
Zusammenfassung
Faszination Forschung: Die Evolutionstheorie trägt zur Entwicklung besserer Grippeimpfstoffe bei
Der Erste Weltkrieg endete im November 1918. Die Zahl der Todesfälle in den vier Kriegsjahren wurde jedoch schon bald übertroffen von den Opfern einer massiven Grippeepidemie, an der weltweit über 50 Mio. Menschen starben – und damit mehr als doppelt so viele wie in den Schlachten des Ersten Weltkriegs.
Die Pandemie von 1918/1919 war insofern bemerkenswert, als die Sterberate unter jungen Erwachsenen, die einer Grippe gewöhnlich mit viel geringerer Wahrscheinlichkeit zum Opfer fallen als Kinder und Greise, um das 20-Fache höher lag als bei den vorherigen und später folgenden Grippeepidemien. Warum erwies sich dieses Grippevirus speziell unter den normalerweise widerstandsfähigsten Menschen als so tödlich? Der Virusstamm von 1918 löste im menschlichen Immunsystem eine besonders starke Reaktion aus. Infolge dieser Überreaktion waren Menschen mit einem leistungsfähigen Immunsystem tendenziell stärker betroffen.
You have full access to this open access chapter, Download chapter PDF
Der Erste Weltkrieg endete im November 1918. Die Zahl der Todesfälle in den vier Kriegsjahren wurde jedoch schon bald übertroffen von den Opfern einer massiven Grippeepidemie, an der weltweit über 50 Mio. Menschen starben – und damit mehr als doppelt so viele wie in den Schlachten des Ersten Weltkriegs.
Die Pandemie von 1918/1919 war insofern bemerkenswert, als die Sterberate unter jungen Erwachsenen, die einer Grippe gewöhnlich mit viel geringerer Wahrscheinlichkeit zum Opfer fallen als Kinder und Greise, um das 20-Fache höher lag als bei den vorherigen und später folgenden Grippeepidemien. Warum erwies sich dieses Grippevirus speziell unter den normalerweise widerstandsfähigsten Menschen als so tödlich? Der Virusstamm von 1918 löste im menschlichen Immunsystem eine besonders starke Reaktion aus. Infolge dieser Überreaktion waren Menschen mit einem leistungsfähigen Immunsystem tendenziell stärker betroffen.
In der Regel können wir uns im Kampf gegen Viren schon auf unser Immunsystem verlassen. Die Immunreaktion bildet auch die Grundlage der Impfung. Seit 1945 haben spezielle Impfprogramme gegen Grippeviren dazu beigetragen, die Anzahl und Schwere der Grippefälle in Grenzen zu halten. Allerdings wirkt der Impfstoff eines bestimmten Jahres vermutlich nicht gegen die Viren des folgenden Jahres. Der Grund: Es entwickeln sich ständig neue Stämme von Grippeviren und sorgen für genetische Variabilität in der Population. Würden diese nicht evolvieren, könnten wir eine dauerhafte Resistenz gegen sie aufbauen. Das würde die jährliche Grippeimpfung überflüssig machen. Da die Viren aber evolvieren, müssen Pharmaunternehmen jedes Jahr einen neuen, anderen Grippeimpfstoff entwickeln und in ausreichender Menge bereitstellen.
Die Immunantwort von Wirbeltieren wird ausgelöst, wenn das Immunsystem Proteine auf der Virenoberfläche erkennt. Demnach kann das Virus durch Veränderungen seiner Oberflächenproteine der Immunabwehr entkommen. Je größer die Anzahl der Veränderungen der Oberflächenproteine ist, desto eher werden die Virenstämme vom Immunsystem nicht erkannt, können ihre Wirte infizieren und haben damit einen Vorteil gegenüber anderen Stämmen. Biologen verfolgen, wie sich die Oberflächenproteine der Grippeviren von Jahr zu Jahr ändern. Dadurch beobachten sie die Evolution in Aktion. Mit diesen Erkenntnissen können sie dann wirkungsvollere Impfstoffe entwickeln.
Durch die Erforschung von rasch evolvierenden Organismen hat man sehr viel über die molekularen Grundlagen der Evolution gelernt. Die Ergebnisse molekularer Evolutionsstudien finden wiederum Anwendung in der Praxis, etwa bei der Entwicklung besserer Strategien zur Bekämpfung tödlicher Krankheiten.
Warum erwies sich die Grippepandemie von 1918/19 als schlimmer als alle anderen zuvor und danach?
In „Experiment: Warum war die Grippepandemie von 1918/19 so schlimm?“ in Abschn. 23.4und in „Faszination Forschung“ am Ende dieses Kapitels finden Sie Antworten auf diese Frage.
1 In den DNA-Sequenzen ist die Evolutionsgeschichte der Gene aufgezeichnet
Das Genom eines Organismus oder Virus setzt sich zusammen aus der Gesamtheit aller seiner Gene sowie sämtlichen nichtcodierenden Abschnitten der Erbsubstanz. Bei Eukaryoten finden sich die meisten Gene auf den Chromosomen im Zellkern, es gibt aber auch Gene in den Mitochondrien und Chloroplasten. Bei Organismen mit sexueller Fortpflanzung vererben sowohl die männlichen als auch die weiblichen Individuen Gene der nucleären DNA, hingegen werden Mitochondrien- und Chloroplastengene in der Regel nur über das Cytoplasma der Eizellen weitergegeben.
Auf den Punkt gebracht
-
Durch Sequenzalignments können Biologen bei einzelnen Individuen oder Arten auftretende Nucleotid- oder Aminosäuresubstitutionen nachweisen.
-
Wird alleine die Anzahl der Nucleotidsubstitutionen oder Aminosäureaustausche zwischen Sequenzen ermittelt, führt dies häufig dazu, dass die tatsächliche Zahl der zugrunde liegenden Veränderungen unterschätzt wird.
Damit Genome von Eltern an die Nachkommen weitergegeben werden können, müssen sie zunächst repliziert werden. Die Replikation der DNA verläuft jedoch, wie Sie bereits wissen, nicht ohne Fehler. Fehler bei der DNA-Replikation – Mutationen – liefern einen Großteil des Ausgangsmaterials für evolutionäre Veränderungen. Mutationen sind eine Grundvoraussetzung für das langfristige Überleben von Organismenpopulationen, denn sie bilden die eigentliche Quelle für die genetische Variabilität, die es Populationen ermöglicht, als Reaktion auf Veränderungen ihrer Umwelt zu evolvieren.
Ein bestimmtes Allel eines Gens kann erst dann an nachfolgende Generationen weitergegeben werden, wenn ein Individuum, das dieses Allel besitzt, überlebt und sich fortpflanzt. Das betreffende Allel muss in Kombination mit zahlreichen anderen Genen des Genoms funktionieren, sonst wird es von der Selektion rasch ausgelesen. Darüber hinaus werden das Ausmaß und der Zeitpunkt der Expression eines Gens streng reguliert. Aus diesem Grund kann man die Gene eines einzelnen Organismus als interagierende Mitglieder einer Gruppe betrachten, unter denen Arbeitsteilung herrscht, aber auch starke wechselseitige Abhängigkeiten bestehen.
Ein Genom ist also nicht einfach eine willkürliche Ansammlung von Genen, die auf den Chromosomen in zufälliger Reihenfolge angeordnet sind. Vielmehr handelt es sich dabei um eine komplexe Zusammenstellung miteinander interagierender Gene, Regulationssequenzen und Strukturelemente. Dazwischen liegen Abschnitte aus nichtcodierender DNA, die vermutlich kaum eine direkte Funktion erfüllen. Die Positionen der Gene unterliegen genau wie ihre Abfolge einem evolutionären Wandel; das Gleiche gilt für den Umfang und die Lage der nichtcodierenden DNA-Abschnitte. All diese Veränderungen können sich auf den Phänotyp eines Organismus auswirken.
Mittlerweile haben Biologen die Genome einer großen Zahl von Organismen einschließlich des Menschen vollständig sequenziert. Die in diesen Sequenzen enthaltenen Informationen tragen dazu bei, dass wir heute besser verstehen, wie und warum sich Organismen unterscheiden, wie sie funktionieren und wie sie sich im Verlauf der Evolution entwickelt haben.
1.1 Durch die Evolution von Genomen entsteht biologische Vielfalt
Das Fachgebiet der molekularen Evolutionsforschung beschäftigt sich mit der Erforschung der Mechanismen und Konsequenzen der Evolution von Makromolekülen, insbesondere von Nucleinsäuren (DNA und RNA) und Proteinen. Auf dieses Gebiet spezialisierte Wissenschaftler erforschen, welche Zusammenhänge zwischen der Struktur von Genen und Proteinen und der Funktionsweise von Organismen bestehen. Außerdem analysieren sie die Variabilität auf Molekülebene und rekonstruieren damit die Stammesgeschichte. Auf diesem Gebiet arbeitende Forscher stellen sich Fragen wie: Was sagt die Variabilität auf molekularer Ebene über die Funktion eines Gens aus? Warum haben die Genome verschiedener Organismen eine so unterschiedliche Größe? Welche evolutionären Kräfte formen die Muster der Variabilität zwischen Genomen? Und eine aus evolutionärer Sicht entscheidende Frage ist: Auf welche Weise übernehmen Gene bzw. Proteine neue Funktionen? Eingehende Analysen der Evolution bestimmter Nucleinsäuren und Proteine sind sehr zweckdienlich, wenn man die Evolutionsgeschichte von Genen rekonstruieren möchte. Letztendlich geht es den auf diesem Gebiet tätigen Wissenschaftlern darum, die molekularen Grundlagen der biologischen Vielfalt aufzuklären. (Zu ergänzen ist, dass Beiträge zu diesen Erkenntnissen nicht nur von Spezialisten für molekulare Evolutionsforschung kommen, sondern praktisch aus allen Bereichen der Biologie, denn fast überall werden für bestimmte Fragestellungen Nucleinsäuren und/oder Proteine untersucht und sequenziert.)
Damit Nucleinsäuren und Proteine evolvieren können, muss zunächst durch Mutationen genetische Variabilität entstehen. Für diese Evolution von Genen gibt es mehrere Möglichkeiten, unter anderem beispielsweise Nucleotidsubstitutionen, also der Austausch von Nucleotiden (die Aufnahme von Punktmutationen in Populationen). Nucleotidaustausche in proteincodierenden Genen führen mitunter dazu, dass Aminosäuren durch andere ersetzt werden. Dadurch können sich die Ladung, die Raumstruktur sowie weitere chemische und physikalische Eigenschaften des codierten Proteins ändern. Solche Veränderungen eines Proteinmoleküls wirken sich häufig auf die Funktionsweise dieses Proteins im Organismus aus. (Ein Großteil von Aminosäureaustauschen ist jedoch funktionell neutral, das heißt ohne Auswirkungen auf die Funktion.)
Erkennen kann man evolutionäre Veränderungen in Genen und Proteinen durch einen Vergleich der Nucleotid- und Aminosäuresequenzen verschiedener Organismen. Je länger zwei Sequenzen separat evolviert sind, desto mehr Unterschiede haben sich angesammelt (wobei man jedoch daran denken sollte, dass verschiedene Gene bei der gleichen Organismenart mit unterschiedlicher Geschwindigkeit evolvieren). Herauszufinden, vor wie langer Zeit Veränderungen der Nucleotid- oder Aminosäuresequenzen erfolgt sind, ist ein nützlicher Schritt in Richtung Ursachenermittlung. Die Muster und die Rate evolutionärer Veränderungen bei einem bestimmten Gen oder Protein zu kennen, ist sehr hilfreich bei der Rekonstruktion der Stammesgeschichte von Organismengruppen.
Um Gene oder Proteine verschiedener Organismen miteinander vergleichen zu können, benötigen Biologen eine Möglichkeit, homologe Sequenzabschnitte identifizieren zu können. (Wie Sie in Abschn. 21.1erfahren haben, treten homologe Merkmale bei zwei oder mehr Arten auf und wurden von einem gemeinsamen Vorfahren geerbt.) Homologe Bestandteile eines Proteins kann man an ihren homologen Aminosäuresequenzen erkennen. Da die Aminosäuresequenzen von Nucleotidsequenzen codiert werden, lässt sich das Konzept der Homologie bis hinunter auf die Ebene einzelner Nucleotidpositionen anwenden. Daher besteht einer der ersten Schritte bei der Erforschung der Evolution von Genen und Proteinen darin, homologe Abschnitte der interessierenden Nucleotid- oder Aminosäuresequenz aneinander auszurichten und miteinander zu „alignieren“ – man spricht vom sogenannten Sequenzalignment. Diese Aufgabe übernehmen spezialisierte Computerprogramme.
1.2 Der Vergleich von Genen oder Proteinen erfolgt durch Sequenzalignment
Sobald homologe Nucleotid- oder Aminosäuresequenzen von verschiedenen Organismen bekannt sind, kann man sie miteinander vergleichen. Als Voraussetzung dafür, homologe Positionen oder Abschnitte zu finden, muss man zunächst Deletionen und Insertionen lokalisieren, die in den betreffenden Molekülen seit der Abspaltung der Organismen von einem gemeinsamen Vorfahren stattgefunden haben. Ein einfaches hypothetisches Beispiel soll Ihnen hier die Methode des Sequenzalignments veranschaulichen. Abb. 23.1zeigt den Abgleich von zwei kurzen Aminosäuresequenzen, die aus zwei verschiedenen Organismenarten stammen sollen. Auf den ersten Blick unterscheiden sich die beiden Sequenzen in der Zahl und Identität ihrer Aminosäuren erheblich. Wenn Sie jedoch nach der ersten Aminosäure in Sequenz 2 eine Lücke einfügen (nach der Aminosäure Leucin), treten die Ähnlichkeiten der beiden Sequenzen deutlicher zutage. Diese Lücke soll das Auftreten eines von zwei möglichen Evolutionsereignissen repräsentieren: einer Insertion einer Aminosäure in dem längeren Protein oder einer Deletion einer Aminosäure in dem kürzeren Protein. Da man in diesem Fall nicht wissen kann, welche von beiden Möglichkeiten zutrifft, spricht man von einem Indel. Nachdem Sie diese Korrektur vorgenommen und dadurch das Indel berücksichtigt haben, zeigt sich in diesem paarweisen Sequenzalignment, dass die beiden Sequenzen nur in einer Aminosäure an Position 6 (Serin bzw. Phenylalanin) voneinander abweichen.

