
Abstract
Polyoxometalates (PMs) as discrete metal-oxide cluster anions with high solubility in water and photochemically and electrochemically active property have a wide variety of structures not only in molecular size from sub-nano to sub-micrometers with a various combination of metals but also in symmetry and highly negative charge. One of the reasons for such a structural variety originates from their conformation change (due to the condensed aggregation and the structural assembly) which strongly depends on environmental parameters such as solution pH, concentration, and coexistent foreign inorganic and/or organic substances. In the course of the application of the physicochemical properties of such PMs to the medical fields, antitumoral, antiviral, and antibacterial activities have been developed for realization of a novel inorganic medicine which provides a biologically excellent activity never replaced by other approved medicines. Several PMs as a candidate for clinical uses have been licensed toward the chemotherapy of solid tumors (such as human gastric cancer and pancreatic cancer), DNA and RNA viruses (such as HSV, HIV, influenza, and SARS), and drug-resistant bacteria (such as MRSA and VRSA) in recent years: [NH3Pri]6[Mo7O24]∙3H2O (PM-8) and [Me3NH]6[H2MoV 12O28(OH)12(MoVIO3)4]∙2H2O (PM-17) for solid tumors; K7[PTi2W10O40]∙6H2O (PM-19), [PriNH3]6H[PTi2W10O38(O2)2]∙H2O (PM-523), and K11H[(VO)3(SbW9O33)2]∙27H2O (PM-1002) for viruses; and K6[P2W18O62]∙14H2O (PM-27), K4[SiMo12O40]∙3H2O (SiMo12), and PM-19 for MRSA and VRSA. The results are discussed from a point of view of the chemotherapeutic clarification in this review.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Human Immunodeficiency Virus
- Herpes Simplex Virus
- Respiratory Syncytial Virus
- Canine Distemper Virus
- Fractional Inhibitory Concentration
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
Polyoxometalates (PMs) as discrete metal-oxide cluster anions with high solubility in water and photochemically and electrochemically active property have a wide variety of structures not only in molecular size from sub-nano to sub-micrometers with a various combination of metals but also in symmetry and highly negative charge (Pope and Müller 1994, 2001; Hill 1998; Yamase and Pope 2002; Borrás-Alamenar et al. 2003; Yamase 2003; Proust et al. 2008; Long et al. 2010). One of the reasons for such a structural variety originates from their conformation change (due to the condensed aggregation and the structural assembly) which strongly depends on environmental parameters such as solution pH, concentration, and coexistent foreign inorganic and/or organic substances. Therefore, PMs under the physiological condition let us expect to induce some modifications in a variety of biological systems such as the adsorption to receptor, the penetration of substances through the cellular membrane, and the multiple enzymes which can work independently or cooperatively. The interaction of PMs with multiple biomolecule targets in a single event or different processes simultaneously in the biological systems will provide a basis for the development of a novel inorganic medicine and greatly improve our understanding of the mechanism and timing of the interacting species, in biological and clinical studies, as well. Both in vitro and in vivo antitumor activities of polyoxomolybdates, especially [Mo7O24]6−, have been investigated along with this line since the finding of in vivo antitumor effect against solid human (breast, lung, and colon) cancer xenografts (MX-1, OAT, and CO-4, respectively) in 1988 (Yamase et al. 1988, 1992a).
Historically, 40 years ago Raynaud and Jasmin first found the biological activity of PMs against various non-retro RNA and DNA viruses in vitro and in vivo and demonstrated the inhibition of Friend leukemia virus and Moloney murine sarcoma virus in vitro by [SiW12O40]4− as a Keggin structure PM and in vivo by [Na(SbW7O24)3(Sb3O7)2]18− (as a French drug [NH4]17Na[Na(SbW7O24)3(Sb3O7)2]∙14H2O historically called HPA-23) (Jasmin et al. 1973, 1974). Also, they showed HPA-23 and other polyoxotungstates to inhibit RNA-dependent DNA polymerases of retroviruses, which let us expect the activity against human immunodeficiency virus (HIV). Although HPA-23 was tested in animal models of lentiviral infections and infected humans, HPA-23 was too toxic to be effective in clinical trials in both France and the USA (Rozenbaum et al. 1985, Moskovitz et al. 1988). In spite of the negativity of in vivo anti-HIV activity of HPA-23, the assay of the antiviral activity for PMs by other groups has been continued, and many polyoxotungstates with Keggin or Wells–Dawson related (lacunary and multiply condensed) structures, in particular, K13[Ce(SiW11O39)2]∙26H2O (JM1590), K6[BGa(H2O)W11O39]∙15H2O (JM2766), [Me3NH]8[(SiNb3W9O37)2O3] (JM2820), K7[PTi2W10O40]∙6H2O (PM-19), K9H5[(GeTi3W9O37)2O3]∙16H2O (PM-504), [PriNH3]6H[PTi2W10O38(O2)2]∙H2O (PM-523), K10[Fe4(H2O)2(PW9O34)2]∙xH2O (HS-058), K7[P2W17NbO62], and K7[P2W17(NbO2)O61], were found to be in vitro active against DNA and RNA viruses (Hill et al. 1990; Inouye et al. 1990; Fukuma et al. 1991; Take et al. 1991; Weeks et al. 1992; Yamamoto et al. 1992; Ikeda et al. 1993; Shigeta et al. 1996, 1997; Barnard et al. 1997; Rhule et al. 1998; Witvrouw et al. 2000). Also, the observation of in vitro anti-HIV-1 activity of [NH4]12H2[Eu4(MoO4)(H2O)16(Mo7O24)4]∙13H2O (PM-104, Naruke et al. 1991) was interesting, since PM-104 is classified in polyoxomolybdates although the attachment of Eu atoms in the molecule seemed to be biologically significant in the anti-HIV-1 activity (Inouye et al. 1993). However, none of these PMs was yet advantageous enough to surpass clinically approved drugs such as 3′-azido-3′-deoxythymidine (AZT) and ribavirin in both antiviral activity and cytotoxicity. For realization of the PM-based inorganic medicines as a novel drug, it is at least necessary to show an excellent activity of PMs against the biological target which is impossible to be replaced by the existing approved drugs, while we have been always confronted with historically conservative (or pessimistic) background for inorganic medicines in medicinal society (Yamase 2005). We have shown a potent activity of the tris(vanadyl)-18-tungsto-2-antimonate(III) anions, [(VIVO)3(SbW9O33)2]12− and [(VVO)(VIVO)2(SbW9O33)2]11−, against a wide variety of the enveloped viruses which infect high-risk individuals such as infants born with prematurity, cardiovascular failure, pulmonary dysplasia, and HIV: their selective index (SI) values ( >104) for HIV are much higher than 3–4 × 103 of AZT and dextran sulfate (in mol. wt. 5,000, DS5000) (Shigeta et al. 2003). In addition, most of the antiviral polyoxotungstates exhibit a great synergistic effect with β-lactam antibiotics against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Staphylococcus aureus (VRSA) strains (Yamase et al. 1996a; Fukuda et al. 1999). Together with their potency against other bacteria such as Streptococcus pneumoniae and Helicobacter pylori (Fukuda and Yamase 1997; Inoue et al. 2005), the synergy effect of PMs lets us investigate the mechanistic details on the antibacterial activity in relevance of a clarification of the antiviral mechanism for the PMs. MRSA, VRSA, and Streptococcus pneumoniae as Gram-positive bacteria and Helicobacter pylori as a microaerophilic Gram-negative bacterium pose serious problems of hospital-acquired infections in surgical intensive care units and global infections of the latter (associated with the development of gastritis, gastric ulcer, duodenal ulcer, peptic ulcer, and gastric cancer) in approximately half of the world population. Also, PMs enhanced nerve growth factor (NGF)-induced neurite growth of PC12 cells with expression of the axonal growth associated protein 43 (GAP-43) (Oda et al. 2007). Since rat neuronal PC12 cells which differentiate upon NGF stimulation are used as the in vitro model for the characterization of neurotoxicants, this finding suggests that PMs may be one of the candidates for a novel drug against the neurodegenerative disorders such as Alzheimer’s disease (Geng et al. 2011). Here we review recent developments on biological (antitumor, antiviral, and anti-MRSA and VRSA bacterial) activities of the PMs selected from a viewpoint of the clinical significance.
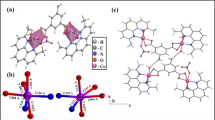
Figure 4.1 shows the structures of anions for some of the bioactive PMs, which will be discussed below. Structures of [MoVI 7O24]6− and [H2MoV 12O28(OH)12(MoVIO3)4]6− are depicted by atom and bond model, as shown in Fig. 4.1a, b where MoVI, MoV, and O atoms are indicated by yellow, red, and white color, respectively (Yamase et al. 1981; Ohashi et al. 1982; Yamase and Ishikawa 2008). The former (with C 2v-symmetry) is the anion for [NH3Pri]6[Mo7O24]∙3H2O (PM-8), and the latter (with T d-symmetry) for [Me3NH]6[H2MoV 12O28(OH)12(MoVIO3)4]∙2H2O (PM-17). Both compounds are antitumor compounds, and the latter as a photoreduction product for the long-term photolysis of PM-8 at pH 5–7 is more active than the former (Yamase et al. 1988). Keggin-structural [PTi2W10O40]7− (Fig. 4.1c) and Keggin-condensed dimer structural [(GeTi3W9O37)2O3]14− (Fig. 4.1d) are anions for antiviral compounds, K7[PTi2W10O40]∙6H2O (PM-19) and K9H5[(GeTi3W9O37)2O3]∙16H2O (PM-504), respectively (Domaille and Knoth 1983; Yamase et al. 1993, 2007). In [PTi2W10O40]7−, two W atoms in T d-symmetric Keggin-structural [PW12O40]3− are substituted by Ti atoms to yield a C 2-symmetric anion (Domaille and Knoth 1983; Yamase et al. 1992b), and [(GeTi3W9O37)2O3]14− can be regarded as a dimeric condensation product (with D 3h-symmetry) for Ti-substituted A-α-[GeTi3W9O40]10− within the T d-Keggin-structural [GeW12O40]4− framework (Yamase et al. 1993). The anion of [(VIVO)3(SbW9O33)2]12− for K11H[(VO)3(SbW9O33)2]∙27H2O (PM-1002) as a potently antiviral (especially anti-SARS) active compound is shown in Fig. 4.1e where (VIVO)3 triangle is sandwiched by two B-α-[SbIIIW9O33]9− (formally as a trilacunary Keggin structure) to yield a D 3h-symmetric anion (Yamase et al. 2001, 2002). Wells–Dawson structural [P2W18O62]6− anion (Lyon et al. 1991) for K6[P2W18O62]∙14H2O (PM-27) is antiviral and also synergistically antibacterial against MRSA and VRSA strains in the coexistence of β-lactam antibody, the structure of which is shown in Fig. 4.1f where D 3h-symmetric structure consists of a dimer of two α-A-[PVW9O31]3− (formally as a trilacunary Keggin structure). Figure 4.1g shows D 4d-symmetric Cl−-encapsulated spherical anion of [VIV 18O42(Cl)]13− for K10H3[V18O42(Cl)]∙12H2O exhibiting a strong antibacterial activity against Streptococcus pneumoniae (Fukuda and Yamase 1997). Also, most of such multiply reduced spherical polyoxovanadates inhibited strongly at their nanomolar levels in vitro the enzymatic activities of specific sialyl/sulfotransferases which are responsible for the biosynthesis of cell-surface carbohydrate chains.

Structures depicted by atom and bond model for anions of some of the bioactive PMs. [MoVI 7O24]6− (a), [H2MoV 12O28(OH)12(MoVIO3)4]6− (b), [PTi2W10O40]7− (c), [(GeTi3W9O37)2O3]14− (d), [(VIVO)3(SbW9O33)2]12− (e), [VIV 18O42(Cl)]13− (f), and [P2W18O62]6− (g). (a) and (b) correspond to the anion for antitumoral [NH3Pri]6[Mo7O24]∙3H2O (PM-8) and [Me3NH]6[H2MoV 12O28(OH)12(MoVIO3)4]∙2H2O (PM-17), respectively, in which MoVI, MoV, and O atoms are indicated by blue, brown, and white color, respectively. (c), (d), (e), and (f) correspond to the anion for antiviral K7[PTi2W10O40]∙6H2O (PM-19), K9H5[(GeTi3W9O37)2O3]∙16H2O (PM-504), K11H[(VO)3(SbW9O33)2]∙27H2O (PM-1002), and K6[P2W18O62]∙14H2O (PM-27), respectively, in which WVI, TiIV (or VIV), PV (GeIV or SbIII), and O atoms are indicated by blue, red, yellow, and white color, respectively. (g) Corresponds to the anion for a strong sialyl/sulfotransferase inhibitor, K10H3[V18O42(Cl)]∙12H2O, which is also antibacterial against Streptococcus pneumoniae. In (f) VIV, Cl, and O atoms are indicated by blue, red, and white color, respectively
4.2 Antitumor Activity of Polyoxomolybdates
In our trial to apply the PMs chemistry to medical fields, C 2v-symmetric V-shaped heptamolybdates (Fig. 4.1a) and C i-symmetric Anderson-structural polyoxomolybdates have been recognized to exhibit antitumor activities against solid tumors at noncytotoxic doses in vivo (Yamase et al. 1988). Especially, [NH3Pri]6[Mo7O24]∙3H2O (PM-8) suppresses significantly the tumor growth in mice bearing methylcholanthrene-induced tumor (Meth-A sarcoma), MM-46 adenocarcinoma, and human cancer xenografts such as MX-1, CO-4, and OAT (Yamase et al. 1992a, b). Its growth suppression is superior to that obtained for 5-fluorouracil (5-FU) and 1-(4-amino-2-methylpyrimidine-5yl)methyl-3-(2-chloroethyl)-3-nitrosourea (ACNU), which are clinically approved drugs showing a good activity against breast, gastrointestinal, and intracranial tumors. Table 4.1 shows results of antitumor activities of PM-8, [NH4]6[Mo7O24]∙4H2O, K6[Mo7O24]∙4H2O, [NH3Pri]Cl, and PM-17 as the brown-colored powder (the photoreduction product of PM-8) against Meth-A sarcoma and MM-46 adenocarcinoma. Five compounds were basically administrated intraperitoneally (i.p.) nine times at 1-day intervals from days 1 to 9 after the subcutaneous (s.c.) or i.p. implantation of tumor cells (1 × 105 cells of Meth-A) into 8–11 mice/group (female Balb/c and C3H/He mice) for Meth-A, respectively on day 0. Tumor weight (mg) for tumor weight inhibition (TWI) was estimated by measuring the length (l) and width (w) of each tumor with vernier caliper (mm) and using a formula of lw 2/2. PM-8 shows a significant inhibition of Meth-A sarcoma with high values of ILS as an increase in life-span (ILS in %) defined by 100(t − c)/c where t and c are mean survival times for PM-treated and untreated (control) groups, respectively. Especially, its i.p. administration of 50 mg/kg provides a remarkable prolongation of ILS (=111 %) for i.p. implants. It is easy to see the dose schedule in which ILS values for PM-8 are higher than for 5-FU and ACNU. The administration of high dose of 5-FU and ACNU leads to toxic deaths of mice, as is indicated by negative changes in body weight. PM-8 is also effective against MM-46 adenocarcinoma (with 5 × 105 cells on day 0) as added in Table 4.1. A dose effect on the inhibition of growth of both Meth-A sarcoma and MM-46 adenocarcinoma is not clear. There is no apparent toxicity of PM-8 in spite of the high dose of 250 mg/kg, as the mice maintain on average their weights throughout 14 days. This excludes the possibility that the tumor growth inhibition is due to a toxic effect on the host. To study the structure-activity relationship of PM-8, the effect of chemical variation against the Meth-A sarcoma is investigated by a use of three different ways (1) [NH3Pri]+ in PM-8 was replaced by [NH4]+ and K+, (2) [Mo7O24]6− was replaced by Cl−, (3) PM-17, the photoreduction product (with brown color) of PM-8, was used, the anion ([H2MoV 12O28(OH)12(MoVIO3)4]6−) of which was X-ray crystallographically characterized for [Me3NH]+ salt (Yamase and Ikawa 1977; Yamase and Ishikawa 2008). Figure 4.1a, b shows anion structures of PM-8 and PM-17, respectively. As shown in Table 4.1, [NH4][Mo7O24]∙4H2O and K4[Mo7O24]∙4H2O are effective as well as PM-8, while [NH3Pri]Cl (100 mg/kg) is hardly effective. The administration of PM-17 of 25 mg/kg gives a significant inhibition of the Meth-A sarcoma growth on day 14. However, the consecutive administration of PM-17 induced a negative change in the body weight. Since PM-17 is a multiply photoreduced species originated from PM-8, the toxicity reflected by the negative change in the body weight seems to be associated with the high condensation structure produced as a result of the 12-electron reduction of [Mo7O24]6−. Because of a small size (about 8 Å) of [Mo7O24]6−, PM-8 will enable to be transported across membranes and undergo enzyme-mediated metabolism or bind to plasma proteins or tissue. The saturation at high concentration of PM-8 through these pathways is likely to occur with a resultant nonlinear relationship between dose of PM-8 and its antitumor activity (Table 4.1).
Figure 4.2 shows the effectiveness of PM-8 and PM-17 against the progressive growth of small xenografts of human breast MX-1 neoplasms. The growth of MX-1 human breast cancers (2 × 2 mm) implanted s.c. in athymic nude mice (6-week-old female Balb/c, 4 mice/group) on day 0 proceeds and can be detected on day 17 when tumor size ranges from 350 to 491 mm3. The tumor volume (V) is determined by measurement of two principal perpendicular diameters (in mm) as V = (length) × (width)2/2. The first administration of PM-8 of 200 mg/kg is i.p. made on day 17. Ten administrations (once a day from days 17–27, except day 19) of PM-8 give a growth inhibition of 73 % on day 46 without any special risk to the mice as long as the mice are appropriately sterilized by filtration. This means that the size of the breast tumor on day 46 is 27 % of the tumor size (7,466 mm3) for the control group. The averaged tumor weights measured after killing (dissection) on day 46 for PM-8-treated and untreated tumor mice were 2.54 ± 0.04 and 8.40 ± 0.33 g, respectively. PM-17 provided apparent signs of toxicity (Fig. 4.1): three i.p. administrations of PM-17 of 100 mg/kg on days 17, 18, and 20 resulted in a death of one mouse on day 22. Subsequent seven i.p. administrations of PM-17 of 25 mg/kg once a day from days 21–27 give toxic deaths of another mouse on day 29 and others on day 34. However, the therapy by PM-17 exhibits a tumor growth inhibition of 75 % compared with the control group on day 29. Thus, it is possible to say that PM-17 exhibits a cancerocidal potency similar to PM-8 but is strongly toxic, indicating that an optimum of the administration schedule should be taken into consideration, as below exemplified for the nude mice loaded by AsPC-1 (human pancreatic cancer ) and MKN-45 (human gastric cancer). The growth suppression by PM-8 was also observed for the nude mice bearing OAT human lung cancer xenograft and CO-4 human colon cancer. The tumor growth suppression of PMs 8 and 17 was also investigated by using the subrenal capsule in kidney (SRC) assay, in which small pieces of CO-4 tumor (approximately 1 mm3) were implanted into subrenal capsules of ICR 5-week-old female mice under anesthesia on day 0 and PMs were i.p. administrated once a day on days 1–4 or 5, thereafter tumor sizes were determined on day 5 or 6. The results for by the SRC assay are shown in Table 4.2, where Na5[IMo6O24]∙34H2O (PM-32), 5-FU, ACNU, and cis-diamminedichloroplatinum(II) (CCDP) are also evaluated for comparison (Fujita et al. 1992; Yamase et al. 1992a, b; Yamase 1994, 1996). The antitumor potency of PM-8 against CO-4 human colon cancer is comparable to that of approved drugs and superior to PM32 (with Anderson structure).

