
Abstract
The N-methyl d-aspartate receptor (NMDAR) is a ligand-gated ion channel that binds the neurotransmitter glutamate. It was pharmacologically identified and differentiated from other ionotropic amino-acid receptors at excitatory synapses in the late 70s for it is activated by NMDA and not kainate. Due to its large calcium conductance, it is involved in many physiological and pathological phenomena, the most notorious of which is synaptic plasticity, considered to be the molecular substrate of learning and memory. During the 40 years that followed their discovery, and owing to other unique properties such as their magnesium-block that makes them key “coincidence detectors”, NMDARs have been mostly studied at synapses. Yet, NMDARs exhibit a great number of other fundamental features that have remained unknown, underappreciated or challenging to study, and that have only become the focus of intense investigation over the past decade. These properties, such as the co-agonist-gating or the subcellular compartmentalization, greatly contribute to the functional diversity of NMDARs and will be the focus of this chapter as they are greatly relevant in the context of their physiological and pathological impact on the central nervous system.
Similar content being viewed by others

Keywords
- NMDA receptor
- Glutamate receptor
- Co-agonist
- d-Serine
- Glycine
- Subcellular localization
- Extrasynaptic
- Excitotoxicity
- Synaptic plasticity
- Subunit composition
- d-Aspartate
- Slow inward currents
- Tonic current
- Astrocytes
- Glia
With over 1500 publications each year since the late 1990s’, the N-methyl d-aspartate receptor (NMDAR) is the most investigated receptor in the field of neurosciences. Nearly 40 years after their pharmacological identification [1, 2], an overwhelming wealth of data has become available about these glutamate-gated ionotropic receptors, from their fine crystallographic structures and electrophysiological properties, to the molecular basis of their functional diversity, their central role in the mechanisms of synaptic plasticity, learning, memory and their direct implication in a variety of diseases.
In this chapter, we will focus on the diversity of NMDARs subtypes and functions in the central nervous system (CNS) in light of the most recent advances in the field. We will focus on a newly discovered source of NMDARs diversity that, similar to their subunit composition, is thought to impact their function: the subcellular location. Indeed, NMDARs are mostly studied at synapses, where glutamate is released, but they also exist at pre-synaptic and extra-synaptic sites where they seem to be engaged in different processes.
As “famous” as they might be, some aspects of these receptors still suffer from a paucity of information. While this is obviously true for most of the properties of extra-synaptic NMDARs which are notoriously hard to study, this is surprisingly true as well for key features of synaptic NMDARs such as their co-agonist gating. Where data are lacking, we will dash this chapter with provocative thoughts that might help shedding a new light on some aspects of NMDARs and offer new perspectives about their roles in the central nervous system (CNS).
NMDARs are distinctive from other glutamate receptors by their remarkable molecular diversity. For the purpose of this introduction, we shall just say that NMDARs assemble as heterotetramers made of two GluN1-subunits combined with two other subunits of the GluN2 (A–D) or GluN3 (A and B) family. Each of these possible combinations provides particular properties to the receptor subtype they make up [3, 4], including fundamental aspects such as the magnesium block, the affinity for agonists and the Ca2+ permeability of the ion pore. Combined with the spatial (brain region-specific) and temporal (developmental) profile of NMDAR subunits expression in the CNS, this results in a very complex and heterogeneous picture of NMDAR properties in a given area of the CNS at a given time of development or adulthood. This is rendered even more complex by the fact that a given NMDAR subtype can be found at diverse subcellular locations at the surface of a single neuron, where it is believed to play distinct roles.
This probably explains why the use of subunit-specific NMDARs antagonists has proven disappointing in a clinical context, considering the plethora of fundamental functions involving NMDARs in the CNS on one hand, and the different, sometimes opposing, roles that a subtype of NMDAR can endorse depending on its brain or cellular location. Indeed, the discovery that many of the properties of NMDARs are highly sensitive to the subunit composition of the receptor generated a great drive to attribute specific functions to particular subtypes of NMDAR and to develop subunit-specific pharmacological agonists, antagonists and modulators, aiming at treating major neuropathological and neurodegenerative diseases (from ischemia to schizophrenia, see following chapters). Besides this great potential and besides the wealth of data available on NMDARs, most of these therapeutic compounds have failed in clinical trial due to adverse side effects and/or lack of efficacy [5]. This type of observations has contributed to make it clear that the cellular and subcellular location, combined with NMDAR subunit composition, could be one of the most important features dictating the functions in which a given subtype of NMDAR is involved. In this chapter, we will focus on neuronal NMDARs, but NMDARs can also be found on blood vessels [6, 7], astrocytes [8] and oligodendrocytes [9].
