
Abstract
The maturation of the SARS coronavirus (CoV) involves the autocleavage of polyproteins 1a and 1ab by a main protease and papain-like protease. The functional unit of the main protease is a dimer in which each subunit has a Cys145–His41 catalytic dyad, with His41 acting as a general base. There is also a close correlation between dimer formation and the enzyme catalytic activity. A flip-flop mechanism is proposed for the main protease, in which the two subunits are used alternately in acylation and deacylation. Both the main protease and the papain-like protease are ideal targets for rational drug design strategies against SARS-CoV.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Severe Acute Respiratory Syndrome
- Quaternary Structure
- Severe Acute Respiratory Syndrome
- Main Protease
- Severe Acute Respiratory Syndrome Coronavirus
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
In 2003, an atypical and highly contagious pneumonia, severe acute respiratory syndrome (SARS), caused a global health crisis. After a worldwide intensive investigation, a specific novel form of human coronavirus (CoV), denoted SARS-CoV, was identified as the pathogenic agent behind this epidemic (Peiris et al. 2004). The maturation and production of the infectious progeny of SARS-CoV involves proteolytic processing of the virus polyproteins by a main protease (Mpro) (also called 3CLpro because of its 3′-proximal chymotrypsin-like catalytic domain) and a papain-like protease (PLpro).
This chapter summarizes the present knowledge of the structure and function of the SARS-CoV Mpro protein. Particular attention is paid to the quaternary structure of this protease, as the catalytically active form of this enzyme is a dimer. The structure and function of PLpro is also touched upon briefly. The current state of play in anti-SARS drug development strategies that target Mpro is discussed in the chapter following (Chap. 9).
2 Molecular Biology of the SARS-CoV Polyproteins
The genomic organization of SARS-CoV is similar to that of other coronaviruses, but phylogenetic analysis and sequence comparisons of the viral proteins indicate that SARS-CoV is in fact different from any of the previously characterized viruses of this type (Tanner et al. 2003; Eickmann et al. 2003). Coronaviruses are enveloped positive-sense, single-strand RNA viruses. The genome length of SARS-CoV is around 30,000 nucleotides and its replicase gene encodes two overlapping polyproteins, polyprotein 1a (pp1a) (486 kDa) and polyprotein 1ab (1a + 1b) (790 kDa) (Fig. 8.1a). These polyproteins are extensively cleaved by the internally encoded SARS-CoV proteases, Mpro and PLpro.


Polyproteins of SARS-CoV. (a) Autoprocessing of polyprotein 1a and 1ab (1a + 1b) by Mpro occurs at 11 sites (green triangles) and of PLpro at three sites (yellow triangles) resulting in the maturation of 16 non-structural proteins (nsp). The locations of the Mpro and PLpro enzymes are at nsp 5 and 3, respectively. (b) Substrate specificity of Mpro and PLpro. Redundant residue positions are labeled with an X
The 33.8-kDa Mpro plays a major role in the proteolytic processing of the virion polyproteins and cleaves pp1a at seven sites and pp1b at four sites (Fig. 8.1a). The 35-kDa protease PLpro cleaves pp1a at three sites. These autoprocessing reactions result in the maturation of 16 nonstructural proteins (nsp), including those that are common to the replication of plus-strand RNA viruses: the RNA-dependent RNA polymerase at nsp12 and helicase at nsp13. The Mpro and PLpro enzymes are themselves located at nsp5 and nsp3, respectively.
These proteases are involved in the viral life cycle, the maturation of the precapsid, and in the production of infectious virions. Viral protease inhibitors would thus be predicted to have great clinical potential in the treatment of the associated infectious diseases (Krausslich and Wimmer 1988; Tong 2002). The structure–function relationship of these proteases has therefore received much recent attention in the search for an effective anti-SARS-CoV agent (Anand et al. 2003; Yang et al. 2005; Wei et al. 2006; Lai et al. 2006; Liang 2006; Bartlam et al. 2008).