Inhibition of tumor growth by PM-8 and PM-17 after implantation of MX-1. PM-8 (open circles) was i.p. administrated by 200 mg/kg/day on days 17–27 except 19 and 100 mg/kg/day on days 17, 18, and 20, and PM-17 (open triangles) was done by 25 mg/kg/day on days 21–27. Control (filled circles) showed no PM treatment. The change in body weights on days 17–32 for PM-17-treated mice was −5.0 g, while there was no significant change on days 17–46 for PM-8-treated mice
The distribution of PM-8 to the organs in the tumor-implanted mice has been investigated by the radioactivation (to 101Mo) analysis of the liver, kidney, brain, plasma, and tumor at 0.5 h after i.p. administration of PM-8 into the MM-46-implanted C3H/HeNCrj mice (Yamase et al. 1992a, b). The tumor mice are prepared by the implantation of 1 × 106 cells of MM-46/mouse on day 0 and PM-8 of 100 mg/kg was administrated i.p. on day 14 when the tumor growth was detectable. Amounts of the Mo atoms uptaken into organs at 0.5 h after i.p. administration of PM-8 and their time profile are shown in Figs. 4.3 and 4.4, respectively. The comparison of the amounts of Mo atoms between PM-8-treated and untreated mice indicates that PM-8 is preferentially distributed to the kidney and tumor but hardly to the brain or liver (Fig. 4.3). The time profile of the amount of Mo atoms in the kidney and tumor indicated a maximum within 1 h after i.p. administration and the excretion by urine during 6 h (Fig. 4.4). A high trapping of Mo atoms in kidney lets us presage a possible damage of the kidney. However, this unpleasant effect may be minimized by maintaining a rapid urine output after the administration through high solubility of PM-8 (1 g/ml at maximum) in water. The antitumor activity of more toxic PM-17 (Tables 4.1 and 4.2 and Fig. 4.2) compared with PM-8 gives us a clue to the mechanism of the antitumor activity of [Mo7O24]6−, provided that PM-17 is generated as one of the metabolites of PM-8 in the biological system as well as the photolysis (Yamase and Ishikawa 2008), as denoted by Eq. (4.1).

Contents of Mo atoms in the liver, kidney, brain, plasma, and tumor for the tumor mice at 0.5 h after i.p. administration of PM-8, which were determined by the radioactivation (to 101Mo) analysis. The contents for PM-8 untreated mice (both tumor mice and normal mice) as backgrounds are also indicated for comparison

Time profile of the Mo contents in the kidney and tumor for the tumor mice after i.p. administration of PM-8
The biological formation of PM-17 is based on the hypothesis that the redox potential of electron-donor sites within tumor cells is negative enough to reduce [Mo7O24]6− to PM-17. The high toxicity of PM-17 seems to be due to the multi-electron redox reaction between PM-17 and host cells in addition to the molecular recognition arising from its T d-symmetric condensed anion structure (Fig. 4.1b). The variety of cations in the [Mo7O24]6− system will modify the residence time of [Mo7O24]6− within the tumor cells (leading to a high amount of Mo atoms) as well as the solubility (or stability) under the physiological condition. The agarose gel electrophoretic patterns for the restriction endonuclease digestion of the pBR322 plasmid DNA treated with PM-8 showed the noncovalent binding of PM-8 with DNA (Tomita et al. 1989), which was different from the case of CDDP showing the covalent attachment to N-7 atoms of DNA guanine bases through the DNA interstrand cross-linking (Pinto and Lippard 1985). This might support low toxicity of PM-8. It is known that the reduction of [Mo7O24]6− is biologically possible through the formation of 1:1 complex with flavin mononucleotide (FMN) with the formation constant (K f) of 8.9 × 103 per molar at pH 5, as shown in Fig. 4.5 where differential pulse polarograms of FMN (2 mM), PM-8 (2 mM), and their mixture in aqueous solutions at pH 5 are shown together with the pseudo-reversible dc polarogram of the mixture (Yamase and Tomita 1990). The one-electron reduction of the 1:1 complex (with half-wave potential (E 1/2) of −0.30 V vs. Ag/AgCl) is approximately reversible and provides a development of the ESR signal due to the formation of both [MoVO5(OH)] site in the [Mo7O24]6− component and FMN semiquinone radical, FMNH• (g = 1.92 with 95,97Mo hyperfine splitting constant of about 57 G and g = 2.00 with 12-line hyperfine structure, respectively) (Yamase 1982; Yamase and Tomita 1990). The analysis of the dc polarograms indicates that the differential pulse polarographic peak of FMN at −0.42 V is shifted to more positive potential at −0.30 V in the presence of PM-8 which gives peak potential at −0.53 V, implying the catalytic reduction of PM-8 through the 1:1 complex with FMN. Since FMN is a prosthetic group in a flavoprotein and acts as an electron carrier for the electron transfer (from NADH to coenzyme Q) coupled with the ATP generation at the site 1 (Stryer 1975), it seems to be reasonable to assume that multi-electron reduction of [Mo7O24]6− to PM-17 starts biologically through the one-electron reduction of the 1:1 [Mo7O24]6−-FMN complex formed on the tumor cell mitochondria with a resultant suppression of the tumor growth due to the inhibition of the ATP generation. The preferential distribution of PM-8 to the tumor (Fig. 4.3) supports the proposed mechanism. Also, the interaction between [Mo7O24]6− and ATP has been investigated using 1H and 31P NMR spectra and has shown that [Mo7O24]6− is a catalysis of the ATP hydrolysis to ADP and phosphate (Ishikawa and Yamase 2006). This implies that the therapy of PM-8 contributes to the elongation of the survival rate, since the ATP hydrolysis is the main source of energy for most biological processes.

Differential pulse polarograms of FMN (2 mM), PM-8 (2 mM), and their mixture in aqueous solutions (at pH 5) containing 0.1 M NaClO4, which are depicted by blue, green, and red curves, respectively. DC polarogram of the mixture is shown by black line for comparison. Top figure shows ESR spectrum observed by the one-electron reduction of the mixture
The crystals of PM-17 isolated from the photolyte of PM-8 at pH 5–7 were X-ray crystallographically characterized, the chemical formula of which is [Me3NH]6[H2MoV 12O28(OH)12(MoVIO3)4]∙2H2O. The structural insight of PM-17 in aqueous solutions has been investigated by using 95Mo NMR, electronic absorption, IR, and electrospray ionization mass (ESI-MS) spectrometries (Yamase and Ishikawa 2008). In the anion of PM-17 (Fig. 4.1b), the ε-Keggin MoV 12 core (showing 12 edge-shared MoVO6 octahedra in the arrangement of 6 diamagnetic MoV 2 pairs with a MoV–MoV bond distance of 2.6 Å) is capped by four MoVIO3 units and remains intact at pH 5–9. A partial detachment of the MoVIO3 unit from PM-17 occurs at high pH level with an accompanying increase in the electronic transition of the MoV 2 pairs. PM-17 was tested for the therapy of more aggressive cancer cells with poor outcome, in order to see the perspective for a novel inorganic drug based on PMs. Expectedly PM-17 showed the cancerocidal potency against AsPC-1 human pancreatic and MKN-45 human gastric cancer cells, with more effective potency than PM-8 (Ogata et al. 2005, 2008; Mitsui et al. 2006). In vivo cell killing by PM-17 for both AsPC-1 and MKN-45 cells is based on the apoptosis in parallel with the autophagy (Ogata et al. 2008). When the cells were suspended in RPMI-1640 culture medium (GIBCO Carlbad, CA. USA) containing PM-17, IC50 values of PM-17 against AsPC-1 cells and MKN-45 cells were 175 μg/ml (63 μM) and 40 μg/ml (14 μM), respectively, which are much smaller than IC50 values of PM-8, 1.65 mg/ml (1.12 mM) and 0.90 mg/ml (0.62 mM). Figure 4.6 shows survival rates of AsPC-1(a) and MKN-45(b) cells (1.5 × 105 cells/5ml) treated with PM-17 for 24 h. In vivo potentiality of PM-17 against the proliferation of AsPC-1 and MKN45 cells was also recognized. The results for the AsPC-1implanted mice are exemplified in Fig. 4.7, where 2 × 106 AsPC-1 cells were transplanted into the back of nude mice, and after 10 days, intratumoral injections of PM-17 (125 μg or 500 μg dissolved in 100 μl of saline) were performed for 10 days with 2-day intermission on day 6. The control mice were treated with 100 μl of saline per day under the same conditions. PM-17 inhibited tumor growth: on 41 days after implantation of the tumor, for example, it showed rates of 33.5 and 68.5 % at the doses of 125 μg and 500 μg/body/day compared with controls, respectively (Fig. 4.7a). The tumors observed at 41 day for five mice/group administrated by PM-17 500 μg/body/day are pictured (at the right side) in comparison with those of the control (at the left side), which were measured by a micrometer caliper (Fig. 4.7b). There was no loss of body weight for mice injected with PM-17 in this experiment, implying that the cytotoxicity of PM-17 is not significant as far as the present therapy of 500 μg/body/day at least was employed (Fig. 4.7c). Little cytotoxicity of PM-17 through the therapy was supported also by the hematological examination and blood chemistry assay for the mice treated with PM-17 (of 1.0 mg/body/day) which indicated no damage of kidney and hepatic functions as described below. Thus, PM-17 exhibits the antitumor activity which inhibits the proliferation of AsPC-1 human pancreatic cancer cells in a dose-dependent manner in vivo. The thin sections of tumors on day 10 after a single intratumoral injection of PM-17 (500 μg/100 μl saline) or PM-8 (4 mg/100 μl saline) into the AsPC-1 transplanted nude mice showed a pathological hyalinization on both hematoxylin and eosin (H&E) staining and terminal deoxynucleotidyl transferase-mediated “nick-end” labeling (TUNEL) staining. Figure 4.8 shows the results of H&E (a) and TUNEL (b) for PM-17 in comparison with the control (with the injection of 100 μl 0.9 % NaCl saline). The genotoxicity of PM-17 and PM-8 for the AsPC-1 and MKN-45 cells during the growth indicated the formation of apoptotic small bodies for the cells due to the DNA fragmentation which was revealed by Hoechst dye 33342 (Sigma) staining of the suspension containing the cells treated with PM-17 and PM-8 (with IC50) for 24 h (Ogata et al. 2005, 2008; Mitsui et al. 2006). The morphological analysis by Hoechst staining indicated a sharp DNA laddering after the PM treatment on the agarose gel electrophoresis of cell DNA extract, which allowed us to confirm the induction of apoptosis by PM-17 and PM-8.

Effect of PM-17 on in vitro survival of AsPC-1 (a) and MKN-45 (b) cells (1.5 × 105 cells/5 ml for each cell) for 24 h. Each point indicates the percentage of living cell in three independent experiments (bars depict s.d.)

