
Abstract
This chapter provides an overview of the current knowledge on aerosols in the marine atmosphere and the effects of aerosols on climate and on processes in the oceanic surface layer. Aerosol particles in the marine atmosphere originate predominantly from direct production at the sea surface due to the interaction between wind and waves (sea spray aerosol, or SSA) and indirect production by gas to particle conversion. These aerosols are supplemented by aerosols produced over the continents, as well as aerosols emitted by volcanoes and ship traffic, a large part of it being deposited to the ocean surface by dry and wet deposition. The SSA sources, chemical composition and ensuing physical and optical effects, are discussed. An overview is presented of continental sources and their ageing and mixing processes during transport. The current status of our knowledge on effects of marine aerosols on the Earth radiative balance, both direct by their interaction with solar radiation and indirect through their effects on cloud properties, is discussed. The deposition on the ocean surface of some key species, such as nutrients, their bioavailability and how they impact biogeochemical cycles are shown and discussed through different time and space scales approaches.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- High-nutrient Low-chlorophyll (HNLC)
- Cloud Droplet Concentration
- BVOC Emissions
- Organic Aerosol (OA)
- Dust Deposition
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Introduction
An aerosol consists of a suspension of particles and its surrounding medium. For atmospheric aerosols the surrounding medium is the air in which the aerosol particles (or droplets) are suspended. Often the term aerosol is used to refer to only the particles or droplets. In this contribution we follow this convention and refer to aerosol particles whether they occur as particles or as droplets, i.e. chemicals in their liquid phase, or dissolved in a liquid, are also referred to as particles. Aerosol sources are numerous and can be of natural or anthropogenic origin, they can enter the atmosphere directly as particles (primary aerosol) or form in the atmosphere from their precursors in the gas phase through physical and chemical reactions (secondary aerosol formation).
Aerosols are an important constituent of the atmospheric boundary layer. Aerosol particles provide surfaces for heterogeneous chemical processes, they also act as a condensation sink for atmospheric trace gases. Hygroscopic particles serve as cloud condensation nuclei. The chemical and physical properties of aerosol particles are very variable in both space and time and depend on the proximity of sources and sinks and the chemical and physical transformation during their atmospheric lifetime. Particle sizes may vary from a few nanometres to some tens of micrometres. Particles at the high end of this size range are sufficiently heavy that their atmospheric residence time is very short and hence their concentrations are negligible, although in hurricane conditions such large sea spray particles may be important in the transfer of ocean–atmosphere transfer of heat and water vapour (e.g., Andreas et al. 2008). Physical processes, in particular the vertical transport and removal of particles by dry deposition to the surface, depend on particle size. Very small particles are subject to growth by condensation and coagulation. Transport is determined by turbulence and Brownian diffusion. Very large particles having sufficient mass are subject to gravitational forces resulting in rapid sedimentation. These processes, in addition to formation by direct emission and secondary processes, chemical transformations and in-cloud processing, determine the number concentrations of aerosol particles which may vary by 10 orders of magnitude depending on size.
The particle size distribution describes the variation of the aerosol number concentration as function of particle size (radius or diameter, specified for a certain relative humidity to account for differences due to hygroscopic growth). Figure 4.1 shows a schematic representation of a hypothetical particle size distribution, presented as number size distribution (bottom) or mass size distribution (top) with five different modes (cluster, nucleation, Aitken, accumulation and coarse modes). The predominant origin of particles contributing to a certain size range is indicated and will be further discussed in Sect. 4.2. As a result of various interacting processes, the most abundant aerosol particles in the atmosphere are those with a radius of a few tenths of microns. Particles in this size range experience a minimum in the efficiency of dry deposition and they are often referred to as accumulation mode particles. The atmospheric lifetime of these particles is relatively long, on the order of a few days to a few weeks depending on the surface roughness and related deposition velocity, and their main removal mechanism is wet deposition.


Schematic representation of a characteristic particle size distribution showing the different modes and the origin of particles which predominantly contribute to a certain size range (see text for further explanation of sources). The bottom figure shows a number size distribution and the top figure shows the associated mass size distributions. Mass is related to number by m = πDp3ρ/6, where Dp is the dry particle diameter (m) and ρ is the particle density (g m-3) (Figure courtesy of Dr. Aki Virkkula, Finnish Meteorological Institute, Helsinki, Finland)
Aerosols in the marine atmosphere originate from a variety of production and transformation processes. Sea spray aerosol is directly produced at the sea surface through the interactions between wind and surface waves. Ship emissions and volcanoes also contribute to primary aerosol in the marine atmosphere. Secondary aerosol formation from gases released from the sea surface also contribute significantly to marine atmosphere aerosol loading. In addition, aerosols formed over land by either primary or secondary formation processes are transported over the oceans and contribute substantially to the aerosol concentrations over most of the world’s oceans. Estimates of the mass concentrations show that the largest aerosol contributions on a global scale are from sea spray aerosol and desert dust (Andreae and Rosenfeld 2008; Jickells et al. 2005). A comprehensive review of marine aerosols was published by Lewis and Schwartz (2004). An update on the status of sea spray aerosol production was published by de Leeuw et al. (2011).
Aerosol particles are important both because they affect atmospheric processes and, after deposition to the sea surface, because they affect processes in sea water. Their effects in the atmosphere are very diverse and depend on the chemical and physical properties of the aerosol particles. Aerosols have a strong impact on climate both due to scattering and absorption of incoming solar radiation (direct effect) and through their effects on cloud properties and associated cloud albedo (first indirect effect) and precipitation (second indirect effect). Optical properties of aerosol particles are determined by their size relative to the wavelength of incident light and their chemical composition (which determines their complex refractive index). The optical properties such as angular scattering (the aerosol phase function) and absorption for a given aerosol size distribution and chemical composition can be computed using a Mie code (Mie 1908). For non-spherical particles more sophisticated codes need to be applied. The scattering and absorption efficiency are near zero for very small particles and near two for very large particles, where small and large are relative to the wavelength, λ, of the incident radiation, i.e. determined by the size parameter 2πr/λ (r is particle in situ radius). Therefore, the particles most important for climate, as determined by the product of the particle size distribution and the scattering efficiency, are in the accumulation mode.
When aerosols are deposited at the ocean surface, a number of processes between dissolved forms and particles (such as dissolution, adsorption, and aggregation) take place on various timescales. Many of these processes impact surface ocean chemistry and in particular the cycles of elements of biogeochemical interest (macro and micronutrients such as nitrogen, phosphorus, iron etc.). New atmospheric nutrients entering the surface layer of the ocean may fertilise phytoplankton growth and as such may affect CO2 draw-down and production of other gases such as DMS, that may modify global climate on long time scales. The complex interactions between atmospheric deposition, marine biogeochemistry, carbon export and sequestration in the deep ocean and their feedbacks on climate have been recognised as an important field of research over the past 20 years and a synthesis of current knowledge is presented here.
In Sect. 4.2 we discuss the occurrence of aerosol particles in the marine atmosphere through their production, transport and transformation and deposition. The discussion on production includes the direct production of sea spray aerosol, its organic enrichment and indirect production of aerosols in the marine atmosphere. Non-marine sources are discussed as well as their transport and transformation in the marine atmosphere and removal by deposition. In Sects. 4.3 and 4.4 effects of aerosols in the marine atmosphere, i.e. direct and indirect effects on climate and effects of atmospheric aerosol deposition on marine biology are discussed. Section 4.5 discusses atmospheric deposition and its biogeochemical impacts into the ocean.
4.2 Aerosol Production and Transport in the Marine Atmosphere
In this section an overview of marine sources of aerosol is presented, starting with the direct production of sea spray aerosol and the enrichment of sea spray aerosol in organic material, followed by secondary production in the marine atmosphere. Subsequently non-marine aerosol sources are discussed.
4.2.1 Sources of Aerosol in the Marine Atmosphere
Aerosols are produced directly at the sea surface from the interaction between wind and waves, or indirectly through secondary processes involving gases exchanged between ocean and atmosphere or gases produced from anthropogenic activities over the ocean (for instance, shipping producing extensive amounts of NOx). The aerosol load in the marine atmosphere is further augmented by the direct production of aerosols over land and their subsequent transport over the ocean. Most particles in the marine atmosphere are deposited to the ocean where they may affect biological processes. Particles which are produced from the ocean and transported over land may affect regional air quality (visibility and corrosion) and climate, and they may play a role in cycling from the marine to the terrestrial environments.
4.2.1.1 Sea Spray Aerosol Production
Sea spray aerosol (SSA) consists of a suspension, in air, of particles that are directly produced at the sea surface (de Leeuw et al. 2011a). Sea spray aerosols may be enriched in certain substances, i.e. their chemical composition may deviate from that of sea salt. These particles exist mainly in the liquid phase (i.e. as drops). The radii of these particles vary from around 10 nm to at least several millimeters. The atmospheric residence times of the particles vary from seconds to minutes for larger particles, for which gravitational sedimentation is the principal removal mechanism, to days for smaller particles, for which removal is primarily by precipitation. The size of an SSA particle is commonly specified by its equilibrium radius at a relative humidity (RH) of 80 %, r80.
Many measurements indicate that the relative concentrations of the major solutes in sea spray particles are similar to their relative concentrations in bulk seawater, although this may not be the situation for some substances as a consequence of the formation process, or of exchange with the atmosphere subsequent to formation. SSA particles are said to be enriched or depleted in such substances, and the enrichment factor, defined as the ratio of the concentration of a substance to the concentration of one of the major constituents of bulk seawater (typically sodium) in the particle to the same ratio for bulk seawater, may be less (depletion) or greater than unity (enrichment).
The aerosol consisting of sea spray particles in the atmosphere has traditionally been termed “sea salt aerosol”, but in de Leeuw et al. (2011a) it was denoted “sea spray aerosol” in recognition that the composition of the particles may differ from that of bulk seawater. One consequence of this difference is that the hygroscopic and cloud droplet activation properties of sea spray particles may differ from those calculated under the assumption that the particles are composed only of sea salt. In particular, in biologically active waters, sea spray aerosol has been observed to be enriched in organic matter (OM) and the contribution of OM to sea spray aerosol has been an important area of recent research, as discussed in Sect. 4.2.2.2.
The production of sea spray aerosol (SSA) was recently reviewed by de Leeuw et al. (2011a) who critically examined laboratory and field experimental results on sea spray production, on the enrichment in organic matter, and on the measurement and parameterisation of whitecap coverage, and placed it in the context of previous understanding which was comprehensively reviewed by Lewis and Schwartz (2004). The review by de Leeuw et al. (2011a) included material published in the peer-reviewed literature until early 2010. These authors considered 13 production flux formulations, as well as fluxes measured by Norris et al. (2008) using the eddy correlation method, and these formulations are provided in Fig. 4.1 and in the Appendix of de Leeuw et al. (2011a). Below we briefly summarise some of that material to provide background and context for an overview of more recent work.
SSA particles are formed at the sea surface mainly by breaking waves via bubble bursting and by the tearing off of wave crests at elevated wind speeds (>9 ms−1). When a wave breaks, air is entrained into the water and dispersed into a cloud of bubbles (Thorpe 1992). The resulting white coloured area of the sea surface is often denoted a “whitecap” on account of enhanced, wavelength-independent scattering of visible radiation by the interfaces between water and bubbles. The fraction of the sea surface covered by white area is defined as the whitecap fraction, W.
The bubbles rise to the surface and float, where water drains off. The film cap of the bubble becomes unstable and when it opens it fragments into many small droplets, the so-called film droplets, and the water jet rising in the remaining cavity breaks up into a stream of 1–6 ‘jet’ droplets.
The production flux of SSA particles can be specified as either the interfacial flux, i.e. the flux of those particles leaving the sea surface, or as the effective flux, which is defined as the flux of those particles produced at the sea surface that attain a given height, typically 10 m, above mean sea level. They thus remain in the atmosphere for a sufficiently long time to participate in processes such as the scattering and absorption of solar radiation, cloud formation and atmospheric chemistry. For small SSA particles (i.e. those with r80 smaller than about 1 μm), the effective flux can, for all practical purposes, be considered to be the same as the interfacial flux. For medium SSA particles (those with r80 between about 1 and 25 μm), the effective flux becomes increasingly less than the interfacial flux with increasing r80. For larger SSA particles, which have short atmospheric residence times and typically do not attain heights more than a few meters above the sea surface, the effective flux is essentially zero.
The SSSF (Sea Spray aerosol Source Function) is a numerical representation of the size-dependent production flux of SSA particles:
where f(r80) denotes the number of particles in a given infinitesimal range of the common logarithm of r80, dlog10r80, introduced into the atmosphere per unit area per unit time, and F(r80) is the total number flux of particles of size less than r80.
An expression for the SSSF required as input to models would represent the size-dependent production flux expressed by Eq. 4.1 as a function of the controlling ambient variables a, b, …; i.e. f(r80; a, b,…). The near-surface wind speed, commonly measured and expressed at a reference height of 10 m, U10, is thought to be the dominant factor affecting sea spray production. Other factors that are expected to affect the SSA production flux are those affecting sea state, such as fetch (the upwind distance over the water of nearly constant wind velocity) and atmospheric stability (often parameterised by the air-sea temperature difference), which also affects vertical transport; seawater temperature and salinity; and the presence, amount, and nature of surface-active substances.
As discussed in de Leeuw et al. (2011a), the effect of water temperature on the resultant size distribution was investigated by Mårtensson et al. (2003) (at 2 °C, 5 °C, 15 °C, and 25 °C) and by Sellegri et al. (2006) (at 4 °C and 23 °C), while effects of salinity were investigated by Mårtensson et al. (2003) and Tyree et al. (2007). Nilsson et al. (2007) compared the Mårtensson et al. (2003) parameterisation with production fluxes derived from eddy covariance measurements at Mace Head (assumed water temperature of 12 °C) and the Clarke et al. (2006) parameterisation derived from profile measurements at the coast of Hawaii (water temperature ca. 25 °C). Both comparisons provided favourable results thus confirming the effect of water temperature on the SSA source flux from two independent types of measurements.
The Mårtensson et al. (2003) experimental data was used by Sofiev et al. (2011) to derive a modification of the Monahan et al. (1986) SSSF formulation which resulted in a temperature and salinity dependent SSSF. This modified SSSF was implemented in the dispersion model SILAM (Sofiev et al. 2006) and applied to compute the distribution of sea salt over the North Atlantic and Western Europe, as well as globally. The influence of sea surface temperature and salinity were evaluated using data from several campaigns, long-term in situ and satellite data (MODIS AOD).
An approach combining satellite observations, in situ data from six cruises and model results was presented by Jaeglé et al. (2011). These authors compared model results (GEOS-Chem, with the Gong (2003) formulation for the SSSF) with MODIS and AERONET AOD observations and in situ aerosol measurements. Modelled mass concentrations of coarse mode sea salt aerosol (SS) were overestimated at high wind speeds over the Southern, North Pacific and North Atlantic Oceans, but underestimated over warm tropical waters of the Central Pacific, Atlantic and Indian Oceans. The in situ observations were used to derive an empirical SS source function depending on both wind speed and SST. This resulted in a correction to the Gong (2003) source function. Using Gong (2003) with this correction, the model results for AOD agree significantly better with the MODIS and AERONET observations and provide an explanation for the high AOD observed over the tropical oceans.
In contrast, Witek et al. (2007a, b) did not find a water temperature dependence of the difference between modelled and measured mass concentrations, where the NAAPS model was compared with measurements from five open-ocean shipboard campaigns covering a range of water temperatures from less than 10 °C to about 30 °C. The Mårtensson et al. (2003) data show a size dependent effect of water temperature which crosses over at r80 of about 30–40 nm. One may argue that in the mass concentration the water temperature effect would cancel out, but because the mass is dominated by larger particles one would expect that the mass increases with increasing water temperature. Witek et al. included particles with aerodynamic radius at 55 % RH of up to 5 μm in their calculation of the sea salt aerosol mass. In NAAPS the sea salt dry mass emission flux is simply parameterised as a function of wind speed only (F = 1.37 × 10−13 U103.41 [kg m−2 s−1]) and has no SST dependence.
Hultin et al. (2011) conducted wave tank experiments using fresh Baltic Sea water with a salinity of 6–7, much lower than oceanic sea water (salinity ~33). These authors observed a clear dependence of aerosol production (r80 between 0.01 and 0.9 μm) on water temperature, i.e. a distinct decrease for all particle sizes with increasing water temperature accompanied by a decrease of dissolved oxygen. As discussed by Hultin et al. (2011) several authors have studied the effect of dissolved gases on the production of sea salt aerosol. For instance, Stramska et al. (1990) observed that more sea salt aerosol particles are produced in water in which dissolved oxygen is super-saturated than in water where dissolved oxygen is sub-saturated. Dissolved oxygen affects the bubble size distribution and thus also the resulting aerosol spectral flux (Lewis and Schwartz 2004). However, Hultin et al. (2011) conclude that the range of dissolved oxygen encountered during their measurements is too small to significantly affect the bubble size distributions in the size range (>2 mm) of importance for their measurements and speculate that the biological activity responsible for the decreased dissolved oxygen concentrations also alters the surface chemistry and the surfactant concentrations which in turn reduce particle production.
Norris et al. (2012) used micrometeorological measurements of SSA fluxes at the open North Atlantic to formulate a source function in terms of only U10. This source function lies within the range of earlier formulations for particles with r80 < 1 μm but decreases more rapidly for larger particles.
4.2.1.2 Organic Enrichment of Particulate Organic Matter in Sea Spray Aerosol
In biologically rich seawater, accumulation of organic substances at the sea surface can result in enrichment of organic matter in sea spray particles, especially for submicron particles (Blanchard 1964; Middlebrook et al. 1998; O’Dowd et al. 2004). As far back as 1948, Woodcock (1948) showed that drops produced by bubbles bursting in areas with high concentrations of plankton could carry irritants across the air-sea interface. Blanchard (1963, 1964) extended research into enrichment of organic matter in sea spray and its subsequent transfer into the atmosphere.
Surface-active OM of biogenic origin (such as lipidic and proteinaceous material and humic substances), enriched in the oceanic surface layer and transferred to the atmosphere by bubble-bursting processes, are the most likely candidates to contribute to the observed organic fraction in marine aerosol (Gershey 1983; Mochida et al. 2002).
The observed organic aerosol characteristics are consistent with laboratory studies on aerosol generated from Atlantic sea water (Gershey 1983) that showed a peak in organic aerosol concentration, and a concomitant increase in WIOC (water insoluble organic carbon) and high-molecular-mass surface-active fractions, during periods of phytoplankton blooming. Moreover, the increasing enrichment of the aerosol organic fraction with decreasing size is consistent with thermodynamic predictions (Oppo et al. 1999) of bubble-bursting processes under conditions in which the ocean surface layer becomes concentrated with surfactant material that can be incorporated into sea spray drops in addition to inorganic salts.
In 2004, O’Dowd et al. (2004) and Cavalli et al. (2004) reported significant organic mass enrichment in submicron aerosol (Fig. 4.2) that possessed a strong seasonality following the chlorophyll a seasonal pattern. The organic matter comprised both water soluble and water insoluble organic matter (WSOM/WIOM). These studies, and a more extended study by Yoon et al. (2007) suggested that the WIOM was primary in origin. This suggestion was corroborated by gradient flux measurements (Ceburnis et al. 2008) at Mace Head which demonstrated that the WIOM had a gradient similar to sea salt, indicating a surface (i.e. primary) source while WSOM possessed a gradient identical to non-sea-salt (nss) sulphate, indicating transfer from gas phase to aerosol surfaces (i.e. secondary aerosol production).


Average mass concentration of total particulate matter (white line, right axis) and mass fraction (colours, left axis) of sea salt, NH4, nss-SO4, NO3, water-soluble organic matter (WSOM), water-insoluble organic matter (WIOM), and black carbon (BC) in several size ranges for North Atlantic marine aerosol sampled at Mace Head, Ireland, in clean marine air during periods of (a) low biological activity, November (2002) January (2003) and February (2003); and (b) high biological activity, March-October, (2002). Radius corresponds to relative humidity of approximately 70 %. For low biological activity mass concentrations of aerosol constituents other than sea salt were below detection limits for the size range 0.03–0.06 μm. Oceanic chlorophyll a concentrations over the North Atlantic for periods of (c) low and (d) high biological activity are 5 year averages (1998–2002) over the same months as for the composition measurements, based on satellite measurements of ocean colour (Courtesy of SeaWiFS Project, NASA/Goddard Space Flight Center and ORBIMAGE) (Adapted from O’Dowd et al. (2004))
These N.E. Atlantic results were corroborated by measurements in the South Atlantic at Amsterdam Island (Sciare et al. 2009). Since 2008, an Aerodyne high resolution Time of Flight aerosol mass spectrometer has been continuously deployed at Mace Head and this continuous database of real-time chemical composition has led to further elucidation of sea spray aerosol chemical properties. In particular, Ovadnevaite et al. (2011a) report the regular occurrence of significant primary organic aerosol plumes at concentrations often exceeding those reported in heavily polluted air (e.g. 4 μg m−3) and extending for periods exceeding 24 h. They also reported a unique primary organic mass spectral fingerprint hitherto unreported. In that study, it was also reported that the organic aerosol was 55 % oxygenated and 45 % hydrocarbon – like. The oxygenated component must have a very low solubility to be consistent with the previous off-line WSOM/WIOM ratios reported. The correlation coefficient between the AMS hydrocarbon-like and oxygenated organics was 0.97, pointing to a common source, and a degree of chemical ageing of sea spray. The large enrichment of hydrocarbon-like organics suggest low water uptake, as shown in Fig. 4.3 (Ovadnevaite et al. 2011b) where hygroscopic growth factors for highly enriched organic particles have a growth factor of 1.3; however, these aerosols have almost a 100 % CCN activation efficiency. So, while water uptake is low at subsaturated conditions (negatively influencing the direct radiative effect), it can be high in supersaturated conditions (positively influencing the indirect aerosol radiative effect).


(a and b) CCN 0.75 % activity (CCN/CN) as a function of GF (at 90 % RH), chemical composition (colour scale) and weighted average particle size (size of the circle). CN is the total particle number above 20 nm in diameter; the colour scale represents the dominance of a given chemical species. Measurement periods: 02nd – 27th May 2009, 11th – 28th August 2009 and 14th July – 12th August 2010. Note, the measurement periods cover periods much longer than individual plume events. In Figure (a), the boxed region highlights particles dominated by primary organic matter while Figure (b) highlights the particles dominated by sulphate. Particles to the extreme right of both figures are dominated by sea salt mass. (c) Two organic-dominated size distributions (on 00:00 UTC – 22:00 UTC 16th August 2009 and 13:30 UTC-16:30 UTC 05th August 2010) and their resultant weighted diameters and (d) the same for sulphate-dominated distributions (on 21:00 UTC – 22:00 UTC 02nd August 2010 and 06:00 UTC – 10:00 UTC 09th August 2010) (Reproduced from Ovadnevaite et al. (2011b) by permission of the American Geophysical Union)
It has also been speculated that the observed properties of marine primary organic aerosols may be driven by the peculiar physico-chemical properties of marine hydrogels transferred into the atmosphere through the bubble bursting process. Evidence of the transfer of biogenic mucus-like exopolymers in sea spray aerosol have been provided, for instance, by Leck and Bigg (2005) from transmission electron microscopy analyses.
Russell et al. (2010) observed an ocean derived primary organic aerosol component in marine aerosol dominated by carbohydrate-like material, based on FTIR measurements of submicron marine aerosol over the North Atlantic and Arctic Oceans and on Positive Matrix Factorisation data elaboration. According to the authors, the primary marine signal in submicron marine aerosol is made on average for 88 % of hydroxyl groups. The apparent solubility of the carbohydrate-like components in an aqueous phase suggests that DOC provides the source of most primary organics, although the authors recognise that in bloom conditions in productive waters POC could also contribute, as shown by Facchini et al. (2008).
More recently, Decesari at al. (2011) presented the results of a multi-technique investigation of the chemical properties of marine organic aerosol collected during a cruise in the NE Atlantic Ocean that downsized the role of hydroxyl groups in favour of carboxyls and carbonyls. Moreover, the work of Decesari et al. (2011) pointed out that both primary and secondary processes contribute to the observed organic aerosol load over remote oceanic regions, with secondary products comprising both the atmospheric evolution of primary organics and the gas-to-particle conversion processes of volatile organic precursors emitted by marine biota.
4.2.1.2.1 Laboratory Studies
Laboratory studies have partially elucidated the properties of organic enriched sea spray. For example, Sellegri et al. (2006) investigated the impact of the artificial surfactant sodium dodecyl sulphate (SDS) on bubble-mediated spray production. In particular, they examined the physical size distribution using online spectrometers and found that the sub-micron sea salt distribution was tri-modal with modal diameters of 50, 110 and 350 nm resulting in a distribution which peaks at a (dry) diameter of 100 nm. With the addition of SDS, however, the peak diameter reduced to 50–80 nm, depending on the type of bubbling. The mode at 350 nm became more prominent when SDS was introduced and when the foam was artificially burst by blowing air over the foam, this mode dominated. The prominence of the large-diameter mode in the presence of a surfactant is consistent with the suggested increase in mean spray size when enriched in organics (O’Dowd et al. 2004; Yoon et al. 2007). In contrast, Tyree et al. (2007) investigated spray size distributions produced from natural sea water for winter and summer DOC concentrations and found little difference regardless of whether or not artificial, filtered or unfiltered sea water was used. Changes were seen, however, in number concentration as 20–40 % more spray droplets were observed for the winter sample compared to that of the summer. More recent studies by Fuentes et al. (2011) focussed on evaluating the impact of nanogel and DOC plankton exudates both in the laboratory and during research cruises. They found an increase in the production of particles smaller than 100 nm for organic carbon concentrations >175 μM. The sea spray produced contained a volume fraction of organic carbon 8–37 % which was somewhat lower than the maximum enrichment fraction observed in the field. Fuentes et al. (2011) suggest that the observed shift to larger mean sizes for enriched sea spray aerosol observed by Yoon et al. (2007) is inconsistent with their results. However, Fuentes et al. (2011) conducted experiments in natural seawater enriched with organics released by algal laboratory cultures which were subjected to 0.2 μm filtration in order to remove bacteria and avoid biodegradation of the organic matter, which may have caused the discrepancy.
Keene et al. (2007) conducted bubble bursting experiments using highly oligotrophic seawater from near the Bermuda coast and found enrichment at all sizes with the enrichment factor increasing with reducing particle size. The most detailed off-line chemical laboratory study was conducted by Facchini et al. (2008a) who produced sea spray in plankton rich North East Atlantic waters amidst a large bloom. They found that the mass fraction of organic matter approached 75 % (Fig. 4.4) for the smallest sizes (down to 0.062 μm diameter) to 20 % at sizes less than 1 μm. Supermicron particles contained less than a few percent organic mass fraction. The majority of the enriched organic matter was water insoluble organic matter (WIOM) and the mass fraction of WIOM and sea salt, as a function of size, replicated very closely the mass fraction observed in air (during the same cruise) and that sampled previously at Mace Head. This comparison suggests that the vast majority of the WIOM observed in clean air samples are primary in origin. Facchini et al. (2008a) reported that the WIOM consisted of colloids and aggregates exuded by phytoplankton.


(a) Mass fraction of sea salt, water-soluble organic matter (WSOM), and water-insoluble organic matter (WIOM) as a function of particle radius sampled at approximately 70 % RH, (a) for seawater bubble-bursting chamber experiments with fresh seawater, conducted in a shipboard laboratory in a plankton bloom over the N.E. Atlantic (May–June 2006), (b) for clean marine air at Mace Head, Ireland, May-June 2006, and (c) for clean marine air 200–300 km offshore west–northwest of Mace Head in a plankton bloom coincident in time with aforementioned samples (Adapted from Facchini et al. (2008a))
4.2.1.2.2 Global Distribution of Organic Enrichment
A number of international studies have expanded the measurement picture emerging from NE Atlantic waters, corroborating the findings of OM enrichment in seaspray. Namely, the following cruises: MAP (Marine Aerosol Production, e.g., Facchini et al. 2008a), OOMPH (Organics over the Ocean Modifying Particles; Zorn et al. 2008), ICEALOT (International Chemistry Experiment in the Arctic LOwer Troposphere; Russell et al. 2010; Frossard et al. 2011), and RHaMBLe (Reactive Halogens in the Marine Boundary Layer; Lee et al. 2010), all demonstrating the enrichment of OM in sea spray, albeit to different degrees. O’Dowd et al. (2008) integrated the studies of O’Dowd et al. (2004), Cavalli et al. (2004) and Yoon et al. (2007) with the eddy correlation microphysical flux measurements of Geever et al. (2005) to produce the first combined organic–inorganic sea spray source function and applied it to the REMOTE (REgional MOdel with Tracer Extension) regional climate model. This “chemical” parameterisation for organic enrichment could be applied to any sea spray physical source function and was indeed applied to global budgets by Langmann et al. (2008), Vignati et al. (2010) and Myriokefalitakis et al. (2010). Figure 4.5 illustrates the global distribution of submicron sea salt and water insoluble organic matter using the parameterisation of Vignati et al. (2010) with the TM5 (Tracer Model 5; Krol et al. 2005) chemical transport model.


Global distribution of mass flux of sea salt (upper panel) and water-insoluble organic matter WIOM (lower panel) in sea spray, with 0.1 μm < r80 < 1 μm averaged over a 1-year period in 2002–2003 using the TM5 chemical transport model as described in Vignati et al. (2010) (E. Vignati, private communication 2010)
The studies by Lapina et al. (2011) and Gantt et al. (2011) are more advanced in that Lapina et al. (2011) apply a water temperature dependent source function, while Gantt et al. (2011) extended the scheme to include a wind speed dependency of organic enrichment – that is, the OM enrichment decreases with wind speed, but still the net OM increased with wind speed. Using this new scheme, Gantt et al. (2011) improved significantly the agreement between measured and predicted OM mass, as illustrated in Fig. 4.6.


Scatterplot of predicted versus observed organic mass fraction of sea spray aerosol < 1.5 μm in diameter for the Mace Head Atmospheric Research Station (53.33°N, 9.90°W) in red and of sea spray aerosol < 2.5 μm in diameter for the Point Reyes National Seashore IMPROVE site (38.12°N, 122.91°W) in blue, with the 1:1 line as a black dotted line (Adapted from Gantt et al. 2011)
Meskhidze et al. (2011) applied the Gantt et al. (2011) scheme and the Vignati et al. (2010) parameterisation, modified using Facchini et al. (2008a) to include size dependence, with the NCAR Community Atmosphere Model CAM5. The findings of Meskhidze et al. (2011) are that different mechanisms contribute to the marine OM fluxes, with a major contribution of marine organic aerosols to the submicron organic aerosol mass over the tropical and mid-latitudes, while methane sulphonate dominates at high latitudes. The Gantt et al. (2011) parameterisation yields a more accurate representation of the seasonal cycle of marine organic aerosol mass concentrations than Vignati et al. (2010).
One question, however, continues to arise given that the OM enrichment scheme is based on chlorophyll a concentration fields and that is: “Is chlorophyll a the best surrogate for OM enrichment?” Russell et al. (2010) suggest there was little or no relationship. A similar conclusion was reached by Lapina et al. (2011). However, Gantt et al. (2011) found that the OM enrichment was best correlated to chlorophyll a rather than to DOM or POM. The original parameterisation was based on correlating satellite-derived chlorophyll a concentrations, in a box 1,000 km × 1,000 km west of Mace Head, with the measured OM enrichment fraction. While the correlation coefficient was significant, it was still quite low (r = 0.3). A sensitivity study shows the effect of the formulation of this parameterisation and various modifications thereof (Albert et al. 2012). Rinaldi et al. (2013) reassessed the relationship between OM enrichment and chlorophyll a using analysis chlorophyll a data from the ESA Data Users Element (DUE) project GLOBCOLOUR (http://www.globcolour.info/). The reanalysis combined SeaWifs, MODIS and MERIS platforms, interpolated to reduce data loss due to clouds. The results are impressive with r increasing from 0.3 to 0.75–0.8. This study also reports that chlorophyll a is the best surrogate for OM enrichment in sea spray.
4.2.1.3 Secondary Aerosol Formation in the Marine Atmospheric Boundary Layer
Secondary aerosol formation is the production of aerosol via gas-to-particle conversion processes. Such processes include homogeneous nucleation of stable clusters, condensation processes, aqueous phase chemical reactions converting dissolved gasses into aerosol mass, and heterogeneous chemical reactions on the surface of particles. Historically, nss-sulphate (i.e. the sulphate not associated with primary sea spray) has been considered the dominant secondary marine aerosol species (Shaw 1983; Charlson et al. 1987) and is one of the main oxidation products of dimethylsulphide (DMS), a plankton waste gas. Ammonium can form a significant contribution to marine secondary aerosol; however, it does not form aerosol on its own, more so, it requires the presence of an acid aerosol which it can neutralise. Typically, the most abundant acid is sulphuric acid; however, a range of organic acids, including methane sulphonic acid (MSA), also an oxidation product of DMS, can also be present in significant amounts, as will be discussed later.
4.2.1.3.1 Secondary Inorganic Aerosol Formation
In a cloudy marine boundary layer, the submicron marine aerosol size distribution is generally bimodal with an Aitken mode at sizes less than 100 nm and an accumulation mode at sizes larger than 100 nm (cf. Fig. 4.1). This bimodality has been shown to result from chemical processing (aqueous phase oxidation of SO2) in non-precipitating clouds (Hoppel et al. 1986). This was corroborated by O’Dowd et al. (1999a, b) who illustrated through airborne measurements of progressive cloud cycling in marine stratocumulus that the accumulation mode modal diameter increased from ~158 to ~194 nm. This growth was observed to occur over four cloud cycles, each taking approximately 40 min. During simulations of this case study, 30 % nss-sulphate production occurred in droplets activated on sulphate nuclei, with the remainder being produced on droplets activated on sea salt.
Detailed cloud parcel modelling studies of the heterogeneous oxidation of SO2 to aerosol sulphate were conducted by O’Dowd et al. (2000) both for activated droplets and un-activated haze particles. The simulations showed that dissolved ozone and hydrogen peroxide were the dominant oxidants, with the ozone oxidation pathway dominant on sea salt activated droplets. The production of sulphate across the aerosol size distribution is non-linear and a significant amount of sulphate production (75–90 %) occurred in sea salt based droplets. The number concentration of activated salt nuclei also significantly influences the total amount of sulphate produced. Below cloud, the amount of sulphate produced in sea salt aerosol is limited by the carbonate buffering capacity (Sievering et al. 1992). However, once activated, in-cloud production can exceed by many times the cloud-free production due to the transient buffering capacity of activated droplets (O’Dowd et al. 2000). Up to 2 μg m−3 nss-sulphate could be produced at SO2 concentrations of the order of 500 ppt. Organic acids and nitric acid tend to be more associated with the sea salt modes and reduces alkalinity and consequently the amount of sulphate produced.
In terms of sulphate aerosol, nss-sulphate is typically partially neutralised by ammonia to different degrees, leading to sulphuric acid, ammonium bisulphate or ammonium sulphate. In Polar Regions, nss-sulphate is typically in the form of sulphuric acid (O’Dowd et al. 1997) with the ammonium to sulphate molar ratio increasing at lower latitudes.
4.2.1.3.2 Secondary Organic Marine Aerosol
The second most abundant aerosol sulphur species is methane sulphonic acid (MSA), an organic acid which is also an oxidation product of DMS. It is the single most dominant secondary organic species (Facchini et al. 2008b). MSA in the aerosol phase results from condensation of gas phase MSA; however, it appears to be semi-volatile as gas phase concentrations have been observed to be inversely correlated with dew point, reflecting a sensitive equilibrium partitioning (Berresheim et al. 2002).
Over the past few years studies over marine remote regions have allowed identification of typical marine SOA components, other than MSA and DMS oxidation products. The presence of monomethylammonium (MMA+), dimethylammonium (DMA+) and trimethylammonium (TMA+) salts in marine aerosol particles was reported for the first time by Gibb et al. (1999). Their presence was attributed to secondary production, suggesting the condensation of volatile alkyl amines, degassed from the sea, through acid–base reactions, in analogy with NH4+. This hypothesis has been strengthened, in recent years, by the observation that alkyl amines participate in SOA formation in different environments reacting with acids (Murphy et al. 2007; Angelino et al. 2001; Tan et al. 2002).
Facchini et al. (2008b) report DMA+ and diethyl-ammonium (DEA+) salt concentrations ranging, together between <0.4 and 56 ng m−3 in submicron marine aerosol particles collected over the North Atlantic Ocean during Spring and Summer. The authors highlight the importance of alkylammonium salts in marine aerosols, observing that they are the most abundant organic species, after MSA, in submicron marine particles. Alkyl-ammonium salts represent on average 11 % of the marine SOA and 35 % of the aerosol water soluble organic nitrogen (WSON). Facchini et al. (2008b) present also considerable evidence that DMA+ and DEA+ are secondary aerosol components, originating from biogenic precursors emitted by the ocean. The maxima in the accumulation mode, as is the case for other well-known secondary components (nssSO4, NH4, MSA), supports the hypothesis of a gas-to-particle conversion process responsible for the accumulation of alkyl-ammonium salts in the fine aerosol fraction. Moreover, DMA+ and DEA+ concentrations measured at Mace Head were always higher in clean marine samples than in polluted air masses, as for MSA, therefore a natural biogenic source is very likely.
Confirming the findings of Facchini et al. (2008b), Müller et al. (2009) reported non-negligible monomethylammonium (MA+), DMA+ and DEA+ in submicrometer particles at Cape Verde, during the season of enhanced oceanic biological activity. Also, Sorooshian et al. (2009) observed DEA+ in submicron particles over the North Pacific Ocean, reporting a certain correlation with chlorophyll a sea surface concentrations, further supporting the hypothesis of a biogenic origin of marine aerosol amines.
Besides alkylammonium salts and MSA, carboxylic and di-carboxylic acids are commonly reported in marine aerosol (Kawamura and Sakaguchi 1999; Claeys et al. 2010; Crahan et al. 2004; Sorooshian et al. 2007; Aggarwal and Kawamura 2008), accounting for less than 10 % of total particulate organic carbon. Usually, a secondary origin is attributed to the detected di-carboxylic acids (Kawamura et al. 2010), of which oxalic acid is often reported as the most abundant (Kawamura et al. 1996a, b). However, oxidised organics, such as C5–C10 carboxylic or di-carboxylic acids, can also be produced by the oxidative degradation of primary particles generated by sea spray and rich in fatty acids (Kawamura and Sakaguchi 1999). Recently, Rinaldi et al. (2011) presented convincing evidence that an important fraction of marine aerosol oxalic acid may derive from the oxidation, in clouds, of glyoxal.
Recent instrumental advances have allowed a deeper insight into the chemical composition of marine organic aerosols. Using liquid chromatography/negative ion electrospray ionisation mass spectrometry, Claeys et al. (2010) investigated marine organic aerosol chemical composition at Amsterdam Island (Southern Indian Ocean). They managed to characterise about 25 % of the analysed marine aerosol WSOC, which is a remarkable result. MSA (17–21 %), oxalate (5 ± 2 %), malonate (1.8 ± 0.9 %) and organosulphates (0.8 ± 1.5 %) were the major identified components. The organosulphates characterised in Claeys et al. (2010) can be considered tracers for an SOA formation process that is specific to the marine environment, that is, oxidation of marine biomass. More specifically, the organosulphates correspond to sulphate esters of C9–C13 hydroxyl carboxylic acids, which are attributed to oxidation of unsaturated fatty acid of phytoplanktonic origin.
In addition, Decesari et al. (2011) demonstrated, through an ensemble of NMR and LC-MS analyses, that marine aerosol WSOC, the fraction traditionally associated to SOA, is the combination of a less hydrophilic fraction, consisting of a distribution of C8–C9 alkanoic acids and diacids, and of a more oxidised, more hydrophilic fraction, where sulphate esters of C6–C11 hydroxycarboxylic acids were found together with MSA and low-molecular weight amines and acids. These results highlight the complexity of the chemical composition of marine SOA and provide evidence for the coexistence in marine SOA of the products of the atmospheric oxidation of primary biogenic materials emitted within sea spray and of compounds deriving from the gas-to-particle conversion of volatile organic compounds emitted by marine biota.
Further, Zhou et al. (2008) found evidence that primary organic matter emitted within sea spray is a dominant sink for the OH radical, with its consequent degradation and the likely production of a series of low-molecular weight organic compounds. These can partition into the gas phase and contribute to SOA formation.
The picture emerging from these results is a complex one, in which primary and secondary aerosol sources interact to generate the observed organic aerosol burden in the MBL. These interactions are schematically shown in Fig. 4.7. Notwithstanding recent improvements, current knowledge of the chemical composition of marine SOA and on their formation mechanisms remains limited and further research is required to address the many unresolved issues.


