
Abstract
Despite remarkable progress in the elucidation of energy balance and regulation, the development of new antiobesity drugs is still at the stage of infancy. This review describes the MCH and MCH receptor system with regard to its involvement in energy homeostasis and summarizes the pharmacological profiles of selected small molecule MCH-R1 antagonists that are relevant for their development as antiobesity drugs. Although their clinical value still has to be demonstrated, and challenges with regard to unwanted side effects remain to be resolved, MCH-R1 antagonists may provide an effective pharmacotherapy for the treatment of obesity in the near future.
Similar content being viewed by others

Keywords
1 Introduction
Chronic inappropriate balance between energy intake and energy expenditure often results in the development of obesity, which is commonly associated with the secondary complications of the metabolic syndrome including non-insulin-dependent diabetes, cardiovascular disease, and certain cancers (Kopelman 2000). Furthermore, obese patients often suffer from some psychosocial discrimination causing depression and anxiety. Due to the western lifestyle (low activity and high caloric food), the epidemic of obesity is rapidly progressing, and up to 30% of adult US population is classified as obese (Ogden et al. 2006). The increasing incidence of obese children and adolescents is of even greater concern (Hedley et al. 2004) because poor eating habits are often established during childhood. Thus, there is an urgent need for an effective pharmacotherapy at present.
Obesity is usually defined as a body mass index (BMI, calculated as body weight in kilograms divided by height in meters squared) bigger than 30 kg/m2, although the BMI alone does not sufficiently predict its detrimental effects. In addition to lifestyle modifications such as diet and exercise, pharmacotherapy is necessary to obtain sufficient weight loss in order to reduce the incidence of secondary complications of the metabolic syndrome. As a guideline for antiobesity drugs suggested by the US Food and Drug Administration (FDA), at least 5% weight loss over a year versus placebo should occur upon drug treatment (Hanif and Kumar 2002). In parallel, other comorbid conditions present in obese patients should be improved without long-term safety issues (Staten 2007). Until recently, only two drugs were approved in the United States for the long-term treatment of obesity: orlistat (Xenical, Roche) and sibutramine (Meridia, Abbott) (McNeely and Goa 1998; Hvizdos and Markham 1999). Orlistat, an inhibitor of gastric and pancreatic lipases, plus a low-calorie diet produced a loss of 3–4% of body weight as compared with diet alone in a 2-year period. The weight reduction was sustained as long as the treatment was continued (Foxcrogt and Milne 2000), but side effects such as fecal urgency/incontinence, flatulence, and steatorrhea limited its use. Sibutramine is a centrally acting appetite suppressant that inhibits reuptake of noradrenaline, serotonin, and dopamine in central neuronal synapses. Chronic treatment with sibutramine resulted in an average reduction of 4 kg over 44–54 weeks treatment, a value comparable to that seen with orlistat (Arterburn et al. 2004). However, because of several cardiovascular side effects (tachycardia and hypertension) induced by its peripheral action, sibutramine is contraindicated in patients with elevated cardiovascular risk such as hypertension (Finer 2002). In Europe and the United States, the drug was withdrawn because it increased mortality in the SCOUT trial designed to investigate its long-term efficacy and safety. Thus, development of more efficacious and safer antiobesity drugs than current pharmacotherapy is needed. Among the many targets investigated for the development of antiobesity drugs, this review will solely focus on the action of melanin-concentrating hormone (MCH) and on the development of small molecule antagonists of melanin-concentrating hormone receptor 1 (MCH-R1) as potential new antiobesity drugs.
2 Importance of MCH in Energy Homeostasis
Over the past decade, a number of neuropeptides have been identified as regulators of food intake and energy metabolism, most of them originating from the hypothalamus. Examples include alpha-melanocyte stimulating hormone (α-MSH) and cocaine- and amphetamine-regulated transcript (CART) as anorexigenic peptides, and neuropeptide Y (NPY), agouti-gene-related peptide (AgRP), orexin, and melanin-concentrating hormone (MCH) as orexigenic ones. Among these, MCH drew great attention as a major player in feeding and energy homeostasis based on numerous studies.
Following discovery of MCH from teleost fish as a skin-paling factor (Kawauchi et al. 1983), mammalian MCHs have also been identified and showed 100% sequence identity between mouse, rat, rabbit, and human (Vaughan et al. 1989; Presse et al. 1990). Whereas fish MCH consisted of 17 amino acids, mammalian MCH is a 19-amino-acid cyclic peptide synthesized from precursor protein, prepro-MCH, by posttranslational cleavage along with the production of two additional peptides named neuropeptide E-I (NEI) and neuropeptide G-E (NGE) (Breton et al. 1993). It was found that residues Arg6, Met8, Arg11, and Tyr13 as well as a disulfide bond between cysteine residues are critical for the biological activity of MCH (MacDonald et al. 2000). MCH is almost exclusively expressed in hypothalamic neurons located in the zona incerta and in the lateral hypothalamic area (LHA), from which these neurons project extensively throughout the brain (Bittencourt et al. 1992; Skofitsch et al. 1985). MCH is also expressed in the peripheral nervous system and in other tissues and cells such as the intestine, the reproductive system, and immune cells (Hervieu and Nahon 1995; Viale et al. 1997).
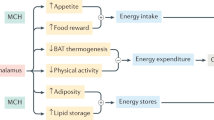
In mammals, MCH has been implicated in a variety of physiological functions including sensory processing (Miller et al. 1993), stress response by regulating hypothalamic–pituitary–adrenal axis (Jezova et al. 1992; Presse et al. 1992), and learning (McBride et al. 1994). Recent evidence also points to a key role of MCH in the regulation of feeding behavior and associated pathologies such as obesity. The lateral hypothalamic area where MCH neurons are located has been known as an important center regulating feeding behavior, as stimulation produces overeating, whereas lesions result in hypophagia and body weight reduction (Anand and Brobeck 1951). In addition, either acute or chronic administration of MCH increased body weight along with higher food intake. For example, acute intracerebroventricular injection of MCH, or direct injection of MCH into the paraventricular nucleus (PVN), arcuate nucleus (ARC), or dorsomedial nucleus (DMH) (Abbott et al. 2003; Rossi et al. 1999), produces a transient increase in food intake in a dose-dependent manner. In parallel with increased food intake, MCH administration increased the expression of the orexigenic peptides NPY and AgRP, but decreased expression of anorexic peptides α-MSH and CART from hypothalamic explants. These results suggest that increase in food intake by MCH is at least in part mediated by the regulation of these peptides. Chronic effects of MCH also demonstrated that central infusion of MCH increases food intake, body weight, and adiposity (Della-Zuana et al. 2002; Ito et al. 2003). Importantly, beyond its role in feeding, MCH appears to affect energy expenditure and thermogenesis since animals exposed to MCH reduced their core body temperature in parallel with a decrease of the expression of genes involved in thermogenesis (Pereira-da-Silva et al. 2003).
Further evidence that MCH acts as a major factor in the control of energy balance comes from the animal studies with genetic manipulation of the MCH gene. Targeted deletion of MCH in mice resulted in hypophagia, increased metabolic activity, and weight loss (Shimada et al. 1998); conversely, overexpression of MCH in transgenic mice led to an increased body weight with hyperphagia, hyperleptinemia, hyperinsulinemia, and hyperglycemia when mice were fed a high-fat diet (Ludwig et al. 2001). Furthermore, it was found that MCH mRNA and pro-MCH-derived peptide levels were upregulated in various obese rodents including ob/ob mice (Qu et al. 1996), db/db mice (Huang et al. 1999), fat/fat mice (Rovere et al. 1996), Ay/a (agouti) mice (Hanada et al. 2000), and fa/fa rat (Stricker-Krongrad et al. 2001), suggesting that the level of MCH expression may be linked to feeding behavior and obesity. Similarly, a threefold increased expression of MCH mRNA and peptide was observed in obese humans as compared to lean subjects, implicating the involvement of MCH in human feeding disorders (Zhang et al. 1999).
