
Abstract
Clustering functionally similar genes helps in understanding the mechanism of a biological pathway. It also provides information of those genes whose biological importance is previously not known. Clustering of genes is highly dependent on the similarity or dissimilarity criterion. Usually, microarray gene expression data is used to cluster genes. However, a microarray data may contain noise that may lead to undesired results. Therefore, incorporating gene ontology information may improve the clustering solutions. In this regard, an integrated dissimilarity measure is introduced for grouping functionally similar genes. It is comprised of city block distance and gene ontology based semantic dissimilarity. While, the city block distance is used to compute distance between two gene expression vectors, gene ontology based semantic dissimilarity measure is used for incorporating biological knowledge. The importance of the integrated dissimilarity measure is shown by incorporating it in different c-means clustering algorithms including rough-fuzzy clustering algorithms. In this work it has been shown that incorporation of integrated dissimilarity measure increases the functional similarity of cluster of genes as compared to the methods that are based on either type of dissimilarity measure. It is also observed that the rough-fuzzy clustering algorithm performs better with the new dissimilarity measure compared to different c-means clustering algorithms.
Similar content being viewed by others


Keywords
1 Introduction
Clustering functionally similar genes is important to understand the role of individual gene in a pathway. It also reveals about the co-expression and co-regulation patterns of genes. A lot of insight can be gained for the genes whose information is previously unknown. Functionally similar genes often are organized in clusters in the genome. Expression analyses showed strong positive correlations among the closely located genes, indicating that they may be controlled by common regulatory element(s). In fact, experimental evidence demonstrated that clustered gene loci form an operon-like gene structure and that they are transcribed from common promoter. Existence of co-expressed genes is also demonstrated using expression profiling analysis in [10]. Several gene clusters have been experimentally shown by RT-PCR [4]. These findings suggest that members of a gene cluster, which are at a close proximity on a chromosome, are highly likely to be processed as co-transcribed units. Expression data of genes can be used to detect clusters of genes as it is suggested that co-expressed genes are co-transcribed, so they should have similar expression pattern. However, a microarray gene expression data may contain noise that may lead to misleading results.
It has been demonstrated in [15] that the quality of generated clusters is always relative to a certain distance measure. Different distance measures may lead to different clustering results. Several similarity or dissimilarity measures such as Euclidean distance, Jaccard index, Pearson correlation coefficient, and city block distance (CBD) are used in various clustering algorithms based on expression vector of genes. To avoid the problem of noise in microarray gene expression data one may integrate prior biological knowledge information so that more functionally relevant genes get grouped. In this regard, several studies have developed or used microarray gene expression data and gene ontology (GO) based semantic dissimilarity. In those works Pearson correlation distance [9] is computed using microarray expression data and it is integrated with GO based semantic dissimilarity. However, the Pearson correlation only measures linear relationship. This integrated dissimilarity measure is used with different c-means clustering algorithms but not with rough-fuzzy clustering algorithms. It has been shown that rough-fuzzy clustering algorithm can perform better than K-means [8], fuzzy c-means algorithms [5] as they can handle the issue of noise as well as overlapping boundary [13]. However, no work has been conducted using integrated dissimilarity measure in rough-fuzzy clustering algorithm.
In this regard, this paper presents a new integrated dissimilarity measure for grouping functionally similar genes. It is developed by integrating city block distance (CBD) and gene ontology (GO) semantic dissimilarity. While, the CBD measure is used in this paper for computing gene expression vector based dissimilarity, GO based dissimilarity measure is used to incorporate biological knowledge in the clustering algorithm. It has been shown that rough-fuzzy clustering algorithm can overcome the issues of noise and overlapping boundaries but no work has demonstrated the impact of integrated dissimilarity measure with rough-fuzzy c-means algorithms. The effectiveness of integrated approach along with different types of c-means clustering algorithms is shown on several gene expression microarray data. It has been observed that the rough-fuzzy c-means algorithm along with proposed integrated dissimilarity measure generates more clusters of functionally related genes.
