
Abstract
Strand displacement and tile assembly systems are designed to follow prescribed kinetic rules (i.e., exhibit a specific time-evolution). However, the expected behavior in the limit of infinite time—known as thermodynamic equilibrium—is often incompatible with the desired computation. Basic physical chemistry implicates this inconsistency as a source of unavoidable error. Can the thermodynamic equilibrium be made consistent with the desired computational pathway? In order to formally study this question, we introduce a new model of molecular computing in which computation is driven by the thermodynamic driving forces of enthalpy and entropy. To ensure greatest generality we do not assume that there are any constraints imposed by geometry and treat monomers as unstructured collections of binding sites. In this model we design Boolean AND/OR formulas, as well as a self-assembling binary counter, where the thermodynamically favored states are exactly the desired final output configurations. Though inspired by DNA nanotechnology, the model is sufficiently general to apply to a wide variety of chemical systems.
D. Doty—Supported by NSF grant CCF-1619343.
T.A. Rogers—Supported by the NSF Graduate Research Fellowship Program under Grant No. DGE-1450079, NSF Grant CAREER-1553166, and NSF Grant CCF-1422152.
D. Soloveichik—Supported by NSF grants CCF-1618895 and CCF-1652824.
C. Thachuk—Supported by NSF grant CCF-1317694.
D. Woods—Part of this work was carried out at California Institute of Technology. Supported by Inria (France) as well as National Science Foundation (USA) grants CCF-1219274, CCF-1162589, CCF-1317694.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Notes
- 1.
That is, we assume like-unlike binding such as that found in DNA Watson-Crick base-pairing, as opposed to like-like binding such as hydrophobic molecules with an affinity for each other in aqueous solution, or base stacking between the blunt ends of DNA helices [6, 13]. It is not clear the extent to which this choice affects the computational power of our model.
- 2.
Because a monomer collection is a multiset of monomer types, each of which is itself a multiset, we distinguish them typographically with an arrow.
- 3.
For instance, the monomer collection shown in Fig. 1 has 2 domains of type a, 2 domains of type b, and 1 domain of type \(a^*\) and \(b^*\) each.
- 4.
A matching of a graph is a subset of edges that share no vertices in common. In our case this enforces that a domain is bound to at most one other domain.
- 5.
We are assuming bonds are of equal strength (although the definition can be naturally generalized to bonds of different strength).
- 6.
Our use of the terms “enthalpy” and “entropy”, and notation H and S is meant to evoke the corresponding physical notions. Note, however, that there are other contributions to physical entropy besides the number of separate complexes. Indeed, the free energy contribution of forming additional bonds typically contains substantial enthalpic and entropic parts.
- 7.
We are generalizing the convention for the word “polymer” in the chemistry literature. We have no requirement that a polymer be linear, nor that it consist of repeated subunits. We chose “polymer” rather than “complex” to better contrast with “monomer”.
- 8.
In typical DNA nanotechnology applications, the Gibbs free energy \(\varDelta G(\alpha )\) of a configuration \(\alpha \) can be estimated as follows. Bonds correspond to domains of length l bases, and forming each base pair is favorable by \(\varDelta G^\circ _{\text {bp}}\). Thus, the contribution of \(H(\alpha )\) to \(\varDelta G(\alpha )\) is \((\varDelta G^\circ _{\text {bp}} \cdot l) H(\alpha )\). At 1 M, the free energy penalty due to decreasing the number of separate complexes by 1 is \(\varDelta G^\circ _{\text {assoc}}\). At effective concentration C M, this penalty increases to \(\varDelta G^\circ _{\text {assoc}} + RT \ln (1/C)\). As the point of zero free energy, we take the configuration with no bonds, and all monomers separate. Thus, the contribution of \(S(\alpha )\) to \(\varDelta G(\alpha )\) is \((\varDelta G^\circ _{\text {assoc}} + RT \ln (1/C))(|\alpha |-S(\alpha ))\), where \(|\alpha |\) is the total number of monomers. To summarize,
$$\begin{aligned} \varDelta G(\alpha )&= (\varDelta G^\circ _{\text {bp}} \cdot l) H(\alpha ) + (\varDelta G^\circ _{\text {assoc}} + RT \ln (1/C))(|\alpha |-S(\alpha )). \end{aligned}$$Note that, as expected, this is a linear combination of \(H(\alpha )\) and \(S(\alpha )\), and that increasing the length of domains l weighs \(H(\alpha )\) more heavily, while decreasing the concentration C weighs \(S(\alpha )\) more heavily. Typically \(G^\circ _{\text {bp}} \approx -1.5\) kcal/mol, and \(G^\circ _{\text {assoc}} \approx 1.96\) kcal/mol [9].
- 9.
Note that the other limiting case, where entropy is infinitely more favorable, is degenerate: the most favorable configuration in that case always has every monomer unconnected to any other.
- 10.
Observations 6, 7, and 8 are not really necessary for our technique, but simplify the description of the conditions under which
 and
and 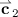 would be saturated: specifically, that if
would be saturated: specifically, that if 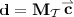 is in the nonnegative orthant, then so are
is in the nonnegative orthant, then so are  and
and  . If we did not use relabeling (thus could not guarantee that \({\mathbf {d}}\) is in the nonnegative orthant) then the requisite condition to apply Lemma 2 would be that \({\mathbf {d}}\), \({\mathbf {d}}_1\), and \({\mathbf {d}}_2\) all occupy the same orthant; i.e., for all \(i \in \{1,\ldots ,d\}\), if any of \({\mathbf {d}}(i)\), \({\mathbf {d}}_1(i)\), or \({\mathbf {d}}_2(i)\) are negative, then the other two are not positive.