Conceptual picture of the interactions of organic aerosol sources within the marine boundary layer
4.2.1.3.3 New Particle Formation in the Marine Boundary Layer?
While the majority of the secondary marine aerosol mass is thought to be produced via heterogeneous and/or condensation processes, the number concentration of secondary marine aerosol is determined by homogeneous nucleation, or new particle formation. New particle production may occur in the marine boundary layer (Russell et al. 1994; Pandis et al. 1994) or in the free troposphere after which it can be entrained into the marine boundary layer (Raes 1995), although the relative importance of these two sources has been an issue of debate. Over the open oceans, there have only been a few recorded observations of new particle formation (Covert et al. 1992, 1996a, b; Clarke et al. 1998; Hoppel et al. 1994; Ehn et al. 2010). Observations of a significant nucleation event by Kollias et al. (2004) in the southeastern Pacific appear to be due to emissions from South America (Tomlinson et al. 2007).
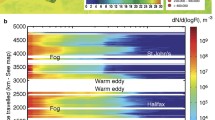
Examination of some of the experimental data indicates that under typical marine conditions, nucleation does not occur. Ultrafine particles observed over the Pacific by Covert et al. (1992) could be explained by entrainment from the free troposphere, while ultrafine particle concentrations observed in the tropics were considered to result from in situ particle nucleation relating to the natural DMS cycle (Clarke et al. 1998). However, ultra-fine particles appear to occur in polar marine air masses in the Antarctic region (O’Dowd et al. 1997) and in a very recent analysis of nucleation and Aitkin mode aerosol in North East Atlantic air sampled at Mace Head, O’Dowd et al. (2010) report the regular appearance of a recently formed nucleation mode (D ~ 10–15 nm) followed by subsequent growth to sizes of 50 nm over 24–48 h timescales (O’Dowd et al. 2010), as shown in Fig. 4.8.


(Top) N3–20 concentrations in clean marine air at Mace Head for a 12 day period in August 2009, illustrating clean nucleation mode event occurrence. (Middle) Combined nSMPS and standard SMPS-derived aerosol size distributions (3–500 nm diameter) corresponding to selected aerosol growth event on JD 236–237 and (bottom) on JD 240–241. A coastal event is evident and distinct from the open ocean event on JD240.6-JD240.8 (Reproduced from O’Dowd et al. (2010) by permission of the American Geophysical Union)
A detailed aerosol nucleation and dynamics parcel modelling study, with monte carlo simulations, by Pirjola et al. (2000) evaluated the likelihood of new particle formation occurring in the marine boundary layer, taking DMS-derived sulphuric acid as the main nucleating candidate. These authors considered both binary nucleation of sulphuric acid and water and ternary nucleation of sulphuric acid, water and ammonia under a range of realistic aerosol regimes. They concluded that the occurrence of new particles in the unperturbed marine boundary could not be explained by known natural sources of sulphur species or DMS; and the occurrence of new or ultra-fine particles could only be explained by the presence of additional condensible species, which are required to grow newly formed clusters to detectable sizes before they are scavenged by the pre-existing aerosols through coagulation processes.
Iodine oxides have also been implicated in marine new particle formation events, particularly in coastal air (O’Dowd et al. 1998, 1999a, b, 2002). Laboratory experiments (Hoffmann et al. 2001; Jimenez et al. 2003; Burkholder et al. 2004) have pointed to the rapid photolysis and subsequent oxidation by ozone of CH2I2, released from macro-algae. McFiggans et al. (2004), Saiz-Lopez et al. (2005) and Sellegri et al. (2005) demonstrated that I2 was the dominant precursor with Sellegri et al. (2005) reporting a linear relationship between I2 concentrations and 3.0–3.4 nm sized particle concentrations. However, some differences in the exact form of iodine oxides is still an issue of debate (Hoffmann et al. 2001; Jimenez et al. 2003; Saunders and Plane 2005).
Such new particle formation events are related to strong coastal emissions of halogen precursors (Mäkelä et al. 2002), resulting in concentrations of the order of 107 cm−3. O’Dowd et al. (2002b) suggested that significantly lower concentrations of iodine oxide precursor condensible vapours (i.e. away from strong coastal sources) could provide either the nucleation mechanism for embryo formation (typically <1 nm in size) and/or the additional condensible vapours required to grow stable sulphate clusters into stable aerosol sizes of a few nanometers (>5–10 nm). If a particle grows to a size of 6 nm, it has a 100 times greater likelihood to survive coagulation loss as compared to a 1–2 nm sized particle.
While iodine oxides may be involved in open ocean nucleation and/or growth, due to the very short lifetimes of these species, it has proved very difficult to specify their role. For that matter, it has also been impossible to elucidate, from experimental field studies, what species are actually involved in the nucleation process and if additional species are required to explain growth of clusters into aerosol particles. This is because of the inability, until recently, to measure particles in the nucleation size range.
However, recent deployment of mass spectrometers has provided realtime information on condensing vapours as detected in conjunction with the appearance and growth of a nucleation mode (Dall’Osto et al. 2012). While not direct evidence of participation of either nucleation or growth of clusters into sizes larger than 3 nm, the quantification of additional condensible aerosol mass during such an event strongly suggests that the same condensible vapours are also responsible for the initial stages of growth. This study pointed to the coincidence of nitrogenated and aliphatic condensible vapours as being responsible for the observed nucleation modes and subsequent growth. In the same study it was found using quantum-chemistry calculations that nucleation of sulphuric acid, dimethylamine and subsequent condensation of MSA decreases cluster evaporation rates (and hence promotes cluster-to-aerosol formation rates).
4.2.2 Non-Marine Sources
4.2.2.1 Desert Dust
Desert aerosol is composed of mineral crustal particles suspended from surface soils by aeolian erosion. Desert and semi-arid areas are a major source of particles in the global atmosphere (e.g. Prospero et al. 2002) and large oceanic areas are regularly under the direct influence of turbid air masses transported from deserts or semi-arid areas, as shown by Fig. 4.9. These dust transport events cause long-range transfers from continent to the remote surface ocean of huge amounts of matter. It has long been recognised that mineral dust deposition to the remote surface ocean significantly influences trace element biogeochemistry (Buat-Ménard and Chesselet 1979; Graham and Duce 1979), marine productivity (Martin and Fitzwater 1988), and deep-sea sedimentation (Venkatarathnam and Ryan 1971; Loÿe-Pilot et al. 1986). More recently, it has been suggested that suspended dust particles affect the optical properties of clear surface waters (Claustre et al. 2002).


Desert dust on the move over the tropical Atlantic. This colour combination of visible and infrared Meteosat images shows a huge desert dust plume transported from Africa. The total mass of dust transported was several millions of tons (Figure courtesy of X. Schneider, CEA)
Due to the trade wind regimes, dust from the Sahara and Sahel is encountered all year long over the tropical Atlantic (Moulin et al. 1997) and as a consequence dominates the solar extinction by aerosol particles in this region on a yearly time scale (Mahowald et al. 2009). Other regions of the world ocean are subject to dust transport and deposition from more seasonal or episodic dust events. Whereas the Sahara-Sahel region dominates dust deposition in the Mediterranean, in most of the North Atlantic and even in regions of the North tropical Indian and Pacific Oceans, source regions in China dominate dust deposition in the North Pacific and Arctic. Middle East sources are most important for dust deposition in the Arabian Sea and Bay of Bengal. Sources in North America provide dust deposited in the North tropical Pacific and northwestern Atlantic while those in South America dominate dust deposition in the South tropical Pacific, the southern Atlantic and the Indian Ocean. Sources in South Africa contribute dust to the South tropical Atlantic and Indian Oceans and Australian sources dominate dust deposition in the southern Pacific and subtropical South Indian Ocean, (Grousset and Biscaye 2005; Mahowald 2007, Fig. 4.10).


Fine dust particles in the long-range transported aerosol size range (diameter smaller than ~20 μm) are strongly bound and not easily mobilised in arid soils, but tend to form large aggregates of fine particles (e.g. clays) or stick to larger particles (e.g. sand grains) which compose the particle size modes found in arid soils (Chatenet et al. 1996). The production of soil dust aerosols is a two-step process (Gomes et al. 1990) firstly resulting from the mobilisation of those large loose soil grains when the surface wind reaches a sufficient velocity (>5–15 m s−1 depending on surface characteristics; Goudie and Middleton 2006) to lift them and produce the so-called saltation of particles, bouncing downstream as commonly seen on sand beaches. Secondly saltating grains then produce small aerosol particles by a sandblasting effect when they settle out and impact the surface soil, disintegrating aggregates or producing small debris. The most productive source areas combine the presence of fine sand grains of ~60–100 μm in diameter that require the minimum threshold friction velocity for being lifted (Iversen and White 1982) and alluvial deposits of aggregated fine clay materials (Prospero et al. 2002) that have the best potential to produce fine aerosol particles (Marticorena et al. 1997).
According to Alfaro et al. (1998) the particle size distribution of aeolian dust aerosol can be approximated by a sum of three lognormal modes with respective mass-median diameters of the order of 1.5, 6.7, and 14.2 μm. (a similar particle size distribution is also produced when clay aggregates from arid soils are crushed and sieved for a long time, allowing the production of a dust aerosol model for use in experiments; Guieu et al. 2010). The relative proportions of the modes are a function of surface wind speed, with larger wind velocities producing more fine particles (Alfaro et al. 1997). Following settling of the largest particles during transport, the mass-median diameter of desert dust over ocean is generally a few microns (e.g. Arimoto et al. 1985, 1997; Dulac et al. 1989; Dubovik et al. 2002; Reid et al. 2003a). However, a number of authors report observations of “giant” sand-sized (>62.5 μm in diameter) dust particles of aeolian origin at very long distances from sources (e.g. Betzer et al. 1988, see also Goudie and Middleton 2006, p. 31). Compared to other super-micron sized aerosol particles such as sea salt aerosol particles which adsorb water, desert dust aerosol particles are characterised by a non-spherical shape, which can be explained by the laminar structure of the clay minerals that are abundant in desert dust aerosols. This irregular shape results in optical properties, i.e. the angular scattering of solar light, which complicate the inversion of dust properties from passive remote sensing data in the solar spectrum (Mishchenko et al. 1995; Dubovik et al. 2002). The irregular particles cause a relatively high rate of depolarization of the scattered light which is useful for identifying dust layers in aerosol lidar remote sensing (Sassen 2000). Another specific optical property of desert dust, responsible for their colour, is their ability to absorb the shortest (UV-blue) solar wavelengths (Moulin et al. 2001). This absorption is controlled by the presence of iron oxides (Alfaro et al. 2004). It causes radiative heating of the atmospheric turbid layers (Alpert et al. 1998) and reinforces a decrease in incoming UV radiation that impacts dissolved organic matter and phytoplankton organisms in surface waters (Tedetti and Sempéré 2006).
Due to the minimum threshold surface wind speed required for aeolian erosion, and to the fact that the dust emission flux varies with the cube of the surface wind speed (Greeley and Iversen 1985), dust events are episodic and show a very high variability in strength. In terms of desert dust atmospheric concentrations or deposition at a given place, the intensity distribution at yearly or longer time scales generally shows a tail towards infrequent high values that control long-term means (Loye-Pilot and Martin 1996; Mahowald et al. 2009 and references therein on observations series).
Desert dust transport is generally characterised by maximum concentrations of aerosol particles in turbid air layers lifted above the marine atmospheric boundary layer, as was observed in the North Pacific (Kritz et al. 1990), the tropical (Carlson and Prospero 1972; Karyampudi et al. 1999; Dulac et al. 2001) and subtropical (Chazette et al. 2001) Atlantic Ocean and the Mediterranean Sea (Dulac et al. 1996; Hamonou et al. 1999). Desert dust settles from the atmosphere to the surface both through gravitational settling and wet deposition, with precipitation either within or below the clouds. Wet deposition of Saharan dust often produces highly concentrated red rains (Avila et al. 1998); mentioned as ‘blood’ rain in Homer’s Illiad. Dry deposition is generally dominant only close to source regions and is controlled by the small fraction of the largest dust particles (Dulac et al. 1989; Arimoto et al. 1997). It seems that atmospheric processes such as the large-scale vertical upward movement of dust-loaded air masses counteract the gravitational settling velocity of dust particles (Dulac et al. 1992; Maring et al. 2003).
The chemical composition of desert dust aerosol particles reflects that of the average Earth surface rocks with a dominance of SiO2 (~60 %) and Al2O3 (10–16 %) resulting from the dominance of quartz and clay minerals (Goudie and Middleton 2006). Either Si or Al are considered as chemical tracers of the soil dust fraction in aerosols. The relative contributions of various clay minerals or minor constituents such as Fe and Ca can generally be used to identify various dust source regions (Bergametti et al. 1989, Goudie and Middleton 2006). The high carbonate content of desert dust is principally responsible for relatively high pH values of rainfall affected by dust particles, with values sometimes larger than six (Loÿe-Pilot et al. 1986; Avila and Rodà 2002). The deposition of desert dust may be a significant source of limiting nutrients such as Fe (Jickells et al. 2005; Mahowald et al. 2009) to remote surface waters (e.g. Bergametti et al. 1992; Ridame and Guieu 2002), despite the relatively low solubility as compared to that of anthropogenic aerosols (Bonnet and Guieu 2004; Baker et al. 2006a). However, solubility is likely to be enhanced by the condensation/evaporation cycle encountered in cloud formation (Desboeufs et al. 2001). Contrary to previous hypotheses, it has been recently shown that the solubility of iron from desert dust is controlled by the iron content in clay minerals rather than by the abundance of iron oxides (Journet et al. 2008).
The interested reader is referred to Goudie and Middleton (2006) for a more comprehensive review of the literature on desert dust related questions.
4.2.2.2 Volcanic Gases, Aerosols and Ash
Volcanic emissions are important sources of atmospheric gases (e.g. Bardintzeff and McBirney 2000), aerosols and ash (e.g. Mastin et al. 2009). Volcanic gas emissions consist primarily of H2O, followed by CO2, SO2, HCl, HF and other compounds. These gases and their oxidation products (in particular sulphate aerosols) may play an important role in the tropospheric and stratospheric chemistry and can impact terrestrial and oceanic ecosystems and human health. H2O and CO2 are important greenhouse gases, but their atmospheric concentrations are so large that volcanic eruptions have only a negligible effect on their concentrations (Robock 2000), although locally the release of CO2 might have important environment effects.
Volcanic ash is a size class referring to fragmented fine-grained particles with diameters of submicron to less than 2 mm. Tephra is the general term for fragmented volcanic material produced during volcanic eruptions that includes ash particles (<2 mm), lapilli (2–64 mm), and bombs and blocks (>64 mm) (Fisher and Schmincke 1984; Schmincke 2004).
Volcanic emissions can be released continuously by passive degassing or diffusive (soil) degassing into the troposphere. Most of the volcanic SO2 in the atmosphere is released from relatively less explosive continuous volcanic activity compared to episodic large scale eruptions (Andres and Kasgnoc 1998). About 99 % of volcanic SO2 is released continuously, while only 1 % is released during sporadic eruptions (Andres and Kasgnoc 1998) (see Table 4.1 for the frequency of eruptions based on the eruption magnitude).
One of the most important climatic effects of explosive volcanic eruptions is through their emission of sulphur species to the stratosphere, mainly in the form of SO2 which reacts with OH and H2O to form sulfate aerosols on a timescale of weeks, producing one of the dominant radiative effects from volcanic eruptions (Robock 2000). Volcanic SO2 release into the atmosphere on a 100 year scale (between 1900 and 2000) is estimated to be 8–11 × 1012 g S year−1, contributing 8–11 % of the total global sulphur emissions of 100 × 1012 g S year−1, which includes emissions from biomass burning, other anthropogenic sources and the marine-derived dimethylsulphide (Halmer et al. 2002).
The global annual direct radiative forcing of sulphate aerosols at the top of the atmosphere by volcanic sulphate is estimated to make up 33 % of the total sulphate forcing (Graf et al. 1997) thereby exceeding the percentage contribution of volcanic SO2 emissions by a factor of about three. Even the silent degassing volcanoes release their emissions into higher atmospheric levels compared to most anthropogenic sulphur emissions, and therefore provide longer atmospheric lifetime of volcanic sulfur species. In particular, volcanic sulphate aerosols from plinian eruptions, like the Pinatubo June 1991 eruption, may influence solar radiation reaching the Earth surface for years, as indicated by the enhanced aerosol optical depth (AOD) after the eruption (Fig. 4.11). Similarly, the Mt. Hudson (Chile) August 1991 eruption may have contributed considerably to the Southern Hemisphere AOD. Reduced solar radiation also affects marine primary productivity (MPP). It should be also considered in satellite retrieval algorithms e.g. for surface ocean chlorophyll a concentration.


Optical depth of stratospheric aerosol during four periods between April 1991 and January 1994 (http://www-sage2.larc.nasa.gov/Introduction.html). The eruption of Pinatubo took place in June 1991, Mt. Hudson erupted in August 1991 (Figure courtesy of NASA)
Volcanic ash and aerosols can be transported over long distances to remote parts of the ocean (Fig. 4.12). Upon deposition in the ocean, volcanic ash can release nutrients as well as toxic substances into the seawater (Frogner et al. 2001; Duggen et al. 2007; Jones and Gislason 2008; Hamme et al. 2010; Langmann et al. 2010; Lin et al. 2011; Olgun et al. 2011). Therefore, volcanic ash may affect marine primary productivity, phytoplankton community structure, atmospheric CO2 concentrations and can eventually (directly or indirectly) impact higher trophic levels the oceanic food-web (e.g. of zooplankton, fish). For the marine ecosystem response related to volcanic eruptions (Chap. 5: Sect. 5.2.2).


Satellite image shows the long distance transport of volcanic eruption plumes that are ejected to high altitudes in the atmosphere as illustrated by the Puyehue eruption in Chile on 6 June 2011 with the white ash plumes reaching more than 10 km altitude and transported across Argentina towards the Atlantic Ocean (Captured by MODIS, NASA)
The importance of volcanic eruptions for the biogeochemistry of the surface ocean, however, has gained limited attention compared to the much better investigated effects of mineral dust. This is despite the fact that an average of about 20 volcanoes erupt at any given time, 50–70 volcanoes erupt every year, and at least one large eruption occurs every year (e.g. Puyehue (Chile) and Grimsvötn (Iceland) in 2011, Table 4.1). Recent estimates based on marine sediment core data show that about 128–221 × 1015 g ka−1 (ka = 1,000 years) of volcanic ash has been deposited into the Pacific Ocean, the largest ocean basin covering 70 % of the iron-limited ocean regions (Olgun et al. 2011). The flux of volcanic ash is of the same order of magnitude as that of mineral dust, which is around 39–519 × 1015 g ka−1 (Rea 1994; Mahowald et al. 2005). On longer time-scales (e.g. during Holocene) the amount of volcanic ash deposition is comparable to that of mineral dust (Olgun et al. 2011), although marine biogeochemical impacts probably differ. Ocean regions with higher likelihood of volcanic ash deposition are shown in Chap. 5: Sect. 5.2.2.
The amount of volcanic ash and bio-available iron attached to the ash surface deposited into the ocean during large episodic volcanic eruptions may exceed the annual dust flux significantly. For example, iron input during the large eruption of Mount Hudson (Chile) between 12th and 15th August 1991 has been found to be equivalent to ~500 years of Patagonian iron dust fallout (Gaiero et al. 2003). Re-mobilization of well-preserved tephra deposits in dry regions can also impact the marine ecosystems after the eruptions (post-eruption impacts). The eruption of Mount Hudson, for example, created several volcanic ash clouds (ash storms) which would have different chemical behaviour compared to mineral dust (Wilson et al. 2011).
4.2.2.3 Global Emissions of Biogenic Volatile Organis Compounds (BVOC’s) from Terrestrial Ecosystems
The term biogenic volatile organic compounds summarises a large number of compounds emitted from terrestrial biota comprising in total an estimated >1,000 Tg C year−1 (Guenther et al. 1995). From an atmospheric chemistry and climate perspective, the isoprenoids (isoprene C5H8, and its monoterpenes and sesquiterpenes derivatives) have been the main focus of attention, reflecting the large mass emitted (isoprene), and/or fast atmospheric reactivity (isoprene, monoterpenes, sesquiterpenes) and related importance for the atmospheric burdens of O3, SOA, OH and CH4 (Atkinson 2000; Atkinson and Arey 2003a, b).
For BVOC emissions no regional or global scale observations exist to provide estimates of their past or present emission strength, distribution and seasonality. Global scale analyses thus have to depend on modelling studies, and these are to date only extensively published and evaluated for isoprene, and to a lesser degree, monoterpenes (Arneth et al. 2008). Current estimates of global isoprene emissions range between approximately 400 and 600 Tg C year−1, while variability in monoterpene emission estimates is larger at ca. 30–130 Tg C year−1 (Arneth et al. 2008). The bottom-up model experiments are complemented by top-down approaches that seek to infer regional emissions of isoprene from remotely sensed formaldehyde column signals (Chance et al. 2000; Palmer et al. 2003; Barkley et al. 2008; Stavrakou et al. 2009), since formaldehyde is one of the chief isoprene oxidation products in the atmosphere. However, linking the formaldehyde retrievals directly to isoprene emissions is hampered by the need to use a chemistry transport model to account for atmospheric isoprene oxidations which are incompletely understood, adding considerable uncertainty to these types of analyses (Barkley et al. 2011, 2012).
Typically, global scale model experiments rely on algorithms that vary diurnal emissions in response to temperature and light (Guenther et al. 1995) that have been found to be the main drivers underlying emission observations in the short-term. Other attempts seek to link emissions and their variability to photosynthetic electron transport rate, reflecting the chloproplastic metabolic pathway of isoprene and monoterpene production and presence or absence of tissue storage of some BVOC, which underlies the observed light and temperature sensitivity (Niinemets et al. 1999; Arneth et al. 2007). Both approaches have to rely on specifying a leaf-level emission capacity which is defined for standard light and temperature conditions. More recently, the use of a canopy-scale emission capacity has been proposed (Guenther et al. 2006) as a model product that relies on the combination of leaf-level measurements and a canopy transfer model. Field observations demonstrate that emission capacities are not constant, but vary strongly during the year and over longer periods, for instance in response to leaf development, previous weather conditions or atmospheric CO2 levels. In a set of recent reviews, emission capacities have been identified as the largest uncertainty in global BVOC emission models (Niinemets et al. 2010a, b, 2011). Emission capacities are species-specific and need to be set for larger plant functional units to be applicable in large-scale models. Emission algorithms are therefore either linked to global vegetation distribution classes derived from remote sensing information, or to dynamic global vegetation models. Only the latter are capable of estimating changes in emissions in future or past environments (see e.g. Arneth et al. 2008 and references therein).
Current models agree on emissions from tropical ecosystems dominating the global totals of isoprene emissions, a combination of tropical vegetation having high emission potential as well as the warm temperatures and high light conditions throughout the year. By contrast, the global monoterpene emission estimates assume also a substantial contribution from the coniferous and evergreen deciduous forests of mid- to high latitudes, even though the period of high emissions in these regions is restricted to few months of favourable weather (Arneth et al. 2011; Guenther et al. 2006; Lathière et al. 2005). While regional differences in emission seasonal patterns and overall strength for BVOC emissions appear to be large, probably much larger than for emission and uptake of CO2, their interannual variability seems small, around 5–10 % of the mean (Arneth et al. 2011).
In the absence of regional to global observational data, model outputs from BVOC simulation experiments are severely hampered by lack of evaluation possibilities. At present, canopy-atmosphere flux measurements by micrometeorological techniques are the sole possibility to provide constraints and reality-checks on the scale of the ecosystem and with the potential to cover a period of months to years. Flux measurements from aircraft could be used to extrapolate to larger regions, although these are restricted to short-term campaigns. On canopy scale, only for isoprene are sensors sufficiently robust to allow long-term observations. But still, only one study location reported measurements covering several years (Pressley et al. 2005). Hence questions on the magnitude of interanual variability essentially remain unanswered. For other BVOC species, flux measurements rely on techniques using gas chromatography or proton transfer reaction spectrometers as sensors. A few short-term campaign studies from a very limited number of ecosystems have been published to-date (a list of example data are provided in Rinne et al. 2009, Pacifico et al. 2011, Arneth et al. 2007, Lathière et al. 2006) which is insufficient for model evaluation.
For future emission estimates large uncertainties exist with respect to how changes in climate, atmospheric CO2, N deposition, tropospheric O3 and human land use/land cover changes interact to alter BVOC emissions in direct and indirect ways (Arneth et al. 2010; Niinemets et al. 2010a, b). In addition to improved understanding of the processes of BVOC production and emissions in response to these various environmental changes, observation and modelling efforts clearly also have to move towards substances beyond isoprene and monoterpenes to reflect the multiple open questions on BVOC-atmosphere and climate interactions (Goldstein and Galbally 2007; Holzinger et al. 2005; Lelieveld et al. 2008).
BVOC directly or via their atmospheric oxidation products, contribute to the formation and growth of secondary organic aerosol (Kulmala et al. 2004). Similar to BVOC emissions, the total mass and number concentration of SOA particles that are formed from BVOCs are highly uncertain. Current estimates are around 12–70 Tg year−1, but these numbers have also been challenged as being too small, perhaps by up to a factor of 10 (Kanakidou et al. 2004; Carslaw et al. 2010; Hallquist et al. 2009). Most modelling work to-date has estimated SOA based on monoterpene emissions only using a constant mass yield of typically 10 % of emissions. Yet, both sesquiterepenes (Bonn and Moortgat 2003) and isoprene (Claeys et al. 2004) have been identified as important precursor sources. While the SOA yield from isoprene may be low, its source strength and the gas-particle partitioning characteristics of its oxidation products are efficient to the point where it may promote SOA growth at higher altitudes and enhance the SOA formation from other sources (Claeys et al. 2004; Henze and Seinfeld 2006).
Quantification of the future direct climate impact of SOA in terms of radiative forcing have so far only considered the case of increasing BVOC emissions over the twenty-first century; these model scenarios result in a substantial cooling (up to −24 W m−2; Carslaw et al. 2010) due to the increased scattering and reflection of radiation by the larger SOA mass. In addition, SOA can grow to particle size classes that act as cloud condensation nuclei, with associated indirect climate effects if cloud albedo and lifetime are affected (Lohmann and Feichter 2005). The full chain of processes from emissions, particle nucleation and subsequent growth to aerosol direct and indirect effects is only just now beginning to be included in global climate models. Initial results indicate large differences between past, present-day and future SOA number concentration and SOA radiative forcing if these processes are treated explicitly (Makkonen et al. 2012).
4.2.2.4 Anthropogenic Emissions
Emission inventories of reactive gases and aerosols are needed as input for climate and atmospheric chemistry and transport models (CTMs) to be able to assess and predict the climate impacts, air pollution concentrations or deposition of elements to ecosystems (Sect. 4.5.1). Emissions can be separated into natural emissions and anthropogenic emissions. The latter implies that the emissions are produced as a result of human activities and can be influenced by changes in technologies and/or emission reduction policies. Emissions need to be spatially distributed, e.g. on a grid, to be suitable as (deposition) model input because the location of emissions is important for their impact, especially for reactive and/or short-lived species. Examples of such gridded emissions can be found at the GEIA/ACCENT emissions portal (http://geiacenter.org).
The most important precursors of secondary anthropogenic aerosol are NOx, SOx, NH3 and Non-Methane Volatile Organic Compounds (NMVOC). For a detailed review of aerosol formation we refer to Seinfeld and Pandis (2006). To quantify the impact of anthropogenic emissions on the ocean (through deposition of nutrients and/or particles) it is necessary to quantify both the primary aerosol emissions and the aerosol precursor emissions. In this section we distinguish between land-based anthropogenic emissions, biomass burning emissions and emissions from international shipping. The latter will in almost all circumstances directly influence the marine ecosystem, although a substantial part of the emissions may also deposit on land surfaces.
4.2.2.4.1 Anthropogenic Land-Based Emissions
Recently Lamarque et al. (2010) developed a new emissions dataset covering the 1850–2000 period, based on the combination and harmonisation of published and publicly available datasets, in support of the Intergovernmental Panel on Climate Change (IPCC) Fifth Assessment Report (AR5). This so-called ACCMIP emission data set also acts as a starting point for the emission projections up to the year 2100 given by different representative concentrations pathways (RCPs) as used in the AR5 (Van Vuuren et al. 2011). The ACCMIP output is the most recent and widely used global emission data. It is distributed on a 0.5° × 0.5° degree grid and made available through ftp://ftp-ipcc.fz-juelich.de/pub/emissions/. Lamarque et al. (2010) provide emission estimates for the precursors of secondary anthropogenic aerosol and for primary carbonaceous particulate matter (BC and OC) (Table 4.2) but not for total PM10.
4.2.2.4.2 Uncertainty in Global Anthropogenic Emissions
Antropogenic uncertainty in emission estimates arise from uncertainty in the activity data, the fuel composition and the emission factors for all individual sources. The resulting uncertainty leads to a range of possible emissions for a given process and base year that varies strongly between regions, sectors, and pollutants. A consistent uncertainty analysis for all pollutants in Table 4.2 is a complex and laborious task, as it would have to be done for all air pollutant/source/technology/country combinations separately and has only been performed for a few species e.g. BC and OC (Bond et al. 2004)/SO2 (Smith et al. 2011). To get an impression of the uncertainties Granier et al. (2011) compiled all currently available consistent global and regional emission inventories and calculated the ratio between the lowest and highest emissions for each species and each region for selected base years. The ratios for the global year 2000 estimates for NOx, SO2, and BC were 1.17, 1.40 and 1.13, respectively (Granier et al. 2011). The ratios for the global inventories were small compared to the variation in the regional inventories which usually ranged between 1.5 and 3.0. The spread gives an impression of consensus but is not an uncertainty analysis. For example, Bond et al. (2004) estimated global fossil black carbon emissions in 1996 as 3.0 Tg C, with an uncertainty range of 2.0–7.4 Tg C, or +150 % and −30 %; this is in strong contrast with the ratio of 1.13 between highest and lowest global inventory for this pollutant. Smith et al. (2011) performed an uncertainty analysis for global and regional sulphur dioxide emissions and concluded that the overall global uncertainty is relatively small: 6–10 % over the twentieth century, but regional uncertainties ranged up to 30 %. For the SO2 year 2000 and values presented in Table 4.2, the uncertainty, based on Smith et al. (2011), would be ~10 % and ~30 % for anthropogenic land-based sources and shipping, respectively. The calculated global SO2 uncertainty bounds are relatively small: the low value is due to cancellation between source categories and regions. This uncertainty level would appear to be unrealistically low given that a number of previous global sulphur dioxide emissions estimates do not fall within this estimated uncertainty bound (the ratio high-low inventories was 1.40; see Granier et al. (2011) for a compilation). The reason is that additional, essentially correlated uncertainties are present that add to the uncertainty value estimated above. Examples include reporting or other biases in global data sets for energy, sulfur removal, and other driver data, methodological assumptions, and the use of common default assumptions for sources where little data exists. Sulphur emissions are less uncertain than emissions of the other air pollutants listed in Table 4.2 because emissions depend largely on sulphur contents in fuels. Uncertainty for other air pollutants is controlled more by combustion conditions and installed technologies and their uncertainty is considerably larger, as quoted above for BC. A qualitative indication is that for VOC, NH3 and OC the uncertainty range will be similar to the BC ranges of Bond et al. (2004) while NOx will be in between the SO2 and BC ranges.
4.2.2.4.3 Global Biomass Burning Emissions
Biomass burning emissions are highly variable from year to year as a result of different environmental and human factors. Schultz et al. (2008) provided a detailed literature review on continental scale estimates of biomass burning emissions and constructed a global emissions data set with monthly time resolution for the period 1960–2000. The previously discussed ACCMIP historical dataset (Lamarque et al. 2010) provides decadal monthly mean biomass burning emissions, mostly based on Schulz et al. (2008). As can be seen from Table 4.2, biomass burning emissions contribute significantly to total global emissions of aerosols and their precursors but its relevance differs by substance, from being quite modest for SO2 to dominant for OC. Over the past 20 years estimates of biomass burning emissions, including their spatial location, have greatly improved due to the availability of earth observation data from satellites. The activity data detected from space (burned area or fire radiative power) includes all major grassland, savanna, and forest fires (including deforestation fires) (e.g. Van der Werf et al. 2006, 2010; Kloster et al. 2010). To estimate emissions, satellite-derived burned areas (Giglio et al. 2010) drive the fire module of a biogeochemical model that calculates fuel loads for each month and grid cell, which are then combined with emission factors (Andreae and Merlet 2001). A good example of this methodology is the Global Fire Emissions Database (GFED) which contains emissions from open fires for the 1997–2004 period (van der Werf et al. 2006). A new version of the inventory that covers the 1997–2009 period, called GFED-v3, was made available at the beginning of 2010 (van der Werf et al. 2010).
4.2.2.4.4 International Shipping Emissions
The ACCMIP emissions dataset (Table 4.2) provides ship emissions including international shipping, domestic shipping and fishing, but excluding military vessels, based on Eyring et al. (2009). These authors used data from the International Maritime Organization (IMO) study discussed in Buhaug et al. (2008), while non-CO2 emission totals were derived as a mean of previous studies (Corbett and Köhler 2003; Eyring et al. 2005; Endresen et al. 2003, 2007). Ship emissions are distributed over the globe using the International Comprehensive Ocean–atmosphere Data Set (ICOADS; Wang et al. 2008), which provides changing shipping patterns on a monthly basis. Eyring et al. (2009) estimated that in 2000, the emissions released by the ocean-going registered fleet for nitrogen oxides (NOx), sulphur oxides (SOx), and particulate matter (PM) were 11.6 Tg NO, 11 Tg SO2, and 1.4 Tg PM, respectively, within a bounded range of 6–21 Tg NO, 6–20 Tg SO2 and 0.4–3.4 Tg PM.
4.2.2.4.5 Comparison and Evaluation of Different Emission Datasets
A detailed comparison and evaluation of different emission datasets of anthropogenic and biomass burning emissions for the 1980–2010 period was made by Granier et al. (2011). They identified large discrepancies between the global and regional emission data sets, showing that there is still no consensus on the best estimates for surface emissions of atmospheric compounds. Hence the data presented in Table 4.2 have a substantial uncertainty; about 10–30 % for land-based anthropogenic emissions and shipping and 50–80 % for biomass burning (Granier et al. 2011). For a full description of the various global and regional emission datasets currently available we refer to Granier et al. (2011). To understand the impact of anthropogenic emissions on marine ecosystems it is important to know not only the absolute emission in a certain base year but also the trend over time. As an example we show the historic development of NOx emissions for the period 1970–2000 in Fig. 4.13.


Clearly all emissions have increased over the past decades, with the land-based emission slowing down somewhat in recent years but shipping emissions showing a steep increase. The significant increase in the emissions from wildfires throughout the period from 1960 to 2000 due to the increasing importance of forest and peat soil burning is remarkable (Schultz et al. 2008). Annual global carbon emissions averaged at 1,660 Tg C year−1 during the 1960s and rose to an average of 2,560 Tg C year−1 during the 1990s. The most important contribution to the trend comes from enhanced deforestation in the tropical regions. The implication of Fig. 4.13 is that the input from the atmosphere to the ocean will have significantly increased over the past decades and that this is bound to have influenced the cycling of elements and nutrients as well as other processes. Trends in other substances may vary somewhat from the NOx trend depicted here but overall the pattern is consistent. For a full description of the various global and regional emission datasets currently available and emission trends for 1980–2010 we refer to Granier et al. (2011); historic emission trends since 1850 are provided by Lamarque et al. (2010).
4.2.3 Ageing and Mixing of Aerosols During Transport
Aerosol particles produced over the continents are present in remote oceanic regions as a result of long-range transport from high-emission regions. From the moment an aerosol particle forms or is emitted until its removal from the atmosphere by deposition, it has undergone various physical and chemical transformations. This aerosol ageing is due to coagulation, condensation/evaporation of semi-volatile components, adsorption/absorption of volatile components and chemical reactions. The dynamic exchange of semi-volatile substances facilitates the exchange of molecules between particles of different origin. As a consequence, ambient aerosols are often complex internal and external mixtures of both inorganic and organic components. The degree to which aerosol particles are transformed controls their chemical composition, size, shape, and surface area and hence determines the aerosol chemical evolution as well as the particles hygroscopic and optical properties.
Recent studies on aerosol ageing have been focussed on dust and organic aerosol (OA). Current understanding of secondary organic aerosol (SOA) formation is incomplete (e.g. de Gouw et al. 2005; Heald et al. 2005, O’Dowd and de Leeuw 2007) and models underpredict SOA concentrations, notably in aging plumes (Alvarado and Prinn 2009; Grieshop et al. 2009). Moreover, despite extensive work on dust evolution during transport, in particular on Asian dust, the chemical mechanisms involved in dust aging are still in question, implying large uncertainties in estimation of dust cycling and impact (Jickells et al. 2005; Su et al. 2008; Formenti et al. 2011).
Recent studies show an explosion of identified organic species in the aerosol phase, notably due to the improvement of analytical techniques. In contrast to the clear evolution of sulphate which is irreversibly oxidised and condensed, the OA presents versatile transformation highly dependent on meteorological and chemical conditions. The chemistry of these species is not well-known and there is a large need for further studies.
4.2.3.1 Chemical Ageing of Organic Aerosols
The organic composition of aerosols continues to change long after initial particle formation as a result of chemical reactions between particle constituents, reactive uptake of gaseous semivolatile molecules, direct photochemical processes inside the particle, and condensation and evaporation of water on the particle (Pöschl 2005; Robinson et al. 2007; Rinaldi et al. 2010). The additional gas-phase oxidation during transport appears to be principally responsible for most of the aging of organic aerosol particles (e.g. Lambe et al. 2009). However, other dominant ageing mechanisms such as condensational ageing (uptake of oxidised organic vapors) or homogeneous ageing (condensed-phase chemistry such as oligomerisation) has also been identified (Volkamer et al. 2007; Jimenez et al. 2009). These processes are schematically shown in Fig. 4.14.


