
Abstract
For the assessment of Fukushima Daiichi Nuclear Power Plant accident, the applicability of the thermal ionization mass spectrometry (TIMS), which is a type of mass spectrometry, was studied. For the study of the recovery/analysis method of cesium and strontium, at first, the radioactive cesium and strontium were generated by the irradiation of natural uranium at KUR. After this study, the applicability of this method to the environmental samples obtained in Fukushima prefecture was verified.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Fukushima Dai-ichi Nuclear Power Plant accident
- Strontium
- Cesium
- Chromatography
- Mass spectrometry
- Isotopic ratio
1 Introduction
On the accident of Fukushima Daiichi Nuclear Power Plant (FDNPP), fission products (FP) such as radioactive Cs and Sr were widely released. The amounts of FP generated in each reactor were calculated by using ORIGEN code [1]. Many studies of radioactive Cs and Sr were performed to estimate external and internal exposures and to analyze the source of radioactive nuclides. These studies were typically performed by γ-ray spectrometry of 134Cs (T1/2 = 2.06 y) and 137Cs (T1/2 = 30.2 y) for the analysis of radioactive Cs and by β-spectrometry of 90Sr (T1/2 = 28.9 y) for that of radioactive Sr.
In addition to 134Cs and 137Cs, radioactive 135Cs (T1/2 = 2.3 × 106) is also generated during the operation of reactors. Because of the difference in the generation process and the half-life of radioactive Cs, the isotopic ratios of 134Cs/137Cs and 135Cs/137Cs have been used for analyzing the operations of nuclear facilities [2–6]. Naturally occurring Sr has four stable isotopes (84Sr, 86Sr, 87Sr, and 88Sr), on the other hand, and the isotopic composition of Sr generated in reactors [1] are totally different from the natural abundance [7]. From the analysis data of the isotopic compositions, thus, the information on the origin of radioactive nuclide release would be obtained. The mass spectrometry provides the isotopic compositions of elements. Although mass spectrometry has been used for the analysis of radioactive nuclides and actinides, few studies have reported the analysis of radioactive Cs and Sr.
The purpose of the present study is to analyze Cs and Sr isotopes in environmental samples in Fukushima prefecture for source analysis and safety assessment. Although the amounts of radioactive Cs and Sr released in this accident were very huge, the contaminated environmental samples show the small radioactivity per unit weight of the contaminated environmental samples, since the contaminated area is very wide. For the study of the recovery/analysis method of cesium and strontium, at first, the radioactive Cs and Sr were generated by the irradiation of natural uranium at KUR. After this study, the applicability of this method to the environmental samples obtained in Fukushima prefecture was verified.
2 Experimental
2.1 Irradiation of UO2 for Study of Radioactive Cs and Sr
10 mg of UO2 of natural uranium was irradiated for 3 h at the Kyoto University Research Reactor with the neutron flux 5.5 × 1012 n/s cm2. From the calculation with ORIGEN-II code [8], the amounts of the major radionuclide of Cs and Sr were estimated as 7.4 × 10−11 g (137Cs) and 4.5 × 10−11 g (90Sr), respectively. After standing for ca. 2 days, radioactive Cs and Sr were recovered and analyzed.
2.2 Recovery of Cs and Sr
2.2.1 Isolation of TRU Elements
Cs and Sr were recovered with UTEVATM-resin (100–150 μm, Eichrom Technologies), Sr-resin (100–150 μm, Eichrom Technologies), ammonium phosphomolybdate (AMP), the cation exchange resin DOWEXTM 50WX8 (100–200 mesh), and the anion exchange resin DOWEXTM 1 X 8 (100–200 mesh).
The irradiated UO2 was dissolved in 8 M HNO3 (TAMAPURE-AA-100) and was evaporated to dryness at 403 K. 8 M HNO3 was added and the insoluble residues removed by centrifugation. After centrifugation, H2O2 (TAMAPURE-AA 100) was added for the preparation of 8 M HNO3/0.3 % H2O2 sample solution to isolate TRU elements such as U and Pu by the extraction chromatography with UTEVA-resin [9].
