
Abstract
De novo genome assembly is one of the most important and challenging computational problems in modern genomics; further, it shares algorithms and communication patterns important to other graph analytic and irregular applications. Unlike simulations, it has no floating point arithmetic and is dominated by small memory transactions within and between computing nodes. In this work, we focus on the highly scalable HipMer assembler and identify the dominant algorithms and communication patterns, also using microbenchmarks to capture the workload. We evaluate HipMer on a variety of platforms from the latest HPC systems to ethernet clusters. HipMer performs well on all single node systems, including the Xeon Phi manycore architecture. Given large enough problems, it also demonstrates excellent scaling across nodes in an HPC system, but requires a high speed network with low overhead and high injection rates. Our results shed light on the architectural features that are most important for achieving good parallel efficiency on this and related problems.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

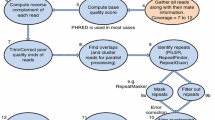

1 Introduction
De novo genome assembly is essential to understanding the genomic structure of plants, animals and microbial communities and has applications in health, the environment, and energy. It involves constructing long genomic sequences from short, overlapping and possibly erroneous DNA fragments produced by modern sequencers. Due to the continued exponential increase in the size (multi-terabyte) and complexity of the sequence datasets, massive parallelism is required to overcome the huge memory and computational requirements, but efficient parallelization is challenging. The genome assembly computation, not unlike other graph analytic and irregular applications, involves graphs and hash tables and is dominated by irregular memory access patterns and fine-grained synchronization. Many assemblers therefore target shared memory hardware, where assembly problems are limited in size and may run for days or even weeks.
In this study we present the first cross-architectural analysis of HipMer [8], an extreme scale distributed memory genome assembler. HipMer produces biologically equivalent results to a serial assembler called Meraculous [3], which has been exhaustively studied for quality and found to excel relative to other assemblers in most metrics [6]. Our HipMer performance evaluation includes a broad range of platforms, ranging from a supercomputer with Intel Xeon Phi processors and a custom HPC network to off-the-shelf Ethernet clusters. HipMer stresses the communication fabric of these systems using communication patterns that are increasingly important for irregular applications. These include all-to-all exchanges, fine-grained lookups, and global atomic operations. Our work presents a detailed analysis of these communication patterns and points to requirements for future architectural designs for scalability on this important class of codes.
2 The Parallel HipMer Assembly Pipeline
In this section we describe the basic algorithms used in the pipeline, our parallelization strategy, and the consequent communication patterns. Although we focus on HipMer, the algorithms are relevant to all de novo assembly pipelines that are based on de Bruijn graphs [14]. We describe four major stages of Hipmer (see Fig. 1-Left), k -mer analysis, contig generation, read-to-contig alignment and scaffolding, as well as gap closing, which is part of the scaffolding stage. Other stages implemented in HipMer assist these main computations and are included in the experimental results. The input to the pipeline is a set of reads, which are short, erroneous sequence fragments of 100–250 letters sampled at random from a genome. The sampling is redundant at a depth of coverage d, so on average each position (base) in the genome is covered by d reads. This redundancy is used to filter out errors in the first stage (k-mer analysis). The k-mer analysis can work with relatively high error rates in the data (2.5%, k = 40+); the user may also choose to decrease k when given data with higher error rates. Sequencers produce reads in pairs with a known distance between them, a fact which is exploited later in the pipeline (scaffolding) to improve the assembly.

Left: the HipMer de novo assembly pipeline. Right: a de Bruijn graph of k-mers with \(k=3\).
