
Abstract
Hemophagocytic lymphohistiocytosis (HLH), or termed macrophage activation syndrome (MAS) when associated with rheumatic disorders, is a frequently fatal complication of infections, rheumatic disorders, and hematopoietic malignancies. Clinically, HLH/MAS is a life-threatening condition that is usually diagnosed among febrile hospitalized patients (children and adults) who commonly present with unremitting fever and a shock-like multiorgan dysfunction scenario. Laboratory studies reveal pancytopenia, elevated liver enzymes, elevated markers of inflammation (ESR, CRP), hyperferritinemia, and features of coagulopathy. In about 60% of cases, excess hemophagocytosis (macrophages/histiocytes engulfing other hematopoietic cell types) is noted on biopsy specimens from the bone marrow, liver, lymph nodes, and other organs. HLH/MAS has been hypothesized to occur when a threshold level of inflammation has been achieved, and genetic and environmental risk factors are believed to contribute to the hyperinflammatory state. A broad variety of infections, from viruses to fungi to bacteria, have been identified as triggers of HLH/MAS, either in isolation or in addition to an underlying inflammatory disease state. Certain infections, particularly by members of the herpesvirus family, are the most notorious triggers of HLH/MAS. Treatment for infection-triggered MAS requires therapy for both the underlying infection and dampening of the hyperactive immune response.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Cytokine storm
- Epstein-Barr virus
- Hemophagocytic lymphohistiocytosis
- Interleukin-1
- Macrophage activation syndrome
Introduction
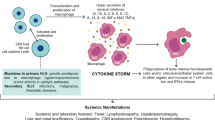
HLH/MAS is thought to be a multisystem inflammatory disorder resulting from a pro-inflammatory “cytokine storm” from excessively activated lymphocytes and macrophages [1]. Hemophagocytic syndromes are divided into primary and secondary forms. Primary cases are rare (1 in 50,000 live births), commonly present in the first year of life, and are often triggered by infection [2]. They include familial, or primary, forms of hemophagocytic lymphohistiocytosis (fHLH ) that have specific genetic homozygous or compound heterozygous loss-of-function mutations in perforin-mediated cytolytic pathway proteins (e.g., PRF1, STX11, UNC13D, UNC18-2) employed by CD8 T cells and natural killer (NK) cells [3,4,5,6]. Children with certain immunodeficiency syndromes, such as Chédiak-Higashi syndrome, type II Hermansky-Pudlak syndrome, and type II Griscelli syndrome [7], have associated genetic defects in cytolysis and are also at risk for developing fHLH. Specific X-linked immunodeficiencies (signaling-lymphocytic-activation-molecule-associated protein (SAP) and X-linked inhibitor of apoptosis (XIAP) deficiencies) are also associated with Epstein-Barr virus (EBV) triggered HLH [8,9,10].
Acquired or secondary forms of HLH (sHLH ) are usually associated with conditions that cause chronic immune dysregulation, such as rheumatologic diseases [e.g., systemic juvenile idiopathic arthritis (sJIA), systemic lupus erythematosus (SLE)] and certain malignancies (e.g., leukemias, lymphomas). Infectious agents, particularly EBV and other herpesvirus family members, may be the sHLH trigger, although identifiable infections are not always present [7]. In addition, up to 40% of sHLH and macrophage activation syndrome (MAS) patients have been found to possess heterozygous (some dominant negative) mutations in known fHLH genes. Thus, some investigators consider MAS, sHLH, and fHLH to lie on a spectrum of disease [11].
In the clinical setting, the distinction between primary and secondary forms of HLH is less clear and even considered artificial by some. It was initially used to differentiate the primary, more fatal, infantile presentations from the secondary forms, which were considered to present later in life and to have better prognoses. However, it is now known that primary genetic forms can present later in life, even during adulthood [12, 13]. Furthermore, in some studies, only 40% of primary HLH cases are found to have recognized genetic mutations. Moreover, both primary and secondary HLH are known to be precipitated by infections [14, 15]. Finally, as mentioned previously, many patients with sHLH have heterozygous mutations in known fHLH-associated genes, thus blurring the distinction between fHLH and sHLH. Regardless of terminology, the individual patient needs to be treated appropriately. At present, most clinicians would agree that clear-cut infantile cases of fHLH will need bone marrow transplantation, typically preceded by an aggressive chemotherapeutic regimen which includes etoposide and corticosteroids. In addition, identified infectious triggers should also be treated appropriately. For all other children and adults with HLH, the most appropriate treatment remains unclear. Etoposide is quite toxic, often leading to pancytopenia itself and increasing the risk of secondary sepsis as well as increased risk of secondary malignancies [14, 16]. Novel approaches have been anecdotally reported to dampen the overly exuberant immune response and control the cytokine storm and associated multiorgan dysfunction. Most notably, targeting of specific pro-inflammatory cytokines [e.g., interleukin-1 (IL-1) and interferon-gamma (IFN-γ)] seems promising and lacks the toxicity associated with traditional chemotherapeutic approaches [17, 18].
Despite advances in the current treatment protocols, the cure rate for HLH is low. Untreated cases of fHLH have a median survival of less than 2–6 months after diagnosis [19]. In a nationwide registry of pediatric patients with HLH in Korea, the 5-year overall survival rate was 68% (38% in the familial group and 81% in the presumed secondary group) [20]. The prognosis for cases of sHLH varies depending on the underlying etiology, for example, the mortality rate is reported to be lower in cases associated with rheumatic diseases (8–22%) and greater when it is associated with malignancy. The median overall survival is about 36–67% [21,22,23].
Pathophysiology and Cytokine Storm
MAS/HLH develops as a “cytokine storm” which is often triggered by infectious, rheumatologic, and oncologic diseases [24]. Although not well defined, the pro-inflammatory cytokines associated with MAS/HLH likely include IL-1, IL-6, IL-12, IL-18, IFN-γ, and tumor necrosis factor (TNF) [25, 26] (Table 14.1). Also IL-27, macrophage colony-stimulating factor (M-CSF), and granulocyte-macrophage colony-stimulating factor (GM-CSF) may be increased [27,28,29]. Furthermore, chemokines , such as IL-8/CXCL8, MIG/CXCL9, IP10/CXCL10, I-TAC/CXCL11, MCP-1/CCL2, MIP-1α/CCL3, and MIP-1β/CCL4, have been reported to be increased [26, 30,31,32]. Both cytokines and chemokines activate the immune system, perpetuating the ongoing cytokine storm. On the other arm, levels of anti-inflammatory cytokines , such as IL-10 and IL-18-binding protein (IL-18BP), are also increased but might not be sufficient to terminate the ongoing inflammation [33, 34]. Mazodier et al. described a discrepancy between the increase in IL-18 and its antagonist IL-18BP that lead to abnormally high levels of free IL-18 [33]. Similarly, the natural antagonist to IL-1, IL-1 receptor antagonist (IL-1Ra), has been noted to be elevated during MAS/HLH, and a recombinant form of IL-1Ra has been reported by several groups to be an effective therapy for MAS/HLH/cytokine storm syndrome [35,36,37].
The etiology of the cytokine storm is not entirely clear. Since fHLH is associated with biallelic defects in gene products involved in the perforin-mediated cytolytic pathway used by NK cells and CD8 T lymphocytes [48, 49], the inability to clear the antigenic stimulus and thus turn off the inflammatory response has been hypothesized to result in hypercytokinemia [50]. Recently, the inability of CD8 T cells and NK cells to lyse antigen-presenting cells (APCs) via the perforin-mediated cytolytic pathway was shown to prolong (by fivefold) the engagement time between the lytic lymphocyte and the APC. This prolonged interaction resulted in increased levels of pro-inflammatory cytokines . For up to 40% of sHLH cases, single-copy mutations in these same perforin pathway genes have been reported. Some of the mutants have been demonstrated to act as complete or partial dominant-negative mutants [51,52,53], resulting in sHLH in older children and adults [53, 54], the oldest reported case being a 62-year-old patient [55]. One heterozygous mutation in RAB27A identified in two unrelated sHLH patients was shown to act in a partial dominant-negative fashion and delayed cytolytic granule polarization to the immunologic synapse between NK cells and their target cells. This was also associated with an increase in IFN-γ production , mimicking the situation described for homozygous defects in perforin or granzyme B. Moreover, increased IL-6 production has been shown to decrease cytolytic activity of NK cells, further exacerbating the lytic defect and resultant production of pro-inflammatory cytokines.
In a healthy individual, exposure of most cells to many intracellular pathogens will normally initiate an inflammatory cascade, frequently leading to release of Th1 cytokines (IFN-γ, TNF) that will activate macrophages, NK cells, and cytolytic T cells. NK and cytolytic T cells release granules that contain perforin and granzymes [49]. Perforin is a key cytolytic protein that causes osmotic lysis of the target cell [56] and is also necessary for the uptake of granzymes by the target cell that will then catalyze cleavage of multiple protein substrates, including caspases which then trigger cell apoptosis. All the genetic defects described in fHLH involve either inadequate levels of perforin itself or improper granule exocytosis leading to impaired apoptosis of the target cell, improper removal of the stimulating antigen, and ultimately ongoing inflammation.
However, other pathways lacking cytolytic pathway gene defects can lead to the final endpoint of HLH or MAS. In a murine model of MAS, it was shown that repeated stimulation of toll-like receptor 9 (TLR9) produced MAS on a normal genetic background, without exogenous antigen. Interestingly, the TLR9-induced MAS model was IFN-γ dependent in some aspects of disease; however, lymphocytes were not required for the pathogenesis [57]. On the other hand, a state of inflammation may also reduce the lytic capacity of NK cells and CD8 T cells [58,59,60], resulting in cytokine storm due to frustrated phagocytosis. This was illustrated in a study where T-cell-directed immunotherapy for refractory leukemia resulted in cytokine storm and an MAS-like presentation [61]. All the aforementioned pathways have led to the proposal that MAS is due to a combination of genetic predisposition and a hyperinflammatory state reducing cytolytic function, put into action by a trigger (e.g., infection, cancer, immunodeficiency, autoimmunity, and autoinflammation) [19, 53, 62,63,64]. At some point a threshold level of hypercytokinemia is reached at which the body is incapable of balancing the cytokine storm with anti-inflammatory products, such as IL-10, IL-1Ra, IL-18BP, and others. This is then believed to trigger the multiorgan dysfunction resulting in clinical HLH.
Clinical Picture
Initial symptoms of HLH/MAS are usually nonspecific. The cardinal feature is unremitting high fever. However, therapeutic targeting of pro-inflammatory cytokines (e.g., IL-1, IL-6) to treat underlying rheumatic diseases that often result in MAS (e.g., sJIA) makes fever not an absolute finding in all cases. This is attributed to the powerful antipyretic effect of biologics such as inhibitors of IL-1, IL-6, and TNF. On examination , many patients have hepatomegaly, splenomegaly, or both, and up to 50% of MAS/HLH patients have central nervous system involvement ranging from mild confusion to seizures or frank coma [65]. Different forms of rash can occur, often erythematous or purpuric. Patients can have progressive hepatic dysfunction and ultimately multiorgan failure . DIC-like features are often present and are partly explained by liver dysfunction, fibrinogen consumption, and thrombocytopenia [66]. This highlights the challenges in distinguishing microbial sepsis-induced DIC from HLH in the intensive care unit (ICU). Despite the similarities these two conditions share in clinical presentations, they are frequently treated differently: broad spectrum antibiotics (sepsis) versus immunosuppression (HLH), respectively [67]. As HLH is not a diagnosis of exclusion, and infections are common triggers of HLH, it is important to treat both infection and, if present, the associated cytokine storm of HLH.
Classification Criteria
MAS/HLH can be difficult to diagnose, especially in the early stages where it can be easily misdiagnosed as shock or multiorgan dysfunction due to sepsis. In addition, MAS may be confused with an underlying disease flare, as in the case of sJIA. Because MAS can have a high mortality rate in children with sJIA (∼8–22%) [68,69,70], sensitive diagnostic criteria are needed to assist with early detection, allowing for appropriate and timely therapy. Because of the different diseases associated with MAS/HLH, different diagnostic and classification criteria have been proposed over the years, such as HLH-04, SLE-MAS criteria, the HScore, and the novel 2016 criteria for MAS complicating sJIA (Table 14.2). Some of these criteria are disease specific (e.g., sJIA, SLE) and can be both sensitive and specific, whereas others encompass all potential HLH-associated diagnoses, but tend to have lower sensitivities overall. Clinically, and outside of clinical trials, the various criteria are useful for clinicians to strongly consider MAS/HLH diagnostically so that appropriate therapy can be initiated as soon as possible to result in optimal outcomes.
MAS as a Part of sJIA (sJIA-MAS)
HLH-2004 Diagnostic Guidelines
Due to the fact that MAS resembles fHLH in its clinical presentation, HLH-2004 diagnostic guidelines were initially used to diagnose MAS. Those guidelines were developed to diagnose genetic homozygous/compound heterozygous cases of fHLH [11]. The main deficiencies regarding the HLH-2004 criteria for diagnosing MAS in patients with sJIA are due to the underlying inflammatory nature of sJIA versus fHLH. In active sJIA, one would expect elevated levels of white blood cell counts, platelets, and fibrinogen as part of the inflammatory process. Accordingly, a drop in their levels, which could still be in the normal limits as regards to the HLH-2004 criteria, should raise the suspicion of MAS. Also the underlying inflammatory process leads to elevated levels of ferritin [74]; therefore, the cutoff of ferritin >500 ng/ml in the HLH-2004 guidelines makes it difficult to distinguish MAS complicating sJIA from an sJIA flare. Adding to the shortcomings of HLH-2004 guidelines overall are the lack of availability and timely results of certain criteria, such as NK cell activity or sCD25 levels in many centers [25, 75, 76].
Preliminary Diagnostic Criteria for MAS Complicating sJIA
Eventually, preliminary diagnostic criteria were introduced for sJIA-MAS comparing it to sJIA flare [71], which yielded better results in identifying MAS among sJIA patients when compared to HLH-2004 diagnostic guidelines [77]. However, these new criteria had their own shortcomings. The study that led to the criteria development was lacking in some important laboratory parameters, including some of the important MAS markers such as ferritin, lactate dehydrogenase, and triglycerides [25, 75, 76]. Moreover, they were based on a relatively small sampling of patients, and they were not validated. These shortcomings were an impetus to develop new sJIA-specific MAS criteria.
2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis
The deficiencies in the previous guidelines have pushed for the development of more accurate criteria for sJIA-MAS. Recently, novel classification criteria have been introduced. These were the result of an international collaborative effort combining expert consensus, evidence compiled from the medical literature, and analysis of real patients. The development of the 2016 criteria was conducted under the auspices of the European League Against Rheumatism, the American College of Rheumatology, and the Paediatric Rheumatology International Trials Organization [73]. Based on the common consensus that clinical criteria of MAS are often delayed and/or difficult to distinguish from an underlying disease flare, the 2016 sJIA-MAS criteria are based primarily on laboratory parameters with fever as the only clinical criterion [76]. These criteria are relatively simple yet proved to be both highly sensitive and specific. However, the criteria are not ideal in the setting of children with sJIA who are being actively treated with IL-1 or IL-6 blockade. Ultimately, simple criteria that are not necessarily disease specific but maintain high sensitivity and specificity for establishing an MAS/HLH diagnosis are needed.