2 Proposed Dissimilarity Measure
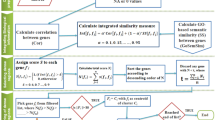
This section describes the proposed integrated dissimilarity measure. It is developed by integrating microarray gene expression data and gene ontology (GO) based semantic dissimilarity. The distance between two gene expression vectors is calculated using city block distance (CBD). While, Du et al. [6] GO based semantic dissimilarity is used for incorporating biological knowledge.
City Block Distance. The CBD, also known as the Manhattan distance or taxi distance, is closely related to the Euclidean distance. Whereas the Euclidean distance corresponds to the length of the shortest path between two points, the CBD is the sum of distances along each dimension. The distance between two objects \(x_{i}\) and \(x_{j}\) is defined as follows:
where m is the number of features of the objects \(x_i\) and \(x_{j}\). As for the Euclidean distance, the expression data are subtracted directly from each other, and therefore should be made sure that they are properly normalized. There are many variants of the CBD. The normalized range-normalized CBD (NRNCBD) is defined as follows:
where \(k_{max}\) and \(k_{min}\) denote the maximum and minimum values along the kth feature, respectively.
Gene Ontology Based Semantic Dissimilarity. The gene ontology (GO) project aims to build tree structures and controlled vocabularies, also called ontologies, which describe gene products in terms of their associated biological processes (BPs), molecular functions (MFs), or cellular components (CCs).
When biological entities are described using a common schema such as an ontology, they can be compared by means of their annotations. This type of comparison is called semantic similarity since it assesses the degree of relatedness between two entities by the similarity in meaning of their annotations. To quantify similarity between two genes, many information content-based measures have been developed [11, 17]. In the present work, the Du et al. semantic similarity measure [6] is used to measure the functional dissimilarity between a pair of genes. The functional similarity between a pair of genes based on gene annotation information from heterogeneous data sources is computed as follows.
Definition 1
Given two genes \(x_{1}\) and \(x_{2}\), and their annotated GO terms \(GO_{1}=\{ go_{11},go_{12},\cdots ,go_{1m}\}\) and \(GO_{2}= \{ go_{21},go_{22},\cdots ,go_{2\acute{m}}\}\), respectively, for the ontology O, the functional similarity between \(x_{1}\) and \(x_{2}\) is defined as follows:
The value of similarity \(Sim(x_{1},x_{2})\) ranges from zero to one, nearer the value to one higher the functional similarity between the pair of genes.
In this work, functional dissimilarity is computed as follows: \(DISSim(x_{1},x_{2})=1-Sim(x_{1},x_{2})\). Nearer the value to zero higher the functional similarity between the pair of genes. Whereas, if its one that means no similarity between the genes.
Integrated Dissimilarity Measure. To generate group of functionally similar genes average of both distance measures is computed. The integrated distance between two genes \(x_{i}\) and \(x_{j}\) can be calculated as follows:
The following properties can be derived for the proposed dissimilarity measure:
-
1.
\(0 \le d(x_i,x_j) \le 1\).
-
2.
\(d(x_i,x_j)=d(x_j,x_i)\).
-
3.
\(d(x_i,x_i)=0\).
-
4.
\(d(x_i,x_j)\le d(x_i,x_k)+ d(x_k,x_j)\).
The proposed integrated dissimilarity measure is incorporated into different c-means clustering algorithm including rough-fuzzy clustering algorithm.
2.1 Selection of Initial Cluster Prototypes
A limitation of any c-means algorithm is that it can only achieve a local optimum solution that depends on the initial choice of the cluster prototypes. Consequently, computing resources may be wasted in that some initial centers get stuck in regions of the input space with a scarcity of data points and may therefore never have the chance to move to new locations where they are needed. To overcome this limitation, the proposed study begins with the selection of c distinct genes from the given gene expression data set using the NRNCBD, which enables the algorithm to converge to an optimum or near optimum solutions [15].