2.1 k-mer Analysis
In this step, the input reads are processed to exclude errors. Each processor reads a portion of the reads and chops them into k-mers, which are formed by a sliding window of length k. A deterministic function is used to map each k-mer to a target processor, assigning all the occurrences of a particular k-mer to the same processor, thus eliminating the need for a global hash table. The k-mers are communicated among the processors using irregular all-to-all communication, which is performed when each processor fills up out of its outgoing buffers and is repeated until all k-mers have been redistributed. A total of \(\varTheta (\frac{Gd}{L}(L-k+1))\) k-mers need to be communicated, where G is the genome size (number of characters in the output) and L is the read length (number of characters in the input). Next, all the k-mers are counted, and those that appear fewer times than a threshold are discarded as erroneous. This filtering is enabled by the redundancy d in the read data set: k-mers that appear close to d times are likely error-free, whereas k-mers that appear infrequently are likely erroneous. k-mer counting is challenging for large datasets because an error in just a single base creates k erroneous k-mers, and it is not uncommon to have over 80% of all distinct k-mers erroneous; as a result the memory footprint increases substantially. We address this problem [10] through the use of Bloom filters, which results in irregular all-to-all communication. Also, highly complex plant genomes, such as wheat, are extremely repetitive and it is not uncommon to see some k-mers occurring millions of times. Such high-frequency k-mers create a significant load imbalance problem, since the processors assigned to high-frequency k-mers require significantly more memory and processing times. We deal with these “heavy hitters” using a streaming algorithm, described further in [8] that does not require any additional communication since it is merged into the initialization of the Bloom filters. Additionally, for each k-mer, the extensions are recorded: these are the two left and right neighboring bases in the original reads. If multiple extensions occur, the most likely one is used; if there is no obvious agreement then none is recorded. The result of k-mer analysis is a set of k-mers and their extensions that with high probability include no errors. This set contains \(\varTheta (G)\) k-mers, and is a compressed representation of the original read dataset because multiple occurrences of a k-mer have been collapsed to a single instance.
2.2 Contig Generation
The k-mers are assembled into longer sequences called contigs, which are error-free (with high probability) sequences that are typically longer than the original reads. In HipMer, Contig generation utilizes a de Bruijn graph, which is a special graph that represents overlaps in sequences. The k-mers are the vertices in the graph and two k-mers are connected by an edge if they overlap by \(k-1\) consecutive bases and have corresponding extensions that are compatible (see Fig. 1-Right for a de Bruijn graph example with \(k=3\)).
A hash table is used to store a compact representation of the graph: A vertex (k-mer) is a key in the hash table and the incident vertices are stored implicitly as a two-letter code [ACGT][ACGT] that indicates the unique bases that immediately precede and follow the k-mer in the read dataset. By combining the key and the two-letter code, the neighboring vertices in the graph can be identified. These graphs can require terabytes of memory for storing large genomes (e.g. pine or wheat [4]), and traditionally have required specialized, very large shared-memory machines. We overcome this limitation by employing the global address space of Unified Parallel C [11] (UPC) in order to transparently store the hash table in distributed memory, thereby utilizing the memory of many individual machines in a unified address space.
During the parallel hash table construction, the input k-mers are hashed and sent to the proper (potentially remote) bucket of the hash table by leveraging the one-sided communication capabilities of UPC. We avoid fine-grained communication and excessive locking on the hash table buckets with a dynamic aggregation algorithm [10]. This algorithm dynamically aggregates the k-mers in batches before they are sent to the appropriate processors. The pattern here is similar to k-mer analysis but is done asynchronously, where a single processor will send an aggregation of remote hash table inserts without waiting for other processors. Unlike k-mer analysis, the total number of k-mers that have to be communicated is \(\varTheta (G)\), since multiple occurrences of k-mers have been collapsed during the k-mer analysis stage and this condensed k-mer set has size proportional to the genome size.
The resulting de Bruijn subgraph is traversed in parallel to identify the connected components, which are linear chains of k-mers, since we have excluded all the vertices that do not have unique neighbors in both directions. Traditional parallelization strategies of the traversal would not scale to large concurrencies due to the size and shape of this high diameter graph (extremely long chains). First, the de Bruijn subgraph is sparse (e.g. for human the de Bruijn graph would be a \(3\cdot 10^9 \times 3\cdot 10^9 \) adjacency matrix with 2–8 eight non-zeros per row). Second, the de Bruijn graph has also extremely high diameter (the connected components in theory can have size up to the length of chromosomes, which is order tens of millions of bases). In our specialized parallel traversal algorithm [10], a processor \(P_i\) chooses a random k-mer as seed and initializes with it a new subcontig. Then \(P_i\) attempts to extend the subcontig towards both of its endpoints by performing lookups for the neighboring vertices in the distributed hash table. The extending process continues until no more new edges can be found, or there are forks in the graph (e.g. the vertex GAA in Fig. 1-Right represents a fork). The access pattern in the distributed hash table consists of irregular, fine-grained lookup operations. If two processors work on the same connected component (i.e. both selected seed k-mers from the same contig), race conditions are avoided via a lightweight synchronization scheme [10] based on remote atomics and we have proved that our synchronization scheme scales to massive concurrencies (thousands of compute nodes). The parallel traversal is terminated when all the connected components in the de Bruijn graph are explored. Since the size of the de Bruijn graph is proportional to the genome size, the traversal involves accessing \(\varTheta (G)\) vertices via atomics and irregular lookup operations.