Three milliliter of the UTEVA-resin conditioned with diluted nitric acid was filled into a column of 54 mm in length and 6.5 mm in diameter and pretreated with 10 mL of 8 M HNO3/0.3 % H2O2 before loading the solution. After loading the solution, the UTEVA-resin was rinsed with 8 M HNO3 to elute alkaline earth metal elements [10]. The effluent was evaporated to dryness and dissolved in 10 mL of 3 M HNO3 solution for the extraction chromatography with Sr-resin.
2.2.2 Recovery of Strontium
The solution was loaded to the Sr-resin conditioned with diluted nitric acid and filled into a column of 54 mm in length and 6.5 mm in diameter up to 3 mL. This effluent was evaporated at 403 K and the residue dissolved in 0.05 M HNO3 for the recovery of Cs. After washing of the Sr-resin with 3 M HNO3, Sr was recovered with 20 mL of 0.05 M HNO3, evaporated to dryness, and dissolved in 10 μL of 1 M HNO3.
2.2.3 Recovery of Cesium
After adding of 0.1 g of AMP to the Cs solution and stirring for several hours, the supernatant was removed from the mixed solution by centrifugation. A 20 mL 3 M ammonium hydroxide (TAMAPURE-AA 100) solution was used to dissolve the residue for subsequent anion-exchange ion chromatography.
After the final conditioning [11], a 3 mL portion of the anion-exchange resin was added to a column of 54 mm in length and 6.5 mm in diameter. The sample solution was added to the column, and the resulting eluate was collected and heated to dryness. The residue was dissolved in 20 mL of 0.1 M HNO3 for the final purification with the cation-exchange ion chromatography.
The cation-exchange resin conditioned with hydrofluoric acid (TAMAPURE-AA-100), etc. [12] was filled into a column of 42 mm in length and 5.0 mm in diameter up to 1.5 mL. After loading the sample solution, the resin was washed with diluted nitric acid followed by 20 mL of 1.5 M HCl (TAMAPURE-AA 100) to recover Cs. The effluent was heated to dryness, and the residue was dissolved in 20 μL of 1 M HNO3 for the analysis of the isotopic composition of Cs.
2.3 Analysis of Isotopic Composition of Cesium and Strontium
Isotopic compositions of Cs and Sr were measured with a TIMS (Triton-T1, Thermo Fisher Scientific). A 1 μL aliquot of each solution was loaded onto a rhenium filament with a TaO activator [13]. The standard material of SRM987 [14] was used as a reference material of mass spectrometry of Sr. The mass spectra of radioactive Cs and Sr were obtained with a secondary electron multiplier detector (SEM) because of the low total amounts of radionuclide loaded onto the filament.
2.4 Analysis of Environmental Samples
The plant samples were obtained from the south area of Iitate village, the northeast area of Okuma town, the southeast area of Futaba town, and southwest area of Futaba town in Fukushima prefecture from November 2012 and May 2013 (Table 4.1). The samples were washed three times with pure water and dried at 373 K. About 2.5 g of the dried samples was incinerated with a ring furnace at 873 K and dissolved in concentrated HNO3 at 403 K and evaporated to dryness. 20 mL of 8 HNO3 was added and the insoluble residues removed by centrifugation for the preparation of recovery of Cs and Sr. Recovery of Cs and Sr from environmental samples was also carried out by the same manner described above.
The concentration of 88Sr was measured with an inductively coupled quadrupole mass spectrometer (ICP-QMS, HP-4500, Yokoagawa) and radioactivity of 90Sr by Cherenkov counting [15]. The total concentration of radioactive Cs was measured by γ-spectrometry. The sample solutions were prepared as 50 ppm of 88Sr in 1 M HNO3 for the analysis of Sr and 5000 Bq/mL for 137Cs in 1 M HNO3 for the analysis of Cs. The mass spectra of radioactive Cs and Sr were obtained with a SEM, while those of stable Cs and Sr were obtained with Faraday cup detector, since the amounts of stable nuclide were much larger than those of radionuclide.