3 Structure of the SARS-CoV Main Protease
3.1 Three-Dimensional Structure of the SARS-CoV Main Protease
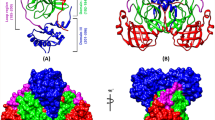
Mpro was the first of the SARS-CoV proteins to have its three-dimensional structure solved by crystallography (Fig. 8.2) (Yang et al. 2003; Bartlam et al. 2005, 2007). This protease is a homodimer in which the two subunits are arranged perpendicularly to each other (Fig. 8.3a). Each protomer of SARS-CoV Mpro comprises 13 strands and 11 helices distributed among three distinct structural domains. The first two domains (residues 8–101 for domain I and 102–184 for domain II) have an antiparallel β-barrel structure, which has a folding scaffold similar to other viral chymotrypsin-like proteases (Anand et al. 2002; Hegyi et al. 2002; Ziebuhr et al. 2003). Each subunit also has its own substrate binding site with a His41–Cys145 catalytic dyad located at the interface between domains I and II. However, unlike chymotrypsin, the active site of SARS-CoV Mpro contains a catalytic cysteinyl residue instead of a seryl residue. Furthermore, SARS-CoV Mpro contains an extra domain (III) consisting of five α-helices (residues 201–306), which is a specific feature of coronavirus main proteases. This helical domain III is linked to domain II by a long loop (residues 185–200) (Fig. 8.2a).


Structure of SARS-CoV Mpro. (a) Ribbon presentation of the SARS-CoV Mpro dimer (pdb code: 1Z1J). Domains I (blue) and II (green) constitute a chymotrypsin-like folding scaffold with a catalytic dyad comprising His41 and Cys145 (alanine in 1Z1J) shown using a bond-and-ball model (red). This catalytic domain is linked to an extra domain III (red) by a long loop (yellow). The N-finger (magenta) of subunit A protrudes into the active site region of subunit B, which is shown in gray. (b) Structural analysis of Mpro. The primary amino acid sequence is displayed along with secondary structural elements, crystallographic contact, and hydropathy. Panel (a) was generated using MacPymol (DeLano 2002) and panel (b) with ENDscript (Gouet et al. 2003)


Quaternary structure of SARS-CoV Mpro. (a) Surface model of Mpro. (b) Subunit contacts at the Mpro dimer interface. Hydrogen bonds (cyan), non-bonded interactions (orange), and the interacting amino acid residues are shown by residue conservation. The width of the striped line is proportional to the number of atomic contacts. (c) Surface potential of the contacting regions. Upper panels show the interfacial region within a 10 Å distance of subunits A and B. In the lower panels, the hollow regions represent the direct contact regions in subunits A and B. Positively charged areas are shown in blue and negatively charged areas in red. Panel (a) was generated using MacPymol (DeLano 2002), panel (b) with PdbSum (Laskowski et al. 2005), and panel (c) with Spock (Christopher 1998)
The catalytic N-terminal domain (I + II) and C-terminal domain III of SARS-CoV Mpro can fold independently. The N-terminal domain (I + II) without domain III folds into a structure that is indistinguishable from the intact chymotrypsin-like fold but is enzymatically inactive (Chang et al. 2007). The extra domain III of SARS-CoV Mpro increases the structural stability of the catalytic domain. This may be achieved by increasing the folding rate of domains I and II, which would thus increase the overall stability of the protein. Furthermore, domain III is related to the quaternary structure of Mpro, which has important functional implications for this enzyme.
3.2 Quaternary Structure of the SARS-CoV Main Protease
A coronavirus Mpro (from transmissible gastroenteritis virus) was the first viral protease shown to be dimeric, both in its crystal form and in solution (Anand et al. 2002). Significantly, SARS-CoV Mpro also exists as a dimer in solution (Chou et al. 2004) and both its N- and C-terminal residues are involved in dimer formation. The N-terminal finger (N-finger, containing residues 1–7) of subunit A extends from domain I toward domain III and forms intensive interactions with subunit B. The side chain of Arg4 at the N-finger fits into a pocket of subunit B and forms a salt bridge with Glu290 that constitutes one of the major interactions between the two subunits (Chou et al. 2004). In addition, the subunit interfacial region of the enzyme contains many hydrophobic interactions and hydrogen bonds (Fig. 8.3b). The interactions between the N-terminus and the other monomer play an important role in maintaining the active site integrity of the dimer (Lin et al. 2008; Chen et al. 2008b; Zhong et al. 2008). Importantly, the dimeric form of SARS-CoV Mpro is the biologically functional form of this enzyme (Anand et al. 2002; Shi et al. 2008) and dissociation of the subunits yields enzymatically inactive monomers (Shi et al. 2004; Fan et al. 2004; Chang et al. 2007).