2.3 Read-to-Contig Sequence Alignment
For the alignment phase, we do not use alternative aligners, because, unlike other aligners, HipMer’s parallel alignment scales to extreme concurrencies. It also outputs all possible alignments, rather than a pruned subset, as input to the scaffolding phase. HipMer’s alignment phase [9] maps the original reads onto the contigs to provide information about the relative ordering and orientation of the contigs, which is used in the final step of the assembly pipeline. First, each processor stores a distinct subset of the contigs in the global address space so that any other processor can access them. Then, substrings of length k, called seeds, are extracted in parallel from the contigs and stored in the seed index, which is a distributed hash table. Although seeds are conceptually the same as k-mers, the value of k may be different than in earlier phases, and have a somewhat different purpose. Each hash table entry has a seed as the key and a pointer to the corresponding source contig as the value. There are \(\varTheta (G)\) seeds in total, because the contigs constitute a fragmented version of the genome. The seed index is constructed via an irregular all-to-all communication step similar to the hash table construction in the contig generation phase. The seed index is then used to align reads onto contigs. Each read of length L contains \(L-k+1\) seeds of length k. For each seed s in a read, a fine-grained lookup in the global seed index produces a set of candidate contigs that contain s. Although an exhaustive lookup of all possible seeds would require a total of \(\varTheta (\frac{Gd}{L}(L-k+1))\) lookups, in practice we perform \(\varTheta (\frac{Gd}{L}\cdot a)\) lookups where \(a < L-k+1\), through the use of optimizations that identify properties in the contigs during the seed index construction [9]. Finally, after locating a candidate contig that has a matching seed with the read under consideration, the Smith-Waterman algorithm [17] is executed in order to perform local sequence alignment between the contig and the read. The output of this stage is a set of reads-to-contig alignments.
2.4 Scaffolding and Gap Closing
The scaffolding step aims to “stitch” together contigs into sequences called scaffolds by assessing the paired-end information from the reads and the reads-to-contigs alignments. Figure 2(a) shows three pairs of reads that map onto the same pair of contigs i and j, creating a link that connects contigs i and j. A graph of contigs can be created by generating links for all the contigs that are supported by pairs of reads (see Fig. 2(b)). The contig graph is stored in a distributed hash table, which requires irregular all-to-all communication for construction. The graph of contigs (and consequently the number of links among them) is orders of magnitude smaller that the k-mer de Bruijn graph because the connected components in the k-mer graph are contracted to single vertices in the contig-graph. According to the Lander-Waterman statistics [5], the expected number of contigs is \(\varTheta (dG/L\cdot e^{-d})\). A parallel traversal of the contig graph is then performed to identify and remove “bubbles”, which are localized structures involving divergent paths. This requires irregular lookups and global atomics. A final traversal is done by selecting start vertices in order of decreasing contig length (this heuristic tries to first stitch together “long” contigs) and therefore it is inherently serial. At smaller scales, this will not have much of an impact since the contig graph is relatively small compared to the k-mer graph. At larger scales, the serial component will become the bottleneck. It is likely that there will be gaps between the contigs within a scaffold (see Fig. 2(b)). An attempt is made to close these gaps using the read-to-contig alignments, which are processed in parallel and projected into the gaps. A distributed hash table is used to localize the unassembled reads onto the appropriate gaps. Construction of the table uses an irregular all-to-all communication pattern, but accessing the information in the table requires irregular lookups. Assuming that a fraction \(\gamma \) of the genome is not assembled into contigs, this communication step involves \(\varTheta (\gamma Gd/L)\) reads. Finally, the gaps are divided into subsets and each set is processed by a separate thread, in a parallel phase. The localized reads are used to attempt to close the gaps via a mini-assembly algorithm (an algorithm that performs only k-mer analysis and contig generation on a strict subset of the reads). The outcome of this step is a set of scaffolds (possibly with some remaining gaps), constituting the result of the HipMer assembly pipeline. For simplicity, we do not go into further detail on HipMer’s algorithms for diploid assembly.

(a) A link between contigs i and j that is supported by three read pairs. (b) Two scaffolds formed by traversing a graph of contigs.
