
Abstract
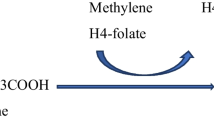
Nonketotic hyperglycinaemia (NKH) or glycine encephalopathy is an autosomal recessive disorder characterised by a rapidly progressive course in the neonatal period or early infancy. Symptoms include muscular hypotonia, seizures, apnoeic attacks, lethargy and coma. Most patients die within a few weeks, whilst survivors show severe psychomotor retardation. Increased glycine concentrations in plasma, urine, and cerebrospinal fluid are biochemical features of the disorder. The primary lesion is a defect in the glycine cleavage system (GCS) (◘ Fig. 24.1). Although this was first demonstrated in the liver, involvement within the brain is responsible for the clinical expression. No specific treatment is available. Prenatal diagnosis is possible by determining the activity of GCS in chorionic villi and by molecular analysis.
Chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
References
[1] Tada K, Kure S (1993) Non-ketotic hyperglycinaemia: molecular lesion, diagnosis and pathophysiology. J Inherit Metab Dis 16:691–703
[2] Dobyns WB (1989) Agenesis of the corpus callosum and gyral malformations are frequent manifestations of nonketotic hyperglycinemia. Neurology 39:817–820
[3] Paupe A, Bidat L, Sonigo P et al. (2002) Prenatal diagnosis of hypoplasia of the corpus callosum in association with non-ketotic hyperglycinemia. Ultrasound Obstet Gynecol 20:616–619
[4] Ozyürek H, Turanli G, Aliefendioglu D, Coskun T (2005) Repetitive EEG recordings are necessary for the diagnosis of early myoclonic encephalopathy. Neurol India 53:235–237
[5] Mourmans J, Majoie CB, Barth PG et al. (2006) Sequential MR imaging changes in nonketotic hyperglycinemia. AJNR Am J Neuroradiol 27:208–211
[6] Hoover-Fong JE, Shah S, Van Hove JL et al. (2004) Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 63:1847–1853
[7] Huisman TA, Thiel T, Steinmann B et al. (2002) Proton magnetic resonance spectroscopy of the brain of a neonate with nonketotic hyperglycinemia: in vivo-in vitro (ex vivo) correlation. Eur Radiol 2002 12:858–861
[8] Huisman TA, Thiel T, Steinmann B et al. (2002) Proton magnetic resonance spectroscopy of the brain of a neonate with nonketotic hyperglycinemia: in vivo-in vitro (ex vivo) correlation, Eur Radiol 12:858–861
[9] Cataltepe S, Marter v LJ, Kozakewich H et al. (2000) Pulmonary hypertension associated with nonketotic hyperglycinaemia. J Inherit Metab Dis 23:137–144
[10] Chien YH, Hsu CC, Huang A et al. (2004) Poor outcome for neonatal- type nonketotic hyperglycinemia treated with high-dose sodium benzoate and dextromethorphan. J Child Neurol 19:39–42
[11] Luder AS, Davidson A, Goodman SI, Greene CL (1989) Transient nonketotic hyperglycinemia in neonates. J Pediatr 114:1013–1015
[12] Zammarchi E, Donati MA, Ciani F (1995) Transient neonatal nonketotic hyperglycinemia: a 13-year follow-up. Neuropediatrics 26:328–330
[13] Manley BJ, Sokol J, Cheong JL (2010) Intracerebral blood and MRS in neonatal nonketotic hyperglycinemia. Pediatr Neurol 42:219–222
[14] Korman SH, Boneh A, Ichinohe A et al. ( 2004) Persistent NKH with transient or absent symptoms and a homozygous GLDC mutation. Ann Neurol 56:139–143
[15] Kure S, Kojima K, Ichinohe A et al. (2002) Heterozygous GLDC and GCSH gene mutations in transient neonatal hyperglycinemia. Ann Neurol 52:643–646
[16] Lang TF, Parr JR, Matthews EE et al. (2008) Practical difficulties in the diagnosis of transient non-ketotic hyperglycinaemia. Dev Med Child Neurol 50:157–159
[17] Flusser H, Korman SH, Sato K et al. (2005) Mild glycine encephalopathy (NKH) in a large kindred due to a silent exonic GLDC splice mutation. Neurology 64:1426–1430
[18] Suzuki Y, Kure S, Oota M et al. (2010) Nonketotic hyperglycinemia: proposal of a diagnostic and treatment strategy. Pediatr Neurol 43:221–224
[19] Boneh A, Allan S, Mendelson D et al. (2008) Clinical, ethical and legal considerations in the treatment of newborns with nonketotic hyperglycinaemia. Mol Genet Metab 94:143–147