Various conditions can regulate the expression of MCH. For example, starvation increases MCH mRNA levels in rodents (Herve and Fellmann 1997; Tritos et al. 1998), whereas leptin negatively regulates MCH expression. Leptin replacement in ob/ob mice and fasted rats reduced increased MCH mRNA levels to normal, and leptin injection abolished hyperphagia elicited by the central administration of MCH (Sahu 1998). This effect may be mediated by a direct action on MCH neurons in the LHA or by an indirect action on projections from areas such as the ARC (Elias et al. 1998, 1999). In contrast, MCH expression was shown to be positively regulated by leptin in dietary obese rats (Elliott et al. 2004). Interestingly, MCH mRNA was significantly elevated upon feeding of a palatable diet. These data suggest that MCH may have a specific role in stimulating appetite for palatable food, which can override mechanisms counteracting the development of obesity.
Recently, peripheral effects of MCH were described in endocrine pancreas and adipose tissue. MCH appears to stimulate leptin release in adipocytes (Bradley et al. 2000) and to modulate insulin secretion (Tadayyon et al. 2000). These findings indicate that MCH also acts within the periphery via regulation of other hormones.
3 Characterization of MCH Receptors
Two receptors for MCH in humans have recently been characterized. The first MCH receptor (MCH-R1) was identified as the orphan G-protein-coupled receptor SLC-1 (Bachner et al. 1999; Chambers et al. 1999; Saito et al. 1999) with high affinity for human MCH in the low nanomolar range and which is not activated by any other known peptide (Chambers et al. 1999). MCH-R1 consists of 353 amino acids with ~32% amino acid identity with members of the somatostatin receptor family. The gene is localized on chromosome 22q13.3. MCH-R1 has a high sequence identity (higher than 95%) and a similar tissue distribution between species (Tan et al. 2002), with particularly high abundance in the hypothalamus, thalamus, olfactory cortex, amygdala, and hippocampus. This pattern of expression corresponds with the areas of MCH immunoreactive implicated in feeding regulation (Hervieu et al. 2000). Some peripheral tissues including the adipose tissue are also reported to express MCH-R1 (Bradley et al. 2000; Saito et al. 1999). It appears that MCH-R1 signaling pathways are diverse via multiple G proteins (Bachner et al. 1999; Chambers et al. 1999; Hawes et al. 2000; Lembo et al. 1999; Shimomura et al. 1999). For example, MCH increased calcium concentration via both pertussis toxin-sensitive and pertussis toxin-insensitive GTP-binding proteins including Gαi, Gα0, and Gαq (Hawes et al. 2000). In addition, MCH inhibited adenylyl cyclase more potently than stimulating calcium mobilization (Lembo et al. 1999).
Similar to the effects on MCH expression, fasting also increased the expression of MCH-R1. Likewise, ob/ob mice and dietary obese rats were shown to have higher level of MCH-R1 (Elliott et al. 2004). To the contrary, MCH-R1 expression is unchanged in MCH knockout mice (Kokkotou et al. 2001), which appears to be distinguishable from other G protein-coupled receptors regulated by their own ligands. Importantly, targeted disruption of the MCH-R1 gene results in resistance to diet-induced obesity and hyperphagia (Marsh et al. 2002), leanness, hyperactivity, and increased metabolic rate (Chen et al. 2002), a phenotype similar to that of MCH knockout mice. These results suggest that an increased metabolic activity as well as a decreased food intake may mediate reduced weight gain in MCH-R1 KO mice (Marsh et al. 2002; Chen et al. 2002) and that the effects of MCH on energy balance are mediated by MCH-R1. The recent reports of the anorectic and antiobesity effects of the MCH-R1 antagonists further confirmed the importance of MCH in the regulation of body weight.
A second MCH receptor referred to as MCH-R2 was found independently by several laboratories (An et al. 2001; Mori et al. 2001; Rodriguez et al. 2001; Sailer et al. 2001; Wang et al. 2001). Overall sequence of MCH-R2 displays an unusually low sequence identity with MCH-R1 (~38%) but a significant homology to the core region of MCH-R1. MCH-R2 is a 340-amino-acid membrane protein which binds MCH in the nanomolar range and has a similar but distinct expression profile in human brain and peripheral tissues. Levels of MCH-R2 mRNA in adipose tissue are significantly higher than those of MCH-R1 (Hill et al. 2001). The coupling mechanism of MCH-R2 is different from that of MCH-R1: MCH-R2 preferentially activates Gq, whereas MCH-R1 activates both Gi/o and Gq (Hawes et al. 2000). Uniquely, MCH-R2 is not expressed in rodent species including mice, rats, rabbits, hamsters, or guinea pigs (Tan et al. 2002), which renders it difficult to determine the functional importance of MCH-R2. On the other hand, nonhuman species such as dogs, ferrets, and rhesus monkeys are known to have a functional MCH-R2 (Tan et al. 2002).
4 Dysregulation of MCH and MCH-R1 in Obesity
Although many genetic analyses have been carried out to elucidate the role of genetic variants of MCH and MCH-R1, no conclusive evidence for an association with obesity has been obtained so far. For example, a single amino acid substitution (R248Q) in the MCH-R1 gene co-segregated with obesity across more than one generation (Gibson et al. 2004), but this mutation was also present in lean subjects. Two other nonconservative missense variations (R317Q and T305M) cosegregated with obesity, but there was no evidence for a functional relevance (Gibson et al. 2004; Wermter et al. 2005). Another mutation in the promoter region of the MCH-R1 gene is associated with a reduced risk of obesity (Bell et al. 2005). At present, it is not conclusive that certain sequence variations in MCH-R1 are associated with obesity, although a combination with other minor variations in different genes may contribute to the susceptibility to obesity. Further studies in large populations and/or in genetically modified animal models would clarify the relationship of genetic variants of MCH and/or MCH-R1 to obesity.
5 MCH-R1 Antagonists in Discovery and Development
After the discovery of MCH-R1, numerous reports on the identification and optimization of small molecule and peptide antagonists of MCH-R1 for the potential treatment of obesity have been published (Carpenter and Hertzog 2002; Collins and Kym 2003; Handlon and Zhou 2006; Luthin 2007; McBriar 2007; Rokosz 2007; Rivera et al. 2008). However, despite extensive research efforts to develop small molecule MCH-R1 antagonists, so far, only four compounds entered human clinical trials as shown in Table 1, and only ALB-127158 is currently under active development.
In this section, the review will focus on the small molecule antagonists of MCH-R1, categorized with regard to companies and/or frequently encountered substructures, and will describe their pharmacological characteristics. Based on the patent literature and to other publications reported so far, common structural characteristics of the MCH-R1 antagonists included a central scaffold to which an aryl or heteroaryl group and a basic amino group are attached, which appears to be essential for antagonistic activity on MCH-R1. On the other hand, these structural characteristics are also common in hERG blockers, which can induce QT prolongation frequently associated with potentially lethal arrhythmias in clinical use. Therefore, interaction with hERG channel has been often a problem observed with MCH-R1 antagonists.
Neurogen researchers have reported several small molecule antagonists of MCH-R1, and among those, a piperazine compound 1 (NGD-4715) is most advanced to phase 1 study. Based on the phase 1 study, NGD-4715 seemed to be safe and well tolerated at all doses studied in the trial. In addition, Neurogen disclosed diaminoethane (compound 2) and 8-azabicyclo[3.2.1]octane (compounds 3 and 4) derivatives as variants of the piperazine-based compounds (Fig. 1).