2.1 Synaptic Versus Extra-Synaptic NMDARs: Location, a Source of Functional Diversity
2.1.1 What Is Synaptic and What Is Not?
While the vision we often have of synapses is very ‘graphic’ owing to their specialized morphology, the definition of the synaptic space remains highly functional. Along those lines, post-synaptic NMDARs are conventionally considered as ‘synaptic’ if they are recruited during low frequency presynaptic activity, which includes low frequency stimulation of axon terminals and miniature/spontaneous vesicular release events [10–14]. Such a definition has the advantage of being very intuitive: An NMDAR is synaptic if it is activated by synaptic activity. However, it provides no insight about the actual localization of receptors participating to synaptic transmission, in particular because the geometry of the cleft and adjacent extra-synaptic space, the rate of glutamate uptake, the presynaptic site of vesicular release, the concentration of transmitter in vesicles, and many other parameters greatly influence the probability that a receptor anchored at a given location will bind glutamate [15].
From the morphological point of view, a receptor is often considered synaptic if it lays no more than 100 nm from the edge of the post-synaptic density (PSD) [16, 17]. This often implies that the receptor faces the presynaptic terminal. In a similar manner that the functional definition is satisfying at the electrophysiological level, this anatomical definition fulfills our need to visualize a synaptic receptor at the synapse. Unfortunately, it is very unlikely that these two definitions encompass the same synaptic space and the same pool of receptors, and the delineation of the synaptic vs. extra-synaptic space is probably a case by case matter given the diversity of synaptic features throughout the different regions of the CNS. Meanwhile, we are still lacking a clear definition of the boundaries of the synaptic space that would best account for both functional and morphological considerations and this would certainly need to be addressed.
2.1.2 How Are NMDARs Organized at Synapses?
The precise organization of NMDARs within the synapses is not yet fully resolved and seems to be specific for each type of synapse. For instance, the density of NMDARs was observed to peak at the edge, or past the edge, of PSD at ganglion cell synapses in the retina. This seems particularly true for GluN2B-containing receptors [18]. However, this finding does not seem to be the case at other synapses, such as in the hippocampus, and this could be due to the nature and distribution of the post-synaptic intracellular partners present at each synapse (see Sect. 2.1.4). NMDARs also exist at pre-synaptic locations [17, 19, 20]. Though they are, strictly speaking, extra-synaptic given their distance to the PSD and their location on a different cell, they are often considered synaptic because they are thought to contribute to synaptic function. Whether pre-synaptic NMDARs are controlled by synaptic glutamate spill-over, by surrounding glial processes or by both, is not clear. Examples of either situation have been documented [19, 20] and it seems that the function of these receptors mostly consists in regulating the release probability of the pre-synaptic element, as could be expected given the Ca2+ permeability of NMDARs.