The main steps for selection of initial genes as mentioned in [15] are as follows:
-
1.
For each gene \(x_i\), calculate \({\mathcal N}(x_i,x_j)\) between itself and the gene \(x_j\), \(\forall _{j=1}^{n}\).
-
2.
Calculate similarity score between two genes \(x_i\) and \(x_j\) as follows:
$$\begin{aligned} {\mathrm {S}}(x_i,x_j)= \left\{ \begin{array}{ll} 1 &{} \text {if}\ {\mathcal N}(x_i,x_j)\le \lambda \\ 0 &{} \text {otherwise}.\\ \end{array} \right. \end{aligned}$$(4) -
3.
For each gene \(x_i\), calculate total number of similar genes of \(x_i\) as
$$\begin{aligned} {\mathrm {N}}(x_i)=\sum _{j=1}^{n} {\mathrm {S}}(x_i,x_j). \end{aligned}$$(5) -
4.
Sort n genes according to their values of \({\mathrm {N}}(x_i)\) such that \({\mathrm {N}}(x_1)> {\mathrm {N}}(x_2)> \cdots > {\mathrm {N}}(x_n)\).
-
5.
If \({\mathrm {N}}(x_i)>{\mathrm {N}}(x_j)\) and \({\mathcal N}(x_i,x_j)\le \lambda \), then \(x_j\) cannot be considered as an initial cluster center, resulting in a reduced set of genes to be considered for c initial cluster centers \(v_i\), \(i=1,2,\cdots ,c\). Also, the \(\lambda \) is a user defined parameter.
-
6.
Stop.
3 Gene Expression Data Sets Used
In this work, publicly available three gene expression data sets are used to compare the performance of different clustering methods along with proposed dissimilarity measure. This section gives a brief description of the following three gene expression data sets, two of which are downloaded from Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/).
-
1.
Cho Data Set: This data set contains gene expression profile of yeast genome during mitotic cell cycle. The number of genes and time points of this data are 5575 and 17, respectively [2].
-
2.
GDS759: This data set is related to analysis of gene expression in temperature sensitive pre-mRNA splicing factor mutants prp17 null, prp17-1, and prp22-1 at various time points following a shift from the permissive temperature of 23 \(^\circ \)C to the restrictive temperature of 37 \(^\circ \)C. The number of genes and time points of this data are 6350 and 24, respectively [18].
-
3.
GDS2347: It contains the analysis of wild type W303 cells across two cell cycles, a length of 2 h after synchronization with alpha factor. The number of genes and time points are 6228 and 13, respectively [16].
4 Results and Discussions
In this section, the performance of the proposed dissimilarity measure is demonstrated. The dissimilarity measure is incorporated with hard c-means (HCM) [8], fuzzy c-means (FCM) [5], rough-fuzzy c-means (RFCM) [12], and robust rough-fuzzy c-means (rRFC-M) [13], The performance of the proposed dissimilarity measure over only normalized range normalized city block distance or Gene ontology based dissimilarity measure is also presented. The results are reported on three microarray gene expression data sets, namely, Cho data set, GDS759, and GDS2347. Gene expression vectors are used for only those genes whose gene ontology information is also available. For each data set, the number of clusters c is decided by using the CLICK [19] algorithm. Each gene data set is pre-processed by standardizing each feature or time point to zero mean and unit variance. The values of two fuzzifiers are set to 2.0, that is, \(\acute{m}_1=2.0\) and \(\acute{m}_2=2.0\). All the results are reported using gene ontology based functional annotation ratio. This evaluation criterion quantify the functional similarity among a set of genes.
4.1 Optimum Clustering Solutions
The threshold \(\lambda \) [15] plays an important role to generate the initial cluster centers. It controls the degree of dissimilarity among the genes present in microarray data. In effect, it has a direct influence on the performance of the initialization method used. Also, the performance of the rough-fuzzy clustering algorithm depends on the weight parameter \(\omega \) [12, 13].
