
Abstract
The usage of biomarkers reflecting atherosclerosis progression is important for preventing serious incidence of cardiovascular events. To elucidate clinically relevant molecular determinants in atherosclerosis, we have taken a comprehensive approach to combine mass spectrometry-based differential proteomics using both clinical and animal model specimens. Clinical plasma samples were collected from patients with acute myocardial infarction (AMI), stable angina (SA), and healthy/low-risk individuals (H-LR). We also obtained plasma and arterial tissue samples from apolipoprotein E-deficient and wild-type mice at various pathognomonic points of age. Cleavable isotope-coded affinity tags were used for differential mass spectrometry. Differential proteomics of clinical plasma samples revealed that more than 10 proteins appeared to be upregulated (relative abundance AMI/H-LR or SA/H-LR >1.5) and 5 proteins downregulated (AMI/H-LR or SA/H-LR <1/1.5). These trends associated with the disease progression are not always coincident with those of mouse ortholog proteins, suggesting a pathophysiological difference between humans and the mouse model. Among the downregulated proteins, the complement factor D (CFD) showed monotonic decrease that was in good agreement with the enzyme-linked immunosorbent assay. These results suggest that the comprehensive and systematic proteomic approach may be promising in terms of the selection and evaluation of biomarker candidates.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
Atherosclerosis is a chronic inflammatory disease and often causes subsequent thrombotic complication. It is well known as a main etiology of critical diseases such as acute coronary syndrome including myocardial infarction, sudden cardiac death, and stroke [1]. To detect the signs of severe incidences, it is important to accurately evaluate the progression stages of atherosclerosis, vulnerability, and rupture risk of atherosclerotic plaques. Therefore, a variety of approaches to establish novel biomarkers directly reflecting the status of atherosclerosis have been conducted to date [2]. However, due to the complexity of the disease, discovery and clinical validation of such biomarkers are still underway in many institutions.
Disease proteomics is a profiling technology using mass spectrometry that makes it possible to identify disease-related alteration of protein expression comprehensively in a wide variety of biological specimens including serum or plasma or extracts from tissues of interests [3]. Numerous studies have been conducted to develop biomarkers for early diagnosis, selection of appropriate therapy, and prognosis assessment. A decade of progress to determine changes in disease-related protein expression has achieved accurate and high-throughput quantification of the individual proteins within mixtures by using differential stable isotopic labeling, such as cleavable isotope-coded affinity tags (cICATs) [4]. Either light or heavy cICATs are labeled at the free thiol groups of cysteine residues of which proteins are isolated from two distinct samples (i.e., disease and healthy states). The ratio of protein amount between these states is estimated by comparing MS signal intensity of the corresponding proteolytic peptide fragments [4].
To elucidate clinically relevant molecular determinants in atherosclerotic development, we have taken a comprehensive approach by using mass spectrometry-based differential proteomics. First, we conducted differential human plasma proteomic studies using clinical samples of patients with atherosclerosis-related cardiovascular disease and healthy (or low-risk) volunteers. Second, to validate the results we obtained in the studies, a biomarker candidate, the complement factor D (CFD) level of patients with atherosclerosis-related cardiovascular disease, was determined by carrying out an enzyme-linked immunosorbent assay (ELISA). Then, we tried combining differential proteome of plasma and arterial tissue obtained from both wild-type (WT) and apoED mice at four pathognomonic time points of age. Finally, we compared identified proteins in humans with mouse plasma proteome.
2 Materials and Methods
This study was approved by the Ethics Committee of Hokkaido University Hospital (no. 010–0118) and the institutional ethics committees of Hitachi, Ltd. Clinical plasma samples were collected prospectively for 48 acute myocardial infarction (AMI) patients, 73 stable angina (SA) patients, and 69 healthy/low-risk (H-LR) individuals.
Animal care and animal experiments were conducted at Japan SLC, Inc. (Hamamatsu, Japan), under the approval of the company’s animal care committee. The apoED (C57BL/6.KOR/StmSlc-Apoeshl) and WT (C57BL/6) mice were fed a high-fat diet from 8 weeks of age. Plasma and arterial tissues were collected from each group of male or female mice at ages of 12, 18, 25, and 35 weeks.
Proteins prepared from plasma or arterial tissue as described above were labeled with isotope-coded affinity tags using the Cleavable ICAT® Reagent Kit (Applied Biosystems, Foster City, USA) according to the manufacturer’s instructions with minor modifications. Tryptic peptides were applied to the strong cation exchange (SCX) column, and the eluted solution was fractionated into 25 fractions. The SCX separated fractions were analyzed using NanoFrontier LD, a liquid chromatograph mass spectrometry (LC-MS) system (Hitachi High-Technologies Corporation, Tokyo, Japan). Original MS/MS peak lists (PLs) were generated using built-in PL generation software in the mass spectrometer, and further protein/peptide identification and cICAT quantitative analyses were conducted using a custom developed software platform with a relational database.