2.5 Summary of Communication Patterns
Table 1 summarizes the main communication patterns along with the corresponding volume of communication for each stage. These communications patterns govern the efficiency of the parallel pipeline at large scale, where most of the stages are communication bound. The different communication patterns have, however, vastly different overheads. For example, the all-to-all communication exchange is typically bounded by the bisection bandwidth of the system, assuming that the partial messages are large enough and there is enough concurrency to saturate the available bandwidth. Conversely, fine-grained, irregular lookups and global atomics are typically latency-bound. Although conventional wisdom would suggest that these sorts of communication patterns are prohibitive for distributed memory systems, we have shown that HipMer can strong scale effectively [7], because there are fewer communication operations on the critical path as concurrency increases.
3 Experimental Results and Analysis
Our experiments are conducted on 5 computing platforms, including the Cori II Cray XC40 and Edison Cray XC30 supercomputers at NERSC, the Cray XK7 MPP at the Oak Ridge National Lab (CPU only), the Genepool heterogenous Mellanox InfiniBand NERSC cluster, and an Ethernet Cluster consisting of 3 SunFire x4600 servers networked via 1 Gb shared switch as well as 10 Gb fiber optic patch. Architectural details are presented in Table 2.
For the experimental evaluation, we used 2 datasets. The first dataset, referred to as chr14, consists of 36.5 million paired-end reads from the fragment library of human chromosome 14, also used in the GAGE [15] evaluation. The reads are 101 bp (base pair) in length and the fragment library has mean insert size 155 bp. This relatively small dataset will be used to investigate the single node performance and scalability at small scales. The second dataset, referred as human, is a member of the CEU HapMap population (identifier NA12878) sequenced by the Broad Institute. The genome contains 3.2 Gbp assembled from 2.9 billion reads, which are 101 bp in length, from a paired-end insert library with mean insert size 395 bp. This dataset which is two orders of magnitude bigger than chr14 will be used for the evaluation of the pipeline at larger scales, although it is still relatively small compared to the genome size of some plants and microbial communities.
3.1 Single-Node Performance Analysis
First, we examine the on-node scalability of HipMer on Cori II (our largest multicore node with 68 cores). HipMer attains perfect single node scaling (see Fig. 3) between 1 and 68 threads (1 thread per core) on the chr14 dataset (37,609.7 s on a single thread and 556.5 s on 68 threads, yielding a 67.6\(\times \) speedup). If we enable hyper-threading and use 2 threads per core on 64 cores, we observe a reduction in the execution time by 19%. If we further use 4 threads per core we observe an additional 3% reduction in the execution time. These results suggest that hyper-threading can help on a single node. However, our benchmarking revealed that the increased concurrency due to hyper-threading on a single node affects severely the efficiency of the off-node communication operations. Therefore we configure all the experiments in this paper with 1 thread per core (no hyper-threading).

Cori II single KNL node speedup up to 68 cores for the small chr14 dataset.

Cross-architecture single-node and two-node performance by stages.
Figure 4 displays the total runtime per stage on the chr14 dataset for one and two nodes of each machine utilizing all cores. For now, we consider only the performance bars that correspond to the single node experiments. Examining single node total runtimes, shows that the ratio between the slowest (Titan, AMD Opteron) and fastest (Cori II, Intel KNL) systems is a factor of 2.4\(\times \). Across architectures, each stage of the pipeline takes similar portion of the respective total execution time. The most time consuming part is k-mer analysis, followed by the sequence alignment stage, confirming our analysis in Sect. 2.
These results also highlight the idiosyncrasies of the genome assembly workload; it does not include any arithmetic computations, instead it heavily relies on irregular memory accesses and string and integer operations. As such, the modern trends in multicore processor design with wider vectors accommodating higher arithmetic throughput do not result in substantial performance improvements (e.g. the single node Cori II execution is only slightly faster than the single node Edison experiment). Efficient vectorization of the key string computations can increase performance, but the major improvements on a single node come from the increased concurrency/parallelism and the ability of the memory subsystem to facilitate concurrent irregular memory accesses. At the same time, the simpler core design in conjunction with the decreased clock frequency results in worse single core performance for Knights Landing compared to the other processors.