Neurogen’s representative compounds
GlaxoSmithKline disclosed various scaffolds as MCH-R1 antagonists as summarized in Fig. 2. After an activity of the biphenyl carboxamide skeleton was identified in a high-throughput screening, compound 5 (SB-568849) was synthesized, and its pharmacological characteristics were investigated. SB-568849 exhibited good affinity (pK i = 7.7) in the FLIPR assay with >30-fold selectivity over a wide range of monoamine receptors. In an effort to discover more rigid analogues of the compound SB-568849, the thienopyrimidinone compound 6 (GW-803430, GW-856464) was developed as a candidate for clinical trials. Compound 6 was found to be a potent MCH-R1 antagonist (pIC50 = 9.3) and was highly selective (>100×) as tested with a battery of G protein-coupled receptors, ion channels, and enzymes (Hertzog et al. 2006). Its maleate salt exhibited good pharmacokinetic properties (F = 31%, t 1/2 = 11 h) in mice, with high brain penetration (brain/plasma concentration = 6:1). Oral administration of the compound at 0.3, 3, and 15 mg/kg once daily during a 12-day treatment caused a sustained dose-dependent weight loss of −6.2%, −12.1%, and −13.1%, respectively, in high-fat-diet-induced obesity of AKR/J mice (Hertzog et al. 2006). Preclinical studies with compound 6 revealed that it also produced an antidepressant response in the mouse forced-swim test and in the tail suspension test. Thus, compound 6 (GW-803430) was proven to be active as both antiobesity agent and antidepressant (Gehlert et al. 2009). However, the agent exhibited some hERG activity. Through further optimization study, the compound 7 was identified to be a potent antagonist (IC50 = 3.1 nM) with excellent oral bioavailability (F = 98%, t 1/2 = 7.7 h, Cltotal = 39.7 mL/min/kg) (Tavares et al. 2006a). Compound 7 had no significant off-target activity including hERG channel as determined by patch clamp assay (pIC50 = 4.66) and was selective over human MCH-R2 (Tavares et al. 2006a). Consistent with its pharmacokinetic profile, oral administration of the compound 7 at 1, 3, and 10 mg/kg once daily during a 26-day treatment caused a sustained dose-dependent weight loss of −4.8%, −9.4%, and −16.9%, respectively, in high-fat-diet-induced obesity of AKR/J mice, with good brain partitioning properties (brain/serum = 31) (Tavares et al. 2006a). The benzothiophene compound 8, an analogue of compound 7, was also found to be a potent antagonist (IC50 = 5.7 nM) and had a good systemic exposure (F = 66%, t 1/2 = 9.1 h) with moderate clearance (Cltotal = 39 mL/min/kg) and good solubility (Tavares et al. 2006b). Oral administration of compound 8 at 1, 3, and 10 mg/kg once daily during a 21-day treatment caused a dose-dependent weight loss of −1.1%, −3.2%, and −11.8%, respectively, in high-fat-diet-induced obesity of AKR/J mice, comparable to the effect of a CB1 receptor inverse agonist rimonabant (−8.4%) (Tavares et al. 2006b). Furthermore, compound 8 was shown to have negligible hERG activity, but no further progress on this compound was reported.

GSK’s representative compounds
The biaryl carboxamide moiety in compound 5 (SB-568849) in Fig. 2 is often encountered in many other representative compounds. For example, the tetrahydronaphthalene compound 9 (T-226296, IC50 = 5.5 and 8.6 nM at human and rat MCH-R1), the first small molecule MCH-R1 antagonist reported by Takeda (Takekawa et al. 2002), exhibited selectivity over a host of other receptors including MCH-R2 (>100 nM affinity) and was shown to be efficacious in suppressing food intake induced by intracerebroventricular injection of MCH in rats when orally administered at the dose of 30 mg/kg (Takekawa et al. 2002; Kowalski and McBriar 2004). Procter & Gamble pharmaceuticals reported compound 10 (DABA-821) as a potent MCH-R1 antagonist (K i = −39.3 nM and IC50 = 14.0 nM). The compound showed ~25-fold selectivity over 5HT2C receptor and caused significant body weight and fat mass reduction in rodents (Hu et al. 2008). Further optimization of the compound by decreasing hERG activity while retaining potent MCH-R1 antagonist activity (K i = 16 nM) was achieved (compound 11) but with concurrent loss of in vivo activity due to poor brain penetration (brain/plasma = 0.3). Neurocrine reported that the 3-aminopyrrolidine compound 12 was a potent antagonist (K i = 2.3 nM) with good oral bioavailability in rats (F = 32%, t 1/2 = 2.7 h). In vivo efficacy of the compound was examined, and the orally, either acutely or chronically, administered compound decreased food intake as well as body weight in rats (Huang et al. 2005). In an effort to improve hERG activity of the Neurocrine compounds, compound 13 was identified. The compound exhibited hERG selectivity (MCH-R1 K i = 7.3 nM versus hERG IC50 = 2.6 μM), but other undesirable characteristics such as inhibition of cytochrome P450 CYP2D6 (IC50 = 3.3 μM) and poor brain penetration (brain/plasma ≪1) hampered further studies on this compound (Fig. 3).

Biaryl carboxamide derivatives
After the compounds 7 and 8 had shown improved hERG selectivity, in vivo activity on food intake, and desirable pharmacokinetic properties, analogous compounds have often been disclosed in related patents by other companies including Neurocrine (Fig. 4). The thienopyridazinone compound 14 (NBI-845) comprising a 3-aminopyrrolidine residue was identified by Neurocrine to be a potent MCH-R1 antagonist (K i = 3.3 nM, and IC50 = 7.7 nM) and to be metabolically stable as assessed with human liver microsomes (Dyck et al. 2006). The agent showed a good pharmacokinetic profile (F = 24%, t 1/2 = 11 h, brain/plasma = 1.4 ~ 3.1), inhibited food intake in vivo, and induced weight loss in DIO male Sprague–Dawley rats (Dyck et al. 2006). Compound 14 was further evaluated in a selectivity screen (only modest cross-reactivity with M1R K i = 260 nM; M3R K i = 160 nM), CYP2D6/3A4 inhibition (<10% at 5 μM), hERG affinity (IC50 = 1.8 μM), and preliminary toxicological studies in rats (Dyck et al. 2006). Similarly, the chromone-based compound 15 was reported to be a potent MCH-R1 antagonist (K i = 1.6 nM, and IC50 = 20 nM) and was further characterized in a pharmacokinetic study with Sprague–Dawley rats (F = 66%, t 1/2 = 5.0 h, brain/plasma = 9.5 ~ 25) (Dyck et al. 2006).

Bicyclic analogues
Biaryl ether and the related compounds were shown to have antagonistic effects on MCH-R1, and their structures are represented in Fig. 5. The indazole compound 16 comprising an ureido linkage was reported to be a potent MCH-R1 antagonist (K i = 12.0 nM, and IC50 = 104 nM) and was also evaluated in pharmacokinetic and oral efficacy studies in mice fed a high-fat diet (Souers et al. 2005). Cerep reported that a representative compound 17 with a hydantoin skeleton was a MCH-R1 ligand (K i = 220 nM). Intraperitoneal administration of the compound 17 (30 mg/kg) reduced cumulative food intake compared to the vehicle group and also showed antidepressant activity comparable to the effect of a clinically used antidepressant, imipramine (Alavoine et al. 2007). However, the compound inhibited the hERG channel (73% current amplitude inhibition at 1 μM in a standard in vitro electrophysiology assay) (Alavoine et al. 2007).