Main pathways of OA aging. OOA corresponds to oxygenated organic aerosol
The ageing takes place on a time scale ranging from minutes to days (e.g. Dunlea et al. 2009), and may significantly change the molecular composition of particles before their removal from the atmosphere. For example, in biomass burning plumes, the unreacted primary organic aerosol, traced by levoglucosan concentrations, represents only 17 % of the OA mass after 3.5 h (Hennigan et al. 2011), suggesting that the majority of OA is transformed via photo-oxidation. Fu et al. (2011) show that over tropical regions, atmospheric oxidation products can account for 47–59 % of the total organics. Typically, field and laboratory measurements indicate that organic material changes its carbon number and becomes increasingly oxidised, less volatile, and more hygroscopic as a result of continuous ageing in the atmosphere (Zhang et al. 2007; Capes et al. 2008; DeCarlo et al. 2008; Jimenez et al. 2009; Morgan et al. 2010, Ng et al. 2010; Kroll et al. 2011). The extent of oxidation of OA, generally estimated from Aerosol Mass Spectrometer (AMS) measurements, increases as the aerosol is exposed to atmospheric oxidants, notably OH radicals (Grieshop et al. 2009; Sage et al. 2008). Thus, diurnal oxidant cycling involves a significant daily variation of OA composition with more oxidized OA in the afternoon (Aiken et al. 2008; Hildebrandt et al. 2010; Adler et al. 2011).
As a result of the large number of reaction pathways, intermediates and products, aerosol particles sampled from ambient air contain thousands of chemically distinct organic compounds (e.g. Fu et al. 2011). Ageing processes in the atmosphere, resulting in further oxidation and oligo-polymerisation of the organic compounds, ultimately transform them to highly oxidised macromolecular material such as humic-like substances (HULIS) (Holmes and Petrucci 2006; Hallquist et al. 2009). Moreover, due to their semi-volatile properties and high atmospheric reactivity, organic species are often observed in internal mixing with inorganic particles. Thus, inorganic sulphate or nitrate aerosols are usually coated or aggregated with organic particles, forming organic sulphate or organic nitrate aerosols (Surratt et al. 2007; Froyd et al. 2010; Hawkins et al. 2010; Li and Shao 2010). The detailed characterisation of the huge set of OA compounds is beyond the capabilities of most analytical techniques (Kroll et al. 2011). As a consequence, several recent studies aim to identify organic species or find a metric which can be used as a tracer of organic ageing, such as the determination of molecular markers (e.g. Claeys et al. 2010), the mass spectral diagnostic (e.g. via f44 increase, Ng et al. 2010) or the use of the average carbon oxidation state coupled with carbon number (Kroll et al. 2011).
Despite this extreme chemical complexity, it seems that organic matter produced from pollutants in urban atmospheres, from biogenic emissions, or from terpenes exposed to photochemical reactions in smog chambers, is processed to particulate organic matter of similar oxidation state, hygroscopicity, volatility, and molecular mass (Jimenez et al. 2009). In other words, organic aerosols from very diverse origin are observed to evolve in the atmosphere to particles with similar chemical and physical properties (Jimenez et al. 2009; Ng et al. 2010).
4.2.3.2 Internal Mixing
4.2.3.2.1 Dust/Inorganic Species
The transformation of dust during atmospheric transport leads mainly to coating by sulphates or nitrates (Formenti et al. 2011). First observations of internal mixing have been made in Asia where desert sources are located close to highly polluted areas (e.g. Iwasaka et al. 1988; Okada et al. 1990; Yamato and Tanaka 1994; Zhou et al. 1996; Fan et al. 1996). Strong internal mixing of dust and sulphate or nitrate was also reported for the eastern Mediterranean (e.g. Falkovich et al. 2001; Sobanska et al. 2003; Putaud et al. 2004; Koçak et al. 2007; Coz et al. 2009) and over the Atlantic Ocean (Kandler et al. 2007; Dall’Osto et al. 2010) in polluted European and North African air masses. The sulphate/nitrate coating is the result of different heterogeneous chemical processes, such as the uptake of gaseous SO2 or HNO3 (and/or NOx) onto the particle surface and their subsequent conversion to SO42− and NO3− (Dentener et al. 1996; Kim and Park 2001; Usher et al. 2002) and collision/coalescence between dust and aerosol ammonium sulphate or nitrate (Mori et al. 1998; Sullivan et al. 2007; Suzuki et al. 2010). The heterogeneous processes responsible for sulphate mixing with dust may be season-dependent with predominance of coagulation process in summer and SO2 uptake in spring (Suzuki et al. 2010). Recent observations of strong internal mixing of dust with nitrate or sulphate in mesoscale convective clouds during the AMMA campaign (Crumeyrolle et al. 2008; Matsuki et al. 2010a) suggest in-cloud processing involving aqueous phase oxidation of NOx/SO2, as previously assumed by Levin and Ganor (1996) and Liu et al. (2005). Carbonate minerals in dust particles, notably calcite (CaCO3), have been identified as the main substrate for adsorbing sulfur and nitrogen gaseous species (Dentener et al. 1996; Krueger et al. 2004). Thus, the heterogeneous reactions involve the conversion of calcium carbonate to other calcium salts such as calcium nitrate (Ca(NO3)2), calcium sulphate (gypsum, CaSO4) or their ammonium salts ((NH4)NO3 and (NH4)HSO4 or (NH4)2SO4) as first demonstrated in laboratory experiments (Krueger et al. 2004) and then observed in recent field studies (Laskin et al. 2005; Matsuki et al. 2005, 2010b; Sullivan and Prather 2007; Shi et al. 2008; Huang et al. 2010; Suzuki et al. 2010).
The reactivity of inorganic acids with dust is determined by several factors: chemical mineralogy of dust (Sullivan et al. 2007), transport pathways, the extent to which dust is transported across polluted sources (Sullivan et al. 2007; McKendry et al. 2008) and meteorological and chemical processing. In particular, sulphate and nitrate coating on dust surfaces is favoured in the marine atmosphere where the relative humidity is high (Hanisch and Crowley 2001; Usher et al. 2002; Trochkine et al. 2003; Zhang et al. 2003a; Okada and Kai 2004; Ooki and Uematsu 2005; Matsuki et al. 2005). Dall’osto et al. (2010), who compare African dust mixing state at different distances from the emission source, demonstrate a continuous chemical evolution of dust particle composition during atmospheric transport, consistent with the relatively slow atmospheric oxidation of sulphur dioxide. Moreover, measurements on individual particles show evidence of the mineralogy-dependent formation of sulphate/nitrate on dust particles (Matsuki et al. 2005; Laskin et al. 2005; Sullivan and Prather 2007). Sulphate formation is favoured on aluminosilicate-rich particles (Laskin et al. 2005; Shi et al. 2008) while preferential nitrate formation on carbonate-rich dust is observed (Ro et al. 2005; Sullivan et al. 2007; Matsuki et al. 2010a; Fairlie et al. 2010). It was proposed that the preferential association of sulphate with Al-rich dust is partly due to the oxidation of SO2 to H2SO4 catalysed by transition-metals, mainly iron, present in aluminosilica minerals (Sullivan et al. 2007; Sullivan and Prather 2007). Yet, the opposite behavior of sulphate and nitrate formation on carbonates could be explained by their difference of hygroscopicity (Sullivan et al. 2009; Formenti et al. 2011). Calcium sulphate is poorly water soluble, preventing further uptake of water and other gaseous species, and hence suppressing the transformation of sulphur dioxide to sulphate in these particles. In contrast, calcium nitrate is highly hydrophilic, enhancing uptake of water and resulting in a positive feedback, transforming all calcium in the particles to calcium nitrate. The calcium-rich spherical particles observed in Asian dust plumes in Japan and in polluted urban air masses in China (Fig. 4.15; Matsuki et al. 2005; Okada et al. 2005), as well as in the Eastern Mediterranean (Laskin et al. 2005) or in convective systems over the Sahel during the monsoon period (Matsuki et al. 2010a) provide field evidence of this feedback process. This mineralogy-dependent salt formation could imply dust source dependence.


Illustration of different mixing states between dust (Si-rich and K-rich), organic species and soot particles by Transmission Electron Microscopy (TEM) images and Energy-Dispersive X-ray (EDX) spectra. The circles show the sites of EDX measurements. The carbon peaks from EDX spectra were compared between the background (grey spectrum from an area without particles) and the particle (black spectrum). (a) S-rich particle with organic coating aggregated with an organic particle. (b) K-rich particle aggregated with an organic particle and soot, including a fine Fe-rich particle. (c) S-rich particle with organic inclusions. (d) S-rich particles with organic coating (Re-produced from Li and Shao (2010) by permission of the American Geophysical Union)
In addition, mixing between dust and chloride has also been observed close to the Asian coast, over the North Pacific (Zhang and Iwasaka 2001; Murphy et al. 2006; Sullivan et al. 2007; Tobo et al. 2009, 2010) and over the North Atlantic (Sullivan et al. 2007). The absorption of hydrogen chloride (HCl) seems to be responsible for the chloride coating, forming calcium chloride CaCl2 (Kelly and Wexler 2005; Tobo et al. 2009). This heterogeneous pathway could be predominant in the remote marine boundary layer with respect to sulphate and nitrate formation (Ma and Choi 2007; Tobo et al. 2009, 2010). The main source of gaseous HCl is volatilization from sea salt particles during heterogeneous reaction of sea salt with HNO3 or H2SO4 (Tobo et al. 2009).
The mixing of dust in polluted air masses also favors mixing with other anthropogenic compounds, such as metals (Cu, As, Ni, Cd, Zn, Pb) (Sun et al. 2005; Zhang et al. 2005b; Erel et al. 2006; Huang et al. 2010; Wang et al. 2011), probably playing a role on the atmospheric deposition of nutrients to the marine biosphere (Sun et al. 2005).
4.2.3.2.2 Dust/Organic Species
Internal mixing between dust and organic carbon has been observed in African biomass burning plumes (Hand et al. 2010) and in polluted Asian air masses (Fig. 4.15, Leaitch et al. 2009; Geng et al. 2009; Li and Shao 2010; Stone et al. 2011). Recent studies show that ATOFMS (Aerosol Time of Flight Mass Spectrometry) in providing aerosol particles composition as a function of particle size, is a pertinent technology to identify organic species mixed with dust (Sullivan and Prather 2007; Yang et al. 2009; Dall’Osto et al. 2010). Thus, Sullivan and Prather (2007) found from their experiments during ACE-Asia that oxalic acid and malonic acid were predominantly internally mixed with mineral dust and aged sea salt particles. They also observed a diurnal enrichment of oxalic acid in mineral dust, indicating a probable gas-phase photochemical production of dicarboxylic acid followed by partitioning into the particle phase (Sullivan et al. 2007). Yang et al. (2009) also observed oxalate-containing dust and sea salt particles in Shangai but they associated oxalate formation to oxalic acid production by heterogeneous reaction occurring in hydrated/deliquesced aerosol. Leaitch et al. (2009) reported enrichment of dust particles by various organic species, notably formate and acetate in Asian dust collected on the North American coast, probably due to uptake of organic particle precursors by dust nearer Asian anthropogenic sources. Internal mixing between dust and carboxylic acid has also been suggested for African dust in source areas during monsoon periods by in-cloud processes in convective systems (Desboeufs et al. 2010) and in zones of transport in Israel (Falkovich et al. 2001, 2004) and over the French Alps (Aymoz et al. 2004). Due to the numerous observations of mixing between dust and carboxylic acids, which are tracers of SOA, dust is suspected to play a role in SOA formation (Mochida et al. 2007; Duvall et al. 2008; Wang et al. 2009; Stone et al. 2011). Nevertheless, there is no clear evidence of the formation of secondary organic aerosol on dust in these studies. If carboxylic acids are the most common identified organic species in mixing with dust, internal mixing of dust has been observed with MSA (Dall’Osto et al. 2010); PAH (Falkovich et al. 2004; Stone et al. 2011); pesticides (Falkovich et al. 2004); fatty acids (Mochida et al. 2007); hopane and levoglucosan (Stone et al. 2011) and organic nitrogen (Dall’Osto et al. 2010). The abundant organic matter found on dust surfaces suggests that dust is an excellent medium for long-range transport of pollution in the troposphere (Falkovich et al. 2004). The extent organic compounds coat onto dust depends on the transport pathway, reactivity of organic species, ambient concentration and ambient humidity, notably for high water-soluble species like carboxylic acids (Falkovich et al. 2004; Dall’Osto et al. 2010).
4.2.3.2.3 Sea Salt
Sea salt aerosols can also participate in heterogeneous reactions with nitric and sulphuric acids, leading to chloride (and also other halogens like Br) depletion (notably Cl-depletion) through HCl volatilisation and the production of halogen radicals, particularly in relatively polluted marine air (Sturges and Shaw 1993; Johansen et al. 1999; Kumar et al. 2008). The release of reactive chloride is considered to be an important intermediate in the oxidation reactions associated with the removal of light hydrocarbons and ozone in the marine atmosphere (Singh and Kasting 1988; Vogt et al. 1996). The magnitude of Cl-depletion of marine aerosols has been demonstrated to usually increase with decreasing sea salt particle size (Mouri and Okada 1993; Kerminen et al. 1998; Yao et al. 2003; Hsu et al. 2007). This reaction produces sea salt particles coated with sulphate and nitrate over Asian and Pacific areas (Matsumoto et al. 2004; Matsuki et al. 2005; Yang et al. 2009) and in the Mediterranean region (Tursic et al. 2006). In the clean atmosphere, methanesulphonate is the major species involved in chloride depletion as observed in Finland (Kerminen et al. 1998) and in the Arctic (Maskey et al. 2011). The S-containing sea salt particles are generated by reactions of the sea salt particles with MSA and/or H2SO4 from biogenic sources rather than anthropogenic ones. Mixing between sea salt and oxalate is also observed (Kerminen et al. 1998; Yang et al. 2009). A number of studies have reported large variations in the magnitude of Cl-depletion (ranging from few percent to 100 %) over different oceanic regions (Graedel and Keene 1995; Song and Carmichael 1999; Maxwell-Meier et al. 2004; Quinn and Bates 2005; Hsu et al. 2007). This variability has been attributed to the complex interplay of several factors, including variable wind-field, turbulence in the vicinity of the sampling site and the influence of anthropogenic components. Thus, the primary mechanisms governing differential Cl loss from sea salt particles in diverse oceanic regions are now well understood (e.g. Song and Carmichael 1999; Maxwell-Meier et al. 2004; Quinn and Bates 2005).
Sea salt mixing with dust has also been reported: more than 60 % of the total particle population collected in Japan was found to be modified by sea salt as concluded from individual particle analysis (Okada et al. 1990; Niimura et al. 1998; Zhang et al. 2003b; Ma et al. 2005) and the interaction of dust with sea salt was likely an important process in size and composition changes of dust aerosols during their long-range transport (Zhang and Iwasaka 2004, 2006). In contrast, mixtures of dust and sea salt were reported to occur only to a minor degree in the African dust plume outflow (e.g. Reid et al. 2003b; Niemi et al. 2005; Kandler et al. 2007). The mechanisms responsible for the mixing of dust particles and sea salt have not been elucidated in detail (Andreae et al. 1986; Zhang et al. 2005a; Andreae and Rosenfeld 2008), while in-cloud processing was suggested as a major route for agglomerate formation (Andreae et al. 1986; Niimura et al. 1998).
4.2.3.2.4 Future Directions
Since the first models of aerosol ageing (e.g. Song and Carmichael 1999), progress in the understanding of chemical multiphase processes enabled, at best, taking into account the chemical reactivity of aerosols. However, recent studies show an explosion of identified organic species in the aerosol phase, notably due to the improvement in analytical techniques. In contrast to the clear evolution of sulphate which irreversibily oxidised and condensed, the OA presents versatile transformations which are highly dependent on meteorological and chemical conditions. The chemistry of the OA species is not well-known and there is a large need for further studies.
While the convergence of physico-chemical properties with ageing is observed for OA, this is not the case for dust and sea salt particles. Thus, even while numerous studies focussed on the chemical evolution of these aerosol particles, the challenge remains to improve our understanding of the link between aerosol ageing and their properties, notably size, ability to release nutrients and hygroscopicity. This point also needs to extend to measurements, up to now largely situated in Asian regions and the Pacific, to other oceanic regions, notably regions where high climate change is expected such as the Mediterranean basin (Giorgi 2006; IPCC 2007).
4.2.4 Dust-Mediated Transport of Living Organisms and Pollutants
A major argument for studying inter-regional and inter-continental transport of dust and its impacts is the recognition of ecological and human health damage risks. Owing to advances in meteorology, analytical instrumentation, satellite technology and image interpretation, more precise information on source areas, transport patterns and depositional zones for dust can now be obtained.
As an example, fungi transported by Saharan dust have been found to damage Caribbean corals (Colarco 2003a, b). Several health studies in urban and suburban environments have shown a higher risk of mortality from high exposure to fine (particles with diameter <10 μm or PM10) particulate matter (e.g. Lippmann 2007). Airborne particles emitted from geological strata also pose threats to human health and the environment worldwide due to expansion of infrastructure development to serve the increasing population. After generation, dust can be carried by wind into sensitive environments and adverse health effects of respiratory “dust” on human health are well documented (Love et al. 1997).
Two misconceptions exist: desert soils are too inhospitable to accommodate a diverse microbial community and if present, the microorganisms will not be able to withstand the physical stresses (UV, desiccation, temperature) of atmospheric transport. However, dust events have been shown to introduce a significant pulse of microorganisms (Griffin 2007; Schlesinger et al. 2006; Polymenakou et al. 2008) and other microbiological materials (e.g. cellular fragments, fungal spores) into the atmosphere (Jaenicke 2005). Dust-borne transport of microorganisms, mainly over the marine environment, should be enhanced due to tolerable humidity levels and attenuation of UV by the particle load of the various dust clouds. Polymenakou et al. (2008) examined the microbial quality of aerosols over the Eastern Mediterranean region (Island of Crete, Greece) during an African dust storm. Bacterial communities associated with aerosol particles of six different size ranges were characterised using molecular culture-independent methods (analysis of 16S rRNA genes). Spore forming bacteria such as Firmicutes were found to be present in all aerosol particles and dominated the large particle sizes. Besides the dominance of Firmicutes in dust particles, phylogenetic neighbours to human pathogens associated with the respirable particles were also detected. These pathogens have been linked to several diseases such as pneumonia, meningitis, and bacteremia or suspected to induce pathologic reactions such as endocarditis (i.e.S. pneumoniae, S. mitis, S. gordonii, H. parainfluenzae, A. lwoffi, A. johnsonii, P. acnes).
The amount of toxic waste stemming from obsolete pesticides in Africa is higher than previously estimated (Mandavilli 2006). In Africa alone the amounts of toxic wastes are estimated at ca. 120,000 tonnes, with more than 500,000 tonnes worldwide. An estimated 30 % of the waste is believed to be persistent organic pollutants (POPs). The POPs family includes dioxins (polychlorinated dibenzodioxins and dibenzofurans), polychlorinated biphenyls (PCBs) and several organochlorine pesticides such as dichlorodiphenyltrichloroethane (DDT) and hexachlorobenzene. It should be pointed out that the POPs composition of dust in air masses changes in relation to modifications in land use, intensity of pesticide use, and burning of synthetic materials and biomass in the dust source regions and in areas swept by dust air masses (Garrison et al. 2003). There are few studies of the transport of POPs from Africa to the Mediterranean. It has been shown that the air masses arriving in the Eastern Mediterranean from Africa contained levels of polychlorinated biphenyls (PCBs) as high (>100 pg m−3) as those in the air masses coming from industrialised Western Europe (Mandalakis and Stephanou 2002). Concurrently, in another study (Garrison et al. 2006) aimed at elucidating the potential role that Saharan dust might play in the degradation of Caribbean ecosystems, a series of persistent organic pollutants (POPs), trace metals and viable microorganisms were identified in the atmosphere over dust source areas of West Africa and in the Caribbean.
Microbes and fungi present on dust can survive a transcontinental journey and stay alive for centuries (Gorbushina et al. 2007). It was shown that Saharan dust collected over the Atlantic by Charles Darwin in the nineteenth century contained members of the spore-forming bacteria Bacillales (Firmicutes) and fungi attached to the particles. By combining geochemical, microbiological and microscopic methods to analyse their almost 200-year-old samples, Gorbushina et al. (2007) were able to show beyond doubt that dust, which clearly originated from West Africa, transported viable microorganisms across the Atlantic Ocean.
4.3 Direct Radiative Effects (DRE)
The direct radiative effect (DRE) is the change in net downward radiative flux, measured in Wm−2, due to aerosol scattering and absorption of radiation (Forster et al. 2007). The DRE exerted by marine aerosols has received less attention that the anthropogenic DRE, which is a climate forcing. However, there are good reasons for quantifying the marine DRE. Firstly, it gives insight into pre-industrial radiative effects of aerosols since the distinction between natural and anthropogenic aerosols is important in the study of the climate system. Secondly, the decrease in sea-ice extent due to climate change leads to an increase in sea salt generation (Bellouin et al. 2011). Climate change also impacts the activity of ocean biogeochemistry and the associated DMS emissions into the atmosphere (Halloran et al. 2010). These changes impact the DRE of marine aerosols and the Earth’s radiative budget.
In the shortwave spectrum and at the top of the atmosphere, the sign of the DRE depends on the balance between the decrease in net flux due to aerosol scattering and the increase due to aerosol absorption. Marine aerosols lack elemental carbon and iron oxides and are therefore weakly absorbing in the shortwave spectrum (Irshad et al. 2009). Consequently, their shortwave DRE at the top of the atmosphere is negative, and more negative per unit mass than absorbing aerosols of similar sizes. The marine aerosol DRE is exerted predominantly in cloud-free conditions, except where clouds are too thin to mask it. Aerosols are more efficient at exerting a DRE when their radius is comparable to the wavelength of the radiation. The size distribution of marine aerosols, especially sea salt, covers both the sub- and super-micron ranges (Dubovik et al. 2002). Their DRE therefore covers both the shortwave and longwave spectra. In the longwave spectrum, marine DRE is positive at the top of the atmosphere because the aerosol layer is typically colder than the surface (Reddy et al. 2005). In addition, since aerosol size is a key parameter for the DRE, hygroscopic growth is important, especially at the high relative humidity experienced by marine aerosols, and is a strong function of chemical composition (Randles et al. 2004).
Four aerosol characteristics are needed to quantify the DRE: optical depth, single-scattering albedo, size distribution, and vertical profile (Yu et al. 2006). Among those parameters, aerosol optical depth is better constrained, with dedicated ground-based and satellite instruments providing routine retrievals (Holben et al. 2001; Remer et al. 2008; Kokhanovsky and de Leeuw 2009; de Leeuw et al. 2011b). Retrievals are however limited to cloud-free conditions, a restriction detrimental to the sampling of marine aerosols, especially at high latitudes of the southern hemisphere where near-surface wind speeds, cloud cover, and sea salt optical depths are large. Satellite retrievals are more accurate over dark ocean surfaces, where marine aerosols are located, than over the relatively brighter land surfaces (King et al. 1999). Satellite products also include information on the aerosol size distribution, through the Ångström coefficient or optical depth of the fine mode (Anderson et al. 2005). Ground-based sun-photometer networks provide retrievals of the single-scattering albedo (Schuster et al. 2005) and size distribution (Dubovik et al. 2002). Coastal and island sites and ship cruises are useful to characterise marine aerosols locally (Smirnov et al. 2011). The aerosol vertical profile is available from ground-based and spaceborne lidars (Winker et al. 2010).
DRE estimates from observations use retrieved aerosol properties coupled to radiative transfer calculations (e.g. Bellouin et al. 2008) or attribute a fraction of broadband radiative fluxes measured by radiometers to aerosols (e.g. Loeb and Manalo-Smith 2005). The shortwave cloud-free top-of-atmosphere total (natural and anthropogenic) DRE is estimated to be in the range −4 to −6 Wm–2 over global oceans and around −5 Wm–2 over land (Yu et al. 2006). In the southern ocean where marine aerosols are expected to contribute most to the total cloud-free DRE, the latter is estimated at −4 to −6 Wm–2 (Yu et al. 2006). Aerosol observations characterise the ambient aerosol. The contribution of different aerosol types to the total DRE is therefore difficult to quantify from observations alone. Aerosol size retrievals have been used as a useful but imperfect proxy for this task (Kaufman et al. 2005; Bellouin et al. 2008). Marine aerosols are found to be the dominant contributor to the global direct effect. Bellouin et al. (2008) estimated that the sea salt shortwave DRE for the year 2006 is −3.9 Wm−2 (Fig. 4.16a) at the cloud-free top of the atmosphere, half of the total estimated DRE. Zhao et al. (2011) mix observation with modelling to attribute the total DRE to individual species. They find a global-averaged sea salt shortwave DRE at the cloud-free top of the atmosphere of −2.2 ± 0.6 Wm−2, one third of the total DRE. Further speciation into the DRE exerted by nss- sulphate from ocean DMS, and marine organics has not been attempted.


(a) Shortwave direct radiative effect, in Wm−2, of marine aerosols at the cloud-free top of atmosphere as estimated from MODIS collection five aerosol retrievals by Bellouin et al. (2008) for the year 2006. (b) Same but for sea salt aerosols modelled in the Hadley Centre climate model HadGEM2 for present-day conditions
Modelling estimates of the DRE involve the conversion of simulated dry masses of various aerosol types into optical properties. A realistic conversion requires a good simulation of size distribution (Vignati et al. 2004), mixing state (Stier et al. 2007), and hygroscopic growth. In a comparison of global numerical aerosol modelling (Kinne et al. 2006), the median sea salt optical depth is 0.030 at 0.55 μm on a global average, 24 % of the total aerosol optical depth. Sea salt is the second largest contributor after sulphate, which itself includes an unspecified marine component. Diversity among participating models is large and the sea salt fraction of the optical depth varies between 18 % and 50 %. Reddy et al. (2005) report a sea salt and natural sulphate (mostly contributed by the ocean) optical depth at 0.55 μm of 0.040, a third of the global total aerosol optical depth. In their model, marine aerosols exert a shortwave DRE of −0.9 Wm−2 in cloud-free conditions and −0.5 Wm−2 in all-sky conditions. Those numbers represent nearly half the total shortwave DRE. In the longwave spectrum and cloud-free sky, the simulated marine DRE is +0.2 Wm−2, 40 % of the total DRE in that spectrum, the remainder being exerted by mineral dust. The sea salt contribution is larger in simulations with the Hadley Centre climate model described by Bellouin et al. (2011). Sea salt optical depth at 0.55 μm is 0.052, 44 % of the total optical depth. Its shortwave DRE in cloud-free sky at the top of the atmosphere is −1.7 Wm−2 (Fig. 4.16b) on a global average, 40 % of the total DRE. In all-sky, the sea salt DRE is estimated at −0.8 Wm−2 for a total DRE of −2.2 Wm−2. In cloud-free longwave, sea salt DRE is +0.4 Wm−2, half of the total DRE in that spectrum. Modelled estimates are typically weaker than observational estimates, although the incomplete coverage of the Earth’s surface by satellite aerosol retrievals complicates the comparison.
The results above show that observation-based and modelling estimates of the total and marine DRE remain associated with large ranges of values. Uncertainties in the aerosol optical properties, mixing, and identification of different components need to be reduced in order to improve our knowledge of the total and marine DRE. Progress is being made in aerosol remote-sensing techniques (Dubovik et al. 2011), with remote-sensing of aerosols in cloudy sky becoming possible (Waquet et al. 2009; Omar et al. 2009). Improving the sampling of remote oceanic regions through a synergy of satellite instruments would be beneficial to observations of marine aerosols. On the modelling side, the diversity in the simulations of aerosol dry mass and optical properties contributes to the large range of simulated DRE. There is a factor 5 among model simulations of sea salt dry mass (Kinne et al. 2006), and improvements in the parameterisation of sea salt production as a function of wind speed could reduce that range, although differences in simulated wind speed will certainly remain. Sea salt and sulphate from DMS oxidation are the only aerosols produced at the ocean–atmosphere interface in current atmosphere–ocean general circulation models. However, evidence for a major role of organic compounds in the atmospheric aerosol cycle is growing (Kirkby et al. 2011). Parameterisations should be developed and included in global numerical models in order to follow on recent observational evidence of the importance of marine organic emissions (O’Dowd et al. 2004).
4.4 Effects on Cloud Formation and Indirect Radiative Effects
Marine clouds, particularly stratiform clouds, contribute significantly to the global albedo for two reasons: firstly, they contribute a large fraction of global cloud coverage and secondly, they comprise reflecting layers over the dark, absorbing, ocean surface. Furthermore, given that marine clouds are typically clean clouds (i.e. low cloud droplet number concentration), they are far more susceptible to perturbations in cloud nuclei availability compared to continental clouds. For example, Slingo (1990) estimated that a 10–15 % decrease in effective radius, corresponding to increase in cloud droplet concentration of 30–45 % would be sufficient to off-set global warming increase relating to a doubling of CO2.
Although in the early days of cloud physics, sea salt was considered the dominant marine cloud condensation species (Mason 1957), by the 1980s, sea salt was more or less dismissed as having little or no role in cloud formation (Charlson et al. 1987) on the basis that the number concentration of sea salt CCN rarely exceeded 1 cm−3 and was typically too large to mix up to cloud base. Despite this, Latham and Smith (1990) postulated that sea salt could be involved in a wind-speed related negative feedback system whereby with increasing global temperatures, zonal wind speeds would increase, leading to increased sea salt CCN generation, increased cloud droplet concentrations and ultimately increased cloud albedo. Their measurements were based on the existence of sea salt CCN down to sizes of ~0.2 μm radius. O’Dowd and Smith (1993) later demonstrated that sea salt nuclei extended into sizes as small as 0.05 μm radius and, under moderately high wind speeds, could account for ~70 % of the nuclei larger than this size. Furthermore, O’Dowd et al. (1993) demonstrated through combined measurement and modelling studies that there was significant competition between sea salt and nss-sulphate nuclei and that under certain conditions, the addition of a small number of sea salt nuclei under moderately low updraft velocity conditions could lead to a reduction in cloud droplet concentrations as these nuclei cloud be preferentially activated over sulphate nuclei, leading to a suppression of the peak supersaturation achieved in clouds.
Korhonen et al. (2010a), taking observations of the accelerating tropospheric westerly jet, estimated an increase in wind speed of 0.45 m s−1 decade−1 at 50–65°S since 1980 and that this wind speed increase has produced an increase in cloud condensation nuclei of 22 % on average and up to 85 % in some regions, leading to increased cloud albedo. The recognition of the importance of sea salt nuclei in marine cloud processes has led to the interesting suggestion that global warming could be ameliorated somewhat through controlled enhancement of albedo and lifetime in low level marine clouds (Latham 2002), in other words, geo-engineering, whereby artificial floating sea spray generators are deployed in regions of persistent stratocumulus clouds. While intriguing as a geo-engineering solution to global warming, Korhonen et al. (2010b), using a global model, and simulating emissions from a fleet of spray-emitting vessels in four regions of persistent stratocumulus fields found increases in cloud droplet concentration of maximum 20 %, and even a reduction was predicted in one region.
The above studies focussed on sea salt as opposed to sea spray nuclei, the latter which can comprise varying degrees of organic matter enrichment. As discuseed in Sect. 4.2.1.2, O’Dowd et al. (2004) quantified the relative contributions of marine aerosol in terms of sea salt, sulphate, WSOM and WIOM during seasons of low and high biological activity over the North East Atlantic. They found that organic matter dominated the sub-micron mass fraction during the high biological activity period and suggested that a significant fraction of the organic matter was of primary, sea spray, origin. This was later corroborated through laboratory and gradient flux studies which demonstrated that the WIOM was almost exclusively produced from bubble bursting at the ocean surface. Ovadnevaite et al. (2011a) further identified, using aerosol mass spectrometry, significant primary organic aerosol plumes approaching 4 μg m−3 for extended periods, although the mass spectrometry revealed significant oxygenated organic matter in the plume. It should be noted that solubility is a relative definition and the detection of oxygenated organic matter in the plumes is not inconsistent with the previous identification of almost exclusively WIOM. The question is whether or not this primary organic matter which is apparently water insoluble, will have a negative or positive impact on cloud droplet concentration? In a follow-on study, Ovadnevaite et al. (2011b) found that the aerosol particles dominating these so-called primary organic plumes have a low hygroscopicity (~1.2–1.25), they have almost a 100 % activation efficiency at 0.25 % supersaturation even for Aitken mode particles.
Ovadnevaite et al. (2011b) calculated the weighted number of (organically-enriched) sea spray and nss-sulphate nuclei and compared the number concentration of these nuclei with CCN and cloud droplet concentration (Fig. 4.17) for the cloud forming on the plume. The total (combined sulphate and sea spray) calculated nuclei concentration agreed almost perfectly with the CCN concentration at 0.75 % supersaturation, while the sea spray concentration agreed very closely to the cloud droplet concentration, leading to a correlation coefficient of r = 0.76, while nss-sulphate was anti-correlated to the cloud droplet concentration. Not only were these results surprising in that what was apparently non- or very low-solubility organic spray aerosol acting as highly efficient cloud nuclei, but the number concentration and resultant cloud droplet concentration exceeded 350 cm−3 which can be regarded as a very high droplet concentration for a maritime stratiform cloud, and more typical of polluted or continental clouds. It was postulated that the organic matter in the sea spray could be a marine hydrogel resulting in such behaviour. To summarise, sea salt aerosol is likely to have an important impact on the indirect effect, and organic enrichment, in certain regions, may have an even more important impact.


(a) CDNC, measured CCN0.75 %, calculated sea spray, sulphate and total nuclei concentration. (b) CDNC as a function of sea spray particle concentration (Reproduced from Ovadnevaite et al. (2011b) by permission of the American Geophysical Union)
4.5 Deposition of Aerosol Particles to the Ocean Surface and Impacts
Studying the key interactions between the atmosphere and the ocean is essential to understand the present functioning of biogeochemical cycles in the ocean and to predict their evolution in the future. Over the past two decades a considerable effort has been made to improve our understanding of the relevant processes involved in the delivery of bioavailable atmospheric nutrients to the surface of the ocean, their impact on marine biogeochemical cycles, biota response and carbon export. As shown in the following sections, different temporal and spatial scales have been explored from the large-scale experiments to the microcosm studies and through modelling approaches, in both high-nutrient low-chlorophyll (HNLC) and low-nutrient-low chlorophyll (LNLC) areas of the ocean. Recent progress is presented in the following section showing that we are beginning to understand the links between atmospheric deposition and global/regional biogeochemical cycles.
4.5.1 Deposition
Atmospheric deposition of aerosols can impact the biogeochemical cycles of several important nutrients in the ocean, especially iron, nitrogen and phosphorus (Martin et al. 1991) (Duce 1986; Callaghan et al. 2008; Falkowski et al. 1998; Fung et al. 2000) (Krishnamurthy et al. 2010), although deposition of other species may also be important (e.g. Nozaki 1997). Some atmospheric inputs (e.g. toxic metals or dissociation products of strong acids and bases) may actually reduce ocean productivity (Paytan et al. 2009; Doney et al. 2007). The most important constituent for ocean biogeochemistry is likely iron, with nitrogen, phosphorus, toxic metals and acidic species also important in some regions (Doney et al. 2007; Hunter et al. 2011; Krishnamurthy et al. 2010; Okin et al. 2011; Paytan et al. 2009).
4.5.1.1 Iron
Iron is a micronutrient, and required in small quantities by biota. In some regions there is insufficient iron, and thus iron limitations occurs (Martin et al. 1991). Therefore, atmospheric deposition can play a critical role in supplying new iron to the surface ocean (Fung et al. 2000). Most of the iron deposited onto the ocean surface comes from atmospheric mineral aerosols which is approximately 3.5 % iron, with small contributions from combustion sources (Luo et al. 2008); because of the dominance of mineral aerosols to the iron budget, the largest deposition of iron to the oceans occurs downwind of the large desert regions (Fig. 4.18a).


