
Abstract
Progenitor cells, derived from the cardiac neural crest (CNC) and the second heart field (SHF), play key roles in development of the cardiac outflow tract (OFT), and their interaction is essential for establishment of the separate pulmonary and systemic circulation in vertebrates. 22q11.2 deletion syndrome (22q11DS) or Takao syndrome is the most common human chromosomal deletion syndrome that is highly associated with OFT defects. Historically, based on the observations in animal models, OFT defects implicated in the 22q11/Takao syndrome are believed to result primarily from abnormal development of CNC that populate into the conotruncal region of the heart. In the twenty-first century, elegant efforts to model 22q11/Takao syndrome in mice succeeded in the identification of T-box-containing transcription factor, Tbx1, as an etiology of OFT defects in this syndrome. Subsequent investigations of the Tbx1 expression pattern revealed that Tbx1 was surprisingly not detectable in CNC but was expressed in the SHF and provided a new concept of molecular and cellular basis for OFT defects associated with 22q11/Takao syndrome. More recently, it was reported that mutations in the gene encoding the transcription factor GATA6 caused CHD characteristic of OFT defects. Genes encoding the neurovascular guiding molecule semaphorin 3C (SEMA3C) and its receptor plexin A2 (PLXNA2) appear to be regulated directly by GATA6. Elucidation of molecular mechanism involving GATA6, SEMA3C, PLXNA2, and TBX1 in the interaction between the CNC and the SHF would provide new insights into the OFT development.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Cardiac outflow tract (OFT) defects account for approximately 30 % of congenital heart disease (CHD) and usually require an intervention during the first year of life [1, 2]. A variety of OFT defects results from disturbance of the morphogenetic process for the establishment of separated systemic and pulmonary circulation. Despite their clinical importance, the etiology of most OFT defects remains unknown because of the multifactorial nature of the diseases.
Progenitor cells derived from the cardiac neural crest (CNC) and the second heart field (SHF) play key roles in development of the OFT. The SHF cells give rise to the OFT myocardium along with subpulmonary conus, and CNC cells give rise to the OFT septum during development. Defects of these progenitor cells may lead to a variety of OFT defects, including tetralogy of Fallot (TOF), characterized by malalignment of the major vessels with the ventricular chambers; interrupted aortic arch type B (IAA-B), resulting from maldevelopment of the left fourth pharyngeal arch artery; and persistent truncus arteriosus (PTA), resulting from failure of septation of the OFT into the aorta and pulmonary artery [3, 4].
2 The 22q11.2 Deletion Syndrome (Takao Syndrome)
The 22q11.2 deletion syndrome (22q11DS) is the most common genetic cause of a spectrum of OFT defects with an incidence of 1 in 4000–5000 births [5, 6]. Most are sporadic in origin, while 10–20 % of deletions are inherited as an autosomal dominant trait. 22q11DS involves three distinct syndromes, namely, DiGeorge syndrome (DGS; OMIM#188400), velocardiofacial syndrome (VCFS; OMIM#192430), and conotruncal anomaly face syndrome (CAFS; OMIM#217095) which is so-called Takao syndrome. Historically, DGS was originally characterized by CHD, hypoparathyroidism, and immune deficiency reported in 1965 from the field of immunology [7]; VCFS was associated with cleft palate, CHD, a distinct facial appearance, and learning difficulties reported in 1978 from the field of plastic surgery [8]; and CAFS or Takao syndrome was characterized by conotruncal CHD (OFT defects), a distinct facial appearance and hyper-nasal voice reported in 1976 (in Japanese) from the field of pediatric cardiology [9]. In 1993, clinical genetics revealed that these syndromes shared a common heterozygous deletion of 22q11.2 region and thus had overlapping phenotype [10–12].
Although the acronym “CATCH22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia, and 22q11 deletions)” was proposed to encompass these syndromes in 1993 [13], clinical use of this term is restricted today, because (1) the term “CATCH22” has a negative meaning which represents a situation where it is impossible for you to do anything, originally from a novel entitled “Catch-22” by Heller [14]; (2) the term “abnormal facies” represented by “A” is difficult to be accepted by patients and their family; and (3) the clinical spectrum associated with 22q11DS is much wider than was previously recognized as “CATCH” [15].
Approximately 75 % of patients with 22q11 DS have CHD. The type of CHD are characterized as OFT defects including TOF, estimated about 30 %; IAA-B, estimated about 15 %; ventricular septal defect (VSD), estimated about 15 %; PTA, estimated about 10 %; and others, estimated about 5 %. Alternatively, 22q11.2 deletion is present in approximately 60 % of patients with IAA-B, 35 % of patients with PTA, and 15 % of patients with TOF. Specifically, it is detected in 55 % of patients with TOF plus pulmonary atresia and major aortopulmonary collateral arteries (MAPCA) [16–18].