Biaryl ether and the related compounds
Amgen researchers reported several small molecule antagonists of MCH-R1 as represented in Fig. 6. In addition to a series of polycyclic compounds based on the indole moiety as exemplified in compound 18, they disclosed the tetrazole compound 20 and biaryl ether compounds 21 and 22. Compound 21 was reported to have potent antagonistic activity (IC50 = 1.0 nM, t 1/2 1.5 h, C max in brain 31.5 ng/g). The compound AMG-076 was reported to enter into phase 1 study in 2004 (Mendez-Andino and Wos 2007), with undisclosed structure. Little information on the biological profile of these compounds is available. Recently, novel pyrrolidine MCH-R1 antagonists with reduced hERG inhibition were reported (Fox et al. 2011).

Amgen’s representative compounds
Researchers at Schering-Plough/Pharmacopeia claimed many variants of biaryl urea derivatives, with the highly mutagenic biarylamine unit. To avoid the mutagenic risk of diphenyl aniline moiety, bicyclo[3,1,0]hexyl urea compound 23 (SCH-A, K i = 2.7 nM, K b = 1.9 nM, F = 27%) was synthesized and tested. This compound exhibited low nanomolar potency for MCH-R1, with good selectivity against MCH-R2 and other receptors, enzymes, and transporters. With 27% of oral bioavailability, in vivo administration of the compound 23 (30 mg/kg, po) to obese mice resulted in a significant reduction of food intake as well as body weight, primarily due to the loss of fat mass (Guo et al. 2005; McBriar et al. 2006). In contrast, no effects of SCH-A on energy expenditure were observed. The inhibitory action of SCH-A on hERG channel (IC50 = 52 nM in patch clamp assay) led to attempts to improve hERG selectivity. The bicyclo[4,1,0]heptyl aminobenzimidazole derivative 24 was shown to be a potent MCH-R1 ligand (K i = 2.2 nM), to have good ex vivo binding (>81%) with good pharmacokinetic properties (AUC = 965 ng ċ h/mL at 10 mg/kg po dose in rat), and to be efficacious in fasted obese mice (9–19% inhibition of cumulative food intake at 30 mg/kg po dose) (Sasikumar et al. 2006). However, the activity on hERG channel was not reported (Fig. 7).

Representative compounds of Schering-Plough
Abbott has disclosed several MCH-R1 antagonists with 4-aminopiperidine scaffold as summarized in Fig. 8. From structure–activity relationship (SAR) investigation on a screening hit, benzamides 25 (binding IC50 = 3 nM, functional IC50 = 90 nM) and 26 (binding IC50 = 2 nM, functional IC50 = 16 nM), an indazole 27 (binding IC50 = 20 nM, functional IC50 = 260 nM), and a coumarin 28 (binding IC50 = 2 nM, functional IC50 = 28 nM) were identified as potent antagonists. Compound 28 showed a high brain/plasma concentration ratio and produced a decrease in body weight without significant loss of lean body mass in obese mice (30% body weight reduction at 30 mg/kg, po) (Vasudevan et al. 2005; Kym et al. 2005). However, the compounds showed adverse hemodynamic effects as revealed in an anesthetized dog model of cardiovascular safety (Souers et al. 2007). In an attempt to reduce the effects on hERG, a series of chromone derivatives such as 29–31 was synthesized and was shown to possess in vivo efficacy in diet-induced obese mice with a high brain/plasma distribution ratio. These compounds still showed unacceptable affinity for hERG channel and caused QT prolongation in dogs at low doses (Lynch et al. 2005). On the other hand, compounds 32 and 33 produced a 7% weight loss in obese mice after 2 weeks of treatment (3 mg/kg, po) while the compounds exhibited good hERG selectivity (Iyengar et al. 2007; Souers et al. 2007).

Abbott’s representative compounds
Arena Pharmaceuticals and Taisho Pharmaceuticals collaborated in a discovery program of MCH-R1 antagonists and described the aminoquinazoline derivative, N-(cis-4-{[4-(dimethylamino)quinazolin-2-yl]amino}cyclohexyl)-3,4-difluorobenzamide hydrochloride (ATC-0175, compound 34) to exhibit potent antagonistic activities on MCH-R1 (IC50 = 7 nM). ATC-0175 showed selectivity against MCH-R2 but had a moderate affinity for 5-HT2B and 5-HT1A receptors. In the diet-induced obesity mouse model, ATC-0175 reduced body weight significantly without loss in lean body mass (Semple et al. 2004). Interestingly, ATC-0175 exhibited anxiolytic effects in numerous animal models of anxiety (Chaki et al. 2005), with adequate pharmacokinetic profile and well-tolerated toxicity, suggesting potential utility for the treatment of depression and/or anxiety disorders. Recently, Taisho has disclosed additional compounds with this scaffold, although there have been no reports of cardiovascular effects of these compounds (Fig. 9).

Taisho and Lundbeck’s representative compounds
Lundbeck/Synaptic reported an arylpiperidine derivative, SNAP-7941 (compound 35), which is one of the earliest examples of efficacious MCH-R1 antagonists. SNAP-7941 showed high affinity to MCH-R1 (K i of 15 nM), >1,000× selectivity against other receptors, and was able to inhibit MCH-induced food intake when injected intraperitoneally but not when administered orally. Chronic intraperitoneal treatment with the compound produced sustained weight loss in obese rat (26% reduction versus vehicle treatment), accompanied by a modest reduction of food intake, suggesting that energy expenditure may also contribute to the reduction of body weight in this model. In addition to its effects on food intake, SNAP-7941 improved anxiety and depression in animal models (Borowsky et al. 2002). The follow-up compound SNAP-94847 (compound 36) exhibited improved bioavailability, resulting in the inhibition of body weight gain in normal rats after oral application. However, relatively moderate selectivity over other targets was described with no published data on hERG selectivity (Fig. 9).
Based on the results reported so far, a large number of small molecule MCH-R1 antagonists were discovered. Some of those showed good in vivo oral efficacy (weight loss accompanied by reduced food intake and/or increased energy expenditure) in animal models, with good selectivity against a panels of receptors, enzymes and transporters. So far, limited success to obtain hERG selectivity was achieved (Judd et al. 2008), and major safety issues on cardiovascular toxicity due to hERG binding still need to be resolved in order to proceed to clinical studies. Since structural characteristics of many MCH-R1 antagonists are shared by classical hERG binding agents, novel scaffolds need to be identified and tested.
6 Perspectives
Besides the effects on food intake and regulation of energy balance, a number of diverse functions of MCH have been reported, which is expected by the broad distribution of MCH fibers in the CNS. Examples of the effects of MCH include (1) reduction of pentylenetetrazole- and kainic acid-induced seizure (Knigge and Wagner 1997), (2) enhancement of luteinizing hormone-releasing hormone (Pissios et al. 2006), (3) reduction of thyroid hormone (Kennedy et al. 2003), and (4) increase in NMDA receptor-mediated long-term potentiation (Varas et al. 2003), all of which may cause adverse effects when using MCH-R1 antagonists. Among many effects elicited by MCH, the effects of MCH on corticosterone levels appear to be very interesting with respect to the development of MCH-R1 antagonists as antiobesity agents. MCH modulates the hypothalamic–pituitary–adrenal axis, resulting in increased plasma corticosterone levels, thus producing anxiety-related responses (Smith et al. 2006). In addition, the findings that MCH reduces serotonin levels in discrete hypothalamic nuclei (Gonzalez et al. 1998), and that 5-HT receptors are expressed in MCH containing neurons, suggest that a reciprocal interaction between MCH and serotonergic pathways may exist (Collin et al. 2002). Indeed, an anxiolytic and antidepressant effect of MCH-R1 antagonists was reported in animal models (Lucki 1997; Borowsky et al. 2002). Since obesity patients are often accompanied with depression, the possibility that MCH-R1 antagonists have anxiolytic and antidepressant properties will definitely offer an additional benefit for the pharmacotherapy of obesity. Furthermore, since rimonabant, a cannabinoid CB1 receptor antagonist, was discontinued due to the side effects of depression (Despres et al. 2005), the potential of anxiolytic and antidepressant effects of MCH-R1 antagonists will provide an exciting clinical utility as antiobesity drugs, although clinical outcome needs to be proven.