3 Results and Discussion
3.1 Isotopic Analysis of Radioactive Cs and Sr from Irradiated UO2
Figure 4.1a shows the mass spectra of Cs recovered from the irradiated UO2. In this measurement, 135Cs, 136Cs, and 137Cs were detected: 134Cs was not detected, because of the difference in the generation scheme. The observed isotopic ratios of 135Cs/137Cs and 136Cs/137Cs were obtained as 0.9103 ± 0.0008 and 0.00022 ± 0.00001. From our calculation with ORIGEN-II code [8], the loading amounts of 135Cs, 136Cs, and 137Cs in this time were about 3.5, 0.03, and 3.7 pg respectively. This means that the femtogram level of Cs is detectable by TIMS.

Mass spectra of Cs. (a) Recovered from UO2 irradiated in KUR. (b) Recovered from environmental sample from Fukushima prefecture (Reproduced from Ref [11])
Figure 4.2 shows the mass spectra of Sr both of stable (a) and radioactive (b) isotopes. At the measurement of 2.6 days later, 89Sr, 90Sr, and 91Sr were detected. From our calculation with ORIGEN-II code [8], the loading amounts of 89Sr, 90Sr, and 91Sr in this time were about 3, 4, and 0.04 pg respectively. This means that the femtogram level of Sr is also detectable by TIMS.

Mass spectra of Sr. Stable isotopes (a) were obtained by measurement of Sr of SRM987 with a Faraday cup detector, and radioactive isotopes (b) were obtained by measurement of Sr recovered from UO2 irradiated in KUR with a secondary electron multiplier detector
The measured isotopic ratios were 0.80 for 89Sr/90Sr and 0.01 for 91Sr/90Sr showing the agreement with the calculated value (0.79 for 89Sr/90Sr and 0.01 for 91Sr/90Sr). Because the half-life of 91Sr is 9.5 h, the mass spectrum of 91Sr disappeared at the measurement of 31 days later. The measured isotopic ratio of 89Sr/90Sr is 0.53 showing the agreement with the calculated value of 0.54. At the measurement of 574 days later, only the mass spectrum of 90Sr was observed because the half-life of 89Sr is 50.5 days. This means that 89Sr/90Sr could not be analyzed by using a typical mass spectrometer after Sep. 2012, if we obtain the sample containing femtogram level of 90Sr. The isotopic ratio of 90Sr/stableSr would be therefore needed for our purpose.
3.2 Analysis of Isotopic Compositions of Cs and Sr from Environmental Samples
3.2.1 Analysis of Cs
Figure 4.1b shows three peaks, representing 134Cs, 135Cs, and 137Cs, were observed on the typical mass spectra of Cs recovered from environmental samples obtained in Fukushima prefecture [11], while the peak representing 136Cs was not observed because of the half-life (T1/2 = 13.2 d). From the calculation with ORIGEN-II code [1], the isotopic ratio of 136Cs/137Cs in the fuel was estimated as ca. 0.00032. This value shows the same order compared with that of the irradiated UO2, suggesting that we could obtain the three isotopic ratios of 134Cs/137Cs, 135Cs/137Cs, and 136Cs/137Cs until July 2011. Since there are three reactors in FDNPP, three isotopic ratios would bring the important information for the source analysis of radioactive Cs in the contaminated area in Fukushima prefecture.
Although we could not obtain the isotopic ratio of 136Cs/137Cs after July 2011, we can obtain the two-dimensional map with the isotopic ratios of 134Cs/137Cs and 135Cs/137Cs as shown in Fig. 4.3. All of the isotopic ratios of 135Cs/137Cs showed less than 0.4. This value was also much smaller than reported isotopic ratios of global fallout (∼0.5 for Chernobyl accident and ∼2.7 for nuclear weapon testing, corrected to March 11, 2011 [11]) and the long half-life of 135Cs (T1/2 = 2.3 × 106 y), meaning that only the isotopic ratio of 135Cs/137Cs would also provide the information for the origin of radioactive Cs among Chernobyl accident, nuclear weapon testing, and FDNPP accident for the long term.