Although CHD are the major cause of mortality in 22q11DS, survivors have an exceptionally high incidence of psychiatric illness, including schizophrenia and bipolar disorder, in adolescents and adults, making del22q11 the most frequent genetic cause of such psychiatric disorders [16, 19, 20]. In our experience of 18 adults with 22q11DS, common school and employment were observed in 11 of 18 cases, and 2 females got married; however, difficulties with social interaction and employment were observed in 7 cases. The main reason of difficulties for social interaction and employment was incomplete repair of CHD in four cases, and all of them had TOF with pulmonary atresia and MAPCA. One case was also diagnosed as schizophrenia. Other three cases had repaired VSD and are away from hospital care. Taken together, lifelong comprehensive evaluation and management of patients with 22q11DS, like as shown in Table 26.1, are required for multisystem disorders [6]. The primary care physician, a pediatric cardiologist in most cases, has an important role in the follow-up for the patients and their families and needs to collaborate with many specialists for the associated abnormalities.
3 Identification of TBX1
Because of the high incidence and association with OFT defects, 22q11DS has attracted attention as a model for investigating the genetic basis for OFT defects [21, 22]. The structures primarily affected in patients with 22q11DS are derivatives of the embryonic pharyngeal arches, or neural crest cells, suggesting that haploinsufficiency of the gene(s) on the 22q11.2 deleted region is essential for pharyngeal arch and/or CNC development [1, 2, 21, 22]. Extensive gene searches have been successful in identifying more than 30 genes in the deleted segment. Although standard positional cloning has failed to demonstrate a role for any of these genes in the syndrome, elegant efforts by several groups to model 22q11DS in mice by creating orthologous chromosomal deletions were successful in revealing the T-box-containing transcription factor, Tbx1, as the etiology of OFT defects associated with 22q11DS [23–26]. Heterozygosity of Tbx1 in mice alone also caused aortic arch defects, while homozygous mutation of Tbx1 in mice resulted in most main clinical presentations of 22q11DS, including OFT defects, abnormal facial features, cleft palate, and hypoplasia of the thymus and parathyroid glands.
4 Expression of TBX1
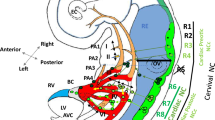
The delineation of the expression pattern of Tbx1 provided a new concept on the molecular and cellular basis of normal and abnormal development of the OFT. We and other group found that Tbx1 was expressed in the SHF but not in the CNC [27–29]. This finding was surprising because CHD associated with 22q11DS had been believed to result primarily from abnormal development of CNC as mentioned above. Interestingly, in mouse and chick embryos, Tbx1 is preferentially expressed in the pharyngeal arches, in the ventral half of the otic vesicle, and in the head (Fig. 26.1) [27, 28]. Within the pharyngeal arch region, Tbx1 is expressed in the pharyngeal mesoderm, including the SHF, the pharyngeal endoderm, and the head mesenchyme. These results suggest that defects of neural crest-derived tissues in 22q11DS may occur in a non-cell autonomous fashion. Our cre-mediated murine transgenic system revealed that Tbx1-expressing descendants representing a subset of cells derived from the SHF contribute predominantly to the pulmonary infundibulum (Fig. 26.1) [30].

Expression patterns of Tbx1. (a–d) RNA in situ hybridizations for whole mount (a–c) and section (b, d) in mouse (a, b) and chick (c, d) embryos demonstrate Tbx1 expression (purple or white signals) in the mesodermal core (arrows) and endodermal epithelium (arrowheads) of pharyngeal arches, head mesenchyme (hm), and otic vesicle (ov). Asterisks indicate pharyngeal arch arteries. (e, f). Transverse sections demonstrate Tbx1-lacZ expression (blue signals) in the myocardial layer of the cardiac outflow tract (oft) but not in the oft cushion (cu) which is mainly contributed by cardiac neural crest cells. (g) Tbx1- descendant cells marked by Tbx1-Cre/Rosa26R mouse system are localized in the anterior portion (oft) of the right ventricle (rv) and the main trunk of the pulmonary artery (mpa). ao Aorta, h head, ht heart, la left atrium, ra right atrium
Although precise embryological mechanisms underlying OFT defects remain uncertain, the anatomical defects in TOF are believed to result from malrotation of the OFT that leads to misalignment of the outlet and trabecular septum and consequent overriding of the aorta above the malaligned ventricular septum [2, 3]. Contribution of CNC is thought to be essential for proper rotation and septation of the OFT. Alternatively, hypoplasia and underdevelopment of the pulmonary infundibulum may also be responsible for the infundibular obstruction and malalignment of the outlet septum [2, 3]. Accordingly, our data suggest that developmental defects of the SHF may cause hypoplasia of the pulmonary infundibulum, resulting in TOF [30]. More severe decreased number or absence of this subset of cells may affect development and/or migration of CNC, resulting in PTA. This hypothetical model is supported by the observation that the OFT defects ranging from TOF to TA are highly associated with 22q11DS (Fig. 26.2).