In vivo tumor growth inhibition by PM-17 for AsPC-1 implanted mice. 2 × 106 AsPC-1 cells were transplanted into the back of Balb/c nude mice, and after 10 days, intratumoral injections of PM-17 (125 μg or 500 μg dissolved in 100 μl of saline) were performed for 10 days with 2-day intermission on day 6. The control mice were treated with 100 μl of saline per day under the same conditions. Changes in tumor volume (a) and body weight (c) for five mice per group are depicted with photographs (b) for the mice on day 41after the tumor implantation

Hematoxylin and eosin (H&E) (a) and terminal deoxynucleotidyl transferase-mediated “nick-end” labeling (TUNEL) (b) stainings for thin sections of the tumor on the 10th day after a single intratumoral injection of PM-17 (500 μg/100 μl saline) into the AsPC-1 transplanted nude mice, in comparison with the control (with the injection of 100 μl 0.9 % NaCl saline)
Transmission electron microscope (TEM) was employed for the analysis of ultrastructural alterations in AsPC-1 and MKN-45 cells treated with PM-17 (with IC50) for 12 h. Figure 4.9 shows the TEM images of the AsPC-1 cells treated with and without PM-17 (Ogata et al. 2008). The average dimensions of control AsPC-1 cells ranged from 13 to 15 μm in diameter, with a nucleus ranging from 7 to 10 μm. In the PM-17 untreated AsPC-1 cells (control), a number of round-shaped mitochondria make the predominant cytoplasmic feature observed along with the external microvilli (Fig. 4.9a, b). In comparison with the control, the morphological alterations of the cells treated with PM-17 were distinguished by the occurrence of both chromatin/chromosome condensation and damage of mitochondria (Clarke 1990). The former in nucleus leads to the apoptotic cell death (Fig. 4.9c), and the latter in the cytoplasm indicates the formation of smaller round organelles which results from damage throughout swelling of mitochondria (Fig. 4.9d). Some of PM-17-treated AsPC-1 cells contained a number of autophagosomes/autolysosomes in the cytoplasm (Fig. 4.9e), which are a characteristic feature of autophagy (Ogata et al. 2008). No significant decrease in the number of microvilli on the surfaces of the treated cells suggests little alteration in membrane permeability on the exposure of the cells to PM-17. To visualize the autophagy, the green fluorescent protein (GFP)-tagged light chain3 (LC3) was expressed in the AsPC-1 cells: during the autophagic process LC3 is concentrated in autophagosomes, and thereby the punctate fluorescence emitted by GFP-LC3 is a good indicator of autophagy (Takeuchi et al. 2005). The result is shown in Fig. 4.10 (Ogata et al. 2008). While the diffuse cytoplasmic localization of GFP-LC3 was observed for the control, the PM-17 (with IC50 and for 24 h)-treated AsPC-1 cells transfected by GFP-LC3 expression plasmid showed the punctate fluorescence (Fig. 4.10a). The expression of LC3 (I and II) proteins for the PM-17-treated AsPC-1 cells indicated the autophagy in contrast to the control showing no expression of LC3 on a Western blotting picture (Fig. 4.10b). Similar result was also obtained for the PM-17-treated MKN-45 cells (Ogata et al. 2008). Since LC3-II, as a truncated form of LC3, is necessary to bind with the seclusion membrane at the primary stage of the autophagy (Takeuchi et al. 2005), the above results reveal that PMs induce the activation of not only apoptosis but also autophagy as cell death pathways. The determination factor of the magnitude of both apoptosis and autophagy in the PM-17-induced cell death remains still unclear, although the activation of caspase-3 and caspase-12 as a regulatory factor could be identified by using a Caspase-Glo™ 3/7 assay kit (Lamparska-Przybysz et al. 2005; Golstein and Kroemer 2007).

TEM images of the AsPC-1 cells treated with (c–e) and without (a and b) PM-17

In vitro expression of autophagy by the green fluorescent protein (GFP)-tagged light-chain 3 (LC3) in the PM-17-treated AsPC-1 cells. 2 × 105 AsPC-1 cells were cultured with the BRMI-1640 growth medium for 24 h in 24-wells plate, transfected with GFP-LC3 expression plasmid (0.8 μg/well) with lipofectamine 2000 transfection reagent (2 μg/well). After 24 h the cells were treated with and without PM-17 (175 μg/ml saline) for 24 h, and the distribution (a) of fluorescence of GFP-LC3 under fluorescence microscope and a Western blotting picture (b) were investigated
After 24 h from the last intratumoral injection of PM-17 (500 μg or 1.0 mg/100 μl saline per day) through the same procedure employed in the tumor growth inhibition test for the AsPC-1 transplanted mice (Fig. 4.7), the mice were anesthetized and blood samples were taken. Whole blood samples were used for the hematological test. Blood sera were assessed for blood urea nitrogen (BUN), creatinine, total bilirubin, total protein, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP). Table 4.3 shows the results for both control mice (with 100 ml of saline/day under the same conditions used for the animals treated with PM-17) and negative control mice (received neither tumor transplantation nor intratumoral injection) are added. The results of the hematological examination and blood chemistry showed no significant difference between negative control (neither tumor transplantation nor PM-17 injection) and PM-17-treated groups, indicating that there was neither impaired kidney nor hepatic function in the above administration model of PM-17 (Ogata et al. 2008).
Thus, the photochemically 12-electron-reduced species of [Mo7O24]6−, [H2MoV 12O28(OH)12(MoVIO3)4]6−, which may be a candidate of metabolites of PM-8, is in vitro and in vivo more inhibitory against solid cancer growth than [Mo7O24]6−. Intratumoral injection of PM-17 showed a significant efficacy in an experimental mice inoculation model of AsPC-1 and MKN-45, indicating a therapeutic usefulness of PM-17 as alternative chemotherapy of PM-8. Together with the fact that the pancreatic cancer is an aggressive form of cancer and has one of the poorest outcomes of all cancers, the development of the drug-delivery system for PM-17 is required for the realization of PM-17 as a novel anticancer drug.
4.3 Antiviral Activity of Polyoxotungstates
In the course of in vitro antiviral assay of a variety of PMs, especially polyoxotungstates, PM-19 (the anion of which is shown in Fig. 4.1c) exhibited a potent inhibition both in vitro and in vivo against a broad spectrum of DNA viruses including herpes simplex virus (HSV) type 1 (HSV-1) and HSV type 2 (HSV-2), thymidine kinase-deficient (TK−) HSV mutant, and human cytomegalovirus (HCMV) (Fukuma et al. 1991; Dan et al. 2003). PM-19 inhibited the viral penetration of the host cells and the second round of infection without its direct interaction with HSV virions (Dan et al. 1998, 2003). PM-19-pretreated cells provided approximately 10-fold enhancement of the anti-HSV potency of PM-19 compared with the cells treated with PM-19 only after infection (Dan et al. 2002). Together with the fact that PM-19 inhibited the interaction between HSV envelope protein (gD, glycosylated ectodomain) and cell-surface membrane proteins (HVEM) (Dan and Yamase 2006), the pretreatment of the cell with PM-19 suggests a strong binding of PM-19 with HVEM. Dose response of PM-19 to its in vivo activity for the mice (12–24/group) infected i.p. with 2.8 × 102 pfu of HSV-2 is shown in Table 4.4, where PM-19 at different dose was i.p. administrated to the HSV-2-infected mice once daily for 3 days by starting immediately after virus challenge. As shown in Table 4.4, a significant protection was achieved with doses of PM-19 at the range of 0.3–50 mg/kg. Acyclovir (ACV) as an approved drug (viral DNA-polymerase inhibition by ACV-triphosphate formed in the cell) against HSV (Elion et al. 1977), tested in parallel with PM-19, showed only weak anti-HSV activity even if administrated at doses of 100 mg/kg/day for 6 days. 50 % effective dose (ED50) values on the survival rates (on day 14 after infection) were 0.25 mg/kg/day for PM-19 and >100 mg/kg/day for ACV. 50 % lethal dose (LD50) values of PM-19 and ACV on i.p. administration were 384 and 739 mg/kg/day, respectively. The mechanism of in vivo anti-HSV activity of PM-19 is attributed to an interference with the virus uptake by the cells and/or a clearance of HSV and also partially to the activation of macrophages (Ikeda et al. 1994). The latter effect was recognized by measuring the phagocytic activity of peritoneal macrophages, which was increased by i.p. administration of PM-19. The marked in vivo efficacy of PM-19 due to such multiple mechanisms involving a direct inhibitory effect on virus replication (inhibition of virus adsorption) as well as host-mediated antiviral response (activation of macrophage activity) was also observed for the HSV-2-infected immunosuppressed mice (Ikeda et al. 1993).
CC50 and EC50 (median cytotoxic and effective concentrations, respectively) values for seven PMs including four V/W-mixed PMs (PM-43, PM-47, PM-1001, and PM-1002) and three Ti/W-mixed PMs (PM-518, PM-520, and PM-523) against RNA viruses are listed in Tables 4.5 and 4.6, respectively (Shigeta et al. 2003). In Table 4.5, CC50 values of PMs for Madin-Darby canine kidney (MDCK), Vero, HEp-2, and MT-4 cells were determined by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetry for the cells cultured in the presence of PMs (with a variety of concentrations) at 35 °C during 5 days (Pauwels et al. 1988; Watanabe et al. 1994; Inouye et al. 1992, 1995; Mori and Shigeta 1995). All V/W- and Ti/W-mixed Keggin or Wells–Dawson PMs (Fig. 4.1c–f) show little toxicity for MDCK, Vero, and HEp-2 cells in CC50 values more than 200 μM, while they are slightly toxic for MT-4 cell: Ti/W-mixed PMs show IC50 > 100 μM and V/W-mixed PMs show CC50 = 41.9–47.4 μM. The listed PMs were inhibitory to the replication of HIV-1(IIIb) in EC50 values at the range of 0.03–2.0 μM and were selectively inhibitory against other RNA viruses such as dengue fever virus (DFV), influenza virus A (FluV-A), respiratory syncytial virus (RSV), parainfluenza virus type 2 (PfluV-2), and canine distemper virus (CDV) with more inhibitive values than ribavirin as an inhibitor of the viral RNA synthesis due to its inhibition of inosine monophosphate dehydrogenase activity in cells (Gilbert and Knight 1986): PM-43, PM-518, and PM-523 were not inhibitory against PfluV-2, CDV, and DFV, respectively, as indicated by EC50 values more than 50 μM. PM-1001 and PM-1002 with the same anion structure (Fig. 4.1e), in which the former anion is the one-electron oxidation species of the latter anion consisting of (VIVO)3 triangle sandwiched by two α-B [SbW9O33]9− ligands, were significantly potent inhibitors of HIV-1. These PMs were also inhibitory against DFV (which is causative of dengue fever recently migrated from the tropical zone to the mild zone) and RSV (which causes bronchitis and/or pneumonia through a respiratory infection among infants) in EC50 values of 0.45–1.95 μM and 0.16–0.75 μM, respectively. DFV and RSV have been known as clinically important pathogens. A difference in the antiviral activity of PMs among a variety of the RNA viruses may be associated with the dependence on the composition of the target amino acid sequence of the viral envelope glycoproteins, as indicated for the different inhibition of dextran sulfate compounds (with a variety of molecular weight) (Hosoya et al. 1991).
K13[Ce(SiW11O39)2]∙26H2O (JM1590) and K6[BGa(H2O)W11O39]∙15H2O (JM2766), which inhibited HIV-1 and simian immunodeficiency viruses at concentrations as low as 0.008–0.8 μM, seem to be the most potent anti-HIV compounds (Yamamoto et al. 1992). They also inhibited the formation of giant cells (syncytium) in co-cultures of HIV-infected HUT-78 cells and uninfected Molt-4 cells. The anti-HIV activity for most of PMs could be based on inhibition of virus-to-cell binding (due to a high affinity for gp120 as the viral glycoprotein involved in virus adsorption to the cells), since there was a good correlation between the inhibitory effects on both HIV-1-induced cytopathicity and syncytium formation and also a close correlation between their inhibitory effects on both the syncytium formation and the interaction with the viral envelope glycoprotein gp-120. Selective indices, SI (=CC50/EC50), of the V/W-mixed Keggin PMs (especially PM-1001 and PM-1002) against HIV-1 in MT-4 cell line were higher than those of AZT (as the inhibitor for the reverse transcriptase) (Mitsuya et al. 1985) and dextran sulfate in mol. wt. 5,000 (DS5000) (as the inhibitor for the syncytium formed by the cell-to-cell infection) (Mitsuya et al. 1988, Baba et al. 1988, Callahan et al. 1991). Table 4.7 shows the result of the multinuclear activation of the galactosidase indicator (MAGI) assay for HeLa CD4/LTR-β-Gal cell line (Kimpton and Emerman 1992), which indicates SI values (>10,000 and >5,500 for PM-1001 and PM-1002, respectively) are higher than for DS5000 (>3,000) and AZT (>2,700), strongly supports their high antiviral activity against HIV-1. The fact that the cocultivation of Molt-4 cells and HIV-1-infected Molt-4/IIIb cells with PM-1001 (in 10 μM) and PM-1002 (in 2 μM) provides a perfect inhibition of the syncytium formation (the multinuclear giant cell formation) assists the plausible binding of PMs with HIV-1 gp120, in contrast to the binding of AMD3100, a bicyclam derivative of octahydrochloride dihydrate of 1,1′-[1,4-phenylenebis(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane, to X4 receptor (CD4 and/or CXCR4) (Donzella et al. 1998). This lets us predict clinically synergistic anti-HIV activity by a use of a combination of PMs with AMD3100.
In vivo anti-HIV activity was carried out by using recombinant vaccinia viruses (rVV), vPE16 expressing HIV-1 env gene (vPE16-env), vP1206 expressing HIV-IIIb gag/pol gene (vP1206-gag/pol), and wild WR strain (WR, control rVV). After Balb/c mice (8-week-old, female, five mice/group) were infected with 5.0 × 106 pfu of vPE16-env and vP1206-gag/pol on day 0, 12.5 or 125 mg/kg/day of each of PM-523, PM-1002, and K12[(VO)3(BiW9O33)2]∙27H2O (PM-1208, Botar et al. 2001) was i.p. once daily administrated for 3 days from day 2. On day 5 after infection, ovaries of mice were removed, homogenized, and the virus activity was evaluated by the β-galactosidase activity in the supernatants. In vivo results of the cytotoxicity (a) and the antiviral activity (b) of three PMs are shown in Fig. 4.11 (Matsui K, Yamase T, Kobayashi K, Saha S, Xin Q, Takeshita F, Okuda K Unpublished results in paper preparation). As shown in Fig. 4.11a, the PM dose for the challenge was determined along with the result of in vivo cytotoxicity of PMs: each of PM-523, PM-1002, and PM-1208 provided a100 % survival rate on the 4th day after its i.p. administration of less than 125 mg/kg/day. All PM-523, PM-1002, and PM-1208 suppressed the viral activity of both vPE16-env and vP1206-gag/pol in vivo with almost the same activity for administrations of 12.5 and 125 mg/kg/day/mouse. Especially, PM-523 and PM-1208 exhibited a potent antiviral activity against vPE16-env (with p < 0.01) and vP1206-gag/pol (with p < 0.05), respectively (Fig. 4.11b). Since no HIV antigen-specific immune responses were detected in the mice treated with PMs after virus infection, it was inferred that the PMs interact with HIV antigen to lead to the suppression of viral replication along with above the in vitro results.