[20] Dinopoulos A, Kure S, Chuck G et al. (2004) Atypical non-ketotic hyperglycinemia: 3 cases with GLDC mutations (poster). J Inherit Metab Dis 27 [Suppl 1]:62
[21] Del Toro M, Macaya A, Moreno et al. (2004) Rapidly progressive infantile leukoencephalopathy associated with nonketotic hyperglycinemia and pulmonary hypertension (poster). J Inherit Metab Dis 27 [Suppl 1]:61
[22] Morrison PF, Sankar R, Shields WD (2006) Valproate-induced chorea and encephalopathy in atypical nonketotic hyperglycinemia. Pediatr Neurol 35:356–358
[23] Bekiesiniska-Figatowska M, Rokicki D, Walecki J (2001) MRI in nonketotic hyperglycinaemia: case report. Neuroradiology 43:792–793
[24] Chiong MA, Procopis P, Carpenter K, Wilcken B (2007) Late-onset nonketotic hyperglycinemia with leukodystrophy and an unusual clinical course. Pediatr Neurol 37:283–286
[25] Lin FY, Gascon GG, Hyland K et al. (2006) Transient nonketotic hyperglycinemia and defective serotonin metabolism in a child with neonatal seizures. J Child Neurol 21:900–903
[26] Ellaway CJ, Mundy H, Lee PJ ( 2001) Successful pregnancy outcome in atypical hyperglycinaemia. J Inherit Metab Dis 24:599–600
[27] Hasegawa T, Shiga Y, Matsumoto A et al. (2002) Late-onset nonketotic hyperglycinemia: a case report. No To Shinkei 54:1068–1072
[28] Toone JR, Applegarth DA, Coulter-Mackie MB, James ER ( 2000) Biochemical and molecular investigations of patients with nonketotic hyperglycinemia. Mol Genet Metab 70:116–121
[29] Wasterlain CG, Shirasaka Y (1994) Seizures, brain damage and brain development. Brain Dev 16:279–295
[30] Sato K, Yoshiada S, Fujiwara K et al. (1991) Glycine cleavage system in astrocytes. Brain Res 567:64–70
[31] Ben Ari Y, Khazipov R, Leinekugel X et al. (1997) GABA-A, NMDA and AMPA receptors: a developmentally regulated »menage à trois«. Trends Neurosci 20:523–529
[32] Molinari F, Raas-Rothschild A, Rio M et al. (2005) Impaired mitochondrial glutamate transport in autosomal recessive neonatal myoclonic epilepsy. Am J Hum Genet 76:334–339
[33] Milh M, Becq H, Villeneuve N et al. (2009) Inhibition of glutamate transporters results in suppression-burst pattern and partial seizures in the newborn rat. Epilepsia 48:169–174
[34] Busanello EN, Moura AP, Viegas CM et al. (2010) Neurochemical evidence that glycine induces bioenergetical dysfunction. Neurochem Int 56:948–954
[35] Aprison MH, Werman R (1965) The distribution of glycine in cat spinal cord and roots. Life Sci 4:2075–2083
[36] Kure S, Narisawa K, Tada K (1991) Structural and expression analyses of normal and mutant mRNA encoding glycine decarboxylase: three-base deletion in mRNA causes nonketotic hyperglycinemia. Biochem Biophys Res Commun 174:1176–1182
[37] Hyasaka K, Nanao K, Takada G et al. (1993) Isolation and sequence determination of cDNA encoding human T-protein of the glycine cleavage system. Biochem Biophys Res Commun 192:766–771
[38] Applegarth DA, Toone JR (2004) Glycine encephalopathy (nonketotic hyperglycinaemia): review and update. J Inherit Metab Dis 27:417–422
[39] Applegarth DA, Toone JR (2001) Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab 74:139–146
[40] Kure S, Takayanagi M, Kurihara Y et al. (1999) Nonketotic hyperglycinemia: mutation spectra of the GLDC and AMT gene in Finnish and non-Finnish populations. Am J Hum Genet 65:A2406
[41] Toone JR, Applegarth DA, Kure S et al. (2002) Novel mutations in the P-protein (glycine decarboxylase) gene in patients with glycine encephalopathy (non-ketotic hyperglycinemia). Mol Genet Metab 76:243–249
[42] Hove v JLK, Mahieu V, Schollen E (2004) Prognosis in nonketotic hyperglycinemia. J Inherit Metab Dis 26:71
[43] Kanno J, Hutchin T, Kamada F et al. (2007) Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J Med Genet 44:e69
[44] Kure S, Narisawa K, Tada K(1992) Enzymatic diagnosis of nonketotic hyperglycinemia with lymphoblasts. J Pediatr 120:95–98
