAC formation and stability depends on the presence of a CEN protein B-binding sequence (CENP-B box) and, to some extent, on the chromosomal origin of the alphoid template and the longer length of the alphoid array (>100 bp).
Established HACs can be either in a linear or a circular state. PAC-based constructs carrying ~70 kb of alphoid DNA array with or without telomeric sequences and in circular or linearized state were used to transfect by lipofection HT1080 cells. Circular alphoid DNA vectors established effectively as minichromosomes in any condition, demonstrating that TEL are not required for the circular conformation. However, capped TEL were essential for establishment of linear PAC vectors because these vectors showed poor chromosome formation in their absence.
1.2.5.1 Advantages and Uses of HAC
Human artificial chromosomes (HACs) represent another extrachromosomal gene delivery and gene expression vector system. Although this technology is less advanced than virus derived vectors, HACs have several potential advantages over currently used episomal viral vectors for gene therapy applications. The presence of a functional CEN provides a long-term stable maintenance of a HAC as a single copy episome without integration to the host chromosomes. There is no upper size limit to DNA that should be cloned in HAC that allows the use of complete genomic loci, including the upstream and downstream regulatory elements. Additionally, being solely human in origin, HAC vectors cannot evoke adverse host immunogenic responses or induce any risk of cellular transformation.
HAC-based vectors offer a promising system for delivery and expression of full-length human genes of any size.
1.3 Conclusion
A vector is a DNA molecule used as a vehicle to transfer foreign genetic material into another cell. All engineered vectors have an origin of replication, a multicloning site, and a selectable marker. Genome size varies among different organisms and the cloning vector must be selected accordingly. For a large genome, a vector with a large capacity is chosen so that a relatively small number of clones are sufficient for coverage of the entire genome. However, it is often more difficult to characterize an insert contained in a high capacity vector. The development of extrachromosomal large-capacity cloning vectors for mammalian cells represents a powerful tool for functional genomic studies. Further, the advances in genome library construction and DNA sequencing are mainly due to the development of high capacity vectors such as cosmids, BACs, PACs, YACs, and HACs.
References
Ish-Horowicz D, Burke JF (1981) Rapid and efficient cosmid cloning. Nucleic Acids Res 9(13):2989–2998
Murray AW, Szostak JW (1983) Construction of artificial chromosomes in yeast. Nature 305(5931):189–193
Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, Simon M (1992) Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Natl Acad Sci 89:8794–8797
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer India
About this chapter
Cite this chapter
Bajpai, B. (2014). High Capacity Vectors. In: Ravi, I., Baunthiyal, M., Saxena, J. (eds) Advances in Biotechnology. Springer, New Delhi. https://doi.org/10.1007/978-81-322-1554-7_1
Download citation
DOI: https://doi.org/10.1007/978-81-322-1554-7_1
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-1553-0
Online ISBN: 978-81-322-1554-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)