In vivo cytotoxicity (a) and antiviral activity (b) for three PMs, 523, 1,002, and 1,208. Survival time indicates the time (in h) after i.p. administration of PM of 125 (open square), 250 (filled square), 500 (open circle), and 1,000 (filled circle) mg/kg/mouse. The activity of the infected virus (vPE 16, vP 1,206, or WR) in mice was evaluated on day 5, by the β-galactosidase activity for ovaries of mice which were i.p. administrated by 12.5 and 125 mg/kg/day/mouse of each PM (PM -523, PM -1002, or PM -1208) for 3 days from days 2 to 4 after the virus infection on day 0
HIV-1 inhibition based on the protease inhibition has been proposed for Wells–Dawson structural PMs containing one peroxoniobium or oxoniobium unit substituted for one tungsten unit in the parent framework, K7[P2W17(NbO2)O61] and K7[P2W17NbO62]: the Nb-substitution in the parent [P2W18O62]6− (Fig. 4.1f) provided the high stability in the physiological pH level (Judd et al. 2001). These compounds exhibited EC50 = 0.17–0.83 μM and CC50 = 50 to >100 μM (for peripheral blood mononuclear cells) and showed the noncompetitive inhibition of purified HIV-1 protease (with inhibition constants of 1.1 ± 0.5 and 4.1 ± 1.8 nM in 0.1 and 1.0 M NaCl, respectively). Unfortunately, the estimated EC50 values are inferior to those (0.3-0.5 nM) of the peptidemimetic PI containing a bis-tetrahydrofuranyl urethane and 4-methoxybenzenesulfonamide (TMC 126) as a clinically developed protease inhibitor (Yoshimura et al. 2002; DeClerq 2005).
It is well known that the initial steps of influenza virus replication start with a very unique interaction between the virus particle and the cellular membrane (Shigeta 1999; Blumenthal et al. 2003) Influenza virus acquires its infectivity after cleavage of the hemagglutinin (HA) peptide in its envelope into HA1 and HA2 by the action of the host cell protease. Under conditions of low endosome pH, the cleaved HA molecule changes its stereoscopic conformation, and the N-terminus of the HA2 cleaved site comes in contact with the cellular membrane which fuses with the virus envelope. Simultaneously, influx of protons through the matrix protein (M2) ion channels occurs and results in uncoating of the inner coat of ribonucleoprotein (M1) sheath. The liberated ribonucleoprotein (RNP) then moves to the nucleus. Influenza viruses are classified to three types (A, B, and C) based on the differences in their RNP antigenicity. Influenza A virus is further subdivided into subtypes based on differences in the antigenicity of both components of HA and neuraminidase (NA) for the virus. The current nomenclature system for human influenza virus takes the geographical location of the first isolation, strain number, year of isolation, and antigenic description of HA and NA, for example, A/Ishikawa/7/82/(H3N2) or B/Singapore/222/79 (Shigeta 1999). Most of PMs are inhibitory only at the virus adsorption stage, namely, the administration of PMs from 1.5 h after the virus infection shows little efficacy. Interestingly, PM-523 (as Keggin structure), a Ti/W-mixed PM, did not inhibit the adsorption of the influenza A virus (FluV-A) onto the cell membrane (0–1.5 h after infection) but inhibits the fusion between the FluV-A envelope and the cellular membrane (1.5–120 h after infection) (Shigeta et al. 1996). Such a different mechanism of anti-influenza activity for PM-523 led to both in vitro and in vivo synergistic efficacies in combination with ribavirin (as a viral RNA synthesis inhibitor) or zanamivir (NA inhibitor). The synergistic therapeutic effect in mice infected with FluV-A (H1N1/PR8) using PM-523 and ribavirin was examined, as shown in Fig. 4.12 (Shigeta et al. 1997, 2006). All of the control mice (inbred 8-week-old female Balb/c) were infected with 10 lethal dose of FluV-A, which corresponds to 1.6 × 104 median tissue culture infection dose (TCID50) titers in MDCK cell line. From 8 h after infection, the mice (10 mice/group) were treated with aerosols of PM-523 and ribavirin either singly or in combination every 12 h for 4 days. Compound solutions were prepared in phosphate-buffered saline (PBS) solutions (at pH 7.2), and the infected mice were exposed to these compounds by using a continuous aerosol generator for 2 h at a rate of distribution of 120 μl of solution/h to each chamber. Each infected 20-g mouse in 10-mice/group was placed in a chamber separately. The average diameter of the aerosol particle was set to be 2.1 μm. In this experiment, the dose of the compound given to the mice was expressed in terms of the concentration of compound in the stock solution used for the aerosol exposure. The compound-untreated mice died on day 9 after infection. Similarly, all the 2.4-mM PM-523 and 40-mM ribavirin-treated mice died on days 9 and 8, respectively. 50 and 60 % of the mice treated with 4.8-mM PM-523 and 40-mM ribavirin, respectively, survived on day 9 after infection. EC50 values of PM-523 and ribavirin on day 9 after infection could be evaluated to be 4.8 and 70 mM, respectively. When a 2.4-mM PM-523/40-mM ribavirin combination was treated for the infected mice, 80 % of the mice survived on day 9 after infection, and 60 % of the mice survived still at the end of the experiment (on day 14 after infection), as shown in Fig. 4.12. 4.8-mM PM-523 and 80-mM ribavirin-treated mice showed 30 and 20 % survivals on day 14 after infection, respectively. Thus, the survival rate for the mice treated with the combination of PM-523 and ribavirin at a ratio of 1:16 was significantly high compared with the one with either 4.8-mM PM-523 or 80-mM ribavirin. The lungs of both the infected and untreated mice and the mice treated with either 2.4-mM PM-523 or 40-mM ribavirin became swollen and reddened due to congestion. On the other hand, the lungs of the infected mice treated with the combination of 2.4 mM PM-523 and 40-mM ribavirin remained normally grayish white and were not congested. The viral titer in the lungs for the infected mice treated with and without PM-523 and/or ribavirin was measured in every 24 h after infection, and the lungs for the measurement were removed from three mice for each group prepared by the same procedure of both infection and administration as the therapeutic effect experiment. The change of the viral titer in the lung is shown in Fig. 4.13 where the results for the group of the uninfected and untreated mice are added into the results for four groups of the infected mice with and without compounds for comparison (Shigeta et al. 1997). While there was no detection of virus for the uninfected mice, the viral titers in the infected mice lungs increased to 105.4 TCID50/g at 48 h after infection and decreased to 103.5 and 103.2 TCID50/g at 72 and 96 h, respectively. The viral titers in the lungs for the infected mice treated with the combination of 2.4-mM PM-523 and 40-mM ribavirin were consistently lower than for the infected mice treated with either 2.4-mM PM-523 or 40-mM ribavirin. To identify the target of the antifusion activity for PM-523, PM-523-resistant FluV-A strains were selected after several passages of FluV-A H3N2/Ishikawa in MDCK cells in the presence of an effective dose of PM-523. The mutations conferring resistance to PM-523 indicated that isoleucine 202 and lysine 189, which were located at the interface edges of the trimer molecules of HA1, were substituted with threonine and asparagine, respectively. This strongly implies that PM-523 binds to the interface edges of HA trimers and inhibits the opening of HA1 trimers to lead to the inhibition of the fusion of viral envelope to cellular membrane by HA2 hydrophobic amino acids at the edge of the cleavage site (Shigeta 2001; Shigeta et al. 2006). The fusion inhibition by PM-523 is unique and the synergistic anti-influenza virus activity of PM-523 with ribavirin or zanamivir is promising as a novel therapeutic drug for the influenza virus infection, as exemplified in Figs. 4.12 and 4.13. PM-523 and its congeners Ti/W-mixed PMs (e.g., PM-19 and PM-504) and V/W-mixed PMs have a broad antiviral activity against enveloped RNA viruses (Table 4.6) and may be developed as broad-spectrum drugs for acute respiratory infection (ARI) caused by orthomyxoviruses (influenza viruses A, B, and C) and paramyxoviruses (human parainfluenza viruses 1, 3, 2, and 4; measles virus; and RSV).

Synergistic therapeutic effect in mice infected with FluV-A (H1N1/PR8) using PM-523 and ribavirin. All of the control mice (inbred 8-week-old female Balb/c) were infected with ten lethal doses of FluV-A, which corresponds to 1.6 × 104 median tissue culture infection dose (TCID50) titers in MDCK cell line. From 8 h after infection, the mice (ten mice/group) were treated with aerosols of PM-523 and ribavirin either singly or in combination every 12 h for 4 days. Compound solutions were prepared in phosphate-buffered saline (PBS) solutions (at pH 7.2), and the infected mice were exposed to the compounds by using a continuous aerosol generator for 2 h at a rate of distribution of 120 μl of solution/h to each chamber. Each infected 20-g mouse was placed in a chamber separately; no compound (filled circle)-treated, 40 mM ribavirin (filled triangle)-treated, 80 mM ribavirin (filled square)-treated, 2.4 mM PM-523 (open triangle)-treated, 4.8 mM PM-523 (open square)-treated, and 40 mM ribavirin/2.4 mM PM-523 combination (open circle)-treated mice. Levels of significance of p < 0.001 for 2.4-mMPM-523/40-mM ribavirin combination, p < 0.01 for 4.8-mM PM-523, and p < 0.05 for 80-mM ribavirin-treated mice on day 14 after infection

Change of viral titers in the lungs of the infected mice with and without treatment of PM-523 and/or ribavirin after infection (1.6 × 104 TCID50 of FluV/A/PR8/mouse). The values for the uninfected and untreated mice are plotted for comparison. Uninfected and untreated (open triangle), infected and untreated (filled circle), 40-mM ribavirin-treated (filled square), 2.4-mMPM-523-treated (open square), and 40 mM ribavirin/2.4 mM PM-523 combination-treated (open circle) mice
Since most of the antiviral PMs inhibit the adsorption of the enveloped RNA viruses toward the host cells, these PMs are expected also to inhibit severe acute respiratory syndrome coronavirus (SARS-V) (Shigeta and Yamase 2005). SARS is severe and highly contagious, which first emerged in China in 2002 and spread within a few months from its origin to all over the world. In vitro antiviral activities of Ti/W- and V/W-mixed PMs were assayed for SARS-V together with other coronaviruses of transmissible gastroenteritis virus of swine (TGEV) and feline infectious peritonitis virus (FIPV) (Yamase 2005; Shigeta et al. 2006). As shown in Table 4.8, all of the PMs exhibited anti-TGEV activities. The V/W-mixed PMs displayed the anti-SARS-V activity, while they showed no apparent relationship between the structure and the anti-FIPV activity. Overall, all (PM-1002, PM-1207, PM-1208, and PM-1213) of tris(vanadyl)-18-tungstate anion, [(VIVO)3(XW9O33)2]12− (X = AsIII, SbIII, BiIII) and [(VIVO)3(PVW9O34)2]12− (which consists of (VIVO)3 ring sandwiched by two A-α-[PVW9O34]9− ligands), in addition to PM-1001 as the one-electron oxidation species ([(VIVO)2(VVO)(XW9O33)2]11−) of PM-1002, exhibit potent and selective activities (Table 4.8). Since [{Mn(H2O)}3(SbW9O33)2]12−, [Co3(AsW9O33)2]12−, [Cu3(AsW9O33)2]12−, and [Co4(H2O)2(PW9O34)2]10−, as similar derivative anions, did not exhibit any efficacy against the coronaviruses (data not shown), the structural significance of tris(vanadyl) ring moiety is pointed out for observation of in vitro antiviral activity against the coronaviruses.
The first step of the infection of host cells by influenza virus is the binding (adsorption) of the influenza virus HA to sialic acid residues of the carbohydrate side chains of cellular proteins projecting from the plasma membrane that the virus exploits as receptors. The second step is the conformation change of HA as a membrane fusogen at the acidic pH (pH 5.0 at 37 °C for optimal fusogenic activity) which induces the fusion of the viral envelope membrane with the membrane of the endosome. This expels the viral RNA into the cytoplasm, where it can begin to replicate (Shigeta 1999; Blumenthal et al. 2003a). Together with the specific binding of PM-19 with HVEM as a TNF receptor superfamily (which was suggested by the investigation of anti-HSV activity of PM-19) (Dan and Yamase 2006), therefore, the antiviral mechanism of the PMs, which inhibit the adsorption of the enveloped RNA (or DNA) viruses toward the host cells, is likely to involve the functional quenching of the sialic acid residues of the carbohydrate side chains of cellular proteins, if we considered that binding between multiple HA ligands of influenza virus and sialic acid surface receptors of an erythrocyte for the cell during viral infection occurs with extremely strong affinity of 1015 per molar, while the association constant for a single sialic acid-HA interaction is only 103 per molar (Mammén et al. 1998). Sialyl/sulfotransferases are responsible for the biosynthesis of these carbohydrate chains: sialyltransferases transfer sialic acid residues from cytidine-5-monophosphato-sialic acid, CMP-Neu5Ac, to specific acceptor sugar chains, whereas sulfotransferases transfer sulfate groups from adenosine 3′-phosphate 5′-phosphosulfate (PAPS) to specific acceptor sugar chains. Most of Ti/W- and V/W-mixed PMs and multiply reduced spherical polyoxovanadates such as K5[H6KV13O31(MePO3)3]∙16.5H2O (Yamase et al. 2000), Na12H2[H2V18O44(N3)]∙30H2O (Yamase et al. 1997), K10[H2V18O42(H2O)]∙16H2O, and K10H3[V18O42(Cl)]∙12H2O (Fig. 4.1g, Yamase et al. 1996b) inhibited in vitro enzymatic activities of specific sialyl/sulfotransferases such as α2,3-sialyltransferases (ST3Gal-I and ST3Gal-III), α2,6-sialyltransferases (ST6GalNAc-I and ST6Gal-I), Gal 3-O-sulfotransferase-2 (Gal3ST-2), GlcNAc 6-O-sulfotransferase-1 (Gn6ST-1), Gal 6-O-sulfotransferase (Gal6ST), heparan sulfate 6-O-sulfotransferase, β1,4-galactosyltransferase-I (β4Gal-TI), β1,3-GlcNAc-transferase-2 (β3Gn-T2), and α1,3-fucosyltransferase-III (FUT3), at sub-nano and nanomolar concentrations with the noncompetitive inhibitory mode for both donor and acceptor substrates (Seko et al. 2009): for example, PM-504 inhibited ST3Gal-1 activity with inhibition constant (K i) ~ 0.5 nM (0.47 and 0.53 nM for core 1 as an acceptor and CMP-Neu5Ac as a donor, respectively) which were much lower than K i = 29 nM for the previously reported lowest inhibitors of glycosides analogues (Schwörer and Schmidt 2002). The inhibition mode of PM-504 on ST3Gal-1 is shown in Fig. 4.14, where plots of the reciprocal velocity versus the reciprocal substrate concentration with and without PM-504. The steady-state kinetics results indicate the noncompetitive inhibition of PM-504 against ST3Gal-1, suggesting the interaction of PM-504 with a variety of the binding sites of ST3Gal-1. 50 % inhibitory concentration (IC50) values of a variety of PMs for two sialyl/sulfotransferases, ST3Gal-1 and Gal3ST-2, are listed in Table 4.9 (Seko et al. 2009). IC50 values of PM-19, PM-43, PM-518, PM-523, and PM-1002 for ST3Gal-1 were extremely small, 0.4, 0.2, 0.3, 0.6, and 0.4 nM, respectively. These values were 2–3 orders of magnitude smaller than for Gal3ST-2 (40, 50, 20, 100, and 60 nM, respectively). On the other hand, the spherical polyoxovanadates inhibited both enzymatic activities in the same order of magnitude of nanomolar concentrations: IC50 values of K5[H6KV13O31(MePO3)3]∙16.5H2O, Na12H2[H2V18O44(N3)]∙30H2O, K10[H2V18O42(H2O)]∙16H2O, and K10H3[V18O42(Cl)]∙12H2O were 7, 9, 7, and 7 nM, respectively, for ST3Gal-1 and were all 3 nM for ST3Gal-1. It should be noted that the inhibition of the above PMs for in vitro enzymatic activities of sialyl/sulfotransferases is at least 100- to 1,000-fold stronger than any other known inhibitors (Schwörer and Schmidt 2002; Rath et al. 2004). The result for the inhibitory effect of PM-504 on the mutated ST3Gal-1 enzymes implied that the inhibition of PMs against the adsorption of the enveloped RNA viruses toward the host cells is due to the strong interaction of the PMs not only with 335Arg residue in the C-terminal region of ST3Gal-1 but also with other basic amino acids residue (Seko et al. 2009). Thus, some types of PMs have the ability to inhibit specific sialyl/sulfotransferases, indicating that the PMs directly inhibit the activities of enzymes involved in carbohydrate metabolism to lead to the inhibition of the binding of the HA ligands of influenza virus with sialic acid surface receptors. As shown in Table 4.9, no activity of PM-8 and PM-17 to the sialyl/sulfotransferases enzymes corresponds to missing of the antiviral activity for these polyoxomolybdates.