. If we did not use relabeling (thus could not guarantee that \({\mathbf {d}}\) is in the nonnegative orthant) then the requisite condition to apply Lemma 2 would be that \({\mathbf {d}}\), \({\mathbf {d}}_1\), and \({\mathbf {d}}_2\) all occupy the same orthant; i.e., for all \(i \in \{1,\ldots ,d\}\), if any of \({\mathbf {d}}(i)\), \({\mathbf {d}}_1(i)\), or \({\mathbf {d}}_2(i)\) are negative, then the other two are not positive. - 11.
In particular, the proof of [7] upper bounds the size of \({\mathbf {x}}\) in terms of the entries of both \({\mathbf {A}}\) and \({\mathbf {b}}\). However, the naïve way to solve a linear inequality \({\mathbf {A}}{\mathbf {x}}\ge {\mathbf {0}}\) using an equality, by introducing slack variables \({\mathbf {b}}\) and asking for solutions \({\mathbf {x}}\in {\mathbb {N}}^m\), \({\mathbf {b}}\in {\mathbb {N}}^n\) such that \({\mathbf {A}}{\mathbf {x}}= {\mathbf {b}}\), allows for the possibility that \(\Vert {\mathbf {b}}\Vert \) is very large compared to \(\Vert {\mathbf {A}}\Vert \), in which case upper bounding \(\Vert {\mathbf {x}}\Vert \) in terms of both \({\mathbf {A}}\) and \({\mathbf {b}}\) does not help to bound \(\Vert {\mathbf {x}}\Vert \) in terms of \({\mathbf {A}}\) alone.
 and
and 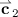 would be saturated: specifically, that if
would be saturated: specifically, that if 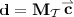 is in the nonnegative orthant, then so are
is in the nonnegative orthant, then so are  and
and  . If we did not use relabeling (thus could not guarantee that
. If we did not use relabeling (thus could not guarantee that References
Barish, R.D., Schulman, R., Rothemund, P.W.K., Winfree, E.: An information-bearing seed for nucleating algorithmic self-assembly. Proc. Natl. Acad. Sci. 106(15), 6054–6059 (2009)
Bennett, C.H.: The thermodynamics of computation—a review. Int. J. Theor. Phys. 21(12), 905–940 (1982)
Breik, K., Prakash, L., Thachuk, C., Heule, M., Soloveichik, D.: Computing properties of stable configurations of thermodynamic binding networks (2017, in preparation)
Dirks, R.M., Bois, J.S., Schaeffer, J.M., Winfree, E., Pierce, N.A.: Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev. 49(1), 65–88 (2007)
Genot, A.J., Zhang, D.Y., Bath, J., Turberfield, A.J.: Remote toehold: a mechanism for flexible control of DNA hybridization kinetics. J. Am. Chem. Soc. 133(7), 2177–2182 (2011)
Gerling, T., Wagenbauer, K.F., Neuner, A.M., Dietz, H.: Dynamic DNA devices and assemblies formed by shape-complementary, non-base pairing 3D components. Science 347(6229), 1446–1452 (2015)
Papadimitriou, C.H.: On the complexity of integer programming. J. ACM (JACM) 28(4), 765–768 (1981)
Phillips, A., Cardelli, L.: A programming language for composable DNA circuits. J. R. Soc. Interface 6(Suppl 4), S419–S436 (2009)
SantaLucia Jr., J., Hicks, D.: The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 33, 415–440 (2004)
Schulman, R., Winfree, E.: Programmable control of nucleation for algorithmic self-assembly. SIAM J. Comput. 39(4), 1581–1616 (2009)
Thachuk, C., Winfree, E., Soloveichik, D.: Leakless DNA strand displacement systems. In: Phillips, A., Yin, P. (eds.) DNA 2015. LNCS, vol. 9211, pp. 133–153. Springer, Cham (2015). doi:10.1007/978-3-319-21999-8_9
Winfree, E.: Algorithmic self-assembly of DNA. Ph.D. thesis, California Institute of Technology (1998)
Woo, S., Rothemund, P.W.K.: Programmable molecular recognition based on the geometry of DNA nanostructures. Nat. Chem. 3, 620–627 (2011)
Zhang, D.Y., Turberfield, A.J., Yurke, B., Winfree, E.: Engineering entropy-driven reactions and networks catalyzed by DNA. Science 318(5853), 1121–1125 (2007)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Doty, D., Rogers, T.A., Soloveichik, D., Thachuk, C., Woods, D. (2017). Thermodynamic Binding Networks. In: Brijder, R., Qian, L. (eds) DNA Computing and Molecular Programming. DNA 2017. Lecture Notes in Computer Science(), vol 10467. Springer, Cham. https://doi.org/10.1007/978-3-319-66799-7_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-66799-7_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66798-0
Online ISBN: 978-3-319-66799-7
eBook Packages: Computer ScienceComputer Science (R0)