The issue of cardiovascular safety became important because several classes of the MCH-R1 antagonists interacted with the hERG channel (Lynch et al. 2005), and early screening of the compounds for hERG liability was necessary in the discovery programs. To avoid the interaction with hERG channel, unique structural motifs that possess the desired pharmacological characteristics such as potency and good pharmacokinetic and safety profiles should be identified. In addition, the fact that the MCH-R2 gene is nonfunctional or is not expressed in rodents may impede the development of selective MCH-R1 antagonists for human therapies, since any actions of MCH-R1 antagonists on MCH-R2 may have adverse effects not detected in studies on rodents. Alternative animal models such as primate, dog, and ferret species with functional MCH-R2 receptors may be amenable to development.
7 Conclusion
Obesity represents a key disease area with considerable therapeutic needs and market potential. However, currently, only few drugs are available for its pharmacotherapy. Therefore, there are considerable opportunities for pharmaceutical companies in the development of effective antiobesity drugs to treat the worldwide obesity epidemic. Since MCH is a central anabolic regulator of appetite and energy balance, MCH-R1 antagonists are among the most thoroughly investigated agents for their efficacy to reduce food intake and increase energy expenditure in animal models. This review has presented an overview on the small molecule MCH-R1 antagonists, based on the published agents and data. While substantial challenges such as the validation in clinical trials and the issues of drug safety remain, it is anticipated that the development of small molecule MCH-R1 antagonists may provide key therapeutic options for the treatment of human obesity and mood disorders.
References
Abbott CR, Kennedy AR, Wren AM, Rossi M, Murphy KG, Seal LJ, Todd JF, Ghatei MA, Small CJ, Bloom SR (2003) Identification of hypothalamic nuclei involved in the orexigenic effect of melanin-concentrating hormone. Endocrinology 144:3943–3949
An S, Cutler G, Zhao JJ, Huang SG, Tian H, Li W, Liang L, Rich M, Bakleh A, Du J, Chen JL, Dai K (2001) Identification and characterization of a melanin-concentrating hormone receptor. Proc Natl Acad Sci U S A 98:7576–7581
Anand BK, Brobeck JR (1951) Localization of a feeding center in the hypothalamus of the rat. Proc Soc Exp Biol Med 77:323–324
Arterburn DE, Crane PK, Veenstra DL (2004) The efficacy and safety of sibutramine for weight loss: a systemic review. Arch Intern Med 164:994–1003
Bachner D, Kreienkamp H, Weise C, Buck F, Richter D (1999) Identification of melanin concentrating hormone (MCH) as the natural ligand for the orphan somatostatin-like receptor 1 (SLC-1). FEBS Lett 457:522–524
Balavoine F, Malabre P, Alleaume T, Rey A, Cherfils V, Jeanneton O, Seigneurin VS, Revah F (2007) Design and synthesis of novel hydantoin-containing melanin concentrating hormone receptor antagonists. Bioorg Med Chem Lett 17:3754–3759
Bell CG, Meyre D, Samson C, Boyle C, Lecoeur C, Tauber M, Jouret B, Jaquet D, Levy-Marchal C, Charles MA, Weill J, Gilson F, Mein CA, Froquel P, Walley AJ (2005) Association of melanin-concentrating hormone receptor 1 5′polymorphism with early onset extreme obesity. Diabetes 54:3049–3055
Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE (1992) The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol 319:218–245
Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C (2002) Antidepressant, anxiolytic and anorectic effects of a melanin concentrating hormone-1 receptor antagonist. Nat Med 8:825–830
Bradley RL, Kokkotou EG, Matatos-Flier E, Cheatam B (2000) Melanin concentrating hormone (MCH) regulates leptin synthesis and secretion in rat adipocytes. Diabetes 49:1073–1077
Breton C, Schorpp M, Nahon JL (1993) Isolation and characterization of the human melanin concentrating hormone gene and a variant gene. Brain Res Mol Brain Res 18:297–310
Carpenter AJ, Hertzog DL (2002) Melanin-concentrating hormone receptor antagonists as potential antiobesity agents. Expert Opin Ther Patents 12:1639–1646
Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM (1999) Melanin-concentrating hormone is the cognate ligand for the orphan G protein-coupled receptor SLC-1. Nature 400:261–265
Chaki S, Yamaguchi J, Yamada H, Thomsen W, Tran TA, Semple G, Sekiguchi Y (2005) ATC0175; an orally active melanin-concentrating hormone receptor 1 antagonist for the potential treatment of depression and anxiety. CNS Drug Rev 11:341–352
Chen Y, Hu C, Hsu CK, Zhang Q, Bi C, Asnicar M, Hsiung HM, Fox N, Slieker LJ, Yang DD, Heiman ML, Shi Y (2002) Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology 143:2469–2477
Collin M, Backberg M, Onnestam K, Meister B (2002) 5-HT1A receptor immunoreactivity in hypothalamic neurons involved in body weight control. Neuroreport 13:945–951
Collins CA, Kym PR (2003) Prospects for obesity treatment: MCH receptor antagonists. Curr Opin Investig Drugs 4:386–394
Della-Zuana O, Presse F, Ortola C, Duhault J, Nahon JL, Levens N (2002) Acute and chronic administration of melanin-concentrating hormone enhances food intake and body weight in Wistar and Sprague–Dawley rats. Int J Obes Relat Metab Disord 26:1289–1295
Despres JP, Golay A, Sjostrom L (2005) Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med 353:2121–2134
Dyck B, Markison S, Zhao L, Tamiya J, Grey J, Rowbottom MW, Zhang M, Vickers T, Sorensen K, Norton C, Wen J, Heise CE, Saunders J, Conlon P, Madan A, Schwarz D, Goodfellow VS (2006) A thienopyridazinone-based melanin concentrating hormone receptor 1 antagonist with potent in vivo anorectic properties. J Med Chem 49:3753–3756
Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK (1999) Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron 23:775–786
Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK (1998) Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol 402:442–459
Elliott JC, Harrold JA, Brodin P, Enquist K, Backman A, Bystrom M, Lindgren K, King P, Williams G (2004) Increases in melanin-concentrating hormone and MCH receptor levels in the hypothalamus of dietary-obese rat. Brain Res Mol Brain Res 128:150–159
Finer N (2002) Sibutramine: its mode of action and efficacy. Int J Obes Relat Metab Disord 26(Suppl 4):S29–S33
Fox BM, Natero R, Richard K, Connors R, Roveto PM, Beckmann H, Haller K, Golde J, Xiao SH, Kayser F (2011) Novel pyrrolidine melanin-concentrating hormone receptor 1 antagonists with reduced hERG inhibition. Bioorg Med Chem Lett 21:2460–2467