Inhibition mode of PM-504 on the enzymatic activity of ST3Gal-1 at various concentrations of core 1 (a) and CMP-Neu5Ac (b) in the absence and presence of PM-504. (a): 0 (filled circle), 0.15 nM (open circle), and 0.3 nM (filled square) of PM-504 in the presence of 1.6-μM CMP-Neu5Ac. (b): 0 (filled circle), 0.1 nM (open circle), and 0.2 nM (filled square) of PM-504 in the presence of 1-mM core 1
4.4 Antibacterial Activity Against Gram-Positive Methicillin-Resistant Staphylococcus aureus and Vancomycin-Resistant Staphylococcus aureus
Most polyoxotungstates show a synergistic effect with β-lactam antibiotics against methicillin-resistant Staphylococcus (S.) aureus (MRSA) as Gram-positive bacterium which was first isolated in 1961 after the introduction of methicillin into clinical use against S. aureus strains (Jevons 1961) and have induced clinically serious problems due to their strong resistance to many antibiotics. The inhibition by methicillin against S. aureus strains is based on the binding of β-lactam antibiotics to penicillin-binding proteins (PBPA, PBP2, PBP3, and PBP4) as the peptidoglycan-synthetic enzyme on the membrane surface. Different from such a methicillin-susceptible Staphylococcus aureus (MSSA), MRSA possesses PBP2′ (sometimes called PBP2a or MecA) as an additional PBP on the membrane surface, which is coded by mecA gene and exhibits low affinity to the β-lactams. Concerning the existence of PBP2′ on the surface, MRSA strain can be classified into two, constitutive (PBP2′ exists irrespective of the β-lactams) and inducible (PBP2′ is induced on the surface in the presence of the β-lactams) (Ubukata et al. 1985; Matsuhashi et al. 1986). Vancomycin as a glycopeptide has become the antibiotic of last resort for the hard-to-treat infection of MRSA infection. However, eventfully bacteria developed resistance to vancomycin, too, in some cases by changing an amide to an ester in a bacterial glycopeptide cell wall precursor. Since Mu50, as a vancomycin-resistant S. aureus (VRSA) strain, was first isolated in 1997 (Hanaki et al. 1998a, b), other VRSA strains have emerged which produce structurally unusually complicated cell wall precursor with few interaction with vancomycin (Boneca and Chiosis 2003). Thus, novel chemotherapeutics against MRSA and VRSA strains are strongly required.
The effect of PMs against both MRSA strains of SR3605 (constitutive) and ATCC43300 (inducible) has been investigated (Yamase et al. 1996a, b, c), as shown in Table 4.10 where both the minimum inhibitory concentration (MIC in μM unit) and the fractional inhibitory concentration (FIC) index for PMs were measured by the agar dilution checkerboard method for the evaluation of the effect in combination with oxacillin (Hallander et al. 1982). As shown in Table 4.10, all of the polyoxotungstates tested in this study exhibited a synergistic effect with oxacillin, as a FIC index less than 1/2 is defined as a synergistic effect. In particular, the synergistic potential of Keggin-structural polyoxotungstates and their lacunary species, [XW11O39]n− and [XW9O34]n−, were high: FIC indices of these compounds against SR3605 and ATCC43300 were 0.010–0.156. No synergistic activity was observed for Na2WO4∙2H2O. Dodeca- and nanotungstosilicates such as Na4[SiW12O40]∙nH2O, A-β-Na9[SiW9O34H]∙23H2O (PM-30), and A-α-Na10[SiW9O34]∙18H2O were highly synergistic (with FIC = 0.019, 0.018, and 0.010, respectively) against ATCC43300, suggesting a significance of the Si atom as a hetero atom in polyoxotungstates against this strain. Most of the PMs of molybdenum (with an exception of [SiMo12O40]4− as discussed below) and vanadium showed little effect against SR3605, as shown in Table 4.11 (Yamase et al. 1996a, b, c). A Keggin-structural polyoxomolybdate, Na3[PMo12O40]∙nH2O, provided FIC = 0.516, although this compound exhibited lower toxicity compared with the same structural polyoxotungstates such as K5[BW12O40]∙15H2O (PM-1), K5[PVW11O40]∙6H2O (PM-44), and K6[BVW11O40]∙nH2O (PM-46) (Table 4.10). Similarly, most of the polyoxovanadates seemed to exhibit no significant synergism, although K7[MnV13O38]∙18H2O with FIC = 0.28 was synergistic. Since neither MnCl2 nor CoCl2 showed any synergistic effect (data not shown), the synergistic activity of polyoxotungstates indicates that the location of W atoms in the PM lattice is an important factor in its combination with oxacillin (Yamase et al. 1996a, b, c; Fukuda et al. 1999). The sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of the membrane proteins separated from MRSA revealed that the synergism of the polyoxotungstates is due to the inhibition of PBP2′ expression on the membrane surface by the PMs (Yamase et al. 1996a, b, c; Fukuda et al. 1999).
Mechanistic details on the synergistic activity of PMs against MRSA and VRSA strains have been investigated by using K6[P2W18O62]∙14H2O (PM-27) as a Wells–Dawson-structural polyoxotungstate (Fig. 4.1f), K4[SiMo12O40]∙3H2O (SiMo12) as a Keggin-structural polyoxomolybdate, and K7[PTi2W10O40]∙6H2O (PM-19) as a Ti/W-mixed Keggin-structural polyoxotungstate (Inoue et al. 2005). The three PMs were tested against five strains of two constitutive MRSA (SR3605 and MRS394-1), an inducible MRSA (ATCC43300), and two constitutive VRSA (Mu3 and Mu50) strains. The observable enhancement of the antibacterial activity of β-lactam antibiotic, oxacillin, with a help of three kinds of PMs, PM-27, SiMo12, and PM-19, is summarized in Table 4.12, where MIC values of the PMs determined by the microdilution method are also added. The synergism between the PMs and oxacillin with the antibacterial activity was investigated by the oxacillin-incorporating disk method using Müller-Hinton (MH) agar medium where the PMs were included at tested concentrations. An observable growth-inhibitory zone around the oxacillin disk indicates the susceptibility of the tested strains to β-lactam. Figure 4.15 shows typical images of the growth-inhibitory zone around the disk for the Mu50 strain treated with PM-19. No appearance of the growth-inhibitory zone indicates the “homo-resistance of the strain” (Fig. 4.15a), the observation of the growth-inhibitory zone with colonies inside “hetero-resistance of the strain” (Fig. 4.15b), and the appearance of growth-inhibitory zone without colonies indicates “oxacillin-susceptible strain” (Fig. 4.15c). All of the tested strains but ATCC43300 were homo-resistant to β-lactam in the absence of the PMs: ATCC43300 was hetero-resistant. At the concentration of less than MIC value, PM-27 and PM-19 showed the growth-inhibitory zone against all of the strains except for Mu3, indicating the transformation of MRSA and VRSA-Mu50 from β-lactam resistance into β-lactam susceptibility (Table 4.12). Also, PM-27 and PM-19 caused Mu3 to be changed from homo- to hetero-resistance with a decrease of resistance to β-lactam (phenotype change). Interestingly, SiMo12 enabled to enhance the antibacterial activity of β-lactam against all the strains, since the growth-inhibitory zone without colonies was observed at the concentration of less than MIC. This is in a strong contrast to the case of the same Keggin-structural Na3[PMo12O40]∙nH2O, which did not enhance the antibacterial activity of β-lactam against MRSA (Table 4.11). The difference in the synergism between PM-27 and Na3[PMo12O40]∙nH2O is associated with the structural stability of the anion at physiological pH level, which was verified by the cyclic voltammetric measurements shown below.

Growth-inhibitory zone around the disk for the Mu50 strain treated with PM-19 at concentrations of 50 (a), 100 (b), and 500 (c) μM
Does the inhibition of PBP2′ expression by PMs result from missing of mecA gene? When the MRSA strains were cultured for 24 h at 37 °C in MH broth containing the PMs at MIC, and thereafter an aliquot of the broth was applied onto MH agar medium in the absence of the PMs according to the standard oxacillin-disk method and was incubated again for 24 h at 37 °C, however, there was no observation of the growth-inhibitory zone around the oxacillin disk on the MH agar medium. This result excludes the possibility of the missing of the mecA gene for the PMs-treated MRSA strains, since the PM-treated strain would exhibit the oxacillin susceptibility if the mecA gene was missed by PMs. As a next step, the possibility of the inhibition of the transcription from mecA gene to mRNA by PMs with and without oxacillin was investigated by using the reverse transcription polymerase chain reaction (RT-PCR) analysis of the MRSA and VRSA strains which were cultured until the initial stage of logarithmic growth phase (OD660 = 0.1–0.2) both in the presence of PM (PM-27, SiMo12, or PM-19) alone (at less than MIC) and in the coexistence of PM and oxacillin (at 1/4 MIC for each). Figures 4.16 and 4.17 show effects of PM alone (a) and the coexistence of PM and oxacillin (b) on the electrophoresis results of the RT-PCR products for MRSA SR3605 and VRSA Mu50, respectively: mecA (258 bp)-, pbpA (411 bp)-, pbp2 (618 bp)-, pbp3 (814 bp)-, and pbp4 (1,122 bp)-induced mRNAs, together with 16SrRNA (174 bp) as a control, in which the PCR primer for each of mecA-pbp genes was designed and the fragments of genes were amplified by the procedures of 40 cycles of the amplification consisting of 15-s denaturation (at 94 °C), 30-s primer annealing (at 55 °C), 60-s primer extension (at 68 °C), and 300-s final extension (Ryffel et al. 1990; Zhao et al. 2001). The inhibitions of the transcription to mecA-, pbpA-, and pbp2-4-induced mRNAs and 16S rRNA were quantitatively evaluated from the band intensities analyzed with image-analysis software. As shown in Figs. 4.16a and 4.17a, each of PM-27, SiMo12, and PM-19 showed a decrease in the band intensities of the mecA-induced mRNA at concentrations less than MIC (1/2 MIC PM-27 displayed the inhibition of 30 % for SR3605 and 10 % for Mu50, although the depression of the band intensities by PM-27 alone at its concentrations less than 1/4 MIC), and the extent of the decrease increased with an increase in its concentration in comparison with that of 16S rRNA, indicating that the transcription process from mecA to mRNA was inhibited by the PMs. Also the RT-PCR results showed that PMs depressed also the expression for pbpA- and pbp2-4-induced mRNAs, and the extent of the depression depends on both PM and pbp genes (Inoue et al. 2005). As exemplified by the fact that the inhibition of the expression of the pbp3-induced mRNA was observed for all of the above PMs at their 1/4 MIC concentrations. Such inhibition of mRNAs by the PMs demonstrates nicely the morphological change (swelling and aggregation) of the strain cells during the cultivation in the presence of the PMs as observed on the microscope (Fukuda et al. 1999). It is notable that the inhibition of the transcription from mecA and pbp genes to mRNA by the PMs was enhanced by β-lactams. Figures 4.16b and 4.17b show the electrophoresis results of the RT-PCR products for MRSA SR3605 and VRSA Mu50, respectively, in the coexistence of oxacillin of 1/4 MIC and PMs of 1/32–1 MIC (Hino K, Inoue M, Oda M, Nakamura Y, Yamase T Unpublished results in paper preparation). The coexistence of 1/4 MIC oxacillin in MRSA SR3601 enhanced the inhibition of the transcription to mecA-induced mRNA by PMs, approximately 16-fold (from 1/2 to 1/32 MIC) for PM-27 and 8-fold (from 1/4 to 1/32 MIC) for SiMo12 and PM-19. Similarly, 1/4 MIC oxacillin in VRSA Mu50 enhanced 8-fold (1/2 to 1/16 MIC) for PM-27 and 4-fold (1/4 to 1/16 MIC) for SiMo12 and PM-19. The enhancement of the PM inhibition of the transcription of mecA for the SR3605 strain by a help of 1/4 MIC oxacillin was approximately 2-fold high compared with the case for the Mu50 strain.

Effects of PM alone (a) and the coexistence of PM and oxacillin (b) on the electrophoresis results of the RT-PCR products for MRSA SR3605. SR3605 strain cells were incubated in Muller–Hinton broth in the presence (a) of PM (PM-27, SiMo12, or PM-19) at various concentrations indicated by MIC units or in the coexistence (b) of PM (at various concentrations less than MIC) and oxacillin (at 1/4 MIC). Lane M indicates 100 bp DNA ladder as molecular weight marker

Effects of PM alone (a) and the coexistence of PM and oxacillin (b) on the electrophoresis results of the RT-PCR products for VRSA Mu50. Mu50 strain cells were incubated in MH broth in the presence (a) of PM (PM-27, SiMo12, or PM-19) at various concentrations indicated by MIC units or in the coexistence (b) of PM (at various concentrations less than MIC) and oxacillin (at 1/4 MIC). Lane M indicates 100 bp DNA ladder as molecular weight marker
The treatment (for 15 min) of both the cells of MRSA SR3605 and VRSA Mu50 with PM-27 (or SiMo12) induced the blue coloration of the cells, in a strong contrast with the case (no coloration) of the (dead) cells treated with ethanol for 1 day. Figure 4.18 shows the blue coloration by PM-27 for live cells of both SR3605 and Mu50. The protoplast of such blue-stained cells, obtained by the treatment with lysostaphin and mutanolysin as cell wall lysis enzymes in hypertonic solution for 12 h, remained still blue. The fact that the biological reduction of PM-27 or SiMo12 occurred not only for the MRSA cells but also for the VRSA cells with much thicker cell walls indicates that the PMs penetrating through the cell wall are uptaken into the electron transfer system of the cellular membrane probably into the respiration system involving NADH/ubiquinone/cytochrome-c, as supported by the dominant distribution of W atoms at the periphery in the elemental spectrum analysis of the PM-27-treated MRSA cells (Fukuda et al. 1999). Redox potentials of NADH, ubiquinone, and cytochrome-c (−0.96, −0.54, and −0.39 V vs. Ag/AgCl, respectively) are negative enough to reduce PM-27 and SiMo12, since the cyclic voltammograms of PM-27 and SiMo12 at pH 7.4 showed their first half potentials (E 1/2) for the reversible one-electron reduction at +0.058 V and +0.237 V vs. Ag/AgCl, respectively (Inoue et al. 2005). The biological reduction of SiMo12 was noted also in the photosystem II of the photosynthesis, where the electron transfer from photoreduced pheophytin to primary plastoquinone acceptor occurred through SiMo12 (Giaquinta and Dilley 1975; Zilinskas and Govindjee 1975). Thus, both PM-27 and SiMo12 are biologically easily reduced to the blue color showing the broad absorption band around 700 nm which is assigned to the intervalence charge transfer of MV–O–MVI ↔ MVI–O–MV (M = W and Mo) (Fukuda et al. 1999). Figure 4.19 shows the TEM images of the MRS396-1 cells treated with and without PM-19 and the elemental spectra of local points of the cells. The observation of the W/Ti atomic ratio of 5.3 (which is close to the one (5) of PM-19) at the periphery of the cell treated with PM-19 strongly implies that the PMs enter the cells and are localized at the cell periphery to keep their structure of the anion to be intact. This is in strong contrast to the case of the PM-19 untreated cells, which indicates no significant intensity of peaks due to W and Ti atoms. That the anion structure of PM is kept intact inside the cell was also pointed out for K10[Co4(H2O)2(PW9O34)2]∙nH2O uptaken into HIV cells (Cholewa et al. 1994). The cyclic voltammograms of PM-19 at pH 7.4 showed the first half potentials (E 1/2) for pseudo-reversible one-electron reduction at −1.02 V vs. Ag/AgCl, indicating that PM-19 was biologically redox inactive. The cyclic voltammogram of PMo12 showed the structural decomposition at physiological pH level of 7.4, whereas at pH 1.0 it provided the reversible two-electron reduction at +0.186 V vs. Ag/AgCl (Inoue et al. 2005). The decomposition of PMo12 at pH 7.4 rationalizes the lack of its synergistic activity. The bacterial ATP within the cytoplasm membrane is produced during the cell growth by the electron transfer from NADH to dissolved oxygen through cytochrome-c, which is triggered by the proton gradient between inside and outside of the cell (Inoue et al. 2005). Therefore, the fact that PM-27 and SiMo12 had two orders of magnitude lower MIC than PM-19 (Table 4.12) is associated with the blocking of the electron transfer system for the respiration leading to the inhibition of the ATP production. Namely, the biologically redox-active PM-27 and SiMo12 uptaken in the cytoplasm membrane inhibits the electron transfer system of MRSA and VRSA to lead to lowering of MIC values.