The functional role of the N-terminus and C-terminus of Mpro has been evaluated by truncation and mutation studies. Both N-terminal and C-terminal regions are involved in the activity of this enzyme as well as in its dimerization. N-terminal truncation of the whole N-finger results in almost complete loss of enzymatic activity (Hsu et al. 2005b). Critical N-terminal amino acid residues to Arg4 and C-terminal to Gln299 have been identified as those involved in dimerization, thus generating the correct conformation of the active site (Hsu et al. 2005b; Lin et al. 2008). The C-terminal helical domain interacts with the active site of another protomer in the dimer and switches the enzyme molecule from the inactive form to the active form (Shi et al. 2004). Hsu et al. (2005a) have proposed an autocleavage mechanism, which explains the dimeric nature of the mature enzyme.
4 Enzyme Activity-Assay for the SARS-CoV Main Protease
The hydrolytic activity of Mpro can be assayed by its ability to cleave a peptide substrate. A procedure to separate the substrate and product peptides by high performance liquid chromatography has now been developed (Fan et al. 2004). The reaction is monitored by the formation of products peaks from the substrate peak and this method is thus very useful in the identification of cleaved peptide products. However, this procedure is very labor-intensive and thus not suitable for high throughput screening protocols.
Various fluorescence-based methods have also been developed for Mpro in which the enzyme activity can be assayed using an internally quenched fluorogenic substrate peptide (e.g., ortho-aminobenzoic acid-peptide-2,4-dinitrophenyl amide) (Chou et al. 2004). Enhanced fluorescence due to cleavage of the peptide can be monitored at 420 nm with excitation at 362 nm using a luminescence spectrometer. For precise quantitation, a calibration curve under identical conditions should be constructed with equal amounts of hydrolytic products.
Alternatively, colorimetric methods (e.g., p-nitroanilide-peptide based assays) have been adopted for use in an Mpro assay (Huang et al. 2004). The chromophore p-nitroanilide has a known absorbance that is conductive to quantitation. In addition, colorimetric assay methods do not require inner filter effect corrections, which are essential for fluorimetric assays, and photometric devices are less expensive.
Fluorimetric analysis is generally at least 10-fold more sensitive than a colorimetric-based assay. However, its intrinsic insensitivity is in fact a distinct advantage of using colorimetric analysis for Mpro. In the case of Mpro activity, to confirm a direct correlation between the quaternary structure of the protease and its enzyme activity, a method that can simultaneously monitor protein dissociation and enzyme inactivation is highly desirable. Analytical ultracentrifugation with a band-forming centerpiece is ideal for this task (Harding and Rowe 1996). The substrate is diluted in buffer and loaded into the ultracentrifuge cell. Deuterium oxide, sucrose, or glycerol can be used to increase the density of this substrate solution. The band-forming centerpiece has small drilled-out holes to accommodate the enzyme solution, which is separated from the substrate before the application of the centrifugal force. Upon commencing centrifugation, the enzyme solution, which is less dense than the substrate solution, will form a thin layer on top of the separation column. The enzymatic reaction will start at the bound interface. During ultracentrifugation, the separation of monomer and dimer bands can be monitored by UV absorption, whereas the enzyme activity levels can be assessed by the absorption at 405 nm if p-nitroanilide-peptide is used. The advantage of then using a colorimetric detector is that the k cat value of Mpro is low enough to allow large amounts of protein to be used. If fluorogenic substrates are used, the sensitivity of enzymatic reaction will be such that the protein levels may be below the threshold of detection.