Sequenzalignment von Aminosäuresequenzen. Die Methode des Sequenzalignments bietet eine Möglichkeit, die Aminosäuresequenzen von Proteinen so miteinander abzugleichen, dass man bei beiden Sequenzen homologe Abschnitte gut erkennen kann. Durch das Einfügen von Lücken kann man übereinstimmende Abschnitte übereinander ausrichten. Multiple Sequenzalignments (mit mehr als zwei Sequenzen) sind die Regel. Unterschiede (Anzahl der Aminosäureunterschiede plus Indels) und Ähnlichkeiten (Zahl identischer Aminosäuren) zwischen jeweils alignierten Sequenzpaaren werden dann in einer Ähnlichkeitsmatrix zusammengefasst. In ähnlicher Weise kann man auch ein Sequenzalignment homologer DNA-Abschnitte durchführen
Durch das Einfügen nur einer einzigen Lücke – also das Erkennen eines Indels (Deletion oder Insertion) – lassen sich die beiden Sequenzen in Abb. 23.1gut aneinander ausrichten (alignieren). Auf ähnliche Weise kann man dem Alignment nun noch weitere Sequenzen hinzufügen und kommt so zu einem multiplen Sequenzalignment. Bei längeren Sequenzen und solchen, die stärker voneinander divergieren, sind umfangreichere Anpassungen erforderlich. Daher wurden komplexe Modelle entwickelt (integriert in Computeralgorithmen), um den relativen Wahrscheinlichkeiten des Auftretens von Deletionen, Insertionen und bestimmten Aminosäureaustauschen Rechnung zu tragen. Ein wichtiges Ziel ist dabei, die Anzahl der eingeführten Lücken möglichst gering zu halten: Je mehr Lücken man braucht, desto unsicherer wird das Alignment. (Sie könnten nämlich durch Einführen entsprechend vieler Lücken zwei völlig beliebige Sequenzen miteinander alignieren.)
Nach dem Alignment der Sequenzen kann man diese miteinander vergleichen. Dazu wird einfach gezählt, in wie vielen Nucleotiden oder Aminosäuren sie voneinander abweichen. Durch Summierung der Zahl übereinstimmender und unterschiedlicher Positionen für ein jeweiliges Sequenzpaar kann man dann eine Ähnlichkeitsmatrix oder Distanzmatrix erstellen. Damit hat man dann ein Maß für die Mindestzahl an Veränderungen, die seit der Auseinanderentwicklung zweier Organismen aufgetreten sind (Abb. 23.1).
1.3 Mithilfe von Modellen zur Evolution von Sequenzen lässt sich die stammesgeschichtliche Divergenz berechnen
Durch die in Abb. 23.1veranschaulichte Methode des Sequenzvergleichs erhalten Sie einfach nur die Anzahl der Ähnlichkeiten und Unterschiede zwischen den Proteinen zweier Arten. Bei einem Alignment von zwei DNA-Sequenzen können Sie die Zahl der Unterschiede an homologen Nucleotidpositionen zählen; diese Zahl gibt an, wie viele Nucleotidveränderungen mindestens stattgefunden haben müssen, seit die beiden Sequenzen sich von einer gemeinsamen Ausgangssequenz abgespalten haben.
Diese bei einem Sequenzalignment ermittelte Zahl ist zwar durchaus von Nutzen, um die Mindestzahl der Veränderungen zwischen zwei DNA-Sequenzen zu ermitteln, mit ziemlicher Sicherheit wird die Zahl der tatsächlichen Veränderungen seit der Divergenz der beiden Sequenzen dadurch aber erheblich unterschätzt. Jede in einer Ähnlichkeitsmatrix von DNA-Sequenzen gezählte Abweichung kann genauso gut die Folge mehrerer Substitutionsereignisse an einer bestimmten Nucleotidposition im Laufe der Zeit sein. Wie aus Abb. 23.2zu ersehen ist, kann jedes der folgenden Ereignisse an einer bestimmten Nucleotidposition stattgefunden haben; bei einer einfachen Zählung der Übereinstimmungen und Unterschiede zwischen zwei DNA-Sequenzen würde sich dies jedoch nicht zeigen:

Multiple Substitutionen treten bei paarweisen Sequenzalignments nicht zutage. Zwei analysierte DNA-Sequenzen gingen durch eine Reihe von Substitutionen aus einer gemeinsamen Ursprungssequenz (Mitte) hervor. Die beiden analysierten Sequenzen unterscheiden sich zwar nur in drei Nucleotiden (farbig hervorgehobene Buchstaben), doch diese drei Unterschiede gehen auf insgesamt neun Substitutionen zurück (Pfeile)
-
Multiple (mehrfache) Substitutionen: An einer bestimmten Position ist zwischen der Ausgangssequenz und mindestens einer der überprüften Sequenzen mehr als ein Austausch erfolgt.
-
Gleichzeitige Substitutionen: An einer bestimmten Position treten zwischen der Ausgangssequenz und jeder überprüften Sequenz unterschiedliche Substitutionen auf.
-
Parallele Substitutionen: Zwischen der Ausgangssequenz und jeder überprüften Sequenz tritt unabhängig voneinander die gleiche Substitution auf.
-
Rücksubstitutionen (auch als Reversionen bezeichnet): Es handelt sich um eine Variante der multiplen Substitution, bei der nach einer Änderung an einer bestimmten Position durch die darauffolgende Substitution an dieser Position wieder der ursprüngliche Zustand hergestellt wird.
Damit die Zahl der Substitutionen nicht zu niedrig eingeschätzt wird, wurden in der molekularen Evolutionsforschung mathematische Modelle entwickelt, die beschreiben, wie DNA- und Proteinsequenzen evolvieren. Diese Modelle berücksichtigen die relative Veränderungsrate von Nucleotid zu Nucleotid. So treten beispielsweise Transitionen – der Austausch einer Purin- gegen eine Purinbase (\(\mathrm{A}{\leftrightarrow}\mathrm{G}\)) oder einer Pyrimidin- gegen eine Pyrimidinbase (\(\mathrm{C}{\leftrightarrow}\mathrm{T}\)) – häufiger auf als Transversionen – der Austausch einer Purin- gegen eine Pyrimidinbase oder umgekehrt. Diese Modelle enthalten auch Parameter wie die unterschiedlichen Substitutionsraten verschiedener Abschnitte eines Gens und die Anteile der einzelnen Nucleotide in einer bestimmten Sequenz. Nach Abschätzen solcher Parameter kann man mithilfe des Modells Korrekturen vornehmen, die multiple, gleichzeitige und parallele Substitutionen wie auch Rücksubstitutionen berücksichtigen. Diese revidierte Einschätzung gibt also die Gesamtzahl der Substitutionen an, die wahrscheinlich zwischen zwei Sequenzen erfolgt sind; diese ist fast immer größer als die beobachtete Zahl der Unterschiede.
Da für immer mehr Gene und Proteine Sequenzinformationen verfügbar werden und die internationalen Gen- bzw. Proteindatenbanken ständig erweitert werden, kann man das Sequenzalignment auch auf mehrere homologe Sequenzen ausdehnen – man spricht in diesem Fall, wie oben erwähnt, vom multiplen Sequenzalignment (Abb. 23.1). Damit kann man die Mindestzahl an Insertionen, Deletionen und Substitutionen der homologen Gene oder Proteine einer ganzen Organismengruppe ermitteln. Abb. 23.3zeigt die Daten eines Alignments von Proteinsequenzen von Cytochrom \(c\) (einem sehr kleinen Protein) von 33 Tier-, Pflanzen- und Pilzarten. Solche Informationen macht man sich umfassend zunutze, um die evolutionären Verwandtschaftsbeziehungen zwischen Organismengruppen herauszufinden. (Cytochrom \(c\) wurde in der Pionierzeit der Sequenzanalysen wegen seiner geringen Größe, und weil es in den meisten Lebewesen vorkommt, sehr oft für Stammbaumanalysen verwendet.)


Aminosäuresequenzen von Cytochrom c. Die in der Tabelle aufgeführten Aminosäuresequenzen stammen aus Analysen des Enzyms Cytochrom \(c\) von insgesamt 33 Pflanzen-, Pilz- und Tierarten. Beachtenswert ist die fehlende Variabilität der Sequenzen an den Positionen 70–80. Vermutlich unterliegt dieser Abschnitt einer starken negativen (reinigenden) Selektion, und eine Veränderung dieser Aminosäuresequenz würde die Funktion des Proteins stark beeinträchtigen. Die beiden Molekülmodelle oben links wurden anhand von Sequenzen und Röntgenstrukturanalysen erstellt und zeigen die sehr ähnliche dreidimensionale Struktur von Cytochrom \(c\) aus Thunfisch und Reispflanze. Die \(\upalpha\)-Helices des Proteins sind rot dargestellt, die übrige Polypeptidkette dunkelgelb, die Hämgruppe überwiegend grau, das Eisenion orange
1.4 Mit Laborexperimenten lässt sich die molekulare Evolution direkt beobachten
Biologen sind meist an der natürlichen Evolution von Genen und Proteinen interessiert; allerdings kann man molekulare Evolution auch im Labor ablaufen lassen und dabei direkt beobachten. Da Substitutionsraten eher mit der Generationszeit korrelieren als mit der absoluten Zeit, werden für die meisten Evolutionsexperimente Mikroorganismen oder Viren mit kurzer Generationszeit verwendet. Viren, Bakterien und einzellige Eukaryoten (wie Hefepilze) lassen sich in großen Populationen im Labor züchten. Viele dieser Viren bzw. Organismen können rasch evolvieren. Im Falle einiger RNA-Viren kann die natürliche Substitutionsrate mit einer Substitution pro 1000 Nucleotide und Generation recht hoch liegen. Daher sind bei einem aus wenigen Tausend Nucleotiden bestehenden Virus in jeder Generation (im Schnitt) eine oder mehrere Substitutionen zu erwarten. Diese Veränderungen lassen sich durch Sequenzierung des vollständigen Genoms aufgrund seiner geringen Größe problemlos feststellen. Die Generationszeit beträgt bisweilen unter 1 h und nicht Wochen wie bei Mäusen oder Jahrzehnte wie bei Großtieren. Daher können Biologen über einen Zeitraum von Tagen, Wochen oder Monaten in einer kontrollierten Population von Einzellern oder Viren direkt eine beträchtliche molekulare Evolution beobachten. (Wie Sie in Abschn. 20.2erfahren haben, sind auf diese Weise sogar Evolutionsexperimente über Zehntausende von Generationen möglich, die dann allerdings fast ein Jahrzehnt oder länger laufen.)
Molekulare Evolution im Laborexperiment wird zu vielerlei Zwecken durchgeführt und hat Biologen in die Lage versetzt, Konzepte und Prinzipien der Evolution weitaus besser überprüfen zu können. Mittlerweile beobachten Biologen Evolution routinemäßig im Labor und produzieren, wie Sie später in diesem Kapitel noch erfahren werden, mit einer in vitro-Evolution neuartige Nucleinsäuren und Proteine, die für industrielle und pharmazeutische Zwecke genutzt werden können.
23.1 Wiederholung
Die Genome aller Organismen evolvieren im Laufe der Zeit. Evolutionäre Veränderungen lassen sich durch einen Vergleich der Nucleotid- und Aminosäuresequenzen verschiedener Arten nachweisen. Durch Analyse der molekularen Evolution im Laborexperiment unter kontrollierten Bedingungen können Biologen viele Evolutionsprozesse direkt erforschen.
Lernziele
Sie sollten …
-
ein Sequenzalignment durchführen und dazu eine Matrix erstellen können, in der die Ähnlichkeiten und Unterschiede der Sequenzen miteinander vergleichend gegenübergestellt sind.
-
zeigen können, warum anhand der ermittelten Zahl der Nucleotid- oder Aminosäureunterschiede zwischen zwei Sequenzen häufig unterschätzt wird, wie viele Veränderungen tatsächlich zwischen den Sequenzen stattgefunden haben.
?
-
1.
Führen Sie ein Sequenzalignment der folgenden Sequenzen durch und erstellen dann eine Distanzmatrix, in der die Zahl der identischen Nucleotide und der Unterschiede (einschließlich Insertions- und Deletionsereignissen) verglichen werden.
$$\begin{array}[]{l@{\quad}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c@{\;\,}c}\text{Sequenz }a\quad&\text{A}\;\,&\text{A}\;\,&\text{T}\;\,&\text{G}\;\,&\text{C}\;\,&\text{A}\;\,&\text{G}\;\,&\text{G}\;\,&\text{G}\;\,&\text{T}\;\,&\text{A}\;\,&\text{T}\;\,&\text{A}\;\,&\text{C}\;\,&\text{G}\;\,&\\ \text{Sequenz }b\quad&\text{A}\;\,&\text{T}\;\,&\text{T}\;\,&\text{C}\;\,&\text{A}\;\,&\text{G}\;\,&\text{G}\;\,&\text{G}\;\,&\text{T}\;\,&\text{A}\;\,&\text{T}\;\,&\text{A}\;\,&\text{C}\;\,&\text{C}\;\,&\;\,&\\ \text{Sequenz }c\quad&\text{A}\;\,&\text{T}\;\,&\text{T}\;\,&\text{G}\;\,&\text{C}\;\,&\text{A}\;\,&\text{G}\;\,&\text{C}\;\,&\text{G}\;\,&\text{T}\;\,&\text{A}\;\,&\text{T}\;\,&\text{A}\;\,&\text{A}\;\,&\text{C}\;\,&\text{C}\\ \text{Sequenz }d\quad&\text{A}\;\,&\text{T}\;\,&\text{T}\;\,&\text{G}\;\,&\text{C}\;\,&\text{A}\;\,&\text{G}\;\,&\text{G}\;\,&\text{G}\;\,&\text{T}\;\,&\text{A}\;\,&\text{T}\;\,&\text{A}\;\,&\text{C}\;\,&\text{G}\;\,&\\ \quad\end{array}$$ -
2.
Erklären Sie, warum durch einfaches Zählen der abweichenden Nucleotide zweier Sequenzen die tatsächliche Zahl der Nucleotidsubstitutionen seit der Auseinanderentwicklung der Sequenzen häufig zu gering eingeschätzt wird. Verwenden Sie dazu als Beispiel einen Vergleich Ihres Sequenzalignments der Sequenzen \(a\) und \(b\) aus Aufgabe 1.
Wie Sie gesehen haben, befasst sich die molekulare Evolutionsforschung mit der Evolution von Genen und Proteinen, vergleicht Nucleotid- und Aminosäuresequenzen verschiedener Organismen miteinander und rekonstruiert, welche Veränderungen während der Stammesgeschichte stattgefunden haben. Als Nächstes werden Sie erfahren, wie sich Genome verändern, und einige der Folgen dieser Veränderungen näher betrachten.
2 An Genomen lassen sich sowohl neutrale als auch selektive Evolutionsprozesse ablesen
Wie Sie in Abschn. 12.2erfahren haben, versteht man unter einer Mutation jegliche Veränderung des genetischen Materials. Eine Mutationsform, die sich in einer Population etablieren kann, ist die Punktmutation, der Austausch eines einzelnen Nucleotids. Viele solcher Nucleotidsubstitutionen in der DNA haben keine Auswirkung auf ein Protein – selbst dann nicht, wenn die Veränderung an einem proteincodierenden Gen erfolgt, denn für die meisten Aminosäuren gibt es mehr als ein Codon (Abb. Abb. 14.4). Eine Substitution, die nicht zu einer anderen Aminosäure führt, bezeichnet man als synonyme, neutrale oder stille Substitution (Abb. 23.4a). Synonyme Substitutionen wirken sich nicht auf die Struktur und Funktion eines Proteins aus (können allerdings andere Auswirkungen haben, wie in Abschn. 15.1beschrieben) und unterliegen daher wahrscheinlich weniger dem Einfluss der natürlichen Selektion als andere Formen der Substitution.