Blue coloration of live cells of both SR3605 and Mu50 treated with PM-27. Photographs indicate the centrifuged cells of SR3605 or Mu50 strain in saline suspension containing 1 mM PM-27. Dead cells were prepared by the treatment of the cells with ethanol for 1 day

TEM images of the MRS396-1 cells treated with and without PM-19 and elemental spectra of local points of the cells (A–D for PM-treated and A and B for untreated ones). Inoculum (108 cfu, colony-forming unit/ml) of MRS394-1 was inoculated in MH broth containing 40 mM PM-27 for 5 h at 34 °C. The cells harvested by centrifugation (with 5,000 × g for 15 min at 4 °C) and washed twice with PBS were fixed with 2.5 % glutaraldehyde in PBS and followed by dehydration with a graded series of ethanol, embedding into a resin, and sectioning by conventional method. The spectrum observed on JEOL JEM-2100 F was examined by the distribution of W and Ti atoms in the cells with a help of energy dispersive X-ray analysis (JEOL JED-2300 T)
The cell wall of MRSA is constructed from the mesh-like structural peptidoglycan as polymer chains which comprise alternate sugar units of N-acetylglucosamine and N-acetylmuramic acid. L-Ala residue of the tetrapeptide (l-Ala-d-Glu-l-Lys-d-Ala) of the peptidoglycan is bound to the N-acetylmuramic acid, and the terminal d-Ala residue of the tetrapeptide is linked to l-Lys residue of the tetrapeptide of the neighboring polymer chain through pentaglycine peptide. The PMs with 1–2 nm size are small enough to pass through the void of the peptidoglycan layer and reach the cytoplasm membrane. Thus, it is reasonable to say that the inhibition of the transcription from the mecA and pbp genes to mRNA is exerted by the PMs uptaken into the cytoplasm membrane, the amounts of which are strongly associated with the synergistic activity. The enhancement of the PM-induced inhibition of the transcription from mecA and pbp genes to mRNA by β-lactams (Figs. 4.16b and 4.17b) gives us a clue to the mechanism of the synergistic activity of PMs. Amounts of W atoms uptaken into the cells in the presence of PMs and/or oxacillin were measured by an inductively coupled plasma atomic emission spectroscopy (ICP) analysis of the MRSA SR3605 (or VRSA Mu50) cells treated with 1/4 MIC PM-27, SiMo12, or PM-19 with and without 1/4 MIC oxacillin. Inoculum of 1 × 107 cfu (colony-forming unit)/ml bacteria and 1/4 MIC additive in MHB medium were cultured at 37 °C until the stage of a logarithmic growth phase (OD660 ≈ 0.2), harvested by the centrifugation, washed three times in ultra pure water, and then freeze-dried. The weighted dry samples were dissolved in 50 % nitric acid, boiled, and filtrated. The resulting filtrates were used for the ICP analysis. Both MRSA SR3605 and VRSA Mu50 provided the highest amounts of W (or Mo) atoms for the culture with PM and oxacillin compared with the case with PM alone. The most highlighted result was obtained for PM-19, as shown in Fig. 4.20 where the cultivation of the bacteria cells in the coexistence of oxacillin and PM-19 (at 1/4 MIC for each) enhanced the uptake of W atoms (of PM-19) into both MRSA SR3605 (a) and VRSA Mu50 (b) cells with p < 0.01 for n = 3 (Hino K, Inoue M, Oda M, Nakamura Y, Yamase T Unpublished results in paper preparation). Interestingly, when 1 × 107 cfu/ml Mu50 cells with 1/4 MIC oxacillin alone were at first cultured at 37 °C in MHB until the initial stage of a logarithmic growth phase and then cultured again with 1/4 MIC PM for 24, the amounts of the uptaken PM were extremely small compared with other three culture modes with 1/4 MIC PM alone until the initial stage of a logarithmic growth phase, at first 1/4 MIC PM until the logarithmic growth phase and subsequent with 1/4 MIC oxacillin for 24 h, and a combination of 1/4 MIC PM and 1/4 MIC oxacillin. There was no significant difference in the amounts of the uptaken PM between modes of the latter two. Figure 4.21 exemplified amounts of W atoms (of PM-27) uptaken into the Mu50 cells for a variety of cultivation modes of the Mu50 cells (Hino K, Inoue M, Oda M, Nakamura Y, Yamase T Unpublished results in paper preparation). As shown in Fig. 4.21, the amount of uptaken W atoms for the mode with at first 1/4 MIC PM until the logarithmic growth phase and subsequent with 1/4 MIC oxacillin for 24 h is higher than with 1/4 MIC PM alone until the initial stage of a logarithmic growth phase, implying that the increase of the amount of the PMs uptaken into the cells by oxacillin strongly associated with the enhancement of the PM inhibition of the transcription from the mecA and pbps genes to mRNA by oxacillin (Figs. 4.16b and 4.17b). It provides a mechanistic key that the enhancement of the PM uptake into the membrane by β-lactam antibiotics which will be captured by PBP proteins on the membrane surface was transient and diminished within 24 h until the initial stage of the logarithmic growth (OD660 ≈ 0.2) of bacteria. One should remark that an ATP-binding cassette (ABC) transporter as a family of membrane transporter systems is encoded by the transcription of abcA gene and contributes to the import or export of a wide range of substances of proteins, peptides, polysaccharides, vitamins, and drugs through a use of ATP (Higgins 1992; Maupin-Furlow et al. 1995; Grunden and Shanmugam 1996). It is well known that the methicillin (or other β-lactam antibiotics such as oxacillin and cefoxitin) stimulates the transcription of abcA gene: abcA expression was induced by increasing concentrations of methicillin, and its induction was obtained only after 3 h of exposure to methicillin (since the transcription of abcA reached its maximum later in the exponential growth phase) (Schrader-Fischer and Berger-Bächi 2001). Therefore, the difference in PMs-uptake patterns among the above different culture modes (Fig. 4.21) seems to be associated with the maximum observed for the transcription of abcA induced by oxacillin. The oxacillin-induced increase of the PMs uptaken in the cytoplasm membrane makes a base of the synergistic activity of the PMs, since the PMs within the cytoplasm membrane indirectly inhibit the mecA and pbps transcription to lead to the inhibition of PBP2′, PBPA, and PBP2-4 expressions on the membrane surface (Figs. 4.16 and 4.17). The ICP analysis of the MRSA SR3605 (or VRSA Mu50) cells treated with 1/4 MIC PMs also revealed the preferential uptake of Na+ (with ratios more than ten, compared with the case of the absence of PMs) into the cells as counter cations of PM anions, although Mg2+, K+, and Ca2+ as other cations were detected (Hino K, Inoue M, Oda M, Nakamura Y, Yamase T Unpublished results in paper preparation). Together with the location of the PMs within the cytoplasmic membrane, thus, the electric field arising from the electrochemical PMs/Na+ double layer over the cell inside, which is enhanced by β-lactams, contributes to the inhibition of the mecA and pbps transcription with a resultant depression of the transcription process. This reflects the synergy between the PMs and β-lactams against MRSA and VRSA strains, when Keggin- and Wells–Dawson-structural PMs are employed. (NH4)6[Mo7O24]∙4H2O, which was little synergistic (Table 4.11), exhibited no significant uptake into the SR3605 and Mu50 cells irrespective of oxacillin, supporting the above synergistic mechanism of the PMs. However, other mechanisms of the synergy are not necessarily excluded, since the RT-PCR result for a double Keggin-structural PM, K9H5[(GeTi3W9O37)2O3]∙16H2O (PM-504) with a larger ionic size suggests the inhibition of the posttranscription process of either translation from mRNA to PBP2′ or posttranslations of the folding of the translated polypeptide chain (Inoue et al. 2005).

Amounts of W atoms uptaken in the cells for the bacteria cells of SR3605 (a) and Mu50 (b) strains, cultured with 1/4 MIC oxacillin, 1/4 PM-19, or a combination of PM-19 and oxacillin (1/4 MIC for each). **p < 0.01 for n = 3. Compound-untreated cells (control) showed no observable amount of W atoms in the cells