MAS as Part of Systemic Lupus Erythematosus (SLE-MAS)
Childhood SLE (cSLE) cases complicated by MAS have been reported with increasing frequency in the recent years. As an SLE disease flare itself often results in pancytopenia, diagnostic criteria for MAS in the setting of SLE are complicated. Accordingly, it has been suggested that cSLE-MAS may be under-recognized [78]. Preliminary guidelines for SLE-MAS were proposed in 2009. A study was conducted based on a multinational survey and data analyzing 38 patients with cSLE-MAS [72]. Patients who had evidence of macrophage hemophagocytosis on bone marrow aspiration were considered to have definite MAS, and those who did not were considered to have probable MAS. The sensitivity, specificity, and the area under the receiver operating characteristic curve of various clinical and laboratory parameters were compared in SLE patients with MAS versus patients with active juvenile SLE without MAS. The best diagnostic performance was obtained using the simultaneous presence of any one or more clinical criteria and any two or more laboratory criteria, which had a sensitivity of 92.1% and a specificity of 90.9% (Table 14.2). The demonstration of macrophage hemophagocytosis in the bone marrow aspirate was considered necessary for confirmation of doubtful cases only. Those results have led to the practical recommendation that in the clinical setting, MAS should be suspected in a patient with cSLE presenting with unexplained fever and cytopenia associated with hyperferritinemia. Both HLH-2004 criteria and preliminary diagnostic guidelines for sJIA-MAS were tested in the study but were found to be inaccurate for detecting cSLE-MAS. Interestingly, about two-thirds of the patients with cSLE-MAS developed it within 1 month of SLE diagnosis. The frequency of ICU admission was 43.7%, and the mortality rate was 11.4%.
Generic MAS Criteria/HScore
In 2014, Fardet et al. developed and partially validated a diagnostic score for the broader category of reactive hemophagocytic syndrome (HS), called the HScore [79], which can be used to estimate an individual’s risk of having reactive hemophagocytic syndrome, or HLH. This score was created and tested using a multicenter retrospective cohort of 312 patients scrutinizing 10 explanatory variables that were issued from a previous Delphi survey involving 24 HLH experts from 13 countries [80]. After showing positive associations of each variable with an HLH diagnosis, multivariate logistic regression was used to assess their independent contributions to the outcome. Following calculating each variable’s threshold value, the coefficients resulting from multiple logistic regression analysis were used to assign score points to each one. The performance of the score was assessed using developmental and validation data sets. The HScore revealed excellent diagnostic performance and discriminative ability in both developmental and validation data sets. The probability of having HLH ranged from <1% with an HScore of ≤90 to >99% with an HScore of ≥250.
The HScore has some limitations including the heterogeneity of the underlying diseases (a high proportion had cancer-associated HLH), the retrospective manner of the data collection, and the small sample size (only 10% of the entire study population) of the validation data set. As the study included only adults with reactive HLH, the applicability in children, particularly those with sJIA-MAS, is questionable. Adding to its limitations in pediatric cases, some of the criteria in the HScore might not be practically applicable in children. For example, the definition of the item, “Known underlying immunosuppression,” lists some medications that are used infrequently in children with sJIA, such as cyclosporine A and azathioprine, and at the same time it does not mention the newer more widely used cytokine antagonists that have been associated with the occurrence of MAS [25, 81, 82]. Moreover, bone marrow aspirates in a search for hemophagocytosis are not frequently performed in children with sJIA-MAS, as it is not considered mandatory in either the HLH-2004 guidelines [11] or the preliminary MAS guidelines [71]. In fact, absence of hemophagocytosis does not rule out MAS, and the procedure should not delay appropriate therapy. Furthermore, the underlying inflammatory nature of sJIA that is associated with marked thrombocytosis makes the threshold level for the platelet count (110,000/mm3) too low for identifying MAS in the setting of sJIA. It is the relative drop in platelets count, rather than an absolute decrease below a certain threshold, that is more useful to make an early diagnosis [77]. Thus, the HScore is likely more valuable in diagnosing adults with HLH, particularly those with associated leukemias and lymphomas.
New Biomarkers
A new promising laboratory marker of MAS is soluble CD163 (sCD163). Its expression is restricted to the macrophage/monocyte lineage only, unlike ferritin and soluble CD25 (IL-2 receptor α-chain), which are produced by a number of tissues and cell types, including the liver, spleen, heart, kidney, and T cells under a variety of nonspecific inflammatory conditions. sCD163 has been mainly evaluated in MAS, where combination testing of sCD25 and sCD163 identified patients with subclinical MAS [83]. Further studies to evaluate its role in HLH not associated with autoimmune diseases are required. Moreover, like sCD25, the testing is not currently available in a timely fashion in most centers around the world.
Another novel biomarker, follistatin-related protein 1 (FSTL-1 ) was reported by Gorelik et al. to be elevated in active sJIA with higher levels during MAS. FSTL-1 levels correlated with sCD25 and ferritin levels, and FSTL-1 normalized after treatment. Perhaps more importantly, Gorelik et al. also reported that in their small cohort (28 sJIA patients) a ferritin to ESR ratio > 80 had the highest sensitivity and specificity (100% and 100%, respectively) in distinguishing between MAS and new-onset sJIA disease flare [84]. As ferritin rises due to inflammation in MAS/HLH, and the ESR tends to drop as fibrinogen (an important driver of high ESRs) is consumed during coagulopathy, a simple ratio of ferritin to ESR may prove to be a simple and valuable tool in getting clinicians to consider a diagnosis of MAS/HLH in their febrile hospitalized patients.
Although the serum IL-18 level is also not routinely available clinically, it may also serve as a distinguishing biomarker for sJIA patients who develop MAS. Comparing cytokine patterns between sJIA-MAS patients, EBV-HLH, Kawasaki disease, and healthy age-matched controls, Shimizu et al. reported that IL-18 concentrations during sJIA-MAS were significantly higher compared to the others, and they correlated with measures of disease activity (CRP, ferritin, LDH, and other cytokines). In addition, serum neopterin and sTNF-RII levels were significantly higher during MAS compared to sJIA flares [85]. Other reports also showed that IL-18 levels were significantly elevated in sJIA [86, 87] and the patients with high levels were more likely to develop MAS [88]. Furthermore, sphingomyelinase was found to be elevated in HLH cases [89]. Thus, a variety of new biomarkers may help identify MAS among sJIA patients.
Genetic Associations
Genetic HLH has been commonly classified into two groups : fHLH which are a group of autosomal recessive disorders, and immunodeficiency syndromes related HLH. Of the immunodeficiency syndromes, Chediak-Higashi, Griscelli, and Hermansky-Pudlak are associated with a variable degree of albinism/hypopigmentation of the skin or hair and platelet dysfunction which can assist in identifying potential cases of HLH (Table 14.3) [2, 90,91,92]. Interestingly, up to 40% of sHLH cases possess heterozygous mutations in these same fHLH-associated gene products. Thus, the overall underlying genetic risk for sHLH may be rather striking.
In addition to underlying inflammatory states (e.g., sJIA, leukemia) and genetic predispositions (e.g., perforin deficiency), infections (some from the commensal human microbiome) are frequently significant contributing factors in lowering the threshold required to develop a cytokine storm syndrome capable of resulting in HLH/MAS (Table 14.4). HLH has been associated with a vast variety of infections, with EBV as the most commonly reported trigger. Both familial (fHLH) and sporadic or secondary (sHLH) cases of HLH are often precipitated by acute infections. It is also important to note that an underlying precipitating infection for HLH can be masked, as the HLH clinical picture can mimic an infectious process or an overwhelming septicemia. It is important nonetheless to detect and remedy any underlying treatable infection in the setting of HLH.
Virus-Associated Hemophagocytic Syndrome
EBV
Epidemiology
As previously mentioned, EBV is the most commonly reported trigger of HLH [93], with the highest incidence in East Asia [202]. This could be explained by the more pathogenic strains of EBV in this part of the world [203] and also by the higher prevalence of EBV and EBV-infected T cells in Asians [204]. EBV-associated HLH (EBV-HLH) cases have also been described in the USA and Europe [205]. Most EBV-HLH cases occur in apparently immunocompetent children and adolescents [206]; however, it can also occur in the setting of primary/genetic forms (fHLH) [207], immunodeficiency disorders (e.g., XLP) [208], and secondary forms, including acute infections (e.g., infectious mononucleosis) [209] and lymphoproliferative disorders (e.g., NK cell and T-cell leukemias and lymphomas) [210].
Pathophysiology
The mechanism by which EBV induces HLH has not been fully explained. During primary infection, EBV typically infects and replicates in B cells, whereas a function of EBV-specific cytotoxic T cells is the regulation of the infected B cells and the production of memory cells. On rare occasions, EBV may infect T cells and NK cells via CD21. CD21 is expressed on the surface of these cells and induces persistent EBV infection, with monoclonal or oligoclonal proliferation resulting in chronic active EBV infection, lymphoproliferative disorders, and fulminant EBV-HLH [211,212,213]. Infection of CD8 T cells with EBV results in a cytokine storm with the release of pro-inflammatory and Th1-type cytokines [214], including TNF and IFN-γ, leading to widespread lymphohistiocytic activation [215]. The resultant cytokine storm tends to be more prominent than those observed in non-EBV-HLH [216]. In addition, impaired function of T cells or NK cells is thought to provide a phenotypic presentation of HLH resulting from EBV via any genetic mutation involved in the T-cell and/or NK cell activation pathways [217, 218].
Diagnosis
Serologic testing can help differentiate primary EBV infection from a reactivation process, although they have limitations such as delay in positivity and difficulty in result interpretation. Real-time PCR is used to measure the EBV viral load which can help predict prognosis and response to treatment [219]. EBV PCR levels are usually higher than those seen in uncomplicated cases of EBV infectious mononucleosis [220]. Other techniques are available to determine the involvement of T cells or NK cells in helping to confirm the diagnosis. T-cell receptor (TCR) gene rearrangement is detectable in half of the patients with EBV-HLH using Southern blotting and/or PCR analyses. It is hypothesized that the presence and change of TCR gene clonality probably plays a prognostic role for EBV-HLH [90]. Sandberg et al. [221] recently reported that Southern blot analysis could be replaced by BIOMED-2 multiplex PCR in routine testing of T-cell clonality. The EuroClonality (BIOMED-2) consortium developed a uniform reporting system for the description of the results and conclusions of Ig/TCR clonality assays to help improve the general performance level of clonality assessment and interpretation in cases with suspected lymphoproliferations [222]. It was reported that TCR gene clonality with BIOMED-2 multiplex PCR [223] is highly sensitive for detecting T-cell clonality and is useful in predicting response to treatment in EBV-HLH cases [223]. Interestingly, it was found that male patients with EBV-HLH may have mutations in the SH2D1A gene which is classically associated with X-linked lymphoproliferative syndrome (XLPS). XLPS is a syndrome of immunodeficiency to EBV virus. Therefore, it is recommended to test for XLPS in male patients with EBV-HLH [224]. It is also recommended to test for other genetic conditions such as fHLH, especially in male patients under 1 year of age, and in those with HLH in a sibling or with consanguineous parents, or when HLH is recurrent or unresponsive to treatment.
Prognosis
Of all the viruses associated with HLH, EBV-HLH carries one of the worst prognoses. In a nationwide survey in Japan to identify prognostic factors in children with EBV-HLH, Kogawa et al. [225] found that most of the clinical and laboratory parameters including EBV load, NK cell activity against EBV-infected cells, and the presence of clonality at the onset of disease were not associated with a poor outcome. Nevertheless, Matsuda et al. showed that change of clonality can be a good marker of disease activity in childhood EBV-HLH [223]. It is also reported that hyperbilirubinemia and hyperferritinemia at the time of diagnosis were significantly associated with a poor outcome. Henter et al. also reported that hyperbilirubinemia and hyperferritinemia at diagnosis, and thrombocytopenia and hyperferritinemia 2 weeks after the initiation of treatment, adversely affect the outcome of HLH [226]. Better outcome is speculated to be associated with going into remission within 8 weeks of treatment initiation [225]. Huang et al. reported that hypoalbuminemia is an independent predictor for HLH in childhood EBV-associated disease [227].
Treatment
Antiviral therapy with acyclovir, ganciclovir, or cidofovir is generally ineffective as monotherapy in infectious mononucleosis and EBV-HLH [228]. However, aggressive therapy including immunochemotherapy and allogenic stem cell transplantation has radically improved the prognosis. The optimal treatment strategy [229] for EBV-HLH consists of immunosuppressive medications that inhibit overactive T-cell and NK cell responses [i.e., corticosteroids, cyclosporine A, intravenous immunoglobulin (IVIg), antithymocyte globulins, etoposide, and plasma or blood exchange transfusions] [229, 230]. Hematopoietic stem cell transplantation (HSCT) is the last treatment resort for refractory forms of EBV-HLH, and in the case of EBV infection occurring in genetic forms of HLH [231]. Despite the fact that reports have shown that HSCT is effective in treating patients with refractory EBV-HLH [232], it should be compared to immunochemotherapy in a randomized study to provide evidence for which approach is superior and/or safer [233].
In 2007, Balamuth et al. [230] reported that adding rituximab to the HLH-2004 treatment protocol improves its efficacy. Rituximab is a monoclonal antibody against CD20 on the surface of B cells. Because EBV targets B cells in the initial phase of the disease, rituximab’s elimination of B cells is thought to inhibit the extent of the infection. In addition, B cells may be a target in EBV-HLH, and rituximab may reduce morbidity and mortality by reducing the circulating B-cell population and the EBV load [234]. Rituximab seems to be most effective in the setting of XLPS patients infected with EBV but is likely less effective when EBV is capable of infecting the T-cell pool. Nonetheless, the addition of rituximab to the treatment repertoire of EBV-HLH provides an opportunity to tailor therapy specific to the patient (personalized medicine approach).
Other Herpes Viruses
Following EBV, cytomegalovirus (CMV) and human herpes virus (HHV) 8 are the next most common of the herpesviruses to be associated with HLH. CMV infection has been associated with HLH in otherwise healthy patients [96, 235], premature infants [97], patients with inflammatory bowel disease [236, 237], rheumatological diseases [238, 239], cancer [240], and in transplant recipients [241, 242]. In a series of 171 patients undergoing HSCT, HLH was observed in 7 (4%) of them and was triggered by CMV in 3 cases [243]. In a Japanese registry with CMV-HLH diagnosed at less than a year of age (1986–2002), four of the five infants died, suggesting that younger age may be associated with a worse prognosis [244]. The use of specific anti-CMV therapy, such as CMV hyperimmune globulin, foscarnet, or ganciclovir, has been associated with recovery in selected cases [96, 236,237,238, 242].
Human Herpes Virus 8
HHV-8 has been associated with HLH, mostly in the setting of Kaposi sarcoma [245], multicentric Castleman disease [246], or lymphoproliferative disorder [247], as well as in immunocompromised hosts (HIV) [99], transplant recipients [248], and, rarely, in immunocompetent hosts [100, 101]. In a prospective cohort of 44 patients with Castleman disease and human immunodeficiency virus (HIV), 4 (9%) had HLH [246]. Intriguingly, in this series, the levels of IL-8 and IFN-γ were increased, though the cytokine levels of many known inflammatory markers were not. In this study, all patients recovered after treatment with splenectomy, etoposide, and rituximab [249]. Ganciclovir and foscarnet have also been associated with recovery in some HHV8-HPS cases. Finally, all other herpes viruses [102,103,104, 250] with the exception of HHV-7 have been associated with HLH.