Wann wirkt sich die Substitution eines Nucleotids aus und wann nicht? a Synonyme (stille) Substitutionen führen nicht zu einer Veränderung der betreffenden Aminosäure und wirken sich daher nicht auf die Proteinfunktion aus; solche Substitutionen unterliegen wahrscheinlich nicht der natürlichen Selektion. b Nichtsynonyme Substitutionen bewirken eine Änderung der Aminosäuresequenz; sie haben dadurch möglicherweise einen (häufig nachteiligen) Einfluss auf die Proteinfunktion. Solche Substitutionen bilden dann Ziele für die natürliche Selektion. Man unterscheidet zwischen Missense-Mutationen, wenn durch den Basenaustausch ein Codon für eine andere Aminosäure entsteht (links und rechts im Bild), und Nonsense-Mutationen, wenn durch den Austausch ein Stoppcodon entsteht, das zum Kettenabbruch führt (Mitte)
Auf den Punkt gebracht
-
Eine neutrale Evolution unterscheidet sich von einer negativen (reinigenden) und positiven Selektion dadurch, dass sie sich nicht auf die Überlebensfähigkeit und Fortpflanzung des betreffenden Organismus auswirkt.
-
Die Fixierungsrate neutraler Nucleotidsubstitutionen innerhalb von Populationen ist unabhängig von der Populationsgröße.
-
Durch Vergleiche der Raten synonymer und nichtsynonymer Substitutionen kann man eine positive und negative Selektion in Proteingenen erkennen.
-
Die Genome von Organismen zeichnen sich durch eine sehr unterschiedliche Größe aus, dagegen ist die Zahl der proteincodierenden Gene deutlich weniger variabel.
Querverweis
Der genetische Code legt fest, welches Codon welche Aminosäure codiert (Abb. Abb. 14.4).
Eine Nucleotidsubstitution, die zu einer Veränderung der von einem Gen codierten Aminosäuresequenz führt, bezeichnet man als nichtsynonyme Substitution (Abb. 23.4b). Bei den solchen Substitutionen unterscheidet man Missense-Substitutionen, die zu einer Änderung der betreffenden Aminosäure führen, und Nonsense-Substitutionen, die in einem Stoppcodon und damit einem Kettenabbruch resultieren. Im Allgemeinen wirken sich nichtsynonyme Substitutionen eher nachteilig für den Organismus aus. Aber nicht jeder Aminosäureaustausch ändert auch die Form und Ladung eines Proteins (und beeinflusst somit seine funktionellen Eigenschaften), sodass nichtsynonyme Substitutionen durchaus auch ganz (oder fast) neutral sein können und daher nicht von der Selektion ausgelesen werden. Umgekehrt wird ein Aminosäureaustausch, der dem Organismus einen Vorteil verschafft, zu einer positiven Selektion für die entsprechende nichtsynonyme Substitution führen.
Für einige hoch konservierte proteincodierende Gene haben Wissenschaftler die Rate nichtsynonymer Nucleotidsubstitutionen ermittelt: Sie liegt bei etwa 0,9 Substitutionen pro Position und Milliarde Jahre. Synonyme Substitutionen sind in diesen Genen etwa fünfmal häufiger erfolgt als nichtsynonyme. Mit anderen Worten, die Substitutionsraten sind an jenen Nucleotidpositionen am höchsten, die zu keiner Veränderung der exprimierten Aminosäure führen (Abb. 23.5). Noch höher ist die Substitutionsrate bei Pseudogenen, also bei durch Duplikation entstandenen Genkopien, die ihre Funktion nicht mehr erfüllen und somit der Selektion nicht unterliegen.

Die Substitutionsraten sind unterschiedlich. Bei nichtsynonymen Substitutionen ist die Mutationsrate viel geringer als bei synonymen Substitutionen und Substitutionen in Pseudogenen. Diese Unterschiede spiegeln das unterschiedliche Ausmaß funktioneller Einschränkungen wider
Die meisten natürlichen Organismenpopulationen weisen mehr genetische Variabilität auf, als zu erwarten wäre, wenn diese Variabilität hauptsächlich durch die natürliche Selektion beeinflusst würde. Diese Feststellung hat zusammen mit der Erkenntnis, dass viele Mutationen die Funktion von Proteinen nicht verändern, zur Entwicklung der Neutraltheorie der molekularen Evolution geführt.
2.1 Evolution verläuft größtenteils neutral
Im Jahr 1968 formulierte der Japaner Motoo Kimura die Neutraltheorie der molekularen Evolution. Nach Kimuras mittlerweile als Theorie etablierten Hypothese ist auf molekularer Ebene die überwiegende Zahl der in den meisten Populationen beobachteten Varianten hinsichtlich der Selektion neutral – das heißt, sie sind für ihre Träger weder vorteilhaft noch nachteilig. Deshalb sammeln sich diese neutralen Varianten eher durch Gendrift an als durch positive Selektion.
Die Fixierungsrate neutraler Mutation durch Gendrift ist unabhängig von der Populationsgröße. Um zu verdeutlichen, warum dies so ist, dient hier eine Population der Größe \(N\) mit einer Rate neutraler Mutationen an einem Genort von \(\mu\) pro Gamet und Generation. Die Zahl der Neumutationen würde in diesem Fall im Schnitt 2 \(N\mu\) betragen, weil \(2N\) Genkopien für Mutationen zur Verfügung stehen. Die Wahrscheinlichkeit, dass eine bestimmte Mutation allein durch Gendrift fixiert wird, ist gleich ihrer Frequenz; diese ist für eine neu entstandene Mutation gleich \(1/(2N)\). Durch Multiplizieren dieser beiden Terme erhalten Sie dann die Fixierungsrate neutraler Mutationen in einer gegebenen Population von \(N\) Individuen:
Somit hängt die Fixierungsrate neutraler Mutationen \(m\) lediglich von der Rate der neutralen Mutationen \(\mu\) ab und ist unabhängig von der Populationsgröße. Eine bestimmte Mutation taucht eher in einer großen als in einer kleinen Population auf, aber in einer kleinen Population wird jede auftauchende Mutation mit höherer Wahrscheinlichkeit fixiert. Diese beiden Auswirkungen der Populationsgröße gleichen einander aus. Somit ist die Fixierungsrate neutraler Mutationen gleich der Mutationsrate (d. h. \(m=\mu\)).
Solange die zugrunde liegende Mutationsrate konstant ist, sollten sich demnach in verschiedenen Populationen entstehende Makromoleküle durch neutrale Veränderungen mit konstanter Rate auseinanderentwickeln. Wie Forschungen bestätigt haben, bleibt die Evolutionsrate bestimmter Gene und Proteine im Laufe der Zeit tatsächlich relativ konstant und kann daher als „molekulare Uhr“ dienen. Wie Sie in Abschn. 21.3erfahren haben, kann man mithilfe von molekularen Uhren berechnen, zu welchem Zeitpunkt sich Aufspaltungen von Arten ereignet haben.
Auch wenn ein Großteil der in Populationen beobachteten genetischen Variabilität das Ergebnis einer neutralen Evolution ist, besagt die Neutraltheorie dennoch nicht, dass sich die meisten Mutationen nicht auf den Organismus auswirken. Viele Mutationen werden in Populationen gar nicht bemerkt, weil sie für die Organismen letal oder sehr nachteilig sind und deswegen rasch durch natürliche Selektion aus der Population beseitigt werden. Ähnlich werden Mutationen, die einen Selektionsvorteil bieten, in Populationen meist rasch fixiert und führen daher ebenfalls nicht zu einer Variabilität auf Populationsebene. Dennoch bleiben in sämtlichen Populationen einige Aminosäurepositionen unter negativer (reinigender) Selektion konstant, andere variieren durch neutrale Gendrift, und wieder andere werden infolge einer positiven Selektion für Veränderungen unterschiedlich sein. Wie lassen sich diese Evolutionsprozesse unterscheiden?
Experiment: Konvergente molekulare Evolution
Blick in die Daten: Konvergente molekulare Evolution

2.2 Positive und negative Selektion lassen sich im Genom nachweisen
Wie Sie gerade erfahren haben, kann man bei den Basenaustauschen in einem proteincodierenden Gen zwischen synonymen und nichtsynonymen Substitutionen unterscheiden, je nachdem, ob sie zu einer Veränderung der Aminosäuresequenz des resultierenden Proteins führen oder nicht. Es ist zu erwarten, dass die relative Rate synonymer und nichtsynonymer Substitutionen in Genabschnitten unterschiedlich sein wird, die neutral evolvieren oder einer positiven Selektion für Veränderungen unterliegen; unter negativer Selektion wird sie unverändert bleiben.
-
Wenn es für eine bestimmte Aminosäure in einem Protein mehrere Alternativen gibt (ohne dass sich dadurch die Proteinfunktion ändert), dann wirkt sich ein solcher Austausch auf die biologische Fitness eines Organismus neutral aus. In diesem Fall ist zu erwarten, dass die Raten synonymer und nichtsynonymer Substitutionen in der entsprechenden DNA-Sequenz sehr ähnlich sein werden, sodass das Verhältnis der beiden Raten nahe bei 1 liegt.
-
Unterliegt eine bestimmte Aminosäureposition einer positiven Selektion für Veränderungen, so ist davon auszugehen, dass die Rate nichtsynonymer Substitutionen die Rate synonymer Substitutionen in den entsprechenden DNA-Sequenzen übersteigt.
-
Unterliegt eine bestimmte Aminosäureposition einer negativen Selektion (oder reinigenden Selektion), dann wird die beobachtete Rate synonymer Substitutionen sehr viel höher sein als die Rate nichtsynonymer Substitutionen in den entsprechenden DNA-Sequenzen.
Durch einen Vergleich der Gensequenzen, die homologe Proteine vieler Arten codieren, können Wissenschaftler die Entstehungsgeschichte und den Zeitpunkt synonymer und nichtsynonymer Substitutionen nachvollziehen. Diese Informationen können dann, wie in Kap. 21erläutert, in einen evolutionären Stammbaum (phylogenetischen Baum) eingetragen werden. Identifizieren kann man Genabschnitte, die sich unter neutraler, negativer oder positiver Selektion entwickeln, indem man die Form der Substitutionen und die Substitutionsraten in einem solchen Stammbaum vergleicht.
Durch die Erforschung der Genomsequenzen synonymer und nichtsynonymer Substitutionen haben Biologen herausgefunden, welche Aminosäurepositionen der Oberflächenproteine von Influenzaviren unter positiver Selektion evolvieren (und damit der Immunerkennung ihrer Wirtsorganismen entgehen). Mittels dieser Informationen können Wissenschaftler herausfinden, welche der Viren gegenwärtiger Grippeepidemien am ehesten nicht vom menschlichen Immunsystem erkannt werden. Diese Viren werden mit ziemlicher Wahrscheinlichkeit die nächste Grippeepidemie auslösen und stellen somit die besten Kandidaten für die Produktion von Impfstoffen dar. Wie in der Einleitung zu diesem Kapitel („Faszination Forschung: Die Evolutionstheorie trägt zur Entwicklung besserer Grippeimpfstoffe bei“) erläutert, konnten die Auswirkungen der Grippeepidemien in den letzten Jahrzehnten durch die Herstellung wirksamer Grippeimpfstoffe erheblich reduziert werden.
An einer Studie zur Evolution von Lysozym lässt sich gut veranschaulichen, wie und warum bestimmte Aminosäurepositionen verschiedenen Formen der Selektion unterliegen („Experiment: Konvergente molekulare Evolution“). Das Enzym Lysozym (Abb. Abb. 3.9) kommt bei fast allen Tieren vor. Es ist Bestandteil der Tränenflüssigkeit, des Speichels und der Milch von Säugetieren sowie des Eiklars (Albumen) von Vogeleiern. Lysozym kann bestimmte Bestandteile der Bakterienzellwand spalten und so die Bakterien schädigen und abtöten. Infolgedessen spielt es eine wichtige Rolle als erste Abwehrlinie gegen bakterielle Infektionen. Die meisten Tiere bekämpfen Bakterien, indem sie diese verdauen; das ist auch der Grund, weshalb die meisten Tiere Lysozym als Abwehrprotein besitzen. Manche Tiere nutzen Lysozym jedoch auch zur Verdauung ihrer Nahrung.
Innerhalb der Säugetiere hat sich zweimal eine Form der Verdauung entwickelt, bei der die Fermentation der Nahrung in einer Gärkammer im vorderen Bereich des Verdauungstrakts erfolgt. Bei diesem Verdauungsmodus wurden Teile des vorderen Verdauungskanals – der hintere Abschnitt der Speiseröhre oder der Magen – in eine Gärkammer umgewandelt, in der Bakterien das aufgenommene Pflanzenmaterial durch Gärung zersetzen. Tiere mit einer solchen Gärkammer können auch die ansonsten unverdauliche Cellulose, aus der pflanzliche Gewebe zu großen Teilen bestehen, noch verwerten (indem Bakterien die Cellulose zu Glucose abbauen und dadurch wachsen und die Tiere dann den Überschuss an Bakterien verdauen). Diese Form der Verdauung mit einer Gärkammer entstand unabhängig voneinander bei Wiederkäuern (einer Gruppe von Huftieren, zu der unter anderem Rinder gehören) und bei bestimmten blattfressenden Affen wie Languren. Es war klar, dass diese Entwicklungen unabhängig voneinander erfolgten, denn sowohl Languren als auch Wiederkäuer haben relativ nahe Verwandte, die keine solchen Gärkammern zur Verdauung besitzen.
In beiden Entwicklungslinien mit einer Gärkammer zur Verdauung wurde Lysozym so modifiziert, dass es nicht mehr nur der Abwehr dient und ihm nun eine neue Rolle zukommt. Dieses Lysozym zerstört die Zellwand eines Teils der Bakterien, die in der Gärkammer leben, und setzt dabei deren Nährstoffe frei, welche die Säugetiere dann verdauen. Wie viele Veränderungen am Lysozymmolekül waren erforderlich, damit es inmitten der Verdauungsenzyme und der sauren Bedingungen in der Gärkammer dieser Säugetiere seine neue Funktion erfüllen konnte? Um diese Frage zu beantworten, verglichen Stewart und ihre Mitarbeiter die lysozymcodierenden Sequenzen von Säugetieren mit einer Gärkammer mit denen einiger ihrer Verwandten, die keine solche Kammer besitzen. Dabei ermittelten sie, welche Aminosäuren bei den jeweiligen Arten unterschiedlich und welche ihnen gemeinsam sind, sowie die Raten synonymer und nichtsynonymer Substitutionen in den Lysozymgenen im Verlauf der Stammesgeschichte der untersuchten Arten.
Für viele der Aminosäurepositionen von Lysozym lag die Rate der synonymen Substitutionen in der entsprechenden Gensequenz deutlich höher als die Rate der nichtsynonymen Substitutionen. Diese Beobachtung lässt darauf schließen, dass viele Aminosäuren, aus denen Lysozym aufgebaut ist, unter Einwirkung von negativer Selektion evolvieren. Mit anderen Worten, es erfolgt eine Selektion gegen eine Veränderung des Proteins an diesen Positionen. Daher müssen die betreffenden Aminosäuren entscheidend für die Funktion von Lysozym sein. An anderen Positionen funktionieren mehrere unterschiedliche Aminosäuren gleich gut; die entsprechenden Gensequenzen zeichneten sich durch ähnliche Raten an synonymen und nichtsynonymen Substitutionen aus. Am verblüffendsten war jedoch die Beobachtung, dass in der zu den Languren führenden Linie mit viel höherer Rate Aminosäureaustausche im Lysozym stattgefunden hatten als in allen anderen Primatenlinien. Die hohe Rate an nichtsynonymen Substitutionen im Lysozymgen von Languren zeigt, dass Lysozym bei der Anpassung an den speziellen Verdauungstrakt der Languren eine Periode rascher Veränderungen durchlief. Darüber hinaus sind die Lysozyme von Languren und Rindern jeweils durch fünf gemeinsame, einzigartige Aminosäureaustausche gekennzeichnet. All diese Aminosäuren befinden sich an der Oberfläche des Lysozymmoleküls, weit entfernt von dessen aktivem Zentrum. Bei zwei dieser gemeinsamen Austausche ersetzte jeweils Lysin ein Arginin. Dies macht die Proteine widerstandsfähiger gegen Angriffe des Magenenzyms Pepsin. Indem sie die funktionelle Bedeutung der Aminosäureaustausche nachvollziehen, können Wissenschaftler die beobachteten Veränderungen der Aminosäuresequenzen mit der erfolgten Funktionsänderung des Proteins erklären.
Zahlreiche Fossilbelege, morphologische Befunde und molekulare Daten zeigen, dass Languren und Wiederkäuer in jüngerer Zeit keinen gemeinsamen Vorfahren hatten. Doch die Lysozyme von Languren und Wiederkäuern haben mehrere Aminosäuren gemeinsam, die jeweils bei keinem der näheren Verwandten dieser beiden Säugetiere vorkommen. Trotz der unterschiedlichen Herkunft der beiden Säugetiergruppen haben ihre Lysozyme an einigen Aminosäurepositionen eine konvergente Evolution durchlaufen. Die beiden Tiergruppen gemeinsamen Aminosäuren verleihen diesen speziellen Lysozymen die Fähigkeit, auch unter den in der Gärkammer herrschenden Umweltbedingungen Bakterien abzubauen.
Ein weiteres bemerkenswertes Beispiel für die konvergente Evolution von Lysozym liefern die auch als Schopfhühner bezeichneten Hoatzins. Diese ungewöhnlichen blattfressenden Vögel aus Südamerika sind die einzigen bekannten Vögel mit einer Gärkammer zur Verdauung. Zwar besitzen viele Vögel eine als Kropf bezeichnete Aussackung der Speiseröhre, doch nur bei Hoatzins enthält der Kropf Lysozym und Bakterien und fungiert als Gärkammer. Viele Aminosäureaustausche, die im Laufe der Anpassung des Lysozyms im Kropf der Hoatzins auftraten, sind mit jenen identisch, die bei Wiederkäuern und Languren entstanden. Obwohl also die Hoatzins und die Säugetiere mit Gärkammer in den letzten paar Hundert Millionen Jahren keinen gemeinsamen Vorfahren hatten, haben sie ähnliche Anpassungen ihres Lysozyms entwickelt, die es ihnen ermöglichen, ihren der Gärung dienenden Bakterien Nährstoffe zu entziehen.
2.3 Die Größe des Genoms evolviert ebenfalls
In der Größe der Genome gibt es bei verschiedenen Organismen bekanntlich erhebliche Unterschiede. Betrachtet man die großen taxonomischen Kategorien, so ist eine gewisse Korrelation zwischen Genomgröße und Komplexität der Organismen zu erkennen. Das Genom des winzigen Bakteriums Mycoplasma genitalium umfasst nur 470 Gene. Aus 634 Genen besteht das Genom des Bakteriums Rickettsia prowazekii, dem Erreger des Fleckfiebers. Hingegen besitzt Homo sapiens ungefähr 21.000 proteincodierende Gene. Abb. 23.6zeigt die Zahl der Gene einer Auswahl von Organismen, deren Genome bereits vollständig sequenziert wurden, angeordnet nach ihren bekannten evolutionären Verwandtschaftsbeziehungen. Wie Ihnen die Abbildung zeigt, bedeutet ein größeres Genom nicht eine höhere Komplexität des Phänotyps. (Vergleichen Sie beispielsweise den Reis mit den anderen Pflanzen.) Es überrascht nicht, dass für den Bau und die Aufrechterhaltung der Funktionen eines großen, vielzelligen Organismus mehr und komplexere genetische Informationen erforderlich sind als bei einem kleinen, einzelligen Bakterium. Überraschend ist jedoch, dass einige Organismen, etwa Lungenfische, manche Schwanzlurche und Lilien, rund 40-mal mehr DNA im Zellkern aufweisen als beispielsweise der Mensch. Natürlich ist ein Lungenfisch oder eine Lilie nicht 40-mal komplexer aufgebaut als ein Mensch. Warum variiert die Genomgröße dann so stark?