Amounts of W atoms (of PM-27) uptaken into the Mu50 cells for six cultivation modes of the Mu50 cells. Six cultivation modes are (1) cultivation without substrate (control), (2) 1/4 MIC oxacillin alone until the stage of a logarithmic growth phase (OD660 ≈ 0.2), (3) 1/4 MIC PM-27 alone until the stage of a logarithmic growth phase (OD660 ≈ 0.2), (4) at first 1/4 MIC oxacillin until the logarithmic growth phase and subsequent 1/4 MIC PM-27 for 24 h, (5) at first 1/4 MIC PM-27 until the logarithmic growth phase and subsequent 1/4 MIC oxacillin for 24 h, and (6) a combination of PM-19 and oxacillin (1/4 MIC for each) until the stage of a logarithmic growth phase (OD660 ≈ 0.2). **p < 0.01 for n = 3
4.5 Conclusions
The antitumor activity of polyoxomolybdates, in particular, PM-8 (containing [Mo7O24]6−) and PM-17 (containing [H2MoV 12O28(OH)12(MoVIO3)4]6−) against solid tumors such as human (breast, lung, and colon) cancer xenografts (MX-1, OAT, and CO-4, respectively) has been extended to both AsPC-1 human pancreatic cancer and MKN-45 human gastric cancer. [H2MoV 12O28(OH)12(MoVIO3)4]6− as 12-electron-reduced species of [Mo7O24]6− was in vitro and in vivo more inhibitory against solid cancer growth than [Mo7O24]6−, due to the activation of both apoptosis and autophagy though the activation of caspase-3 and caspase-12. Intratumoral injection of PM-17 showed a significant efficacy in an experimental mice inoculation model of AsPC-1 and MKN-45, which indicates a therapeutic usefulness of PM-17 as an alternative chemotherapy of PM-8. Together with the fact that the pancreatic cancer is an aggressive form of cancer and has one of the poorest outcomes of all cancers, the development of the drug-delivery system for PM-17 is required for the realization of PM-17 as a novel anticancer drug. It is assumed that the multi-electron reduction of [Mo7O24]6− to [H2MoV 12O28(OH)12(MoVIO3)4]6− is biologically possible (namely, PM-17 is one of metabolites of PM-8) through the one-electron reduction of the 1:1 [Mo7O24]6−-FMN complex formed on the tumor cell mitochondria with a resultant suppression of the tumor growth due to the inhibition of the ATP generation. Many polyoxotungstates with Keggin or Wells–Dawson related (lacunary and multiply condensed) structures, in particular, K13[Ce(SiW11O39)2]∙26H2O (JM1590), K6[BGa(H2O)W11O39]∙15H2O (JM2766), [Me3NH]8[(SiNb3W9O37)2O3] (JM2820), K7[PTi2W10O40]•6H2O (PM-19), K9H5[(GeTi3W9O37)2O3]∙16H2O (PM-504), [PriNH3]6H[PTi2W10O38(O2)2]∙H2O (PM-523), K10[Fe4(H2O)2(PW9O34)2]∙xH2O (HS-058), K7[P2W17NbO62], K7[P2W17(NbO2)O61], and K11H[(VO)3(SbW9O33)2]∙27H2O (PM-1002), were in vitro active against DNA and RNA viruses. PM-19 showed a potent inhibition both in vitro and in vivo against a broad spectrum of DNA viruses including herpes simplex virus (HSV-1 and HSV-2), TK-HSV, and HCMV. In vivo anti-HSV activity of PM-19 was attributed to an interference with the virus uptake by the cells and/or a clearance of HSV and also partially to the activation of macrophages. Ti/W- and V/W-mixed PMs have a broad antiviral activity against enveloped RNA viruses such as HIV, FluV-A, and SARS-V, the infections of which in high-risk individuals cause severe manifestations with often resultant fatalities. A significant in vivo potentiality against HIV was recognized for PM-523 and PM-1002. While most of PMs are inhibitory only at the virus adsorption stage (e.g., due to the binding of PMs with HIV-1 gp120), PM-523 did not inhibit the adsorption of the influenza A virus (FluV-A) onto the cell membrane (0–1.5 h after infection) but inhibited the fusion between the FluV-A envelope and the cellular membrane (1.5–120 h after infection). Such a different mechanism of anti-influenza activity for PM-523 led to both in vitro and in vivo synergistic efficacies in combination with ribavirin (as a viral RNA synthesis inhibitor) or zanamivir (NA inhibitor). The antifusion activity of PM-523 was investigated by using PM-523-resistant FluV-A strains and was attributed to the binding of PM-523 to the interface edges of HA trimers, which inhibits the opening of HA1 trimers of FluV-A. It is found that some of above PMs inhibited specific sialyl/sulfotransferases with in vitro enzymatic activities of at least 100- to 1,000-fold stronger than any other known inhibitors, implying that the PMs directly inhibit the activities of enzymes involved in carbohydrate metabolism to lead to the inhibition of the binding of the HA ligands of influenza virus with sialic acid surface receptors.
The synergistic effect of PMs with β-lactam antibiotics is the basis of the development of a novel chemotherapy for MRSA/VRSA-infected disease. Keggin-structural polyoxotungstates and their lacunary species, [XW11O39]n− and [XW9O34]n−, exhibited a synergistic effect with oxacillin, with FIC indices of 0.010–0.156. Mechanistic details on the synergistic activity of PMs against five strains of two constitutive MRSA (SR3605 and MRS394-1), an inducible MRSA (ATCC43300), and two constitutive VRSA (Mu3 and Mu50) strains were investigated by using K6[P2W18O62]∙14H2O (PM-27), K4[SiMo12O40]∙3H2O (SiMo12), and PM-19. The ABC transporter, which participates in the transport of PMs into the cytoplasm membrane with an accompanying preferential flow of Na+ as counter cations, is activated by β-lactams captured on the membrane surface, and thereby the uptaken PMs within the membrane make an electrochemical double layer with Na+ cations inside of the cytoplasm. The transcription of mecA and pbps genes to mRNA will be depressed within such PM/Na+ electrical double layer to lead to depression of the formation of the PBP enzymes on the membrane surface. Together with the PBP binding with β-lactams on the membrane surface, the β-lactams-induced enhancement of the PM-induced suppression of the PBPs expression provides a base of the synergistic activity in combination of PMs and β-lactams. In vivo experiments are required for both the development of a novel chemotherapy for MRSA/VRSA-infected disease and the certification of the above mechanism.
Abbreviations
- 5-FU:
-
5-Fluorouracil
- ABC transporter:
-
ATP-binding cassette transporter
- ACNU:
-
1-(4-amino-2-methylpyridine-5yl)methyl-3-(2-chloroethyl)-3-nitrosourea
- ACV:
-
Acyclovir
- ALP:
-
Alkaline phosphatase
- ALT:
-
Alanine aminotransferase
- AMD3100:
-
Octahydrochloride dihydrate of 1,1′-[1,4-phenylenebis(methylene)]-bis-1,4,8,11-tetraazacyclotetradecane
- AST:
-
Aspartate aminotransferase
- AZT:
-
3′-azido-3′-deoxythymidine
- BUN:
-
Blood urea nitrogen
- CC50 :
-
Median cytotoxic concentration
- CDDP:
-
cis-Diamminedichloroplatinum(II)
- CDV:
-
Canine distemper virus
- DFV:
-
Dengue fever virus
- EC50 :
-
Median effective concentration
- ED50 :
-
50 % Effective dose
- FIC:
-
Fractional inhibitory concentration index
- FIPV:
-
Feline infectious peritonitis virus
- FluV-A:
-
Influenza virus A
- FMN:
-
Flavin mononucleotide
- FUT3:
-
α1,3-fucosyltransferase-III
- Gal3ST-2:
-
Gal:3-O-sulfotransferase-2
- Gal6ST:
-
Gal 6-O-sulfotransferase
- GFP:
-
Green fluorescent protein
- Gn6ST-1:
-
GlcNAc 6-O-sulfotransferase-1
- H&E:
-
Hematoxylin and eosin staining
- HA:
-
Hemagglutinin peptide
- HCMV:
-
Human cytomegalovirus
- HPA-23:
-
[NH4]17Na[Na(SbW7O24)3(Sb3O7)2]∙14H2O
- HS-058:
-
K10[Fe4(H2O)2(PW9O34)2]∙xH2O
- HSV-1:
-
Herpes simplex virus type 1
- HSV-2:
-
Herpes simplex virus type 2
- i.p. :
-
Intraperitoneal or intraperitoneally
- IC50 :
-
50 % Inhibitory concentration
- ILS:
-
Increase in life-span
- JM1590:
-
K13[Ce(SiW11O39)2]∙26H2O
- JM2766:
-
K6[BGa(H2O)W11O39]∙15H2O
- JM2820:
-
[Me3NH]8[(SiNb3W9O37)2O3]
- K i :
-
Inhibition constant
- LC3:
-
Light-chain3
- LD50 :
-
50 % Lethal dose
- M1:
-
Ribonucleoprotein sheath
- M2:
-
Matrix protein ion channel
- MAGI:
-
Multinuclear activation of the galactosidase indicator assay
- MDCK:
-
Madin–Darby canine kidney
- MIC:
-
Minimum inhibitory concentration
- MRSA:
-
Methicillin-resistant Staphylococcus aureus
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide colorimetry
- NA:
-
Neuraminidase
- PBP:
-
Penicillin-binding protein as the peptidoglycan-synthetic enzyme on the membrane surface
- PBS:
-
Phosphate-buffered saline
- PfluV-2:
-
Parainfluenza virus type 2
- PM-1:
-
K5[BW12O40]∙15H2O
- PM-1001:
-
K10Na[(VO)3(SbW9O33)2]∙26H2O
- PM-1002:
-
K11H[(VO)3(SbW9O33)2]∙27H2O
- PM-104:
-
[NH4]12H2[Eu4(MoO4)(H2O)16(Mo7O24)4]∙13H2O
- PM-1207:
-
K12[(VO)3(AsW9O33)2]∙12H2O
- PM-1208:
-
K12[(VO)3(BiW9O33)2]∙29H2O
- PM-1213:
-
K12[(VO)3(PW9O34)2]∙nH2O
- PM-17:
-
[Me3NH]6[H2MoV 12O28(OH)12(MoVIO3)4]∙2H2O
- PM-19:
-
K7[PTi2W10O40]∙6H2O
- PM-27:
-
K6[P2W18O62]∙14H2O
- PM-30:
-
A-β-Na9[SiW9O34H]∙23H2O
- PM-32:
-
Na5[IMo6O24]∙34H2O
- PM-43:
-
K5[SiVW11O40]∙nH2O
- PM-44:
-
K5[PVW11O40]∙6H2O
- PM-46:
-
K6[BVW11O40]∙nH2O
- PM-47:
-
K7[BVW11O40]∙nH2O
- PM-504:
-
K9H5[(GeTi3W9O37)2O3]∙16H2O
- PM-518:
-
[Et2NH2] 7[PTi2W10O40]∙4H2O
- PM-520:
-
[Pri 2NH2]5[PTiW11O40]∙4H2O
- PM-523:
-
[PriNH3]6H[PTi2W10O38(O2)2]∙H2O
- PM-8:
-
[NH3Pri]6[Mo7O24]∙3H2O
- RNP:
-
Ribonucleoprotein
- RSV:
-
Respiratory syncytial virus
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- S. aureus :
-
Staphylococcus aureus
- s.c.:
-
Subcutaneous or subcutaneously
- SARS-V:
-
Severe acute respiratory syndrome coronavirus
- SARS-V:
-
Severe acute respiratory syndrome coronavirus
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SI:
-
Selective index
- SiMo12 :
-
K4[SiMo12O40]∙3H2O
- SRC:
-
Subrenal capsule in kidney assay
- ST3Gal-I and -III:
-
Gal:α2,3-sialyltransferase-1 and Gal:α2,3-sialyltransferase-1II
- ST6Gal-I:
-
Gal:α2,6-sialyltransferase-1
- ST6GalNAc-I:
-
GalNAc:α2,6-sialyltransferase-1
- TCID50 :
-
Median tissue culture infection dose
- TEM:
-
Transmission electron microscope
- TGEV:
-
Transmissible gastroenteritis virus of swine
- TK− :
-
Thymidine kinase deficient
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated “nick-end” labeling staining
- TWI:
-
Tumor weight inhibition
- VRSA:
-
Vancomycin-resistant Staphylococcus aureus
- β3Gn-T2:
-
β1,3-GlcNAc-transferase-2
- β4Gal-TI:
-
β1,4-galactosyltransferase-I
References
Baba M, Snoeck R, Pauwels R, DeClerq E (1988) Sulfated polysaccharides are potent and selective inhibitors of various enveloped viruses including herpes simplex viruses, cytomegalovirus, vesicular stomatitis viruses, and human immunodeficiency virus. Antimicrob Agents Chemother 32:1742–1745
Barnard DL, Hill CL, Gage T, Matheson JE, Huffmann JH, Sidwell RW, Otto MI, Schinazi RF (1997) Potent inhibition of respiratory syncytial virus by polyoxometalates of several structure classes. Antiviral Res 34:27–37
Blumenthal R, Clgue MJ, Durell SR, Epand RM (2003) Membrane fusion. Chem Rev 103:53–69
Boneca IG, Chiosis G (2003) Vancomycin resistance: occurrence, mechanism and strategies to combat it. Expert Opin Ther Targets 7:311–328
Borrás-Alamenar JJ, Coronado E, Müller A, Pope MT (eds) (2003) Polyoxometalate molecular science, NATO science series. Kluwer, Dordrecht
Botar B, Yamase T, Ishikawa E (2001) Synthesis and crystal structure of a novel vanadium-containing tungstobismutate(III) K12[(VO)3(BiW9O33)2]∙30H2O. Inorg Chem Commun 4:551–554
Callahan LN, Phelan M, Mallinson M, Norcross MA (1991) Dextran sulfate blocks antibody binding with gp120-CD4 interactions J. Virol 65:1543–1550
Cholewa M, Legge GJF, Weigold H, Holan G, Birch CJ (1994) The use of a scanning proton microprobe to observe anti-HIV drugs within cells. Life Sci 54:1607–1612
Clarke PGH (1990) Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol 181:195–213
Dan K, Miyashita K, Seto Y, Yamase T (1998) Quantitation of herpes simplex viral DNA in Vero cells for evaluation of an antiviral agent using the polymerase chain reaction. J Virol Methods 76:73–79
Dan K, Miyashita K, Seto Y, Fujita H, Yamase T (2002) The memory effect of heteropolyoxotungstate (PM-19) pretreatment on infection by herpes simplex virus at the penetration stage. Pharmacol Res 46:357–362
Dan K, Miyashita K, Seto Y, Fujita H, Yamase T (2003) Mechanism of the protective effect of heteropolyoxotungstate (PM-19) against herpes simplex virus type 2. Pharmacology 67:83–89
Dan K, Yamase T (2006) Prevention of the interaction between HVEM, herpes virus entry mediator, and gD, HSV envelope protein, by a Keggin polyoxotungstate, PM-19. Biomed Pharmacother 60:169–173
DeClerq E (2005) New approaches toward anti-HIV chemotherapy. J Med Chem 48:1313–1297
Domaille PJ, Knoth WH (1983) Ti2W10PO40 7- and (CpFe(CO)2Sn)2 W10PO38 5-. Preparation properties and structure determination by tungsten-183 NMR. Inorg Chem 22:818–822
Donzella GA, Schols D, Lin SW, Esté JA, Nagashima A, Maddon PJ, Allaway GP, Sakmar TP, Henson G, DeClerq E, Moore JP (1998) AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med 4:72–77
Elion GB, Furman PA, Fyfe JA, Maranda P, Beauchamp L, Schaeffer HJ (1977) Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl)guanine. Proc Natl Acad Sci USA 74:5716–5720
Fujita H, Fujita T, Sakurai T, Yamase T, Seto Y (1992) Antitumor activity of new antitumor substance, polyoxomolybdate, against several human cancers in athymic nude mice. Tohoku J Exp Med 168:421–426
Fukuda N, Yamase T (1997) In vitro antibacterial activity of vanadate and vanadyl compounds against Streptococcus pneumoniae. Biol Pharm Bull 20:927–930
Fukuda N, Yamase T, Tajima Y (1999) Inhibitory effect of polyoxotungstates on the production of penicillin-binding proteins and β-lactamase against methicillin-resistant Staphylococcus aureus. Biol Pharm Bull 22:463–470, In this ref. all the MIC values in Tables 1-3 should be corrected to μM unit instead of μg/ml unit
Fukuma M, Seto Y, Yamase T (1991) In vitro antiviral activity of polyoxotungstate (PM-19) and other polyoxometalates against herpes simplex viruses. Antiviral Res 16:327–339
Geng J, Li M, Ren J, Wang E, Qu X (2011) Polyoxometalates as inhibitors of the aggregation of amyloid β peptides associated with Alzheimer’s disease. Angew Chem Int Ed 50:4184–4188
Giaquinta RT, Dilley RA (1975) A partial reaction in photosystem II: reduction of silicomolybdate prior to the site of dichlorophenyldimethylurea inhibition. Biochim Biophys Acta 387:288–305
Gilbert BE, Knight V (1986) Biochemistry and clinical application of ribavirin. Antimicrob Agents Chemother 30:201–205
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32:37–43
Grunden AM, Shanmugam KT (1996) Molybdate transport and regulation in bacteria. Arch Microbiol 168:345–354
Hallander HO, Dornbusch K, Gezelius L, Jacobson K, Karlsson I (1982) Synergism between aminoglycosides and cephalosporins with antipseudomonal activity: interaction index and killing curve method. Antimicrob Agents Chemother 22:743–752
Hanaki H, Kuwahara-arai K, Boyle-Vavra S, Daum RS, Labischinski H, Hiramatsu K (1998a) Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-resistant Staphylococcus aureus strains Mu3 and Mu50. J Antimicrob Chemother 42:199–209
Hanaki H, Labischinski H, Inaba Y, Kondo N, Murakami H, Hiramatsu K (1998b) Increase in glutamine-non-amidated muropeptides in the peptidoglycan of vancomycin-resistant Staphylococcus aureus strain Mu50. J Antimicrob Chemother 42:315–320
Higgins CF (1992) ABC transporters: from microorganism to man. Annu Rev Cell Biol 8:67–113
Hill CL (ed) (1998) Polyoxometalates. Chem Rev 98:1–387
Hill CL, Weeks MS, Shinazi RF (1990) Anti-HIV activity, toxicity, and stability studies of representative structural families of polyoxometalates. J Med Chem 33:2767–2772
Hosoya M, Balzarini J, Shigeta S, DeClerq E (1991) Differential inhibitory effect of sulfated polysaccharides and polymers on the replication of various myxoviruses and retroviruses, depending on the composition of the target amino acid sequences of the viral envelope glycoproteins. Antimicrob Agents Chemother 35:2515–2520
Ikeda S, Nishiya S, Yamamoto A, Yamase T, Nishimura C, DeClercq E (1993) Activity of the Keggin polyoxotungstate PM-19 against herpes simplex virus type 2 infection in immunosuppressed mice; role of peritoneal macrophage activation. J Med Virol 41:191–195
Ikeda S, Nishiya S, Yamamoto A, Yamase T, Nishimura C, DeClercq E (1994) Antiviral activity of a keggin polyoxotungstate PM-19 against herpes simplex virus in mice. Antivir Chem Chemother 5:47–50
Inoue M, Segawa K, Matsunaga S, Matsumoto N, Oda M, Yamase T (2005) Antimicrobial activity of highly negative charged polyoxotungstates, K27[KAs4W44O140] and K18[KSb9W21O86], and Keggin-structural polyoxotungstates against Helicobacter pylori. J Inorg Biochem 99:1023–1031