Let \(\varPhi =\{\lambda ,\omega \}\) be the set of parameters and \(\varPhi ^\star =\{\lambda ^\star ,\omega ^\star \}\) is the set of optimal parameters. To find out the optimum set \(\varPhi ^\star \), containing optimum values of \(\lambda ^\star \) and \(\omega ^\star \), the Davies-Bouldin (DB) cluster validity index [3] is used here. DB index [3] is designed to identify sets of clusters that are compact and well separated. DB index minimizes
for \(1 \le i,k \le c\). The DB index minimizes the within-cluster distance \(S(v_i)\) and maximizes the between-cluster separation \(d(v_i,v_k)\). Therefore, for a given data set and c value, the higher the similarity values within the clusters and the between-cluster separation, the lower would be the DB index value. A good clustering procedure should make the value of DB index as low as possible.
For three gene microarray data sets, the value of \(\lambda \) is varied from 0.0 to 0.15, while the value of \(\omega \) is varied from 0.51 to 0.99. The optimum values of \(\lambda ^\star \) and \(\omega ^\star \) for each microarray data set and for two rough-fuzzy clustering algorithms [12, 13] are obtained using the following relation:
While for HCM and FCM only \(\lambda \) parameter is considered. The optimum values of \(\lambda \) and \(\omega \) for the rough-fuzzy clustering algorithms [12, 13] as well as HCM and FCM with NRNCBD distance for three data sets, namely, Cho data set, GDS759, and GDS2347 are mentioned in Table 1.
4.2 Importance of Integrated Dissimilarity Measure
This section describes about the importance of the integrated dissimilarity measure in terms of functional consistency of a gene cluster. In order to evaluate the functional consistency of the gene clusters produced by different algorithms, the biological annotations of the gene clusters are considered in terms of the GO. The annotation ratios of each gene cluster in three GO ontologies are calculated using the GO Term Finder [1]. The GO term is searched in which most of the genes of a particular cluster are enriched. The annotation ratio, also termed as cluster frequency, of a gene cluster is defined as the number of genes in both the assigned GO term and the cluster divided by the number of genes in that cluster. A higher value of annotation ratio indicates that the majority of genes in the cluster are functionally more closer to each other, while a lower value signifies that the cluster contains much more noises or irrelevant genes. After computing the annotation ratios of all gene clusters for a particular ontology, the sum of all annotation ratios is treated as the final annotation ratio. A higher value of final annotation ratio represents that the corresponding clustering result is better than other, that is, the genes are better clustered by function, indicating a more functionally consistent clustering result [20].
Here, the importance of proposed integrated dissimilarity measure is shown over dissimilarity measure based on only either type of dissimilarity measure. Table 2 presents the comparative results of different types of dissimilarity measures, in term of final annotation ratio or cluster frequency, for the MF, BP, and CC ontologies on three data sets. From the table it is seen that the integrated dissimilarity measure performs better than city block distance alone and gene ontology based distance alone. The dissimilarity measures are incorporated in to different c-means clustering algorithms. Out of 36 cases the integrated dissimilarity measure performs better in 24 cases. On the other hand, the HCM and FCM algorithms generates more functionally consistent clustering results with integrated dissimilarity measure in most of the cases. Only in three cases in each of the HCM and FCM algorithms the ontology based dissimilarity measure performs better. The RFCM algorithm performs better with GO based dissimilarity measure in most of the cases. The sign ‘-’ indicates that the algorithm could not generate desired number of clusters. Using only GO based distance in few cases the clustering algorithms generate co-incident clusters. Therefore, they are not further studied. The rRFCM algorithm always generates better result with integrated dissimilarity measure. From the results it is seen that incorporation of integrated dissimilarity measure drastically improves the performance of clustering algorithms.