The Mpro protease is an ideal model for the analysis of the correlation between quaternary structure and enzyme activity (Barrila et al. 2006; Shi and Song 2006). This is due partly to the fact that it is a relatively simple dimeric system and that it has only a moderate catalytic efficiency. To date, no activity has been detected for the Mpro monomer.
5 Catalytic Mechanism of the SARS-CoV Main Protease
SARS-CoV Mpro cleaves pp1a at 11 sites containing the canonical Leu–Gln–↓–(Ala/Ser) sequence (Fig. 8.1b). The first step in this process involves binding of the substrate at the enzyme active center, which forms a Michaelis complex. An electrostatic trigger mediated by Cys145 at the susceptible peptide bond initiates the chemical reaction. Acylation of the sulfhydryl group of this cysteine results in a covalent link between the C-terminal moiety of the substrate and the SH group and the release of the N-terminal moiety (Fig. 8.4a). Finally, deacylation and release of the C-terminal moiety completes the reaction (Solowiej et al. 2008).


Reaction mechanism of SARS-CoV Mpro. (a) Catalytic mechanism of Mpro. The binding of a peptide substrate to the active site forms a Michaelis complex. The peptide substrate is then cleaved at the Gln–Ala (or Gln–Ser) peptide bond. The N-terminal half peptide is released as the first product whereas the C-terminal half acylates the active site Cys145 residue. The acylated intermediate is then deacylated, releasing the C-terminal peptide, and this completes the catalytic cycle. His41 acts as a catalytic general base in the deacylation step. (b) A proposed flip-flop mechanism for the possible role of the Mpro quaternary structure in the regulation of its activity. The active subunit is indicated by the circle, and the inactive subunit is shown as a square. Only one of the two subunits is catalytically active at any one time and the two subunits thus exist in an alternate active–inactive cycle. Substrate binding at one subunit induces the deacylation of the other
The –SH group of Cys145 is ion-paired with a nearby histidine residue (His41). This forms the catalytic dyad (Cys145–His41), which differs from most serine proteases that have a catalytic Ser–His–Asp triad in their active sites. In Mpro, a stable water molecule occupies the Asp position of the typical serine protease triad and this molecule might play a role in stabilizing the imidazolium ring during catalysis (Bartlam et al. 2005). Mutations at the catalytic dyad residues (H41A and C145A) almost completely abolish enzymatic activity and these mutant enzymes exist exclusively as dimers (Huang et al. 2004; Chang et al. 2007). However, mutation of Cys145 to Ser results in a partially active enzyme. These results are consistent with the notion that in the chemical mechanism underlying Mpro activity, His41 acts as a general base during the deacylation step and that the catalytic dyad involving Cys145 and His41 is left uncharged (Huang et al. 2004; Solowiej et al. 2008).
The rate-limiting step for Mpro hydrolysis is the covalent deacylation step. There is a close correlation here between the kinetic parameters and subunit dissociation constant. Mpro subunit dissociation affects catalysis but not substrate binding (Lin et al. 2008). Molecular dynamic simulations have also demonstrated an asymmetric dimer and inactivation of the enzyme after dissociation (Tan et al. 2005; Chen et al. 2006; Zheng et al. 2007). Crystal structures of the monomeric Mpro provide direct structural evidence for the catalytic incompetence of the dissociated monomer (Chen et al. 2008a; Shi et al. 2008).
An association–activation–catalysis–dissociation mechanism has been proposed for Mpro enzyme activity control (Chen et al. 2005). The catalytically competent conformation in one protomer is induced only upon dimer formation. Under physiological conditions, Mpro exists as an asymmetric dimer that might have a half-site acylation–deacylation catalytic cycle; i.e., when one subunit is in the active acylated form, the other is in the deacylated form. The dimer is the essential functional unit of this protease that regulates catalytic turnover.
The proposed flip-flop mechanism for Mpro is shown in Fig. 8.4b, which may account for the kinetics and structural information available for this enzyme. The two subunits are used alternately in acylation and deacylation reactions whereby binding at subunit A induces the deacylation at subunit B and vice versa. Mpro is thus proposed to be regulated by negative cooperativity.