Inoue M, Suzuki T, Fujita Y, Oda M, Matsumoto N, Yamase T (2005) Enhancement of antibacterial activity of β-lactam antibiotics by [P2W18O62]6-, [SiMo12O40]4-, and [PTi2W10O40]7- against methicillin-resistant and vancomycin-resistant Staphylococcus aureus. J Inorg Biochem 100:1225–1233
Inoue M, Suzuki T, Fujita Y, Oda M, Matsumoto N, Iijima J, Yamase T (2006b) Synergistic effect of polyoxometalates in combination with oxacillin against methicillin-resistant and vancomycin-resistant Staphylococcus aureus: a high initial inoculum of 1 x 108 cfu/ml for in vivo test. Biomed Pharmacother 60:220–226
Inouye Y, Take Y, Tokutake Y, Yoshida T, Yamamoto A, Yamase T (1990) Inhibition of replication of human immunodeficiency virus by a heteropolyoxotungstate (PM-19). Chem Pharm Bull 38:285–287
Inouye Y, Tokutake Y, Kunihara J, Yoshida T, Yamase T, Nakata A, Nakamura S (1992) Suppressive effect of polyoxometalates on the cytopathogenicity of human immunodeficiency virus type 1 (HIV-1) in vitro and their inhibitory activity against HIV-1 reverse transcriptase. Chem Pharm Bull 40:805–807
Inouye Y, Tokutake Y, Yoshida T, Seto Y, Fujita H, Dan K, Yamamoto A, Nishiya S, Yamase T, Nakamura S (1993) In vitro antiviral activity of polyoxomolybdates. Mechanism of inhibitory effect of PM-104 (NH4)12H2(Eu4(MoO4)(H2O)16(Mo7O24)4)∙13H2O on human immunodeficiency virus type 1. Antiviral Res 20:317–331
Inouye Y, Fujimoto Y, Sugiyama M, Yoshida T, Yamase T (1995) Structure-activity correlationship and strain specificity of polyoxometalates in anti-human immunodeficiency virus activity. Biol Pharm Bull 18:996–1000
Ishikawa E, Yamase T (2006) 31P NMR and isothermal titration calorimetry studies on polyoxomolybdates-catalyzed hydrolysis of ATP. J Inorg Biochem 100:344–350
Jasmin C, Raybaud N, Chermann JC, Haapala D, Sinoussi F, Boy Loustau C, Bonissol C, Kona P, Raynaud M (1973) In vivo effects of silicotungstate on some RNA viruses. Biomedicine 18:319–327
Jasmin C, Chermann JC, Hervé G, Teze A, Souchay P, Boy Loustau C, Raybaud N, Sinoussi F, Raynaud M (1974) In vivo inhibition of murine leukemia and sarcoma viruses by the heteropolyanion 5-tungsto-2-antimonate. J Natl Cancer Inst 53:463–474
Jevons MP (1961) “Celbenin”-resistant Staphylococci. Br Med J 1:124–125
Judd DA, Nettles JH, Nevins N, Snyder JP, Liotta DC, Tang J, Ermolieff J, Schinazi RF, Hill CL (2001) Polyoxometalate HIV-1 protease inhibitors. A new mode of protease inhibition. J Am Chem Soc 123:886–897
Kimpton J, Emerman M (1992) Detection of replication-component and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J Virol 66:2232–2239
Lamparska-Przybysz M, Gajkowska B, Motyl T (2005) Cathepsins and BID are involved in the molecular switch between apoptosis and autophagy in breast cancer MCF-7 cells exposed to camptothecin. J Physiol Pharmacol 56:159–179
Long DL, Thusnashima R, Cronin L (2010) Polyoxometalates: building blocks for functional nanoscale systems. Angew Chem Int Ed 49:1736–1758
Lyon DK, Miller WK, Novet T, Domaille PJ, Evitt E, Johnson DC, Finke RG (1991) Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. I. The synthesis and characterization of aqueous-soluble potassium salts of α2-P2W17O61(Mn+∙OH2)(n-10) and organic solvent soluble tetra-n-butylammonium salts of α2-P2W17O61(Mn+∙OH2) (n-11) (M = Mn3+, Fe3+, Co2+, Ni2+, Cu2+). J Am Chem Soc 113:7209–7221
Mammén M, Choi SK, Whitesides GM (1998) Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed 37:2754–2794
Matsuhashi M, Dong Song M, Ishino F, Washi M, Doi M, Inoue M, Ubukata K, Yamashita N, Konno M (1986) Molecular cloning of the gene of a penicillin-binding protein supported to cause high resistance to β-lactam antibiotics in Staphylococci aureus. J Bacteriol 167:975–980
Maupin-Furlow JA, Rosentel JA, Lee J, Deppenmeier U, Gunsalus RP, Shanmugam KT (1995) Genetic analysis of the modABCD (molybdate transport) operon of Escherichia coli. J Bacteriol 177:4851–4856
Mitsui S, Ogata A, Yanagie H, Kasano H, Hisa T, Yamase T, Eriguchi M (2006) Antitumor activity of polyoxomolybdate, [NH3Pri]6[Mo7O24]∙3H2O against human gastric cancer model. Biomed Pharmacother 60:353–358
Mitsuya H, Weinhold KJ, Furman FA, St Clair MH, Lehrman SN, Gallo RC, Bolognesi D, Barry DW, Broder S (1985) 3’-Azido-3’deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci USA 82:7096–7100
Mitsuya H, Looney DJ, Kuno S, Ueno R, Wong-Staal F, Broder S (1988) Dextran sulfate suppression of viruses in the HIV family: inhibition of virion binding to CD4+ cells. Science 240:646–649
Mori S, Shigeta S (1995) A colorimetric LDH assay for the titration of infectivity and the evaluation of anti-viral activity against ortho- and paramyxoviruses. Tohoku J Exp Med 177:315–325
Moskovitz B, HPA-23 Cooperative Study Group (1988) Clinical trial of tolerance of HPA-23 in patients with acquired immune deficiency syndrome. Antimicrob Agents Chemother 32:1300–1303
Naruke H, Ozeki T, Yamase T (1991) Structure of photoluminescent polyoxomolybdoeuropate (NH4)12H2[Eu4(MoO4)(H2O)10(Mo7O24)4]∙13H2O. Acta Crystallogr C47:693–696
Oda M, Inoue M, Hino K, Nakamura Y, Yamase T (2007) Enhancement effect of polyoxometalates on NGF-induced neurite-outgrowth for PC-12 cells. Biol Pharm Bull 30:787–790
Ogata A, Mitsui S, Yanagie H, Kasano H, Hisa T, Yamase T, Eriguchi M (2005) A novel anti-tumor agent, Polyoxomolybdate induces apoptotic cell death in AsPC-1 human pancreatic cancer cells. Biomed Pharmacother 59:240–244
Ogata A, Yanagie H, Ishikawa E, Morishima Y, Mitsui S, Yamashita A, Hasumi K, Takamoto S, Yamase T, Eriguchi M (2008) Anti-tumoral effect of polyoxomolybdates: induction of apoptotic cell death and autophagy in in vitro and in vivo models. Br J Cancer 98:399–409
Ohashi Y, Yanagi K, Sasada Y, Yamase T (1982) Crystal structure and photochemistry of isopolymolybdates. I. The crystal structures of hexakis(propylammonium) hept Hexakis(propylammonium)heptamolybdate(VI) trihydrate and hexakis(isopropylammonium)heptamolybdate(VI) trihydrate. Bull Chem Soc Jpn 55:1254–1260
Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijin P, Desmyter J, DeClercq E (1988) Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 20:309–321
Pinto AL, Lippard SJ (1985) Binding of the antitumor drug cis-diamminedichloroplatinum(II) (cisplatin) to DNA. Biochim Biophys Acta 780:167–180
Pope MT, Müller A (eds) (1994) Topics in molecular organization and engineering. Polyoxometalates: from platonic solids to anti-retroviral activity. Kluwer, Dordrecht
Pope MT, Müller A (eds) (2001) Polyoxometallates: from topology to industrial applications. Kluwer, Dordrecht
Proust A, Thouvenot R, Gouzerh P (2008) Functionalization of polyoxometalates: towards advanced applications in catalysis and material science. Chem Commun: 1837–1852
Rath VL, Verdugo D, Hemmerich S (2004) Sulfotransferase structural biology and inhibitor discovery. Drug Discov Today 9:1003–1011
Rhule JT, Hill CL, Judd DA (1998) Polyoxometalates in medicine. Chem Rev 98:327–357
Rozenbaum W, Dormont D, Spire B, Vilmer E, Gentilini M, Griscelli C, Montagnier L, Barr-Sinoucci F, Chermann JC (1985) Antimoniotungstate (HPA 23) treatment of three patients with AIDS and one with prodrome. Lancet 23:450–451
Ryffel C, Tesch W, Birch-Machin I, Reynolds PE, Barberis-Maino L, Kayser FH, Berger-Bächi B (1990) Sequence comparison of mecA genes isolated from methicillin-resistant Staphylococcus aureus and Staphylococcus epidermidis. Gene 94:137–138
Schrader-Fischer G, Berger-Bächi B (2001) The AbcA transporter of Staphylococcus aureus affects cell autolysis. Antimicrob Agents Chemother 45:407–412
Schwörer R, Schmidt RR (2002) Efficient sialyltransferase inhibitors based on glycosides of N-acetylglucosamine. J Am Chem Soc 124:1632–1637
Seko A, Yamase T, Yamashita K (2009) Polyoxometalates as effective inhibitors for sialyl- and sulfotransferases. J Inorg Biochem 101:1061–1066
Shigeta S (1999) Recent progress in anti-influenza chemotherapy. Drugs R D 2:153–164
Shigeta S (2001) Targets of anti-influenza chemotherapy other than neuraminidase and proton pump. Antivir Chem Chemother 12(Suppl):179–188
Shigeta S, Yamase T (2005) Current status of anti-SARS agents. Antivir Chem Chemother 16:23–32
Shigeta S, Mori S, Watanabe J, Yamase T, Schinazi RF (1996) In vitro antimyxovirus activity and mechanism of anti-influenza virus activity of polyoxometalates PM-504 and PM-523. Antivir Chem Chemother 7:346–352
Shigeta S, Mori S, Watanabe J, Soeda S, Takahashi K, Yamase T (1997) Synergistic anti-influenza virus A (H1N1) activities of PM-523 (polyoxometalate) and ribavirin in vitro and in vivo. Antimicrob Agents Chemother 41:1423–1427
Shigeta S, Mori S, Kodama E, Kodama J, Takahashi K, Yamase T (2003) Broad spectrum anti-RNA virus activities of titanium or vanadium substituted polyoxotungstates. Antiviral Res 58:265–271
Shigeta S, Mori S, Yamase T, Yamamoto N, Yamamoto N (2006) Anti-RNA virus activity of polyoxometalates. Biomed Pharmacother 60:211–219
Stryer L (1975) Biochemistry. Freeman, San Francisco, CA
Take Y, Tokutake Y, Inouye Y, Yoshida T, Yamamoto A, Yamase T, Nakamura S (1991) Inhibition of proliferation of human immunodeficiency virus type 1 by novel heteropolyoxotungstates in vitro. Antiviral Res 15:113–124
Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S (2005) Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res 65:3336–3346
Tomita K, Yamase T, Shishido K (1989) Medical chemistry of polyoxometalates. Part 2. Enzymatic study of binding of heptamolybdate to DNA. Inorg Chim Acta 157:L167–L169
Ubukata K, Yamashita N, Konno M (1985) Occurrence of a β-lactam-inducible penicillin-binding protein in methicillin-resistant Staphylococci. Antimicrob Agents Chemother 27:851–857
Watanabe W, Konno K, Ijichi K, Inoue H, Yokota T, Shigeta S (1994) MTT colorimetric assay system for the screening of anti-orthomyxo- and anti-paramyxoviral agents. J Virol Methods 48:257–265
Weeks MS, Hill CL, Schinazi RF (1992) Synthesis, characterization, and anti-human immunodeficiency virus activity of water-soluble salts of polyoxotungstate anions with covalently attached organic groups. J Med Chem 35:1216–1221
Witvrouw M, Weigold H, Pannecouque C, Schols D, DeClercq E, Holan G (2000) Potent anti-HIV (type 1 and type 2) activity of polyoxometalates: structure-activity relationship and mechanism of action. J Med Chem 43:778–783
Yamamoto N, Schols D, DeClercq E, Debyser Z, Pauwels R, Barlzarini J, Nakashima H, Baba M, Hosoya M, Snoeck R, Neyts J, Andrei G, Murrer BA, Theobalt B, Bossard G, Henson G, Abrams M, Picker D (1992) Mechanism of anti-human immunodeficiency virus action of polyoxometalates, a class of broad-spectrum antiviral agents. Mol Pharmacol 42:1109–1117
Yamase T (1982) Photochemical studies of the alkylammonium molybdates. Part 6. Photoreducible octahedron site of [Mo7O24]6- as determined by electron spin resonance. JCS Dalton Trans: 1987–1991
Yamase T (1994) Polyoxometalates for molecular devices: antitumor activity and luminescence. In: Pope MT, Müller A (eds) Topics in molecular organization and engineering. Polyoxometallates: from platonic solids to anti-retroviral activity. Kluwer, Dordrecht
Yamase T (1996) Antitumoral and antiviral polyoxometalates (inorganic discrete polymers of metal oxide). In: Salamone JC (ed) Polymeric materials encyclopedia: synthesis, properties, and applications. CRC, Boca Raton, FL
Yamase T (2003) Photoredox chemistry of polyoxometalates as a photocatalyst. Catal Surv Asia 7:203–217
Yamase T (2005) Anti-tumor, antiviral, and antibacterial activities of polyoxometalates for realizing an inorganic drug. J Mater Chem 15:4773–4782
Yamase T, Ikawa T (1977) Photochemical studies of the alkylammonium molybdates. III. Preparation and properties. Bull Chem Soc Jpn 50:746–749
Yamase T, Ishikawa E (2008) Photoreductive self-assembly of [MoVI 7O24]6- to anti-tumoral [H2MoV 12O28(OH)12(MoVIO3)4]10- in aqueous media. Bull Chem Soc Jpn 81:983–991
Yamase T, Pope MT (eds) (2002) Polyoxometalate chemistry for nanocomposite design. Kluwer, Dordrecht
Yamase T, Tomita K (1990) Medical chemistry of polyoxometalates. Part 3. Electrochemical study of a 1:1 polyoxomolybdate-flavin mononucleotide complex in aqueous solutions. Inorg Chim Acta 169:147–150
Yamase T, Sasaki R, Ikawa T (1981) Photochemical studies of the alkylammonium molybdates. Part 5. Photolysis in weak acid solutions. JCS Dalton Trans: 628–634
Yamase T, Fujita H, Fukushima K (1988) Medical chemistry of polyoxometalates. Part 1. Potent antitumor activity of polyoxomolybdates in animal transplantable tumors and human cancer xenograft. Inorg Chim Acta 151:L15–L18
Yamase T, Tomita K, Seto Y, Fujita H (1992a) Antitumor and antiviral activities of certain polyoxometalates. In: Ottenbrite RM, Chiellini E (eds) Polymers in medicine: biomedical and pharmaceutical applications. Technomic Publishing Company Inc., Lancaster, PA, pp 187–212
Yamase T, Ozeki T, Motomura S (1992b) 183W NMR and x-ray crystallographic studies on the peroxo complexes of the Ti-substituted α-Keggin typed tungstophosphates. Bull Chem Soc Jpn 65:1453–1459
Yamase T, Ozeki T, Sakamoto H, Nishiya S, Yamamoro A (1993) Structure of hexatitanooctadecatungstodigermanate. Bull Chem Soc Jpn 66:103–108
Yamase T, Fukuda N, Tajima Y (1996a) Synergistic effect of polyoxotungstates in combination with β-lactam antibiotics on antibacterial activity against methicillin-resistant Staphylococcus aureus. Biol Pharm Bull 19:459–465, In this ref. all the MIC values in Tables 1-3 should be corrected to μM unit instead of μg/ml unit
Yamase T, Ohtaka K, Suzuki M (1996b) Structural characterization of spherical octadecavanadates encapsulating Cl− and H2O. JCS Dalton Trans: 283–289
Yamase T, Suzuki M, Ohtaka K (1997) Structures of photochemically prepared mixed-valence polyoxovanadate clusters: oblong [V18O44(N3)]14-, superkeggin [V18O42(PO4)]11- and doughnut-shaped [V12B32O84Na4]15- anions. JCS Dalton Trans: 2463–2472
Yamase T, Makino H, Naruke H, San José Wéry AM (2000) A spherical potassium-capped vanadium methylphosphonate as another ε-Keggin fragment, [H6KV12O27(VO4)(PO3CH3)3]5-. Chem Lett: 1350–1351
Yamase T, Botar B, Ishikawa E, Fukaya K (2001) Chemical structure and intramolecular spin-exchange interaction of [(VO)3(SbW9O33)2]12-. Chem Lett: 56–57
Yamase T, Botar B, Ishikawa E, Fukaya K, Shigeta S (2002) Magnetic exchange coupling and potent antiviral activity of [(VO)3(SbW9O33)2]12-. In: Yamase T, Pope MT (eds) Polyoxometalate chemistry for nanocomposite design. Kluwer, Dordrecht
Yamase T, Cao X, Yazaki S (2007) Structure of double keggin-Ti/W-mixed polyanion, [A-β-GeTi3W9O37]2O3]14- and multielectron-transfer-based photocatalytic H2-generation. J Mol Catal A 262:119–127
Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H (2002) A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in protease active site. J. Virol 76:1349–1358
Zhao W, Hu ZQ, Okubo S, Hara Y, Shimamura T (2001) Mechanism of synergy between epigallocatechin gallate and β-lactam against methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 45:1737–1742
Zilinskas BA, Govindjee (1975) Silicomolybdate and silicotungstate mediated dichlorophenyl dimethylurea-insensitive photosystem II reaction: electron flow, chlorophyll a fluorescence and delayed light emission changes. Biochim Biophys Acta 387: 306–319
Acknowledgments
I would like to thank Professors Masazumi Eriguchi and Hironobu Yanagië (University of Tokyo), Professor Kenji Okuda and Dr. Kiyohiko Matsui (Yokohama City University), Professors Katsuko Yamashita and Akira Seko (Tokyo Institute of Technology), Professor Shiro Shigeta (Fukushima Medical University), and Dr. Yutaka Tajima (Juntendo University) for their valuable discussions and help in my research to date. Also, I acknowledge Grants-in-Aid for Scientific Research, No. 14204067 from the Ministry of Education, Science, Sports, and Culture and No. 99P01201 from RFTF/JSPS, and “Creation of bio-devices and bio-systems with chemical and biological molecules for medical use,” CREST, Japan Science and Technology Agency (JST).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Yamase, T. (2013). Polyoxometalates Active Against Tumors, Viruses, and Bacteria. In: Müller, W., Wang, X., Schröder, H. (eds) Biomedical Inorganic Polymers. Progress in Molecular and Subcellular Biology, vol 54. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-41004-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-642-41004-8_4
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-41003-1
Online ISBN: 978-3-642-41004-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)