4.3 Comparative Performance Analysis of Different Clustering Algorithms
In this section performance of different c-means clustering algorithm is shown in terms of annotation ratio or cluster frequency. Table 3 represents the performance of different clustering algorithms along with integrated dissimilarity measure. From the table it is seen that the rRFCM generates more number of functionally consistent clusters. Out of nine cases the rRFCM algorithm along with integrated dissimilarity measure performs better than other clustering algorithms in six cases. Only in one case and two cases the FCM and HCM, respectively performs better.

Eisen plots of different clusters for Cho yeast data set generated by HCM, FCM, RFCM, and rRFCM
4.4 Qualitative Performance Analysis
The Eisen plot gives a visual representation of the clustering result. In Eisen plot [7], the expression value of a gene at a specific time point is represented by coloring the corresponding cell of the data matrix with a color similar to the original color of its spot on the microarray. The shades of red color represent higher expression level, the shades of green color represent low expression level and the colors towards black represent absence of differential expression values. In the present representation, the genes are ordered before plotting so that the genes that belong to the same cluster are placed one after another. The cluster boundaries are identified by white colored blank rows.
The gene clusters produced by the HCM, FCM, RFCM, SOM, and rRFCM algorithms on Cho yeast data set are visualized by TreeView software, which is available at http://rana.lbl.gov/EisenSoftware and the plots for one data sets are reported in Fig. 1 as examples. From the Eisen plots presented in Fig. 1, it is evident that the expression profiles of the genes in a cluster are similar to each other and they produce similar color pattern, whereas the genes from different clusters differ in color patterns. Also, the results obtained by both RFCM and rRFCM algorithms are more promising than that by both HCM and FCM algorithms.
4.5 Performance of Clustering Algorithms in Terms of Cluster Validity Indices
The expression data of clustering solutions is used to evaluate the performance of different clustering algorithms in terms of some standard cluster validity indices. Table 4 presents the performance of different c-means algorithms for optimum values of \(\lambda \) and \(\omega \) in terms of Davies Bouldin Index [3] and \(\beta \) Index [14].
The \(\beta \) index [14] is defined as the ratio of total variation and within-cluster variation, and is given by
\(n_i\) is the number of objects in the ith cluster (\(i=1,2,\cdots ,c\)), n is the total number of objects, \(x_{ij}\) is the jth object in cluster i, \(v_i\) is the mean or centroid of ith cluster, and \(\overline{v}\) is the mean of n objects. For a given data set and c value, the higher the homogeneity within the clusters, the higher would be the \(\beta \) value. The value of \(\beta \) also increases with c.
The results and subsequent discussions are presented with respect to DB index and Beta index. The results establish the fact that the rRFCM algorithm performs equal or better than other c-means clustering algorithms.
5 Conclusion
In this paper importance of dissimilarity measure in a clustering algorithm has been demonstrated. Application of only one type of distance measure may not generate desired results as that of integrated dissimilarity measure. Here, microarray gene expression data based distance as well as Gene Ontology based distance measures are integrated and their importance over individual type of distance measure is shown. It has been observed that integrated dissimilarity measure generates more functionally similar gene clusters. The effectiveness of the integrated dissimilarity measure is shown on different gene expression data sets. It has been observed that the integrated dissimilarity measure along with rRFCM algorithm performs better than any other clustering algorithm.