Neonatal Infection-Associated HLH
Due to the lack of disease awareness among many physicians, HLH presenting within the first 4 weeks of life is rarely recognized. It could either pass undiagnosed or be diagnosed late in the course, or even at autopsy. Neonatal HLH differs from HLH in older children in etiology, manifestations, and laboratory findings. In a nationwide survey in Japan published in 2009, 20 neonates were diagnosed with HLH within 4 weeks after birth; 6 (30%) of them were diagnosed with fHLH, and 6 (30%) were diagnosed with herpes simplex virus-associated HLH (HSV-HLH) [251]. The overall survival rate of these 20 patients was 28.6% for fHLH and severe combined immunodeficiency (SCID)-HLH, and 33.3% for HSV-HLH, despite acyclovir treatment. Although uncommon in HLH of older children, enterovirus (echovirus and coxsackievirus) and HSV have been associated with fatal or fulminant neonatal HLH [252, 253]. This mandates early treatment with high-dose acyclovir in suspected cases without awaiting viral studies results.
HIV
HLH can be associated with HIV [117] infection [the etiology of acquired immunodeficiency syndrome (AIDS)] in different settings. HLH can occur either with HIV alone or with a variety of underlying associated disorders. HLH has been reported in acute or late stages of HIV infection, in the setting of immune reconstitution inflammatory syndromes (IRIS), and in association with HIV-associated malignancies or infections (both opportunistic and non-opportunistic) [254]. HLH has even been reported as the initial presentation of HIV infection [118], which suggests a direct role for HIV in triggering HLH [195].
Due to the fact that both HIV and HLH share many similar clinical and biological findings, it is likely that this association is also underdiagnosed. In one study, hemophagocytosis was observed in 20% of 56 autopsy cases of HIV-positive patients [255]. Around 10% of bone marrow biopsies in HIV patients before highly active antiretroviral therapy (HAART) initiation revealed hemophagocytosis [256]. In adult cases with acute HIV infection, HLH was associated with low CD4 T-cell counts (<200 cells/μL) in almost two-thirds of the cases. In addition, a lower CD4 T-cell count was associated with a worse prognosis [257].
Influenza
The association of HLH with influenza has been described with seasonal [122, 258,259,260,261,262], avian [263, 264], and swine (non-pandemic) influenza [265]. It has also been associated with both immunocompromised [258,259,260,261] and otherwise healthy children [122, 266]. In a prospective pediatric study, which included 32 children hospitalized with seasonal influenza, one case had a fatal outcome [262]. Interestingly, patients with severe H5N1 avian influenza have clinical pictures and laboratory findings similar to HLH; these consist mainly of encephalitis [267], organ dysfunction with hemophagocytosis [268], bone marrow suppression [268, 269], and cytokine storm [270, 271]. The most common characteristic pathological picture seen on autopsy and biopsy in such cases is hemophagocytosis [264, 271, 272].
Clinical studies have found that mutations in some viral genes (NS1, PB2, HA, and NA) are significantly related to cytokine release, and it has been shown that recombinant hemagglutinin (H5) from H5N1 virus may suppress perforin expression and reduce the cytotoxicity of CD8 T cells, including their ability to kill H5-bearing cells leading to marked lymphoproliferation and IFN-γ hyperproduction with macrophage overactivation [273]. Considering the high mortality caused by H5N1-HLH, the resistance to many antivirals by H5N1, and the similarity between HLH and severe flu infections have led some to suggest the use of a modified HLH-94 protocol [274] with a shorter course of etoposide and dexamethasone in such cases. However, in a randomized study from Vietnam, all patients with H5N1-HLH died despite receiving corticosteroids [275].
Recently, a study was done on 16 cases of fatal influenza A (H1N1) infection who met 44 and 81% of modified HLH-2004 and MAS criteria, respectively. Five subjects (36%) carried one of three different heterozygous LYST mutations, two of whom also possessed the relatively common p.A91V PRF1 mutation, which was shown to mildly decrease NK cell cytolytic function. Several patients also carried rare variants in other genes previously observed in patients with MAS. The high percentage of HLH gene mutations suggests they are risk factors for mortality among individuals with influenza A (H1N1) infection [276].
Parvovirus
HLH has been reported in approximately 30 cases of parvovirus B19 infection; most of them had hereditary spherocytosis as the underlying disease, and less than 50% were children [106, 109, 110, 277, 278]. Of these patients, 22 survived, of whom 16 did not receive any treatment. This suggests a better prognosis of parvovirus-HLH than that with other viral infections.
Hepatitis Viruses
Fulminant viral hepatitis may mimic and even cause HLH. Hepatitis A virus (HAV) is more frequently associated with HLH than other hepatotropic viruses, including HBV and HCV. Fifteen cases (including children) have been described in the literature, mainly in Asia; three of these patients also had a concurrent rheumatological disease (sJIA or the related adult onset Still disease), and two also had hepatitis C. Four patients survived without specific treatment. The others received corticosteroids with or without IVIg. Overall, 11 of the 15 had a good outcome [112, 113, 279].
Enterovirus
Twelve cases of pediatric enterovirus-related HLH have been described. Four patients had an underlying disease (Hodgkin and non-Hodgkin lymphoma, acute lymphoblastic leukemia, JIA) and had a higher mortality rate (75%). Ten patients received IVIg (six in combination with corticosteroids), but only seven patients survived [132]. Other viruses associated have been associated with HLH (Table 14.4), for most of which varying courses of corticosteroids and IVIg have been used. Interestingly, HLH may contribute to the high mortality rate associated with certain hemorrhagic fever viruses, such as those causing Dengue fever and Ebola.
Bacteria-Associated Hemophagocytic Syndrome
Of the bacterial pathogens, intracellular organisms have most commonly been the precipitating bacterial agents of HLH. The pathophysiology is probably related to the host lymphocytes and monocytes producing high levels of activating cytokines. Defective NK cell and cytotoxic T-cell function is also hypothesized to play a role in the pathophysiology [2].
HLH has been reported with disseminated Mycobacterium tuberculosis (TB) infection . It can occur in otherwise healthy patients [280], in end-stage renal disease patients receiving hemodialysis [281], in those who had undergone renal transplantation [164], or who had malignancy [282], HIV/AIDS [283], or sarcoidosis [284]. In a review of 36 cases by Brastianos et al., 83% of cases had evidence of extrapulmonary tuberculosis. The mortality rate was approximately 50% which portrays the poor outcome of TB-HLH, although antituberculous and immunomodulatory therapy (consisting of high-dose corticosteroids, IVIg, antithymocyte globulin, cyclosporine A, epipodophyllotoxin, or plasma exchange) may lead to better outcomes [155]. Early diagnosis and timely administration of antituberculous treatment are crucial in these patients. Moreover, one reported case of HLH occurred after childhood vaccination with the bacillus Calmette-Guérin [157].
HLH has also been described in association with brucellosis, with Brucella melitensis being the most frequently isolated species [168]. Leptospirosis has been reported with HLH and has required treatment with corticosteroids, IVIg, or etoposide, in addition to antibiotic treatment [179]. Reports have also related Rickettsia and Ehrlichia to HLH, and the prognosis seems to be influenced by the specific Rickettsia species, patient’s immunologic status, and delay in antibiotic therapy or corticosteroid therapy [184]. Also, MAS following urinary tract infection with Acinetobacter baumannii was reported for the first time in a previously healthy 3-year-old child; recovery occurred without any cytotoxic treatment or immunotherapy, using only multiple doses of GCSF and red blood cell/platelet transfusions [159]. Just like viruses, a large array of additional bacterial infections has been associated with HLH (Table 14.4), but a propensity for intracellular invasion is a common theme to many of these triggers.
Parasitic and Fungal Infection-Associated Hemophagocytic Syndrome
Leishmania infection has been associated with HLH (particularly Leishmania donovani and Leishmania infantum), but considering that it presents with organomegaly and pancytopenia, it can also just mimic the syndrome of HLH. This is of importance in non-endemic areas, where visceral leishmaniasis is unlikely considered as a differential diagnosis, and repeated bone marrow smears are often required to identify Leishmania species by means of PCR with species-specific probes [285]. While specific treatment with amphotericin B is usually sufficient to control HLH, fatal outcomes have been seen with undiagnosed Leishmania cases treated as HLH [188]. History of travel to endemic areas is also of utmost importance for suspecting HLH secondary to certain parasitic infections such as malaria, toxoplasma, babesia, and strongyloides.
Yeast infections (Candida sp., Cryptococcus sp., and Pneumocystis sp.) and molds (Histoplasma sp., Aspergillus sp., and Fusarium sp.) have been associated with HLH, most commonly during HIV infection, malignancy, prolonged corticosteroid administration, and transplantation [196, 286, 287]. Disseminated Penicillium marneffei infection is common among HIV-infected patients in Southeast Asia. The first case of penicilliosis-HLH was reported in a Thai HIV-infected child in 2001, with complete recovery after antifungal and IVIg therapy [200].
Treatment Options for MAS/HLH
In addition to treating any underlying infection, treatment designed to dampen the cytokine storm associated with MAS/HLH is critical for improving survival (Table 14.5). Some researchers reported the success of high-dose corticosteroids alone [25, 288] in treating sJIA-MAS. While the fundamental role of corticosteroids in the therapy of this disease is not doubted [25, 69, 70], current regimens usually add more aggressive treatment to corticosteroids including cyclophosphamide [289], which has not gained wide use in this condition; cyclosporine A (CsA), which is currently the most commonly added medicine to corticosteroids [290, 291]; and etoposide-based regimens, such as HLH-94 and HLH-2004 [11], which have their not insignificant mortality rates during the pre- and post-bone marrow transplant periods [292]. Of the biologics, IVIg has been used, particularly for infection-associated sHLH, but IVIg must be given early in disease to be effective [293]. Furthermore, IVIg has been shown to be ineffective in some reports [17]. Antithymocyte globulin has been used successfully in two patients with probable MAS [294], but it carries a significant risk of serious infections and mortality.
Recently, newer more targeted biologic therapies have provided a more targeted and effective option in treating sJIA-MAS. While there was some initial excitement about TNF blockers with reports on their success in treating many cases of MAS including several children with sJIA [295,296,297,298,299,300,301,302,303], reports of TNF inhibitors triggering MAS diminished enthusiasm for this therapy [304,305,306,307,308,309,310]. Knowing that cause and effect is certainly difficult to prove in these situations, the fact that MAS did develop in the setting of TNF inhibition is concerning [311, 312]. This has led to focusing on therapy directed at two other pro-inflammatory cytokines, IL-1 and IL-6.
The IL-1 receptor antagonist anakinra was shown to be highly effective for sJIA [313, 314]. MAS and sJIA flare share many clinical and laboratory features. Moreover in addition to the 10% risk of developing overt MAS as part of sJIA, another 30–40% of sJIA patients may have occult or subclinical MAS during flare that can eventually lead to overt MAS [68, 83]. This suggested that anakinra would also be a valuable treatment for sJIA-MAS. There are several reports of dramatically successful use of anakinra in cases of sJIA-MAS after failing to respond to corticosteroids and CsA [313, 315,316,317].
Anakinra is regarded as a generally safe drug because it is a recombinant human protein with a short half-life (approximately 4 h) [318] and a wide therapeutic window (1–48 mg/kg/day) [35, 317]. However, cases of hepatitis attributed to anakinra in children with sJIA have been reported [319]. Moreover, though cause and effect are difficult to establish, there has been a suggestion that anakinra triggered MAS in two children with sJIA [320, 321]. In a large case series of 46 sJIA patients treated with anakinra at disease onset, anakinra was a potential MAS trigger in five children at doses of 1–2 mg/kg/day [313]. However, dose escalation of anakinra often seemed to help control MAS, and none of the children had to permanently stop the anakinra [313]. More research is needed to define the role of anakinra in sJIA-MAS and other forms of sHLH.
IL-6 blockade , via an anti-IL-6 receptor monoclonal antibody (tocilizumab ), has also proven successful in treating sJIA [322]. However, a case report of MAS attributed to IL-6 blockade [323] underscores the need for further studies to define its role in the treatment of sJIA-MAS . IL-6 blockade, however, has been successfully used in treating cytokine storm syndrome associated with chimeric antigen receptor (CAR) T-cell-directed therapy against resistant leukemia [324]. Other biologic therapies are being explored. Co-stimulatory blockade with cytotoxic T-lymphocyte-associated protein 4-immunoglobulin (CTLA-4-Ig) has been anecdotally beneficial in children with severe sJIA [317], but its role in treating MAS is unknown. Nevertheless, there is building evidence that biologic therapies, particularly IL-1 inhibitors, are a welcome addition to corticosteroids and CsA in treating MAS associated with sJIA [17]. Finally, rituximab has recently been reported to lead to remission in a sizable percentage of children with refractory sJIA [325], in addition to its effectiveness in treating EBV-HLH [230, 326].
HSCT
The first report of successful HSCT in HLH was reported in 1986 [327]. HSCT tends to be more frequently used in familial cases of HLH, but it is used in secondary cases as well. While several studies have shown that HSCT is the best option for permanent disease control or cure [328,329,330,331,332], the overall transplant morbidity and mortality is still high (~45%) and it is not uncommon for patients to develop recurrence of HLH before a suitable donor is identified. Moreover, autologous and allogeneic HSCT have been reported to induce HLH in transplanted patients, probably due to the increased risk of infection imposed by the immunosuppressive conditioning regimen with an estimated risk of 4% in a recent cohort [333]. This risk appeared to be reduced in etoposide-containing conditioning regimens [333]. The current era of biologic therapies has reduced the need for HSCT.
In the future, autologous HSCT combined with gene therapy to correct the genetic defects might be applicable. Carmo et al. have shown that transfer of a functional perforin gene (Prf1) into autologous hematopoietic stem cells from perforin-deficient mice restored perforin expression, partially repaired the cytotoxic defect, and attenuated HLH symptoms after viral challenge, provided that at least 30% engraftment was attained [334]. Similarly, Rivat et al. showed that in a mouse model of XLP, gene transfer also restored SAP expression and normalized cytotoxic function [335].
IFN-γ Blockade
IFN-γ has been shown to play a pivotal role in several HLH models. Its neutralization has substantially improved survival in HLH animal models [336, 337] and an MAS model using IL-6 transgenic mice [338]. Levels of both IFN-γ and IFN-γ -induced chemokines such as CXCL10 and CXCL9 are elevated in children with HLH [339]. Moreover, sJIA-MAS is commonly triggered by viral infections which are known to activate IFN-γ-associated pathways. Furthermore, numerous IFN-γ-producing T cells were found in close proximity to activated hemophagocytic histiocytes in a study of inflammatory infiltrates in tissues affected by MAS [340], and children with MAS show increased levels of neopterin, which is known to be released by interferon-stimulated macrophages [26]. A longitudinal study of the cytokine changes in patients with sJIA showed that levels of IFN-γ and IFN-γ-induced chemokines (particularly, CXCL9) markedly increased with the beginning of clinical MAS and returned to normal with its resolution. Furthermore, they were strongly correlated with many laboratory features associated with MAS [338]. These findings highlighted IFN-γ as an appealing targeted and potentially less toxic therapeutic option in HLH, and a clinical trial evaluating NI-0501, which is an anti-IFN-γ monoclonal antibody that binds to and neutralizes human IFN-γ, is underway [341]. Recently, a report on this trial showed promising results [18].