In der Anzahl der Gene pro Genom gibt es enorme Unterschiede. Diese Abbildung zeigt die Zahl der Gene einer Auswahl von Organismen, deren Genome schon vollständig sequenziert wurden, angeordnet nach ihren bekannten evolutionären Verwandtschaftsbeziehungen. Bakterien und Archaeen weisen im Normalfall weniger Gene auf als die meisten Eukaryoten. Unter den Eukaryoten besitzen vielzellige Organismen mit organisierten Geweben (Pflanzen und Tiere; die dunkelgrünen und blauen Zweige) mehr Gene als einzellige Organismen (türkisfarbene Zweige) oder vielzellige Lebewesen ohne eine auffällige Organisation der Gewebe (gelbe Zweige)
Die Unterschiede in der Genomgröße sind nicht so groß, wenn man nur jenen Anteil der DNA betrachtet, der tatsächlich Proteine codiert oder Sequenzen anderer RNAs als mRNAs festlegt. Die Organismen mit der größten Menge an Kern-DNA (einige Farne und Blütenpflanzen) weisen zwar 80.000-mal so viel DNA auf wie die Bakterien mit den kleinsten Genomen, aber keine der Arten besitzt mehr als 100-mal so viele proteincodierende Gene wie ein Bakterium. Daher beruhen die meisten Unterschiede in der Genomgröße nicht auf der Zahl funktioneller Gene, sondern auf der Menge an nichtcodierender DNA (Abb. 23.7).