Plasma CFD levels were measured with the use of ELISA kits according to the manufacturer’s instructions (Quantikine, DFD00, R&D Systems, Inc., Minneapolis, USA). Plasma levels of known markers including high-sensitive C-reactive protein (hsCRP), amino-terminal pro-brain natriuretic peptide (NT-pro BNP), and adiponectin were also determined (SRL, Inc., Tokyo, Japan).
3 Results and Discussion
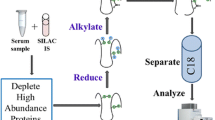
Atherosclerosis is a chronic inflammatory disease, in which atherosclerotic lesions are formed in blood vessels through complicated molecular processes occurring between the circulating blood stream and lesions. To identify proteins showing changes in expression levels associated with atherosclerotic plaque progression, we conducted mass spectrometry-based proteomic analyses. Figure 10.1 shows an overall picture of the proteomic experiments we have done. Most previous proteomic studies have focused on only one point of the disease. However, these strategies would not be adequate to indentify proteins showing serial quantitative change in accordance with progression of atherosclerosis. In this study, we conducted a differential proteome study at several characteristic points of disease stage and compared the results of identified proteins. The proteins we identified that showed changes in plasma level according to the severity of the atherosclerosis-related disease (e.g., ischemic heart disease) or atherosclerosis progression of the mouse model might be diagnostic or predictive biomarker candidates for blood testing. On the other hand, the proteins that showed changes, especially an increase tendency, in arterial tissues might be biomarkers for diagnostic imaging.

Entire research workflow of stage-dependent, mass spectrometry-based differential proteomics. (a) Clinical proteomic study using human plasma samples. (b) Animal model proteomic study using plasma and arterial tissue samples from WT and atherosclerotic model mice
3.1 Disease Stage-Dependent Differential Proteome in Human Plasma
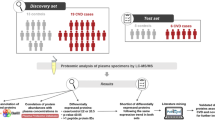
As shown in Fig. 10.1, we conducted disease stage-dependent differential human plasma proteomic experiments using clinical samples. We selected three clinical conditions, H-LR, SA, and AMI (Table 10.1). Two independent age-matched plasma pools from each group (n = 7) were used for the experiments with combinations of H-LR vs. SA, H-LR vs. AMI, and SA vs. AMI. Table 10.2 shows the summary of identified proteins by differential proteomics of clinical plasma samples. We identified about 110 proteins in each trial of differential proteome and found that 8–13 proteins appeared to be upregulated (relative abundance AMI/H-LR or SA/H-LR >1.5), and 2–8 proteins were downregulated (AMI/H-LR or SA/H-LR <1/1.5). Finally, five identical proteins were identified in common between trials 1 and 2. Three were blood coagulation related, such as von Willebrand factor, and the other two were categorized as structural-integrity-related or immuno-response (immunoglobulin)-related proteins. The downregulated proteins identified in common between trials were found to be apolipoproteins involving cholesterol homeostasis, blood coagulation, and CFD.
To validate the results we obtained in the differential proteome experiments as described above, we determined the CFD level of plasma of clinical samples by ELISA as a representative example. We found that CFD levels in plasma of the AMI group were the lowest among the three clinical groups. The statistical difference of AMI to H-LR was the most pronounced (H/LR vs. AMI, p = 2.4 × 10−10; H/LR vs. SA, p = 0.54). The diminished plasma levels of CFD in AMI patients showed that the results we obtained in our proteomic study were basically verified. Furthermore, the plasma CFD levels showed no correlation with body mass index (BMI) (r = 0.03, P < 0.05), hsCRP (r = −0.09, P < 0.05), NT-proBNP (r = −0.03, P < 0.05), and adiponectin (r = 0.11, P < 0.05). In recent years, it has been suggested that the activation of the complement system plays a role in atherosclerosis progression [5]. Among the components of the complement pathway, CFD is known as a key regulatory serine protease of the alternative complement pathway. CFD mRNA and protein are abundantly expressed in adipose tissue, and the protein is found at high levels in serum [6]. CFD expression levels in rodent models of obesity are reduced [7]. Our results may suggest some kind of CFD implication in atherosclerosis.
3.2 Disease Stage-Dependent Differential Proteome in Atherosclerosis Mouse Model
As shown in Fig. 10.1, we conducted differential proteome experiments using plasma and arterial tissues of WT and apolipoprotein E-deficient (apoED) mice. Table 10.3 shows phenotypic characteristics of atherosclerotic lesions of WT and apoED at each point of age. As we previously demonstrated, on a high-fat diet, early lesions were observed in apoED at 12–18 weeks of age, and advanced lesions were prominent at 25–35 weeks. No atherosclerotic lesions were found in WT mice throughout four points of age [8]. At 25 weeks of age, vulnerable atheromatous lesions were more abundant, but fibroatheromatous lesions were less at 25 weeks than those at 35 weeks [8]. Table 10.4 shows a summary of identified proteins. In plasma, the total number of identified proteins and the ratio of their upregulated proteins (expression ratio, apoED/WT; > 1.5) did not significantly change throughout the time course (Table 10.4). However, the ratio of downregulated proteins (apoED/WT; <1/1.5) increased on and after 25 weeks of age (Table 10.4). We finally found 100 proteins in the plasma, and 390 proteins in the arterial tissues were detected throughout all four time points: 29 were identified in common between plasma and arterial tissues [data not shown, 9]. Interestingly, we found that disease stage-dependent quantitative variation patterns did not always correspond between plasma and arterial tissues. Furthermore, proteins showed characteristic change in abundance in plasma, and/or arterial tissues were found to be components of inflammation, thrombus formation, and vascular remodeling [9].