However, some recent studies question the potential of IFN-γ blocking therapy in MAS. First, Tesi et al. reported two cases of HLH in children with novel IFN-γ receptor mutations associated with IFN-γ deficiency, findings which highlight the significance of IFN-γ-independent mechanisms in the immune pathology of HLH and warrant that other novel therapies, beside anti-IFN-γ therapy, be investigated [342]. Additionally, sJIA patients were found to have normal levels of IFN-γ independent of disease activity [343], which suggests that this cytokine does not always play an essential role in disease pathogenesis.
Janus Kinase Inhibition
Because targeting individual cytokines might be insufficient during severe hypercytokinemia, cytokine signaling pathways can be targeted to avoid an imbalance in the cytokine network. Janus kinases control the signaling of many cytokines, notably IFN-γ, IL-2, and IL-6. Thus, inhibition of Janus kinases via ruxolitinib for example may serve this purpose. Das et al. reported that in rodent models of primary and secondary HLH, treatment with the JAK1/2 inhibitor ruxolitinib significantly lessened the clinical and laboratory manifestations, including weight loss, organomegaly, anemia, thrombocytopenia, hypercytokinemia, and tissue inflammation. Importantly, ruxolitinib treatment also significantly improved survival in this model [344]. Similarly, Maschalidi et al. reported that JAK1/2 inhibition in Prf1−/− and Rab27a−/− mice with full-blown HLH syndrome has led to recovery in both models [345].
Other Targets
Peroxisome proliferator-activated receptor-γ agonists have also been presented as potential agents. They interfere with the activation of the NFκB pathway and exert both a broad anti-inflammatory effect and antiviral capacities [346, 347]. Based on research in HLH animal models, other targets for future therapy have been proposed. These include the induction of T-cell exhaustion through the stimulation of inhibitory receptors like programmed cell death 1 (PDCD1/PD-1), restoring cytokine balances by the anti-inflammatory IL-10 or IL-18BP, halting chronic TLR activation by TLR antagonists or blocking TLR signaling pathways, and targeting dendritic cells as the main drivers of ongoing antigen stimulation or suppressing antigen presentation itself [348, 349].
Over the past few years, more has been revealed about the role of IL-18 in the pathogenesis of sJIA and MAS. It has been shown that patients with sJIA have significantly higher levels of IL-18 [85,86,87] as opposed to other rheumatic diseases such as SLE or rheumatoid arthritis [350, 351]. sJIA patients with high levels of IL-18 are more likely to develop systemic features of the disease and are more prone to develop MAS [85]. Interestingly, the development of MAS in these patients is associated with a further rise in IL-18 levels [85]. IL-18 has also been found to correlate with ferritin in adult onset Still disease [86]. Therefore, IL-18 has been suggested as a promising biomarker for MAS. Another interesting field of growing research is in the role of IL-18-binding protein (IL-18BP) which is a naturally occurring protein that counter-regulates the activity of IL-18. Imbalance between IL-18 and IL-18BP, leading to higher levels of unbound IL-18, has been found in patients with increased disease severity [33, 86, 352]. The administration of synthetic IL-18BP in perforin-deficient mice infected with murine CMV ameliorated liver damage but has not shown an effect on the pro-inflammatory cytokine levels or on the overall survival [353]. Based on these findings, further work is needed to demonstrate the role of IL-18 and IL-18BP in MAS. Along these lines, IL-18BP was shown to be beneficial in treating refractory MAS in a child with an NLRC4 mutation [354].
Recently, neutralizing antibodies and antagonists targeting the alarmin HMGB1 (high mobility group box 1) were proposed as potential therapeutic options, aiming to reduce the immunostimulatory load of necrosis- and pyroptosis-derived danger signals. Models of systemic sterile and infectious inflammation have demonstrated the efficacy of this strategy [355]. Moreover, blocking the alarmin IL-33, via its receptor ST2/IL-1RL1, is also a potential therapy [356] [43]. Overall, the future is looking brighter for a variety of potential therapeutics to treat HLH/MAS in a patient-specific fashion.
Abbreviations
- AIDS:
-
Acquired immunodeficiency syndrome
- APC:
-
Antigen-presenting cell
- CsA:
-
Cyclosporine A
- CTL:
-
Cytotoxic T lymphocyte
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DIC:
-
Disseminated intravascular coagulopathy
- EBV:
-
Epstein-Barr virus
- fHLH:
-
Familial hemophagocytic lymphohistiocytosis
- FSTL-1:
-
Follistatin-like 1
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HAV:
-
Hepatitis A virus
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- HLH:
-
Hemophagocytic lymphohistiocytosis
- HS:
-
HScore
- HSCT:
-
Hematopoietic stem cell transplant
- ICU:
-
Intensive care unit
- IFN-γ:
-
interferon-gamma
- IL:
-
Interleukin
- IL-18BP:
-
Interleukin-18-binding protein
- IL-1Ra:
-
Interleukin-1 receptor antagonist
- IVIg:
-
Intravenous immunoglobulin
- MAS:
-
Macrophage activation syndrome
- NK cell:
-
Natural killer cell
- sCD163:
-
Soluble haptoglobin receptor
- sCD25:
-
Soluble interleukin-2 receptor alpha chain
- sHLH:
-
Secondary hemophagocytic lymphohistiocytosis
- sJIA:
-
Systemic juvenile idiopathic arthritis
- SLE:
-
Systemic lupus erythematosus
- Th1:
-
T-helper 1
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- USA:
-
United States of America
- XLP:
-
X-linked lymphoproliferative disease
References
Canna SW, Behrens EM. Making sense of the cytokine storm: a conceptual framework for understanding, diagnosing, and treating hemophagocytic syndromes. Pediatr Clin N Am. 2012;59(2):329–44.
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–46.
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–73.
Zur Stadt U, Rohr J, Seifert W, Koch F, Grieve S, Pagel J, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–92.
zur Stadt U, Schmidt S, Kasper B, Beutel K, Diler AS, Henter J-I, et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14(6):827–34.
Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–9.
Emmenegger U, Schaer D, Larroche C, Neftel K. Haemophagocytic syndromes in adults: current concepts and challenges ahead. Swiss Med Wkly. 2005;135(21–22):299–314.
Arico M, Imashuku S, Clementi R, Hibi S, Teramura T, Danesino C, et al. Hemophagocytic lymphohistiocytosis due to germline mutations in SH2D1A, the X-linked lymphoproliferative disease gene. Blood. 2001;97(4):1131–3.
Marsh RA, Satake N, Biroschak J, Jacobs T, Johnson J, Jordan MB, et al. STX11 mutations and clinical phenotypes of familial hemophagocytic lymphohistiocytosis in North America. Pediatr Blood Cancer. 2010;55(1):134–40.
Rigaud S, Fondaneche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110–4.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31.
Allen M, De Fusco C, Legrand F, Clementi R, Conter V, Danesino C, et al. Familial hemophagocytic lymphohistiocytosis: how late can the onset be? Haematologica. 2001;86(5):499–503.
Ueda I, Kurokawa Y, Koike K, Ito S, Sakata A, Matsumora T, et al. Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol. 2007;82(6):427–32.
Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82(4):369–72.
Henter JI, Elinder G, Lubeck PO, Ost A. Myelodysplastic syndrome following epipodophyllotoxin therapy in familial hemophagocytic lymphohistiocytosis. Pediatr Hematol Oncol. 1993;10(2):163–8.
Imashuku S. Etoposide-related secondary acute myeloid leukemia (t-AML) in hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):121–3.
Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology. 2011;50(2):417–9.
Jordan M, Locatelli F, Allen C, De Benedetti F, Grom AA, Ballabio M, et al. editors. A novel targeted approach to the treatment of hemophagocytic lymphohistiocytosis (HLH) with an anti-interferon gamma (IFN gamma) monoclonal antibody (mAb), NI-0501: first results from a pilot phase 2 study in children with primary HLH. Blood; 2015. AMER SOC HEMATOLOGY 2021 L ST NW, SUITE 900, WASHINGTON, DC 20036 USA.
Zhang K, Filipovich AH, Johnson J, Marsh RA, Villanueva J. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. Hemophagocytic Lymphohistiocytosis, familial. Seattle: GeneReviews((R)); 1993.
Koh KN, Im HJ, Chung NG, Cho B, Kang HJ, Shin HY, et al. Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea histiocytosis working party. Eur J Haematol. 2015;94(1):51–9.
Lehmberg K, Sprekels B, Nichols KE, Woessmann W, Muller I, Suttorp M, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol. 2015;170(4):539–49.
Celkan T, Berrak S, Kazanci E, Özyürek E, Ünal S, Uçar C, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in pediatric cases: a multicenter study from Turkey. Turk J Pediatr. 2009;51(3):207.
Veerakul G, Sanpakit K, Tanphaichitr VS, Mahasandana C, Jirarattanasopa N. Secondary hemophagocytic lymphohistiocytosis in children: an analysis of etiology and outcome. J Med Assoc Thailand. 2002;85:S530–41.
Grom AA, Mellins ED. Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol. 2010;22(5):561.
Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–98.
Put K, Avau A, Brisse E, Mitera T, Put S, Proost P, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin-18 and interferon-γ. Rheumatology. 2015;54(8):1507–17.
Akashi K, Hayashi S, Gondo H, Mizuno S, Harada M, Tamura K, et al. Involvement of interferon-γ and macrophage colony-stimulating factor in pathogenesis of haemophagocytic lymphohistiocytosis in adults. Br J Haematol. 1994;87(2):243–50.
Kuriyama T, Takenaka K, Kohno K, Yamauchi T, Daitoku S, Yoshimoto G, et al. Engulfment of hematopoietic stem cells caused by down-regulation of CD47 is critical in the pathogenesis of hemophagocytic lymphohistiocytosis. Blood. 2012;120(19):4058–67.
Nold-Petry CA, Lehrnbecher T, Jarisch A, Schwabe D, Pfeilschifter JM, Muhl H, et al. Failure of interferon γ to induce the anti-inflammatory interleukin 18 binding protein in familial hemophagocytosis. PLoS One. 2010;5(1):e8663.
Bracaglia C, Marafon DP, Caiello I, de Graaf K, Guilhot F, Ferlin W, et al. High levels of interferon-gamma (IFNγ) in macrophage activation syndrome (MAS) and CXCL9 levels as a biomarker for IFNγ production in MAS. Pediatr Rheumatol. 2015;13(1):1.
Tamura K, Kanazawa T, Tsukada S, Kobayashi T, Kawamura M, Morikawa A. Increased serum monocyte chemoattractant protein-1, macrophage inflammatory protein-1β, and interleukin-8 concentrations in hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(5):662–8.
Teruya-Feldstein J, Setsuda J, Yao X, Kingma DW, Straus S, Tosato G, et al. MIP-1alpha expression in tissues from patients with hemophagocytic syndrome. Lab Investig. 1999;79(12):1583–90.
Mazodier K, Marin V, Novick D, Farnarier C, Robitail S, Schleinitz N, et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood. 2005;106(10):3483–9.
Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89(11):4100–3.
Fisher CJ, Dhainaut J-FA, Opal SM, Pribble JP, Balk RA, Slotman GJ, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome: results from a randomized, double-blind, placebo-controlled trial. JAMA. 1994;271(23):1836–43.
Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: reanalysis of a prior phase III trial. Crit Care Med. 2016;44(2):275–81.
Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997;25(7):1115–24.
Créput C, Galicier L, Buyse S, Azoulay E. Understanding organ dysfunction in hemophagocytic lymphohistiocytosis. Intensive Care Med. 2008;34(7):1177–87.
Henter J, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78(11):2918–22.
Lachmann HJ, Quartier P, So A, Hawkins PN. The emerging role of interleukin-1β in autoinflammatory diseases. Arthritis Rheum. 2011;63(2):314–24.
Sieni E, Cetica V, Mastrodicasa E, Pende D, Moretta L, Griffiths G, et al. Familial hemophagocytic lymphohistiocytosis: a model for understanding the human machinery of cellular cytotoxicity. Cell Mol Life Sci. 2012;69(1):29–40.
Avau A, Put K, Wouters CH, Matthys P. Cytokine balance and cytokine-driven natural killer cell dysfunction in systemic juvenile idiopathic arthritis. Cytokine Growth Factor Rev. 2015;26(1):35–45.
Brisse E, Matthys P, Wouters CH. Understanding the spectrum of haemophagocytic lymphohistiocytosis: update on diagnostic challenges and therapeutic options. Br J Haematol. 2016;174(2):175–87.
De Kerguenec C, Hillaire S, Molinié V, Gardin C, Degott C, Erlinger S, et al. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96(3):852–7.
Dinarello C, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289.
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19(1):683–765.
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166(2):95–109.
Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med. 2015;212(3):307–17.
Janka G. Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klin Padiatr. 2009;221(05):278–85.
Grom AA. Natural killer cell dysfunction: a common pathway in systemic-onset juvenile rheumatoid arthritis, macrophage activation syndrome, and hemophagocytic lymphohistiocytosis? Arthritis Rheum. 2004;50(3):689–98.
Saltzman RW, Monaco-Shawver L, Zhang K, Sullivan KE, Filipovich AH, Orange JS. Novel mutation in syntaxin-binding protein 2 (STXBP2) prevents IL-2-induced natural killer cell cytotoxicity. J Allergy Clin Immunol. 2012;129(6):1666.
Spessott WA, Sanmillan ML, McCormick ME, Patel N, Villanueva J, Zhang K, et al. Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood. 2015;125(10):1566–77.
Zhang M, Behrens EM, Atkinson TP, Shakoory B, Grom AA, Cron RQ. Genetic defects in cytolysis in macrophage activation syndrome. Curr Rheumatol Rep. 2014;16(9):1–8.
Zhang X-Y, Ye X-W, Feng D-X, Han J, Li D, Zhang C. Hemophagocytic lymphohistiocytosis induced by severe pandemic influenza A (H1N1) 2009 virus infection: a case report. Case Rep Med. 2011;2011:1–3.
Nagafuji K, Nonami A, Kumano T, Kikushige Y, Yoshimoto G, Takenaka K, et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica. 2007;92(7):978–81.
Lichtenheld MG, Olsen KJ, Lu P, Lowrey DM, Hameed A, Hengartner H, et al. Structure and function of human perforin. Nature. 1988;335(6189):448–51.
Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome–like disease in mice. J Clin Investig. 2011;121(6):2264–77.
Sullivan KE, Delaat CA, Douglas SD, Filipovich AH. Defective natural killer cell function in patients with hemophagocytic lymphohistiocytosis and in first degree relatives. Pediatr Res. 1998;44(4):465–8.
Villanueva J, Lee S, Giannini EH, Graham TB, Passo MH, Filipovich A, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2004;7(1):1.
Caiello I, Minnone G, Holzinger D, Vogl T, Prencipe G, Manzo A, et al. IL-6 amplifies TLR mediated cytokine and chemokine production: implications for the pathogenesis of rheumatic inflammatory diseases. PLoS One. 2014;9(10):e107886.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Zhang L, Zhou J, Sokol L. Hereditary and acquired hemophagocytic lymphohistiocytosis. Cancer Control. 2014;21(4):301–12.