Not all forms of iron are thought to be equally bioavailable (Jickells and Spokes 2001; Mahowald et al. 2009). Most soils have relatively insoluble iron forms, while atmospheric aerosols appear to be more soluble, arguing that atmospheric processing may be occurring (Jickells and Spokes 2001) and plausible mechanisms based on acidity have been proposed (e.g. Jickells and Spokes 2001; Meskhidze et al. 2005; Zhu et al. 1997), although this can be very sensitive to the mineralogical composition of aerosol (Journet et al. 2008). Smaller particles are slightly more soluble than larger particles (e.g. Baker and Jickells 2006; Chen and Siefert 2004; Hand et al. 2004), which can largely be explained by the longer residence time of smaller particles (Hand et al. 2004). Recent studies have suggested a role for combustion sources of soluble iron (Chuang et al. 2005; Guieu et al. 2005; Sedwick et al. 2007), however estimates from observations and models still suggest that the largest deposition of soluble iron occurs downwind of the main desert dust source areas (Fig. 4.18b). Extrapolations of the limited data on soluble iron from combustion and atmospheric processing suggest that soluble iron deposition may have doubled over the last century, assuming constant mineral aerosol composition because of increased pollution (Luo et al. 2008; Mahowald et al. 2009). More recent estimates of changes in desert dust over the past century based on observations suggest there may have been almost a doubling in desert dust between 1900 and 2000 (Mahowald et al. 2010), suggesting almost a quadrupling of soluble iron inputs to the oceans over this time period.
The importance of atmospheric iron was probably initially overestimated (e.g. Ridgwell and Watson 2002), because of an underestimate of the ocean sediment sources of iron (Lam and Bishop 2008) and a lack of understanding of the role of colloids in the ocean in maintaining iron supply (Parekh et al. 2004). However, current understanding suggests that changes in iron between glacial and interglacial times can play an important, if secondary, role in facilitating the drawdown of atmospheric carbon dioxide (Kohfeld et al. 2005), and changes in soluble iron deposition over the anthropocene may have driven small (5 ppm) changes in atmospheric carbon dioxide (Mahowald et al. 2010).
4.5.1.2 Phosphorus
On longer time scales, ocean productivity is limited by phosphorus (Falkowski et al. 1998), and in some regions on shorter time scales (Mills et al. 2004; Moore et al. 2006; Wu et al. 2000). While the source of phosphorus to the ocean from rivers is thought to be much larger than from atmospheric deposition (11 Tg P year−1 vs. 0.6 Tg P year−1) (Seitzinger et al. 2005; Mahowald et al. 2008), much of the riverine inputs may be sequestered in estuaries, and not be available to open ocean biota. Thus, atmospheric deposition of phosphorus can be important. Similar to iron, most atmospheric phosphorus is thought to be in the form of aerosols (Graham and Duce 1979), predominately mineral aerosol particles (83 %) (Mahowald et al. 2008), since crustal material is on average 700 ppm phosphorus. Thus, atmospheric phosphorus deposition is similar to iron in being largest downwind of desert regions (Fig. 4.18e). Other sources of phosphorus include primary biogenic emission of aerosol particles and aerosol emission from biomass burning, fossil and bio-fuel burning, volcanoes, as well as sea salt aerosol (Mahowald et al. 2008). Not all phosphorus is likely to be soluble in the oceanic mixed layer; soluble P or phosphate is measured to be between 7 % and 100 % of total phosphorus in aerosols (e.g. Graham and Duce 1979; Mahowald et al. 2008). It is likely that mineral aerosols are less soluble than other sources of phosphorus (e.g. Mahowald et al. 2008), but there may be atmospheric processing by acids of phosphorus to make it more soluble (Baker et al. 2006b; Nenes et al. 2011). Estimates of phosphate deposition are limited by shortage of measurements of phosphorus and phosphate, as well as limitations in the understanding of the atmospheric phosphorus cycling (e.g. Mahowald et al. 2008). Because of the large reservoir of phosphorus in the oceans, atmospheric deposition is not thought to be a dominant control on global ocean productivity (Krishnamurthy et al. 2010).
4.5.1.3 Nitrogen
The two known sources of new nitrogen to the ocean are biological N2 fixation and atmospheric deposition. Although molecular nitrogen gas (N2) is only available for diazotrophs organisms, nitrogen fixation can be an important process induced by atmospheric deposition of other limiting nutrient and is discussed elsewhere (see Sect. 4.5.4.2). Nitrogen aerosol deposition comes predominately from combustion sources (NOx) and agricultural sources (NH4). The atmospheric reactive nitrogen flux (NOx, NH4, organic nitrogen compounds) has dramatically increased over the last century (Duce et al. 2008; Galloway et al. 2008) and most of it is bioavailable (Duce et al. 2008). While released into the atmosphere as a gas, about half of the reactive nitrogen is deposited as aerosols (Adams et al. 1999). Nitrogen aerosols tend to have a relatively short lifetime in the atmosphere (few days), which is seen in the estimated distribution of reactive nitrogen deposition (Fig. 4.18c, d). Because the pool of nitrogen in the ocean is so large, it is unlikely that anthropogenic new nitrogen is important to net ocean uptake of carbon (Krishnamurthy et al. 2010; Okin et al. 2011). However, large parts of the ocean are thought to be nitrogen limited, additional N deposition could locally lead to additional productivity, especially because the effects of increasing atmospheric nitrogen deposition are expected to continue to grow in the future (Duce et al. 2008; Krishnamurthy et al. 2010).
4.5.1.4 Deposition of Other Species
While iron, nitrogen and phosphorus deposition are thought to provide nutrients which increase ocean productivity, deposition of toxic metals (e.g. Cu) or acidic species are thought to reduce ocean productivity. While Cu is deposited into the ocean predominately by mineral aerosols, deposition is thought to be enhanced by anthropogenic activity (Paytan et al. 2009). Atmospheric acids are deposited to the oceans as acid rain in the form of sulphates and nitrates occurring in aerosols: thus nitrate aerosols both increase productivity by adding nitrogen, and potentially reduce productivity by increasing acidity (Doney et al. 2007) (Fig. 4.18g).
4.5.2 Elements of Biogeochemical Interest and Their Chemical Forms
As shown in the previous section, atmospheric particles from both natural and anthropogenic sources contain chemical elements which participate to marine biogeochemistry when deposited to the surface ocean. Among them, iron (Fe) has received particular attention, especially when associated with mineral dust which dominates the external input of this key element to the surface open ocean (Jickells et al. 2005). Dust-derived Fe has been proposed to be responsible of the glacial-interglacial differences in atmospheric CO2 due to the strong Fe limitation of biological activity in high-nutrient low chlorophyll oceanic regions (Martin 1990). This has boosted the literature on the chemical forms of dust-derived Fe and the multiple factors controlling its solubility both in the atmosphere and in the marine environment (see Sect. 4.5.3). Other sources of atmospheric Fe such as biomass burning (Guieu et al. 2005) or volcanic ash (Sect. 4.2.2.2) can partially contribute to the marine Fe pool at a regional scale although their overall contribution remains low compared to aeolian dust (Mahowald et al. 2005).
As opposed to HNLC oceanic regions, in LNLC areas biological activity is often limited or co-limited by phosphorus and/or nitrogen. This has motivated interest in atmospheric sources of these two elements and their contribution to the marine pool, particularly in two LNLC areas: the Mediterranean Sea (e.g.Markaki et al. 2010) and the Atlantic Ocean (e.g. Baker et al. 2010). At a global scale, mineral dust appears as the major source of P followed by primary biogenic particles and combustion sources (Mahowald et al. 2008). Indeed, dust deposition events can transiently increase the concentration of dissolved inorganic P in the surface waters of the LNLC Mediterranean Sea (Pulido-Villena et al. 2010). The partitioning between dissolved and total P in atmospheric deposition varies widely between 7 % and 100 % (Migon and Sandroni 1999; Mahowald et al. 2008). As for Fe, numerous factors such as the aerosol source and physico-chemical transformation during transport control the dissolved fraction of atmospheric P deposition. Recent results highlight the importance of the atmospheric acidification of aerosols resulting from the mixing of polluted and dust-laden air masses as a source of dissolved P to the oceans (Nenes et al. 2011).
As opposed to Fe and P, N in the atmosphere is mainly present in gaseous form which implies a different behaviour. N deposition is dominated at a global scale by anthropogenic emissions which have significantly increased since the mid-1800s and for which future increases are expected (Dentener et al. 2006; Duce et al. 2008). Atmospheric fluxes of N are also very important in remote dusty regions. Modelling and laboratory studies have indicated that mineral dust particles can take up acids resulting in increased coarse mode nitrate (NO3−) (Usher et al. 2003 and references therein). When secondary NO3− is accumulated in the coarse mode, it can be removed more rapidly by dry or wet deposition (Dentener et al. 1996).
The bioavailable fraction of nutrients has been traditionally associated with the inorganic forms. This may explain the scarcity of data concerning atmospheric deposition of organic nutrients. Today, we are aware that not only heterotrophic organisms but also autotrophic phytoplankton is able to take up organic forms of either phosphorus or nitrogen. Organic N and P contained in atmospheric deposition can thus exert a biogeochemical effect on the surface ocean the magnitude of which remains poorly explored. Although organic nitrogen has been measured in atmospheric deposition (e.g. Cornell et al. 1995), it is only recently that a real interest on its contribution to biogeochemical cycles at a global scale has begun to emerge (Cornell 2011; Lesworth et al. 2010). Indeed, organic nitrogen can constitute 30 % of total N deposition and an important fraction is of anthropogenic origin (Duce et al. 2008 and references therein).
Virtually nothing is known about the significance of organic P in total P deposition. A few studies have been conducted in the Mediterranean Sea showing a contribution of organic P to total P deposition of 34 % and 38 % in the western and eastern basins respectively (Migon and Sandroni 1999; Markaki et al. 2010). These values are in good agreement with the results of Chen et al. (2007) who estimated that, on average, organic P constituted 31 % of total P deposition in the Gulf of Aqaba. These few reported values suggest that atmospheric inputs can be a significant source of organic phosphorus and highlight the need of evaluating the role of this external source in marine biogeochemistry.
Current models of the carbon cycle, either at regional or global scale, do not account for atmosphere–ocean exchanges of organic carbon, particularly, atmospheric inputs of organic carbon to the surface ocean. And yet, the few existing data indicate that the magnitude of these deposition processes are far from being negligible (Fig. 4.19). Willey et al. (2000) estimated the input of dissolved organic carbon (DOC) to the ocean to be 90 Tg C year−1, equivalent to the magnitude of riverine input to the open ocean. Accounting not only for rain, but also for dry deposition, Jurado et al. (2008) derived a global estimate of 245 × 1012 Tg C year−1, a value largely dominated (76 %) by wet deposition of gaseous species (not including CO2). This atmospheric source may be particularly important for the open ocean areas less affected by riverine influences.


Comparison between the only existing estimate of the global atmospheric flux of organic carbon to the ocean and selected global oceanic carbon fluxes typically considered in carbon cycle models. The aim of this figure is to represent visually the relative magnitudes, rough values are given and no error bars are included. (1) Global atmospheric deposition of organic carbon (Jurado et al. 2008). (2) Global oceanic downward flux of particulate organic carbon of which 45 % actually reaches the deep sea floor (Jahnke 1996). (3) Global oceanic downward flux of dissolved organic matter of which 10 % survives to depths >500 m (Hansell et al. 2009). (4) Current estimate of the oceanic sink of anthropogenic carbon (Denman et al. 2007). (5) Global riverine input of organic carbon to the ocean (Seitzinger et al. 2010). The atmospheric input of organic carbon is of the same order of magnitude as the POC and DOC deep downward flux and represents more than 10 % of the oceanic carbon sink. Moreover, it is similar to the riverine input, which has been so far the main allochthonous source of organic carbon represented in carbon cycle models
In summary, our current knowledge of the atmospheric fluxes of nutrients to the surface ocean suffers from a disequilibrium between the organic and inorganic fractions. Further data on atmospheric deposition of organic phosphorus, nitrogen and carbon will certainly constitute a step forward towards the quantification and prediction of the impact of atmospheric deposition on marine biogeochemistry.
4.5.3 Dissolution- Scavenging Processes
In recent years, an abundant literature has been published on the effect of dust deposition on the biogeochemical functioning of the surface ocean (e.g. Landing and Paytan 2010). Even if the assumption is rough, it can be considered that microorganisms preferentially uptake elements in the dissolved form. Therefore, from a (bio)-geochemical point of view, for a given chemical element, a key issue is to assess the impact of atmospheric particle deposition on the dissolved pool of such elements in the ocean. Atmospheric deposition can transport a large amount of essential macro and micro-nutrients to the ocean (see Sect. 4.5.1). Due to the large scientific debate on the role of atmospheric dust iron deposition to the surface ocean on the climate variability at millennium time scales (Martin 1990), an extensive set of studies have been produced on the impact of dust deposition on iron stocks. Due to its importance in oligotrophic oceanic areas, an abundant literature has also been produced for phosphorus in the last two decades. However, some recent studies suggest a potential control of biological activity by other trace elements of atmospheric origin (Paytan et al. 2009; Ridame et al. 2011) in large oceanic areas. It is therefore important to emphasise that, even if most of the examples hereafter are based on iron or phosphorus chemistry, the concepts introduced have a wider importance than just for these elements.
This section will focus on the “post-depositional” processes, which include all processes that occur at the surface of aerosol particles once they have deposited at the ocean surface. The impact of atmospheric particles on elemental stocks in the ocean will be controlled by the balance between dissolution and scavenging at the surface of the atmospheric particles which settle through the water column. For a given elemental stock, dissolution will constitute a source whereas scavenging will be a sink.
A large set of literature exists on the dissolution of nutrients or trace metals from atmospheric particles, with an important emphases on iron (Baker and Croot 2010). A number of these studies focus on atmospheric processes. They have demonstrated that the solubility of different elements from atmospheric particles is controlled by the nature, origin, size and mineralogy of the particles and by the complex processes that affect particles during atmospheric transport (see Sect. 4.2.3).
Once atmospheric particles deposit in seawater, the solvent where the solubility processes occur changes drastically from atmospheric conditions, shifting from low pH value and low ionic strength in cloud droplets to high ionic strength and a slightly basic pH value for seawater. Recent studies have demonstrated that solubility of different trace elements is lower in seawater than in ultrapure deionized water (Buck et al. 2010; Chen et al. 2006). The main driving force for these differences is pH.
In the ocean, the thermodynamic solubility of ionic species can be very low. For example, the solubility of inorganic iron in oxic seawater is between 100 and 200 pM (Liu and Millero 2002). Higher concentrations of dissolved iron are possible in seawater through complexation by organic iron binding ligands. Indeed, more than 99 % of dissolved iron is in the form of organic complexes (Hunter and Boyd 2007). In this case, the seawater binding ligand concentration may control the amount of a specific element that can dissolve in seawater. For iron, this control by specific binding ligand has been demonstrated in batch dissolution experiments with dust end-member particles (Wagener et al. 2008, 2010). Moreover, the potential of free iron binding ligand to keep iron from dust particles dissolved was demonstrated in situ during a cruise in the tropical Atlantic after a Saharan dust event (Rijkenberg et al. 2008). Other elements, such as cobalt (Saito and Moffet 2001) and copper (Buck and Bruland 2005) are also found predominantly in the form of organic complexes in seawater and the same control mechanism on dissolution as for iron could occur.
The importance of the particulate concentration in seawater on the relative solubility has been discussed in earlier studies. Bonnet and Guieu (2004) have demonstrated that the relative solubility of iron decreases when the particulate concentration increases. This trend might be explained by the iron binding capacity of seawater which limits the iron that can dissolve and therefore decreases the relative amount of dissolved iron for higher particulate concentrations (Baker and Croot 2010). However, similar trends have been demonstrated for phosphorus dissolution (Ridame and Guieu 2002) whereas dissolved phosphorus concentrations are not demonstrated to be controlled by the solubility capacity of seawater.
Scavenging of dissolved elements on particles in the water column has been extensively studied on settling particles because this process is essential to explain the deep concentrations of different chemical elements. For some elements, scavenging on particles is even used to trace fluxes of biological particles in the water column: thorium-234 deficit in the water column is used to assess the export of organic carbon (Buesseler et al. 1992). Moreover, in the field of geochemistry, a rich literature exists on rare earth elements scavenging on pure mineral phases because of the importance of this geochemical process during sediment formation.
Only few studies exist on scavenging processes for atmospheric particles in the ocean. Zhuang and Duce (1993) performed a set of scavenging experiments with radiolabelled 59Fe on natural aerosols in seawater and demonstrated significant scavenging on dust particles. A similar type of experiments with radiolabelled 33P demonstrated scavenging of phosphorus on Saharan dust end-member particles (Ridame et al. 2003). In earlier studies, based on pure haematite particles, Honeyman and Sanchi (1991) demonstrated the scavenging of colloidal iron when freshly produced haematite particles coagulate to form larger aggregates. This points to the importance of the particle size (and certainly chemical composition) for scavenging processes.
Even fewer studies have taken into account the balance between scavenging and dissolution processes when atmospheric particles deposit to the surface ocean. In a set of batch reactor iron dissolution experiments in seawater, Bonnet and Guieu (2004) considered the importance of scavenging to suggest that the net result of dust dissolution is lower than the effective dissolution from dust particles. In a recent artificial dust seeding experiment in a large mesocosm, it was demonstrated that the net effect of dust addition was a decrease of the dissolved iron stock in the first 10 m of the water column due to scavenging on the settling dust particles (Wagener et al. 2010). The mesoscoms experiment was simulated with a 1D model which takes into account dissolution and scavenging of iron on dust particles. The equilibrium between both processes allowed definition of a critical Fe concentration above which the balance between dissolution and scavenging is in favour of scavenging, resulting in a net sink of dissolved iron (Ye et al. 2011).
One of the major challenges in future studies on abiotic processes occurring when atmospheric particles deposit at the ocean surface will be to investigate the above process in a more realistic way. This implies studying the processes involved (scavenging and dissolution) at a higher temporal and vertical resolution. So far, most of the studies on the biogeochemical impact of atmospheric deposition, have assumed that once they deposit at the ocean surface, atmospheric particles are perfectly mixed through the surface mixed layer (SML) (Fig. 4.20 – Left panel). It is considered that after a certain time, a fraction of a soluble element is dissolved (or scavenged) and will increase (or decrease) the dissolved stock of the element. Based on this postulate, and in order to asses the impact of atmospheric particles, batch reactor experiments need to be conducted where atmospheric particles are in contact with seawater at the expected concentration in the SML. The net effect of atmospheric deposition for an element would then be assessed by measuring the increase or decrease of the element after a certain amount of time. However, in order to understand the atmospheric particle deposition process in a more realistic way, two main issues (among others) need to be considered:


Conceptual view of the fate of atmospheric particles after their deposition. Left panel: “Batch reactor” point of view with atmospheric particles perfectly mixed over the SML. Right panel: Hypothetical dynamics of atmospheric particles in the water column. At T0 particles are in the surface microlayer. After T0, bigger particles (orange) settle faster than smaller particles (black) which form aggregates with organic matter (green) at an intermediate depth
-
1.
When they deposit at the surface of the ocean, particles will first encounter the sea surface micro-layer. This micro-layer is enriched in organic matter with a specific chemical composition (Frew 1997), which might greatly impact processes at the surface of atmospheric particles directly after deposition.
-
2.
Once particles cross the micro-layer, the physico-chemical processes that occur during settling through the water column must be considered. Through the formation of aggregates (between atmospheric particles but also with organic particles), the size distribution of the particles will change when they settle through the water column. Moreover the influence of organic matter, which may play the role of a glue during these coagulation and aggregation processes, will also change the chemical nature of the particles and in particular the surface properties (Verdugo et al. 2004). These changes will influence the settling rate of the particles, but also the scavenging or dissolution processes that may occur at the particle surface.
One challenge for future research is thus to take into account the dynamics of the particles which settle through the water column. As illustrated in Fig. 4.20 (right panel), this new approach might lead to a much more complex image of the impact of atmospheric particles in the ocean. However, to tackle these concepts, new tools able to investigate at such scales need to be developed. In particular, two major technological issues have to be solved:
-
1.
The settling of atmospheric particles through the water column and their change in size and composition must be followed in situ with high temporal resolution. Promising results have been recently obtained in a mesocosm experiment with a combination of optical in situ measurements (Bressac et al. 2012).
-
2.
Chemical techniques must be fast and reliable enough to investigate at these scales. Even though the recent development of new sensors for biogeochemical parameters on board new platforms (gliders, profilers,…) is promising, there is a crucial need of chemical sensors for relevant micro and macro nutrients.
In conclusion, a major perspective in assessing the impact of atmospheric deposition on the ocean is to investigate the competition between scavenging and dissolution within a dynamical process of the settling of the particles through the water column. The debate is largely open on the relevant scales that need to be investigated.
4.5.4 Atmospheric Impacts in HNLC and LNLC Areas
4.5.4.1 Experimental: Large Scale Fertilisation Experiments (Fe, P)
Direct assessment of the fate of atmospheric dust deposition and the subsequent biogeochemical impact on HNLC or LNLC waters has been extremely limited due to the episodic nature of such events and the need for researchers to ‘be in the right place at the right time’. Such assessments have therefore been indirect, and mainly confined to a comparison of remotely-sensed datasets from different satellite sensors (Boyd et al. 2010). The use of such indirect approaches has in many cases led to mis-attribution of cause (aerosol deposition) and effect (altered ocean biogeochemistry via an aerosol-iron mediated phytoplankton bloom) (Boyd et al. 2010).
Due to the lack of direct information on the ‘dust-nutrients-biota-biogeochemistry’ linkages, we have had to rely on experimental approaches in which a known quantity of nutrients (either iron or phosphate, and in a few cases iron and phosphate, Boyd et al. 2007) are added to an area (generally 50–100 km2) of the surface ocean. Such experiments, referred to as in situ mesoscale ocean enrichment, provide valuable insights into the wide range of processes that are influenced by an episodic nutrient addition, and as such provide a ‘biogeochemical timeline’ of how the nutrients released from dust deposition may have myriad effects on both surface and subsequently subsurface or atmospheric processes.
Trends from over ten of these experiments reveal convergences but also differences, which in many cases are due to regional differences between the environmental characteristics of HNLC waters in tropical to polar waters (de Baar et al. 2005). Convergent results provide confidence in compiling a biogeochemical timeline of the many processes that may be altered by aerosol deposition. In the case of iron enrichment, rapid initial changes (i.e. hours to days) include altered iron chemistry, up-regulation of cellular machinery in pelagic microbes such as photosynthetic competence (Fv/Fm), increased growth rates by all phytoplankton groups which is first manifested as an increase in stocks of cyanobacteria followed by those of nanophytoplankton (such as haptophytes). This initial increase in cyanobacterial abundances is truncated within a day or so as they are grazed down to initial levels as nano-grazers respond to higher pico-prey concentrations. A similar increase in haptophyte abundances lags the cyanobacteria, and then they are also brought under grazer control. In contrast the diatom stocks take longer to reveal an increase in abundances, and in many cases they escape grazing pressure resulting in an iron-mediated bloom (de Baar et al. 2005; Boyd et al. 2007).
This biological activity results in distinct biogeochemical signatures that produce feedbacks that are important for ocean–atmosphere interactions. For example, at some but not all experimental sites, the grazing of the haptophytes by microzooplankton may result in increased ocean DMSP inventories (Boyd et al. 2007) that consequently may in some cases result in elevated DMS concentrations in the upper ocean. However, in other studies such as SERIES in the NE subarctic Pacific no such increase in DMS was observed by the end of the bloom (Boyd et al. 2007). Concurrently there is widespread evidence of significant decreases in oceanic CO2 concentrations due to enhanced C fixation by the blooming diatoms (de Baar et al. 2005) which may result, in time, in a drawdown of atmospheric CO2. There are also knock-on effects on ocean physics (warming due to more absorption of incoming solar radiation by the higher phytoplankton stocks) and chemistry (uptake of added iron, and decrease of much of the inventories (with in some cases alteration of the stoichiometry of nutrient stocks – Boyd et al. 2004) of other plant nutrients such as silicate and nitrate). In some studies, the imprint of the iron-mediated blooms is eventually recorded (1–2 weeks) in the waters underlying the surface mixed layer, such varying degrees of enhancement of downward export of algal carbon from the bloom (Boyd et al. 2004; c.f. Smetacek et al. 2012), and elevated concentrations of other greenhouse gases such as N2O and CH4 probably due to remineralisation of the sinking particles (Boyd et al. 2007). However, in many experiments there were major uncertainties in estimating the ratio of iron added to additional carbon exported to depth from the base of the surface mixed layer.
Although fewer in situ nutrient enrichment studies have taken place in LNLC waters, relative to those in HNLC waters, they too provide evidence of the many effects of episodic nutrient enhancement, for example resulting in widespread foodweb effects such as the putative mechanisms of ‘ecological tunnelling’ reported for the CYCLOPS experiment in the E. Mediterranean by Thingstad et al. (2005). As there have been so few studies in LNLC waters, it is premature to comment on whether more effects are observed when an episodic pulse of nutrients is added to either HNLC or LNLC waters.
The benefits of what we have learned from these in situ mesoscale experiments for better understanding the biogeochemical effects of aerosol nutrient deposition into the ocean are twofold. Firstly, they provide us with a timeline of conspicuous changes in upper ocean properties that may help detection and attribution of the effects of episodic dust deposition events on the upper ocean using a combination of shipboard and/or satellite observations. Second, they have increased our conceptual understanding of how atmospheric processes influence those in the ocean, and how in turn those in the ocean may feedback on the lower atmosphere. However, when the magnitude, resulting elemental stoichiometry of the upper ocean, and chemistry of added nutrients of mesoscale ocean nutrient enrichment is compared with that resulting from an aerosol deposition event it is clear that the former is much greater than the latter (see Fig. 4.21). This prevents the ready extrapolation of the results from such experiments to the natural world, and suggests that some cautious downscaling – via modelling simulations – of the observed biogeochemical signatures might provide some bounds on how an aerosol deposition event might influence the upper ocean.


A comparison for Southern Ocean waters of mechanisms responsible for perturbations in Fe supply. Numbers in each panel: (1) Fe*, the relative magnitude of Fe supply relative to macronutrient supply (Parekh et al. 2005); (2) the mode of Fe supply; (3) the time scale over which surface waters receive increased Fe supply; and (4) the length scales of Fe supply events. (a) Satellite image of a purposeful in situ Southern Ocean FeAX (Mesoscale iron addition experiments) [SOIREE (Boyd et al. 2000)]. (b) An FeNX (Fe natural enrichment experiments) near Crozet within the HNLC Southern Ocean, where naturally occurring blooms are evident from remote sensing (Boyd et al. 2007 and references given therein). (c) An atmospheric dust deposition event (dust units are g m–2 year–1) in the modern Southern Ocean [e.g. from Patagonia (Jickells et al. 2005)]. (d) Fe supply to the Southern Ocean during the last glacial maxima from direct [i.e. higher dust deposition (Martin 1990; Wolff et al. 2006)] and/or indirect [i.e. upwelling of waters with higher Fe concentrations (Lefevre and Watson 1999)] sources. The magnitude of this supply is unknown; hence, Fe* is expressed as < 0. Fe* is defined as Fe* = [Fe]–{(Fe/P) algal uptake ratio × [PO43–]} (Parekh et al. 2005). If Fe* > 0, primary production is ultimately macronutrient-limited; if Fe* < 0, production is ultimately Fe-limited. The width of red arrows denotes the relative magnitude of changes in Fe supply; the hatched arrows in (d) denote uncertainties about whether Fe supply in the geological past was episodic or sustained. In (b) to (d), downward- and upward-pointing arrows represent atmospheric and oceanic (upwelling) supply, respectively (Reproduced from Boyd et al. 2007 by permission of American Association for the Advancement of Science)
4.5.4.2 Experimental: Microcosms
Microcosm experiments involve the enclosure of a water sample in a container, typically 1–10 l in volume, and its incubation under simulated in situ conditions. Microcosm experiments are often used in the study of the effects of atmospheric nutrient deposition on natural plankton communities, as they represent an easy experimental approach to monitor a wide range of chemical and biological properties and their response to controlled perturbations. Microcosm experiments have the same limitations as any approach that involves in vitro confinement of water samples, including sampling bias and the difficulty of precisely simulating in situ conditions. The composition of the community can change markedly after bottle enclosure (Massana et al. 2001; Calvo-Díaz et al. 2011) and therefore most bioassay experiments have a duration that does not exceed 3–4 days (e.g. Bonnet et al. 2005; Mills et al. 2004; Moore et al. 2008), which means the recorded changes represent short-term responses to the simulated perturbation. For all these reasons, the results obtained from bioassay experiments are best interpreted in conjunction with in situ observations collected during oceanographic cruises and time-series monitoring programmes.
Published reports show a large degree of variability in the responses of surface plankton communities during microcosm fertilisation experiments intended to simulate the effects of atmospheric deposition. Part of this variability stems from the fact that the materials used to amend the water samples differ widely among studies, as they may consist of collected aerosols (Herut et al. 2005), collected rainwater (Klein et al. 1997), unprocessed desert soils (Mills et al. 2004), and atmospherically processed soils (Ternon et al. 2011). In addition to the geographical variability in the composition and solubility of natural aerosols, the relative abundance of anthropogenic particles varies widely in both space and time, and plays a crucial role in determining the biological effects of atmospheric deposition. An additional source of variability is the fact that different communities inhabiting the surface ocean have distinct community structures and experience different types and degrees of nutrient limitation (Moore et al. 2013), all of which result in diverging responses to a given perturbation. In spite of all these sources of variability, the microcosm experiments conducted during the last decade have yielded consistent patterns regarding the responses of surface plankton to atmospheric inputs in terms of biomass and abundance, community structure, and metabolic activity.
4.5.4.2.1 Main Results Obtained from the Microcosm Approach
Phytoplankton biomass, estimated from chlorophyll a concentration, typically increases after the addition of aerosols, although the reported increases are usually modest, e.g. around 50 % or less (Mills et al. 2004; Bonnet et al. 2005; Marañón et al. 2010). In LNLC waters, the potential of atmospheric deposition to cause phytoplankton blooms (e.g. resulting in chlorophyll a concentrations of 1 μg L−1 or more) is limited because, after taking into account nutrient content and solubility as well as dilution over the upper mixed layer, even large deposition events such as strong dust storms are unlikely to increase nitrate and phosphate concentrations by more than approximately 0.2 μM (Guieu et al. 2002; Ridame and Guieu 2002; Mills et al. 2004). As an example, if in a nitrogen-limited system nitrate concentration is increased by 0.2 μM and one assumes a molar C:N ratio of 6.6 for phytoplankton biomass production and a carbon to chlorophyll a ratio (g:g) of 100, the resulting maximum increase in chlorophyll a concentration will be only 0.16 μg L−1. It must be taken into account, however, that LNLC waters cover vast expanses of the ocean, such as the subtropical gyres, which means that even small areal increases in biomass and productivity can have significant global impacts. The situation is different in the case of HNLC regions, where iron limits both phytoplankton standing stocks and primary production rates, and an excess of macronutrients is available. A strong dust deposition event (delivering 2 mg L−1 of aerosols to the upper mixed layer) can release around 2 nM Fe (Bonnet and Guieu 2004; Mills et al. 2004). If we assume that 10 % of this dissolved iron is bioavailable (Wu et al. 2001) and take a molar C:Fe ratio of 105 for surface phytoplankton (Boyd et al. 2007), the resulting estimated chlorophyll a increase would be 2.4 μg L−1. Thus, stoichiometric constraints and the different nutrient limitation conditions dictate that dust deposition events have a much larger potential for stimulation of phytoplankton biomass and production in HNLC regions than they do in LNLC regions.
Bacterial abundance and biomass have also been reported to increase after the addition of atmospheric aerosols (Herut et al. 2005; Pulido-Villena et al. 2008; Marañón et al. 2010). It is now well established that bacterial metabolism can be limited by inorganic nutrients in very oligotrophic regions (Rivkin and Anderson 1997; Mills et al. 2008) and atmospheric inputs therefore have the potential to stimulate bacterial growth. In most cases, however, the observed increases of bacterial abundance in the amended treatments, compared with the controls, are modest (<20 %). A recurrent pattern in microcosm experiments is that the response of phytoplankton and bacterioplankton to the addition of aerosols or individual nutrients is much stronger in terms of metabolic activity that in terms of abundance or biomass (see Fig. 4.22). In oligotrophic environments, a tight trophic coupling exists between picoplankton and their unicellular grazers, which means that a stimulation of production rates translates only partially, if at all, into an increase of population abundance.


Relative change (%) of different variables in response to dust addition during eight bioassay experiments in the tropical Atlatic Ocean. Relative change was calculated as 100 × (D–C)/C, where D and C are the mean value of the variable in the dust and the control treatments, respectively. Boxes and bars enclose the 25th–75th and 5th–95th percentiles, respectively, the vertical dotted line is the mean, and the vertical continuous line is the mode (n = 8). Variables shown are total chlorophyll a concentration; the abundance of Synechococcus, Prochlorococcus, picoeukaryotes and total heterotrophic bacteria; the abundance of bacteria belonging to the groups Roseobacter, SAR11, bacteroidetes and gammaproteobacteria; and the metabolic rates primary production, N2 fixation, bacterial production, and community respiration (Figure modified from Marañón et al. (2010))
In addition to considering the bulk biomass of bacterioplankton and phytoplankton, it is also important to take into account community structure and how the relative abundance of different taxonomic groups changes in response to atmospheric deposition. There is growing evidence that within both the phytoplankton and the bacterioplankton different taxonomic groups respond differently to a given input. In particular, the abundance of the cyanobacterium Prochlorococcus has been shown to decrease in response to the addition of Saharan soils and collected aerosols (Hill et al. 2010; Marañón et al. 2010) and also during natural deposition events (Herut et al. 2005). Adverse effects of some aerosols have also been reported for Synechococcus and the picoeukaryotes (Paytan et al. 2009; Marañón et al. 2010). The toxicity of some aerosols for certain groups of phytoplankton may be related to the presence of high levels of copper (Paytan et al. 2009). A negative response by some groups can be compensated by an increase in other groups, such that the bulk biomass does not change markedly. For instance, during a set of eight dust addition experiments conducted in the Atlantic Ocean it was found that both Prochlorococcus and Synechococcus decreased in the presence of dust, whereas the opposite was true for the picoeukaryotes (Marañón et al. 2010) (Fig. 4.22). Less is known about the response of bacterioplankton community structure to atmospheric deposition, but again contrasting changes in different groups have been observed. In the Atlantic Ocean, the abundance of the SAR11 lineage, which is dominant in ultraoligotrophic environments (Morris et al. 2002), tends to decrease in treatments amended with Saharan dust (Hill et al. 2010; Marañón et al. 2010), whereas the opposite is true for gammaproteobacteria, a group known to respond to increased nutrient concentrations (Horňák et al. 2006).
Metabolic rates are usually more responsive than standing stocks to aerosol additions and nutrient amendments. Phytoplankton primary production and N2 fixation can show marked increases, by a factor of 2 or more, which are due to the fertilising effect of N, P and Fe (Mills et al. 2004; Bonnet et al. 2005; Herut et al. 2005; Blain et al. 2004; Marañón et al. 2010; Law et al. 2011; Ternon et al. 2011). Factorial nutrient addition experiments have demonstrated that N is the primary limiting nutrient for carbon fixation in LNLC waters (Moore et al. 2008), whereas P and Fe co-limit N2 fixation in the Atlantic Ocean (Mills et al. 2008). Thus, desert dust has the potential to stimulate autotrophic productivity in the ocean through an increase in both carbon and N2 fixation, therefore favouring CO2 sequestration by the biological pump. However, recent evidence indicates that heterotrophic processes such as bacterial production and bacterial respiration are in fact stimulated by atmospheric deposition to a larger degree than autotrophic processes. In the NW Mediterranean Sea, it has been reported that bacterial respiration can increase by a factor of 3 in response to Saharan dust inputs (Pulido-Villena et al. 2008). Similarly, bacterial production was shown to increase by a factor of 8 during microcosms dust addition experiments in the Eastern Mediterranean Sea (Herut et al. 2005). In a set of eight dust addition experiments conducted in the Atlantic Ocean, the largest increases in metabolic rates (up to 700 %) were reported for bacterial production and community respiration (Marañón et al. 2010) (Fig. 4.22). Whether phytoplankton or bacterioplankton dominate the metabolic response to atmospheric inputs has important biogeochemical implications. If bacteria outcompete phytoplankton in using the newly supplied nutrients, and increase their production and respiration rates, the net result may be the remineralisation of dissolved organic carbon, the release of CO2 and a weakening of the potential for biological CO2 sequestration. This is a paradoxical outcome if we consider that atmospheric deposition is commonly regarded as a process that fertilises the open ocean. However, only a handful of studies have addressed concurrently the metabolic rates of both phytoplankton and bacteria to atmospheric inputs, and therefore more studies are needed in order to understand how competitive interactions within the planktonic community modulate the overall response in terms of the balance between CO2 fixation and respiration. Another priority for future studies is to conduct repeated microcosm addition experiments over time and in different locations, in order to obtain general patterns that relate the initial characteristics of the community, in terms of taxonomic composition and type and degree of limitation by different nutrients, to its response to a given perturbation.
4.5.4.3 Experimental: In Situ Mesocosms
Although in situ mesocosms have been widely used to study biological responses to changes in nutrient conditions (e.g. Duarte et al. 2000) or environmental conditions such as increasing levels of atmospheric carbon dioxide on carbon uptake import or export from the upper ocean (eg Riebesell et al. 2007), impacts of actual atmospheric deposition on ecosystems was only performed recently (Guieu et al. 2010). In situ marine manipulation, although being a technical challenge, represents a significant progress compared to the approach using microcosms. Indeed, mesocosms have the potential to allow controlling physical and chemical forcing, including atmospheric deposition, and the advantage of following simultaneously the whole ecosystem and the export of matter (Fig. 4.23) the biogeochemical modelling in a 1-D model taking into account the role of atmospheric inputs on the biological carbon pump can thus be done. Such experiments remain challenging first because of the difficulty to reproduce an actual atmospheric deposition event, meaning that enough representative atmospheric material has to be produced and second because contamination has to be avoided during the different steps of the experiments. As a matter of fact, the water tested for their response to atmospheric inputs has usually low concentrations of (micro)nutrients (typically iron and phosphorus are at nanomolar levels) and the induced changes in the biogeochemical cycle of those elements is one of the objectives of such large mesocosm experiments. In the recent experiment reported by Guieu et al. 2010, large (52 m3) mesocosms containing no metallic part and with as little as possible induced perturbation during the sampling sequence have been used and the required conditions for biogeochemical studies in oligotrophic environments have been reproduced (Fig. 4.24). Such large clean mesocosms have been thus proven to be highly relevant tools to study in situ the response of an oligotrophic ecosystem to atmospheric deposition.