3.3 Comparison Between Human and Mouse Plasma Proteome
In this study, we conducted disease stage-dependent differential proteome experiments in two identical studies: one for human plasma proteome using clinical samples and one for plasma/arterial tissue proteome using an atherosclerotic mouse model, apoED. Importantly, we found that the changes associated with the disease progression in the amount of identified proteins were not always coincident with those of mouse ortholog proteins in plasma or arterial tissues determined by proteomic studies (data not shown). In case of CFD, the plasma level of CFD in humans determined in the proteomic study or ELISA decreased in accordance with the severity of the clinical conditions. On the other hand, the plasma level of CFD in mice showed a slight but not significant decreasing tendency determined in differential proteome between WT mice and the apoED mouse model. We preliminarily determined the plasma level of CFD in mice by ELISA and detected no statistical significance between WT mice and apoED throughout all four time points. CD5L, a soluble member of the scavenger receptor cysteine-rich domain superfamily protein, did not show any significant change in abundance as determined by human plasma differential analyses. However, the expression ratios (apoED/WT) of CD5L in mouse plasma were high throughout the four time points [9]. In addition, we found that CD5L accumulated in advanced lesions of apoED in accordance with macrophage infiltration by immunohistochemistry. These results suggest that fundamental mechanisms of atherosclerosis development are similar to each other; however, there would be a pathophysiological difference between humans and the mouse model.
3.4 Limitation
We failed to detect several known atherosclerotic plaque-related proteins such as cytokines, monocyte chemoattractant protein-1 (MCP-1), and intercellular adhesion molecule-1(ICAM-1). We assume that there were several technical reasons for this. For example, the practical detection limit in our mass spectrometry system was assumed to be 1–10 ng/mL, which is much higher than the normal concentration of cytokines. We used cICAT labeling reagents, which covalently bind only to cysteine residues of targeted proteins. Therefore, it becomes difficult to detect proteins with trypsin-digested peptides with few or no cysteine residues.
4 Conclusion
To identify proteins showing changes in expression levels associated with atherosclerotic plaque progression, we conducted mass spectrometry-based proteomic analyses. We adopted two separate study designs: one for human plasma differential proteome using clinical samples and one for mouse plasma/arterial tissue differential proteome using samples obtained from wild-type mice and an atherosclerotic mouse model apoED. Then several proteins showing quantitative changes in accordance with disease were found, including the complement factor D (CFD). The diminished plasma levels of CFD in acute myocardial infarction patients were verified in an enzyme-linked immunosorbent assay. These results suggest that CFD might be a potential biomarker for atherosclerosis. The comprehensive and systematic proteomic approach using different states of samples is promising in terms of the selection of biomarker candidates.
References
Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–26.
Wang X, Connolly TM. Biomarkers of vulnerable atheromatous plaques: translational medicine perspectives. Adv Clin Chem. 2010;50:1–22.
Hanash S. Disease proteomics. Nature. 2003;422:226–32.
Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–9.
Torzewski M, Bhakdi S. Complement and atherosclerosis-united to the point of no return? Clin Biochem. 2013;46:20–5.
Cook KS, Min HY, Jonson D, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237:402–5.
Flier JS, Cook KS, Usher P, et al. Severely impaired adipsin expression in genetic and acquired obesity. Science. 1987;237:405–8.
Zhao Y, Kuge Y, Zhao S, et al. Prolonged high-fat feeding enhances aortic 18F-FDG and 99mTc-Annexin A5 uptake in apolipoprotein E-deficient and wild-type C57BL/6J Mice. J Nucl Med. 2008;49:1707–14.
Hanzawa H, Sakamoto T, Kaneko A, et al. Combined plasma and tissue proteomic study of atherogenic model mouse: Approach to elucidate molecular determinants in atherosclerosis development. J Proteome Res. 2015;14:4257–69.
Acknowledgments
This study was supported in part by the grant “The matching program for innovations in future drug discovery and medical care” from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to Tamaki, N.). The authors thank Megumi Hikichi, Yuko Komori, and Yumi Yanagiya for their technical assistance. Without their help and support, we could never have done this study.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this paper
Cite this paper
Sakamoto, T. et al. (2016). Discovery and Evaluation of Biomarkers for Atherosclerosis. In: Kuge, Y., Shiga, T., Tamaki, N. (eds) Perspectives on Nuclear Medicine for Molecular Diagnosis and Integrated Therapy. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55894-1_10
Download citation
DOI: https://doi.org/10.1007/978-4-431-55894-1_10
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55892-7
Online ISBN: 978-4-431-55894-1
eBook Packages: MedicineMedicine (R0)