6 Structure and Function of the SARS-CoV Papain-Like Protease
In addition to Mpro, SARS-CoV expresses a papain-like protease (PLpro) that cleaves polyprotein 1a at three sites harboring the canonical Leu–(Lys/Asn)–Gly–Gly–↓–(Ala/Lys) sequence (Fig. 8.1b). The tertiary structure of PLpro reveals a distant relationship to the papain family of cysteine proteases (Ratia et al. 2006). The catalytic triad of this enzyme (Cys112–His273–Asp287) also has a broad range of pH optima that is characteristic of the thiolate–imidazolium ion pair that exists also in other papain-like cysteine proteases (Storer and Ménard 1994; Han et al. 2005).
The functional unit of PLpro is a monomer comprising four structural domains (Fig. 8.5). A zinc atom is bound at the finger domain and the active site is located at the interface of the palm and thumb domains. A special feature of PLpro is its ubiquitin-like domain, and indeed SARS-CoV PLpro has been shown to possess deubiquitination activity (Barretto et al. 2005; Lindner et al. 2005, 2007). This dual-functional role makes PLpro another viable target for the development of anti-SARS drugs.


Structure of SARS-CoV PLpro. (a) Ribbon diagram of SARS-CoV PLpro (pdb code: 2FE8) is shown in rainbow colors from the N-terminus (blue) to C-terminus (red). The catalytic triad (Cys112–His273–Asp287) and the Zn atom are highlighted using a sphere model. (b) Amino acid sequence and other structural annotations. The key for these structural features of PLpro is shown below the panel. Panel (a) was generated using MacPymol (DeLano 2002) and panel (b) with PdbSum (Laskowski et al. 2005)
Thiocarbonyl-containing analogs (6-mercaptopurine and 6-thioguanine) have been demonstrated to be PLpro active site-directed compounds (Chou et al. 2008) that bind with high affinity, block the essential sulfhydryl group after binding, and thereby prevent subunit acylation and block enzyme activity. These thiopurine compounds are currently used clinically to treat children with acute lymphoblastic or myeloblastic leukemia (Pui and Evans 1998; Elion 1989) and the adverse toxicities of these drugs are well documented. These thiopurine analogs are important potential lead compounds for the development of anti-SARS-CoV agents in the near future.
7 Conclusions
The maturation of SARS-CoV involves two viral proteases, Mpro and PLpro. Mpro has a Cys145–His41 catalytic dyad at its active center with His41 acting as a general base. In addition, the functional unit of Mpro is a dimer and there is a close correlation between dimer formation and catalytic activity. A flip-flop mechanism is proposed for Mpro in which its two subunits are alternately used in acylation and deacylation steps. The subunit interfacial region of the main protease is an ideal target for rational drug design in the future treatment of SARS-CoV. Inhibitors of PLpro are also potential avenues for developing anti-SARS therapies.
Abbreviations
- AUC:
-
Analytical ultracentrifuge
- CoV:
-
Coronavirus
- Mpro:
-
Main protease
- nsp:
-
Nonstructural proteins
- PLpro:
-
Papain-like protease
- pp:
-
Polyprotein
- SARS:
-
Severe acute respiratory syndrome
- WT:
-
Wild type
References
Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R (2002) Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J 21:3213–3224
Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R (2003) Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767
Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC (2005) The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol 79:15189–15198
Barrila J, Bacha U, Freire E (2006) Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 45:14908–14916
Bartlam M, Yang H, Rao Z (2005) Structural insights into SARS coronavirus proteins. Curr Opin Struct Biol 15:664–672
Bartlam M, Xu Y, Rao Z (2007) Structural proteomics of the SARS coronavirus: a model response to emerging infectious diseases. J Struct Funct Genomics 8:85–97