Stocks (in green) and fluxes (in blue) that can be measured in a mesocosm experiment after the simulation of a realistic atmospheric input in a water body large enough to be representative of natural processes. As the particles are naturally sinking, those changes are closer to the ‘real’ processes occurring in the open ocean, compared to microcosms approaches where the particles are homogenised and not allowed to sink. Sampling in quick succession at different depths of the mesocosms allows for parameterising the complex processes involved. Sedimentation, including carbon export, can be quantified and this represents a major step forward of that methodology


The different steps to reproduce a realistic atmospheric dust deposition and subsequently follow the biogeochemical changes induced near the surface of the oligotrophic ocean illustrated by experiment 2 (2010) of the DUNE project: (a) sampling soils in a region of South Tunisia where the Saharan aerosol is produced and is typical of inputs to the North-Western Mediterranean, (b) Soil collected on the three stages of the dry sieving column at the end of the fine particle production process. (c) Artificial ageing of the atmospheric particles in a clean room to reproduce the processes taking place during aerosols transport (d) a group of three mesocosms from below, (e) a view of the seven mesocosms during the DUNE2 campaign in June 2010 (f) a view above the surface of a group of three mesocosms showing the artificial seeding. (Photos a, b : F. Dulac, CEA; photo c: LISA; photos d, e, f : D. Luquet, OOV). Methodology fully described in Guieu et al. 2010
Mesocosms present the advantage of enabling studies of processes both as a function of time and depth while the atmospheric particles are sinking. According to what was pointed out in Sect. 4.5.3, this is a very important consideration, in particular regarding adsorption/desorption processes and aggregation mediated by the introduction of lithogenic particles (Bressac et al. 2012 and Sect. 4.5.5). Mesocosms are thus much more representative of the reality of the atmospheric deposition to the surface of the ocean and results obtained can be more easily extrapolated than results obtained in microcosms that rely on a fixed and homogeneously-distributed concentration of particles that can alter the dust dissolution kinetics and does not account for particle migration through the water column, precluding any estimation of C export induced by atmospheric deposition.
Those recent mesocosm experiments confirm that significant changes in the cycling of chemical elements are induced by the deposition of dust particles, and that there are strong responses of the ecosystem at different trophic levels. Pulido-Villena et al. (2010) provided a quantitative confirmation of the fertilising potential of mineral dust as a significant increase in phosphate concentration was observed soon after seeding. The dissolved inorganic phosphorus (DIP) released was completely lost after 24 h and no further increase was observed during the experiment. DIP loss might thus have been dominated by biological uptake, in agreement with the observed rapid response of both bacteria and phytoplankton. The iron cycle was also profoundly affected by the dust input to the surface seawater, as a dissolved iron (DFe) scavenging was observed rapidly after the seeding, withdrawing almost 1 nM DFe from the whole water column inside the three mesocosms amended with dust while the atmospheric particles are settling (Wagener et al. 2010). Those results were satisfactorily simulated with a one-dimensional model of the Fe biogeochemical cycle, coupled with a simple ecosystem model (Ye et al. 2011). When a second dust wet deposition was simulated, iron dissolution from the dust particles was then evidenced due to the excess Fe binding ligand concentrations produced by the enhanced biological activity (Wuttig et al. 2013). When simulating dust wet deposition at least a doubling of Chla (mainly attributed to small phytoplankton (<3 μm); Giovagnetti et al. 2013) was observed but the Chla concentrations remained very low (maximum values 0.22 μg L-1) maintaining the oligotrophic status of the tested waters (Guieu et al. 2013). Laghdass et al. (2011) investigated the effect of dust deposition on the diversity of the heterotrophic bacterial community. Combining several molecular biology approaches, the results indicate that besides a natural temporal trend observed inside and outside the mesocosms, dust deposition affected the particle-attached bacterial community. In particular, Alteromomas macleodii made a higher contribution to the active particle-attached bacterial community in the dust-amended than in the control mesocosms. Bacterial respiration and primary production both increased very rapidly after the seeding (by a factor of 2 and 3, respectively), showing a competition for the new nutrients between heterotrophic bacteria and phytoplankton (Guieu et al. 2012). Among phytoplankton, diazotrophs – although only responsible for few percent of the induced new production – were strongly stimulated by the atmospheric input (Ridame et al. 2013). Coupling geochemical and optical measurements have demonstrated that rapid particulate carbon export was in part due to aggregation processes between organic matter and lithogenic particles (Bressac et al. 2012). Results from mesocosm approaches indicate that the role of atmospheric deposition on oligotrophic areas cannot be seen solely as a simple fertilisation effect because (1) both autotrophs and heterotrophs are stimulated (confirming on longer time scales the findings from microcosm experiments described in Sect. 4.5.4.2) and (2) a significant export of particulate organic carbon to the deep ocean is attributed to aggregation processes (Bressac et al. 2013).
Although they represent a powerful tool to handle the complex question of the role of atmospheric deposition on ecosystem functioning, the use of such large, clean and pelagic mesocosm in the open ocean remains a difficult task for obvious logistical reasons. So far, the only two experiments conducted have been done in coastal areas chosen for being representative of waters with comparable chemical and biological characteristic to open waters. Future development could be the feasibility of identical experiments in the open ocean. Another disadvantage of the mesocosm approach is that only few treatments can be handled (during the DUNE (A DUst experiment in a low Nutrient, low chlorophyll Ecosystem) experiment, triplicates of controls and triplicates of dust deposition were the only treatments considered), contrary to microcosm approach where a lot of different treatments can easily be used.
4.5.4.4 Modelling
In the past two decades, ocean biogeochemical models (e.g. Maier-Reimer 1993) have been used to study the response of marine biogeochemistry to a variety of forcings. Many studies have focussed in particular on the response of ocean carbon storage (Sarmiento and Le Quéré 1996) and marine productivity (Bopp et al. 2001) to anthropogenic climate change. Among the various forcings employed, the impact of changes in atmospheric deposition on marine biota and marine biogeochemistry has received much less attention. One reason for this relative lack of attention is linked to the very large uncertainties in future changes of atmospheric nutrient deposition to the ocean due to climate change, as exemplified by simulated future changes in dust deposition (e.g. Mahowald and Luo 2003; Mahowald et al. 2006; Tegen et al. 2004).
Early studies have focussed on the role of changes in dust deposition on the marine carbon pump and atmospheric CO2 during glacial times. Watson et al. (2000) used a box model of the ocean carbon cycle forced with atmospheric iron fluxes derived from the Vostok ice-core dust record (Petit et al. 1999). They showed that changes in the Southern Ocean biota caused by iron deposition could explain only a maximum of 40 ppmv drawdown of atmospheric CO2 at the glacial maximum, half of the observed glacial-interglacial CO2 change. Other modelling studies, however, using general circulation models and more complex biogeochemistry (including a explicit representation of the ocean iron cycle) converge towards a more moderate effect; the impact of changes in dust deposition on marine biota at glacial maximum could drive a ~15 ppm reduction in atmospheric CO2 (e.g. Archer et al. 2000; Bopp et al. 2003).
Similar models have been used to quantify or predict the effect of changing dust deposition on marine biota and the global carbon cycle in the recent past and in the near future. Over the twentieth century, increase in dust deposition inferred from observations has been estimated to be responsible for an increase in marine productivity and an additional drawdown of carbon into the ocean (Mahowald et al. 2010). This additional drawdown could amount to 8 Pg C, more than 5 % of total anthropogenic carbon stored in the ocean over the industrial period.
While future projections of desert dust deposition over the ocean are still largely uncertain, even as regards the sign of changes (Tegen et al. 2004; Mahowald et al. 2009), several ocean modelling studies have addressed its potential impact on marine biogeochemistry. Changes in dust deposition may force changes in ocean productivity, as large as the changes in productivity forced by CO2 increases and the resulting climate change (Mahowald et al. 2011). But there is only relatively little impact of varying aeolian Fe input on cumulative ocean CO2 fluxes and atmospheric pCO2 over 2000–2100 (Tagliabue et al. 2008).
The simulated sensitivity of marine biota to iron deposition is, however, largely dependent on processes that are or not explicitly included in models. Complex iron chemistry in the ocean (Tagliabue et al. 2009), adsorptive scavenging of dissolved iron and solubilisation of particulate iron (Ye et al. 2011), varying iron content of dust particles as well as varying iron solubility in seawater (Luo et al. 2006) have been investigated in individual modelling studies but an explicit representation of these processes is still lacking in most of the biogeochemical models that do include an explicit representation of the iron cycle. In addition, Krishnamurthy et al. (2009) have shown that increasing iron deposition could not only lead to increasing marine productivity in HNLC regions, but also significantly enhance N-fixation in subtropical regions if iron limitation of N-fixation is included. Finally, iron mobilisation from sediments has been suggested to be of the same magnitude as dust deposition and its inclusion in models significantly reduces the response to changes in atmospheric iron deposition (Moore and Braucher 2008).
Apart from the effect of iron deposition, few studies have focussed on the role of atmospheric deposition of other elements on marine biota and ocean biogeochemical cycles. Using a coupled ocean general circulation/biogeochemical model, Krishnamurthy et al. (2010) have shown that atmospheric Si and P depositions have only a weak effect on marine productivity and biogeochemical cycles, as these depositions are small relative to the flux of these nutrients from below. They would contribute only to a very small fraction of export production (Fig. 4.25). Even if atmospheric nitrogen inputs have only a weak effect on marine biota and productivity at the global scale, they can contribute significantly (up to 25 %) to export production at the regional scale, especially in subtropical oligotrophic gyres (Krishnamurthy et al. 2009, 2010). The impacts of changes in N, P and Si deposition in the near future on marine biota are largely unknown.


Simulated ratios between atmospheric inputs of N, Fe, Si, and P and corresponding sinking particulate export for modern era climate conditions. Note the change in scale between upper and lower panels (Reproduced from Krishnamurthy et al. (2010) by permission of the American Geophysical Union)
In addition to the direct effect of nutrient deposition on the marine biota through a fertilisation effect, Doney et al. (2007) have shown that atmospheric inputs of reactive sulphur and nitrogen could potentially alter seawater alkalinity and pH. They show that inputs of strong acids such as HNO3 and H2SO4 could lead to a decrease in pH. The alterations in surface water chemistry however are only a few percent of the acidification due to the oceanic uptake of anthropogenic CO2. The impacts could be more substantial in coastal waters.
4.5.5 Particulate Matter and Carbon Export
Depending on the source of the aerosols, atmospheric deposition to the ocean is of very variable intensity, the most spectacular atmospheric fluxes being associated with lithogenic particles from the different deserts of the continents. Indeed, strong dust events can bring large amounts of particulate matter to the ocean surface in a few hours and fluxes as high as 25 g m−2 per event have been reported in the Mediterranean Sea (Ternon et al. 2010), see Fig. 4.26. This lithogenic material is transferred to the deep ocean and contributes significantly to sedimentation in areas such as the tropical North Pacific, the North Atlantic and the Mediterranean Sea, which are well impacted by dust deposition (Uematsu et al. 1985; Prospero et al. 2002; Loÿe-Pilot et al. 1986). But not only lithogenic export is associated with those important atmospheric depositions.