[45] Tan ES, Wiley V, Carpenter K, Wilcken B (2007) Non-ketotic hyperglycinemia is usually not detectable by tandem mass spectrometry newborn screening. Mol Genet Metab 90:446–448
[46] Vigevano F, Maccagnani F, Bertini E et al. (1982) Enceflopatia mioclonica precoce associata ad alti livelli di acido propioico nel siero. Boll Lega It Epil 39:181–182
[47] Dreyfus-Brisac C, Cukier F (1969) Le tracé paroxystique: sa valeur pronostique selon le degré de prématurité. Rev Neuropsychiatr Infant 17:795–802
[48] Pampiglione G (1962) Electroencephalographic studies after cardiorespiratory resuscitation. Proc R Soc Med 55:653–657
[49] Lombroso CT (1990) Early myoclonic encephalopathy, early infantile epileptic encephalopathy, and benign and severe infantile myoclonic epilepsies: a critical review and personal contributions. J Clin Neurophysiol 7:380–408
[50] Schlumberger E, Dulac O, Plouin P (1992) Syndrome of neonatal epilepsy. Epilepsy syndromes in childhood and adolescence. Libbey, London Paris Rome, pp 35–42
[51] Maeda T, Inutsuka M, Goto K, Izumi T (2000) Transient nonketotic hyperglycinemia in an asphyxiated patient with pyridoxinedependent seizures. Pediatr Neurol 22:225–227
[52] Aukett A, Bennett MJ, Hosking GP (1988) Molybdenum cofactor deficiency: an early missed inborn error of metabolism. Dev Med Child Neurol 30:531–535
[53] Dalla Bernardina B, Dulac O, Fejerman N et al. (1983) Early myoclonic epileptic encephalopathy (EMEE). Eur J Pediatr 140:248–252
[54] Wolf NI,Garcia-Cazorla A, Hoffmann GF (2009) Epilepsy and inborn error of metabolism in children. J Inherit Metab Dis 32:609–617
[55] Ohtahara S (1978) Clinico-electrical delineation of epileptic encephalopathies in childhood. Asian Med J 21:499–509
[56] Knaap v d MS, Wevers RA, Kure S et al. (1999) Increased cerebrospinal fluid glycine: a biochemical marker for a leukoencephalopathy with vanishing white matter. J Child Neurol 14:728–731
[57] Sener RN(2003) The glycine peak in brain diseases. Comput Med Imaging Graph 27:297–305
[58] Applegarth DA, Toone JR, Rolland MO et al. (2000) Non-concordance of CVS and liver glycine cleavage enzyme in three families with non-ketotic hyperglycinaemia (NKH) leading to false negative prenatal diagnoses. Prenat Diagn 20:367–370
[59] Vianey-Saban C, Chevalier-Porst F, Froissart R, Rolland MO (2003) Prenatal Diagnosis of nonketotic Hyperglycinemia: a 13-year experience, from enzymatic to molecular analysis. J Inherit Metab Dis 26 [Suppl 2]:164-P
[60] Garcia-Munoz MJ, Belloque J, Merinero B et al. (1989) Non-ketotic hyperglycinaemia: glycine/serine ratio in amniotic fluid -- an unreliable method for prenatal diagnosis. Prenat Diagn 9:473–476
[61] Palekar A (2000) Effect of panthotenic acid on hippurate formation in sodium benzoate-treated HepG2 cells. Pediatr Res 48:357–359
[62] Wiltshire EJ, Poplawski NK, Harrison JR, Flechter JM (2000) Treatment of late-onset nonketotic hyperglycinaemia: effectiveness of imipramine and benzoate. J Inherit Met Dis 23:15–21
[63] Matsuo S, Inoue F, Takeuchi Y et al. (1995) Efficacy of tryptophan for the treatment of nonketotic hyperglycinemia: a new therapeutic approach for modulating the N-methyl-d-aspartate receptor. Pediatrics 95:142–146
[64] Tekgul H, Serdaroğlu G, Karapinar B et al. (2006) Vigabatrin caused rapidly progressive deterioration in two cases with early myoclonic encephalopathy associated with nonketotic hyperglycinemia. J Child Neurol 21:82–84
[65] Tsao CY (2010) The efficacy of vagus nerve stimulation in intractable epilepsy associated with nonketotic hyperglycinemia in two children. J Child Neurol 25:375–378
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Dulac, O., Rolland, MO. (2012). Nonketotic Hyperglycinaemia (Glycine Encephalopathy). In: Saudubray, JM., van den Berghe, G., Walter, J.H. (eds) Inborn Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-15720-2_24
Download citation
DOI: https://doi.org/10.1007/978-3-642-15720-2_24
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-15719-6
Online ISBN: 978-3-642-15720-2
eBook Packages: MedicineMedicine (R0)