Bartlam M, Xue X, Rao Z (2008) The search for a structural basis for therapeutic intervention against the SARS coronavirus. Acta Crystallogr A 64:204–213
Chang HP, Chou CY, Chang GG (2007) Reversible unfolding of the severe acute respiratory syndrome coronavirus main protease in guanidinium chloride. Biophys J 92:1374–1383
Chen S, Chen L, Tan J, Chen J, Du L, Sun T, Shen J, Chen K, Jiang H, Shen X (2005) Severe acute respiratory syndrome coronavirus 3C-like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization. Biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J Biol Chem 280:164–173
Chen H, Wei P, Huang C, Tan L, Liu Y, Lai L (2006) Only one protomer is active in the dimer of SARS 3C-like proteinase. J Biol Chem 281:13894–13898
Chen S, Hu T, Zhang J, Chen J, Chen K, Ding J, Jiang H, Shen X (2008a) Mutation of Gly-11 on the dimer interface results in the complete crystallographic dimer dissociation of severe acute respiratory syndrome coronavirus 3C-like protease: crystal structure with molecular dynamics simulations. J Biol Chem 283:554–564
Chen S, Zhang J, Hu T, Chen K, Jiang H, Shen X (2008b) Residues on the dimer interface of SARS coronavirus 3C-like protease: dimer stability characterization and enzyme catalytic activity analysis. J Biochem 143:525–536
Chou CY, Chang HC, Hsu WC, Lin TZ, Lin CH, Chang GG (2004) Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry 43:14958–14970
Chou CY, Chien CH, Han YS, Prebanda MT, Hsieh HP, Turk B, Chang GG, Chen X (2008) Thiopurine analogues inhibit papain-like protease of severe acute respiratory syndrome coronavirus. Biochem Pharmacol 75:1601–1609
Christopher JA (1998) The SPOCK program manual. http://quorum.tamu.edu/. Cited 14 May 2008
DeLano WL (2002) The PyMOL molecular graphics system, DeLano Scientific, Palo Alto, CA, USA, http://www.pymol.org/. Cited 14 May 2008
Eickmann M, Becker S, Klenk HD, Doerr HW, Stadler K, Censini S, Guidotti S, Masignani V, Scarselli M, Mora M, Donati C, Han JH, Song HC, Abrignani S, Covacci A, Rappuoli R (2003) Phylogeny of the SARS coronavirus. Science 302:1504–1505
Elion GB (1989) The purine path to chemotherapy. Science 244:41–47
Fan K, Wei P, Feng Q, Chen S, Huang C, Ma L, Lai B, Pei J, Liu Y, Chen J, Lai L (2004) Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J Biol Chem 279:1637–1642
Gouet P, Robert X, Courcelle E (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31:3320–3323; http://espript.ibcp.fr//ESPript/ENDscript/. Cited 14 May 2008
Han YS, Chang GG, Juo CG, Lee HJ, Yeh SH, Hsu JT, Chen X (2005) Papain-like protease 2 (PLP2) from severe acute respiratory syndrome coronavirus (SARS-CoV): expression, purification, characterization, and inhibition. Biochemistry 44:10349–10359
Harding SE, Rowe AJ (1996) Active enzyme centrifugation. In: Engel PC (ed) Enzymology LabFax. Bios Scientific, Oxford, UK, pp 66–75
Hegyi A, Friebe A, Gorbalenya AE, Ziebuhr J (2002) Mutational analysis of the active centre of coronavirus 3C-like proteases. J Gen Virol 83:581–593
Hsu MF, Kuo CJ, Chang KT, Chang HC, Chou CC, Ko TP, Shr HL, Chang GG, Wang AH, Liang PH (2005a) Mechanism of the maturation process of SARS-CoV 3CL protease. J Biol Chem 280:31257–31266
Hsu WC, Chang HC, Chou CY, Tsai PJ, Lin PY, Chang GG (2005b) Critical assessment of important regions in the subunit association and catalytic action of the severe acute respiratory syndrome coronavirus main protease. J Biol Chem 280:22741–22748
Huang C, Wei P, Fan K, Liu Y, Lai L (2004) 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 43:4568–4574
Krausslich HG, Wimmer E (1988) Viral proteinases. Annu Rev Biochem 57:701–754
Lai L, Han X, Chen H, Wei P, Huang C, Liu S, Fan K, Zhou L, Liu Z, Pei J, Liu Y (2006) Quaternary structure, substrate selectivity and inhibitor design for SARS 3C-like proteinase. Curr Pharm Des 12:4555–4564
Laskowski RA, Chistyakov VV, Thornton JM (2005) PDBsum more: new summaries and analyses of the known 3D structures of proteins and nucleic acids. Nucleic Acids Res 33:D266–D268; http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/. Cited 14 May 2008