Ein großer Anteil der DNA ist nichtcodierend. Der überwiegende Teil der DNA von Bakterien und Hefepilzen codiert Proteine oder legt die Sequenzen anderer RNAs als mRNAs fest, bei vielzelligen Arten ist der größte Teil der DNA hingegen nichtcodierend
Warum enthalten die Zellen der meisten eukaryotischen Organismen so viel nichtcodierende DNA? Wie bereits an früherer Stelle erwähnt, hat ein Teil der nichtcodierenden DNA regulierende Funktion und kontrolliert das Ausmaß oder den Zeitpunkt der Expression codierender Gene. Die Genome vieler Arten enthalten aber weitaus mehr nichtcodierende DNA, als für die Genregulation erforderlich ist. Kommt dieser zusätzlichen nichtcodierenden DNA irgendeine Funktion zu, oder ist sie völlig unnütz (früher wurde sie oft auch als Junk-DNA – „DNA-Müll“ – bezeichnet)? Viele Abschnitte nichtcodierender DNA bestehen aus Pseudogenen (das sind funktionslose Kopien ehemaliger Gene), die einfach im Genom verbleiben, weil der Aufwand dafür so verschwindend gering ist. Diese Pseudogene können zum Ausgangsmaterial für die Evolution neuer Gene mit neuen Funktionen werden. Ein Teil der nichtcodierenden DNA trägt ausschließlich dazu bei, die Struktur der Chromosomen aufrechtzuerhalten. Bei anderen Sequenzen handelt es sich um „egoistische“ transponierbare Elemente, die sich stark ausbreiten, weil sie sich schneller reproduzieren als das Wirtsgenom.
Im Laufe der Zeit sammelt sich in Genomen nicht nur DNA an, es gehen auch unwichtige Nucleotidsequenzen aus ihnen verloren. Manche Arten haben auch deswegen so unterschiedlich große Genome, weil die unbedeutenden Sequenzen mit sehr unterschiedlichen Raten beseitigt werden. Anhand von Retrotransposons können Wissenschaftler abschätzen, mit welcher Rate Arten DNA einbüßen. Retrotransposons sind transponierbare Elemente (Abb. Abb. 17.4), die sich über ein RNA-Zwischenstadium selbst kopieren. Die häufigste Form von Retrotransposons trägt an jedem Ende duplizierte Sequenzen, die man als lange terminale Sequenzwiederholungen (long terminal repeats, LTRs) bezeichnet. Gelegentlich kommt es zu einer Rekombination der LTRs im Wirtsgenom in der Form, dass die dazwischenliegende DNA herausgeschnitten wird. Wenn das geschieht, bleibt ein rekombiniertes LTR zurück. Die Zahl solcher „verwaisten“ LTRs in einem Genom ist ein Maß dafür, wie viele Retrotransposons verloren gegangen sind. Durch einen Vergleich der Zahl der LTRs im Genom hawaiianischer Grillen der Gattung Laupala und in jenem von Taufliegen (Drosophila) fanden Forscher heraus, dass Laupala 40-mal langsamer DNA verliert als Drosophila. Daher überrascht es nicht, dass das Genom von Laupala sehr viel größer ist als das von Drosophila.
Warum gibt es bei verschiedenen Arten so große Unterschiede in der Rate, mit der scheinbar funktionslose DNA-Abschnitte hinzukommen oder verloren gehen? Einer Hypothese zufolge steht die Genomgröße mit der Geschwindigkeit der Individualentwicklung (Ontogenie) eines Organismus in Zusammenhang, die einem Selektionsdruck unterliegen kann. Große Genome können die Entwicklungsrate verlangsamen und dadurch den relativen Zeitpunkt der Expression bestimmter Gene verändern. Wie in Abschn. 19.4erläutert, können Verschiebungen des Zeitpunkts der Genexpression – Heterochronie – bedeutende Veränderungen des Phänotyps nach sich ziehen. Auch wenn manche nichtcodierenden DNA-Sequenzen also vielleicht keine direkte Funktion haben, können sie die Individualentwicklung des Organismus demnach trotzdem beeinflussen.
Eine andere Hypothese besagt, dass der Anteil nichtcodierender DNA in erster Linie mit der Populationsgröße korreliert. Nichtcodierende, für den Organismus nur in geringem Maße nachteilige Sequenzen werden wahrscheinlich bei Arten mit großer Populationsgröße effektiver durch Selektion eliminiert. Bei Arten mit kleinen Populationen können die Auswirkungen der Gendrift stärker sein als die Selektion gegen nichtcodierende Sequenzen, die geringfügig nachteilige Folgen haben. Deshalb erfolgt die Selektion gegen die Anhäufung nichtcodierender Sequenzen am effizientesten bei Arten mit großen Populationen. Daher besitzen solche Arten (wie Bakterien oder Hefepilze) im Vergleich zu Arten mit kleinen Populationen relativ wenig nichtcodierende DNA (Abb. 23.8).
23.2 Wiederholung
Die Neutraltheorie der molekularen Evolution liefert eine Erklärung für die relativ konstante Rate molekularer Veränderungen, wie man sie bei vielen Genen und Proteinen beobachtet. Durch eine Analyse der relativen Raten synonymer und nichtsynonymer Substitutionen von Genen im Laufe der Zeit können Biologen unterscheiden, welche Evolutionsmechanismen auf einzelne Gene einwirken. Der nichtcodierende Anteil des Genoms von Eukaryoten ist bei verschiedenen Arten viel variabler als der codierende Anteil.
Lernziele
Sie sollten …
-
erläutern können, warum Substitutionen bei einem bestimmten Organismus neutral oder selektiv sein können.
-
eine mathematische Erklärung dafür liefern können, warum die Fixierungsrate neutraler Mutationen unabhängig von der Populationsgröße ist.
-
Gensequenzen von proteincodierenden Genen beurteilen und erkennen können, welche Codons unter negativer bzw. positiver Selektion evolvieren; dieses Wissen sollten Sie auf ein biologisches Problem anwenden können.
-
die Evolution von Genomen unter Berücksichtigung der nichtcodierenden DNA beschreiben können.
?
-
1.
Im Laufe der Entwicklungsgeschichte haben viele Gruppen von höhlenbewohnenden Organismen ihre Lichtsinnesorgane eingebüßt. So besitzen beispielsweise nicht in Höhlen lebende Krebsarten funktionelle Augen, während manche Arten, deren Vorkommen auf unterirdische Habitate beschränkt ist, keine Augen haben. Opsine sind lichtempfindliche Proteine, die nachweislich eine wichtige Funktion für das Sehen erfüllen (Kap. 45). Die Opsingene werden in Augengewebe exprimiert. Sie sind aber auch im Genom der augenlosen, höhlenbewohnenden Krebse vorhanden. Das Vorhandensein von Opsingenen bei augenlosen Organismen könnte sich durch zwei Hypothesen erklären lassen: 1. Die Gene wurden von einem Vorfahren geerbt, der Augen besaß, erfüllen nun aber keine Funktion mehr. 2. Die Gene haben eine neue Funktion, die nichts mit dem Sehvermögen zu tun hat. Wie würden Sie diese beiden Hypothesen anhand der Sequenzen der Opsingene verschiedener Krebsarten erforschen?
-
2.
Warum ist die Fixierungsrate neutraler Mutationen unabhängig von der Populationsgröße?
-
3.
Ein Wissenschaftler verglich zahlreiche Sequenzen von Genen, die Oberflächenproteine von Influenzaviren codieren. Die folgende Tabelle zeigt die Ergebnisse.
Codonposition
synonyme
Substitutionen
nichtsynonyme
Substitutionen
12
0
7
15
1
9
61
0
12
80
7
0
137
12
1
156
24
2
165
3
4
226
38
3
-
a.
Welche Positionen codieren Aminosäuren, die sich vermutlich infolge einer positiven Selektion verändert haben? Erläutern Sie Ihre Antwort.
-
b.
Welche Positionen codieren Aminosäuren, die sich vermutlich infolge einer negativen Selektion verändert haben? Erläutern Sie Ihre Antwort.
(Hinweis: Zur Berechnung der Rate der beiden Formen von Substitutionen betrachten Sie die Anzahl der vorhandenen synonymen und nichtsynonymen Substitutionen relativ zur Zahl der möglichen Substitutionen jedes Typs. Es erfolgen ungefähr dreimal mehr mögliche nichtsynonyme Substitutionen als synonyme Substitutionen.)
-
a.
-
4.
Schlagen Sie zwei Hypothesen für die enorme Vielfalt an Genomgrößen verschiedener Organismen vor und stellen diese einander gegenüber.
Sie haben nun einige der Möglichkeiten kennengelernt, wie Organismen DNA verlieren können, ohne dass dadurch die Genfunktionen beeinträchtigt sind. Wie aber entwickeln sich bei Organismen im Laufe der Zeit neue Funktionen?
3 Horizontaler Gentransfer und Genduplikationen können große Veränderungen nach sich ziehen
Wie im vorherigen Abschnitt erwähnt, besitzen die meisten vielzelligen Organismen sehr viel mehr Gene als der größte Teil der einzelligen Arten. Aber die vielzelligen Lebewesen sind aus einzelligen Vorfahren hervorgegangen. Wie kam es zu der Zunahme der Anzahl der Gene innerhalb des Genoms von vielzelligen Organismen im Laufe der Evolution? Eine solche Zunahme kann vor allem durch zwei Mechanismen zustande kommen: Es können Gene von anderen Arten übertragen oder innerhalb einer Art dupliziert werden.
Auf den Punkt gebracht
-
Bisweilen können Gene zwischen entfernt miteinander verwandten Zweigen am Stammbaum des Lebens ausgetauscht werden.
-
Die Duplikation eines Gens bietet Gelegenheiten für die Evolution neuer Funktionen.
-
Manche Gene liegen im Genom in multiplen Kopien vor; diese evolvieren im Laufe der Zeit häufig gemeinsam.
3.1 Horizontaler Gentransfer kann zum Erwerb neuer Funktionen führen
In Kap. 22wurde beschrieben, wie sich durch den Prozess der Artbildung ursprüngliche Linien in Linien von Abkömmlingen aufspalten. Genau diese Artbildungsereignisse sind in den Zweigen am Stammbaum des Lebens festgehalten. Daneben gibt es aber auch den Vorgang des horizontalen Gentransfers (auch lateraler Gentransfer genannt). Durch diesen können einzelne Gene, Organellen oder Genomfragmente horizontal zwischen zwei Linien übertragen werden (im Gegensatz zum vertikalen Gentransfer von einer Generation zur nächsten). Manche Arten können DNA-Fragmente direkt aus der Umgebung aufnehmen. Andere Gene werden in ein Virengenom eingeschleust und dann auf einen neuen Wirtsorganismus übertragen, wenn das Virus in das Genom des Wirts eingebaut wird. Bei der Hybridisierung von Arten kommt es ebenfalls zu einer Übertragung zahlreicher Gene.
Ein horizontaler Gentransfer kann für eine Art, die neue Gene von einem entfernt verwandten Organismus empfängt, ausgesprochen vorteilhaft sein. So werden beispielsweise zwischen verschiedenen Bakterienarten häufig Gene übertragen, die eine Resistenz gegen Antibiotika verleihen. Zudem ist horizontaler Gentransfer neben Mutation und Rekombination eine weitere Möglichkeit, wie Arten ihre genetische Variabilität erhöhen können. Diese genetische Variabilität bildet dann das Rohmaterial, auf das die Selektion einwirken kann, was letztlich zu einer Evolution führt.
Erstellt man anhand eines einzelnen, horizontal übertragenen Genomfragments einen evolutionären Stammbaum, so spiegelt dieser wahrscheinlich ausschließlich die Herkunft dieses Fragments wider und nicht die gesamte Stammesgeschichte des Organismus (Abschn. 25.1). Die meisten Biologen ziehen es daher vor, solche Stammbäume anhand einer großen Auswahl von Genen oder Proteinen zu erstellen, damit sie den zugrunde liegenden Organismenstammbaum (ebenso wie jegliche Ereignisse horizontalen Gentransfers) rekonstruieren können. Werden horizontale Gentransfers in einen solchen Organismenstammbaum eingetragen, wird aus dem phylogenetischen Baum ein phylogenetisches Netzwerk.
In welchem Umfang horizontaler Gentransfer in der Stammesgeschichte des Lebens eine Rolle gespielt hat, ist derzeit Gegenstand zahlreicher Untersuchungen und Debatten. In den meisten Eukaryotenlinien scheint ein horizontaler Gentransfer relativ selten vorzukommen. Allerdings kann man die beiden bedeutenden Endosymbiosen, aus denen Mitochondrien und Chloroplasten hervorgingen, als horizontalen Transfer kompletter Bakteriengenome in die Eukaryotenlinie auffassen. In einigen Gruppen der Eukaryoten, insbesondere bei manchen Pflanzen, kommt es in ziemlich hohem Ausmaß zu einer Hybridisierung zwischen nahe verwandten Arten. Bei einer solchen Hybridisierung werden zwischen zwei Pflanzenlinien, die sich vor nicht allzu langer Zeit aufgespalten haben, zahlreiche Gene ausgetauscht. Im größten Stil tritt ein horizontaler Gentransfer jedoch offenbar zwischen verschiedenen Bakterien auf. Zahlreiche Bakteriengene wurden mehrfach zwischen verschiedenen Linien übertragen. Das geht so weit, dass es oft ziemlich problematisch ist, die Verwandtschaftsbeziehungen zwischen Bakterienarten zu entschlüsseln. Dennoch lassen sich die groben Beziehungen zwischen den großen Bakteriengruppen ermitteln, wie Sie in Teil VII dieses Buches noch sehen werden. Der horizontale Transfer von Genen erschwert es jedoch, die Grenzen von Bakterienarten festzulegen. Dies ist einer der Gründe dafür, warum bislang nicht alle Bakterienarten, die man entdeckt hat, auch wissenschaftlich benannt wurden.
3.2 Die meisten neuen Funktionen entstehen durch Genduplikation
Eine weitere Möglichkeit, wie Genome neue Funktionen erwerben können, ist die Genduplikation. Wird ein Gen dupliziert, so wird eine der beiden Kopien dieses Gens potenziell von ihrer ehemaligen Funktion entbunden. Den identischen Kopien eines duplizierten Gens kann ein unterschiedliches Schicksal bevorstehen, wobei es die vier folgenden Möglichkeiten gibt:
-
Beide Kopien des Gens können ihre ursprüngliche Funktion beibehalten. (Das kann dazu führen, dass der Organismus größere Mengen des Produkts dieses Gens produziert.)
-
Beide Kopien des Gens können die Fähigkeit behalten, das ursprüngliche Genprodukt zu produzieren, aber die Expression der Gene kann in verschiedenen Geweben oder in verschiedenen Entwicklungsstadien abweichen.
-
Eine Kopie des Gens kann durch Anhäufung nachteiliger Mutationen außer Funktion gesetzt und zu einem funktionslosen Pseudogen oder gänzlich aus dem Genom eliminiert werden.
-
Eine Kopie des Gens kann ihre ursprüngliche Funktion beibehalten, während die zweite Kopie durch Mutation und Selektion eine andere Funktion übernimmt.
Wie oft erfolgen Genduplikationen, und welche dieser vier gerade beschriebenen Möglichkeiten tritt am wahrscheinlichsten ein? Wie Forscher herausfanden, ist die Genduplikationsrate bei einer Hefe- oder Drosophila-Population hoch genug, dass sich im Laufe von 1 Mio. Jahre mehrere Hundert duplizierte Gene ansammeln. Wie sie außerdem feststellten, sind die meisten duplizierten Gene dieser Organismen noch sehr jung. Viele überzählige Gene gehen innerhalb von 10 Mio. Jahren aus einem Genom verloren (das ist im zeitlichen Maßstab der Evolution schnell).
Querverweis
In Abschn. 19.1sind Mechanismen der Individualentwicklung beschrieben, die durch spezifische DNA-Sequenzen kontrolliert werden; diese Sequenzen wurden modifiziert und neu gemischt, und sie haben die bemerkenswerte Diversität an Pflanzen, Tieren und anderen Organismen hervorgebracht, die wir heute kennen. Ähnlichkeiten in den Homöoboxsequenzen der Hox-Gene legen nahe, dass die Hox-Gene durch Duplikation eines Vorläufergens entstanden sind und dann divergierten und neue Funktionen übernahmen.
Manche Gene können viele Male dupliziert werden. Das führt dann zu einer großen Zahl von verwandten Pseudogenen, die im ganzen Genom verstreut sind. Im Genom des Menschen liegt die funktionelle Kopie des Gens RPL21, das ein ribosomales Protein codiert, auf Chromosomenpaar 13, doch auf fast allen anderen Chromosomenpaaren finden sich von ihm abstammende Pseudogene (Abb. 23.8). Nicht alle Gene sind durch Pseudogene repräsentiert, aber man kennt im menschlichen Genom nahezu ebenso viele Pseudogene wie funktionelle proteincodierende Gene.

Manche funktionellen Gene werden viele Male dupliziert und liegen dann als nichtfunktionelle Pseudogene vor. a Das funktionelle Gen, das das ribosomale Protein RPL21 codiert, liegt auf Chromosom 13 des Menschen (orange hervorgehoben). Zusätzlich gibt es noch zahlreiche nichtfunktionelle Pseudogene von RPL21 im menschlichen Genom, die durch wiederholte Duplikationsereignisse entstanden sind (blau hervorgehoben). b Zwar stellt RPL21 ein relativ extremes Beispiel für eine Pseudogenduplikation dar, aber man kennt im Genom des Menschen fast genauso viele Pseudogene wie funktionelle Gene
Zwar gehen viele überzählige Gene rasch wieder verloren, aber manche Duplikationsereignisse führen auch zur Evolution von Genen mit neuen Funktionen. Durch mehrere aufeinanderfolgende Duplikationen und Mutationen kann eine Genfamilie entstehen, eine Gruppe homologer Gene mit verwandten Funktionen, die entlang eines Chromosoms oft im Tandem angeordnet sind. Ein Beispiel für diesen Prozess bietet die Globingenfamilie (Abb. Abb. 17.8). Die Globine gehörten zu den ersten Proteinen, deren Sequenzen ermittelt und verglichen wurden. Vergleiche ihrer Aminosäuresequenzen deuten nachdrücklich darauf hin, dass die verschiedenen Globine durch Genduplikationen entstanden sind und eine multigene Proteinfamilie bilden. Durch solche Vergleiche lässt sich auch abschätzen, wie lange sich die Globine getrennt voneinander entwickelt haben, da sich im Laufe der Zeit Sequenzunterschiede zwischen diesen Proteinen angesammelt haben.
Hämoglobin ist im Tierreich weit verbreitet und für den Sauerstofftransport im Blut zuständig. Das Hämoglobin der Wirbeltiere ist (meist) ein Tetramer aus zwei \(\upalpha\)- und zwei \(\upbeta\)-Globin-Untereinheiten. Myoglobin, ein Monomer, ist das primäre Sauerstoffspeicherprotein in der Muskulatur. Die Sauerstoffaffinität von Myoglobin ist viel höher als die von Hämoglobin, wohingegen sich Hämoglobin so entwickelt hat, dass es seine Funktion auf vielfältigere Weise erfüllen kann. Es bindet den Sauerstoff in den Lungen oder Kiemen, wo der \(\mathrm{O_{2}}\)-Partialdruck relativ hoch ist, transportiert ihn in tiefer liegende Gewebe des Körpers mit niedrigem \(\mathrm{O_{2}}\)-Partialdruck und setzt ihn dort frei. Bei dieser reversiblen Sauerstoffbindung arbeiten die vier Untereinheiten zusammen – sie zeigen Kooperativität. Aufgrund seiner komplizierten tetrameren Struktur ist Hämoglobin in der Lage, neben vier \(\mathrm{O_{2}}\)-Molekülen auch Protonen und Kohlenstoffdioxid im Blut zu transportieren.
Um abschätzen zu können, zu welchem Zeitpunkt es zur Duplikation des ursprünglichen Globingens in \(\upalpha\) und \(\upbeta\) kam, kann man einen Genstammbaum erstellen – einen phylogenetischen Baum, der die Evolutionsgeschichte bestimmter Gene oder Genfamilien veranschaulicht, in diesem Fall der verschiedenen globincodierenden Gensequenzen (Abb. 23.9). Die Rate der molekularen Evolution der Globingene wurde mittels anderer Analysen abgeschätzt. Hierzu hat man die Zeitpunkte der Divergenz bestimmter Wirbeltiergruppen herangezogen, die gut in den Fossilbelegen dokumentiert sind. Diese Untersuchungen deuten auf eine durchschnittliche Divergenzrate der Globingene von etwa einer Nucleotidsubstitution alle 2 Mio. Jahre hin. Wendet man diese Rate auf den Globingenstammbaum an, so lässt sich abschätzen, dass die Aufspaltung in die beiden Globingencluster \(\upalpha\) und \(\upbeta\) vermutlich vor ungefähr 450 Mio. Jahren stattgefunden hat. Dies war etwas nach dem Zeitpunkt des Auftretens der ersten Wirbeltiere in den Fossilfunden.
?
Frage zu Abb. 23.9: Wann erfolgte die Genduplikation, durch die die \(\updelta\)- und \(\upbeta\)-Ketten entstanden sind?