Cellular interaction of the second heart field (SHF) and cardiac neural crest (CNC) for the outflow tract (OFT) development and diseases. Progenitor cells derived from the SHF and the CNC give rise to the OFT myocardium and septum, respectively. TBX1 is exclusively expressed in the SHF cells. TBX1 deletion in 22q11DS may affect not only the SHF cells but also the interaction between the SHF cells and CNC, resulting in OFT defects ranging from TOF, which is characterized by malalignment of the OFT septum, to PTA, which results from aplasia of the OFT septum. GATA6-SEMA3C (ligand)-PLXNA2 (receptor) pathway also plays a role in interaction between the SHF and CNC during the OFT development (Modified from [36])
5 Mutations of GATA6
Recently, we identified and characterized mutations of GATA6 in our series of Japanese patients with OFT defects [31]. Mutations in GATA6 disrupted its transcriptional activity on downstream target genes involved in the development of the OFT. We also found that the expression of SEMA3C and PLXNA2 was directly regulated during development of the OFT through the consensus GATA binding sites well conserved across species. Mutant GATA6 proteins failed to transactivate SEMA3C and PLXNA2, and mutation of the GATA sites on enhancer elements of Sema3c and Plxna2 abolished their activity, specifically in the OFT/subpulmonary myocardium and CNC derivatives in the OFT region, respectively. These results indicate that mutations of GATA6 are implicated in genetic causes of OFT defects, as a result of the disruption of the direct regulation of semaphorin-plexin signaling (Fig. 26.2).
6 Future Direction: Elucidating the Interaction Between CNC and SHF
Recent studies have demonstrated that reciprocal epithelial-mesenchymal signaling is essential for proper development of the pharyngeal arches and that the primary impairment of epithelial endoderm may secondarily affect migration or differentiation of neural crest cells during the pharyngeal arch development [32–34]. As for the development of the OFT, clear roles of CNC- and SHF-derived cells have been established [35]. Future direction in this research field is to reveal how the CNC and SHF interact using complex reciprocal signaling essential for precise morphogenesis of the OFT. Importantly, mutations in genes expressed in either CNC or SHF can result in similar OFT defects in mice. For example, Pax3 is expressed in the CNC, and Tbx1 is expressed in the SHF, and both Pax3-null mice and Tbx1-null mice show PTA. Studies are, thus, required to focus on the signals that mediate interactions between CNC and SHF in order to uncover the developmental mechanisms underlying various types of the OFT defect. Our result from the research of mutation of GATA6, described above, is an example that revealed such a molecular mechanism. Our recent preliminary data suggest that a molecular cascade involving Gata6, Foxc1/2, Tbx1, Sema3C, and Fgf8 may play roles in reciprocal signaling between SHF and CNC that are essential for the migration of CNC toward the OFT myocardium derived from the SHF (in revision). Further study utilizing our model system may provide new insights into the OFT development and embryogenesis of OFT defects.
References
Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–900.
Thom T, et al. Heart disease and stroke statistics–2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;14:e85–151.
Siwik ES, Patel CR, Zahka KG. Tetralogy of fallot. In: Allen HD, Gutgesell HP, Clark EB, Driscoll DJ, editors. Moss and Adams’ heart disease in infants, children, and adolescents including the fetus and young adult. Philadelphia: Lippincott: Williams and Wilkins; 2001. p. 880–902.
Yamagishi H, Yamagishi C. Embryology. In: Saremi F, editor. Cardiac CT and MR for adult congenital heart disease. New York: Springer; 2014. p. 7–21.
Scambler PJ. The 22q11 deletion syndromes. Hum Mol Genet. 2000;9:2421–6.
Yamagishi H. The 22q11.2 deletion syndrome. Keio J Med. 2002;51:77–88.