Schulert GS, Grom AA. Macrophage activation syndrome and cytokine-directed therapies. Best Pract Res Clin Rheumatol. 2014;28(2):277–92.
Weaver LK, Behrens EM. Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr Opin Rheumatol. 2014;26(5):562.
Minoia F, Davì S, Horne A, Demirkaya E, Bovis F, Li C, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66(11):3160–9.
Tsuda H, Shirono K. Serum lipids in adult patients with hemophagocytic syndrome. Am J Hematol. 1996;53(4):285.
Prendki V, Stirnemann J, Lemoine M, Lohez M, Aras N, Ganne-Carrié N, et al. Prevalence and clinical significance of Küpffer cell hyperplasia with hemophagocytosis in liver biopsies. Am J Surg Pathol. 2011;35(3):337–45.
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–8.
Sawhney S, Woo P, Murray K. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421–6.
Stephan J, Koné-Paut I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285–92.
Ravelli A, Magni-Manzoni S, Pistorio A, Besana C, Foti T, Ruperto N, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146(5):598–604.
Parodi A, Davì S, Pringe AB, Pistorio A, Ruperto N, Magni-Manzoni S, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum. 2009;60(11):3388–99.
Ravelli A, Minoia F, Davi S, Horne A, Bovis F, Pistorio A, et al. 2016 classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European league against rheumatism/American College of rheumatology/paediatric rheumatology international trials organisation collaborative initiative. Arthritis Rheumatol. 2016;68(3):566–76.
Pelkonen P, Swanljung K, Siimes M. Ferritinemia as an indicator of systemic disease activity in children with systemic juvenile rheumatoid arthritis. Acta Paediatr. 1986;75(1):64–8.
Davì S, Lattanzi B, Demirkaya E, Rosina S, Bracciolini G, Novelli A. Toward the development of new diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Paediatr Rheumatol. 2012;1:1–7.
Kelly A, Ramanan AV. Recognition and management of macrophage activation syndrome in juvenile arthritis. Curr Opin Rheumatol. 2007;19(5):477–81.
Davì S, Minoia F, Pistorio A, Horne A, Consolaro A, Rosina S, et al. Performance of current guidelines for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66(10):2871–80.
Pringe A, Trail L, Ruperto N, Buoncompagni A, Loy A, Breda L, et al. Review: macrophage activation syndrome in juvenile systemic lupus erythematosus: an under-recognized complication? Lupus. 2007;16(8):587–92.
Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–20.
Hejblum G, Lambotte O, Galicier L, Coppo P, Marzac C, Aumont C, et al. A web-based Delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS One. 2014;9(4):e94024.
De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–95.
Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2396–406.
Bleesing J, Prada A, Siegel DM, Villanueva J, Olson J, Ilowite NT, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor α-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(3):965–71.
Gorelik M, Fall N, Altaye M, Barnes MG, Thompson SD, Grom AA, et al. Follistatin-like protein 1 and the ferritin/erythrocyte sedimentation rate ratio are potential biomarkers for dysregulated gene expression and macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2013;40(7):1191–9.
Shimizu M, Yokoyama T, Yamada K, Kaneda H, Wada H, Wada T, et al. Distinct cytokine profiles of systemic-onset juvenile idiopathic arthritis-associated macrophage activation syndrome with particular emphasis on the role of interleukin-18 in its pathogenesis. Rheumatology. 2010;49(9):1645–53.
Kawashima M, Yamamura M, Taniai M, Yamauchi H, Tanimoto T, Kurimoto M, et al. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001;44(3):550–60.
Maeno N, Takei S, Imanaka H, Yamamoto K, Kuriwaki K, Kawano Y, et al. Increased interleukin-18 expression in bone marrow of a patient with systemic juvenile idiopathic arthritis and unrecognized macrophage-activation syndrome. Arthritis Rheum. 2004;50(6):1935–8.
Shimizu M, Nakagishi Y, Inoue N, Mizuta M, Ko G, Saikawa Y, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160(2):277–81.
Jenkins RW, Clarke CJ, Lucas JT, Shabbir M, Wu BX, Simbari F, et al. Evaluation of the role of secretory sphingomyelinase and bioactive sphingolipids as biomarkers in hemophagocytic lymphohistiocytosis. Am J Hematol. 2013;88(11):E265–E72.
Ishii E. Hemophagocytic lymphohistiocytosis in children: pathogenesis and treatment. Front Pediatr. 2016;4:47.
Cron RQ, Davi S, Minoia F, Ravelli A. Clinical features and correct diagnosis of macrophage activation syndrome. Expert Rev Clin Immunol. 2015;11(9):1043–53.
Yanagimachi M, Goto H, Miyamae T, Kadota K, Imagawa T, Mori M, et al. Association of IRF5 polymorphisms with susceptibility to hemophagocytic lymphohistiocytosis in children. J Clin Immunol. 2011;31(6):946–51.
Chen C, Huang Y, Jaing T, Hung I, Yang C, Chang L, et al. Hemophagocytic syndrome: a review of 18 pediatric cases. J Microbiol Immunol Infect. 2004;37(3):157.
Risdall RJ, McKenna RW, Nesbit ME, Krivit W, Balfour HH, Simmons RL, et al. Virus-associated hemophagocytic syndrome A benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979;44(3):993–1002.
Burgio GR, Aricó M, Marconi M, Lanfranchi A, Caselli D, Ugazio AG. Spontaneous NBT reduction by monocytes as a marker of disease activity in children with histiocytosis. Br J Haematol. 1990;74(2):146–50.
Oloomi Z, Moayeri H. Cytomegalovirus infection-associated hemophagocytic syndrome. Arch Iran Med. 2006;9:284–7.
Maruyama K, Koizumi T, Hirato J. Cytomegalovirus associated hemophagocytic lymphohistiocytosis in a premature infant. Pediatr Int. 2006;48(6):648–50.
Hoang MP, Dawson DB, Rogers ZR, Scheuermann RH, Rogers BB. Polymerase chain reaction amplification of archival material for Epstein-Barr virus, cytomegalovirus, human herpesvirus 6, and parvovirus B19 in children with bone marrow hemophagocytosis. Hum Pathol. 1998;29(10):1074–7.
Fardet L, Blum L, Kerob D, Agbalika F, Galicier L, Dupuy A, et al. Human herpesvirus 8-associated hemophagocytic lymphohistiocytosis in human immunodeficiency virus-infected patients. Clin Infect Dis. 2003;37(2):285–91.
Grossman WJ, Radhi M, Schauer D, Gerday E, Grose C, Goldman FD. Development of hemophagocytic lymphohistiocytosis in triplets infected with HHV-8. Blood. 2005;106(4):1203–6.
Re A, Facchetti F, Borlenghi E, Cattaneo C, Capucci M, Ungari M, et al. Fatal hemophagocytic syndrome related to active human herpesvirus-8/Kaposi sarcoma-associated herpesvirus infection in human immunodeficiency virus-negative, non-transplant patients without related malignancies. Eur J Haematol. 2007;78(4):361–4.
van der Werff ten Bosch J, Kollen WJ, Ball LM, Brinkman D, Vossen AC, Lankester AC, et al. Atypical varicella zoster infection associated with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009;53(2):226–8.
Yamada K, Yamamoto Y, Uchiyama A, Ito R, Aoki Y, Uchida Y, et al. Successful treatment of neonatal herpes simplex-type 1 infection complicated by hemophagocytic lymphohistiocytosis and acute liver failure. Tohoku J Exp Med. 2008;214(1):1–5.
Ramasamy K, Lim Z, Savvas M, Salisbury J, Dokal I, Mufti G, et al. Disseminated herpes virus (HSV-2) infection with rhabdomyolysis and hemophagocytic lymphohistiocytosis in a patient with bone marrow failure syndrome. Ann Hematol. 2006;85(9):629–30.
Levy J, Wodell R, August C, Bayever E. Adenovirus-related hemophagocytic syndrome after bone marrow transplantation. Bone Marrow Transplant. 1990;6(5):349–52.
Ardalan M, Shoja M, Tubbs R, Esmaili H, Keyvani H. Postrenal transplant hemophagocytic lymphohistiocytosis and thrombotic microangiopathy associated with parvovirus b19 infection. Am J Transplant. 2008;8(6):1340–4.
Wada Y, Kai M, Tanaka H, Shimizu N, Shimatani M, Oshima T. Computed tomography findings of the liver in a neonate with Herpes simplex virus-associated hemophagocytic lymphohistiocytosis. Pediatr Int. 2011;53(5):773–6.
Yamaguchi K, Yamamoto A, Hisano M, Natori M, Murashima A. Herpes Simplex Virus 2–Associated Hemophagocytic Lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5, Part 2):1241–4.
Yilmaz S, Oren H, Demircioglu F, Firinci F, Korkmaz A, Irken G. Parvovirus B19: a cause for aplastic crisis and hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2006;47(6):861.
Dutta U, Mittal S, Ratho RK, Das A. Acute liver failure and severe hemophagocytosis secondary to parvovirus B19 infection. Indian J Gastroenterol. 2005;24(3):118.
Faurschou M, Nielsen OJ, Hansen PB, Juhl BR, Hasselbalch H. Fatal virus-associated hemophagocytic syndrome associated with coexistent chronic active hepatitis B and acute hepatitis C virus infection. Am J Hematol. 1999;61(2):135–8.
Tuon FF, Gomes VS, Amato VS, Graf ME, Fonseca GHH, Lazari C, et al. Hemophagocytic syndrome associated with hepatitis A: case report and literature review. Rev Inst Med Trop Sao Paulo. 2008;50(2):123–7.
Russo RA, Rosenzweig SD, Katsicas MM. Hepatitis A-associated macrophage activation syndrome in children with systemic juvenile idiopathic arthritis: report of 2 cases. J Rheumatol. 2008;35(1):166–8.
Tierney Jr LM, Thabet A, Nishino H. Case 10-2011: a woman with fever, confusion, liver failure, anemia, and thrombocytopenia. N Engl J Med. 2011;364(13):1259–70.
Akamatsu N, Sugawara Y, Tamura S, Matsui Y, Hasegawa K, Imamura H, et al., editors. Hemophagocytic syndrome after adult-to-adult living donor liver transplantation. Amsterdam: Elsevier; 2006.
Pease DF, Mathew J, Hepatitis C. Virus associated hemophagocytic lymphohistiocytosis: case report and literature review. J Hematol. 2013;2(2):76–8.
Doyle T, Bhagani S, Cwynarski K. Haemophagocytic syndrome and HIV. Curr Opin Infect Dis. 2009;22(1):1–6.
Sun H-Y, Chen M-Y, Fang C-T, Hsieh S-M, Hung C-C, Chang S-C. Hemophagocytic lymphohistiocytosis: an unusual initial presentation of acute HIV infection. J Acquir Immune Defic Syndr. 2004;37(4):1539–40.
Park K-H, Yu H-S, Jung S-I, Shin D-H, Shin J-H. Acute human immunodeficiency virus syndrome presenting with hemophagocytic lymphohistiocytosis. Yonsei Med J. 2008;49(2):325–8.
Harms PW, Schmidt LA, Smith LB, Newton DW, Pletneva MA, Walters LL, et al. Autopsy findings in eight patients with fatal H1N1 influenza. Am J Clin Pathol. 2010;134(1):27–35.
Lai S, Merritt BY, Chen L, Zhou X, Green LK. Hemophagocytic lymphohistiocytosis associated with influenza A (H1N1) infection in a patient with chronic lymphocytic leukemia: an autopsy case report and review of the literature. Ann Diagn Pathol. 2012;16(6):477–84.
Mou SS, Nakagawa TA, Riemer EC, McLean TW, Hines MH, Shetty AK. Hemophagocytic lymphohistiocytosis complicating influenza A infection. Pediatrics. 2006;118(1):e216–9.
Özdemir H, Ciftci E, Ince EÜ, Ertem M, Ince E, Dogru Ü. Hemophagocytic lymphohistiocytosis associated with 2009 pandemic influenza A (H1N1) virus infection. J Pediatr Hematol Oncol. 2011;33(2):135–7.
Shrestha B, Omran A, Rong P, Wang W. Report of a fatal pediatric case of hemophagocytic lymphohistiocytosis associated with pandemic influenza A (H1N1) infection in 2009. Pediatr Neonatol. 2015;56(3):189–92.
Willekens C, Cornelius A, Guerry M-J, Wacrenier A, Fourrier F. Fulminant hemophagocytic lymphohistiocytosis induced by pandemic A (H1N1) influenza: a case report. J Med Case Rep. 2011;5(1):1.
Zheng Y, Yang Y, Zhao W, Wang H. Novel swine-origin influenza A (H1N1) virus-associated hemophagocytic syndrome—a first case report. Am J Trop Med Hyg. 2010;82(4):743–5.
Beffermann N, Pilcante J, Sarmiento M. Acquired hemophagocytic syndrome related to parainfluenza virus infection: case report. J Med Case Rep. 2015;9(1):1.
Xing Q, Xing P. Mumps caused hemophagocytic syndrome: a rare case report. Am J Emerg Med. 2013;31(6):1000.e1–2.
Iaria C, Leonardi MS, Buda A, Toro ML, Cascio A. Measles and secondary hemophagocytic lymphohistiocytosis. Emerg Infect Dis. 2012;18(9):1529.
Otagiri T, Mitsui T, Kawakami T, Katsuura M, Maeda K, Ikegami T, et al. Haemophagocytic lymphohistiocytosis following measles vaccination. Eur J Pediatr. 2002;161(9):494–6.
Koubaa M, Marrakchi C, Maaloul I, Makni S, Berrajah L, Elloumi M, et al. Rubella associated with hemophagocytic syndrome. First report in a male and review of the literature. Mediterranean J Hematol Infect Dis. 2012;4(1):e2012050.
Katsibardi K, Moschovi MA, Theodoridou M, Spanakis N, Kalabalikis P, Tsakris A, et al. Enterovirus-associated hemophagocytic syndrome in children with malignancy: report of three cases and review of the literature. Eur J Pediatr. 2008;167(1):97–102.
Nagao T, Takahashi N, Saitoh H, Noguchi S, Guo Y, Ito M, et al. Adult T-cell leukemia-lymphoma developed from an HTLV-1 carrier during treatment of B-cell lymphoma-associated hemophagocytic syndrome. [Rinsho ketsueki] Jpn J Clin Hematol. 2012;53(12):2008–12.
Takahashi S, Oki J, Miyamoto A, Koyano S, Ito K, Azuma H, et al. Encephalopathy associated with haemophagocytic lymphohistiocytosis following rotavirus infection. Eur J Pediatr. 1999;158(2):133–7.
Ray S, Kundu S, Saha M, Chakrabarti P. Hemophagocytic syndrome in classic dengue fever. J Global Infect Dis. 2011;3(4):399.
Koshy M, Mishra AK, Agrawal B, Kurup AR, Hansdak SG. Dengue fever complicated by hemophagocytosis. Oxford Med Case Rep. 2016;2016(6):121–4.
Ab-Rahman HA, Rahim H, AbuBakar S, Wong P-F. Macrophage activation syndrome-associated markers in severe dengue. Int J Med Sci. 2016;13(3):179.