3.2 Scalability from Single Node to Multiple Nodes
Having examined HipMer’s single node performance, we now examine how it scales to multiple nodes, again using the chr14 dataset - one small enough for single nodes. Figure 4 shows the performance difference by stage as we scale from 1 to 2 nodes. For all machines, we observe speedups well under 2\(\times \). The speedup is between 1.12\(\times \) and 1.18\(\times \) for Cori II, Edison, and Genepool. Titan has the highest speedup at 1.6\(\times \); however note, in absolute runtime, its single node performance is 2.1\(\times \) slower than Edison’s (for example), and due to its relatively limited on-node memory and parallelism (see Table 2), it has the most to benefit from additional node resources. Its relative internode latency is also a significant factor, as we will discuss momentarily. The Ethernet Cluster, ran for 810 s on a single node and with either a 1 Gb or a 10 Gb interconnect, actually has a 18.2\(\times \) and 10.6\(\times \) slowdown respectively (not shown due to scale).
This behavior is justified via a detailed analysis of each stage. The k-mer analysis step typically is computation bound because its communication involves efficient collective all-to-all exchanges with large messages (see Table 1) which effectively utilizes the available bandwidth. For example, on 2 Cori II nodes, 6% of the k-mer analysis time is spent in communication, and we observe almost linear scaling of the k-mer analysis step. On the other hand, the sequence alignment step does not speedup and in some cases actually slows down. The communication pattern necessitated in the alignment stage consists of irregular, fine-grained lookups implemented with get operations. Such operations are latency bound and their efficiency depends on the underlying machine/network. Consequently, we expect the alignment phase to be communication bound. For example, the get latency for small messages on a fully occupied Edison node is 0.75 \(\upmu \)s, while the average latency for two nodes is 2.39 \(\upmu \)s (measured via microbenchmarks). We refer to “average” latency in the latter case because, under such a setting, half the get operations are expected to result in on-node communication and the remaining in off-node communication. Note, the number of lookups on the critical path can be calculated from the number of reads assigned to each processor. Even though the number of threads is increased by a factor of two and the number of irregular lookups on the critical path is decreased by a factor of two, each of those operations is 3.2\(\times \) more expensive, eventually yielding larger overall communication time in the alignment step. However, on Titan the respective get latencies for small messages are 1.10 \(\upmu \)s for a single node and 1.79 \(\upmu \)s for 2 nodes. As a result we expect a speedup in the alignment phase, which is confirmed in Fig. 4. The same scaling argument holds for the remaining parallel algorithms that rely on fine-grained irregular lookups and atomics (see Table 1). For a description of the microbenchmarks used, see [7]; we were not able to include our microbenchmarking data for all machines due to space limitations.
Figure 5 shows the strong scaling results for all machines on the chr14 dataset. Efficiency (the y axis) is calculated as \(T_1 / (T_p \cdot p)\) where \(T_1\) is the total runtime on a single node, p is the number of nodes (x axis), and \(T_p\) is the total runtime on p nodes. From 1 to 2 nodes, Cori II, Edison, and Genepool’s efficiencies decrease down to 55–60%. This behavior is explained in the previous paragraph. As we scale from 2 to 8 nodes, the respective parallel efficiencies drop at most by 26%. At this range of node counts most of the irregular accesses in the parallel algorithms are off-node and as such the efficiency levels should remain the same as we strong scale. This is the regime where we can observe good strong scaling. Titan has the smallest parallel efficiency decrease between 1 and 2 nodes (20%), but it is still the most significant decrease in its series (which continues to decrease roughly by 10% as the number of nodes doubles). While its relative efficiency is higher than other machines, its absolute runtime is much worse, and improves significantly with more memory and compute cores (hence, higher speedups, as discussed in the previous section). The Ethernet Cluster drops in efficiency by 95% or more from 1 to 2 nodes; because the Ethernet cluster has only 3 nodes, we do not present further data. These trends show that parallelizing the computation across some minimum number of nodes is necessary to overcome the overhead incurred by internode communication. This minimum number is dependent on the network and node characteristics. Beyond this minimum number, the application can scale efficiently to large number of nodes. We emphasize here that for realistically large datasets, one might have to use multiple nodes to acquire the necessary aggregate memory. In such scenarios the baseline performance is of that of multiple nodes and as such the strong scaling efficiency is even better as we will see in the next subsection.