135Cs/137Cs (atomic ratio) vs 134Cs/137Cs (activity ratio). Error here means ±2SE. Data of OKM01, FTB01, FTB35R and ITT07 were reproduced from Ref [11]. Both of isotopic ratio was corrected to March 11, 2011. Single asterisk (*) represents calculation results from estimation of radioactive nuclides with ORIGEN-II code [1]. Double asterisk (**) represents values reported for 134Cs/137Cs activity ratio in polluted water [22]
3.2.2 Analysis of Sr
The FP of Sr in each reactor has mainly five isotopes [1]: two stable isotopes of 86Sr and 88Sr and three radioactive isotopes of 89Sr, 90Sr, and 91Sr. The relationship between the isotopic ratio of radioactive Cs and that of Sr estimated by ORIGEN Code calculation [1] is plotted in Fig. 4.4. In addition to the radioactive isotopes, the stable isotopes of Sr generated in each reactor show the characteristic profile. This suggests that the stable isotopes of Sr could be also used for the analysis of the FP of Sr.

Estimated relationship between isotopic ratios of 134Cs/137Cs, 89Sr/90Sr and 87Sr/86Sr and that of 135Cs/137Cs. Isotopic ratios were estimated by calculation results with ORIGEN Code [1]. Open circle means isotopic ratio of 89Sr/90Sr. Closed circle represents isotopic ratio of 87Sr/86Sr
Among the isotopic ratios of stable isotopes, the isotopic ratio of 87Sr/86Sr is important in the field of the geological chronology [16], because 87Sr is generated by the β-decay of 87Rb having the half-life of 4.9 × 1010 y. Thus, the isotopic ratio of stable isotopes, in this study, will be focused on the isotopic ratio of 87Sr/86Sr.
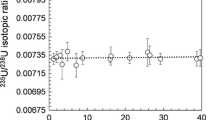
The certified value for SRM987 of the isotopic ratio of 87Sr/86Sr showing the 95 % confidence intervals is 0.71036 ± 0.00026 [14]. The averaged measurement value was obtained as 0.71025 ± 0.00002 (n = 26) showing the agreement with the certified value.
In this study, the variations in the isotopic ratio of 87Sr/86Sr were normalized with that of SRM987; this would be expressed as delta-value (δ87/86) in per mill notation as the following equation:
The samples of ITT01 to ITT07 were prepared by the division of one sample. The δ87/86–values of samples ITT01 to ITT07 in Table 4.1 agreed within the error showing the reproducibility of the isotopic ratio measurement including chemical treatment. From the δ87/86–values of samples ITT01 to ITT07, the averaged δ87/86–value of them was obtained to be δ87/86 = −3.14 ± 0.06 ‰.
The results of the isotopic ratio measurements for all samples are summarized in Table 4.1 and shown in Fig. 4.5a. The result of the measurement for the reference material of IAEA-156: Radionuclides in clover [17] is also included. This reference material contains ca. 0.0075 Bq/g in June 2015. The δ87/86-values of the samples of Okuma range from −1.4 to −4.4, while those of Futaba range from −2.5 to −4.2. It is found that these values have significant difference, by comparison with the δ87/86-value of Iitate samples.

Results of 87Sr/86Sr isotopic ratio measurement for plant samples (a), and isotopic ratio of 87Sr/86Sr as a function of 135Cs/137Cs (b). Bark is results of comparison between OKM03 and FTB01. Grass shows results of comparison between FTB35R and FTB35L. Isotopic ratio of 135Cs/137Cs of OKM01, FTB01, FTB35R and ITT07 were reproduced from Ref [11], and used after time correlation on March 11, 2011. Open square and open circle mean analytical results in this study and results of estimation by calculation results with ORIGEN Code [1]
Though the samples OKM03 and FTB01 are bark samples from the plants of the same family, these showed different magnitudes (Fig. 4.5a and Table 4.1). The isotopic ratio of 87Sr/86Sr has received attention as the indicator of the production region of plants and reported the δ87/86–values ranged from −25.0 to 5.5 [18]. As the reason of the difference in the δ87/86–values among samples OKM03 and FTB01, two origins could be considered: the first is the difference in the δ87/86–values of soils of sampling point (as the supply source of Sr) and the second is the difference in the degree of the isotope fractionation during the translocation process (considered as the reason of the difference in the isotopic ratio between the parts of the identical organism). Because of the comparison of the δ87/86–values of the same parts in this case, the difference in the δ87/86–value among samples OKM03 and FTB01 might be caused by the soils in sampling area.