Sam S-S, Omar SFS, Teoh B-T, Abd-Jamil J, AbuBakar S. Review of dengue hemorrhagic fever fatal cases seen among adults: a retrospective study. PLoS Negl Trop Dis. 2013;7(5):e2194.
Erduran E, Cakir M. Reactive hemophagocytic lymphohistiocytosis and Crimean-Congo hemorrhagic fever. Int J Infect Dis. 2010;14:e349.
Baty V, Schuhmacher H, Bourgoin C, Latger V, Buisine J, May T, et al. Hemophagocytic syndrome and hemorrhagic fever with renal syndrome. Presse Méd. 1998;27(31):1577.
Lee JJ, Chung IJ, Shin DH, Cho SH, Cho D, Ryang DW, et al. Hemorrhagic fever with renal syndrome presenting with hemophagocytic lymphohistiocytosis. Emerg Infect Dis. 2002;8(2):209–10.
Lin L, Xu Y-Z, Wu X-M, Ge H-F, Feng J-X, Chen M-F, et al. A rare fatal case of a novel bunyavirus-associated hemophagocytic lymphohistiocytosis. J Infect Dev Countries. 2016;10(05):533–6.
Kamihira T, Yano K, Tamada Y, Matsumoto T, Miyazato M, Nagaoka S, et al. Case of domestically infected hepatitis E with marked thrombocytopenia. Nihon Shokakibyo Gakkai Zasshi. 2008;105(6):841–6.
Lu M, Xie Z, Gao Z, Wang C, Li N, Li M, et al. Histopathologic study of avian influenza H5N1 infection in humans. Zhonghua bing li xue za zhi. 2008;37(3):145–9.
Pei F, Zheng J, Gao Z, Zhong Y, Fang W, Gong E, et al. Lung pathology and pathogenesis of severe acute respiratory syndrome: a report of six full autopsies. Zhonghua bing li xue za zhi: Chin J Pathol. 2005;34(10):656–60.
Yoshiyama M, Kounami S, Nakayama K, Aoyagi N, Yoshikawa N. Clinical assessment of Mycoplasma pneumoniae-associated hemophagocytic lymphohistiocytosis. Pediatr Int. 2008;50(4):432–5.
Ishida Y, Hiroi K, Tauchi H, Oto Y, Tokuda K, Kida K. Hemophagocytic lymphohistiocytosis secondary to Mycoplasma pneumoniae infection. Pediatr Int. 2004;46(2):174–7.
Non LR, Patel R, Esmaeeli A, Despotovic V. Typhoid fever complicated by hemophagocytic lymphohistiocytosis and rhabdomyolysis. Am J Trop Med Hyg. 2015;93(5):1068–9.
Sniderman JD, Cuvelier GD, Veroukis S, Hansen G. Toxic epidermal necrolysis and hemophagocytic lymphohistiocytosis: a case report and literature review. Clin Case Rep. 2015;3(2):121–5.
Dube R, Kar SS, Mahapatro S, Ray R. Infection associated hemophagocytic lymphohistiocytosis: a case report. Indian J Clin Pract. 2013;24(2).
Tseng Y-T, Sheng W-H, Lin B-H, Lin C-W, Wang J-T, Chen Y-C, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect. 2011;44(3):191–7.
Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7(12):814–22.
Yagi K, Kano G, Shibata M, Sakamoto I, Matsui H, Imashuku S. Chlamydia pneumoniae infection-related hemophagocytic lymphohistiocytosis and acute encephalitis and poliomyelitis-like flaccid paralysis. Pediatr Blood Cancer. 2011;56(5):853–5.
Maheshwari P, Chhabra R, Yadav P. Perinatal tuberculosis associated hemophagocytic lymphohistiocytosis. Indian J Pediatr. 2012;79(9):1228–9.
Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated haemophagocytic syndrome. Lancet Infect Dis. 2006;6(7):447–54.
Wali Y, Beshlawi I. BCG lymphadenitis in neonates with familial hemophagocytic lymphohistiocytosis. Pediatr Infect Dis J. 2012;31(3):324.
Rositto A, Molinaro L, Larralde M, Ranalletta M, Drut R. Disseminated cutaneous eruption after BCG vaccination. Pediatr Dermatol. 1996;13(6):451–4.
Schleinitz N, Bernit E, Harle J-R. Severe hemophagocytic syndrome after intravesical BCG instillation. Am J Med. 2002;112(7):593–4.
Gosh J, Roy M, Bala A. Infection associated with hemophagocytic lymphohistiocytosis triggered by nosocomial infection. Oman Med J. 2009;24(3):223–5.
Chang C-C, Hsiao P-J, Chiu C-C, Chen Y-C, Lin S-H, Wu C-C, et al. Catastrophic hemophagocytic lymphohistiocytosis in a young man with nephrotic syndrome. Clin Chim Acta. 2015;439:168–71.
Saidi W, Gammoudi R, Korbi M, Aounallah A, Boussofara L, Ghariani N, et al. Hemophagocytic lymphohistiocytosis: an unusual complication of leprosy. Int J Dermatol. 2015;54(9):1054–9.
Kiernan TJ, O’Flaherty N, Gilmore R, Ho E, Hickey M, Tolan M, et al. Abiotrophia defectiva endocarditis and associated hemophagocytic syndrome—a first case report and review of the literature. Int J Infect Dis. 2008;12(5):478–82.
Dumler JS. The biological basis of severe outcomes in Anaplasma phagocytophilum infection. FEMS Immunol Med Microbiol. 2012;64(1):13–20.
Karras A, Thervet E, Legendre C. Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation. 2004;77(2):238–43.
Cantero-Hinojosa J, D’iez-Ruiz A, Santos-Perez J, Aguilar-Martinez J, Ramos-Jimenez A. Lyme disease associated with hemophagocytic syndrome. J Mol Med. 1993;71(8):620.
Akbayram S, Dogan M, Akgun C, Peker E, Parlak M, Caksen H, et al. An analysis of children with brucellosis associated with pancytopenia. Pediatr Hematol Oncol. 2011;28(3):203–8.
Erduran E, Makuloglu M, Mutlu M. A rare hematological manifestation of brucellosis: reactive hemophagocytic syndrome. J Microbiol Immunol Infect. 2010;43(2):159–62.
Sari I, Altuntas F, Hacioglu S, Kocyigit I, Sevinc A, Sacar S, et al. A multicenter retrospective study defining the clinical and hematological manifestations of brucellosis and pancytopenia in a large series: hematological malignancies, the unusual cause of pancytopenia in patients with brucellosis. Am J Hematol. 2008;83(4):334–9.
Anstead GM, Jorgensen JH, Craig FE, Blaser MJ, Patterson TF. Thermophilic multidrug-resistant Campylobacter fetus infection with hypersplenism and histiocytic phagocytosis in a patient with acquired immunodeficiency syndrome. Clin Infect Dis. 2001;32(2):295–6.
Tamura A, Matsunobu T, Kurita A, Shiotani A. Hemophagocytic syndrome in the course of sudden sensorineural hearing loss. ORL. 2012;74(4):211–4.
Ramon I, Libert M, Guillaume M-P, Corazza F, Karmali R. Recurrent haemophagocytic syndrome in an HIV-infected patient. Acta Clin Belg. 2010;65(4):276–8.
Chinen K, Ohkura Y, Matsubara O, Tsuchiya E. Hemophagocytic syndrome associated with clostridial infection in a pancreatic carcinoma patient. Pathol Res Pract. 2004;200(3):241–5.
Harris P, Dixit R, Norton R. Coxiella burnetii causing haemophagocytic syndrome: a rare complication of an unusual pathogen. Infection. 2011;39(6):579–82.
Hufnagel M, Niemeyer C, Zimmerhackl LB, Tüchelmann T, Sauter S, Brandis M. Hemophagocytosis: a complication of acute Q fever in a child. Clin Infect Dis. 1995;21(4):1029–31.
Hanson D, Walter AW, Powell J. Ehrlichia-induced hemophagocytic lymphohistiocytosis in two children. Pediatr Blood Cancer. 2011;56(4):661–3.
Burns S, Saylors R, Mian A. Hemophagocytic lymphohistiocytosis secondary to Ehrlichia chaffeensis infection: a case report. J Pediatr Hematol Oncol. 2010;32(4):e142–3.
Abbott KC, Vukelja SJ, Smith CE, McAllister CK, Konkol KA, O’rourke TJ, et al. Hemophagocytic syndrome: a cause of pancytopenia in human ehrlichiosis. Am J Hematol. 1991;38(3):230–4.
Krishnamurthy S, Mahadevan S, Mandal J, Basu D. Leptospirosis in association with hemophagocytic syndrome: a rare presentation. Indian J Pediatr. 2013;80(6):524–5. https://doi.org/10.1007/s12098-012-0863-0.
Niller H. Myelodysplastic syndrome (MDS) as a late stage of subclinical hemophagocytic lymphohistiocytosis (HLH): a putative role for Leptospira infection. A hypothesis. Acta Microbiol Immunol Hung. 2010;57(3):181–9.
Lambotte O, Fihman V, Poyart C, Buzyn A, Berche P, Soumelis V. Listeria monocytogenes skin infection with cerebritis and haemophagocytosis syndrome in a bone marrow transplant recipient. J Infect. 2005;50(4):356–8.
Pellegrin J, Merlio J, Lacoste D, Barbeau P, Brossard G, Beylot J, et al. Syndrome of macrophagic activation with hemophagocytosis in human immunodeficiency virus infection. Rev Med interne. 1992;13(6):438–40.
Yang W, Fu L, Lan J, Shen G, Chou G, Tseng C, et al. Mycobacterium avium complex-associated hemophagocytic syndrome in systemic lupus erythematosus patient: report of one case. Lupus. 2003;12(4):312–6.
Valsalan R, Kosaraju K, Sohanlal T, Kumar PP. Hemophagocytosis in scrub typhus. J Postgrad Med. 2010;56(4):301.
Cascio A, Giordano S, Dones P, Venezia S, Iaria C, Ziino O. Haemophagocytic syndrome and rickettsial diseases. J Med Microbiol. 2011;60(4):537–42.
Gutiérrez-Ravé PV, Luque MR, Ayerza LM, Cañavate IM, Prados MD. Reactive hemophagocytic syndrome: analysis of a series of 7 cases. Med Clin. 1990;94(4):130–4.
Guo X, Chen N, Wang T, Zhou C, Li Q, Gao J. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis: report of four childhood cases. Zhonghua er ke za zhi. 2011;49(7):550–3.
Rajagopala S, Dutta U, Chandra KP, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis–case report and systematic review. J Infect. 2008;56(5):381–8.
Gagnaire M-H, Galambrun C, Stéphan JL. Hemophagocytic syndrome: a misleading complication of visceral leishmaniasis in children—a series of 12 cases. Pediatrics. 2000;106(4):e58.
Arslan F, Batirel A, Ramazan M, Ozer S, Mert A. Macrophage activation syndrome triggered by primary disseminated toxoplasmosis. Scand J Infect Dis. 2012;44(12):1001–4.
Briand P, Gangneux J, Favaretto G, Ly-Sunnaram B, Godard M, Robert-Gangneux F, et al. Hemophagocytic syndrome and toxoplasmic primo-infection. Ann Biol Clin. 2008;66(2):199–205. https://doi.org/10.1684/abc.2008.0209.
Gupta P, Hurley RW, Helseth PH, Goodman JL, Hammerschmidt DE. Pancytopenia due to hemophagocytic syndrome as the presenting manifestation of babesiosis. Am J Hematol. 1995;50(1):60–2.
Bhagat M, Kanhere S, Kadakia P, Phadke V, George R, Chaudhari K. Haemophagocytic lymphohistiocytosis: a cause of unresponsive malaria in a 5-year-old girl. Paediatr Int Child Health. 2015;35(4):333–6. https://doi.org/10.1080/20469047.2015.1109227.
Saribeyoglu ET, Anak S, Agaoglu L, Boral O, Unuvar A, Devecioglu O. Secondary hemophagocytic lymphohistiocytosis induced by malaria infection in a child with Langerhans cell histiocytosis. Pediatr Hematol Oncol. 2004;21(3):267–72.
Ullah W, Abdullah HMA, Qadir S, Shahzad MA. Haemophagocytic lymphohistiocytosis (HLH): a rare but potentially fatal association with Plasmodium vivax malaria. BMJ Case Rep. 2016;2016:bcr2016215366.
Bhatia S, Bauer F, Bilgrami SA. Candidiasis-associated hemophagocytic lymphohistiocytosis in a patient infected with human immunodeficiency virus. Clin Infect Dis. 2003;37(11):):e161–6.
Numata K, Tsutsumi H, Wakai S, Tachi N, Chiba S. A child case of haemophagocytic syndrome associated with cryptococcal meningoencephalitis. J Infect. 1998;36(1):118–9.
Majluf-Cruz A, Hurtado MR, Souto-Meirino C, del Río CC, Simon J. Hemophagocytic syndrome associated with histoplasmosis in the acquired immunodeficiency syndrome: description of 3 cases and review of the literature. Sangre. 1993;38(1):51–5.
Quijano G, Siminovich M, Drut R. Histopathologic findings in the lymphoid and reticuloendothelial system in pediatric HIV infection: a postmortem study. Pediatr Pathol Lab Med. 1997;17(6):845–56.
Keller FG, Kurtzberg J. Disseminated histoplasmosis: a cause of infection-associated hemophagocytic syndrome. J Pediatr Hematol Oncol. 1994;16(4):368–71.
Chokephaibulkit K, Veerakul G, Vanprapar N, Chaiprasert A, Tanphaichitr V, Chearskul S. Penicilliosis-associated hemophagocytic syndrome in a human immunodeficiency virus-infected child: the first case report in children. J Med Assoc Thai. 2001;84(3):426–9.
Chim C, Fong C, Ma S, Wong S, Yuen K. Reactive hemophagocytic syndrome associated with Penicillium marneffei infection. Am J Med. 1998;104(2):196–7.
Elazary AS, Wolf DG, Amir G, Avni B, Rund D, Yehuda DB, et al. Severe Epstein–Barr virus-associated hemophagocytic syndrome in six adult patients. J Clin Virol. 2007;40(2):156–9.
Tabata Y, Hibi S, Teramura T, Kuriyama K, Yagi T, Todo S, et al. Molecular analysis of latent membrane protein 1 in patients with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Leuk Lymphoma. 2000;38(3–4):373–80.
Kawaguchi H, Miyashita T, Herbst H, Niedobitek G, Asada M, Tsuchida M, et al. Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus-associated hemophagocytic syndrome. J Clin Investig. 1993;92(3):1444.
Beutel K, Gross-Wieltsch U, Wiesel T, Stadt UZ, Janka G, Wagner HJ. Infection of T lymphocytes in Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children of non-Asian origin. Pediatr Blood Cancer. 2009;53(2):184–90.
Imashuku S. Clinical features and treatment strategies of Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44(3):259–72.
Cho HS, Park YN, Lyu CJ, Park SM, Oh SH, Yang CH, et al. EBV-elicited familial hemophagocytic lymphohistiocytosis. Yonsei Med J. 1997;38:245–8.
Purtilo D, Yang JS, Cassel C, Harper R, Stephenson S, Landing B, et al. X-linked recessive progressive combined variable immunodeficiency (Duncan’s disease). Lancet. 1975;305(7913):935–41.