Another interesting feature in the data presented in Fig. 5, is the cross-over in efficiency between Edison and Cori II at 64 nodes. Between 1 and 32 nodes, the two machines maintain relatively close levels of efficiency (\(\le \)4% difference). At 64 nodes onwards, Edison maintains a higher level of efficiency by roughly 10%. The key factors here are the higher core count of the Cori II nodes (64 versus 24 on Edison nodes) and the relatively small size of the dataset. At 64 nodes, the workload is parallelized over 4 K cores on Cori II, while Edison has 1.5 K cores at that same node count. Because the data set is relatively small, at the concurrency of 4 K cores, Cori II lacks sufficient work per thread that can be efficiently parallelized, especially during the scaffolding and gapclosing phases.

Strong scaling efficiency for the small chr14 dataset

Execution time for the human dataset
3.3 Large Scale Experimental Results
Finally, we present results from running HipMer at scale on the human dataset. In Fig. 6, we show the total runtime of the pipeline (y axis) over the number of nodes (x axis) for Cori II, Edison, and Titan. Not shown are the Ethernet Cluster results, which ran for 22.56 h on a single node and on two nodes took approximately 280 h and 161 h on the 1 Gb and 10 Gb interconnects respectively a 12.4\(\times \) and 7.1\(\times \) slowdown. Genepool results are also not shown since sufficiently many nodes for this data set were not reservable.
The first thing to observe is the different node count that constitutes the baseline for each machine. Since the memory requirement of the human dataset, and the communication data structures for its effective distribution are quite large, we need at least 32, 64, and 128 nodes on Cori II, Edison, and Titan, respectively, to obtain the minimum required aggregate memory (approximately 4TB, see Table 2). On Cori II we scale up to 512 nodes (32,768 cores) with 47% strong scaling efficiency, on Edison up to 1,024 nodes (24,576 cores) with 49% efficiency and on Titan up to 1024 nodes (16,384 cores) with 37% efficiency. After these levels of parallelism, the parallel efficiency drops substantially because the work per thread is not sufficient. Other factors influencing the pipeline’s scalability is the serial traversal in the scaffolding step and the initial I/O overhead to load the input data. As the scale increases, the percentage of the total runtime spent in the serial scaffolding traversal also increases. For example, on Cori II at 512 nodes 29% of the total execution time is spent in the serial part of the scaffolding while the corresponding serial component takes only 4% of the overall execution time at 32 nodes.
4 Related Work
Our performance study in this paper captures the workload of other assemblers, and here we described the most closely related ones that also use distributed memory parallelism. Ray [2] is an end-to-end parallel de novo genome assembler that utilizes MPI and exhibits strong scaling up to a modest number of nodes. It produces both contigs and scaffolds directly from raw sequencing reads. One drawback of Ray is the lack of parallel I/O support for reading and writing files. ABySS [16] was the first de novo assembler written in MPI that also exhibits strong scaling. Unfortunately, only the first assembly step of contig generation is fully parallelized with MPI, and the subsequent scaffolding steps must be performed on a single shared memory node. Spaler [1] is a contig generating assembler based on Spark and GraphX. Results from Spaler have been given for our smaller data set, chr14, and it shows good scaling. PASHA [12] is another partly MPI based de Bruijn graph assembler, though not all steps are fully parallelized as its algorithm, like ABySS, requires a large-memory single node for the last scaffolding stages. SWAP 2 [13] is a parallelized MPI based de Bruijn assembler that has been shown to assemble contigs efficiently for the human genome, however it does not provide parallel scaffolding modules.
5 Conclusion
This work presents a cross-architectural evaluation of large-scale genome assembly, a first study of its kind. The algorithms described in Sect. 2, are relevant for all de novo assembly pipelines based on de Bruijn graphs [14], and is characterized by a workload dominated by fine-grained irregular memory accesses, with no floating point arithmetic. Nonetheless, as shown in Sect. 3, HipMer attains both excellent single node and distributed multinode scalability. We identified the key computation and communication patterns, and associated architecture and network characteristics, for achieving such effective scalability; namely all-to-all exchanges (bisection bandwidth bounded), fine-grained irregular lookups and global atomics (latency bounded). Further, we find the key to on-node scalability for this type of workload is the available concurrency coupled with the memory subsystems’ performance. We expect that these insights will help impact future implementations of irregularly structured parallel methods and the underlying architectural designs targeting these classes of computations.