Saharan dust event in February 2004 and its consequences for marine lithogenic material and particulate organic carbon exports in the water column. (a) Satellite image showing the transport of Saharan dust across the Mediterranean Sea (SeaWiFS, NASA). (b) Total atmospheric deposition mass flux measured at the Cap Ferrat sampling site (French Riviera) along with fluxes from sediment traps at 200 m at the nearby DYFAMED time series station (43°25′N 07°52′E; http://www.obs-vlfr.fr/sodyf/) showing the huge increase in both lithogenic and POC flux following the event (Adapted from Ternon et al. 2010)
As shown in previous sections, aerosols from different sources do bring to the ocean a significant amount of elements of biogeochemical interest that feed the pool of nutrients and micronutrients in the ocean surface layer. When the body of water impacted by atmospheric deposition is depleted in those (micro)nutrient(s) brought by atmospheric deposition, (micro)nutrient(s) limited organisms such as bacteria and phytoplankton will feed on this new resource. In such conditions, atmospheric deposition has a ‘fertilising’ effect (see references in Sect. 4.5.4) and carbon fixation by autotrophic organisms should be followed by particulate organic carbon fluxes to the deep ocean. Although the link between addition of aerosol iron and export production was emphasised by Cassar et al. 2007, the quantification of such induced export of particulate carbon was difficult to assess in the numerous iron fertilisation experiments reported in de Baar et al. (2005) where it was only firmly proven in two of the reported experiments for which it was shown to be quite modest. Differently, Smetacek et al. (2012) concluded that following the EIFEX ocean iron fertilisation experiment a substantial portion of the induced bloom likely reached the sea floor. According to Blain et al. (2007) carbon export from such artificially induced short-term blooms are at least an order of magnitude lower than carbon export induced by natural iron fertilisation in the Southern Ocean.
On the other hand, several studies have shown the importance of lithogenic particles for the export of organic matter through an aggregation process (Hamm 2002; Passow and De la Rocha 2006; Ploug et al. 2008) and the export in this case is not related to a fertilisation effect. During 4 years of simultaneous measurements of atmospheric deposition and export of material in the water column in the NW Mediterranean Sea, Ternon et al. 2010 found a series of “lithogenic events” corresponding to both high POC and high lithogenic marine fluxes (originating mainly from either recent Saharan fallout events, or from ‘old’ Saharan dust ‘stored’ in the upper water column layer). Such “lithogenic events”, are believed to result in part from organic-dust aggregation inducing a ballast effect. The most remarkable event was in February 2004 when an extreme Saharan event exported ~45 % of the total annual POC (Fig. 4.26), compared to an average of ~25 % for the bloom period, at a time of the year when the organisms are not limited by the concentrations of nutrient in the upper ocean and thus such high POC flux couldn’t be the consequence of a fertilisation by the dust. Zooplankton activity and the incorporation of mineral particles into faecal pellets have also been proposed as a mechanism to explain such lithogenic events (e.g. Fowler et al. 1987; Buat-Menard et al. 1989). Ternon et al. also found another ‘lithogenic event’, in summer when the surface mixed layer is depleted in nutrients, where only 20 % of the POC exported could be explained by fixation of carbon induced by atmospheric fertilisation, the rest being assumed to be due to the incorporation of mineral particles acting as ballast in organic aggregates formed by the dissolved and colloidal organic matter present in the water column prior to the event.
From those few examples, we can stipulate that dust deposition can have two main effects: a POC export directly linked to the carbon fixation induced by the fertilisation via atmospheric nutrients and the POC export ‘mediated’ by the introduction of lithogenic particles with a ballast effect on organic matter, leading to the formation of large aggregates. As mentioned above, both effects can be combined and this was recently observed by Bressac et al. (2012) following an artificial dust addition in a large clean mesocosm: it was observed, with optical measurements, that particulate export following a strong dust deposition (10 g m−2) was a nonlinear multi-step process composed of particle populations with different size distributions and composition as a function of time and depth. Interestingly, it was shown that the pattern of the size distribution and composition of particles in the mesocosm was strongly influenced by the formation of organic-mineral aggregates and that the fresh organic matter produced by the fertilisation induced by the dust deposition lead to organic-mineral aggregates with the fastest settling velocities compared to the other particles populations. Bressac et al. measured organic-mineral aggregate populations (>61.2 μm) having settling velocities of the order of 24~86 m d–1. Such rapid transfer following a dust event with settling velocity of at least 100 m d–1 has been observed for ‘lithogenic events’ from sediment trap data (Ternon et al. 2010). Such fast sinking particles would be less prone to remineralisation while transferring to the deeper layers (Hedges et al. 2000) and this may have important consequences regarding carbon sequestration. Those different recent studies have shown that lithogenic particles do not sink following Stokesian settling calculations and that atmospheric particles may have a very important role in transferring rapidly particulate organic matter to the deep ocean but, so far, no quantification of this role in carbon export and budget has been performed.
The parameterisation of the complex processes involved in the sinking of both lithogenic and organic particles following an intense dust event will only be possible by realising a combination of optical, biological and chemical measurements in the upper water column when dust deposition occurs. This could be done by a combination of in situ measurements (including in situ mesocosms) and in large tanks in the laboratory (both in biotic and abiotic conditions) with approaches allowing the actual sinking of the particles. The characterisation of organic matter is very much needed in such combined approaches as organic-mineral aggregation depends mainly on the quantity and quality of the organic matter (Passow and De La Rocha 2006) meaning that the biogeochemical conditions of the water body in which the deposition occurs may be a crucial factor for the fate of particulate matter following a dust event.
4.6 Summary and Outlook
Aerosol particles in the marine atmosphere are produced either directly at the sea surface, as sea spray aerosol particles, or over land from where they can be subsequently transported over the ocean. Secondary production takes place in the atmosphere from precursor gases which are emitted either from the sea surface or from a variety of sources over land. The direct production of sea spray aerosol was recently reviewed by de Leeuw et al. (2011a) and conclusions presented there still hold. Results in the last decade show the large contribution of organic substances to SSA particles especially in locations of high biological activity, which dominates the chemical composition for particles with r80 < 0.25 μm. The consequences for hygroscopic properties of SSA particles and thus their effect on climate via light scattering and their influence on cloud properties has been recognised and is still an important topic for research. Meskhidze and co-workers (Gantt et al. 2011; Meskhidze et al. 2011) have shown the effect of wind speed on the fraction of organic matter in SSA particles in addition to the dependence on particle size (Facchini et al. 2008a). Ovadnevaite et al. (2011) show the importance of organic matter for the ability of SSA particles to act as CCN.
Production fluxes have been determined for particles with r80 as small as 0.01 μm, which is also important for the assessment of the role of SSA particles on climate using large-scale models. However, de Leeuw et al. (2011a) concluded that the uncertainties in the production fluxes are still very large. For particles with r80 > 1 μm the uncertainty is a multiplicative factor of 4 (Lewis and Schwartz 2004). Recent research by Jaeglé et al. (2011) confirms the large importance of the effect of SST (Mårtensson et al. 2003) on the production fluxes of coarse particles (those with r80 > 1 μm), with consequences for the effect of SSA particles on the direct radiative effect on climate.
For particles with 0.3 < r80 < 1 μm and smaller all flux estimates published after Lewis and Schwartz (2004) appear to be higher than the range of values discussed by these authors. These high production fluxes would result in unrealistically large number concentrations of SSA particles in the marine atmospheric boundary layer (de Leeuw et al. 2011a) but the reason for these high production fluxes is not clear. There is no convincing reason to discard the Lewis and Schartz (2004) production flux estimates and their uncertainties, which are based on numerous observations, but the more recent formulations not only produce larger production fluxes but they also seem to converge to values which differ by about a factor of 2–3 (Clarke et al. 2006).
For SSA particles with r80 < 0.3 μm the production flux estimates rely on the whitecap method, i.e. estimates of the whitecap fraction and on the particle production flux per unit area of whitecap. The latter is mainly obtained from laboratory experiments and comparisons by de Leeuw et al. (2011a) show the large difference in the shape and magnitude of the SSA particle size spectra produced in such experiments, with a large dependence on experimental conditions (e.g. Fuentes et al. 2010). This puts into question the validity of one of the underlying assumptions of the whitecap method, i.e. that the production per unit whitecap area can be independently determined and that the total production flux is given by the product of the production per unit whitecap area and the whitecap fraction. The whitecap fraction in turn varies over a very wide range between different observations (Lewis and Schwartz 2004; Anguelova and Webster 2006) and all the most recent determinations, using novel methods, appear to result in smaller whitecap fractions than the most commonly used function by Monahan and O’Muirchaertaigh (1986) which is based on numerous previous observations. Furthermore the whitecap fraction appears not to be unambiguously determined by wind speed and depends on wind speed history (Callaghan et al. 2008).
Most of these studies use a combination of different techniques and novel technology developments. These techniques include combinations of advanced modelling using different information sources based on results from laboratory and field experiments, long term observations or intensive field campaigns at coastal locations or open ocean, using advanced techniques such as AMS, HTDMA, CCNC and ship-borne eddy covariance measurements. Many of the recent studies rely on the use of earth observation (EO), i.e. satellite-based instruments, to obtain information on aerosol concentrations, forcing parameters and aerosol effects. EO is increasingly used in studies on sea spray aerosols and their effects. Nevertheless, remote sensing results also show rather large ambiguities such as in the determination of the aerosol optical depth (AOD) as determined by different remote sensing methods (Smirnov et al. 2012). The use of EO in air-sea interaction studies and effects on SSA production and their effects on climate and biogeochemical cycles has an important place in the SOLAS community.
In spite of recent progress based on combinations of modelling, EO and in situ observations, a large drawback put forward in the literature is the lack of reliable experimental data for a wide variety of conditions. Another question posed in the literature concerns the reliability and accuracy of EO products which are further developed based on new insights and improved EO instrumentation leading to improvement of data quality and development of new products. However, satellite data alone cannot be used to assess SSA production and needs to be used in combination with other techniques such as modelling and in situ or ground-based remote sensing.
The contribution of land-based aerosol particles and precursor gases is another issue which is extremely important for the assessment of atmospheric inputs into the ocean and their biogeochemical effects (see below). These studies rely to a large extent on models to determine aerosol transport. The models in turn utilise emission data bases as input and they are often based on bottom-up emission inventories for a certain period or year and therefore represent mean emissions. Often the inventories lack detail on seasonal or diurnal variations and do not include actual episodic emission strenghts of species which are important for biogeochemical cycles such as dust, volcanic ash and biomass burning aerosol. Earth Observation (satellites) may contribute to improvement of emission estimates and several studies have recently been published on the use of EO data with advanced modelling techniques to provide this information for emissions of e.g. volcanic ash, VOCs and NO2.
Numerous studies have been conducted to establish links between atmospheric deposition and biogeochemical cycles in the past 20 years, increasing exponentially with recent developments of new sophisticated methodologies (in particular the use of clean sampling techniques in the field and the generalisation of expertise in low levels determination of chemicals in seawater).
Studies on effects of atmospheric iron on biogeochemical processes have been an important research activity for more than 20 years because of, its recognised importance as a limiting nutrient in the large HNLC ocean. These studies no longer focus only on iron in dust but also the biogeochemical impact of other recently recognised important sources – such as volcanic ash – are now considered. Other studies have recently focussed on LNLC areas where microbes can be limited or co-limited by deposition of atmospheric inorganic nitrogen and phosphorus. In terms of biotic response to episodic atmospheric nutrient inputs, the large scale experiments conducted over the past 20 years have mainly focussed on the role of iron in HNLC areas. Although responses differ due to regional differences between the environmental characteristics of HNLC waters, such experiments have allowed simultaneous observation of the wide range of processes involved by such inputs of new nutrients, how these change the upper ocean properties and how the ocean feedbacks on the lower atmosphere. Microcosm experiments show the response of micro-organisms to deposition of aerosol particles to LNLC areas. Such experiments also show consistent patterns in terms of biomass and abundance, community structure, and metabolic activity revealing that both autotrophs (including diazotrophs) and heterotrophs can be significantly stimulated. This is confirmed by mesocosm experiments where both chemical and biological processes can be studied simultaneously over longer time scales while accounting for the vertical dimension as well. In situ observations and mesocosm approaches indicate that atmospheric deposition, in particular dust, can result in carbon export through two types of processes: export directly linked to carbon fixation induced by the fertilization via atmospheric nutrients and export by carbon ballasting by the lithogenic particles. Ocean biogeochemical models to study the response of marine biota and biogeochemistry to atmospheric deposition have mainly focussed so far on dust (iron) deposition over glacial times, recent past and near future, showing moderate effects on reduction of atmospheric CO2. The few modelling studies which recently focussed on the role of atmospheric deposition of other elements than iron have shown that atmospheric N, P and Si deposition have a weak effect on marine productivity and biogeochemical cycles contributing only to a very small fraction of export production.
All those different approaches indicate that extrapolation of the results to the ‘real world’ is not easy and that further studies using a combination of the different approaches are necessary to quantify the present picture and how it will change in the future. A number of research directions have been recommended, such as specific studies to understand the role of the microlayer, to establish the partitioning between organic and inorganic forms of atmospheric nutrients and their link with bioavailability, to consider the balance between scavenging/dissolution taking into account the dynamics of the particles while they settle through the water column (Law et al. 2013). Studies considering a wide range of in situ conditions should be conducted in order to understand the competition for the new nutrient resources among planktonic organisms and how the balance between CO2 fixation and respiration impact the carbon budget. The balance between POC export linked to carbon fixation induced by the atmospheric nutrient fertilisation, and the POC export mediated by the introduction of atmospheric particles through aggregation processes, have to be better constrained. Another challenge is to correctly model the complex biogeochemical processes involved in order to quantify the impacts on biota and global carbon cycle and to predict the changes induced by the evolution of emission/deposition in the future.
References
Adams P, Seinfeld J, Koch D (1999) Global concentrations of tropospheric sulfate, nitrate and ammonium aerosol simulated in a general circulation model. J Geophys Res 104:13791–13823
Adler G, Flores JM, Riziq AA, Borrmann S, Rudich Y (2011) Chemical, physical, and optical evolution of biomass burning aerosols: a case study. Atmos Chem Phys 11:1491–1503. doi:10.5194/acp-11-1491-2011
Aggarwal SG, Kawamura K (2008) Molecular distributions and stable carbon isotopic compositions of dicarboxylic acids and related compounds in aerosols from Sapporo, Japan: implications for photochemical aging during long-range atmospheric transport. J Geophys Res 113, D14301. doi:10.1029/2007JD009365
Aiken AC, DeCarlo PF, Kroll JH et al (2008) O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry. Environ Sci Technol. doi:10.1021/es703009q
Albert MFMA, Schaap M, Scannell C, O’Dowd CD, de Leeuw G (2012) Uncertainties in the determination of the organic fraction of global sub-micron sea-spray emissions. Atmos Environ 57:289–300. doi:10.1016/j.atmosenv.2012.04.009
Alfaro SC, Gaudichet A, Gomes L, Maillé M (1997) Modeling the size distribution of a soil aerosol produced by sandblasting. J Geophys Res 102:11239–11249
Alfaro SC, Gaudichet A, Gomes L, Maillé M (1998) Mineral aerosol production by wind erosion: aerosol particle sizes and binding energies. Geophys Res Lett 25:991–994
Alfaro SC, Lafon S, Rajot JL, Formenti P, Gaudichet A, Maillé M (2004) Iron oxides and light absorption by pure desert dust: an experimental study. J Geophys Res 109, D08208. doi:10.1029/2003JD004374
Alpert P, Kaufman YJ, Shay-El Y, Tanré D, da Silva A, Schubert S, Joseph JH (1998) Quantification of dust-forced heating of the lower troposphere. Nature 395:367–370
Alvarado MJ, Prinn RG (2009) Formation of ozone and growth of aerosols in young smoke plumes from biomass burning: 1. Lagrangian parcel studies. J Geophys Res 114, D09306
Anderson TL, Wu Y, Chu DA, Schmid B, Redemann J, Dubovik O (2005) Testing the MODIS satellite retrieval of aerosol fine-mode fraction. J Geophys Res 110, doi:10.1029/2005JD005978
Andreae MO, Merlet P (2001) Emission of trace gases and aerosols from biomass burning. Global Biogeochem Cycles 15:955–966
Andreae MO, Rosenfeld D (2008) Aerosol-cloud-precipitation interactions. Part 1. The nature and sources of cloud-active aerosols. Earth-Sci Rev 89:13–41
Andreae MO, Charlson RJ, Bruynseels F, Storms H, Van Grieken R, Maenhaut W (1986) Internal mixture of sea salt, silicates, and excess sulfate in marine aerosols. Science 32:1620–1623
Andreas EL, Persson POG, Hare JE (2008) A bulk turbulent air-sea flux algorithm for high-wind, spray conditions. J Phys Oceanogr 38:1581–1596
Andres RJ, Kasgnoc AD (1998) A time-averaged inventory of subaerial volcanic sulphur emissions. J Geophys Res 103:25251–25261
Angelino S, Suess DT, Prather KA (2001) Formation of aerosol particles from reactions of secondary and tertiary alkylamines: characterization by aerosol time-of-flight mass spectrometry. Environ Sci Technol 35(15):3130–3138
Anguelova MD, Webster F (2006) Whitecap coverage from satellite measurements: a first step toward modeling the variability of oceanic whitecaps. J Geophys Res Oceans 111, C03017. doi:10.1029/2005JC003158
Archer D, Winguth A, Lea D, Mahowald N (2000) What caused the glacial/interglacial atmospheric pCO2 cycles? Rev Geophys 38:159–189
Arimoto R, Duce RA, Ray BJ, Unni CK (1985) Atmospheric trace element at Enewetak Atoll: 2. Transport to the ocean by wet and dry deposition. J Geophys Res 90:2391–2408
Arimoto R, Ray BJ, Lewis NF, Tomza U, Duce RA (1997) Mass particle size distribution of atmospheric dust and the dry deposition of dust to the remote ocean. J Geophys Res 102:15867–15874
Arneth A, Niinemets U, Pressley S, Back J, Hari P, Karl T, Noe S, Prentice IC, Serca D, Hickler T, Wolf A, Smith B (2007) Process-based estimates of terrestrial ecosystem isoprene emissions: incorporating the effects of a direct CO2-isoprene interaction. Atmos Chem Phys 7:31–53
Arneth A, Monson RK, Schurgers G, Niinemets U, Palmer PI (2008) Why are estimates of global isoprene emissions so similar (and why is this not so for monoterpenes)? Atmos Chem Phys 8:4605–4620
Arneth A, Sitch S, Bondeau A, Butterbach-Bahl K, Foster P, Gedney N, de Noblet-Ducoudre N, Prentice IC, Sanderson M, Thonicke K, Wania R, Zaehle S (2010) From biota to chemistry and climate: towards a comprehensive description of trace gas exchange between the biosphere and atmosphere. Biogeosciences 7:121–149
Arneth A, Schurgers G, Lathiere J, Duhl T, Beerling DJ, Hewitt CN, Martin M, Guenther A (2011) Global terrestrial isoprene emission models: sensitivity to variability in climate and vegetation. Atmos Chem Phys 11:8037–8052. doi:10.5194/acp-11-8037-2011, 2011
Atkinson R (2000) Atmospheric chemistry of VOCs and NOx. Atmos Environ 34:2063–2101
Atkinson R, Arey J (2003a) Atmospheric degradation of volatile organic compounds. Chem Rev 103:4605–4638
Atkinson R, Arey J (2003b) Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review. Atmos Environ 37(2):197–219
Avila A, Rodà F (2002) Assessing decadal changes in rainwater alkalinity at a rural Mediterranean site in the Montseny mountains (NE Spain). Atmos Environ 36:2881–2890
Avila A, Alarcón M, Queralt I (1998) The chemical composition of dust transported in red rains – its contribution to the biogeochemical cycle of a holm aok forest in Catalonia (Spain). Atmos Environ 32:179–191
Aymoz G, Jaffrezo JL, Jacob V, Colomb A, George C (2004) Evolution of organic and inorganic components of aerosol during a Saharan dust episode observed in the French Alps. Atmos Chem Phys 4:2499–2512
Baker AR, Croot PL (2010) Atmospheric and marine controls on aerosol iron solubility in seawater. Mar Chem 120:4–13
Baker A, Jickells T (2006) Mineral particle size as a control on aerosol iron solubility. Geophys Res Lett 33, doi:10.1029/2006GL026557
Baker AR, Jickells TD, Witt M, Linge KL (2006a) Trends in the solubility of iron, aluminium, manganese and phosphorus in aerosol collected over the Atlantic Ocean. Mar Chem 98:43–58
Baker A, French M, Linge K (2006b) Trends in aerosol nutrient solubility along a west–east transect of the Saharan dust plume. Geophys Res Lett 33, doi:10.1029/2005GL024764
Baker AR, Lesworth T, Adams C, Jickells TD, Ganzeveld L (2010) Estimation of atmospheric nutrient inputs to the Atlantic Ocean from 50°N to 50°S based on large-scale field sampling: fixed nitrogen and dry deposition of phosphorus. Global Biogeochem Cycles 24, GB3006. doi:10.1029/2009GB003634
Bardintzeff J-M, McBirney AR (2000) Volcanology. Second edition, Jones, Bartlett. Andres RJ, Kasgnoc AD (1998) A time-avaraged inventory of subaerial volcanic sulphur emissions. J Geophys Res 103:25252–25261
Barkley MP, Palmer PI, Kuhn U, Kesselmeier J, Chance K, Kurosu TP, Martin RV, Helmig D, Guenther A (2008) Net ecosystem fluxes of isoprene over tropical South America inferred from GOME observations of HCHO columns. J Geophys Res 113(D20), D20304. doi:10.1029/2008jd009863
Barkley MP, Palmer PI, Ganzeveld L, Arneth A, Hågberg D, Karl T, Guenther A, Paulot F, Wennberg PO, Mao J, Kurosu TP, Chance K, Muller J-F, De Smedt I, Van Roozendael M, Chen D, Wang Y, Yantosca RM (2011) Can a ‘state of the art’ chemistry transport model simulate Amazonian tropospehric chemistry? J Geophys Res 116, D16302. doi:10.1029/2011JD015893
Barkley MP, Kurosu TP, Chance K, De Smedt I, Van Roozendael M, Arneth A, Hagberg D, Guenther A (2012) Assessing sources of uncertainty in formaldehyde air mass factors over tropical South America: implications for top-down isoprene emission estimates. J Geophys Res 117, D13304. doi:10.1029/2011JD016827
Bellouin N, Jones A, Haywood J, Christopher S (2008) Updated estimate of aerosol direct radiative forcing from satellite observations and comparison against the Hadley Centre climate model. J Geophys Res 113, doi:10.1029/2007JD009385
Bellouin N, Rae JGL, Jones A, Johnson CE, Haywood JM, Boucher O (2011) Aerosol forcing in the Climate Model Intercomparison Project (CMIP5) simulations by HadGEM2-ES and the role of ammonium nitrate. J Geophys Res 116, D20206. doi:10.1029/2011JD016074
Bergametti G, Gomes L, Remoudaki E, Desbois M, Martin D, Buat-Ménard P (1989) Present transport and deposition patterns of African dusts to the north-western Mediterranean. In: Leinen M, Sarnthein M (eds) Paleoclimatology and paleometeorology: modern and past patterns of global atmospheric transport. Kluwer, Boston, pp 227–251
Bergametti G, Remoudaki E, Losno R, Steiner E, Chatenet B, Buat-Ménard P (1992) Source, transport and deposition of atmospheric phosphorus over the northwestern Mediterranean. J Atmos Chem 14:501–513
Berresheim H, Elste T, Tremmel HG, Allen AG, Hansson H-C, Rosman K, Dal Maso M, Mäkelä JM, Kulmala M, O’Dowd CD (2002) Gas-aerosol relationships of H2SO4, MSA, and OH: observations in the coastal marine boundary layer at Mace Head, Ireland. J Geophys Res 107(D19):8100. doi:10.1029/2000JD000229
Betzer PR, Carder KL, Duce RA, Merrill JT, Tindale NW, Uematsu M, Costello DK, Young RW, Feely RA, Breland JA, Bernstein RE, Greco AM (1988) Long-range transport of giant mineral aerosol particles. Nature 336:568–571
Blain S, Guieu C, Claustre H, Leblanc K, Moutin T, Quéguiner B, Ras J, Sarthou G (2004) Availability of iron and major nutrients for phytoplankton in the northeast Atlantic Ocean. Limnol Oceanogr 49:2095–2104
Blain S, Queguiner B, Armand L, Belviso S, Bombled B, Bopp L, Bowie A, Brunet C, Brussaard C, Carlotti F, Christaki U, Corbiere A, Durand I, Ebersbach F, Fuda J-L, Garcia N, Gerringa L, Griffiths B, Guigue C, Guillerm C, Jacquet S, Jeandel C, Laan P, Lefevre D, Lo Monaco C, Malits A, Mosseri J, Obernosterer I, Park Y-H, Picheral M, Pondaven P, Remenyi T, Sandroni V, Sarthou G, Savoye N, Scouarnec L, Souhaut M, Thuiller D, Timmermans K, Trull T, Uitz J, van Beek P, Veldhuis M, Vincent D, Viollier E, Vong L, Wagener T (2007) Effect of natural iron fertilization on carbon sequestration in the Southern Ocean. Nature 446:1070–1075
Blanchard DC (1963) The electrification of the atmosphere by particles from bubbles in the sea. Prog Oceanogr 1:73–112
Blanchard DC (1964) Sea to air transport of surface active material. Science 146:396–397
Bond TC, Streets DG, Yarber KF, Nelson SM, Woo J-H, Klimont Z (2004) A technology-based global inventory of black and organic carbon emissions from combustion. J Geophys Res 109, D14203. doi:10.1029/2003JD003697
Bonn B, Moortgat GK (2003) Sesquiterpene ozonolysis: origin of atmospheric new particle formation from biogenic hydrocarbons. Geophys Res Lett 30(11):1585. doi:10.1029/2003GL017000
Bonnet S, Guieu C (2004) Dissolution of atmospheric iron in seawater. Geophys Res Lett 31, doi:10.1029/2003GL018423
Bonnet S, Guieu C, Chiaverini J, Ras J, Stock A (2005) Effect of atmospheric nutrients on the autotrophic communities in a low nutrient, low chlorophyll system. Limnol Oceanogr 50:1810–1819
Bopp L, Monfray P, Aumont O, Dufresne J-L, Le Treut H, Madec G, Terray L, Orr JC (2001) Potential impact of climate change on marine export production. Global Biogeochem Cycles 15:81–99
Bopp L, Kohfeld KE, Le Quéré C, Aumont O (2003) Dust impact on marine biota and atmospheric CO2 during glacial periods. Paleoceanography 18:1046. doi:10.1029/2002PA000810
Bory AJ-M, Biscaye PE, Grousset FE (2003) Two distinct seasonal Asian source regions for mineral dust deposited in Greenland (North GRIP). Geophys Res Lett 30:1167. doi:10.1029/2002GL016446
Boyd PW, Watson AJ, Law CS, Abraham ER, Trull T, Murdoch R, Bakker DCE, Bowie AR, Buessler KO, Chang H, Charette MA, Croot P, Downing K, Frew RD, Gall M, Hadfield M, Hall JA, Harvey M, Jameson G, La Roche J, Liddicoat MI, Ling R, Maldonado M, McKay RM, Nodder SD, Pickmere S, Pridmore R, Rintoul S, Safi K, Sutton P, Strzepek R, Tanneberger K, Turner SM, Waite A, Zeldis J (2000) A mesoscale phytoplankton bloom in the polar Southern Ocean stimulated by iron fertilization. Nature 407:695–702
Boyd PW, Law CS, Wong CS, Nojiri Y, Tsuda A, Levasseur M, Takeda S, Rivkin R, Harrison PJ, Strzepek R, Gower J, McKay RM, Abraham E, Arychuk M, Barwell-Clarke J, Crawford W, Hale M, Harada K, Johnson K, Kiyosawa H, Kudo I, Marchetti A, Miller W, Needoba J, Nishioka J, Ogawa H, Page J, Robert M, Saito H, Sastri A, Sherry N, Soutar T, Sutherland N, Taira Y, Whitney F, Wong SE, Yoshimura T (2004) The decline and fate of an iron-induced subarctic phytoplankton bloom. Nature 428:549–553
Boyd PW, Jickells T, Law CS, Blain S et al (2007) A synthesis of mesoscale iron-enrichment experiments 1993–2005: key findings and implications for ocean biogeochemistry. Science 315:612–617
Boyd PW, Mackie DS, Hunter KA (2010) Aerosol iron deposition to the surface ocean — modes of iron supply and biological responses. Mar Chem 120:130–145. doi:10.1016/j.marchem.2009.01.008
Bressac M, Guieu C, Doxaran D, Bourrin F, Obolensky G, Grisoni J-M (2012) A mesocosm experiment coupled with optical measurements to assess the fate and sinking of atmospheric particles in clear oligotrophic waters. Geo-Mar Lett 32(2):153–164
Bressac M, Guieu C, Doxaran D, Bourrin F, Desboeufs K, Leblond N, Ridame C (2013) Quantification of the lithogenic carbon pump following a dust deposition event. Biogeoscience 10, 13639–13677. doi:10.5194/bgd-10-13639-2013, 2013
Buat-Ménard P, Chesselet R (1979) Variable influence of the atmospheric flux on the trace metal chemistry of oceanic suspended matter. Earth Planet Sci Lett 42:399–411
Buat-Ménard P, Davies PJ, Remoudaki E, Miquel J-C, Bergametti G, Lamber CE, Ezat E, Quétel CR, La Rosa J, Fowler SW (1989) Non-steady-state biological removal of atmospheric particles from Mediterranean surface waters. Nature 340:131–133
Buck KN, Bruland KW (2005) Copper speciation in San Francisco Bay: a novel approach using multiple analytical windows. Mar Chem 96:185–198
Buck CS, Landing WM, Resing JA, Measures CI (2010) The solubility and deposition of aerosol Fe and other trace elements in the North Atlantic Ocean: observations from the A16N CLIVAR/CO2 repeat hydrography section. Mar Chem 120(1–4):57–70
Buesseler KO, Bacon MP, Cochran JK, Livingston HD (1992) Carbon and nitrogen export during the JGOFS North Atlantic bloom experiment estimated from 234Th:238U disequilibria. Deep-Sea Res 39(7/8):1115–1137
Buhaug Ø, Corbett JJ, Endresen Ø, Eyring V, Faber J, Hanayama S, Lee DS, Lindstad H, Mjelde A, Palsson C, Wanquing W, Winebrake JJ, Yoshida K (2008) Updated study on greenhouse gas emissions from ships: phase I report. International Maritime Organization (IMO), London, 1 Sept 2008, p 129
Burkholder JB, Curtius J, Ravishankara AR et al (2004) Laboratory studies of the homogeneous nucleation of iodine oxides. Atmos Chem Phys 4:19–34
Callaghan A, de Leeuw G, Cohen L, O’Dowd CD (2008) Relationship of oceanic whitecap coverage to wind speed and wind history. Geophys Res Lett 35, L23609. doi:10.1029/2008GL036165
Calvo-Díaz A, Díaz-Pérez L, Suárez LA, Morán XAG, Teira E, Marañón E (2011) Decrease in the autotrophic-to-heterotrophic biomass ratio of picoplankton in oligotrophic marine waters due to bottle enclosure. Appl Environ Microbiol 77:5739–5746
Capes G, Johnson B, McFiggans G, Williams PI, Haywood J, Coe H (2008) Aging of biomass burning aerosols over West Africa Aircraft measurements of chemical composition microphysical properties and emission ratios. J Geophys Res 113, D00C15. doi:10.1029/2008JD009845
Carlson TN, Prospero JM (1972) The large-scale movement of Saharan air outbreaks over the northern Equatorial Atlantic. J Appl Meteorol 11:283–297
Carslaw KS, Boucher O, Spracklen DV, Mann GW, Rae JGL, Woodward S, Kulmala M (2010) A review of natural aerosol interactions and feedbacks within the Earth system. Atmos Chem Phys 10:1701–1737
Cassar N, Bender ML, Barnett BA, Fan S, Moxim WJ, Levy H II, Tilbrook B (2007) The southern ocean biological response to Aeolian iron deposition. Science 317:1067–1070
Cavalli F, Facchini MC, Decesari S, Mircea M, Emblico L, Fuzzi S, Ceburnis D, Yoon YJ, O’Dowd CD, Putaud J-P, Dell’Acqua A (2004) Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic. J Geophys Res 109, D24215. doi:10.1029/2004JD005137
Ceburnis D, O’Dowd CD, Jennings SG, Facchini MC, Emblico L, Decesari S, Fuzzi S, Sakalys J (2008) Marine aerosol chemistry gradients: elucidating primary and secondary processes and fluxes. Geophys Res Lett 35, L07804. doi:10.1029/2008GL033462
Chance K, Palmer PI, Spurr RJD, Martin RV, Kurosu TP, Jacob DJ (2000) Satellite observations of formaldehyde over North America from GOME. Geophys Res Lett 27:3461–3464
Charlson RJ, Lovelock JE, Andreae MO, Warren SG (1987) Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate. Nature 326:655–661
Chatenet B, Marticorena B, Gomes L, Bergametti G (1996) Assessing the microped size distribution of desert soils erodible by wind. Sedimentology 43:901–911
Chazette P, Pelon J, Moulin C, Dulac F, Carrasco I, Guelle W, Bousquet P, Flamant P-H (2001) Lidar and satellite retrieval of dust aerosols over the Azores during SOFIA/ASTEX. Atmos Environ 35:4297–4304
Chen Y, Siefert R (2004) Sesaonal and spatial distributions and dry deposition fluxes of atmospheric total and labile iron over the tropical and subtropical North Atlantic Ocean. J Geophys Res 109, D09305. doi:09310.01029/02003JD003958
Chen Y, Street J, Paytan A (2006) Comparison between pure-water- and seawater-soluble nutrient concentrations of aerosols from the Gulf of Aqaba. Mar Chem 101:141–152
Chen Y, Mills S, Street J, Golan D, Post A, Jacobson M, Paytan A (2007) Estimates of atmospheric dry deposition and associated input of nutrients to Gulf of Aqaba seawater. J Geophys Res 112, D04309. doi:10.1029/2006JD007858
Chou C, Formenti P, Maille M, Ausset P, Helas G, Harrison M, Osborne S (2008) Size distribution, shape, and composition of mineral dust aerosols collected during the African monsoon multidisciplinary analysis special observation period 0: dust and biomass-burning experiment field campaign in Niger, January 2006. J Geophys Res 113, D00C10. doi:10.1029/2008JD009897
Chuang P, Duvall R, Shafer M, Schauer J (2005) The origin of water soluble particulate iron in the Asian atmospheric outflow. Geophys Res Lett 32, doi:10.1029/2004GL021946
Claeys M, Graham B, Vas G, Wang W, Vermeylen R, Pashynska V, Cafmeyer J, Guyon P, Andreae MO, Artaxo P, Maenhaut W (2004) Formation of secondary organic aerosols through photooxidation of isoprene. Science 303:1173–1176
Claeys M, Wang W, Vermeylen R, Kourtchev I, Chi X, Farhat Y, Surratt JD, Gómez-González Y, Sciare J, Maenhaut W (2010) Chemical characterisation of marine aerosol at Amsterdam Island during the austral summer of 2006–2007. J Aerosol Sci 41:13–22
Clarke AD, Davis D, Kapustin VN, Eisele F, Chen G, Paluch I, Lenschow D, Bandy AR, Thornton D, Moore K, Mauldin L, Tanner D, Litchy M, Carroll MA, Collings J, Albercook G (1998) Particle nucleation in the tropical boundary layer and its coupling to marine sulfur sources. Sciences 282:89–91
Clarke AD, Qwens SR, Zhou J (2006) An ultrafine sea-salt flux from breaking waves: implications for cloud condensation nuclei in the remote marine atmosphere. J Geophys Res 111, D06202. doi:10.1029/2005JD006565
Claustre H, Morel A, Hooker SB, Babin M, Antoine D, Oubelkheir K, Bricaud A, Leblanc K, Quéguiner B, Maritorena S (2002) Is desert dust making oligotrophic waters greener? Geophys Res Lett 29:1469. doi:10.1029/2001GL014056
Colarco PR, Toon OB, Holben BN (2003a) Saharan dust transport to the Caribbean during PRIDE: 1. Influence of dust sources and removal mechanisms on the timing and magnitude of downwind aerosol optical depth events from simulations of in situ and remote sensing observations. J Geophys Res D Atmos 108(19):5–1–5–20
Colarco PR, Toon OB, Reid JS, Livingston JM, Russell PB, Redemann J, Schmid B, Maring HB, Savoie D, Welton EJ, Campbell JR, Holben BN, Levy R (2003b) Saharan dust transport to the Caribbean during PRIDE: 2. Transport, vertical profiles, and deposition in simulations of in situ and remote sensing observations. J Geophys Res D Atmos 108(19):6–1–6–16
Corbett JJ, Köhler HW (2003) Updated emissions from ocean shipping. J Geophys Res 108:4650. doi:10.1029/2003JD003751
Cornell SE (2011) Atmospheric nitrogen deposition: revisiting the importance of the organic component. Environ Pollut 159:2214–2222
Cornell SE, Rendell A, Jickells TD (1995) Atmospheric inputs of dissolved organic nitrogen to the oceans. Nature 376:243–246
Covert DS, Kapustin VN, Bates TS, Quinn PK (1992) New particle formation in the marine boundary layer. J Geophys Res 97:20581–20589
Covert DS, Wiedensohler A, Aalto P et al (1996a) Aerosol number size distributions from 3 to 500 nm diameter in the arctic marine boundary layer during summer and autumn. TELLUS ser B 48(2):197–212
Covert DS, Kapustin VN, Bates TS et al (1996b) Physical properties of marine boundary layer aerosol particles of the mid-Pacific in relation to sources and meteorological transport. J Geophys Res-Atmos 101(D3):6919–6930. doi:10.1029/95JD03068
Coz E, Gómez-Moreno FJ, Pujadas M, Casuccio GS, Lersch TL, Artíñano B (2009) Individual particle characteristics of North African dust under different long-range transport scenarios. Atmos Environ 43:1850–1863
Crahan KK, Hegg D, Covert DS, Jonsson H (2004) An exploration of aqueous oxalic acid production in the coastal marine atmosphere. Atmos Environ 38:3757–3764
Crumeyrolle S, Gomes L, Tulet P, Matsuki A, Schwarzenboeck A, Crahan K (2008) Increase of the aerosol hygroscopicity by cloud processing in a mesoscale convective system: a case study from the AMMA campaign. Atmos Chem Phys 8:6907–6924
Dall’Osto M, Harrison RM, Highwood EJ, O’Dowd C, Ceburnis D, Querol X, Achterberg EP (2010) Variation of the mixing state of Saharan dust particles with atmospheric transport. Atmos Environ 44:3135–3146
Dall-Osto M, Ceburnis D, Monahan C, Worsnop DR, Bialek J, Kulmala M, Kurtén T, Ehn M, Wenger J, Sodeau J, Healy RC, O’Dowd C (2012) Nitrogenated and aliphatic organic vapours as possible drivers for marine secondary organic aerosol growth. J Geophys Res doi:10.1029/2012JD017522
de Baar HJW, Boyd PW, Coale KH, Landry MR, Tsuda A, Assmy P, Bakker DCE, Bozec Y, Barber RT, Brzezinski MA, Buesseler KO, Boyé M, Croot PL, Gervais F, Gorbunov MY, Harrison PJ, Hiscock WT, Laan P, Lancelot C, Law CS, Levasseur M, Marchetti A, Millero FJ, Nishioka J, Nojiri Y, van Oijen T, Riebesell U, Rijkenberg MJA, Saito H, Takeda S, Timmermans KR, Veldhuis MJW, Waite AM, Wong C-S (2005) Synthesis of iron fertilization experiments: from the iron age in the age of enlightenment. J Geoph Res 110, C09S16. doi:10.1029/2004JC002601
de Gouw JA, Middlebrook AM, Warneke C, Goldan PD, Kuster WC, Roberts JM, Fehsenfeld FC, Worsnop DR, Canagaratna MR, Pszenny AAP, Keene WC, Marchewka M, Bertman SB, Bates TS (2005) Budget of organic carbon in a polluted atmosphere: results from the New England air quality study in 2002. J Geophys Res 110, D16305. doi:10.1029/2004jd005623
de Leeuw G, Andreas EL, Anguelova MD, Fairall CW, Lewis ER, O’Dowd C, Schulz M, Schwartz SE (2011a) Production flux of sea spray aerosol. Rev Geophys 49, RG2001. doi:10.1029/2010RG000349
de Leeuw G, Kinne S, Leon JF, Pelon J, Rosenfeld D, Schaap M, Veefkind PJ, Veihelmann B, Winker DM, von Hoyningen-Huene W (2011b) Retrieval of aerosol properties. In: Burrows JP, Platt U, Borrell P (eds) The remote sensing of tropospheric composition from space. Springer, Berlin/Heidelberg, pp 359–313. doi:10.1007/978-3-642-14791-3. ISBN 978-3-642-14790-6
DeCarlo PF, Dunlea EJ, Kimmel JR, Aiken AC, Sueper D, Crounse J, Wennberg PO, Emmons L, Shinozuka Y, Clarke A, Zhou J, Tomlinson J, Collins DR, Knapp D, Weinheimer AJ, Montzka DD, Campos T, Jimenez JL (2008) Fast airborne aerosol size and chemistry measurements above Mexico City and Central Mexico during the MILAGRO campaign. Atmos Chem Phys 8:4027–4048
Decesari S, Finessi E, Rinaldi M, Paglione M, Fuzzi S, Stephanou EG, Tziaras T, Spyros A, Ceburnis D, O’Dowd CD, Dall’Osto M, Harrison RM, Allan J, Coe H, Facchini MC (2011) Primary and secondary marine organic aerosols over the North Atlantic Ocean during the MAP experiment. J Geophys Res 116, D22210. doi:10.1029/2011JD016204
Denman KL, Brasseur G, Chidthaisong A, Ciais P, Cox PM, Dickinson RE, Hauglustaine D, Heinze C, Holland E, Jacob D, Lohmann U, Ramachandran S, da Silva Dias PL, Wofsy SC, Zhang X (2007) Couplings between changes in the climate system and biogeochemistry. In: Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (eds) Climate change 2007: the physical science basis. Contribution of working group I to the fourth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge, UK/New York
Dentener FJ, Carmichael GR, Zhang Y, Lelieveld J, Crutzen PJ (1996) Role of mineral aerosol as a reactive surface in the global troposphere. J Geophys Res 101:22869–22889
Dentener F, Drevet J, Lamarque JF, Bey I, Eickhout B et al (2006) Nitrogen and sulfur deposition on regional and global scales: a multimodel evaluation. Global Biogeochem Cycles 20, GB4003
Desboeufs KV, Losno R, Colin JL (2001) Factors influencing aerosol solubility during cloud processes. Atmos Environ 35:3529–3537
Desboeufs K, Journet E, Rajot JL, Chevaillier S, Triquet S, Formenti P, Zakou A (2010) Chemistry of rain events in West Africa: evidence of dust and biogenic influence in convective systems. Atmos Chem Phys 10:9283–9293
Doney S, Mahowald N, Lima I, Feeley R, Mackenzie F, Lamarque JF, Rasch P (2007) Impact of an-thropogenic atmospheric nitrogen and sulfur depositionon ocean acidification and the inorganic carbon system. PNAS 104, doi:10.1073/pnas.0702218104; 0702214580–0702214585
Duarte CM, Agustí S, Gasol JM, Vaqué D, Vazquez-Dominguez E (2000) Effect of nutrient supply on the biomass structure of planktonic communities: an experimental test on a Mediterranean coastal community. Mar Ecol Prog Ser 206:87–95
Dubovik O, Holben BN, Eck TF, Smirnov A, Kaufman YJ, King MD, Tanré D, Slutsker I (2002) Variability of absorption and optical properties of key aerosol types observed in worldwide locations. J Atmos Sci 59:590–608
Dubovik O, Herman M, Holdak A, Lapyonok T, Tanré D, Deuzé JL, Ducos F, Sinyuk A, Lopatin A (2011) Statistically optimized inversion algorithm for enhanced retrieval of aerosol properties from spectral multi-angle polarimetric satellite observations. Atmos Meas Tech 4:975–1018