Christensson B, Braconier JH, Winqvist I, Relander T, Dictor M. Fulminant course of infectious mononucleosis with virus-associated hemophagocytic syndrome. Scand J Infect Dis. 1987;19(3):373–9.
Akashi K, Mizuno S-I. Epstein-Barr virus-infected natural killer cell leukemia. Leuk Lymphoma. 2000;40(1–2):57–66.
Ohshima K, Suzumiya J, Sugihara M, Nagafuchi S, Ohga S, Kikuchi M. Clinicopathological study of severe chronic active Epstein-Barr virus infection that developed in association with lymphoproliferative disorder and/or hemophagocytic syndrome. Pathol Int. 1998;48(12):934–43.
Su I-J, Wang C-H, Cheng A-L, Chen R-L. Hemophagocytic syndrome in Epstein-Barr virus-associated T-lymphoproliferative disorders: disease spectrum, pathogenesis, and management. Leuk Lymphoma. 1995;19(5–6):401–6.
Lindemann TL, Greene JS. Persistent cervical lymphadenopathy in an adolescent with Epstein–Barr induced hemophagocytic syndrome: manifestations of a rare but often fatal disease. Int J Pediatr Otorhinolaryngol. 2005;69(7):1011–4.
Chuang H-C, Lay J-D, Hsieh W-C, Wang H-C, Chang Y, Chuang S-E, et al. Epstein-Barr virus LMP1 inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome. Blood. 2005;106(9):3090–6.
Kasahara Y, Yachie A. Cell type specific infection of Epstein–Barr virus (EBV) in EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Crit Rev Oncol Hematol. 2002;44(3):283–94.
Imashuku S, Hibi S, Tabata Y, Sako M, Sekine Y, Hirayama K, et al. Biomarker and morphological characteristics of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1998;31(3):131–7.
Nazaruk RA, Rochford R, Hobbs MV, Cannon MJ. Functional diversity of the CD8+ T-cell response to Epstein-Barr virus (EBV): implications for the pathogenesis of EBV-associated lymphoproliferative disorders. Blood. 1998;91(10):3875–83.
Hatta K, Morimoto A, Ishii E, Kimura H, Ueda I, Hibi S, et al. Association of transforming growth factor-β1 gene polymorphism in the development of Epstein-Barr virus-related hematologic diseases. Haematologica. 2007;92(11):1470–4.
Teramura T, Tabata Y, Yagi T, Morimoto A, Hibi S, Imashuku S. Quantitative analysis of cell-free Epstein-Barr virus genome copy number in patients with EBV-associated hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2002;43(1):173–9.
Kimura H, Hoshino Y, Hara S, Nishikawa K, Sako M, Hirayama M, et al. Viral load in Epstein-Barr virus-associated hemophagocytic syndrome. Microbiol Immunol. 2002;46(8):579–82.
Sandberg Y, van Gastel-Mol EJ, Verhaaf B, Lam KH, van Dongen JJ, Langerak AW. BIOMED-2 multiplex immunoglobulin/T-cell receptor polymerase chain reaction protocols can reliably replace Southern blot analysis in routine clonality diagnostics. J Mol Diagn. 2005;7(4):495–503.
Langerak AW, Groenen PJ, Bruggemann M, Beldjord K, Bellan C, Bonello L, et al. EuroClonality/BIOMED-2 guidelines for interpretation and reporting of Ig/TCR clonality testing in suspected lymphoproliferations. Leukemia. 2012;26(10):2159–71.
Matsuda K, Nakazawa Y, Yanagisawa R, Honda T, Ishii E, Koike K. Detection of T-cell receptor gene rearrangement in children with Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis using the BIOMED-2 multiplex polymerase chain reaction combined with GeneScan analysis. Clin Chim Acta. 2011;412(17):1554–8.
Kelesidis T, Humphries R, Terashita D, Eshaghian S, Territo MC, Said J, et al. Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis in Los Angeles county. J Med Virol. 2012;84(5):777–85.
Kogawa K, Sato H, Asano T, Ohga S, Kudo K, Morimoto A, et al. Prognostic factors of Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis in children: report of the Japan Histiocytosis Study Group. Pediatr Blood Cancer. 2014;61(7):1257–62.
Trottestam H, Berglöf E, Horne A, Onelöv E, Beutel K, Lehmberg K, et al. Risk factors for early death in children with haemophagocytic lymphohistiocytosis. Acta Paediatr. 2012;101(3):313–8.
Huang SC, Chen JS, Cheng CN, Yang YJ. Hypoalbuminaemia is an independent predictor for hemophagocytic lymphohistiocytosis in childhood Epstein–Barr virus-associated infectious mononucleosis. Eur J Haematol. 2012;89(5):417–22.
Bakhshi S, Pautu JL. EBV-associated hemophagocytic lymphohistiocytosis with spontaneous regression. Indian Pediatr. 2005;42(12):1253.
Imashuku S, Tabata Y, Teramura T, Hibi S. Treatment strategies for Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH). Leuk Lymphoma. 2000;39(1–2):37–49.
Balamuth NJ, Nichols KE, Paessler M, Teachey DT. Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2007;29(8):569–73.
Imashuku S, Hibi S, Todo S, Sako M, Inoue M, Kawa K, et al. Allogeneic hematopoietic stem cell transplantation for patients with hemophagocytic syndrome (HPS) in Japan. Bone Marrow Transplant. 1999;23(6):569–72.
Ohga S, Kudo K, Ishii E, Honjo S, Morimoto A, Osugi Y, et al. Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein–Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer. 2010;54(2):299–306.
Qin Q, Xie Z, Shen Y, Yang S, Liu C, Huang Z, et al. Assessment of immunochemotherapy and stem cell transplantation on EBV-associated hemophagocytic lymphohistiocytosis in children: a systematic review and meta analysis. Eur Rev Med Pharmacol Sci. 2012;16(5):672–8.
Milone MC, Tsai DE, Hodinka RL, Silverman LB, Malbran A, Wasik MA, et al. Treatment of primary Epstein-Barr virus infection in patients with X-linked lymphoproliferative disease using B-cell–directed therapy. Blood. 2005;105(3):994–6.
Danish EH, Dahms BB, Kumar ML. Cytomegalovirus-associated hemophagocytic syndrome. Pediatrics. 1985;75(2):280–3.
Kohara MM, Blum RN. Cytomegalovirus Ileitis and Hemophagocytic syndrome associated with use of anti—tumor necrosis factor—α antibody. Clin Infect Dis. 2006;42(5):733–4.
Koketsu S-I, Watanabe T, Hori N, Umetani N, Takazawa Y, Nagawa H. Hemophagocytic syndrome caused by fulminant ulcerative colitis and cytomegalovirus infection: report of a case. Dis Colon Rectum. 2004;47(7):1250–5.
Amenomori M, Migita K, Miyashita T, Yoshida S, Ito M, Eguchi K, et al. Cytomegalovirus-associated hemophagocytic syndrome in a patient with adult onset Still’s disease. Clin Exp Rheumatol. 2005;23(1):100–2.
Sakamoto O, Ando M, Yoshimatsu S, Kohrogi H, Suga M, Ando M. Systemic lupus erythematosus complicated by cytomegalovirus-induced hemophagocytic syndrome and colitis. Intern Med. 2002;41(2):151–5.
Devecioglu O, Anak S, Atay D, Aktan P, Devecioglu E, Ozalp B, et al. Pediatric acute lymphoblastic leukemia complicated by secondary hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009;53(3):491–2.
Sato M, Matsushima T, Takada S, Hatsumi N, Kim K, Sakuraya M, et al. Fulminant, CMV-associated, haemophagocytic syndrome following unrelated bone marrow transplantation. Bone Marrow Transplant. 1998;22(12):1219.
Hardikar W, Pang K, Al-Hebbi H, Curtis N, Couper R. Successful treatment of cytomegalovirus-associated haemophagocytic syndrome following paediatric orthotopic liver transplantation. J Paediatr Child Health. 2006;42(6):389–91.
Abdelkefi A, Jamil WB, Torjman L, Ladeb S, Ksouri H, Lakhal A, et al. Hemophagocytic syndrome after hematopoietic stem cell transplantation: a prospective observational study. Int J Hematol. 2009;89(3):368–73.
Imashuku S, Ueda I, Teramura T, Mori K, Morimoto A, Sako M, et al. Occurrence of haemophagocytic lymphohistiocytosis at less than 1 year of age: analysis of 96 patients. Eur J Pediatr. 2005;164(5):315–9.
Uneda S, Murata S, Sonoki T, Matsuoka H, Nakakuma H. Successful treatment with liposomal doxorubicin for widespread Kaposi’s sarcoma and human herpesvirus-8 related severe hemophagocytic syndrome in a patient with acquired immunodeficiency syndrome. Int J Hematol. 2009;89(2):195–200.
Stebbing J, Ngan S, Ibrahim H, Charles P, Nelson M, Kelleher P, et al. The successful treatment of haemophagocytic syndrome in patients with human immunodeficiency virus-associated multi-centric Castleman’s disease. Clin Exp Immunol. 2008;154(3):399–405.
Pastore RD, Chadburn A, Kripas C, Schattner EJ. Novel association of haemophagocytic syndrome with Kaposi’s sarcoma-associated herpesvirus-related primary effusion lymphoma. Br J Haematol. 2000;111(4):1112–5.
Bossini N, Sandrini S, Setti G, Luppi M, Maiorca P, Maffei C, et al. Successful treatment with liposomal doxorubicin and foscarnet in a patient with widespread Kaposi’s sarcoma and human herpes virus 8-related, serious hemophagocytic syndrome, after renal transplantation. G ital Nefrol. 2004;22(3):281–6.
Corbellino M, Bestetti G, Scalamogna C, Calattini S, Galazzi M, Meroni L, et al. Long-term remission of Kaposi sarcoma–associated herpesvirus-related multicentric Castleman disease with anti-CD20 monoclonal antibody therapy. Blood. 2001;98(12):3473–5.
Tanaka H, Nishimura T, Hakui M, Sugimoto H, Tanaka-Taya K, Yamanishi K. Human herpesvirus 6-associated hemophagocytic syndrome in a healthy adult. Emerg Infect Dis. 2002;8(1):87.
Suzuki N, Morimoto A, Ohga S, Kudo K, Ishida Y, Ishii E, et al. Characteristics of hemophagocytic lymphohistiocytosis in neonates: a nationwide survey in Japan. J Pediatr. 2009;155(2):235–8.e1.
Whitley R. Neonatal herpes simplex virus infection. Curr Opin Infect Dis. 2004;17(3):243–6.
Rittichier KR, Bryan PA, Bassett KE, Taggart EW, Enriquez FR, Hillyard DR, et al. Diagnosis and outcomes of enterovirus infections in young infants. Pediatr Infect Dis J. 2005;24(6):546–50.
Albrecht H, Schafer H, Stellbrink H-J, Greten H. Epstein-Barr virus-associated hemophagocytic syndrome. Arch Pathol Lab Med. 1997;121(8):853.
Niedt G, Schinella R. Acquired immunodeficiency syndrome. Clinicopathologic study of 56 autopsies. Arch Pathol Lab Med. 1985;109(8):727–34.
Sailler L, Duchayne E, Marchou B, Brousset P, Pris J, Massip P, et al. Etiological aspects of reactive hemophagocytoses: retrospective study in 99 patients. Rev Med Interne. 1996;18(11):855–64.
Maakaroun NR, Moanna A, Jacob JT, Albrecht H. Viral infections associated with haemophagocytic syndrome. Rev Med Virol. 2010;20(2):93–105.
Potter M, Foot A, Oakhill A. Influenza A and the virus associated haemophagocytic syndrome: cluster of three cases in children with acute leukaemia. J Clin Pathol. 1991;44(4):297–9.
Imashuku S. Differential diagnosis of hemophagocytic syndrome: underlying disorders and selection of the most effective treatment. Int J Hematol. 1997;66(2):135–51.
Cunney RJ, Bialachowski A, Thornley D, Smaill FM, Pennie RA. An outbreak of influenza A in a neonatal intensive care unit. Infect Control Hosp Epidemiol. 2000;21(7):449–54.
Ando M, Miyazaki E, Hiroshige S, Ashihara Y, Okubo T, Ueo M, et al. Virus associated hemophagocytic syndrome accompanied by acute respiratory failure caused by influenza A (H3N2). Intern Med. 2006;45(20):1183–6.
Deerojanawong J, Prapphal N, Poovorawan Y. Prevalence, clinical presentations and complications among hospitalized children with influenza pneumonia. Jpn J Infect Dis. 2008;61:446–9.
To K-F, Chan PK, Chan K-F, Lee W-K, Lam W-Y, Wong K-F, et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J Med Virol. 2001;63(3):242–6.
Zhang Z, Zhang J, Huang K, Li K-S, Yuen K-Y, Guan Y, et al. Systemic infection of avian influenza A virus H5N1 subtype in humans. Hum Pathol. 2009;40(5):735–9.
Kimura K, Adlakha A, Simon PM. Fatal case of swine influenza virus in an immunocompetent host. Mayo Clin Proc. 1998;73(3):243–5.
Watanabe T, Okazaki E, Shibuya H. Influenza A virus-associated encephalopathy with haemophagocytic syndrome. Eur J Pediatr. 2003;162(11):799–800.
de Jong MD, Cam BV, Qui PT, Hien VM, Thanh TT, Hue NB, et al. Fatal avian influenza A (H5N1) in a child presenting with diarrhea followed by coma. N Engl J Med. 2005;352(7):686–91.
Chokephaibulkit K, Uiprasertkul M, Puthavathana P, Chearskul P, Auewarakul P, Dowell SF, et al. A child with avian influenza A (H5N1) infection. Pediatr Infect Dis J. 2005;24(2):162–6.
Peiris J, Yu W, Leung C, Cheung C, Ng W, Nicholls JA, et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet. 2004;363(9409):617–9.
Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, et al. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360(9348):1831–7.
Chan PK. Outbreak of avian influenza A (H5N1) virus infection in Hong Kong in 1997. Clin Infect Dis. 2002;34(Suppl 2):S58–64.
Ng WF, To KF, Lam WW, Ng TK, Lee KC. The comparative pathology of severe acute respiratory syndrome and avian influenza A subtype H5N1—a review. Hum Pathol. 2006;37(4):381–90.
Hsieh SM, Chang SC. Insufficient perforin expression in CD8+ T cells in response to hemagglutinin from avian influenza (H5N1) virus. J Immunol. 2006;176(8):4530–3.
Henter JI, Chow CB, Leung CW, Lau YL. Cytotoxic therapy for severe avian influenza A (H5N1) infection. Lancet. 2006;367(9513):870–3.
Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, et al. Avian influenza A (H5N1) infection in humans. N Engl J Med. 2005;353(13):1374–85.
Schulert GS, Zhang M, Fall N, Husami A, Kissell D, Hanosh A, et al. Whole-exome sequencing reveals mutations in genes linked to hemophagocytic lymphohistiocytosis and macrophage activation syndrome in fatal cases of H1N1 influenza. J Infect Dis. 2016;213(7):1180–8.
Kaya Z, Ozturk G, Gursel T, Bozdayi G. Spontaneous resolution of hemophagocytic syndrome and disseminated intravascular coagulation associated with parvovirus b19 infection in a previously healthy child. Jpn J Infect Dis. 2005;58(3):149–51.