Ein Genstammbaum der Globingenfamilie. Eine Analyse mittels der Methode der molekularen Uhr legt nahe, dass sich die \(\upalpha\)- und \(\upbeta\)-Globingen-Cluster (blau bzw. grün) vor ungefähr 450 Mio. Jahren auseinanderentwickelt haben – also relativ bald nach der Entstehung der Wirbeltiere
Viele Genduplikationen betreffen jeweils nur ein Gen oder wenige Gene, bei polyploiden Organismen (darunter zahlreiche Pflanzen) wurden jedoch komplette Genome dupliziert. Bei einer Duplikation sämtlicher Gene ergeben sich unzählige Möglichkeiten, neue Funktionen zu evolvieren. Genau das ist offenbar bei der Evolution der Wirbeltiere passiert. Das Genom der Gnathostomata (Kiefermünder – das sind alle Wirbeltiere mit einem beweglichem Unterkiefer) scheint von vielen wichtigen Genen vier diploide Sätze aufzuweisen. Aufgrund dieser Erkenntnis gelangten Biologen zu dem Schluss, dass sich bei dem Vorfahren dieser Gruppe zwei Duplikationen des gesamten Genoms ereigneten. Diese Duplikationen ermöglichten eine beträchtliche Spezialisierung einzelner Wirbeltiergene, deren Expression heute in hohem Maße gewebespezifisch erfolgt.
3.3 Einige Genfamilien evolvieren durch konzertierte Evolution
Zwar haben sich die Vertreter der Globingenfamilie in ihrer Form und Funktion diversifiziert, die Mitglieder vieler anderer Genfamilien evolvieren hingegen nicht unabhängig voneinander. So besitzen beispielsweise fast alle Organismen zahlreiche Kopien (bis zu mehrere Tausend) der Gene für ribosomale RNA. Ribosomale RNA (rRNA) bildet das Hauptstrukturelement der Ribosomen und erfüllt als solches eine wichtige Rolle bei der Proteinsynthese. Sämtliche Lebewesen müssen – oft in großen Mengen – Proteine synthetisieren (vor allem während ihrer frühen Entwicklung). Durch den Besitz zahlreicher Kopien der rRNA-Gene wird gewährleistet, dass die Organismen rasch viele Ribosomen produzieren und dadurch eine hohe Proteinsyntheserate aufrechterhalten können.
Die Gene für ribosomale RNA evolvieren wie alle anderen Teile des Genoms, und so sammeln sich in den rRNA-Genen verschiedener Arten mit der Zeit Unterschiede an. Innerhalb einer Art sind sich die zahlreichen Kopien der rRNA-Gene hingegen sowohl strukturell als auch funktionell sehr ähnlich. Diese Ähnlichkeit ist auch sinnvoll, denn im Idealfall sollte jedes Ribosom einer Art auf die gleiche Weise Proteine synthetisieren. Mit anderen Worten, innerhalb einer Spezies evolvieren die vielen Kopien dieser rRNA-Gene gemeinsam. Dieses Phänomen bezeichnet man als konzertierte Evolution.
Wie kommt es zu einer solchen konzertierten Evolution? Offensichtlich liegen ihr zwei verschiedene Mechanismen zugrunde. Einer davon ist ungleiches Crossing-over. Während der Replikation der DNA eines diploiden Organismus im Zuge der Meiose lagern sich die homologen Chromosomenpaare aneinander, und es kommt zu einer Rekombination durch Crossing-over (Abschn. 11.4). Im Falle hochrepetitiver Gene wie der rRNA-Gene passiert es jedoch leicht, dass diese bei der Paarung der Chromosomen gegeneinander versetzt werden, weil so viele Kopien der gleichen Gene auf den Chromosomen vorhanden sind (Abb. 23.10a). Infolgedessen erhält das eine der beiden homologen Chromosomen beim Crossing-over zusätzliche Kopien des rRNA-Gens, während das andere Chromosom entsprechend weniger Kopien abbekommt. Erfolgt in einer der Kopien eine Punktmutation in Form einer Basensubstitution, kann diese durch das ungleiche Crossing-over schrittweise vermehrt und damit allmählich fixiert werden. Umgekehrt kann sie durch das ungleiche Crossing-over auch schrittweise verringert und schließlich eliminiert werden. So oder so bleiben die vielen Kopien des Gens einander sehr ähnlich.

Konzertierte Evolution. Zwei Mechanismen können zu einer konzertierten Evolution hochrepetitiver Gene führen. a Durch ungleiches Crossing-over kommt es zu Deletionen und Duplikationen von Genwiederholungen. b Durch gerichtete Genkonversion kann sich eine neue Variante rasch über viele Kopien eines repetitiven Gens ausbreiten
Den zweiten Mechanismus, der zu einer konzertierten Evolution führt, bezeichnet man als gerichtete Genkonversion. Dieser Mechanismus erfolgt mit viel höherer Geschwindigkeit als ein ungleiches Crossing-over und hat sich als primärer Mechanismus für die konzertierte Evolution von rRNA-Genen erwiesen. In DNA-Strängen kommt es häufig zu Brüchen, welche dann wieder repariert werden (Abschn. 13.4). Die Gene für die ribosomale RNA liegen während des Zellzyklus die meiste Zeit eng beieinander. Kommt es zu einer Beschädigung eines der Gene, kann eine Kopie des rRNA-Gens des homologen Chromosoms als Matrize zur Reparatur der beschädigten Kopie dienen; dabei ersetzt die als Matrize fungierende Sequenz die Originalsequenz (Abb. 23.10b). In vielen Fällen erfolgt diese Reparatur offenbar insofern recht einseitig, als häufig bestimmte Sequenzen als Matrizen verwendet werden. Dadurch kann sich eine solche bevorzugt verwendete Sequenz rasch auf alle Kopien des Gens ausbreiten. Auf diese Weise kann es passieren, dass sich eine Veränderung, die in einer einzelnen Kopie auftritt, rasch auf alle anderen Kopien ausbreitet.
Aber ganz gleich, welcher dieser Mechanismen der konzertierten Evolution im konkreten Fall zugrunde liegt: Durch sie evolvieren die Kopien eines hochrepetitiven Gens nicht unabhängig voneinander. Zwar treten nach wie vor Mutationen auf, aber wenn diese in einer einzelnen Kopie erfolgen, dann breiten sie sich entweder rasch auf sämtliche Kopien aus oder gehen vollständig aus dem Genom verloren. Dieser Prozess ermöglicht es, dass jede Kopie über die Zeit bezüglich Sequenz und Funktion ähnlich bleibt.
23.3 Wiederholung
Durch horizontalen Gentransfer können bestimmte Funktionen von Genen auch zwischen entfernt verwandten Arten übertragen werden. Genduplikation kann zur Evolution neuer Funktionen führen. Manche hochrepetitiven Gene durchlaufen eine konzertierte Evolution, wodurch ihre gleichförmige Funktionalität erhalten bleibt.
Lernziele
Sie sollten …
-
beschreiben können, wie es durch horizontalen Gentransfer zur Übertragung von Genen zwischen verschiedenen Linien kommen kann, insbesondere zwischen Bakterien.
-
anhand eines Genstammbaums einer Genfamilie ableiten können, wann in der Stammesgeschichte einer Gruppe von Arten Genduplikationen stattgefunden haben.
-
erläutern können, inwiefern ein dupliziertes Gen Gelegenheiten für die Evolution neuer Funktionen liefert.
-
die beiden Prozesse, die einer konzertierten Evolution zugrunde liegen, grafisch darstellen und ihre Unterschiede aufzeigen können.
?
-
1.
Nennen Sie einige potenzielle Vorteile eines horizontalen Gentransfers für den Organismus, der durch diesen Mechanismus neue Gene erhält.
-
2.
Betrachten Sie den folgenden Stammbaum einer Genfamilie, zusammengestellt anhand der vollständig sequenzierten Genome von Mensch, Schimpanse und Gorilla. Der Stammbaum umfasst sämtliche Mitglieder der Genfamilie. Wie viele Genduplikationen und Genverluste haben in der Entwicklungsgeschichte dieser Genfamilie wahrscheinlich stattgefunden? Tragen Sie die betreffenden Stellen in den Stammbaum ein.
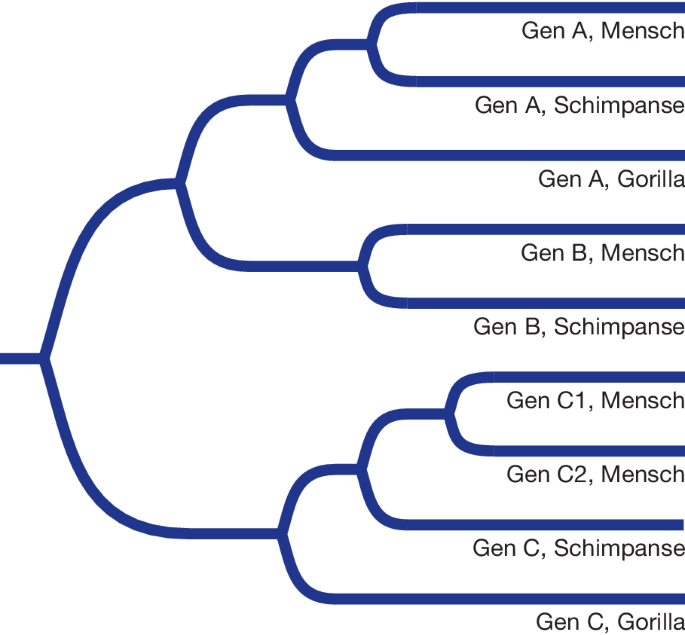
-
3.
Warum wird Genduplikationen für evolutionäre Veränderungen über einen längeren Zeitraum eine große Bedeutung zugemessen?
-
4.
Beschreiben Sie den Verlauf einer konzertierten Evolution hochrepetitiver Gene, und stellen Sie die Prozesse, die dieser konzertierten Evolution zugrunde liegen, grafisch dar.

Sie haben nun erfahren, wie die Prinzipien und Methoden der molekularen Evolution den Evolutionsforschern neue Einblicke verschafft haben. Nun werden Sie einige der praktischen Anwendungsmöglichkeiten auf diesem Gebiet kennenlernen.
4 Die Prinzipien der molekularen Evolution haben viele praktische Anwendungsmöglichkeiten
Für die Ergebnisse von Untersuchungen der molekularen Evolution gibt es in der gesamten Biologie praktische Anwendungsmöglichkeiten, etwa, um grundlegende Aspekte biologischer Funktionen besser zu verstehen, oder für Forschungen zur menschlichen Gesundheit.
Auf den Punkt gebracht
-
Die Evolutionsgeschichte von Genen liefert Informationen zur Funktion von Proteinen.
-
Mithilfe der Prinzipien der in vitro-Evolution lassen sich neue Makromoleküle mit neuen, nützlichen Funktionen herstellen.
-
Untersuchungen zur molekularen Evolution können Erkenntnisse über die Übertragung von Krankheiten und die zuständigen Vektoren liefern. Solche Erkenntnisse sind eine wesentliche Voraussetzung für ein besseres Verständnis und die Bekämpfung von Epidemien.
4.1 Mithilfe von Sequenzdaten lässt sich die Evolution von Genen und Proteinen aufklären
Mit einem Genstammbaum kann man die evolutionären Beziehungen eines einzelnen Gens bei verschiedenen Organismenarten veranschaulichen oder die Evolution der Mitglieder einer Genfamilie zurückverfolgen (wie in Abb. 23.10). Erstellt wird ein solcher Genstammbaum mithilfe der gleichen Methoden, wie sie in Abschn. 21.2für die Konstruktion evolutionärer Stammbäume (phylogenetischer Bäume) von Organismenarten beschrieben wurden. Zunächst müssen Unterschiede zwischen Genen festgestellt werden; anhand dieser Unterschiede lässt sich dann die Evolutionsgeschichte der Gene rekonstruieren. Häufig werden Genstammbäume dazu verwendet, daraus evolutionäre Stammbäume von Arten oder höheren Taxa abzuleiten, aber die beiden Stammbaumtypen sind nicht zwangsläufig äquivalent. Durch Prozesse wie Genduplikation können Unterschiede zwischen den phylogenetischen Bäumen von Genen und Arten entstehen. Mithilfe eines Genstammbaums können Biologen die Geschichte und den Zeitpunkt von Genduplikationen rekonstruieren und so in Erfahrung bringen, wie die Diversifizierung von Genen zur Evolution neuer Proteinfunktionen geführt hat.
Alle Gene einer bestimmten Genfamilie weisen ähnliche Sequenzen auf, weil sie gemeinsamer Abstammung sind. Wie Sie in Abschn. 21.1erfahren haben, bezeichnet man Merkmale, die sich infolge einer gemeinsamen Abstammung ähneln, als homolog. Bei der Besprechung von Genstammbäumen muss man jedoch in der Regel zwischen zwei verschiedenen Formen von Homologie unterscheiden. Bei verschiedenen Organismen vorkommende homologe Gene, die in diesen Arten die gleiche Funktion haben, bezeichnet man als orthologe Gene. Sie sind also eine Folge der Artbildungsereignisse. Hingegen spricht man von paralogen Genen, wenn homologe Gene bei derselben Art vorkommen und dort eine unterschiedliche Funktion haben. Sie gehen auf Genduplikation zurück. Bei der Analyse eines Genstammbaums richtet es sich nach der Fragestellung, ob man orthologe oder paraloge Gene vergleicht. Geht es darum, die Stammesgeschichte der betreffenden Organismenarten zu rekonstruieren, dann sollte sich der Vergleich auf orthologe Gene beschränken (denn diese spiegeln die Geschichte der Artbildungsereignisse wider). Ist man hingegen an Funktionsänderungen interessiert, die durch Genduplikationen entstanden sind, dann eignet sich hierzu ein Vergleich paraloger Gene (weil diese die Geschichte von Genduplikationen widerspiegeln). Gilt das Interesse aber der Diversifizierung einer Genfamilie durch beide Prozesse, dann sollte der Stammbaum natürlich sowohl paraloge als auch orthologe Gene beinhalten.
In Abb. 23.11ist ein Genstammbaum für Mitglieder der engrailed-Genfamilie dargestellt (diese codieren Transkriptionsfaktoren, die Entwicklungsprozesse regulieren). In dieser Familie sind mindestens drei Genduplikationen erfolgt, wodurch bei einigen Wirbeltierarten (wie dem Zebrafisch) bis zu vier verschiedene engrailed-Gene (En) entstanden sind. Bei den engrailed-Genen handelt es sich durchweg um homologe Gene, weil sie auf einen gemeinsamen Vorläufer zurückgehen. In einigen Wirbeltierlinien entstanden durch Genduplikationen paraloge engrailed-Gene (En1 und En2). Um die Entwicklung der Wirbeltiere mit knöchernem Skelett zu rekonstruieren (also aller Arten in Abb. 23.11mit Ausnahme des Neunauges), könnten wir die orthologen Sequenzen der En1-Gengruppe vergleichen oder auch die orthologen Sequenzen der En2-Gengruppe, was das gleiche Ergebnis erwarten ließe (da die zugrunde liegenden Artbildungsereignisse die gleichen sind). Alle Wirbeltiere mit Knochenskelett besitzen beide Gruppen von engrailed-Genen, weil diese beiden Gruppen durch eine Genduplikation beim gemeinsamen Vorfahren dieser Tiergruppe entstanden sind. Würde uns mehr die Diversifizierung interessieren, die im Anschluss an diese Duplikation erfolgte, dann böte sich dazu ein Vergleich der paralogen Gene der En1- und der En2-Gruppe an.
?
Frage zu Abb. 23.11: Wie viele Genduplikationsereignisse fanden nur innerhalb der Linie der Zebrafische statt?