Duce R (1986) The impact of atmospheric nitrogen, phosophorus and iron species on marine biological productivity. In: Buat-Menard P (ed) Geochemical cycling. D. Reidel, Norwell, pp 497–529
Duce RA et al (2008) Impacts of atmospheric nitrogen on the open ocean. Science 320:893–897
Duggen S, Croot P, Schacht U, Hoffmann L (2007) Subduction zone volcanic ash can fertilize the surface ocean and stimulate phytoplankton growth: evidence from biogeochemical experiments and satellite data. Geophys Res Lett 34, doi:10.1029/2006GL027522
Dulac F, Buat-Ménard P, Ezat U, Melki S, Bergametti G (1989) Atmospheric input of trace metals to the western Mediterranean: uncertainties in modelling dry deposition from cascade impactor data. Tellus 41B:362–378
Dulac F, Bergametti G, Losno R, Remoudaki E, Gomes L et al (1992) Dry deposition of mineral aerosol particles in the marine atmosphere: significance of the large size fraction. In: Schwartz SE, Slinn WGN (eds) Precipitation scavenging and atmosphere-surface exchange 2. Hemisphere, Washington, DC, pp 841–854
Dulac F, Moulin C, Lambert CE, Guillard F, Poitou J, Guelle W, Quétel CR, Schneider X, Ezat U (1996) Quantitative remote sensing of African dust transport to the Mediterranean. In: Guerzoni S, Chester R (eds) The impact of African dust across the Mediterranean. Kluwer, Norwell, pp 25–49
Dulac F, Chazette P, Gomes L, Chatenet B, Berger H, Vinicula Dos Santos JM (2001) A method for aerosol profiling in the lower troposphere with coupled scatter and meteorological rawindsondes and first data from the tropical Atlantic off Sahara. J Aerosol Sci 32:1069–1086
Dunlea EJ, DeCarlo PF, Aiken AC, Kimmel JR, Peltier RE, Weber RJ, Tomlinson J, Collins DR, Shinozuka Y, McNaughton CS, Howell SG, Clarke AD, Emmons LK, Apel EC, Pfister GG, van Donkelaar A, Martin RV, Millet DB, Heald CL, Jimenez JL (2009) Evolution of Asian aerosols during transpacific transport in INTEX-B. Atmos Chem Phys 9:7257–7287. doi:10.5194/acp-9-7257-2009
Duvall RM, Majestic BJ, Shafer MM, Chuang PY, Simoneit BRT, Schauer JJ (2008) The water-soluble fraction of carbon, sulfur, and crustal elements in Asian aerosols and Asian soils. Atmos Environ 42:5872–5884
Ehn M, Vuollekoski H, Petäjä T, Kerminen V-M, Vana M, Aalto P, de Leeuw G, Ceburnis D, Dupuy R, O’Dowd CD, Kulmala M (2010) Growth rates during coastal and marine new particle formation in Western Ireland. J Geophys Res. doi:10.1029/2010JD014292
Endresen Ø, Sørgard E, Sundet JK, Dalsøren SB, Isaksen ISA, Berglen TF, Gravir G (2003) Emission from international sea transportation and environmental impact. J Geophys Res 108:4560. doi:10.1029/2002JD002898
Endresen Ø, Sørgard E, Behrens HL, Brett PO, Isaksen ISA (2007) A historical reconstruction of ships’ fuel consumption and emissions. J Geophys Res 112, D12301. doi:10.1029/2006JD007630
Erel Y, Dayan U, Rabi R, Rudich Y, Stein M (2006) Trans boundary transport of pollutants by atmospheric mineral dust. Environ Sci Technol 40:2996–3005
Eyring V, Köhler HW, van Aardenne J, Lauer A (2005) Emissions from international shipping: 1. The last 50 years. J Geophys Res 110, D17305
Eyring V, Isaksen ISA, Berntsen T, Collins WJ, Corbett JJ, Endresen O, Grainger RG, Moldanova J, Schlager H, Stevenson DS (2009) Transport impacts on atmosphere and climate: shipping. Atmos Environ. doi:10.1016/j.atmosenv.2009.04.059
Facchini MC, Rinaldi M, Decesari S, Carbone C, Finessi E, Mircea M, Fuzzi S, Ceburnis D, Flannigan R, Nilsson ED, de Leeuw G, Martino M, Woeltjen J, O’Dowd CD (2008a) Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates. Geophys Res Lett 35, L17814. doi:10.1029/2008GL034210
Facchini MC, Decesari S, Rinaldi M, Carbone C, Finessi E, Mircea M, Fuzzi S, Moretti F, Tagliavini E, Ceburnis D, O’Dowd CD (2008b) An important source of marine secondary organic aerosol from biogenic amines. Environ Sci Technol. doi:10.1021/es8018385
Fairlie TD, Jacob DJ, Dibb JE, Alexander B, Avery MA, van Donkelaar A, Zhang L (2010) Impact of mineral dust on nitrate, sulfate, and ozone in transpacific Asian pollution plumes. Atmos Chem Phys 10:3999–4012
Falkovich AH, Ganor E, Levin Z, Formenti P, Rudich Y (2001) Chemical and mineralogical analysis of individual mineral dust particles. J Geophys Res 106:18029–18036
Falkovich AH, Schkolnik G, Ganor E, Rudich Y (2004) Adsorption of organic compounds pertinent to urban environments onto mineral dust particles. J Geophys Res 109, D02208. doi:10.1029/2003jd003919
Falkowski PG, Barber RT, Smetacek V (1998) Biogeochemical controls and feedbacks on ocean primary production. Science 281:200–206
Fan X-B, Okada K, Niimura N, Kai K, Arao K, Shi G-Y, Qin Y, Mitsuta Y (1996) Mineral particles collected in China and Japan during the same Asian dust-storm event. Atmos Environ 30:347–351
Fisher RV, Schmincke H-U (1984) Pyroclastic rocks. Springer, Berlin/Heidelberg/New York/Tokyo
Formenti P, Schütz L, Balkanski Y, Desboeufs K, Ebert M, Kandler K, Petzold A, Scheuvens D, Weinbruch S, Zhang D (2011) Recent progress in understanding physical and chemical properties of African and Asian mineral dust. Atmos Chem Phys 11:8231–8256
Forster PM, Ramaswamy V et al (2007) Changes in atmospheric constituents and in radiative forcing. In: Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (eds) Climate change 2007: the physical science basis contribution of working group I to the fourth assessment report of the intergovernmental panel on climate change. Cambridge University Press, Cambridge
Fowler SW, Buat-Ménard P, Yokoyama Y, Ballestra S, Holm E, Van Nguyen H (1987) Rapid removal of Chernobyl fallout from Mediterranean surface waters by biological activity. Nature 329:56–58
Frew NM (1997) The role of organic films in air-sea gas exchange. In: Liss PS, Duce RA (eds) The sea surface and global change. Cambridge University Press, Cambridge, pp 121–172
Frogner P, Gislason SR, Óskarsson N (2001) Fertilizing potential of volcanic ash in ocean surface water. Geology 29:487–490
Frossard AA, Shaw PM, Russell LM, Kroll JH, Canagaratna MR, Worsnop DR, Quinn PK, Bates TS (2011) Springtime Arctic haze contributions of submicron organic particles from European and Asian combustion sources. J Geophys Res 116, D05205. doi:10.1029/2010JD015178
Froyd KD, Murphy SM, Murphy DM, de Gouw JA, Eddingsaas NC, Wennberg PO (2010) Contribution of isoprene-derived organosulfates to free tropospheric aerosol mass. Proc Natl Acad Sci. doi:10.1073/pnas.1012561107
Fu PQ, Kawamura K, Miura K (2011) Molecular characterization of marine organic aerosols collected during a round-the-world cruise. J Geophys Res Atmos 116(14), D13302. doi:10.1029/2011jd015604
Fuentes E, Coe H, Green D, de Leeuw G, McFiggans G (2010) Laboratory-generated primary marine aerosol via bubble-bursting and atomization. Atmos Meas Tech 3:141–162
Fuentes E, Coe H, Green D, McFiggans G (2011) On the impacts of phytoplankton-derived organic matter on the properties of the primary marine aerosol – part 2: composition, hygroscopicity and cloud condensation activity. Atmos Chem Phys 11:2585–2602. doi:10.5194/acp-11-2585-2011
Fung I, Meyn SK, Tegen I, Doney S, John J, Bishop J (2000) Iron supply and demand in the upper ocean. Global Biogeochem Cycles 14:281–295
Gaiero DM, Probst JL, Depetris PJ, Bidart SM, Leleyter L (2003) Iron and other transition metals in Patagonian riverborne and windborne materials: geochemical control and transport to the southern South Atlantic Ocean. Geochim Cosmochim Acta 67:3603–3623
Galloway J, Townsend A, Erisman J, Bekunda M, Cai Z, Freney J, Martinelli L, Seitzinger S, Sutton M (2008) Transformation of the nitrogen cycle: recent trends, questions and potential solutions. Science 320:889–892
Gantt B, Meskhidze N, Facchini MC, Rinaldi M, Ceburnis D, O’Dowd CD (2011) Wind speed dependent size-resolved parameterization for the organic enrichment of sea spray. Atmos Chem Phys 11:1–13
Garrison VH, Shinn EA, Foreman WT, Griffin DW, Holmes CW, Kellogg CA, Majewski MS, Richardson LL, Ritchie KB, Smith GW (2003) African and Asian dust: from desert soils to coral reefs. Bioscience 53:469–479
Garrison VH, Foreman WT, Genualdi S, Griffin DW, Kellogg CA, Majewski MS, Mohammed A, Ramsubhag A, Shinn EA, Simonich SL, Smith GW (2006) Saharan dust – a carrier of persistent organic pollutants, metals and microbes to the Caribbean? Rev Biol Trop (Int J Trop Biol ISSN-0034-7744) 54(3):9–21
Geever M, O’Dowd CD, van Ekeren S, Flanagan R, Nilsson DE, de Leeuw G, Rannik Ü (2005) Sub-micron sea-spray fluxes. Geophys Res Lett. doi:10.1029/2005GL023081
Geng H, Park Y, Hwang H, Kang S, Ro CU (2009) Elevated nitrogen-containing particles observed in Asian dust aerosol samples collected at the marine boundary layer of the Bohai Sea and the Yellow Sea. Atmos Chem Phys 9:6933–6947
Gershey RM (1983) Characterization of seawater organic matter carried by bubble-generated aerosols. Limnol Oceanogr 28:309–319
Gibb SW, Mantoura RFC, Liss PS (1999) Ocean atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea. Global Biogeochem Cycles 13:161–178
Giglio L, Randerson JT, van der Werf GR, Kasibhatla PS, Collatz GJ, Morton DC, DeFries RS (2010) Assessing variability and long-term trends in burned area by merging multiple satellite fire products. Biogeosciences 7:1171–1186
Giorgi F (2006) Climate change hot-spots. Geophys Res Lett 33, L08707. doi:10.1029/2006gl025734
Giovagnetti V, Brunet C, Conversano F, Tramontano F, Obernosterer I, Ridame C, Guieu C (2013) Assessing the role of dust deposition on phytoplankton ecophysiology and succession in a low-nutrient low-chlorophyll ecosystem: a mesocosm experiment in the Mediterranean, Sea. Biogeosciences 10:2973–2991. doi:10.5194/bg-10-2973-2013
Goldstein AH, Galbally IE (2007) Known and unexplored organic constituents in the Earth’s at-mosphere. Environ Sci Technol 41:1514–1521
Gomes L, Bergametti G, Coudé-Gaussens G, Rognon P (1990) Submicron desert dusts: a sandblasting process. J Geophys Res 95:13927–13935
Gong SL (2003) A parameterization of sea-salt aerosol source function for sub- and super-micron particles. Global Biogeochem Cycles 17:1097. doi:10.1029/2003GB002079
Gorbushina AA, Kort R, Schulte A, Lazarus D, Schnetger B, Brumsack H-J, Broughton WJ, Favet J (2007) Life in Darwin’s dust: intercontinental transport and survival of microbes in the nineteenth century. Environ Microbiol 9(12):2911–2922
Goudie AS, Middleton NJ (2006) Desert dust in the global system. Springer, Berlin
Graedel TE, Keene WC (1995) Tropospheric budget of reactive chlorine. Global Biogeochem Cycles 9:47–77
Graf HF, Feichter J, Langmann B (1997) Volcanic sulfur emissions: estimates of source strength and its contribution to the global sulfate distribution. J Geophys Res-Atmos 102:10727–10738
Graham WF, Duce RA (1979) Atmospheric pathways of the phosphorus cycle. Geochimica et Cosmo-chimica Acta 43:1195–1208
Granier C, Bessagnet B, Bond T, D’Angiola A, Denier van der Gon H, Frost GJ, Heil A, Kaiser JW, Kinne S, Klimont Z et al (2011) Evolution of anthropogenic and biomass burning emissions of air pollutants at global and regional scales during the 1980–2010 period. Clim Chang. doi:10.1007/s10584-011-0154-1
Greeley R, Iversen J (1985) Wind as a geological process on Earth, Mars, Venus and Titan, vol 4, Cambridge planetary sciences series. Cambridge University Press, Cambridge, p 333
Grieshop AP, Logue JM, Donahue NM, Robinson AL (2009) Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution. Atmos Chem Phys 9:1263–1277
Griffin DW (2007) Atmospheric movement of microorganisms in clouds of desert dust and implications for human health. Clin Microbiol Rev 20(3):459–477. doi:10.1128/CMR.00039-06
Grousset F, Biscaye P (2005) Tracing dust sources and transport patterns using Sr, Nd and Pb isotopes. Chem Geol 222:149–167
Guenther A, Hewitt CN, Erickson D, Fall R, Geron C, Graedel T, Harley P, Klinger L, Lerdau M, McKay WA, Pierce T, Scholes B, Steinbrecher R, Tallamraju R, Taylor J, Zimmermann P (1995) A global model of natural volatile organic compound emissions. J Geophys Res 100(D5):8873–8892
Guenther A, Karl T, Harley P, Wiedinmyer C, Palmer PI, Geron C (2006) Estimates of global terrestrial isoprene emissions using MEGAN (Model of emissions of gases and Aerosols from nature). Atmos Chem Phys 6:3181–3210
Guieu C, Loye-Pilot MD, Ridame C, Thomas C (2002) Chemical characterization of the Saharan dust end-member: some biogeochemical implications for the western Mediterranean Sea. J Geophys Res Atmos 107:4258. doi:10.1029/2001JD000582
Guieu C, Bonnet S, Wagener T, Loÿe-Pilot MD (2005) Biomass burning as a source of dissolved iron to open ocean? Geophys Res Lett 32, doi:L1960810.1029/2005GL022962
Guieu C, Dulac F, Desboeufs K, Wagener T, Pulido-Villena E, Grisoni J-M, Louis F, Ridame C, Blain S, Brunet C, Bon Nguyen E, Tran S, Labiadh M, Dominici J-M (2010) Large clean mesocosms and simulated dust deposition: a new methodology to investigate responses of marine oligotrophic ecosystems to atmospheric inputs. Biogeosciences 7:2765–2784
Guieu C, Ridame C, Pulido-Villena E, Blain S, Bressac M, Desboeufs K, Dulac F, Does dust deposition change the metabolic balance of a typical oligotrophic marine environment? (in preparation)
Guieu C, Dulac F, Ridame C, Pondaven P (2013) Introduction to the project DUNE, a DUst experiment in a low Nutrient, low chlorophyll Ecosystem, Biogeosciences Discuss 10:12491–12527
Halloran PR, Bell TG, Totterdell IJ (2010) Can we trust empirical marine DMS parameterisations within projections of future climate? Biogeosciences 7:1645–1656
Hallquist M, Wenger JC, Baltensperger U, Rudich Y, Simpson D, Claeys M, Dommen J, Donahue NM, George C, Goldstein AH, Hamilton JF, Herrmann H, Hoffmann T, Iinuma Y, Jang M, Jenkin ME, Jimenez JL, Kiendler-Scharr A, Maenhaut W, McFiggans G, Mentel TF, Monod A, Prevot ASH, Seinfeld JH, Surratt JD, Szmigielski R, Wildt J (2009) The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos Chem Phys 9(14):5155–5235
Halmer MM, Scmincke HU, Graf HF (2002) The annual volcanic gas input into the atmosphere, in particular into the stratosphere: a global data set for the past 100 years. J Volcanol Geotherm Res 115:511–528
Hamm CE (2002) Interactive aggregation and sedimentation of diatoms, and clay-sized lithogenic material. Limnol Oceanogr 47:1790–1795
Hamme RC, Webley PW, Crawford WR, Whitney FA, DeGrandpre MD, Emerson SR, Eriksen CC, Giesbrecht KE, Gower JFR, Kavanaugh MT, Peña MA, Sabine CL, Batten SD, Coogan LA, Grundle DS, Deirdre LD (2010) Volcanic ash fuels anomalous plankton bloom in subarctic northeast Pacific. Geophys Res Lett 37, L19604. doi:10.1029/2010GL044629
Hamonou E, Chazette P, Balis D, Dulac F, Schneider X, Galani E, Ancellet G, Papayannis A (1999) Characterization of the vertical structure of Saharan dust export to the Mediterranean basin. J Geophys Res 104:22257–22270
Hand J, Mahowald N, Chen Y, Siefert R, Luo C, Subramaniam A, Fung I (2004) Estimates of soluble iron from observations and a global mineral aerosol model: biogeochemical implications. J Geophys Res 109, D17205, doi:17210.11029/12004JD004574
Hand VL, Capes G, Vaughan DJ, Formenti P, Haywood JM, Coe H (2010) Evidence of internal mixing of African dust and biomass burning particles by individual particle analysis using electron beam techniques. J Geophys Res Atmos 115, D13301. doi:10.1029/2009jd012938
Hanisch F, Crowley JN (2001) The heterogeneous reactivity of gaseous nitric acid on authentic mineral dust samples, and on individual mineral and clay mineral components. Phys Chem Chem Phys 3:2474–2482
Hansell DA, Carlson CA, Repeta DJ, Schlitzer R (2009) Dissolved organic matter in the ocean: a controversy stimulates new insights. Oceanography 22:202–211
Hawkins LN, Russell LM, Covert DS, Quinn PK, Bates TS (2010) Carboxylic acids, sulfates, and organosulfates in processed continental organic aerosol over the southeast Pacific Ocean during VOCALS-REx 2008. J Geophys Res 115, D13201. doi:10.1029/2009jd013276
Heald CL, Jacob DJ, Park RJ, Russell LM, Huebert BJ, Seinfeld JH, Liao H, Weber RJ (2005) A large organic aerosol source in the free troposphere missing from current models. Geophys Res Lett 32, L18809. doi:10.1029/2005gl023831
Hedges IH, Eglinton G, Hatcher PG, Kirchman DL, Arnosti C, Derenne S, Evershed RP, Ogel-Knabner IK, de Leeuw JW, Littke R, Michaelis W, Rullkotter J (2000) The molecularly uncharacterized component of nonliving organic matter in natural environments. Organic Geochem 31:945–951
Hennigan CJ, Miracolo MA, Engelhart GJ, May AA, Presto AA, Lee T, Sullivan AP, McMeeking GR, Coe H, Wold CE, Hao WM, Gilman JB, Kuster WC, de Gouw J, Schichtel BA, Collett JL, Kreidenweis SM, Robinson AL (2011) Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber. Atmos Chem Phys 11:7669–7686
Henze D, Seinfeld JH (2006) Global secondary organic aerosol from isoprene oxidation. Geophys Res Lett 33, L09812. doi:10.1029/2006GL025976
Herut B et al (2005) Response of East Mediterranean surface water to Saharan dust: on-board microcosm experiment and field observations. Deep-Sea Res II 52:3024–3040
Hildebrandt L, Engelhart GJ, Mohr C, Kostenidou E, Lanz VA, Bougiatioti A, DeCarlo PF, Prevot ASH, Baltensperger U, Mihalopoulos N, Donahue NM, Pandis SN (2010) Aged organic aerosol in the Eastern Mediterranean: the Finokalia aerosol measurement experiment 2008. Atmos Chem Phys 10:4167–4186. doi:10.5194/acp-10-4167-2010
Hill PG, Zubkov MV, Purdie DA (2010) Differential responses of Prochlorococcus and SAR11-dominated bacterioplankton groups to atmospheric dust inputs in the tropical Northeast Atlantic Ocean. FEMS Microbiol Lett 306:82–89
Hoffmann T, O’Dowd CD, Seinfeld JH (2001) Iodine oxide homogeneous nucleation: an explanation for coastal new particle production. Geophys Res Lett 28(10):1949–1952. doi:10.1029/2000GL012399
Holben BN, Tanre D, Smirnov A, Eck TF, Slutsker I, Abuhassan N, Newcomb WW, Schafer J, Chatenet B, Lavenue F, Kaufman YJ, Vande Castle J, Setzer A, Markham B, Clark D, Frouin R, Halthore R, Karnieli A, O’Neill NT, Pietras C, Pinker RT, Voss K, Zibordi G (2001) An emerging ground-based aerosol climatology: aerosol optical depth from AERONET. J Geophys Res 106:12067–12097
Holzinger R, Lee A, Paw U KT, Goldstein AH (2005) Observations of oxidation products above a forest imply biogenic emissions of very reactive compounds. Atmos Chem Phys 5:67–75
Holmes BJ, Petrucci GA (2006) Water-soluble oligomer formation from acid catalyzed reactions of levoglucosan in proxies of atmospheric aqueous aerosols. Environ Sci Technol 40:4983–4989
Honeyman BD, Santschi PH (1991) Coupling adsorption and particle aggregation: laboratory studies of “colloidal pumping” using 59Fe-labeled hematite. Environ Sci Technol 25:1739–1747
Hoppel WA, Frick GM, Larson RE (1986) Effect of nonprecipitating clouds on the aerosol size distribution. Geophys Res Lett 13:125–128
Hoppel WA, Frick GM, Fitzgerald J et al (1994) Marine boundary-layer measurements of new particle formation and the effects nonprecipitating clouds have on aerosol-size distribution. J Geophys Res-Atmos 99(D7):14443–14459. doi:10.1029/94JD00797
Horňák K, Jezbera J, Nedoma J, Gasol JM, Simek K (2006) Effects of resource availability and bac-terivory on leucine incorporation in different groups of freshwater bacterioplankton, assessed using microautoradiography. Aquat Microb Ecol 45:277–289
Hsu S-C, Liu SC, Kao S-J, Jeng W-L, Huang Y-T, Tseng C-M, Tsai F, Tu J-Y, Yang Y (2007) Water-soluble species in the marine aerosol from the northern South China Sea: high chloride depletion related to air pollution. J Geophys Res 112, D19304. doi:10.1029/2007jd008844
Huang K, Zhuang GS, Li JA, Wang QZ, Sun YL, Lin YF, Fu JS (2010) Mixing of Asian dust with pollution aerosol and the transformation of aerosol components during the dust storm over China in spring 2007. J Geophys Res Atmos 115, D00k13, doi:10.1029/2009jd013145
Hultin KAH, Krejci R, Pinhassi J, Gomez-Consarnau L, Mårtensson EM, Hagström Å, Nilsson ED (2011) Aerosol and bacterial emissions from Baltic seawater. Atmos Res 99:1–14
Hunter KA, Boyd PW (2007) Iron-binding ligands and their role in the ocean biogeochemistry of iron. Environ Chem 4:221–232. doi:10.1071/EN07012
Hunter K, Liss P, Surapipith V, Dentener F, Duce R, Kanakidou M, Kubilay N, Mahowald N, Okin G, Sarin M, Uematsu M, Zhu T (2011) Impacts of anthropogenic SOx, NOx and NH3 on acidifiation of coastal waters and shipping lanes. Geophys Res Lett 38, doi:10.1029/2011GL047720
IPCC (2007) Climate change 2007: the physical science basis. Contribution of working group I to the fourth assessment report of the intergovernmental panel on climate change. In: Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (eds). Cambridge University Press, Cambridge, UK and New York, NY, p 996
Irshad R, Grainger RG, Peters DM, McPheat RA, Smith KM, Thomas G (2009) Laboratory measurements of the optical properties of sea salt aerosol. Atmos Chem Phys 9:221–230
Iversen JB, White DR (1982) Saltation threshold on Earth, Mars and Venus. Sedimentology 29:111–119
Iwasaka Y, Yamato M, Imasu R, Ono A (1988) Transport of Asian dust (KOSA) particles; importance of weak KOSA events on the geochemical cycle of soil particles. Tellas B Chem Phys Meterol 40B:494–503
Jaeglé L, Quinn PK, Bates TS, Alexander B, Lin J-T (2011) Global distribution of sea salt aerosols: new constraints from in situ and remote sensing observations. Atmos Chem Phys 11:3137–3157
Jaenicke R (2005) Abundance of cellular material and proteins in the atmosphere. Science 308(5718):73. doi:10.1126/science.1106335
Jahnke RA (1996) The global ocean flux of particulate organic carbon: areal distribution and magnitude. Global Biogeochem Cycles 10:71–88
Jickells T, Spokes L (2001) Atmospheric iron inputs to the oceans. In: Turner DR, Hunteger K (eds) Biogeochemistry of iron in seawater. Wiley, Chichester, pp 85–121
Jickells TD, An ZS, Andersen KK, Baker AR, Bergametti G, Brooks N, Cao JJ, Boyd PW, Duce RA, Hunter KA, Kawahata H, Kubilay N, la Roche J, Liss PS, Mahowald N, Prospero JM, Ridgwell AJ, Tegen I, Torres R (2005) Global iron connections between desert dust, ocean biogeochemistry, and climate. Science 308:67–71
Jimenez JL, Bahreini R, Cocker DR III, Zhuang H, Varutbangkul V, Flagan RC, Seinfeld JH, O’Dowd CD, Hoffmann T (2003) New particle formation from photooxidation of diiodomethane (CH2 I2). J Geophys Res 108(D10):4318. doi:10.1029/2002JD002452
Jimenez JL, Canagaratna MR, Donahue NM, Prevot ASH, Zhang Q, Kroll JH, DeCarlo PF, Allan JD, Coe H, Ng NL, Aiken AC, Docherty KS, Ulbrich IM, Grieshop AP, Robinson AL, Duplissy J, Smith JD, Wilson KR, Lanz VA, Hueglin C, Sun YL, Tian J, Laaksonen A, Raatikainen T, Rautiainen J, Vaattovaara P, Ehn M, Kulmala M, Tomlinson JM, Collins DR, Cubison MJE, Dunlea J, Huffman JA, Onasch TB, Alfarra MR, Williams PI, Bower K, Kondo Y, Schneider J, Drewnick F, Borrmann S, Weimer S, Demerjian K, Salcedo D, Cottrell L, Griffin R, Takami A, Miyoshi T, Hatakeyama S, Shimono A, Sun JY, Zhang YM, Dzepina K, Kimmel JR, Sueper D, Jayne JT, Herndon SC, Trimborn AM, Williams LR, Wood EC, Middlebrook AM, Kolb CE, Baltensperger U, Worsnop DR (2009) Evolution of organic aerosols in the atmosphere. Science 326:1525–1529
Johansen AM, Siefert RL, Hoffmann MR (1999) Chemical characterization of ambient aerosol collected during the southwest monsoon and intermonsoon seasons over the Arabian Sea: anions and cations. J Geophys Res 104:26325–26347
Jones MT, Gislason SR (2008) Rapid releases of metal salts and nutrients following the deposition of volcanic ash into aqueous environments. Geochim Cosmochim Acta 72:3661–3680
Journet E, Desboeufs KV, Caquineau S, Colin J-L (2008) Mineralogy as a critical factor of dust iron solubility. Geophys Res Lett 35, L07805. doi:10.1029/2007GL031589
Jurado E, Dachs J, Duarte CM, Simó R (2008) Atmospheric deposition of organic and black carbon to the global oceans. Atmos Environ 42:7931–7939
Kanakidou M, Seinfeld JH, Pandis SN, Barnes I, Dentener FJ, Facchini R, van Dingenen R, Ervens B, Nenes A, Nielsen CJ, Swietlicki E, Putaud JP, Balkanski Y, Fuzzi S, Horth J, Moortgat GK, Winterhalter R, Myhre CEL, Tsigaridis K, Vignati E, Stephanou EG, Wilson J (2004) Organic aerosol and global climate modelling: a review. Atmos Chem Phys 5:1053–1123
Kandler K, Benker N, Bundke U, Cuevas E, Ebert M, Knippertz P, Rodríguez S, Schütz L, Weinbruch S (2007) Chemical composition and complex refractive index of Saharan mineral dust at Izaña, Tenerife (Spain) derived by electron microscopy. Atmos Environ 41:8058–8074
Karyampudi VM, Palm SP, Reagen JA, Fang H, Grant WB, Hoff RM, Moulin C, Pierce HF, Torres O, Browell EV, Melfi SH (1999) Validation of the Saharan dust plume conceptual model using lidar, Meteosat, and ECMWF data. Bull Am Meteorol Soc 80:1045–1075
Kaufman YJ, Boucher O, Tanré D, Chin M, Remer LA, Takemura T (2005) Aerosol anthropogenic component estimated from satellite data. Geophys Res Lett 32, doi:10.1029/2005GL023125
Kawamura K, Sakaguchi F (1999) Molecular distributions of water soluble dicarboxylic acids in marine aerosols over the Pacific Ocean including tropics. J Geophys Res 104:3501–3509
Kawamura K, Kasukabe H, Barrie LA (1996a) Source and reaction pathways ofdicarboxylic acids, ketoacids and dicarbonyls in arctic aerosols at polar sunrise. Atmos Environ 30:1709–1722
Kawamura K, Semèrè R, Imai Y, Fujii Y, Hayashi M (1996b) Water soluble dicarboxylic acids and related compounds in Antarctic aerosols. J Geophys Res 101:18721–18728
Kawamura K, Kasukabe H, Barrie LA (2010) Secondary formation of water-soluble organic acids and a-dicarbonyls and their contributions to total carbon and water-soluble organic carbon: photochemical aging of organic aerosols in the Arctic spring. J Geophys Res 115, D21306. doi:10.1029/2010JD014299
Keene WC, Maring H, Maben JR, Kieber DJ, Pszenny AAP, Dahl EE, Izaguirre MA, Davis AJ, Long MS, Zhou X, Smoydzin L, Sander R (2007) Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface. J Geophys Res 112, D21202. doi:10.1029/2007JD008464
Kelly JT, Wexler AS (2005) Thermodynamics of carbonates and hydrates related to heterogeneous reactions involving mineral aerosol. J Geophys Res 110, D11201. doi:10.1029/2004jd005583
Kerminen VM, Hillamo RE, Wexler AS (1998) Model Simulations on the variability of particulate MSA to non-sea-salt sulfate ratio in the marine environment. J Atmos Chem 30:345–370
Kim B-G, Park S-U (2001) Transport and evolution of a winter-time yellow sand observed in Korea. Atmos Environ 35:3191–3201
King MD, Kaufman YJ, Tanré D, Nakajima T (1999) Remote sensing of tropospheric aerosols from space: past, present, and future. Bull Am Meteorol Soc 80:2229–2259
Kinne S, Schulz M, Textor C et al (2006) An AeroCom initial assessment optical properties in aerosol component modules of global models. Atmos Chem Phys 6:1815–1834
Kirkby J, Curtius J, Almeida J et al (2011) Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 476:429–433
Klein C, Dolan JR, Rassoulzadegan F (1997) Experimental examination of the effects of rainwater on micro-bial communities in the surface of the NW Mediterranean Sea. Mar Ecol Prog Ser 158:41–50
Kloster S, Mahowald NM, Randerson JT, Thornton PE, Hoffman FM, Levis S, Lawrence PJ, Feddema JJ, Oleson KW, Lawrence DM (2010) Fire dynamics during the 20th century simulated by the Community Land Model. Biogeosciences 7:1877–1902
Koçak M, Mihalopoulos N, Kubilay N (2007) Chemical composition of the fine and coarse fraction of aerosols in the northeastern Mediterranean. Atmos Environ 41:7351–7368
Kohfeld K, LeQuere C, Harrison S, Anderson R (2005) Role of marine biology in glacial-integlacial CO2 cycles. Science 308:74–78
Kollias P, Fairall CW, Zuidema P, Tomlinson J, Wick GA (2004) Observations of marine stratocumulus in SE Pacific during the PACS 2003 cruise. Geophys Res Lett 31, L22110. doi:10.1029/2004GL020751
Korhonen H, Carslaw KS, Forster PM, Mikkonen S, Gordon ND, Kokkola H (2010a) Aerosol climate feedback due to decadal increases in Southern Hemisphere wind speeds. Geophys Res Lett 37, L02805. doi:10.1029/2009GL041320
Korhonen H, Carslaw KS, Romakkaniemi S (2010b) Enhancement of marine cloud albedo via controlled sea spray injections: a global model study of the influence of emission rates, microphysics and transport. Atmos Chem Phys 10:4133–4143. doi:10.5194/acp-10-4133-2010
Kokhanovsky AA, de Leeuw G (eds) (2009) Satellite aerosol remote sensing over land. Springer-Praxis, Berlin, p 388. ISBN 978-3-540-69396-3
Krishnamurthy A, Moore JK, Mahowald N, Luo C, Doney SC, Lindsay K, Zender CS (2009) The impacts of increasing anthro- pogenic soluble iron and nitrogen deposition on ocean biogeochemistry. Global Biogeochem Cycles 23, GB3016. doi:10.1029/2008GB003440
Krishnamurthy A, Moore JK, Mahowald N, Luo C, Zender CS (2010) Impacts of atmospheric nutrient inputs on marine biogeochemistry. J Geophys Res 115, G01006. doi:10.1029/2009JG001115
Kritz MA, Le Roulley J-C, Danielsen EF (1990) The China Clipper—fast advective transport of radon-rich air from the Asian boundary layer to the upper troposphere near California. Tellus 42B:46–61
Krol M, Houweling S, Bregman B, van den Broek M, Segers A, van Velthoven P, Peters W, Dentener F, Bergamaschi P (2005) The two-way nested global chemistry-transport zoom model TM5: algorithm and applications. Atmos Chem Phys 5:417–432
Kroll JH, Donahue NM, Jimenez JL et al (2011) Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat Chem 3:133–139
Krueger BJ, Grassian VH, Cowin JP, Laskin A (2004) Heterogeneous chemistry of individual mineral dust particles from different dust source regions: the importance of particle mineralogy. Atmos Environ 38:6253–6261
Kulmala M, Suni T, Lehtinen KEJ, Dal Maso M, Boy M, Reissell A, Rannik U, Aalto P, Keronen P, Hakola H, Back JB, Hoffmann T, Vesala T, Hari P (2004) A new feedback mechanism linking forests, aerosols, and climate. Atmos Chem Phys 4:557–562
Kumar A, Sudheer AK, Sarin MM (2008) Chemical characteristics of aerosols in MABL of Bay of Bengal and Arabian Sea during spring inter-monsoon: a comparative study. Springer, Heidelberg, p 8, ALLEMAGNE
Laghdass M, Blain S, Besseling M, Catala P, Guieu C, Obernosterer I (2011) Impact of Saharan dust deposition on the bacterial diversity and activity in the NW Mediterranean Sea. Aquat Microb Ecol 62:201–213
Lam P, Bishop J (2008) The continental margin is a key sources of iron to the North Pacific Ocean. Geophys Res Lett 35, doi:10.1029/2008GL033294
Lamarque J-F, Bond TC, Eyring V, Granier C, Heil A, Klimont Z, Lee D, Liousse C, Mieville A, Owen B, Schultz MG, Shindell D, Smith SJ, Stehfest E, Van Aardenne J, Cooper OR, Kainuma M, Mahowald N, McConnell JR, Naik V, Riahi K, van Vuuren DP (2010) Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: methodology and application. Atmos Chem Phys 10:7017–7039
Lambe AT, Miracolo MA, Hennigan CJ, Robinson AL, Donahue NM (2009) Effective rate constants and uptake coefficients for the reactions of organic molecular markers (n-Alkanes, Hopanes, and Steranes) in motor oil and diesel primary organic aerosols with hydroxyl radicals. Environ Sci Technol 43:8794–8800
Landing WM, Paytan A (2010) Aerosol chemistry and impacts on the ocean. Mar Chem 120:1–3
Langmann BC, Scannell C, O’Dowd CD (2008) Organic matter contribution to marine aerosols and cloud condensation nuclei. Atmos Environ. doi:10.1016/j.atmosenv.2008.09.002
Langmann B, Zaksek K, Hort M, Duggen S (2010) Volcanic ash as fertiliser for the surface ocean. Atmos Chem Phys 10:3891–3899
Lapina K, Heald CL, Spracklen DV, Arnold SR, Allan JD, Coe H, McFiggans G, Zorn SR, Drewnick F, Bates TS, Hawkins LN, Russell LM, Smirnov A, O’Dowd C, Hind AJ (2011) Investigating organic aerosol loading in the remote marine environment. Atmos Chem Phys 11:8847–8860
Laskin A, Wietsma TW, Krueger BJ, Grassian VH (2005) Heterogeneous chemistry of individual mineral dust particles with nitric acid: a combined CCSEM/EDX, ESEM, and ICP-MS study. J Geophys Res 110, doi:10.1029/2004jd005206
Latham J (2002) Amelioration of global warming by controlled enhancement of the albedo and longevity of low-level maritime clouds. Atmos Sci. doi:10.1006/Asle.2002.0048
Latham J, Smith MH (1990) Effect on global warming of wind-dependent aerosol generation at the ocean surface. Nature 347:372–373
Latham J (2002) Amelioration of global warming by controlled enhancement of the albedo and longevity of low-level maritime clouds. Atmos Sci. doi:10.1006/Asle.2002.0048
Lathière J, Hauglustaine DA, De Noblet-Ducoudré N (2005) Past and future changes in biogenic volatile organic compound emissions simulated with a global dynamic vegetation model. Geophys Res Lett 32, L20818. doi:10.1029/2005GL024164
Lathière J, Hauglustaine DA, Friend A, De Noblet-Ducoudré N, Viovy N, Folberth G (2006) Impact of climate variability and land use changes on global biogenic volatile organic compound emissions. Atmos Chem Phys 6:2129–2146
Law CS, Woodward EMS, Ellwood MJ, Marriner A, Bury SJ, Safic KA (2011) Response of surface nutrient inventories and nitrogen fixation to a tropical cyclone in the southwest Pacific. Limnol Oceanogr 56:1372–1385
Law CS, Brévière E, de Leeuw G, Garçon V, Guieu C, Kieber D, Kontradowitz S, Paulmier A, Quinn P, Saltzman E, Stefels J, von Glasow R (2013) Evolving research directions in Surface Ocean-Lower Atmosphere (SOLAS) Science. Environ Chem 10:1–16, http://dx.doi.org/10.1071/EN12159
Leaitch WR, Macdonald AM, Anlauf KG, Liu PSK, Toom-Sauntry D, Li SM, Liggio J, Hayden K, Wasey MA, Russell LM, Takahama S, Liu S, van Donkelaar A, Duck T, Martin RV, Zhang Q, Sun Y, McKendry I, Shantz NC, Cubison M (2009) Evidence for Asian dust effects from aerosol plume measurements during INTEX-B 2006 near Whistler, BC. Atmos Chem Phys 9:3523–3546
Lee JD, McFiggans G, Allan JD, Baker AR, Ball SM, Benton AK, Carpenter LJ, Commane R, Finley BD, Evans M, Fuentes E, Furneaux K, Goddard A, Good N, Hamilton JF, Heard DE, Herrmann H, Hollingsworth A, Hopkins JR, Ingham T, Irwin M, Jones CE, Jones RL, Keene WC, Lawler MJ, Lehmann S, Lewis AC, Long MS, Mahajan A, Methven J, Moller SJ, Müller K, Müller T, Niedermeier N, O’Doherty S, Oetjen H, Plane JMC, Pszenny AAP, Read KA, Saiz-Lopez A, Saltzman ES, Sander R, von Glasow R, Whalley L, Wiedensohler A, Young D (2010) Reactive Halogens in the Marine Boundary Layer (RHaMBLe): the tropical North Atlantic experiments. Atmos Chem Phys 10:1031–1055. doi:10.5194/acp-10-1031-2010
Leck C, Bigg EK (2005) Source and evolution of the marine aerosol—a new perspective. Geophys Res Lett 32, L19803. doi:10.1029/2005GL023651
Lefevre N, Watson AJ (1999) Modeling the geochemical cycle of iron in the oceans and its impact on atmospheric CO2 concentrations. Global Biogeochem Cycles 13:727–736
Lelieveld J, Butler TM, Crowley JN, Dillon TJ, Fischer H, Ganzeveld L, Harder H, Lawrence MG, Martinez M, Taraborrelli D, Williams J (2008) Atmospheric oxidation capacity sustained by a tropical forest. Nature 452:737–740
Lesworth T, Baker AR, Jickells T (2010) Aerosol organic nitrogen over the remote Atlantic Ocean. Atmos Environ 44:1887–1893
Levin Z, Ganor E (1996) The effect of desert particles on cloud and rain formation in the eastern Mediterranean. In: Guerzoni S, Chester R (eds) The impact of desert dust across the Mediterranean. Kluwer, Dordrecht, pp 77–86
Lewis ER, Schwartz SE (2004) Sea salt aerosol production: mechanisms, methods, measurements and models—a critical review. American Geophysical Union, Washington, DC, p 413
Li WJ, Shao LY (2010) Mixing and water-soluble characteristics of particulate organic compounds in individual urban aerosol particles. J Geophys Res Atmos 115, D02301. doi:10.1029/2009jd012575
Lin II, Hu C, Li YH, Ho TY, Fischer TP, Wong GTF, Wu J, Huang CW, Chu DA, Ko DS, Chen JP (2011) Fertilization potential of volcanic dust in the low-nutrient low-chlorophyll western North Pacific subtropical gyre: Satellite evidence and laboratory study. Global Biogeochem Cycles 25, GB1006. doi:10.1029/2009GB003758
Lippmann M (2007) Health effects of airborne particulate matter. N Engl J Med 357:2395–2397
Liu X, Millero FJ (2002) The solubility of iron in seawater. Mar Chem 77:43–54
Liu X, Zhu J, Van Espen P, Adams F, Xiao R, Dong S, Li Y (2005) Single particle characterization of spring and summer aerosols in Beijing: formation of composite sulfate of calcium and potassium. Atmos Environ 39:6909–6918
Loeb NG, Manalo-Smith N (2005) Top-of-atmosphere direct radiative effect of aerosols over global oceans from merged CERES and MODIS observations. J Climate 18:3506–3526
Lohmann U, Feichter J (2005) Global indirect aerosol effects: a review. Atmos Chem Phys 5:715–737
Love RG, Miller BG, Groat SK, Hagen S, Cowie HA, Johnston PP, Hutchison PA, Soutar CA (1997) Respiratory health effects of opencast coalmining: a cross sectional study of current workers. Occup Environ Med 54(6):416–423
Loÿe-Pilot M-D, Martin J-M (1996) Saharan dust input to the western Mediterranean: an eleven years record in Corsica. In: Guerzoni S, Chester R (eds) The impact of desert dust across the Mediterranean. Kluwer, Dordrecht, pp 191–199
Loÿe-Pilot MM, Martin J-M, Morelli J (1986) Influence of Saharan dust on the rain acidity and atmospheric input to the Mediterranean Sea. Nature 321:427–428
Luo C, Mahowald N, Meskhidze N, Chen Y, Siefert R, Baker A, Johansen A (2006) Estimation of iron solubility from observations and a global aerosol model. J Geophys Res 110, D23307, doi:10.1029/2005JD006059, http://www.agu.org/journals/jd/jd0523/2005JD006059/
Luo C, Mahowald N, Bond T, Chuang PY, Artaxo P, Siefert R, Chen Y, Schauer J (2008) Combustion iron distribution and deposition. Global Biogeochem Cycles 22, doi:10.1029/2007GB002964
Ma C-J, Tohno S, Kasahara M (2005) A case study of the size-resolved individual particles collected at a ground-based site on the west coast of Japan during an Asian dust storm event. Atmos Environ 39:739–747
Ma C-J, Choi K-C (2007) A combination of bulk and single particle analyses for Asian dust, water. Air Soil Pollut 183:3–13. doi:10.1007/s11270-006-9302-z
Mahowald NM, Luo C (2003) A less dusty future? Geophys Res Lett 30, doi:1910.1029/2003GRL017880
Mahowald N, Baker A, Bergametti G, Brooks N, Duce R, Jickells T, Kubilay N, Prospero J, Tegen I (2005) The atmospheric global dust cycle and iron inputs to the ocean. Global Biogeochem Cycles 19, GB4025, doi:4010.1029/2004GB002402
Mahowald NM, Muhs DR, Levis S, Rasch PJ, Yoshioka M, Zender CS, Luo C (2006) Change in atmospheric mineral aerosols in response to climate: last glacial period, preindustrial, modern, and doubled carbon dioxide climates. J Geophys Res 111, D10202. doi:10.1029/2005JD006653
Mahowald N (2007) Anthropocene changes in desert area: sensitivity to climate model predictions. Geophys Res Lett 34, L18817
Mahowald N, Jickells TD, Baker AR, Artaxo P, Benitez-Nelson CR, Bergametti G, Bond TC, Chen Y, Cohen DD, Herut B, Kubilay N, Losno R, Luo C, Maenhaut W, McGee KA, Okin GS, Siefert RL, Tsukuda S (2008) The global distribution of atmospheric phosphorus deposition and anthropogenic impacts. Global Biogeochem Cycles 22, doi:10.1029/2008GB003240
Mahowald N, Engelstaedter S, Luo C, Sealy A, Artaxo P, Benitez-Nelson C, Bonnet S, Chen Y, Chuang PY, Cohen DD, Dulac F, Herut B, Johansen AM, Kubilay N, Losno R, Maenhaut W, Paytan A, Prospero JM, Shank LM, Siefert RL (2009) Atmospheric Iron deposition: global distribution, variability and human perturbations. Ann Rev Mar Sci 1:245–278
Mahowald NM, Kloster S, Engelstaedter S, Moore JK, Mukhopadhyay S, McConnell JR, Albani S, Doney SC, Bhattacharya A, Curran MAJ, Flanner MG, Hoffman FM, Lawrence DM, Lindsay K, Mayewski PA, Neff J, Rothenberg D, Thomas E, Thornton PE, Zender CS (2010) Observed 20th century desert dust variability: impact on climate and biogeochemistry. Atmos Chem Phys 10:10875–10893
Mahowald NM, Lindsay K, Rothenberg D, Doney SC, Moore JK, Thornton P, Randerson JT, Jones CD (2011) Desert dust and anthropogenic aerosol interactions in the Community Climate System Model coupled-carbon-climate model. Biogeosciences 8:387–414
Maier-Reimer E (1993) Geochemical cycles in an ocean general circulation model. Preindustrial tracer distributions. Global Biogeochem Cycles 7(3):645–677. doi:10.1029/93GB01355
Mäkelä JM, Hoffmann T, Holzke C, Väkevä M, Suni T, Mattila T, Aalto PP, Tapper U, Kauppinen EI, O’Dowd CD (2002) Biogenic iodine emissions and identification of end-products in coastal ultrafine particles during nucleation bursts. J Geophys Res 107, doi:10.1029/2001JD000580
Makkonen R, Asmi A, Kerminen V-M, Boy M, Arneth A, Hari P, Kulmala M (2012) Air pollution control and decreasing new particle formation lead to strong climate warming. Atmos Chem Phys 12:1515–1524. doi:10.5194/acp-12-1515-2012
Mandalakis M, Stephanou EG (2002) Study of atmospheric PCB concentrations over the eastern Mediterranean Sea. J Geophys Res 107:4716. doi:10.1029/2001JD001566
Mandavilli A (2006) Health agency backs use of DDT against malaria. Nature 443(7109):250–251
Marañón E et al (2010) Degree of oligotrophy controls the response of microbial plankton to Saharan dust. Limnol Oceanogr 55:2339–2352
Maring H, Savoie DL, Izaguirre MA, Custals L, Reid JS (2003) Mineral dust aerosol size distribution change during atmospheric transport. J Geophys Res 108:8592. doi:10.1029/2002JD002536
Markaki Z, Loÿe- Pilot M-D, Violaki K, Mihalopoulos N (2010) Variability of atmospheric deposition of dissolved nitrogen and phosphorus in the Mediterranean and possible link to the anomalous seawater N/P ratio. Mar Chem 120:187–194
Mårtensson EM, Nilsson ED, de Leeuw G, Cohen LH, Hansson H-C (2003) Laboratory simulations and parameterization of the primary marine aerosol production. J Geophys Res 108:4297. doi:10.1029/2002JD002263
Marticorena B, Bergametti G, Aumont B, Callot Y, N’Doumé C, Legrand M (1997) Modeling the atmospheric dust cycle 2 Simulation of Saharan dust sources. J Geophys Res 102:4387–4404
Martin JH (1990) Glacial-interglacial CO2 change: the iron hypothesis. Paleoceanography 5:1–13
Martin JH, Fitzwater SE (1988) Iron deficiency limits phytoplancton growth in the North-East Pacific subarctic. Nature 331:341–343
Martin J, Gordon RM, Fitzwater SE (1991) The case for iron. Limnol Oceanogr 36:1793–1802
Maskey S, Geng H, Song YC, Hwang H, Yoon YJ, Ahn KH, Ro CU (2011) Single-particle characterization of summertime Antarctic aerosols collected at King George Island using quantitative energy-dispersive electron probe X-ray microanalysis and attenuated total reflection Fourier transform-infrared imaging techniques. Environ Sci Technol 45:6275–6282
Mason BJ (1957) The physics of clouds. Clarendon Press, Oxford, 671
Massana R, Pedrós-Alió C, Casamayor EO, Gasol JM (2001) Changes in marine bacterioplankton phylogenetic composition during incubations designed to measure biogeochemically significant parameters. Limnol Oceanogr 46:1181–1188
Mastin LG, Guffanti M, Servranckx R, Webley P, Barsotti S, Dean K, Durant A, Ewert JW, Neri A, Rose WI, Schneider D, Siebert L, Stunder B, Swanson G, Tupper A, Volentik A, Waythomas CF (2009) A multidisciplinary effort to assign realistic source parameters to models of volcanic ash-cloud transport and dispersion during eruptions. J Volcanol Geotherm Res 186:10–21
Matsuki A, Iwasaka Y, Shi G, Zhang D, Trochkine D, Yamada M, Kim Y-S, Chen B, Nagatani T, Miyazawa T, Nagatani M, Nakata H (2005) Morphological and chemical modification of mineral dust: observational insight into the heterogeneous uptake of acidic gases. Geophys Res Lett 32, L22806. doi:10.1029/2005gl024176
Matsuki A, Quennehen B, Schwarzenboeck A, Crumeyrolle S, Venzac H, Laj P, Gomes L (2010a) Temporal and vertical variations of aerosol physical and chemical properties over West Africa: AMMA aircraft campaign in summer 2006. Atmos Chem Phys 10:8437–8451
Matsuki A, Schwarzenboeck A, Venzac H, Laj P, Crumeyrolle S, Gomes L (2010b) Cloud processing of mineral dust: direct comparison of cloud residual and clear sky particles during AMMA aircraft campaign in summer 2006. Atmos Chem Phys 10:1057–1069
Matsumoto K, Uyama Y, Hayano T, Uematsu M (2004) Transport and chemical transformation of anthropogenic and mineral aerosol in the marine boundary layer over the western North Pacific Ocean. J Geophys Res 109, D21206. doi:10.1029/2004jd004696
Maxwell-Meier K, Weber R, Song C, Orsini D, Ma Y, Carmichael GR, Streets DG, 2004 (2004) Inorganic composition of fine particles in mixed mineral dust– pollution plumes observed from airborne measurements during ACE-Asia. J Geophys Res 109, D19S07. doi:10.1029/2003jd004464