If the difference of δ87/86–values between samples OKM03 and FTB01 originated from a difference of contamination level by the FP of Sr, the isotopic ratio may show a correlation as
According to the relation and the concentrations of Sr; 72 ppm for OKM03 and 24 ppm for FTB03, the amount of the FP of 86Sr contained in the sample OKM03 would be higher than that of FTB01, about 10.3 ng. This is equivalent to ca. 10.5 μg of 90Sr (ca. 5.3 × 107 Bq) according to the averaged isotopic ratio of 90Sr/86Sr of the FP of Sr [1]. 90Sr was not found in the plant samples by TIMS and Cherenkov counting having the detection limit of several ten mBq/g [15], however, suggesting that our samples contain 90Sr < <10 fg and was less than 1 Bq/g.
Sample FTB35R is roots, while FTB35L is leaves, of the same plant. The δ87/86-values (Fig. 4.5a and Table 4.1) showed a significant difference. Sample FTB35R shows higher δ87/86-value compared with sample FTB35L. The isotopic fractionations were observed in some biological processes. For example, the isotopic analysis of Sr [19], Fe [20], and Zn [21] proves that roots are isotopically heavy compared with the aerial parts; the maximum δ87/86-value was ca. −5.0 for Sr, the maximum δ56/54-value was ca. −1.4 for Fe, and the maximum δ66/64-value was ca. −0.26 for Zn, respectively. Since the Cherenkov counting showed the amounts of 90Sr in these samples were under the detection limit, the difference in the δ87/86-value between samples FTB35R and FTB35L might be caused by the isotopic fractionations in the biological processes along with the contamination of sample FTB35R by the soil.
The isotopic ratios of radioactive Cs in samples FTB01, OKM01, FTB35R, and ITT07 measured by TIMS have been reported in our previous study [11], and that in OKM02 was measured in this study. The relationships between the isotopic ratio of 87Sr/86Sr as δ87/86-value of these samples and that of 135Cs/137Cs are plotted in Fig. 4.5b. The isotopic ratios of 135Cs/137Cs show the significant difference from the reported values of the global fallout (ca. 0.5 for Chernobyl accident and ca. 2.7 for nuclear weapon testing corrected on March 11, 2011 [11]), while these values agreed with the estimated values with the results of ORIGEN Code calculation [1]. This means that all of the samples are contaminated by radioactive Cs released from FDNPP. The δ87/86-values of these samples, on the other hand, are far from that of the FP calculated by ORIGEN Code [1]. This suggests that the amount of deposit of 90Sr is very little compared with that of Cs and agrees with our previous report [15].
Although 90Sr was not found in the plant samples suggesting that our samples contain 90Sr < < 10 fg, typical mass spectrometers have the external analytical precision of ppm level. Assumed that this precision could be applied for the isotopic ratio of 90Sr/stablesSr, the isotopic ratio of 90Sr/stableSr must be higher than 10−6. For the natural sample, since the Sr concentration ranges from ppb level to several hundred ppm level (Fig. 4.6), the detectable lower limit of the isotopic ratio of 90Sr/stableSr can be evaluated.

Detectable lower limits of 90Sr in environmental samples with TIMS. Solid line indicates a limit for 90Sr/84Sr, and broken line a limit for 90Sr/88Sr
If 88Sr having the natural abundance ca. 82 % was used as reference isotope, the concentration of 90Sr should be higher than 1 Bq/g in almost any type of sample. When the isotopic ratio of 90Sr/84Sr is used, because the abundance of 84Sr (ca. 0.56 %) is lower than that of 88Sr, the applicable range will become much wider than the case of 88Sr (Fig. 4.5). The improvement in the sensitivity of 90Sr detection and the obtaining of samples including small amounts of natural Sr will also bring wide applicable range.
4 Conclusions
Cs and Sr recovered from samples were analyzed by TIMS to study the applicability of TIMS for safety assessment and source analysis.
For the study of the recovery/analysis method of Cs and Sr, Cs and Sr were recovered from the natural uranium irradiated at KUR. From the measurement of radionuclide recovered from irradiated UO2, it was concluded that several tens of femtogram level of radionuclide is detectable.