Phylogenie der engrailed-Gene. Die engrailed-Gene sind homolog, weil sie auf einen gemeinsamen Vorfahren zurückgehen. Durch Artbildungsereignisse entstanden bei Wirbeltieren mit knöchernem Skelett orthologe engrailed-Gene, durch Genduplikationen (rote Kreise) paraloge engrailed-Gene
Anstatt mit Genstammbäumen arbeiten viele Biologen je nach Fragestellung auch mit Proteinstammbäumen, zum Beispiel um die Verwandtschaftsbeziehungen oder den Funktionswandel homologer Proteine zu analysieren. Entsprechend zu den Genen unterscheidet man orthologe Proteine (gleiche Funktion in unterschiedlichen Organismen) und paraloge Proteine (unterschiedliche Funktion im gleichen Organismus). So sind die \(\upalpha\)- und die \(\upbeta\)-Untereinheit des menschlichen Hämoglobins paralog (Abb. 23.9), die \(\upbeta\)-Untereinheiten von Mensch und Maus dagegen ortholog.
4.2 Anhand der Evolution von Genen kann man die Proteinfunktion analysieren
Weiter vorne in diesem Kapitel haben Sie erfahren, wie Biologen Genabschnitte identifizieren können, die einer positiven Selektion für Veränderungen unterliegen. Wie lassen sich solche Informationen praktisch anwenden? Betrachten Sie die Evolution der Familie von Genen, die spannungsgesteuerte Natriumkanäle codieren. Verschiedene Toxine können Natriumkanäle blockieren, etwa Tetrodotoxin (TTX), ein Neurotoxin (Nervengift), das in den Geweben mancher Kugelfischarten und einiger anderer Tiere vorkommt. Beim Menschen kann der Verzehr von Kugelfischfleisch, das TTX enthält, zu Lähmungen führen oder sogar tödlich sein, weil dieses Nervengift die Natriumkanäle blockiert und dadurch ein normales Funktionieren von Nerven und Muskeln verhindert.
Querverweis
Welche Funktionen die spannungsgesteuerten Ionenkanäle erfüllen, wird ausführlich in Abschn. 44.2behandelt.
Kugelfische besitzen aber auch selbst Natriumkanäle. Warum ruft TTX dann bei ihnen nicht ebenfalls Lähmungen hervor? Die Natriumkanäle von Kugelfischen (und anderen Tieren, die TTX bilden, wie der Rauhäutige Gelbbauchmolch in Abb. Abb. 20.18) haben eine Resistenz gegen dieses Gift entwickelt. Nucleotidsubstitutionen im Genom des Kugelfischs haben Veränderungen der Proteine nach sich gezogen, aus denen die Natriumkanäle aufgebaut sind. Diese Veränderungen verhindern, dass TTX an die Öffnung der Natriumkanäle bindet.
Bei den verschiedenen duplizierten Genen für Natriumkanäle der vielen Kugelfischarten sind mehrere unterschiedliche Substitutionen erfolgt, die zu einer Resistenz gegen TTX geführt haben. In diesen Genen sind aber auch zahlreiche andere Veränderungen aufgetreten, die nichts mit der Evolution der TTX-Resistenz zu tun haben. Biologen, die sich mit der Funktion von Natriumkanälen befassen, können eine Menge über die Funktionsweise dieser Kanäle lernen (und auch über neurologische Krankheiten, die durch Mutationen in den Genen für Natriumkanäle verursacht werden), indem sie in Erfahrung bringen, welche Veränderungen für eine TTX-Resistenz selektiert wurden. Dazu vergleichen sie die Raten synonymer und nichtsynonymer Substitutionen bei den Genen der verschiedenen Linien, in denen sich eine Resistenz gegen TTX entwickelt hat. In ähnlicher Weise versucht man mithilfe der Prinzipien der molekularen Evolution die Funktion sowie die Diversifizierung der Funktion zahlreicher anderer Proteine zu verstehen.
4.3 Mittels in vitro-Evolution werden neue Makromoleküle hergestellt
Bei ihren Untersuchungen der Zusammenhänge zwischen Selektion, Evolution und Funktion von Makromolekülen kamen Biologen schon bald auf die Idee, sich die molekulare Evolution in einer kontrollierten Laborumgebung zunutze zu machen, um neuartige Makromoleküle mit nützlichen Eigenschaften herzustellen. Das war die Geburtsstunde der Anwendungen der in vitro-Evolution („Evolution im Reagenzglas“).
Lebewesen produzieren Tausende von chemischen Verbindungen, die sich für den Menschen als nützlich erwiesen haben. Die Suche nach solchen natürlich vorkommenden Verbindungen mit einem Nutzwert für pharmazeutische, landwirtschaftliche oder industrielle Anwendungen bezeichnet man als Biosprospecting. Diese chemischen Verbindungen sind das Ergebnis einer Jahrmillionen währenden molekularen Evolution bei Millionen Arten von Lebewesen. Allerdings können sich Biologen auch Moleküle vorstellen, die durchaus hätten evolvieren können, aber nie entstanden sind, weil einfach nicht die richtige Kombination von Selektionsdrücken und Gelegenheiten bestanden hat. So könnte man beispielsweise an einem Enzym interessiert sein, das einen bestimmten Umweltschadstoff bindet und umsetzt, sodass man ihn leicht beseitigen kann. Handelt es sich jedoch um einen synthetischen Schadstoff (d. h., der in der Natur nicht vorkommt), so ist es ziemlich unwahrscheinlich, dass irgendein Organismus ein Enzym mit der gewünschten Funktion evolviert hat. Die Lösung dieses Problems ist ein Einsatzbereich der in vitro-Evolution.
Das Verfahren für die in vitro-Evolution eines neuartigen Enzyms aus einem Ausgangsenzym A umfasst folgende Schritte:
-
1.
Zufallsmutagenese des \(A\)-Gens bzw. der \(A\)-cDNA zur Erzeugung Tausender von Mutanten
-
2.
Genexpression aller Mutanten, um die codierten Proteine zu erhalten
-
3.
Massenscreening in einem effizienten System, das die vielen mutierten Proteine auf die gewünschte Eigenschaft selektiert
-
4.
Auswahl der am besten geeigneten Mutante
-
5.
erneute Zufallsmutagenese des Gens dieser Mutante, gefolgt von Expression und Massenscreening
-
6.
beliebig häufige Wiederholung des Vorgangs, bis das gewünschte Endprodukt vorliegt – ein Enzym mit den geforderten neuen Eigenschaften
Ziel der in vitro-Evolution sind oft auch RNA-Moleküle. Betrachten Sie das Beispiel eines neuen RNA-Moleküls, das unter Anwendung von Mutation und Selektion im Labor hergestellt wurde. Die gewünschte Funktion dieses Makromoleküls war das enzymatische Kopplung zweier anderer RNA-Moleküle. Im Labor wurde also ein Ribozym erzeugt – ein RNA-Molekül, das eine biochemische Reaktion katalysieren kann. Als Ausgangspunkt diente ein immens großer Pool zufällig zusammengestellter RNA-Sequenzen (\(10^{15}\) unterschiedliche Sequenzen von jeweils 300 Nucleotiden Länge); diese wurden dann in einem Massenscreening auf Ligaseaktivität selektiert (Abb. 23.12). Unter den Molekülen fanden sich keine besonders effektiven Ligasen, manche eigneten sich jedoch etwas besser als andere. Die besten dieser Ribozyme wurden ausgewählt und ihre mRNA dann (mithilfe des Enzyms Reverse Transkriptase) in cDNA umgeschrieben. Anschließend wurden diese cDNA-Moleküle mittels Polymerasekettenreaktion (PCR; Abb. Abb. 13.16) amplifiziert.

In vitro-Evolution. Ausgehend von einem großen Pool aus zufällig zusammengestellten RNA-Sequenzen stellten Wissenschaftler in mehreren Durchläufen aus Mutation und Selektion auf die Fähigkeit, RNA-Sequenzen miteinander zu verknüpfen, ein neues Ribozym her
Die Amplifikation mittels PCR verläuft nicht perfekt. Dadurch gelangten viele Neumutationen in den Sequenzpool. Diese Sequenzen wurden dann mithilfe von RNA-Polymerase in RNA-Moleküle rücktranskribiert und der ganze Prozess anschließend wiederholt. Die Ligaseaktivität der RNAs evolvierte rasch: Bereits nach zehn Durchläufen der in vitro-Evolution hatte sie um das 7 \(\times\) \(10^{6}\)-Fache zugenommen. Mittels ähnlicher Techniken hat man seither eine Vielzahl verschiedener RNAs und Proteine mit neuen Enzymaktivitäten und Bindungseigenschaften hergestellt.
4.4 Die molekulare Evolution macht man sich auch für die Erforschung und Bekämpfung von Krankheiten zunutze
Viele der problematischsten Krankheiten des Menschen werden von evolvierenden Lebewesen oder Viren verursacht und stellen somit veränderliche Ziele für die moderne Medizin dar. Erinnern Sie sich an das in der Einleitung zu diesem Kapitel („Faszination Forschung: Die Evolutionstheorie trägt zur Entwicklung besserer Grippeimpfstoffe bei“) beschriebene Beispiel des Grippevirus oder an HIV, das in Kap. 21behandelt wurde. Um Grippe, Aids und ähnliche Krankheiten des Menschen unter Kontrolle zu bringen, bedarf es Techniken, welche die Evolution der Pathogene (Krankheitserreger) im Zeitverlauf nachvollziehen.
Die Fortschritte im Transportwesen im vergangenen Jahrhundert haben es dem Menschen ermöglicht, in zuvor unvorstellbarer Geschwindigkeit und Häufigkeit die gesamte Welt zu bereisen. Leider hat diese Mobilität aber auch die zunehmende Übertragung von Krankheitserregern in der menschlichen Bevölkerung sowie zwischen Menschen und verschiedenen Tierarten ermöglicht. Die Übertragung von Viren über Artgrenzen hinweg hat dazu geführt, dass weltweit viele „neue“ Krankheiten auftraten.
In der Einleitung zu diesem Kapitel haben Sie etwas über die verheerende Grippeepidemie von 1918/19 erfahren, die ja eine Pandemie war – eine weltweite Epidemie. Mittels stammesgeschichtlicher Analysen der Influenzagenome haben Michael Worobey und seine Mitarbeiter Ursachenforschung betrieben, warum gerade diese Grippeepidemie so viel schlimmer verlief als jede andere zuvor oder danach („Experiment: Warum war die Grippepandemie von 1918/19 so schlimm?“). Dazu rekonstruierten sie die Entwicklungsgeschichte der Grippeviren bei Vögeln, Schweinen, Pferden und Menschen. Wie sie herausfanden, wurden die Grippeviren wiederholt von Vögeln auf verschiedene Säugetiere übertragen. Die meisten dieser die Artgrenzen überschreitenden Übertragungen hatten eine starke Epidemie bei dem neuen Wirtsorganismus zu Folge. Auch die Epidemie von 1918/19 ereignete sich unmittelbar nach einer solchen Übertragung zwischen verschiedenen Wirten. Weil das menschliche Immunsystem noch keine Erfahrung mit dem neu erworbenen Virus hatte sammeln können, kam es zu einer starken Reaktion mit vielen der schwersten Symptome und Todesfällen. Nachdem der Zusammenhang zwischen einer Übertragung der Erreger über Artgrenzen hinweg und schwerwiegenden Grippeepidemien inzwischen erkannt ist, achten die Verantwortlichen im Gesundheitswesen weltweit verstärkt darauf, ob es bei den Grippefällen irgendwelche Anzeichen für erneute Übertragungen über Artgrenzen hinweg gibt. Sofern es gelingt, neu entstehende Viren frühzeitig zu erkennen, steigen die Chancen, sie durch entsprechende Maßnahmen erfolgreich bekämpfen und auslöschen zu können. Bislang konnten wir dank unserer Erkenntnisse über die molekulare Evolution verhindern, dass sich eine solch verheerende Grippepandemie wie 1918/19 wiederholt hat.
Experiment: Warum war die Grippepandemie von 1918/19 so schlimm?
Blick in die Daten: Warum war die Grippepandemie von 1918/19 so schlimm?
Die Sequenzierung der Genome zahlreicher infektiöser Erreger hat dafür gesorgt, dass wir heute sehr viel mehr über sie wissen und die von ihnen verursachten Krankheiten besser behandeln können. So wurden beispielsweise von Nagetieren stammende Hantaviren als Ursache für verbreitete Erkrankungen der Atemwege identifiziert. Ebenfalls durch evolutionäre Vergleiche von Genen wurde das Virus (wie auch sein Wirt) identifiziert, welches das schwere akute Atemnotsyndrom (severe acute respiratory syndrome, SARS) hervorruft. Untersuchungen zur Entstehung, zum Zeitpunkt des erstmaligen Auftauchens sowie zur weltweiten Diversität vieler Pathogene des Menschen bauen ebenso auf den Prinzipien der molekularen Evolution auf wie die Bemühungen, wirkungsvolle Impfstoffe gegen diese Krankheitserreger zu entwickeln und einzusetzen. So beruhen beispielsweise die Techniken zur Entwicklung von modernen Impfstoffen gegen Kinderlähmung wie auch die Methoden, mit denen man ihre Wirksamkeit in der menschlichen Bevölkerung überprüft, auf den Grundlagen der molekularen Evolution.
In Zukunft wird die molekulare Evolution noch entscheidendere Bedeutung erlangen, um die Erreger menschlicher (und sonstiger) Krankheiten zu identifizieren. Wenn die Biologen erst einmal Daten über die Genome von genügend Organismen zusammengetragen haben, wird es irgendwann möglich sein, den Erreger einer Infektion dadurch zu identifizieren, dass man einen Teil des Genoms dieses Organismus sequenziert und die erhaltene Sequenz mit anderen Sequenzen eines evolutionären Stammbaums vergleicht. Gegenwärtig bereitet es noch Schwierigkeiten, die Erreger vieler verbreiteter Virusinfektionen zu identifizieren (etwa die Erreger von Erkältungen). Mit zunehmender Zahl an Einträgen in Genomdatenbanken und Stammbäumen werden uns jedoch automatisierte Methoden der Sequenzierung und rascher phylogenetischer Sequenzvergleiche ermöglichen, eine viel größere Bandbreite an menschlichen Krankheiten zu identifizieren und zu behandeln.
23.4 Wiederholung
Untersuchungen der molekularen Evolution haben Biologen neue Möglichkeiten eröffnet, nicht nur die Funktion von Makromolekülen zu verstehen, sondern auch, wie sich diese Funktionen im Laufe der Zeit ändern können. Das dabei erworbene Wissen kann dazu genutzt werden, synthetische Moleküle zu entwickeln und menschliche Krankheiten zu erkennen und zu bekämpfen.
Lernziele
Sie sollten …
-
erläutern können, wie Untersuchungen der molekularen Evolution dazu beitragen können, die Funktionen von Proteinen besser zu verstehen.
-
sich einen Ablaufplan für eine in vitro-Evolution ausdenken können, durch die sich Sequenzen mit erwünschten funktionellen Eigenschaften erzeugen lassen.
-
die evolutionsgeschichtlichen Verwandtschaftsbeziehungen zwischen den Sequenzen von Viren beurteilen und daraus die Evolution und Übertragung einer viralen Erkrankung ableiten können.
?
-
1.
Wie kann man anhand der Evolution von Genen die Proteinfunktion erforschen? Beschreiben Sie ein spezifisches Beispiel dafür.
-
2.
Welches sind die entscheidenden Elemente einer in vitro-Evolution und inwiefern entsprechen diese Elemente natürlichen Evolutionsprozessen?
-
3.
Wie kann man unter Anwendung der Prinzipien der Evolutionsbiologie neu aufkommende Krankheiten erkennen?
Nun haben Sie erfahren, wie Organismen, Gene und Proteine evolvieren. Das nächste Kapitel befasst sich mit der umfassenderen Entwicklungsgeschichte der Organismen auf der Erde. In Kap. 24lernen Sie die über weite Zeiträume ablaufenden evolutionären Veränderungen kennen, durch welche die gesamte Vielfalt des Lebens entstanden ist.
Faszination Forschung: Warum erwies sich die Grippepandemie von 1918/19 als schlimmer als alle anderen zuvor und danach?
In der menschlichen Bevölkerung und bei anderen Wirbeltierwirten kursieren alljährlich zahlreiche unterschiedliche Stämme von Influenzaviren, aber nur wenige dieser Stämme überleben und erzeugen Nachkommen. Die Influenzastämme unterscheiden sich unter anderem in der Struktur der Proteine an ihrer Oberfläche. Diese Oberflächenproteine bilden das Ziel für die Erkennung durch das Immunsystem des Wirtsorganismus. Sofern es in den Oberflächenproteinen eines Influenzavirus zu Veränderungen kommt, kann es sein, dass das Immunsystem des Wirts das eingedrungene Virus nicht mehr erkennt – mit der Folge, dass sich dieses mit höherer Wahrscheinlichkeit erfolgreich replizieren kann. Am ehesten entgehen die Virenstämme mit der größten Zahl von Veränderungen ihrer Oberflächenproteine der Erkennung durch das Immunsystem des Wirtsorganismus. Daher breiten sie sich auch am ehesten in der Wirtspopulation aus und verursachen künftige Grippeepidemien. Mit anderen Worten: Es findet eine positive Selektion für Veränderungen der Oberflächenproteine der Influenzaviren statt.
Wie die Daten in „Experiment: Warum war die Grippepandemie von 1918/19 so schlimm?“ zeigen, ereignete sich diese Grippeepidemie kurz nach einer die Artgrenzen überschreitenden Übertragung eines Vogelgrippevirus auf die menschliche Bevölkerung. Für den Menschen war dieses Virus neu, denn es wich von sämtlichen Influenzaviren ab, die zuvor in der menschlichen Bevölkerung in Umlauf waren. Da das Immunsystem des Menschen zuvor noch nicht mit diesem Virus in Kontakt gekommen war, hatte es noch keine wirksame Immunabwehr dagegen entwickelt. Daher führte das Virus zu einer Überstimulation des menschlichen Immunsystems, was sekundäre Komplikationen und zahlreiche Todesfälle zur Folge hatte. Heute überwachen Wissenschaftler ganz genau, welche Influenzaviren weltweit in der menschlichen Bevölkerung kursieren. Neue Übertragungen über Artgrenzen hinweg, etwa von Vögeln oder Schweinen, frühzeitig zu erkennen, ist eine wesentliche Voraussetzung, um diese Viren eliminieren zu können, bevor sie eine neue globale Grippepandemie beim Menschen auslösen können.
Ausblick
Biologen können die Anpassung der Influenzaviren im Laufe der Zeit erforschen. Dazu vergleichen sie die Überlebens- und Vermehrungsraten von Virenstämmen mit unterschiedlichen Gensequenzen, die Oberflächenproteine codieren. Es muss den Biologen gelingen vorherzusagen, welche der gegenwärtig kursierenden Influenzastämme am wahrscheinlichsten der Erkennung durch den Wirt entgehen. Dann nämlich können sie jene Stämme identifizieren, die mit der höchsten Wahrscheinlichkeit an aufkommenden Grippeepidemien beteiligt sein werden, und diese Stämme gezielt für die Produktion von Impfstoffen verwenden.
Wie können Biologen solche Vorhersagen treffen? Dazu müssen sie das Verhältnis an synonymen und nichtsynonymen Substitutionen bei den Genen ermitteln, die die Oberflächenproteine der Viren codieren. Auf diese Weise können sie herausfinden, welche Codonveränderungen (d. h. Mutationen) einer positiven Selektion unterliegen. Anschließend können sie feststellen, welche der gegenwärtig kursierenden Influenzastämme die größte Zahl von Veränderungen in diesen positiv selektierten Codons aufweisen. Diese Influenzastämme werden nämlich am ehesten überleben, sich vermehren und zukünftig Grippeepidemien verursachen – und bilden somit die logischen Ziele für neue Impfstoffe. Diese praktische Anwendung der Prinzipien der Evolutionstheorie führt dazu, dass wirkungsvollere Impfstoffe gegen Grippeviren entwickelt werden – und damit alljährlich weniger Menschen an Grippe erkranken oder gar daran sterben.
Kapitelzusammenfassung
4.4.1 Synapsenfutter: Wenden Sie an, was Sie gelernt haben
Rückblick
-
Wird alleine die Anzahl der Nucleotidsubstitutionen oder Aminosäureaustausche zwischen Sequenzen ermittelt, führt dies häufig dazu, dass die tatsächliche Zahl der zugrunde liegenden Veränderungen unterschätzt wird (Abschn. 23.1).
-
Die Fixierungsrate neutraler Nucleotidveränderungen innerhalb von Populationen ist unabhängig von der Populationsgröße (Abschn. 23.2).
-
Durch Vergleiche der Raten synonymer und nichtsynonymer Substitutionen kann man eine positive und negative Selektion bei Proteinen erkennen (Abschn. 23.2).
Durch die Erforschung der molekularen Evolutionsmuster von Genen ist es Biologen möglich, Rückschlüsse über verschiedene Formen der darauf einwirkenden natürlichen Selektion zu ziehen. Diese Rückschlüsse sind von großer Bedeutung, denn sie ermöglichen uns ein besseres Verständnis der Funktionsweise von Genen und ihrer Evolution im Laufe der Zeit infolge der Selektion für neue Funktionen oder als Anpassung an neue Umweltbedingungen.
Ein hypothetisches Gen codiert bei verschiedenen Drosophila-Arten einen Transkriptionsfaktor. Der Stammbaum dieser Arten ist in dieser Abbildung dargestellt, die Divergenzzeit ist in Millionen Jahren angegeben.