McFiggans G, Coe H, Burgess R et al (2004) Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine. Atmos Chem Phys 4:701–713
McKendry IG, Macdonald AM, Leaitch WR, van Donkelaar A, Zhang Q, Duck T, Martin RV (2008) Trans-Pacific dust events observed at Whistler, British Columbia during INTEX-B. Atmos Chem Phys 8:6297–6307
Meskhidze N, Chameides W, Nenes A (2005) Dust and pollution: a recipe for enhanced ocean fertiliza-tion? J Geophys Res 110, doi:10.1029/2004JD005082
Meskhidze NJX, Xu J, Gantt B, Zhang Y, Nenes A, Ghan SJ, Liu X, Easter R, Zaveri R (2011) Global distribution and climate forcing of marine organic aerosol: 1. Model improvements and evaluation. Atmos Chem Phys 11:11689–11705
Middlebrook AM, Murphy DM, Thomson DS (1998) Observation of organic material in individual particles at Cape Grim during the First Aerosol Characterization Experiment (ACE 1). J Geophys Res 103:16475–16483
Mie G (1908) Beitrge zur Optik trber Medien, speziell kolloidaler Metallsungen. Ann Phys Leipsig 25:377–445
Migon C, Sandroni V (1999) Phosphorus in rainwater: partitioning, inputs and impact on the surface coastal ocean. Limnol Oceanogr 44:1160–1165
Mills MM, Ridame C, Davey M, La Roche J, Geider RJ (2004) Iron and phosphorus co-limit nitrogen fixation in the eastern tropical North Atlantic. Nature 429:292–294
Mills MM et al (2008) Nitrogen and phosphorus co-limitation of bacterial productivity and growth in the oligotrophic subtropical North Atlantic. Limnol Oceanogr 53:824–834
Mishchenko MI, Lacis AA, Carlson BE, Travis LD (1995) Nonsphericity of dust-like tropospheric aerosols: Implications for aerosol remote sensing and climate modelling. Geophys Res Lett 22:1077–1080
Mochida M, Kitamori Y, Kawamura K, Nojiri Y, Suzuki K (2002) Fatty acids in the marine atmosphere: factors governing their concentrations and evaluation of organic films on sea salt particles. J Geophys Res 107:4325. doi:10.1029/2001JD001278
Mochida M, Umemoto N, Kawamura K, Lim HJ, Turpin BJ (2007) Bimodal size distributions of various organic acids and fatty acids in the marine atmosphere: influence of anthropogenic aerosols, Asian dusts, and sea spray off the coast of East Asia. J Geophys Res Atmos 112(13), D15209. doi:10.1029/2006jd007773
Monahan EC, O’Muircheartaigh IG (1986) Whitecaps and the passive remote sensing of the ocean surface. Int J Remote Sens 7:627–642
Monahan EC, Spiel DE, Davidson KL (1986) A model of marine aerosol generation via whitecaps and wave disruption. In: Monahan EC, MacNiochaill G (eds) Oceanic whitecaps. D. Reidel, Norwell, pp 167–193
Moore JK, Braucher O (2008) Sedimentary and mineral dust sources of dissolved iron to the world ocean. Biogeosciences 5:631–656
Moore JK, Doney S, Lindsay K, Mahowald N, Michaels A (2006) Nitrogen fixation amplifies the ocean biogeochemical response to decadal timesclae variations in mineral dust deposition. Tellus 58B:560–572
Moore CM et al (2008) Relative influence of nitrogen and phosphorus availability on phytoplankton physiology and productivity in the oligotrophic sub-tropical North Atlantic Ocean. Limnol Oceanogr 53:291–305
Moore CM, Mills MM, Arrigo KR, Berman-Frank I, Bopp L, Boyd PW, Galbraith ED, Geider RJ, Guieu C, Jaccard SL, Jickells TD, La Roche J, Lenton TM, Mahowald NM, Marañón E, Marinov I, Moore JK, Nakatsuka T, Oschlies A, Saito MA, Thingstad TF, Tsuda A, Ulloa O (2013) Processes and patterns of oceanic nutrient limitation. Nat Geosci. doi:10.1038/ngeo1765
Morgan WT, Allan JD, Bower KN, Highwood EJ, Liu D, McMeeking GR, Northway MJ, Williams PI, Krejci R, Coe H (2010) Airborne measurements of the spatial distribution of aerosol chemical composition across Europe and evolution of the organic fraction. Atmos Chem Phys 10:4065–4083
Mori I, Nishikawa M, Iwasaka Y (1998) Chemical reaction during the coagulation of ammonium sulphate and mineral particles in the atmosphere. Sci Total Environ 224:87–91
Morris RM et al (2002) SAR11 clade dominates ocean surface bacterioplankton communities. Nature 420:806–810
Moulin C, Lambert CE, Dayan U, Dulac F (1997) Control of atmospheric export of dust from North Africa by the North Atlantic Oscillation. Nature 387:691–694
Moulin C, Gordon HR, Banzon VF, Evans RH (2001) Assessment of Saharan dust absorption in the visible from SeaWiFs imagery. J Geophys Res 106:18239–18249
Mouri H, Okada K (1993) Shattering and modification of sea-salt particles in the marine atmosphere. Geophys Res Lett 20:49–52
Müller C, Iinuma Y, Karstensen J et al (2009) Seasonal variation of aliphatic amines in marine sub-micrometer particles at the Cape Verde Islands. Atmos Chem Phys 9:9587–9597
Murphy DM, Cziczo DJ, Froyd KD, Hudson PK, Matthew BM, Middlebrook AM, Peltier RE, Sullivan A, Thomson DS, Weber RJ (2006) Single-particle mass spectrometry of tropospheric aerosol particles. J Geophys Res 111, D23S32. doi:10.1029/2006jd007340
Murphy SM, Sorooshian A, Kroll JH et al (2007) Secondary aerosol formation from atmospheric reactions of aliphatic amines. Atmos Chem Phys 7:2313–2337
Myriokefalitakis S, Vignati E, Tsigaridis K, Papadimas C, Sciare J, Mihalopoulos N, Facchini MC, Rinaldi M, Dentener FJ, Ceburnis D, Hatzianastassiou N, O’Dowd CD, van Weele M, Kanakidou M (2010) Global modelling of the oceanic source of organic aerosols. Adv Meteorol 2010:939171. doi:10.1155/2010/939171
Nenes A, Krom M, Mihalopoulos N, Van Cappellen P, Shi Z, Bougiatioti A, Zarmpas P, Herubt B (2011) Atmospheric acidification of mineral aerosols: a source of bioavailable phosphorus for the oceans. Atmos Chem Phys 11:6265–6272
Newhall CG, Self S (1982) The volcanic explosivity index (VEI): an estimate of explosive magnitude for historical volcanism. J Geophys Res 87:1231–1238
Ng NL, Canagaratna MR, Zhang Q et al (2010) Organic aerosol components observed in Northern Hemispheric datasets from aerosol mass spectrometry. Atmos Chem Phys 10:4625–4641. doi:10.5194/acp-10-4625-2010
Ng NL, Canagaratna MR, Jimenez JL, Chhabra PS, Seinfeld JH, Worsnop DR (2011) Changes in organic aerosol composition with aging inferred from aerosol mass spectra. Atmos Chem Phys 11:6465–6474
Niemi JV, Tervahattu H, Virkkula A, Hillamo R, Teinilä K, Koponen IK, Kulmala M (2005) Continental impact on marine boundary layer coarse particles over the Atlantic Ocean between Europe and Antarctica. Atmos Res 75:301–321
Niimura N, Okada K, Fan X-B, Kenji K, Kimio A, Shi G-Y, Takahashi S (1998) Formation of Asian dust-storm particles mixed internally with sea salt in the atmosphere. J Meteorol Soc Japan 76:275–288
Niinemets U, Tenhunen JD, Harley PC, Steinbrecher R (1999) A model of isoprene emission based on energetic requirements for isoprene synthesis and leaf photosynthetic properties for Liquidambar and Quercus. Plant Cell Environ 22:1319–1335
Niinemets Ü, Arneth A, Kuhn U, Monson RK, Peñuelas J, Staudt M (2010a) The emission factor of volatile isoprenoids: stress, acclimation, and developmental responses. Biogeosciences 7:2203–2223
Niinemets Ü, Monson RK, Arneth A, Ciccioli P, Kesselmeier J, Kuhn U, Noe SM, Penuelas J, Staudt M (2010b) The emission factor of volatile isoprenoids: caveats, model algorithms, response shapes and scaling. Biogeosciences 7:1809–1832
Niinemets Ü, Kuhn U, Harley PC, Staudt M, Arneth A, Cescatti A, Ciccioli P, Copolovici L, Geron C, Guenther A, Kesselmeier J, Lerdau MT, Monson RK, Peñuelas J (2011) Estimations of isoprenoid emission capacity from enclosure studies: measurements, data processing, quality and standardized measurement protocols. Biogeosciences 8:2209–2246
Nilsson ED, Mårtensson EM, Van Ekeren JS, de Leeuw G, Moerman M, O'Dowd C (2007) Primary marine aerosol emissions: size resolved eddy covariance measurements with estimates of the sea salt and organic carbon fractions. Atmos Chem Phys Discuss 7:13345–13400. doi:10.5194/acpd-7-13345-2007
Norris S, Brooks I, de Leeuw G, Smith MH, Moerman M, Lingard J (2008) Eddy covariance measurements of sea spray particles over the Atlantic Ocean. Atmos Chem Phys 8:555–563
Norris SJ, Brooks IM, Hill MK, Brooks BJ, Smith MH, Sproson DAJ (2012) Eddy covariance measurements of the sea spray aerosol flux over the open ocean. J Geophys Res 117, D07210. doi:10.1029/2011JD016549
Nozaki Y (1997) A fresh look at element distribution in the North Pacific. EOS, Am Geophys Union 78(21):221
O’Dowd CD, Smith MH (1993) Physicochemical properties of aerosols over the northeast Atlantic: evidence for wind-speed-related submicron sea-salt aerosol production. J Geophys Res 98, doi:10.1029/92JD02302
O’Dowd CD, Smith MH, Jennings SG (1993) Submicron aerosol, radon and soot carbon characteristics over the northeast Atlantic. J Geophys Res 98:1123–1135
O’Dowd CD, Geever M, Hill MK, Jennings SG, Smith MH (1998) New particle formation: spatial scales and nucleation rates in the coastal environment. Geophys Res Lett 25:1661–1664
O’Dowd C, Lowe JA, Smith MH (1999a) Observations and modelling of aerosol growth in marine stratocumulus – case study. Atmos Environ 33:3053–3062
O’Dowd CD, McFiggens G, Pirjola L, Creasey DJ, Hoell C, Smith MH, Allen B, Plane JMC, Heard DE, Lee JD, Pilling MJ, Kulmala M (1999b) On the photochemical production of new particles in the coastal boundary layer. Geophys Res Lett 26:1707–1710
O’Dowd CD, Lowe JA, Clegg N, Smith MH, Clegg SL (2000) Modeling heterogeneous sulphate production in maritime stratiform clouds. J Geophys Res-Atmos 105(D6):7143–7160
O’Dowd CD, Hämeri K, Mäkelä JM, Pirjola L, Kulmala M, Jennings SG, Berresheim H, Hansson H-C, de Leeuw G, Allen AG, Hewitt CN, Jackson A, Viisanen Y, Hoffmann T (2002) A dedicated study of new particle formation and fate in the coastal environment (PARFORCE): overview of objectives and initial achievements. J Geophys Res 107, doi:10.1029/2001000555
O’Dowd CD, Facchini MC, Cavalli F, Ceburnis D, Mircea M, Decesari S, Fuzzi S, Yoon YJ, Putaud J-P (2004) Biogenically driven organic contribution to marine aerosol. Nature 431:676–680
O’Dowd CD, Langmann B, Varghese S, Scannell C, Ceburnis D, Facchini MC (2008) A combined organic–inorganic sea-spray source function. Geophys Res Letts 35, L01801. doi:10.1029/2007GL030331
O’Dowd CD, Monahan C, Dall’Osto M (2010) On the occurrence of open ocean particle production and growth events. Geophys Res Lett 37, L19805. doi:10.1029/2010GL044679
O’Dowd CD, de Leeuw G (2007) Marine aerosol production: a review of the current knowledge. Philos Trans Royal Soc A: Math Phys Eng Sci 365:1753–1774
O’Dowd CD, Davison B, Lowe JA, Smith MH, Harrison RM, Hewitt CN (1997) Biogenic sulphur emissions and inferred sulphate CCN concentrations in and around Antarctica. J Geophys Res 102:12839–12854
Okada K, Kai K (2004) Atmospheric mineral particles collected at Qira in the Taklamakan Desert, China. Atmos Environ 38:6927–6935
Okada K, Naruse H, Tanaka T, Nemoto O, Iwasaka Y, Wu P-M, Ono A, Duce RA, Uematsu M, Merrill JT, Arao K (1990) X-ray spectrometry of individual Asian dust-storm particles over the Japanese islands and the North Pacific Ocean. Atmos Environ 24:1369–1378
Okada K, Qin Y, Kai K (2005) Elemental composition and mixing properties of atmospheric mineral particles collected in Hohhot, China. Atmos Res 73:45–67
Okin G, Baker A, Tegen I, Mahowald N, Dentener F, Duce R, Galloway J, Hunter K, Kanakidou M, Kubilay N, Prospero J, Sarin M, Surpipith V, Uematsu M, Zhu T (2011) Impacts of atmospheric nutrient deposition on marine productivity: roles of nitrogen, phosphorus and iron. Global Biogeochem Cycles 25, doi:10.1029/2010GB003858
Olgun N, Duggen S, Croot PL, Delmelle P, Dietze H, Schacht U, Óskarsson N, Siebe C, Auer A, Garbe-Schönberg D (2011) Surface ocean iron fertilization: the role of airborne volcanic ash from subduction zone and hot spot volcanoes and related iron fluxes into the Pacific Ocean. Global Biogeochem Cycles 25, GB4001. doi:10.1029/2009GB003761
Omar AH, Winker DM, Vaughan MA, Hu Y, Trepte CR, Ferrare RA, Lee KP, Hostetler CA, Kittaka C, Rogers RR, Kuehn RE, Liu Z (2009) The CALIPSO automated aerosol classification and lidar ratio aelection algorithm. J Atmos Ocean Technol 26(10):1994–2014
Ooki A, Uematsu M (2005) Chemical interactions between mineral dust particles and acid gases during Asian dust events. J Geophys Res 110, D03201. doi:10.1029/2004jd004737
Oppo C, Bellandi S, Degli Innocenti N, Stortini AM, Loglio G, Schiavuta E, Cini R (1999) Surfactant component of marine organic matter as agents for biogeochemical fractionation of pollutants transport via marine aerosol. Mar Chem 63:235–253
Ovadnevaite J, Ceburnis D, Bialek J, Monahan C, Martucci G, Rinaldi M, Facchini MC, Berresheim H, Worsnop DR, O’Dowd C (2011a) Primary marine organic aerosol: a dichotomy of low hygroscopicity and high CCN activity. Geophys Res Lett 38, L21806. doi:10.1029/2011GL048869
Ovadnevaite J, O’Dowd C, Dall’Osto M, Ceburnis D, Worsnop DR, Berresheim H (2011) Detecting high contributions of primary organic matter to marine aerosol: a case study. Geophys Res Lett 38, L02807. doi:10.1029/2010GL046083
Pacifico F, Harrison SP, Jones CD, Arneth A, Sitch S, Weedon GP, Barkley MP, Palmer PI, Serça D, Potosnak M, Fu TM, Goldstein A, Bai J, Schurgers G (2011) Evaluation of a photosynthesis-based biogenic isoprene emission scheme in JULES and simulation of isoprene emissions under modern climate conditions. Atmos Chem Phys 11:4371–4389
Palmer PI, Jacob DJ, Fiore AM, Martin RV, Chance K, Kurosu TP (2003) Mapping isoprene emissions over North America using formaldehyde column observations from space. J Geophys Res 108(D6):4180. doi:10.1029/2002JD002153
Pandis SN, Russell LM, Seinfeld JH (1994) The relationship between DMS flux and CCN concentration in remote marine regions. J Geophys Res 99:16945–16957
Parekh P, Follows MJ, Boyle E (2004) Modeling the global ocean iron cycle. Global Biogeochem Cycles 18, GB1002. doi:10.1029/2003GB002061
Parekh P, Follows MJ, Boyle EA (2005) Decoupling of iron and phosphate in the global ocean. Global Biogeochem Cycles 19, GB2020. doi:10.1029/2004GB002280
Passow U, De la Rocha C (2006) Accumulation of mineral ballast on organic aggregates. Global Biogeochem Cycles 20, GB1013. doi:10.1029/2005GB002579
Paytan A, Mackey KRM, Chen Y, Limac ID, Doneyc SC, Mahowaldd N, Labiosae R, Postf AF (2009) Toxicity of atmospheric aerosols on marine phytoplankton. Proc Natl Acad Sci 106:4601–4605
Petit JR, Jouzel J, Raynaud D, Barkov NI, Barnola J-M, Basile I, Benders M, Chappellaz J, Davis M, Delayque G, Delmotte M, Kotlyakov VM, Legrand M, Lipenkov VY, Lorius C, Pépin L, Ritz C, Saltzman E, Stievenard M (1999) Climate and atmospheric history of the past 420,000 years from the Vostok ice core, Antarctica. Nature 399:429–436
Pirjola L, O’Dowd CD, Brooks IM, Kulmala M (2000) Can new particle formation occur in the clean marine boundary layer? J Geophys Res 105:26531–26546
Ploug H, Hvitfeld Iversen M, Fischer G (2008) Ballast, sinking velocity, and apparent diffusivity within marine snow and zooplankton fecal pellets: implications for substrate turnover by attached bacteria. Limnol Oceanogr 53:1878–1886
Polymenakou PN, Mandalakis M, Stephanou EG, Tselepides A (2008) Particle size distribution of airborne microorganisms and pathogens during an intense African dust event in the Eastern Mediterranean. Environ Heal Perspect 116(3):292–296
Pöschl U (2005) Atmospheric aerosols: composition, transformation, climate and health effects. Angew Chem Int Ed 44:7520–7540. doi:10.1002/anie.200501122
Pressley S, Lamb B, Westberg H, Flaherty J, Chen J, Vogel C (2005) Long-term isoprene flux measurements above a northern hardwood forest. J Geophys Res 110, D07301. doi:10.1029/2004JD005523
Prospero JM, Ginoux P, Torres O, Nicholson SE, Gill TE (2002) Environmental characterization of global sources of atmospheric soil dust identified with the Nimbus 7 Total Ozone Mapping Spectrometer (TOMS) absorbing aerosol product. Rev Geophys 40, doi:10.1029/2000RG000095
Pulido-Villena E, Wagener T, Guieu C (2008) Bacterial response to dust pulses in the western Mediterranean: implications for carbon cycling in the oligotrophic ocean. Global Biogeochem Cycles 22, GB1020. doi:10.1029/2007GB003091
Pulido-Villena E, Rerolle V, Guieu C (2010) Transient fertilizing effect of dust in P-deficient LNLC surface ocean. Geophys Res Lett 37, L01603. doi:10.1029/2009GL041415
Putaud J-P, Van Dingenen R, Dell’Acqua A, Raes F, Matta E, Decesari S, Facchini MC, Fuzzi S (2004) Size-segregated aerosol mass closure and chemical composition in Monte Cimone (I) during MINATROC. Atmos Chem Phys 4:889–902. doi:10.5194/acp-4-889-2004
Quinn PK, Bates TS (2005) Regional aerosol properties: comparisons of boundary layer measurements from ACE 1, ACE 2, Aerosols99, INDOEX, ACE Asia, TARFOX, and NEAQS. J Geophys Res 110, D14202. doi:10.1029/2004jd004755
Raes F (1995) Entrainment of free tropospheric aerosols as a regulating mechanism for cloud condensation nuclei in the remote marine boundary layer. J Geophys Res 100:2893–2903
Randles CA, Russell LM, Ramaswamy V (2004) Hygroscopic and optical properties of organic sea salt aerosol and consequences for climate forcing. Geophys Res Lett 31, doi:10.1029/2004GL020628
Rea DK (1994) The paleoclimatic record provided by eolian deposition in the deep sea: the geologic history of wind. Rev Geophys 32:159–195
Reddy MS, Boucher O, Balkanski Y, Schulz M (2005) Aerosol optical depths and direct radiative perturbations by species and source type. Geophys Res Lett 32, doi:10.1029/2004GL021743
Reid JS, Jonsson HH, Maring HB, Smirnov A, Savoie DL, Cliff SS, Reid EA, Livingston JM, Meier MM, Dubovik O, Tsay S-C (2003a) Comparison of size and morphological measurements of coarse mode dust particles from Africa. J Geophys Res 108:8593. doi:10.1029/2002JD002485
Reid EA, Reid JS, Meier MM, Dunlap MR, Cliff SS, Broumas A, Perry K, Maring H (2003b) Characterization of African dust transported to Puerto Rico by individual particle and size segregated bulk analysis. J Geophys Res 108, doi:10.1029/2002JD002935
Remer LA, Kleidman RG, Levy RC et al (2008) Global aerosol climatology from the MODIS satellite sensors. J Geophys Res 113, doi:10.1029/2007JD009661
Ridame C, Guieu C (2002) Saharan input of phosphorus to the oligotrophic water of the open western Mediterranean. Limnol Oceanogr 47:856–869
Ridame C, Moutin T, Guieu C (2003) Does the absortion process of phosphate onto Saharan dust explain the unusual N/P ratio in the Mediterranean sea? Oceanol Acta 26:629–634
Ridame C, Le Moal M, Guieu C, Ternon E, Biegala I, L’Helguen S, Pujo-Pay M (2011) Nutrient control of N2 fixation in the oligotrophic Mediterranean Sea and the impact of Saharan dust events. Biogeosciences 8:2629–2657
Ridame C et al (2012) Strong stimulation of N2 fixation to Saharan dust events: results from dust fertilizations in large mesocosms (in preparation)
Ridame C, Guieu C, L’Helguen S (2013) Strong stimulation of N2 fixation in oligotrophic Mediterranean Sea: results from dust addition in large in situ mesocosms. Biogeosciences Discuss 10:10581–10613
Ridgwell AJ, Watson A (2002) Feedback between aeolian dust, climate and atmospheric CO2 in glacial time. Paleoceanography 17, doi:10.1029/2001PA000729
Riebesell U, Schulz KG, Bellerby RGJ, Botros M, Fritsche P, Meyerhöfer M, Neill C, Nondal G, Oschlies A, Wohlers J, Zöllner E (2007) Enhanced biological carbon consumption in a high CO2 ocean. Nature 450:545–548
Rijkenberg MJA, Powell C, Dall’Osto M, Nielsdottir M, Patey M, Hill P, Baker AR, Jickells T, Harrison R, Achterberg E (2008) Changes in iron speciation following a Saharan dust event in the tropical North Atlantic Ocean. Mar Chem 110:56–67
Rinaldi M, Decesari S, Finessi E, Giulianelli L, Carbone C, Fuzzi S, Dowd CD, Ceburnis D, Facchini MC (2010) Primary and secondary organic marine aerosol and oceanic biological activity: recent results and new perspectives for future studies. Adv Meteorol. doi:10.1155/2010/310682
Rinaldi M, Decesari S, Carbone C, Finessi E, Fuzzi S, Ceburnis D, O’Dowd CD, Sciare J, Burrows JP, Vrekoussis M, Ervens B, Tsigaridis K, Facchini MC (2011) Evidence of a natural marine source of oxalic acid and a possible link to glyoxal. J Geophys Res 116, D16204. doi:10.1029/2011JD015659
Rinaldi M, Fuzzi S, Decesari S, Marullo S, Santoleri R, Provenzale A, von Hardenberg J, Ceburnis D, Vaishya A, O’Dowd CD, Facchini M (2013) Is chlorophyll-a the best surrogate for organic matter enrichment in submicron primary marine aerosol? J Geophys Res Atmos 118:1–10. doi:10.1002/jgrd.50417
Rinne J, Back J, Hakola H (2009) Biogenic volatile organic compound emissions from the Eurasian tai-ga: current knowledge and future directions. Boreal Environ Res 14:807–826
Rivkin RB, Anderson MR (1997) Inorganic nutrient limitation of oceanic bacterioplankton. Limnol Oceanogr 42:730–740
Ro C-U, Hwang H, Kim H, Chun Y, Van Grieken R (2005) Single-particle characterization of four Asian dust samples collected in Korea, using low-Z particle electron probe X-ray microanalysis. Environ Sci Technol 39:1409–1419
Robinson AL, Donahue NM, Shrivastava MK, Weitkamp EA, Sage AM, Grieshop AP, Lane TE, Pierce JR, Pandis SN (2007) Rethinking organic aerosols: semivolatile emissions and photochemical aging. Science 315:1259–1262
Robock A (2000) Volcanic eruptions and climate. Rev Geophys 38:191–219
Russell LM, Pandis SN, Seinfeld JH (1994) Aerosol production and growth in the marine boundary layer. J Geophys Res 9:20989–21003
Russell LM, Hawkins LN, Frossard AA, Quinn PK, Bates TS (2010) Carbohydrate-like composition of submicron atmospheric particles and their production from ocean bubble bursting. PNAS 107(15):6652–6657. doi:10.1073/pnas.0908905107
Sage AM, Weitkamp EA, Robinson AL, Donahue NM (2008) Evolving mass spectra of the oxidized component of organic aerosol: results from aerosol mass spectrometer analyses of aged diesel emissions. Atmos Chem Phys 8:1139–1152
Saito MA, Moffett JW (2001) Complexation of cobalt by natural organic ligands in the Sargasso Sea as determined by a new high-sensitivity electrochemical cobalt speciation method suitable for open ocean work. Mar Chem 75:49–68
Saiz-Lopez A, Plane JMC, McFiggans G, Williams PI, Ball SM, Bitter M, Jones RL, Hongwei C, Hoffmann T (2005) Modelling molecular iodine emissions in the coastal marine environment: the link to new particle formation. Atmos Chem Phys 5:5405–5439
Sarmiento JL, Le Quéré C (1996) Oceanic carbon dioxide uptake in a model of century-scale global warming. Science 274:1346–1350
Sassen K (2000) Lidar backscatter depolarization technique for cloud and aerosol research. In: Mishchenko ML (ed) Light scattering by nonspherical particles: theory, measurements, and geophysical applications. Academic, San Diego, pp 393–416
Saunders RW, Plane JMC (2005) Formation pathways and composition of iodine oxide ultra-fine particles. Environ Chem 2:199–303. doi:10.1071/EN05079
Schlesinger P, Mamane Y, Grishkan I (2006) Transport of microorganisms to Israel during Saharan dust events. Aerobiologia 22:259–273
Schmincke H-U (2004) Volcanism. Springer, Berlin/Heidelberg/New York, p 324. ISBN 3-540-43650-2
Schultz MG, Heil A, Hoelzemann JJ, Spessa A, Thonicke K, Goldammer J, Held AC, Pereira JM (2008) Global emissions from wildland fires from 1960 to 2000. Global Biogeochem Cycles 22, GB2002. doi:10.1029/2007GB003031
Schuster GL, Dubovik O, Holben BN, Clothiaux EE (2005) Inferring black carbon content and specific absorption from Aerosol Robotic Network (AERONET) aerosol retrievals. J Geophys Res 110, doi:10.1029/2004JD004548
Sciare J, Favez O, Oikonomou K, Sarda-Estève R, Cachier H, Kazan V (2009) Long-term observation of carbonaceous aerosols in the Austral Ocean: evidence of a marine biogenic origin. J Geophys Res 114, D15302. doi:10.1029/2009JD011998
Sedwick P, Sholkovitz E, Church T (2007) Impact of anthropogenic combustion emissions on the frac-tional solubility of aerosol iron: evidence from the Sargasso Sea. Geochem Geophys Geosyst 8, doi:10.1029/2007GC001586
Seinfeld JH, Pandis SN (2006) Atmospheric chemistry and physics: from air pollution to climate change, 2nd edn. Wiley, New York
Seitzinger S, Harrison J, Dumont E, Beusen A, Bouwman A (2005) Sources and delivery of carbon, nitrogen and phosphorus to the coastal zone: an overview of the Global Nutrient Export from Watersheds (NEWS) models and their application. Global Biogeochem Cycles 19, doi:10.1029/2005GB002606
Seitzinger SP, Mayorga E, Bouwman AF, Kroeze C, Beusen AHW, Billen G, Van Drecht G, Dumont E, Fekete BM, Garnier J, Harrison JA (2010) Global river nutrient export: a scenario analysis of past and future trends. Global Biogeochem Cycles 24, doi:10.1029/2009GB003587
Sellegri K, Yoon YJ, Jennings SG, Pirjola L, Cautenet S, O’Dowd CD (2005) Quantification of coastal new ultra-fine particles formation from in-situ and chamber measurements during the BIOFLUX campaign. Environ Chem 2:260–270
Sellegri K, O’Dowd CD, Yoon YJ, Jennings SG, de Leeuw G (2006) Surfactants and submicron sea spray generation. J Geophys Res 111, D22215. doi:10.1029/2005JD006658
Shaw G (1983) Bio-controlled thermostasis involving the sulfur cycle. Clim Chang 5:297–303
Shi Z, Zhang D, Hayashi M, Ogata H, Ji H, Fujiie W (2008) Influences of sulfate and nitrate on the hygroscopic behaviour of coarse dust particles. Atmos Environ 42:822–827
Sievering H et al (1992) Removal of sulphuer from the marine boundary layer by ozone oxidation in sea-salt aerosols. Nature 360:571–573
Simkin T, Siebert L (1994) Volcanoes of the world, 2nd edn. Geoscience Press, Tucson, p 349
Singh HB, Kasting JF (1988) Chlorine-hydrocarbon photochemistry in the marine troposphere and lower stratosphere. J Atmos Chem 7:261–285
Slingo A (1990) Sensitivity of the Earth’s radiation budget to changes in low clouds. Nature 343:49–51
Smetacek V, Klaas C, Strass VH, Assmy P, Montresor M, Cisewski B, Savoye N, Webb A, d’Ovidio F, Arrieta JM, Bathmann U, Bellerby R, Mine Berg G, Croot P, Gonzalez S, Henjes J, Herndl GJ, Hoffmann LJ, Leach H, Losch M, Mills MM, Neill C, Peeken I, Röttgers R, Sachs O, Sauter E, Schmidt MM, Schwarz J, Terbrüggen A, Wolf-Gladrow D (2012) Deep carbon export from a Southern Ocean iron-fertilized diatom bloom. Nature 487:313–319
Smith SJ, van Aardenne J, Klimont Z, Andres RJ, Volke A, Delgado Arias S (2011) Anthropogenic sulfur dioxide emissions: 1850–2005. Atmos Chem Phys 11:1101–1116
Smirnov A, Holben BN, Giles DM, Slutsker I, O’Neill NT, Eck TF, Macke A, Croot P, Courcoux Y, Sakerin SM, Smyth TJ, Zielinski T, Zibordi G, Goes JI, Harvey MJ, Quinn PK, Nelson NB, Radionov VF, Duarte CM, Losno R, Sciare J, Voss KJ, Kinne S, Nalli NR, Joseph E, Krishna Moorthy K, Covert DS, Gulev SK, Milinevsky G, Larouche P, Belanger S, Horne E, Chin M, Remer LA, Kahn RA, Reid JS, Schulz M, Heald CL, Zhang J, Lapina K, Kleidman RG, Griesfeller J, Gaitley BJ, Tan Q, Diehl TL (2011) Maritime aerosol network as a component of AERONET – first results and comparison with global aerosol models and satellite retrievals. Atmos Meas Tech 4:583–597. doi:10.5194/amt-4-583-2011
Smirnov A, Sayer AM, Holben BN, Hsu NC, Sakerin SM, Macke A, Nelson NB, Courcoux Y, Smyth TJ, Croot P, Quinn PK, Sciare J, Gulev SK, Piketh S, Losno R, Kinne S, Radionov VF (2012) Effect of wind speed on aerosol optical depth over remote oceans, based on data from the maritime aerosol network. Atmos Meas Tech 5:377–388, 10.5194/amt-5-377-2012
Sobanska S, Coeur C, Maenhaut W, Adams F (2003) SEM-DEX characterization of tropospheric aerosols in the Negev Desert (Israel). J Atmos Chem 44:299–322
Sofiev M, Siljamo P, Valkama I, Ilvonen M, Kukkonen J (2006) A dispersion modelling system SILAM and its evaluation against ETEX data. Atmos Environ 40:674–685
Sofiev M, Soares J, Prank M, de Leeuw G, Kukkonen J (2011) A regional-to-global model of emission and transport of sea salt particles in the atmosphere. J Geophys Res 116, D021302. doi:10.1029/2010JD014713
Song CH, Carmichael GR (1999) The aging process of naturally emitted aerosol (sea-salt and mineral aerosol) during long range transport. Atmos Environ 33:2203–2218
Sorooshian A, Lu M-L, Brechtel FJ, Jonsson H, Feingold G, Flagan RC, Seinfeld JH (2007) On the source of organic acid aerosol layers above clouds. Environ Sci Technol 41:4647–4654
Sorooshian A, Padrò LT, Nenes A et al (2009) On the link between ocean biota emissions, aerosol, and maritime clouds: airborne, ground, and satellite measurements off the coast of California. Global Biogeochem Cycles 23, GB4007
Stavrakou T, Müller JF, De Smedt I, Van Roozendael M, van der Werf GR, Giglio L, Guenther A (2009) Global emissions of non-methane hydrocarbons deduced from SCIAMACHY formaldehyde columns through 2003–2006. Atmos Chem Phys 9:3663–3679
Stier P, Seinfeld JH, Kinne S, Boucher O (2007) Aerosol absorption and radiative forcing. Atmos Chem Phys 7:5237–5261
Stone EA, Yoon SC, Schauer JJ (2011) Chemical characterization of fine and coarse particles in Gosan, Korea during springtime dust events. Aerosol Air Qual Res 11:31–43
Stramska M, Marks R, Monahan EC (1990) Bubble-mediated aerosol production as a consequence of wave breaking in supersaturated (hyperoxic) seawater. J Geophys Res 95(C10):18281–18288
Sturges WT, Shaw GE (1993) Halogens in aerosols in Central Alaska. Atmos Environ Part A Gen Top 27:2969–2977
Su J, Jianping H, Qiang F, Minnis P, Jinming G, Jianrong B (2008) Estimation of Asian dust aerosol effect on cloud radiation forcing using Fu-Liou radiative model and CERES measurements. Atmos Chem Phys 8:2763–2771
Sullivan RC, Prather KA (2007) Investigations of the diurnal cycle and mixing state of oxalic acid in individual particles in Asian aerosol outflow. Environ Sci Technol 41:8062–8069. doi:10.1021/es071134g
Sullivan RC, Guazzotti SA, Sodeman DA, Prather KA (2007) Direct observations of the atmospheric processing of Asian mineral dust. Atmos Chem Phys 7:1213–1236
Sullivan RC, Moore MJK, Petters MD, Kreidenweis SM, Roberts GC, Prather KA (2009) Effect of chemical mixing state on the hygroscopicity and cloud nucleation properties of calcium mineral dust particles. Atmos Chem Phys 9:3303–3316
Sun Y, Zhuang G, Wang Y, Zhao X, Li J, Wang Z, An Z (2005) Chemical composition of dust storms in Beijing and implications for the mixing of mineral aerosol with pollution aerosol on the pathway. J Geophys Res 110, D24209. doi:10.1029/2005jd006054
Surratt JD, Lewandowski M, Offenberg JH, Jaoui M, Kleindienst TE, Edney EO, Seinfeld JH (2007) Effect of acidity on secondary organic aerosol formation from isoprene. Environ Sci Technol 41:5363–5369
Suzuki I, Igarashi Y, Dokiya Y, Akagi T (2010) Two extreme types of mixing of dust with urban aerosols observed in Kosa particles: ‘after’ mixing and ‘on-the-way’ mixing. Atmos Environ 44:858–866
Tagliabue A, Bopp L, Aumont O (2008) Ocean biogeochemistry exhibits contrasting responses to a large scale reduction in dust deposition. Biogeosciences 5:11–24
Tagliabue A, Bopp L, Aumont O, Arrigo K (2009) Influence of light and temperature on the marine iron cycle: from theoretical to global modeling. Global Biogeochem Cycle 23, GB2017. doi:10.1029/2008GB003214
Tan PV, Evans GJ, Tsai J et al (2002) On-line analysis of urban particulate matter focusing on elevated wintertime aerosol concentrations. Environ Sci Technol 36:3512–3518
Tedetti M, Sempéré R (2006) Penetration of ultraviolet radiation in the marine environmen. A review. Photochem Photobiol 82:389–397
Tegen I, Werner M, Harrison SP, Kohfeld KE (2004) Relative importance of climate and land use in determining present and future global soil dust emission. Geophys Res Lett 31, L05105. doi:10.1029/2003GL019216
Ternon E, Guieu C, Loÿe-Pilot M-D, Leblond N, Bosc E, Gasser B, Martin J, Miquel J-C (2010) The impact of Saharan dust on the particulate export in the water column of the North Western Mediterranean Sea. Biogeosciences 7:809–826
Ternon E, Guieu C, Ridame C, L’Helguen S, Catala P (2011) Longitudinal variability of the biogeochemical role of Mediterranean aerosols in the Mediterranean Sea. Biogeosciences 8:1067–1080
Thingstad TF, Law CS, Krom MD, Mantoura RFC, Pitta P, Psarra S, Rassoulzadegan F, Tanaka T, Wassmann P, Wexels Riser C, Zohary T (2005) Nature of phosphorus limitation in the ultraoligotrophic Eastern Mediterranean. Science 309:1068–1071. doi:10.1126/science.1112632
Thorpe SA (1992) Bubble clouds and the dynamics of the upper ocean. Q J R Meteorol Soc 118:1–22
Tobo Y, Zhang DZ, Nakata N, Yamada M, Ogata H, Hara K, Iwasaka Y (2009) Hygroscopic mineral dust particles as influenced by chlorine chemistry in the marine atmosphere. Geophys Res Lett 36, L05817. doi:10.1029/2008gl036883
Tobo Y, Zhang D, Matsuki A, Iwasaka Y (2010) Asian dust particles converted into aqueous droplets under remote marine atmospheric conditions. Proc Natl Acad Sci 107:17905–17910. doi:10.1073/pnas.1008235107
Tomlinson JM, Li R, Collins DR (2007) Physical and chemical properties of the aerosol within the southeastern Pacific marine boundary layer. J Geophys Res 112, D12211. doi:10.1029/2006JD007771
Trochkine D, Iwasaka Y, Matsuki A, Yamada M, Kim YS, Nagatani T, Zhang D, Shi GY, Shen Z (2003) Mineral aerosol particles collected in Dunhuang, China, and their comparison with chemically modified particles collected over Japan. J Geophys Res 108, doi:10.1029/2002jd003268
Tursic J, Podkrajsek B, Grgic I, Ctyroky P, Berner A, Dusek U, Hitzenberger R (2006) Chemical composition and hygroscopic properties of size-segregated aerosol particles collected at the Adriatic coast of Slovenia. Chemosphere 63:1193–1202
Tyree CA, Hellion VM, Alexandrova OA, Allen JO (2007) Foam droplets generated from natural and artificial seawaters. J Geophys Res 112, D12204. doi:10.1029/2006JD007729
Uematsu M, Duce RA, Prospero JM (1985) Deposition of atmospheric mineral particles in the North Pacific Ocean. J Atmos Chem 3:123–138
Usher CR, Al-Hosney H, Carlos-Cuellar S, Grassian VH (2002) A laboratory study of the heterogeneous uptake and oxidation of sulfur dioxide on mineral dust particles. J Geophys Res 107:4713. doi:10.1029/2002jd002051
Usher CR, Michel AE, Grassian VH (2003) Reactions on mineral dust. Chem Rev 103:4883–4939
Van der Werf GR, Randerson JT, Giglio L, Collatz GJ, Kasibhatla PS, Arellano AF (2006) Interannual variability in global biomass burning emissions from 1997 to 2004. Atmos Chem Phys 6:3423–3441
Van der Werf GR, Randerson JT, Giglio L, Collatz GJ, Mu M, Kasibhatla PS, Morton DC, DeFries RS, Jin Y, van Leeuwen TT (2010) Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997–2009). Atmos Chem Phys 10:11707–11735. doi:10.5194/acp-10-11707-2010
van Vuuren DP, Edmonds J, Kainuma M, Riahi K, Thomson A, Hibbard K, Hurtt GC, Kram T, Krey V, Lamarque JF, Masui T, Meinshausen M, Nakicenovic N, Smith SJ, Rose SK (2011) The representative concentration pathways: an overview. Clim Chang 109:5–31. doi:10.1007/s10584-011-0148-z
Venkatathnam K, Ryan WBF (1971) Dispersal patterns of clay minerals in the sediments of the Eastern Mediterranean Sea. Mar Geol 11:261–282
Verdugo P, Alldredge AL, Azam F, Kirchman DL, Passow U, Santschi PH (2004) The oceanic gel phase: a bridge in the DOM-POM continuum. Mar Chem 92:67–85
Vignati E, Wilson J, Stier P (2004) M7: an efficient size resolved aerosol microphysics module for large-scale aerosol transport models. J Geophys Res 109, doi:10.1029/2003JD004485
Vignati E, Facchini MC, Rinaldi M, Scannell C, Ceburnis D, Sciare J, Kanakidou M, Myriokefalitakis S, Dentener F, O’Dowd CD (2010) Global scale emission and distribution of sea spray aerosol: sea-salt and organic enrichment. Atmos Environ 44, doi:10.1016/j.atmosenv.2009.11.013
Vogt R, Crutzen PJ, Sander R (1996) A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer. Nature 383:327–330
Volkamer R, San Martini F, Molina LT, Salcedo D, Jimenez JL, Molina MJ (2007) A missing sink for gas-phase glyoxal in Mexico city: formation of secondary organic aerosol. Geophys Res Lett 34, L19807. doi:10.1029/2007gl030752
Wagener T, Pulido-Villena E, Guieu C (2008) Dust iron dissolution in seawater: results from a one-year time-series in the Mediterranean Sea. Geophys Res Lett 35, L16601. doi:10.1029/2008GL034581
Wagener T, Guieu C, Leblond N (2010) Effects of dust deposition on iron cycle in the surface Mediterranean Sea: results from a mesocosm seeding experiment. Biogeosciences 7:3769–3781
Wang C, Corbett JJ, Firestone J (2008) Improving spatial representation of global ship emissions inventories. Environ Sci Technol 42:193–199. doi:10.1021/es0700799
Wang G, Kawamura K, Lee M (2009) Comparison of organic compositions in dust storm and normal aerosol samples collected at Gosan, Jeju Island, during spring 2005. Atmos Environ 43:219–227
Wang Q, Zhuang G, Li J, Huang K, Zhang R, Jiang Y, Lin Y, Fu JS (2011) Mixing of dust with pollution on the transport path of Asian dust – revealed from the aerosol over Yulin, the north edge of Loess Plateau. Sci Total Environ 409:573–581
Waquet F, Riédi J, Labonnote LC, Goloub P, Cairns B, Deuzé JL, Tanré D (2009) Aerosol remote sensing over clouds using the A-Train observations. J Atmos Sci 66:2468–2480
Watson AJ, Bakker DCE, Ridgwell AJ, Boyd PW, Law CS (2000) Effect of iron supply on Southern Ocean CO2 uptake and implications for glacial atmospheric CO2. Nature 407:730–733
Willey JD, Kieber RJ, Eyman MS, Avery GB (2000) Rainwater dissolved organic carbon: concentrations and global flux. Global Biogeochem Cycles 14:139–148
Wilson TM, Cole JW, Sreward C (2011) Ash storms: impact of wind-remobilised volcanic ash on rural communities and agriculture following the 1991 Hudson eruption, southern Patagonia, Chile. Bull Volcanol 73:223–239
Winker DM, Pelon J, Coakley JA, Ackerman SA, Charlson RJ, Colarco PR, Flamant P, Fu Q, Hoff R, Kittaka C, Kubar TL, LeTreut H, McCormick MP, Megie G, Poole L, Powell K, Trepte C, Vaughan MA, Wielicki BA (2010) The CALIPSO mission: a global 3D view of aerosols and clouds. Bull Am Meteorol Soc 91:1211–1229
Witek ML, Flatau PJ, Teixeira J, Westphal DL (2007a) Coupling an ocean wave model with a global aerosol transport model: a sea salt aerosol parameterization perspective. Geophys Res Lett 34, L14806. doi:10.1029/2007GL030106
Witek ML, Flatau PJ, Quinn PK, Westphal DL (2007b) Global sea-salt modeling: results and validation against multicampaign shipboard measurements. J Geophys Res 112, D08215. doi:10.1029/2006JD007779
Wolff EW, Fischer H, Fundel F, Ruth U, Twarloh B, Littot GC, Mulvaney R, Röthlisberger R, de Angelis M, Boutron CF, Hansson M, Jonsell U, Hutterli MA, Lambert F, Kaufmann P, Stauffer B, Stocker TF, Steffensen JP, Bigler M, Siggaard-Andersen ML, Udisti R, Becagli S, Castellano E, Severi M, Wagenbach D, Barbante C, Gabrielli P, Gaspari V (2006) Southern Ocean sea-ice extent, productivity and iron flux over the past eight glacial cycles. Nature 440:491–496. doi:10.1038/nature04614
Woodcock AH (1948) Note concerning human respiratory irritation associated with high concentrations of plankton and mass mortality of marine organism. J Mar Res 7:56–62
Wu J, Sunda W, Boyle E, Karl D (2000) Phosphate depletion in the western North Atlantic Ocean. Science 289:759–762
Wu J, Boyle E, Sunda W, Wen LS (2001) Soluble and colloidal iron in the oligotrophic North Atlantic and North Pacific. Science 293:847–849
Wuttig K, Wagener T, Bressac M, Dammshäuser A, Streu P, Guieu C, Croot PL (2013) Impacts of dust deposition on dissolved trace metal concentrations (Mn, Al and Fe) during a mesocosm experiment. Biogeosciences 10:2583–2600. doi:10.5194/bg-10-2583-2013
Yamato M, Tanaka H (1994) Aircraft observations of aerosols in the free marine troposphere over the North Pacific Ocean: particle chemistry in relation to air mass origin. J Geophys Res 99:5353–5377
Yang F, Chen H, Wang X, Yang X, Du J, Chen J (2009) Single particle mass spectrometry of oxalic acid in ambient aerosols in Shanghai: mixing state and formation mechanism. Atmos Environ 43:3876–3882
Yao X, Fang M, Chan CK (2003) The size dependence of chloride depletion in fine and coarse sea-salt particles. Atmos Environ 37:743–751
Ye Y, Wagener T, Volker C, Guieu C, Dieter A, Wolf-Gladrow DA (2011) Dust deposition: iron source or sink? A case study. Biogeosciences 8:2107–2124
Yoon YJ, Ceburnis D, Cavalli F, Jourdan O, Putaud J-P, Facchini MC, Descari S, Fuzzi S, Sellegri K, Jennings SG, O’Dowd CD (2007) Seasonal characteristics of the physicochemical properties of North Atlantic marine atmospheric aerosols. J Geophys Res 112, D04206. doi:10.1029/2005JD007044
Yu H, Kaufman YJ, Chin M, Feingold G, Remer LA, Anderson TL, Balkanski Y, Bellouin N, Boucher O, Christopher S, DeCola P, Kahn R, Koch D, Loeb N, Reddy MS, Schulz M, Takemura T, Zhou M (2006) A review of measurement-based assessment of aerosol direct radiative effect and forcing. Atmos Chem Phys 6:613–666
Zhang D, Iwasaka Y (2001) Chlorine deposition on dust particles in marine atmosphere. Geophys Res Lett 28:3613–3616. doi:10.1029/2001gl013333
Zhang D, Iwasaka Y (2004) Size change of Asian dust particles caused by sea salt interaction: measurements in southwestern Japan. Geophys Res Lett 31, L15102. doi:10.1029/2004gl020087
Zhang DZ, Iwasaka Y (2006) Comparison of size changes of Asian dust particles caused by sea salt and sulphate. J Meteorol Soc Japan 84:939–947. doi:10.2151/jmsj.84.939
Zhang D, Iwasaka Y, Shi G, Zang J, Matsuki A, Trochkine D (2003a) Mixture state and size of Asian dust particles collected at southwestern Japan in spring 2000. J Geophys Res 108:4760. doi:10.1029/2003jd003869
Zhang D, Zang J, Shi G, Iwasaka Y, Matsuki A, Trochkine D (2003b) Mixture state of individual Asian dust particles at a coastal site of Qingdao, China. Atmos Environ 37:3895–3901
Zhang D, Iwasaka Y, Shi G (2005a) Sea salt shifts the range sizes of Asian dust. Eos Trans Am Geophys Union 86, doi:10.1029/2005EO500003
Zhang R, Arimoto R, An J, Yabuki S, Sun J (2005b) Ground observations of a strong dust storm in Beijing in March 2002. J Geophys Res 110, D18S06. doi:10.1029/2004jd004589
Zhang Q, Jimenez JL, Canagaratna MR, Allan JD, Coe H, Ulbrich I, Alfarra MR, Takami A, Middlebrook AM, Sun YL, Dzepina K, Dunlea E, Docherty K, DeCarlo PF, Salcedo D, Onasch T, Jayne JT, Miyoshi T, Shimono A, Hatakeyama S, Takegawa N, Kondo Y, Schneider J, Drewnick F, Borrmann S, Weimer S, Demerjian K, Williams P, Bower K, Bahreini R, Cottrell L, Griffin RJ, Rautiainen J, Sun JY, Zhang YM, Worsnop DR (2007) Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys Res Lett 34, L13801. doi:10.1029/2007gl029979
Zhao XJ, Zhuang GS, Wang ZF, Sun YL, Wang Y, Yuan H (2007) Variation of sources and mixing mechanism of mineral dust with pollution aerosol – revealed by the two peaks of a super dust storm in Beijing. Atmos Res 84:265–279
Zhao TXP, Loeb NG, Laszlo I, Zhou M (2011) Global component aerosol direct radiative effect at the top of atmosphere. Int J Remote Sens 32:633–655
Zhou M, Okada K, Qian F, Wu PM, Su L, Casareto BE, Shimohara T (1996) Characteristics of dust-storm particles and their long-range transport from China to Japan – case studies in April 1993. Atmos Res 40:19–31
Zhou X, Davis AJ, Kieber DJ et al (2008) Photochemical production of hydroxyl radical and hydroperoxides in water extracts of nascent marine aerosols produced by bursting bubbles from Sargasso seawater. Geophys Res Lett 35, L20803
Zhu X, Prospero J, Millero F (1997) Diel variability of soluble Fe(II) and soluble total Fe in North Africa dust in the trade winds at Barbados. J Geophys Res 102:21297–21305
Zhuang G, Duce R (1993) The adsorption of dissolved iron on marine aerosol particles in surface waters of the open ocean. Deep Sea Res 40:1413–1429
Zorn SR, Drewnick F, Schott M, Hoffmann T, Borrmann S (2008) Characterization of the South Atlantic marine boundary layer aerosol using an aerodyne aerosol mass spectrometer. Atmos Chem Phys 8:4711–4728
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2014 The Author(s)
About this chapter
Cite this chapter
de Leeuw, G. et al. (2014). Ocean–Atmosphere Interactions of Particles. In: Liss, P.S., Johnson, M.T. (eds) Ocean-Atmosphere Interactions of Gases and Particles. Springer Earth System Sciences. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-25643-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-642-25643-1_4
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-25642-4
Online ISBN: 978-3-642-25643-1
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)