Cs and Sr were recovered from the environmental samples obtained from Fukushima prefecture and were analyzed by a method based on the results of irradiated UO2. In the case of the analysis of Cs, it was confirmed that the analysis of the radioactive Cs by TIMS would provide important information for the source analysis. The isotopic ratio of 135Cs/137Cs was useful for the precise evaluation of the radioactive Cs from FDNPP apart from that of global fallout after the radioactivity of 134Cs became below the detection limit of γ-ray measurement.
In the case of the analysis of Sr, on the other hand, the presence of 90Sr was not detected in any samples, while the changes in the isotopic ratios of 87Sr/86Sr were observed. From the discussion for the amount of the FP of Sr, it was conjectured that the changes in the isotopic ratios of 87Sr/86Sr might be brought by some isotopic fractionation in the biological processes. The evaluation of the detectable lower limit of the isotopic ratio of 90Sr/stableSr suggests that the isotopic ratio of 90Sr/84Sr is the most suitable index to judge a source of radioactive Sr released during the accident of FDNPP by TIMS.
References
Nishihara K, Iwamoto H, Suyama K (2012) JAEA-data/code 2012–018 [in Japanese]
Karam LR, Pibida L, McMahon CA (2002) Appl Rad Isot 56:369–374
Pibida L, MacMahon CA, Busharw BA (2004) Appl Rad Isot 60:567–570
Taylor VF, Evans RD, Cornett RJ (2008) J Environ Radact 99:109–118
Delmore JE, Snyder DC, Tranter T, Mann NR (2011) J Environ Radact 102:1008–1011
Snyder DC, Delmore JE, Tranter T, Mann NR, Abbott ML, Olson JE (2012) J Environ Radact 110:46–52
Berglund M, Wieser ME (2011) Pure Appl Chem 83:397–410
Ludwig SB, Renier JP (1989) Standard- and extended-Burnup PWR and BWR reactor models for the ORIGEN2 Computer Code. Oak Ridge National Laboratory, ORNL/TM-11018
Shibahara Y, Kubota T, Fujii T, Fukutani S, Ohta T, Takamiya K, Okumura R, Mizuno S, Yamana H (2015) J Radioanal Nucl Chem 303:1421–1424
Lee MH, Park JH, Oh SY, Ahn HJ, Lee CH, Song K, Lee MS (2011) Talanta 86:99–102
Shibahara Y, Kubota T, Fujii T, Fukutani S, Ohta T, Takamiya K, Okumura R, Mizuno S, Yamana H (2014) J Nucl Sci Technol 51:575–579
Yoshikawa M, Nakamura E (1993) J Min Petr Econ Geol 88:548–561
Birck JL (1986) Chem Geol 56:73–83
https://www-s.nist.gov/srmors/view_detail.cfm?srm=987. Accessed 20 Aug 2014
Kubota T, Shibahara Y, Fujii T, Fukutani S, Ohta T, Takamiya K, Okumura R, Mizuno S, Yamana H (2015) J Radioanal Nucl Chem 303:39–46
Faure G, Powell JL (1972) Strontium isotope geology. Springer, New York
Strachnov V, Valkovic V, Dekner R (1991) Report on the intercomparison run IAEA-156: radionuclides in clover. International Atomic Energy Agency, Austria, IAEA/AL/035
Almeida CM, Vasconcelos MTSD (2001) J Anal At Spectrom 16:607–611
de Souza GF, Reynolds BC, Kiczka M, Bourdon B (2010) Geochim Gosmochim Acta 74:2596–2614
Moynier F, Fujii T, Wang K, Foriel J (2013) Compt Rend Geosci 345:230–240
Weiss DJ, Mason TFD, Zhao FJ, Kirk GJD, Coles BJ, Horstwood MSA (2005) New Phytol 165:703–710
Komori M, Shozugawa K, Nogawa N, Matsuo M (2013) Bunseki Kagaku 62:475–483 [in Japanese]
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Shibahara, Y. et al. (2016). Application of Mass Spectrometry for Analysis of Cesium and Strontium in Environmental Samples Obtained in Fukushima Prefecture. In: Takahashi, T. (eds) Radiological Issues for Fukushima’s Revitalized Future. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55848-4_4
Download citation
DOI: https://doi.org/10.1007/978-4-431-55848-4_4
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55847-7
Online ISBN: 978-4-431-55848-4
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)