Ein Exon dieses Gens wurde bei den vier Arten sequenziert. In der Matrix (Abb. 23.13) sind jeweils die Zahl der synonymen (oberhalb der Diagonalen) und der nichtsynonymen (unterhalb der Diagonalen) Substitutionen zwischen Artenpaaren gegenübergestellt. Zwischen D. melanogaster und D. yakuba erfolgten beispielsweise 21 synonyme Substitutionen. Dieses Exon umfasst insgesamt 2000 nichtsynonyme und 600 synonyme Substitutionen. Pro Jahr gibt es fünf Generationen.
Die Antworten auf die folgenden Fragen liefern Informationen darüber, welche Formen der Selektion auf diesen Transkriptionsfaktor einwirken, und potenziell auch über dessen Funktion.

Matrix der synonymen und nichtsynonymen Substitutionen bei Artenpaaren der Gattung Drosophila
Aufgaben
-
1.
Stellen Sie den Zusammenhang zwischen der Zahl der synonymen Substitution und den Jahren der Divergenz zwischen jedem Artenpaar grafisch dar und beschriften Sie die beiden Achsen. Versuchen Sie nun anhand der Grafik die Rate synonymer Substitutionen pro Million Jahre und pro Generation für das gesamte Exon und für die einzelnen Positionen abzuschätzen. Denken Sie dabei daran, dass in beiden Linien eine Evolution stattfindet.
-
2.
Wie hoch läge die erwartete Mutationsrate pro Position unter Berücksichtigung der Lösung von Aufgabe 1, wenn man davon ausgeht, die synonymen Substitutionen seien neutral.
-
3.
Die Linie der Stubenfliege (Musca domestica) spaltete sich vor etwa 90 Mio. Jahren von derjenigen von Drosophila melanogaster ab. Angenommen, zwischen diesen beiden Taxa seien 174 synonyme Substitutionen erfolgt. Nehmen Sie weiterhin an, die Zahl der Generationen sei bei beiden Arten konstant bei fünf pro Jahr geblieben. Berechnen Sie die erwartete Zahl an synonymen Substitutionen zwischen diesen beiden Arten anhand der errechneten Rate synonymer Substitutionen von Aufgabe 1, außerdem die Divergenzzeit und die Anzahl an Generationen pro Jahr. Liefern Sie eine Erklärung dafür, warum eine Diskrepanz besteht zwischen den erwarteten und den tatsächlich beobachteten Werten, weiterhin davon ausgehend, dass die Mutationen völlig neutral sind. (Hinweis: Berücksichtigen Sie dabei, was Sie über Substitutionsmuster wissen.)
-
4.
Berechnen Sie die durchschnittliche Rate nichtsynonymer Substitutionen pro Position und Generation zwischen D. ananassae und den drei anderen Arten. Wenn Sie die Unterschiede zwischen dieser Rate und der in Aufgabe 1 berechneten Rate synonymer Substitutionen berücksichtigen: Welche Form der Selektion wird Ihrer Ansicht nach auf die Positionen mit nichtsynonymen Substitutionen einwirken?
-
5.
Angenommen, ein Biologe behauptet, bei diesem Transkriptionsfaktor handele es sich bei Fliegen in Wirklichkeit um ein Pseudogen. Würden Sie diesem Biologen auf der Grundlage der Informationen, die Sie erhalten haben, zustimmen? Warum oder warum nicht?


4.4 Kapitelzusammenfassung
23.1 In den DNA-Sequenzen ist die Evolutionsgeschichte der Gene aufgezeichnet
-
Als Genom eines Organismus bezeichnet man die Gesamtheit seiner Gene, regulatorischen Sequenzen und Strukturelemente einschließlich der nichtcodierenden DNA.
-
Das Gebiet der molekularen Evolution befasst sich damit, welche Zusammenhänge zwischen der Struktur von Genen und Proteinen und der Funktionsweise von Organismen bestehen.
-
Mittels Sequenzalignment von Nucleinsäuren oder Proteinen verschiedener Organismen lassen sich diese vergleichen und homologe Positionen identifizieren. Siehe Abb. 23.1; Activity 23.1
-
Die minimale Zahl von Veränderungen zwischen zwei Sequenzen kann man mithilfe einer Ähnlichkeitsmatrix (Distanzmatrix) berechnen. Durch Anwendung von Modellen der Evolution von Sequenzen lassen sich Veränderungen erklären, die man nicht direkt beobachten kann. Siehe Abb. 23.2; Activity 23.2
23.2 An Genomen lassen sich sowohl neutrale als auch selektive Evolutionsprozesse ablesen
-
Nichtsynonyme Substitutionen von Nucleotiden führen zu Veränderungen der Aminosäuresequenz von Proteinen, synonyme Substitutionen hingegen nicht. Siehe Abb. 23.4
-
Die Neutraltheorie der molekularen Evolution besagt, dass sich durch einen Großteil der molekularen Veränderungen in Nucleotidsequenzen die Funktion des Genoms nicht ändert. Die Fixierungsrate neutraler Mutationen ist unabhängig von der Populationsgröße und gleich der Mutationsrate.
-
Eine positive Selektion für Veränderungen in einem proteincodierenden Gen kann man anhand der höheren Rate von nichtsynonymen gegenüber synonymen Substitutionen feststellen. Das Umgekehrte gilt für negative (reinigende) Selektion.
-
Verbreitete Selektionszwänge können zu einer konvergenten Evolution von Aminosäuresequenzen bei entfernt verwandten Arten führen. Siehe „Experiment: Konvergente molekulare Evolution“
-
Die Gesamtgröße der Genome von vielzelligen Arten ist durch eine viel größere Variationsbreite gekennzeichnet als die Zahl funktioneller Gene. Siehe Abb. 23.6, 23.7
23.3 Horizontaler Gentransfer und Genduplikationen können große Veränderungen nach sich ziehen
-
Durch horizontalen (lateralen) Gentransfer können rasch neue Funktionen selbst von entfernt verwandten Arten erworben werden.
-
Genduplikationen können zu einer erhöhten Produktion des Genprodukts, zur Entstehung von funktionslosen Pseudogenen oder zu neuen Genfunktionen führen. Durch mehrere wiederholte Genduplikationen können multiple Gene mit verwandten Funktionen entstehen; man fasst diese dann als Genfamilie zusammen. Siehe Abb. 23.8, 23.9
-
Einige hochrepetitive Gene durchlaufen eine konzertierte Evolution. Dabei erhalten die zahlreichen Kopien in einem Organismus die große Ähnlichkeit innerhalb der Art aufrecht, obgleich die Gene zwischenartlich weiter divergieren. Siehe Abb. 23.10; Animation 23.1
23.4 Die Prinzipien der molekularen Evolution haben viele praktische Anwendungsmöglichkeiten
-
Orthologe Gene bzw. Proteine stehen durch Artbildungsereignisse miteinander in Beziehung, paraloge Gene bzw. Proteine dagegen durch Genduplikationen. Siehe Abb. 23.11
-
Die Funktion von Proteinen kann auch anhand der Evolution der codierenden Gene erforscht werden. Durch Feststellung einer positiven Selektion kann man molekulare Veränderungen identifizieren, die Funktionsänderungen bewirkt haben.
-
Mittels in vitro-Evolution kann man synthetische Nucleinsäuren und Proteine mit erwünschten Eigenschaften herstellen. Siehe Abb. 23.12
-
Viele Krankheiten lassen sich durch Untersuchungen identifizieren, analysieren und bekämpfen, denen die Prinzipien der molekularen Evolution zugrunde liegen. Siehe „Faszination Forschung: Warum war die Grippepandemie von 1918/19 so schlimm?“
Change history
28 July 2022
Erratum zu: D. Sadava et al., Purves Biologie, https://doi.org/10.1007/978-3-662-58172-8
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer-Verlag GmbH Deutschland, ein Teil von Springer Nature
About this chapter

Cite this chapter
Markl, J., Sadava, D., Hillis, D.M., Heller, H.C., Hacker, S.D. (2019). Evolution von Genen und Genomen. In: Markl, J. (eds) Purves Biologie. Springer Spektrum, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-58172-8_23
Download citation
DOI: https://doi.org/10.1007/978-3-662-58172-8_23
Published:
Publisher Name: Springer Spektrum, Berlin, Heidelberg
Print ISBN: 978-3-662-58171-1
Online ISBN: 978-3-662-58172-8
eBook Packages: Life Science and Basic Disciplines (German Language)