Larroche C, Scieux C, Honderlick P, Piette AM, Morinet F, Bletry O. Spontaneous resolution of hemophagocytic syndrome associated with acute parvovirus B19 infection and concomitant Epstein-Barr virus reactivation in an otherwise healthy adult. Eur J Clin Microbiol Infect Dis. 2002;21(10):739–42.
Watanabe M, Shibuya A, Okuno J, Maeda T, Tamama S, Saigenji K. Hepatitis A virus infection associated with hemophagocytic syndrome: report of two cases. Intern Med. 2002;41(12):1188–92.
Verma T, Aggarwal S. Childhood tuberculosis presenting with haemophagocytic syndrome. Indian J Hematol Blood Transfus. 2012;28(3):178–80.
Yang C, Lee J, Kim Y, Kim P, Lee S, Kim B, et al. Tuberculosis-associated hemophagocytic syndrome in a hemodialysis patient: case report and review of the literature. Nephron. 1996;72(4):690–2.
Ruiz-argüelles GJ, Arizpe-Bravo D, Garces-Eisele J, Sanchez-Sosa S, Ruiz-argüelles A, Ponce-de-Leon S. Tuberculosis-associated fatal hemophagocytic syndrome in a patient with lymphoma treated with fludarabine. Leuk Lymphoma. 1998;28(5–6):599–602.
Baraldes MA, Domingo P, Gonzalez MJ, Aventin A, Coll P. Tuberculosis-associated hemophagocytic syndrome in patients with acquired immunodeficiency syndrome. Arch Intern Med. 1998;158(2):194–5.
Lam K, Ng W, Chan A. Miliary tuberculosis with splenic rupture: a fatal case with hemophagocytic syndrome and possible association with long standing sarcoidosis. Pathology. 1994;26(4):493–6.
Cascio A, Pernice L, Barberi G, Delfino D, Biondo C, Beninati C, et al. Secondary hemophagocytic lymphohistiocytosis in zoonoses. A systematic review. Eur Rev Med Pharmacol Sci. 2012;16(10):1324–37.
Koduri PR, Chundi V, DeMarais P, Mizock BA, Patel AR, Weinstein RA. Reactive hemophagocytic syndrome: a new presentation of disseminated histoplasmosis in patients with AIDS. Clin Infect Dis. 1995;21(6):1463–5.
De Lavaissière M, Manceron V, Bourée P, Garçon L, Bisaro F, Delfraissy J-F, et al. Reconstitution inflammatory syndrome related to histoplasmosis, with a hemophagocytic syndrome in HIV infection. J Infect. 2009;58(3):245–7.
Hadchouel M, Prieur A-M, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–6.
Wallace CA, Sherry DD. Trial of intravenous pulse cyclophosphamide and methylprednisolone in the treatment of severe systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 1997;40(10):1852–5.
Mouy R, Stephan J-L, Pillet P, Haddad E, Hubert P, Prieur A-M. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996;129(5):750–4.
Ravelli A, De Benedetti F, Viola S, Martini A. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine. J Pediatr. 1996;128(2):275–8.
Henter J-I, Samuelsson-Horne A, Arico M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–73.
Emmenegger U, Spaeth PJ, Neftel KA. Intravenous immunoglobulin for hemophagocytic lymphohistiocytosis? J Clin Oncol. 2002;20(2):599–601.
Coca A, Bundy KW, Marston B, Huggins J, Looney RJ. Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol. 2009;132(1):10–8.
Aeberli D, Oertle S, Mauron H, Reichenbach S, Jordi B, Villiger PM. Inhibition of the TNF pathway: use of infliximab and etanercept as remission-inducing agents in cases of therapy-resistant chronic inflammatory disorders. Swiss Med Wkly. 2002;132(29–30):414–22.
Emmenegger U, Reimers A, Frey U, Fux C, Bihl F, Semela D, et al. Reactive macrophage activation syndrome: a simple screening strategy and its potential in early treatment initiation. Swiss Med Wkly. 2002;132(17/18):230–6.
Henzan T, Nagafuji K, Tsukamoto H, Miyamoto T, Gondo H, Imashuku S, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematol. 2006;81(1):59–61.
Maeshima K, Ishii K, Iwakura M, Akamine M, Hamasaki H, Abe I, et al. Adult-onset Still’s disease with macrophage activation syndrome successfully treated with a combination of methotrexate and etanercept. Mod Rheumatol. 2012;22(1):137–41.
Makay B, Yılmaz Ş, Türkyılmaz Z, Ünal N, Ören H, Ünsal E. Etanercept for therapy-resistant macrophage activation syndrome. Pediatr Blood Cancer. 2008;50(2):419–21.
Prahalad S, Bove KE, Dickens D, Lovell DJ, Grom AA. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol. 2001;28(9):2120–4.
Sellmer A, Stausbøl-Grøn B, Krag-Olsen B, Herlin T. Successful use of infliximab in macrophage activation syndrome with severe CNS involvement. Scand J Rheumatol. 2011;40(2):156–7.
Takahashi N, Naniwa T, Banno S. Successful use of etanercept in the treatment of acute lupus hemophagocytic syndrome. Mod Rheumatol. 2008;18(1):72–5.
Verbsky JW, Grossman WJ. Hemophagocytic lymphohistiocytosis: diagnosis, pathophysiology, treatment, and future perspectives. Ann Med. 2006;38(1):20–31.
Chauveau E, Terrier F, Casassus-Buihle D, Moncoucy X, Oddes B. Macrophage activation syndrome after treatment with infliximab for fistulated Crohn’s disease. Presse Med. 2005;34(8):583–4.
Kaneko K, Kaburaki M, Muraoka S, Tanaka N, Yamamoto T, Kusunoki Y, et al. Exacerbation of adult-onset Still’s disease, possibly related to elevation of serum tumor necrosis factor-alpha after etanercept administration. Int J Rheum Dis. 2010;13(4):e67–e9.
Kimura Y, Pinho P, Walco G, Higgins G, Hummell D, Szer I, et al. Etanercept treatment in patients with refractory systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2005;32(5):935–42.
Ramanan AV, Schneider R. Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003;30(2):401–3.
Sandhu C, Chesney A, Piliotis E, Buckstein R, Koren S. Macrophage activation syndrome after etanercept treatment. J Rheumatol. 2007;34(1):241.
Sterba G, Sterba Y, Stempel C, Blank J, Azor E, Gomez L. Macrophage activation syndrome induced by etanercept in a patient with systemic sclerosis. Isr Med Assoc J. 2010;12(7):443.
Stern A, Riley R, Buckley L. Worsening of macrophage activation syndrome in a patient with adult onset Still’s disease after initiation of etanercept therapy. J Clin Rheumatol. 2001;7(4):252–6.
Agarwal S, Moodley J, Goel GA, Theil KS, Mahmood SS, Lang RS. A rare trigger for macrophage activation syndrome. Rheumatol Int. 2011;31(3):405–7.
Moltó A, Mateo L, Lloveras N, Olivé A, Minguez S. Visceral leishmaniasis and macrophagic activation syndrome in a patient with rheumatoid arthritis under treatment with adalimumab. Joint Bone Spine. 2010;77(3):271–3.
Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, Van Rossum MA, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–55.
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479–86.
Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol. 2011;17(1):23–7.
Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol. 2008;4(11):615–20.
Record JL, Beukelman T, Cron RQ. Combination therapy of abatacept and anakinra in children with refractory systemic juvenile idiopathic arthritis: a retrospective case series. J Rheumatol. 2011;38(1):180–1.
Clark SR, McMahon CJ, Gueorguieva I, Rowland M, Scarth S, Georgiou R, et al. Interleukin-1 receptor antagonist penetrates human brain at experimentally therapeutic concentrations. J Cereb Blood Flow Metab. 2008;28(2):387–94.
Canna S, Frankovich J, Higgins G, Narkewicz MR, Nash SR, Hollister JR, et al. Acute hepatitis in three patients with systemic juvenile idiopathic arthritis taking interleukin-1 receptor antagonist. Pediatr Rheumatol. 2009;7(1):1.
Lurati A, Teruzzi B, Salmaso A, Demarco G, Pontikati I, Gattinara M, et al. Macrophage activation syndrome (MAS) during anti-IL1 receptor therapy (anakinra) in a patient affected by systemic onset idiopathic juvenile arthritis (soJIA): a report and review of the literature. Pediatr Rheumatol Online J. 2005;3(2):79–85.
Zeft A, Hollister R, LaFleur B, Sampath P, Soep J, McNally B, et al. Anakinra for systemic juvenile arthritis: the Rocky Mountain experience. J Clin Rheumatol. 2009;15(4):161–4.
Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371(9617):998–1006.
Mizutani S, Kuroda J, Shimura Y, Kobayashi T, Tsutsumi Y, Yamashita M, et al. Cyclosporine A for chemotherapy-resistant subcutaneous panniculitis-like T cell lymphoma with hemophagocytic syndrome. Acta Haematol. 2011;126(1):8–12.
Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–30.
Alexeeva EI, Valieva SI, Bzarova TM, Semikina EL, Isaeva KB, Lisitsyn AO, et al. Efficacy and safety of repeat courses of rituximab treatment in patients with severe refractory juvenile idiopathic arthritis. Clin Rheumatol. 2011;30(9):1163–72.
Bosman G, Langemeijer SC, Hebeda K, Raemaekers J, Pickkers P, van der Velden W. The role of rituximab in a case of EBV-related lymphoproliferative disease presenting with haemophagocytosis. Neth J Med. 2009;67(8):364–5.
Fischer A, Cerf-Bensussan N, Blanche S, Le Deist F, Bremard-Oury C, Leverger G, et al. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J Pediatr. 1986;108(2):267–70.
Baker K, Filipovich A, Gross T, Grossman W, Hale G, Hayashi R, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2008;42(3):175–80.
Baker KS, DeLaat CA, Steinbuch M, Gross TG, Shapiro RS, Loechelt B, et al. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood. 1997;89(10):3857–63.
Blanche S, Caniglia M, Girault D, Landman J, Griscelli C, Fischer A. Treatment of hemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation: a single-center study of 22 cases. Blood. 1991;78(1):51–4.
Dürken M, Horstmann M, Bieling P, Erttmann R, Kabisch H, Löliger C, et al. Improved outcome in haemophagocytic lymphohistiocytosis after bone marrow transplantation from related and unrelated donors: a single-centre experience of 12 patients. Br J Haematol. 1999;106(4):1052–8.
Dürken M, Finckenstein FG, Janka GE. Bone marrow transplantation in hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2001;41(1–2):89–95.
Kobayashi R, Tanaka J, Hashino S, Ota S, Torimoto Y, Kakinoki Y, et al. Etoposide-containing conditioning regimen reduces the occurrence of hemophagocytic lymphohistiocytosis after SCT. Bone Marrow Transplant. 2014;49(2):254–7.
Carmo M, Risma KA, Arumugam P, Tiwari S, Hontz AE, Montiel-Equihua CA, et al. Perforin gene transfer into hematopoietic stem cells improves immune dysregulation in murine models of perforin deficiency. Mol Ther. 2015;23(4):737–45.
Rivat C, Booth C, Alonso-Ferrero M, Blundell M, Sebire NJ, Thrasher AJ, et al. SAP gene transfer restores cellular and humoral immune function in a murine model of X-linked lymphoproliferative disease. Blood. 2013;121(7):1073–6.
Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–43.
Schmid JP, Ho CH, Chrétien F, Lefebvre JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of IFNγ defeats haemophagocytosis in LCMV-infected perforin-and Rab27a-deficient mice. EMBO Mol Med. 2009;1(2):112–24.
Bracaglia C, Caiello I, De Graaf K, D’Ario G, Guilhot F, Ferlin W, et al. Interferon-gamma (IFNy) in macrophage activation syndrome (MAS) associated with systemic juvenile idiopathic arthritis (sJIA). High levels in patients and a role in a murine mas model. Pediatr Rheumatol. 2014;12(1):1.
Takada H, Takahata Y, Nomura A, Ohga S, Mizuno Y, Hara T. Increased serum levels of interferon-γ-inducible protein 10 and monokine induced by gamma interferon in patients with haemophagocytic lymphohistiocytosis. Clin Exp Immunol. 2003;133(3):448–53.
Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-γ-producing lymphocytes and IL-6-and TNF-α-producing macrophages. Blood. 2005;105(4):1648–51.
Science UnLo. 2016. https://clinicaltrials.gov/ct2/show/NCT01818492.
Tesi B, Sieni E, Neves C, Romano F, Cetica V, Cordeiro AI, et al. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-[gamma] receptor deficiency. J Allergy Clin Immunol. 2015;135(6):1638.
Sikora KA, Fall N, Thornton S, Grom AA. The limited role of interferon-γ in systemic juvenile idiopathic arthritis cannot be explained by cellular hyporesponsiveness. Arthritis Rheum. 2012;64(11):3799–808.
Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127(13):1666–75.
Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. 2016;128(1):60–71. https://doi.org/10.1182/blood-2016-02-700013.
Chuang HC, Lay JD, Hsieh WC, Su IJ. Pathogenesis and mechanism of disease progression from hemophagocytic lymphohistiocytosis to Epstein–Barr virus-associated T-cell lymphoma: nuclear factor-κB pathway as a potential therapeutic target. Cancer Sci. 2007;98(9):1281–7.
Hsieh W-C, Lan B-S, Chen Y-L, Chang Y, Chuang H-C, Su I-J. Efficacy of peroxisome proliferator activated receptor agonist in the treatment of virus-associated haemophagocytic syndrome in a rabbit model. Antivir Ther. 2010;15(1):71–81.
Yang J, Huck SP, McHugh RS, Hermans IF, Ronchese F. Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc Natl Acad Sci U S A. 2006;103(1):147–52.
Brisse E, Wouters CH, Matthys P. Hemophagocytic lymphohistiocytosis (HLH): a heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev. 2015;26(3):263–80.
Favilli F, Anzilotti C, Martinelli L, Quattroni P, De Martino S, Pratesi F, et al. IL-18 activity in systemic lupus erythematosus. Ann N Y Acad Sci. 2009;1173:301–9.
Novick D, Elbirt D, Miller G, Dinarello CA, Rubinstein M, Sthoeger ZM. High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J Autoimmun. 2010;34(2):121–6.
Dinarello CA. Interleukin-18 and the pathogenesis of inflammatory diseases. Semin Nephrol. 2007;27(1):98–114.
Chiossone L, Audonnet S, Chetaille B, Chasson L, Farnarier C, Berda-Haddad Y, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front Immunol. 2012;3:239.
Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017;139(5):1698–701.
Palmblad K, Schierbeck H, Sundberg E, Horne AC, Harris HE, Henter JI, et al. High systemic levels of the cytokine-inducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol Med. 2014;20:538–47.
Rood JE, Rao S, Paessler M, Kreiger PA, Chu N, Stelekati E, et al. ST2 contributes to T-cell hyperactivation and fatal hemophagocytic lymphohistiocytosis in mice. Blood. 2016;127(4):426–35.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Eloseily, E.M., Cron, R.Q. (2018). Macrophage Activation Syndrome. In: Ragab, G., Atkinson, T., Stoll, M. (eds) The Microbiome in Rheumatic Diseases and Infection. Springer, Cham. https://doi.org/10.1007/978-3-319-79026-8_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-79026-8_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-79025-1
Online ISBN: 978-3-319-79026-8
eBook Packages: MedicineMedicine (R0)