Foxcroft DR, Milne R (2000) Orlistat for the treatment of obesity: rapid review and cost effectiveness model. Obes Rev 1:121–126
Gehlert DR, Rasmussen K, Shaw J, Li X, Ardayfio P, Craft L, Coskun T, Zhang HY, Chen Y, Witkin JM (2009) Preclinical evaluation of melanin-concentrating hormone receptor 1 antagonism for the treatment of obesity and depression. J Pharmacol Exp Ther 329:429–438
Gibson WT, Pissios P, Trombly DJ, Luan J, Keogh J, Wareham NJ, Maratos-Flier E, O’Rahilly S, Farooqi IS (2004) Melanin-concentrating hormone receptor mutations and human obesity: functional analysis. Obes Res 12:743–749
Gonzalez MI, Baker BI, Hole DR, Wilson CA (1998) Behavioral effects of neuropeptide E-I(NEI) in the female rat: interaction with alpha-MSH, MCH and dopamine. Peptides 19:1007–1016
Guo T, Shao Y, Qian G, Rokosz LL, Stauffer TM, Hunter RC, Babu SD, Gu H, Hobbs DW (2005) Discovery and SAR of biaryl piperidine MCH1 receptor antagonists through solid-phase encoded combinatorial synthesis. Bioorg Med Chem Lett 15:3696–3700
Hanada R, Nakazato M, Mutsukura S, Murakami N, Yoshimatu H, Sakata T (2000) Differential regulation of MCH and orexin genes in the agouti-related protein/melanocortin-4 (AgRP/MC4) receptor system. Biochem Biophys Res Commun 268:88–91
Handlon AL, Zhou H (2006) Melanin-concentrating hormone-1 receptor antagonists for the treatment of obesity. J Med Chem 49:4017–4022
Hanif MW, Kumar S (2002) Pharmacological management of obesity. Expert Opin Pharmacother 3:1711–1718
Hawes BE, Kil E, Green B, O’Neill K, Fried S, Graziano MP (2000) The melanin concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways. Endocrinology 141:4524–4532
Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM (2004) Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. J Am Med Assoc 291:2847–2850
Hervieu G, Nahon JL (1995) Pro-melanin-concentrating hormone messenger ribonucleic acid and peptides expression in peripheral tissues of the rat. Neuroendocrinology 61:348–364
Herve C, Fellmann D (1997) Changes in rat melanin-concentrating hormone and dynorphin messenger ribonucleic acids induced by food deprivation. Neuropeptides 31:237–242
Hervieu GJ, Cluderay JE, Harrison D, Meakin J, Maycox P, Nasir S, Leslie RA (2000) The distribution of the mRNA and protein products of the melanin-concentrating hormone (MCH) receptor gene, slc-1, in the central nervous system of the rat. Eur J Neurosci 12:1194–1216
Hertzog DL, Al-Barazanji KA, Bigham EC, Bishop MJ, Britt CS, Carlton DL, Cooper JP, Daniels AJ, Garrido DM, Goetz AS, Grizzle MK, Guo YC, Handlon AL, Ignar DM, Morgan RO, Peat AJ, Tavares FX, Zhou H (2006) The discovery and optimization of pyrimidinone-containing MCH R1 antagonists. Bioorg Med Chem Lett 16:4723–4727
Hill J, Duckworth M, Murdock P, Rennie G, Sabido-David C, Ames RS, Szekeres P, Wilson S, Bergsma DJ, Gloger IS, Levy DS, Chambers JK, Muir AI (2001) Molecular cloning and functional characterization of MCH2, a novel human MCH receptor. J Biol Chem 276:20125–20129
Hu XE, Wos JA, Dowty ME, Suchanek PM, Ji W, Chambers JB, Benoit SC, Clegg DJ, Reizes O (2008) Small-molecule melanin-concentrating hormone-1 receptor antagonists require brain penetration for inhibition of food intake and reduction in body weight. J Pharmacol Exp Ther 324:206–213
Huang Q, Viale A, Picard F, Nahon JL, Richard D (1999) Effects of leptin on Melanin-concentrating hormone (MCH) expression in the brain of lean and obese Lep(ob)/Lep(ob) mice. Neuroendocrinology 69:145–153
Huang CQ, Baker T, Schwarz D, Fan J, Heise CE, Zhang M, Goodfellow VS, Markison S, Gogas KR, Chen T, Wang XC, Zhu YF (2005) 1-(4-Amino-phenyl) pyrrolidin-3-yl-amine and 6-(3-amino-pyrrolidin-1-yl)-pyridin-3-yl-amine derivatives as melanin-concentrating hormone receptor-1 antagonists. Bioorg Med Chem Lett 15:3701–3706
Hvizdos KM, Markham A (1999) Orlistat: a review of its use in the management of obesity. Drugs 58:743–760
Ito M, Gomori A, Ishihara A, Oda Z, Mashiko S, Matsushita H, Yumoto M, Ito M, Sano H, Tokita S, Moriya M, Iwaasa H, Kanatani A (2003) Characterization of MCH-mediated obesity in mice. Am J Physiol Endocrinol Metabol 284:E940–E945
Iyengar RR, Lynch JK, Mulhern MM, Judd AS, Freeman JC, Gao J, Souers AJ, Zhao G, Wodka D, Falls HD, Brodjian S, Dayton BD, Reilly RM, Swanson S, Su Z, Martin RL, Leitza ST, Houseman KA, Diaz G, Collins CA, Sham HL, Kym PR (2007) An evaluation of 3,4-methylenedioxy phenyl replacements in the aminopiperidine chromone class of MCHr1 antagonists. Bioorg Med Chem Lett 17:874–878
Jezova D, Bartanusz V, Westergren I, Johansson BB, Rivier J, Vale W, Rivier C (1992) Rat melanin-concentrating hormone stimulates adrenocorticotropin secretion: evidence for a site of action in brain regions protected by the blood-brain barrier. Endocrinology 130:1024–1029
Judd AS, Souers AJ, Kym PR (2008) Lead optimization of melanin concentrating hormone receptor 1 antagonists with low hERG channel activity. Curr Top Med Chem 8:1152–1157
Kawauchi H, Kawazoe I, Tsubokawa M, Kishida M, Baker BI (1983) Characterization of melanin concentrating hormone in chum salmon pituitaries. Nature 305:321–323
Kennedy AR, Todd JF, Dhillo WS, Seal LJ, Ghatei MA, O’Toole CP, Jones M, Witty D, Winborne K, Riley G, Hervieu G, Wilson S, Bloom SR (2003) Effects of direct injection of melanin-concentrating hormone into the paraventricular nucleus: further evidence for a stimulatory role in the adrenal axis via SLC-1. J Neuroendocrinol 15:268–272
Knigge KM, Wagner JE (1997) Melanin-concentrating hormone (MCH) involvement in pentylenetetrazole (PTZ)-induced seizure in rat and guinea pig. Peptides 18:1095–1097
Kokkotou EG, Tritos NA, Mastaitis JW, Slieker L, Maratos-Flier E (2001) Melanin concentrating hormone receptor is a target of leptin action in the mouse brain. Endocrinology 142:680–686
Kopelman PG (2000) Obesity as a medical problem. Nature 404:635–643
Kowalski TJ, Mcbriar MD (2004) Therapeutic potential of melanin-concentrating hormone-1 receptor antagonists for the treatment of obesity. Expert Opin Investig Drugs 13:1113–1122
Kym PR, Iyengar R, Souers AJ, Lynch JK, Judd AS, Gao J, Freeman J, Mulhern M, Zhao G, Vasudevan A, Wodka D, Blackburn C, Brown J, Che JL, Cullis C, Lai SJ, LaMarche MJ, Marsilje T, Roses J, Sells T, Geddes B, Govek E, Patane M, Fry D, Dayton BD, Brodjian S, Falls D, Brune M, Bush E, Shapiro R, Knourek-Segel V, Fey T, McDowell C, Reinhart GA, Preusser LC, Marsh K, Hernandez L, Sham HL, Collins CA (2005) Discovery and characterization of aminopiperidinecoumarin melanin concentrating hormone receptor 1 antagonists. J Med Chem 48:5888–5891