Liang PH (2006) Characterization and inhibition of SARS-coronavirus main protease. Curr Top Med Chem 6:361–376
Lin PY, Chou CY, Chang HC, Hsu WC, Chang GG (2008) Correlation between dissociation and catalysis of SARS-CoV main protease. Arch Biochem Biophys 472:34–42
Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R (2005) The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 79:15199–15208
Lindner HA, Lytvyn V, Qi H, Lachance P, Ziomek E, Ménard R (2007) Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch Biochem Biophys 466:8–14
Peiris JS, Guan Y, Yuen KY (2004) Severe acute respiratory syndrome. Nat Med 10:S88–S97
Pui CH, Evans WE (1998) Acute lymphoblastic leukemia. N Engl J Med 339:605–615
Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD (2006) Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA 103:5717–5722
Shi J, Song J (2006) The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J 273:1035–1045
Shi J, Wei Z, Song J (2004) Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J Biol Chem 279:24765–24773
Shi J, Sivaraman J, Song J (2008) Mechanism for controlling dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J Virol 82:4620–4629
Solowiej J, Thomson JA, Ryan K, Luo C, He M, Lou J, Murray BW (2008) Steady-state and pre-steady-state kinetic evaluation of severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro cysteine protease: development of an ion-pair model for catalysis. Biochemistry 47:2617–2630
Storer AC, Ménard R (1994) Catalytic mechanism in papain family of cysteine peptidases. Methods Enzymol 244:486–500
Tan J, Verschueren KH, Anand K, Shen J, Yang M, Xu Y, Rao Z, Bigalke J, Heisen B, Mesters JR, Chen K, Shen X, Jiang H, Hilgenfeld R (2005) pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: molecular dynamics simulations and multiple X-ray structure analyses. J Mol Biol 354:25–40
Tanner JA, Watt RM, Chai YB, Lu LY, Lin MC, Peiris JS, Poon LL, Kung HF, Huang JD (2003) The severe acute respiratory syndrome (SARS) coronavirus NTPase/helicase belongs to a distinct class of 5′ to 3′ viral helicases. J Biol Chem 278:39578–39582
Tong L (2002) Viral proteases. Chem Rev 102:4609–4626
Wei P, Fan K, Chen H, Ma L, Huang C, Tan L, Xi D, Li C, Liu Y, Cao A, Lai L (2006) The N-terminal octapeptide acts as a dimerization inhibitor of SARS coronavirus 3C-like proteinase. Biochem Biophys Res Commun 339:865–872
Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, Gao GF, Anand K, Bartlam M, Hilgenfeld R, Rao Z (2003) The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA 100:13190–13195
Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z, Pei D, Ziebuhr J, Hilgenfeld R, Yuen KY, Wong L, Gao G, Chen S, Chen Z, Ma D, Bartlam M, Rao Z (2005) Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3:e324
Zheng K, Ma G, Zhou J, Zen M, Zhao W, Jiang Y, Yu Q, Feng J (2007) Insight into the activity of SARS main protease: Molecular dynamics study of dimeric and monomeric form of enzyme. Proteins 66:467–479
Zhong N, Zhang S, Zou P, Chen J, Kang X, Li Z, Liang C, Jin C, Xia B (2008) Without its N-finger, SARS-CoV main protease can form a novel dimer through its C-terminal domain. J Virol 82:4227–4234
Ziebuhr J, Bayer S, Cowley JA, Gorbalenya AE (2003) The 3C-like proteinase of an invertebrate nidovirus links coronavirus and potyvirus homologs. J Virol 77:1415–1426
Acknowledgment
This work was supported by the National Science Council, ROC.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Chang, GG. (2010). Quaternary Structure of the SARS Coronavirus Main Protease. In: Lal, S. (eds) Molecular Biology of the SARS-Coronavirus. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-03683-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-03683-5_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-03682-8
Online ISBN: 978-3-642-03683-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)