DiGeorge AM. Discussion on a new concept of the cellular basis of immunology. J Pediatr. 1965;67:907.
Shprintzen RJ, GoldbergR B, Lewin ML, Sidoti EJ, Berkman MD, Argamaso RV, Young D. A new syndrome involving cleft palate, cardiac anomalies, typical facies, and learning disabilities: velo-cardio-facial syndrome. Cleft Palate J. 1978;15:56–62.
Kinouchi A, Mori K, Ando M, Takao A. Facial appearance of patients with conotruncal anomalies. Shonika (Pediatr Jpn). 1976;17:84 (in Japanese).
Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50:924–33.
Scambler PJ, Kelly D, Lindsay E, Williamson R, Goldberg R, Shprintzen R, Wilson DI, Goodship JA, Cross IE, Burn J. Velo-cardio-facial syndrome associated with chromosome 22 deletions encompassing the DiGeorge locus. Lancet. 1992;339:1138–9.
Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30:822–4.
Wilson DI, Burn J, Scambler P, Goodship J. DiGeorge syndrome: part of CATCH 22. J Med Genet. 1993;30:852–6.
Hellar J. CATCH 22. London: Jonathan Cape; 1962.
Burn J. Closing time for CATCH22. J Med Genet. 1999;36:737–8.
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804.
Goldmuntz E, Clark BJ, Mitchell LE, Jawad AF, Cuneo BF, Reed L, McDonald-McGinn D, Chien P, Feuer J, Zackai EH, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol. 1998;32:492–8.
Maeda J, Yamagishi H, Matsuoka R, Ishihara J, Tokumura M, Fukushima H, Ueda H, Takahashi E, Yoshiba S, Kojima Y. Frequent association of 22q11.2 deletion with tetralogy of Fallot. Am J Med Genet. 2000;92:269–72.
Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J, Gos A, Nestadt G, Wolyniec PS, Lasseter VK, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92:7612–6.
Sugama S, Namihira T, Matsuoka R, Taira N, Eto Y, Maekawa K. Psychiatric inpatients and chromosome deletions within 22q11.2. J Neurol Neurosurg Psychiatry. 1999;67:803–6.
Lindsay EA, Baldini A. Congenital heart defects and 22q11 deletions: which genes count? Mol Med Today. 1998;4:350–7.
Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med. 2003;9:383–9.
Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101.
Lindsay EA. Chromosome microdeletions: dissecting del22q11 syndrome. Nat Rev Genet. 2001;2:858–68.
Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–29.
Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–91.
Garg V, Yamagishi C, Hu T, Kathiriya IS, Yamagishi H, Srivastava D. Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev Biol. 2001;235:62–73.
Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, Meyers EN, Yamagishi C, Srivastava D. Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 2003;17:269–81.
Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–27.
Maeda J, Yamagishi H, McAnally J, Yamagishi C, Srivastava D. Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev Dyn. 2006;235:701–10.
Kodo K, Nishizawa T, Furutani M, Arai S, Yamamura E, Joo K, Takahashi T, Matsuoka R, Yamagishi H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphoring-plexin signaling. Proc Natl Acad Sci U S A. 2009;106:13933–8.
Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–48.
Thomas T, Kurihara H, Yamagishi H, Kurihara Y, Yazaki Y, Olson EN, Srivastava D. A signaling cascade involving endothelin-1, dHAND and msx1 regulates development of neural-crest-derived branchial arch mesenchyme. Development. 1998;125:3005–14.
Wendling O, Dennefeld C, Chambon P, Mark M. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development. 2000;127:1553–62.
Neeb Z, Lajiness JD, Bolanis E, Conway SJ. Cardiac outflow tract anomalies. Dev Biol. 2013;2:499–530.
Yamagishi H. Human genetics of truncus arteriosus. In: Rickert-Sperling S, Kelly RG, Driscoll EJ, editors. Congenital heart diseases: the broken heart. Clinical features, human genetics and molecular pathways. Vienna: Springer; 2016. p. 559–67
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Yamagishi, H. et al. (2016). A History and Interaction of Outflow Progenitor Cells Implicated in “Takao Syndrome”. In: Nakanishi, T., Markwald, R., Baldwin, H., Keller, B., Srivastava, D., Yamagishi, H. (eds) Etiology and Morphogenesis of Congenital Heart Disease. Springer, Tokyo. https://doi.org/10.1007/978-4-431-54628-3_26
Download citation
DOI: https://doi.org/10.1007/978-4-431-54628-3_26
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-54627-6
Online ISBN: 978-4-431-54628-3
eBook Packages: MedicineMedicine (R0)