Lembo PM, Grazzini E, Cao J, Hubatsch DA, Pelletier M, Hoffert C, St-Onge S, Pou C, Labrecque J, Groblewski T, O'Donnell D, Payza K, Ahmad S, Walker P (1999) The receptor for the orexigenic peptide melanin-concentrating hormone is a G protein-coupled receptor. Nat Cell Biol 1:267–271
Lucki I (1997) The forced swimming test as a model for core and component behavioral effects of antidepressant drugs. Behav Pharmacol 8:523–532
Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, Maratos-Flier E (2001) Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 107:379–386
Luthin DR (2007) Anti-obesity effects of small molecule melanin-concentrating hormone receptor1 (MCHR1) antagonists. Life Sci 81:423–440
Lynch JK, Collins CA, Freeman JC, Gao J, Iyengar RR, Judd AS, Kym PR, Mulhern MM, Sham HL, Souers AJ, Zhao G, Wodka D (2005) Preparation of piperidinyl chromenecarboxamides as antagonists of melanin concentrating hormone effects on the melanin concentrating hormone receptor. US 2005/209274
Macdonald D, Mugolo N, Zhang R, Durkin JP, Yao X, Strader CD, Graziano MP (2000) Molecular characterization of the melanin-concentrating hormone/receptor complex: identification of critical residues involved in binding and activation. Mol Pharmacol 58:217–225
Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen ASM, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, Van der Ploeg LH, Qian S (2002) Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A 99:3240–3245
McBride RB, Beckwith BE, Swenson RR, Sawyer TK, Hadley ME, Matsunaga TO, Hruby VJ (1994) The actions of melanin-concentrating hormone (MCH) on passive avoidance in rats: a preliminary study. Peptides 15:757–759
McBriar MD (2007) Melanin concentrating hormone receptor antagonists as antiobesity agents: from M2 to MCHR-1. Curr Top Med Chem 7:1440–1454
McBriar MD, Guzik H, Shapiro S, Paruchova J, Xu R, Palani A, Clader JW, Cox K, Greenlee WJ, Hawes BE, Kowalski TJ, O’Neill K, Spar BD, Weig B, Weston DJ, Farley C, Cook J (2006) Discovery of orally efficacious melanin-concentrating hormone receptor-1 antagonists as antiobesity agents. Synthesis, SAR, and biological evaluation of bicyclo[3.1.0]hexyl ureas. J Med Chem 49:2294–2310
McNeely W, Goa KL (1998) Sibutramine: a review of its contribution to the management of obesity. Drugs 56:1093–1124
Mendez-Andino JL, Wos JA (2007) MCH-R1 antagonists: what is keeping most research programs away from the clinic? Drug Discov Today 12:972–979
Miller CL, Hruby VJ, Matsunaga TO, Bickford PC (1993) Alpha-MSH and MCH are functional antagonists in a Central nervous system (CNS) auditory gating paradigm. Ann NY Acad Sci 680:571–574
Mori M, Harada M, Terao Y, Sugo T, Watanabe T, Shimomura Y, Abe M, Shintani Y, Onda H, Nishimura O, Fujino M (2001) Cloning of a novel G protein-coupled receptor, SLT, a subtype of the melanin-concentrating hormone receptor. Biochem Biophys Res Commun 283:1013–1018
Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM (2006) Prevalence of overweight and obesity in the United States, 1999–2004. J Am Med Assoc 295:1549–1555
Pereira-da-Silva M, Torsoni MA, Nourani HV, Augusto VD, Souza CT, Gasparetti AL, Carvalheira JB, Ventrucci G, Marcondes MC, Cruz-Neto AP, Saad MJ, Boschero AC, Carneiro EM, Velloso LA (2003) Hypothalamic melanin-concentrating hormone is Induced by cold exposure and participates in the control of energy expenditure in rats. Endocrinology 144:4831–4840
Pissios P, Bradley RL, Maratos-Flier E (2006) Expanding the scales: the multiple roles of MCH in regulating energy balance and other biological functions. Endocr Rev 27:606–620
Presse F, Nahon JL, Fischer WH, Vale W (1990) Structure of the human melanin concentrating hormone mRNA. Mol Endocrinol 4:632–637
Presse F, Hervieu G, Imaki T, Sawchenko PE, Vale W, Nahon JL (1992) Rat melanin-concentrating hormone messenger ribonucleic acid expression: marked changes during development and after stress and glucocorticoid stimuli. Endocrinology 131:1241–1250
Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E (1996) A role for melanin-concentrating hormone in the central regulation of feeding behavior. Nature 380:243–247
Rivera G, Bocanegra-Garcia V, Galiano S, Cirauqui N, Ceras J, Pérez S, Aldana I, Monge A (2008) Melanin-concentrating hormone receptor 1 antagonists: a new perspective for the pharmacologic treatment of obesity. Curr Med Chem 15:1025–1043
Rodriguez M, Beauverger P, Naime I, Rique H, Ouvry C, Souchaud S, Dromaint S, Nagel N, Suply T, Audinot V, Boutin JA, Galizzi JP (2001) Cloning and molecular characterization of the novel human melanin-concentrating hormone receptor MCH2. Mol Pharmacol 60:632–639
Rokosz LL (2007) Discovery and development of melanin-concentrating hormone receptor 1 antagonists for the treatment of obesity. Expert Opin Drug Discov 2:1301–1327
Rossi M, Beak SA, Choi SJ, Small CJ, Morgan DG, Ghatei MA, Smith DM, Bloom SR (1999) Investigation of the feeding effects of melanin concentrating hormone on food intake-action independent of galanin and the melanocortin receptors. Brain Res 846:164–170
Rovere C, Viale A, Nahon JL, Kitabgi P (1996) Impaired processing of brain pro-neurotensin and pro-melanin-concentrating hormone in obese fat/fat mice. Endocrinology 137:2954–2958
Sahu A (1998) Leptin decreases food intake induced by melanin-concentrating hormone (MCH), galanin (GAL) and neuropeptide Y (NPY) in the rat. Endocrinology 139:4739–4742
Sailer AW, Sano H, Zeng Z, McDonald TP, Pan J, Pong SS, Feighner SD, Tan CP, Fukami T, Iwaasa H, Hreniuk DL, Morin NR, Sadowski SJ, Ito M, Bansal A, Ky B, Figueroa DJ, Jiang Q, Austin CP, MacNeil DJ, Ishihara A, Ihara M, Kanatani A, Vander Ploeg LH, Howard AD, Liu Q (2001) Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proc Natl Acad Sci U S A 98:7564–7569
Saito Y, Nothacker HP, Wang Z, Lin SH, Leslie F, Civelli O (1999) Molecular characterization of the melanin-concentrating hormone receptor. Nature 400:265–269
Sasikumar TK, Qiang L, Burnett DA, Greenlee WJ, Hawes BE, Kowalski TJ, O'Neill K, Spar BD, Weig B (2006) Novel aminobenzimidazoles as selective MCH-R1 antagonists for the treatment of metabolic diseases. Bioorg Med Chem Lett 16:5427–5431
Semple G (2004) Identification and biological activity of a new series of antagonists of hMCH-R1. Presented at the 228th National American Chemical Society Meeting, Philadelphia, PA, August 22, Medi 007
Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E (1998) Mice lacking melanin concentrating hormone are hypophagic and lean. Nature 396:670–674
Shimomura Y, Mori M, Sugo T, Ishibashi Y, Abe M, Kurokawa T, Onda H, Nishimura O, Sumino Y, Fujino M (1999) Isolation and identification of melanin-concentrating hormone as the endogenous ligand of the SLC-1 receptor. Biochem Biophys Res Commun 261:622–626
Skofitsch G, Jacobowitz DM, Zamir N (1985) Immunohistochemical localization of a melanin concentrating hormone-like peptide in the rat brain. Brain Res Bull 15:635–649
Smith DG, Davis RJ, Rorick-Kehn L, Morin M, Witkin JM, McKinzie DL, Nomikos GG, Gehlert DR (2006) Melanin-concentrating hormone-1 receptor modulates neuroendocrine, behavioral, and corticolimbic neurochemical stress responses in mice. Neuropsychopharmacol 31:1135–1145
Souers AJ, Gao J, Wodka D, Judd AS, Mulhern MM, Napier JJ, Brune ME, Bush EN, Brodjian SJ, Dayton BD, Shapiro R, Hernandez LE, Marsh KC, Sham HL, Collins CA, Kym PR (2005) Synthesis and evaluation of urea-based indazoles as melanin concentrating hormone receptor 1 antagonists for the treatment of obesity. Bioorg Med Chem Lett 15:2752–2757
Souers AJ, Iyengar RR, Judd AS, Beno DW, Gao J, Zhao G, Brune ME, Napier JJ, Mulhern MM, Lynch JK, Freeman JC, Wodka D, Chen CJ, Falls HD, Brodjian S, Dayton BD, Diaz GJ, Bush EN, Shapiro R, Droz BA, Knourek-Segel V, Hernandez LE, Marsh KC, Reilly RM, Sham HL, Collins CA, Kym PR (2007) Constrained 7-fluorocarboxychromone-4-aminopiperidine based melanin concentrating hormone receptor 1 antagonists: the effects of chirality on substituted indan 1-ylamines. Bioorg Med Chem Lett 17:884–889
Staten MA (2007) Challenges in the discovery and development of new agents for the treatment of obesity. Clin Pharmacol Ther 81:753–755
Stricker-Krongrad A, Dimitrov T, Beck B (2001) Central and peripheral dysregulation of melanin concentrating hormone in obese Zucker rats. Brain Res Mol Brain Res 92:43–48
Tadayyon M, Welters HJ, Haynes AC, Cluderay JE, Hervieu G (2000) Expression of melanin concentrating hormone receptors in insulin-producing cells: MCH stimulates insulin release in RINm5F and CRI-G1 cell lines. Biochem Biophys Res Commun 275:709–712
Takekawa S, Asami A, Ishihara Y, Terauchi J, Kato K, Shimomura Y, Mori M, Murakoshi H, Kato K, Suzuki N, Nishimura O, Fujino M (2002) T-226296: a novel, orally active and selective melanin-concentrating hormone receptor antagonist. Eur J Pharmacol 438:129–135
Tan CP, Sano H, Iwaasa H, Pan J, Sailer AW, Hreniuk DL, Feighner SD, Palyha OC, Pong SS, Figueroa DJ, Austin CP, Jiang MM, Yu H, Ito J, Ito M, Guan XM, MacNeil DJ, Kanatani A, Van der Ploeg LHT, Howard AD (2002) Melanin-concentrating hormone receptor subtypes 1 and 2: species-specific gene expression. Genomics 79:785–792
Tavares FX, Al-Barazanji KA, Bigham EC, Bishop MJ, Britt CS, Carlton DL, Feldman PL, Goetz AS, Grizzle MK, Guo YC, Handlon AL, Hertzog DL, Ignar DM, Lang DG, Ott RJ, Peat AJ, Zhou HQ (2006a) Potent, selective, and orally efficacious antagonists of melanin-concentrating hormone receptor 1. J Med Chem 49:7095–7107
Tavares FX, Al-Barazanji KA, Bishop MJ, Britt CS, Carlton DL, Cooper JP, Feldman PL, Garrido DM, Goetz AS, Grizzle MK, Hertzog L, Ignar DM, Lang DG, McIntyre MS, Ott RJ, Peat AJ, Zhou HQ (2006b) 6-(4-Chlorophenyl)-3-substituted-thieno[3,2-d]pyrimidin-4(3H)-one-based melanin concentrating hormone receptor 1 antagonist. J Med Chem 49:7108–7118
Tritos NA, Elmquist JK, Mastaitis JW, Flier JS, Maratos-Flier E (1998) Characterization of expression of hypothalamic appetite-regulating peptides in obese hyperleptinemic brown adipose tissue-deficient (uncoupling protein-promoter-driven diphtheria toxin A) mice. Endocrinology 139:4634–4641
Varas MM, Perez MF, Ramirez OA, de Barioglio SR (2003) Increased susceptibility to LTP generation and changes in NMDA-NR1 and NR-2B subunits mRNA expression in rat hippocampus after MCH administration. Peptides 24:1403–1411
Vasudevan A, Souers AJ, Freeman JC, Verzal MK, Gao J, Mulhern MM, Wodka D, Lynch JK, Engstrom KM, Wagaw SH, Brodjian S, Dayton B, Falls DH, Bush E, Brune M, Shapiro RD, Marsh KC, Hernandez LE, Collins CA, Kym PR (2005) Aminopiperidine indazoles as orally efficacious melanin concentrating hormone receptor-1 antagonists. Bioorg Med Chem Lett 15:5293–5297
Vaughan JM, Fischer WH, Hoeger C, Rivier J, Vale W (1989) Characterization of melanin concentrating hormone from rat hypothalamus. Endocrinology 125:1660–1665
Viale A, Zhixing Y, Breton C, Pedeutour F, Coquerel A, Jordan D, Nahon JL (1997) The melanin-concentrating hormone gene in human: flanking region analysis, fine chromosome mapping, and tissue-specific expression. Brain Res Mol Brain Res 46:243–255
Wang S, Behan J, O’Neill K, Weig B, Fried S, Laz T, Bayne M, Gustafson E, Hawes BE (2001) Identification and pharmacological characterization of a novel human melanin concentrating hormone receptor, MCH-R2. J Biol Chem 276:34664–34670
Wermter AK, Reichwald K, Buch T, Geller F, Platzer C, Huse K, Hess C, Remschmidt H, Gudermann T, Preibisch G, Siegfried W, Goldschmidt HP, Li WO, Price RA, Biebermann H, Krude H, Vollmert C, Wichmann HE, Illig T, Sorensen TI, Astrup A, Larsen LH, Pedersen O, Eberle D, Clement K, Blundell J, Wabitsch M, Schafer H, Platzer M, Hinney A, Hebebrand J (2005) Mutation analysis of the MCHR1 gene in human obesity. Eur J Endocrinol 152:851–862
Wu WL, Burnett DA, Caplen MA, Domalski MS, Bennett C, Greenlee WJ, Hawes BE, O’Neill K, Weig B, Weston D, Spar B, Kowalski T (2006) Design and synthesis of orally efficacious benzimidazoles as melanin-concentrating hormone receptor 1 antagonists. Bioorg Med Chem Lett 16:3674–3678
Zhang, P, Liand, JD, Samdusky, GE (1999) Hypothalamic MCH mRNA and protein are increased in human obesity. Satellite Symposium; 9th International Congress of Obesity France, Proceedings of the Symposium. Int J Obes, p 51
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Cheon, H.G. (2012). Antiobesity Effects of Melanin-Concentrating Hormone Receptor 1 (MCH-R1) Antagonists. In: Joost, HG. (eds) Appetite Control. Handbook of Experimental Pharmacology, vol 209. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-24716-3_18
Download citation
DOI: https://doi.org/10.1007/978-3-642-24716-3_18
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-24715-6
Online ISBN: 978-3-642-24716-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)