References
Abu-Doleh, A., Catalyurek, U.V.: Spaler: Spark and GraphX based de novo genome assembler. In: 2015 IEEE International Conference on Big Data (Big Data), October 2015
Boisvert, S., Laviolette, F., Corbeil, J.: Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comput. Biol. 17(11), 1519–1533 (2010)
Chapman, J.A., Ho, I., Sunkara, S., Luo, S., Schroth, G.P., Rokhsar, D.S.: Meraculous: de novo genome assembly with short paired-end reads. PLoS ONE 6(8), e23501 (2011)
Chapman, J.A., Mascher, M., Buluç, A., Barry, K., Georganas, E., Session, A., Strnadova, V., Jenkins, J., Sehgal, S., Oliker, L., Schmutz, J., Yelick, K.A., Scholz, U., Waugh, R., Poland, J.A., Muehlbauer, G.J., Stein, N., Rokhsar, D.S.: A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome. Genome Biol. 16, 26 (2015)
Deonier, R.C., Tavaré, S., Waterman, M.: Computational Genome Analysis: An Introduction. Springer Science & Business Media, New York (2005). doi:10.1007/0-387-28807-4
Earl, D., Bradnam, K., St John, J., Darling, A., et al.: Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 21(12), 2224–2241 (2011)
Georganas, E.: Scalable parallel algorithms for genome analysis. Ph.D. thesis, EECS Department, University of California, Berkeley (2016)
Georganas, E., Buluç, A., Chapman, J., Hofmeyr, S., Aluru, C., Egan, R., Oliker, L., Rokhsar, D., Yelick, K.: HipMer: an extreme-scale de novo genome assembler. In: International Conference for High Performance Computing, Networking, Storage and Analysis (SC 2015) (2015)
Georganas, E., Buluç, A., Chapman, J., Oliker, L., Rokhsar, D., Yelick, K.: merAligner: a fully parallel sequence aligner. In: Proceedings of the IPDPS (2015)
Georganas, E., Buluç, A., Chapman, J., Oliker, L., Rokhsar, D., Yelick, K.: Parallel de Bruijn graph construction and traversal for de novo genome assembly. In: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis (SC 2014) (2014)
Husbands, P., Iancu, C., Yelick, K.: A performance analysis of the Berkeley UPC compiler. In: Proceedings of International Conference on Supercomputing, ICS 2003, pp. 63–73. ACM, New York (2003)
Liu, Y., Schmidt, B., Maskell, D.L.: Parallelized short read assembly of large genomes using de Bruijn graphs. BMC Bioinform. 12(1), 354 (2011)
Meng, J., Seo, S., Balaji, P., Wei, Y., Wang, B., Feng, S.: Swap-assembler 2: optimization of de novo genome assembler at extreme scale. In: 45th International Conference on Parallel Processing (ICPP), pp. 195–204. IEEE (2016)
Miller, J.R., Koren, S., Sutton, G.: Assembly algorithms for next-generation sequencing data. Genomics 95(6), 315–327 (2010)
Salzberg, S.L., Phillippy, A.M., Zimin, A., Puiu, D., et al.: GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 22(3), 557–567 (2012)
Simpson, J.T., Wong, K., et al.: ABySS: a parallel assembler for short read sequence data. Genome Res. 19(6), 1117–1123 (2009)
Smith, T.F., Waterman, M.S.: Identification of common molecular subsequences. J. Mol. Biol. 147(1), 195–197 (1981)
Acknowledgments
All authors at Lawrence Berkeley National Laboratory (LBNL) were supported by Department of Energy (DOE) Offices of Advanced Scientific Computing Research (ASCR) and Biological and Environmental Research (BER), both under contract number DE-AC02-05CH11231. This includes funding to BER’s Joint Genome Institute, the ASCR-funded Exascale Computing Project, and the ASCR Mathematics and Computer Science Research Programs. This word used resources of ASCR’s National Energy Research Scientific Computing Center (NERSC) under the same LBNL contract and ASCR’s Oak Ridge Leadership Facility (OLCF) under Contract No. DE-AC05-00OR22725.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Ellis, M. et al. (2017). Performance Characterization of De Novo Genome Assembly on Leading Parallel Systems. In: Rivera, F., Pena, T., Cabaleiro, J. (eds) Euro-Par 2017: Parallel Processing. Euro-Par 2017. Lecture Notes in Computer Science(), vol 10417. Springer, Cham. https://doi.org/10.1007/978-3-319-64203-1_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-64203-1_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64202-4
Online ISBN: 978-3-319-64203-1
eBook Packages: Computer ScienceComputer Science (R0)