References
Boyle, E.I., Weng, S., Gollub, J., Jin, H., Botstein, D., Cherry, J.M., Sherlock, G.: GO: term finder open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715 (2004)
Cho, R.J., Campbell, M.J., Winzeler, E.A., Steinmetz, L., Conway, A., Wodicka, L., Wolfsberg, T.G., Gabrielian, A.E., Landsman, D., Lockhart, D.J., Davis, R.W.: A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell 2(1), 65–73 (1998)
Davies, D.L., Bouldin, D.W.: A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 1, 224–227 (1979)
de Jong, S., Boks, M.P.M., Fuller, T.F., Strengman, E., Janson, E., de Kovel, C.G.F., Ori, A.P.S., Vi, N., Mulder, F., Blom, J.D., Glenthj, B., Schubart, C.D., Cahn, W., Kahn, R.S., Horvath, S., Ophoff, R.A.: A gene co-expression network in whole blood of schizophrenia patients is independent of antipsychotic-use and enriched for brain-expressed genes. PLOS One 7(6), 1–10 (2012)
Dembele, D., Kastner, P.: Fuzzy C-means method for clustering microarray data. Bioinformatics 19(8), 973–980 (2003)
Du, Z., Li, L., Chen, C.F., Yu, P.S., Wang, J.Z.: G-SESAME: web tools for go-term-based gene similarity analysis and knowledge discovery. Nucleic Acids Res. 37, W345–W349 (2009)
Eisen, M.B., Spellman, P.T., Patrick, O., Botstein, D.: Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95(25), 14863–14868 (1998)
Heyer, L.J., Kruglyak, S., Yooseph, S.: Exploring expression data: identification and analysis of coexpressed genes. Genome Res. 9(11), 1106–1115 (1999)
Kustra, R., Zagdanski, A.: Incorporating gene ontology in clustering gene expression data. In: 19th IEEE Symposium on Computer-Based Medical Systems (CBMS 2006), pp. 555–563 (2006)
Li, J., Bushel, P.R.: EPIG-Seq: extracting patterns and identifying co-expressed genes from RNA-Seq data. BMC Genomics 17(1), 255 (2016)
Lin, D.: An information-theoretic definition of similarity. In: Proceedings of 15th International Conference on Machine Learning, pp. 296–304 (1998)
Maji, P., Pal, S.K.: RFCM: a hybrid clustering algorithm using rough and fuzzy sets. Fundam. Informaticae 80(4), 475–496 (2007)
Maji, P., Paul, S.: Rough-fuzzy clustering for grouping functionally similar genes from microarray data. IEEE/ACM Trans. Comput. Biol. Bioinform. 10(2), 286–299 (2013)
Pal, S.K., Ghosh, A., Shankar, B.U.: Segmentation of remotely sensed images with fuzzy thresholding and quantitative evaluation. Int. J. Remote Sens. 21(11), 2269–2300 (2000)
Paul, S., Maji, P.: City block distance and rough-fuzzy clustering for identification of co-expressed microRNAs. Mol. BioSyst. 10(6), 1509–1523 (2014)
Pramila, T., Miles, S., GuhaThakurta, D., Jemiolo, D., Breeden, L.L.: Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle. Genes Dev. 16(23), 3034–3045 (2002)
Resnik, P.: Using information content to evaluate semantic similarity in a taxonomy. In: Proceedings of 14th International Joint Conference on Artificial Intelligence, pp. 448–453 (1995)
Sapra, A.K., Arava, Y., Khandelia, P., Vijayraghavan, U.: Genome-wide analysis of pre-mRNA splicing: intron features govern the requirement for the second-step factor, Prp17 in Saccharomyces cerevisiae and Schizosaccharomyces pombe. J. Biol. Chem. 279(50), 52437–52446 (2004)
Shamir, R., Sharan, R.: CLICK: a clustering algorithm for gene expression analysis. In: Proceedings of the 8th International Conference on Intelligent Systems for Molecular Biology (2000)
Wang, H., Wang, Z., Li, X., Gong, B., Feng, L., Zhou, Y.: A robust approach based on Weibull distribution for clustering gene expression data. Algorithms Mol. Biol. 6(1), 14 (2011)
Acknowledgements
The author wants to acknowledge Dr. Pradipta Maji of Indian Statistical Institute, Kolkata, India for his valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Paul, S. (2017). Integration of Gene Expression and Ontology for Clustering Functionally Similar Genes. In: Polkowski, L., et al. Rough Sets. IJCRS 2017. Lecture Notes in Computer Science(), vol 10313. Springer, Cham. https://doi.org/10.1007/978-3-319-60837-2_47
Download citation
DOI: https://doi.org/10.1007/978-3-319-60837-2_47
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60836-5
Online ISBN: 978-3-319-60837-2
eBook Packages: Computer ScienceComputer Science (R0)