2.1.3 How Are NMDARs Organized at Extra-Synaptic Sites?
NMDARs were discovered at non-synaptic locations in the mid-1990s [21–31] and they were initially thought to be very similar to synaptic NMDARs, because the channel behavior in excised patches (extra-synaptic NMDARs) was directly related to the macroscopic properties of the excitatory post-synaptic current (EPSC) recorded from synapses (synaptic NMDARs) [23]. Subsequent functional comparison of synaptic NMDAR-mediated currents and outside-out patches, however, has since revealed that “different NMDAR subtypes are expressed in sub-synaptic and extra-synaptic compartments” [23, 25, 27]. For 20 years, our approach of extra-synaptic NMDARs as remained bound to this idea [25] and the first study actually dedicated to the organization of NMDARs at extra-synaptic sites, rather than to their subunit composition, was only published in 2010 [17]. It revealed that extra-synaptic NMDARs are found in cultures as well as on brain sections, both at early developmental stage and in adulthood. This was concordant with electrophysiological evidence that extra-synaptic NMDARs represent an estimated 1/3 of the total NMDAR population on hippocampal neurons in young adult rats [12] and as high as ¾ of all NMDARs in the immature hippocampus [14]. At extra-synaptic sites, NMDARs form clusters that are not evenly distributed on dendrites. Instead, they are preferentially associated with specific portions of the dendritic shaft that are contacted by other cells such as by glial cell processes (~30 %), axon-like processes (~50 %) or other dendrites (~20 %). This is in remarkable agreement with earlier observations [24] that found extra-synaptic GluN1 immuno-gold labeling consistently localized between dendrites and astrocytic processes in adult somatosensory cortex. This very specific localization supports a role for NMDARs in reciprocal neuron–glia communication (Fig. 2.1). At synapses, the PSD, a dense and intricate ensemble of intracellular partners held together with scaffolding proteins that play a key role in regulating glutamate receptors signaling and synaptic architecture, typically spreads over 195 nm on average in the area CA1 of the hippocampus [32] or over 260 nm at the particular CA3-CA1 synapse [33]. Intriguingly, extra-synaptic NMDARs were found to gather over an area of less than 100 nm width on average with no clear PSD-like sub-membrane electron-dense structure. This could suggest that the sites where extra-synaptic NMDARs accumulate are comprised of no (or few) other types of receptors, thus requiring limited sub-membrane partners and anchoring proteins, and are dedicated to NMDAR-mediated extra-synaptic signaling only. This would explain why they are found in a narrower region, and why NMDAR-containing extra-synaptic sites lack a clear PSD-like electron density. None the less, much like their synaptic homologues, clusters of NMDARs at extra-synaptic sites seem to associate with intercellular adhesion molecules such as β-catenins and with the scaffolding proteins SAP-102 (synapse associated protein-102) and PSD95 (post-synaptic density protein 95). Therefore, it seems that the molecular organization of synaptic and extra-synaptic NMDARs at the plasma membrane follow the same elementary rules, even though the molecular details of the intracellular partners involved in anchoring NMDARs at extra-synaptic sites remain to be elucidated.

Immuno-gold labelling of GluN1 antibody in the area CA1 of adult hippocampus. Note the presence of GluN1 labeling at remote distance from the pre-synaptic terminal (p) but in close proximity with a glial (g) process (arrowhead, panel F). On panel I, GluN1 subunits are clearly clustered at the surface of a dendrite (d) where a glial process makes a “synapse-like” contact (arrowhead). Scale bar: 100 nm. Adapted from [17] Figure 5
Taken altogether, these evidence about their organization strongly suggest that extra-synaptic NMDARs represent a functionally distinct pool of receptors, specifically tethered to this location, and not a pool of rogue receptors escaped from synaptic trapping or a mere reserve pool of receptors waiting to be recruited to synapses. Instead, these observations could be taken as evidence that extra-synaptic receptors, much like their synaptic counterpart, are engaged in cell-to-cell signaling. The finding that these receptors are clustered to specific cell contact areas, such as specialized neuron–glia appositions, indeed suggests that they may be engaged in a separate signaling function independent from their synaptic homologues. This view is also supported by the functional demonstration that synaptic and extra-synaptic NMDARs form distinct and stable pools of receptors at the surface of CA1 neurons [12]. These data also challenge our primitive vision of extra-synaptic NMDARs as an ensemble of highly mobile and randomly distributed receptors as was suggested by work in cultures [34–36].
2.1.4 NMDARs Are Mobile
All of these views, and most of the work performed on NMDARs, are somewhat based on the assumption that NMDARs are mostly static over short periods of time, in particular at the time-scale of synaptic transmission. Surprisingly, this may not be true. Indeed, recent work carried out in cultures has demonstrated the high motility of glutamate receptors. Although initially reported decades ago [37, 38], membrane diffusion of neurotransmitter receptors only recently emerged as a cellular pathway involved in the regulation of synaptic receptor content and distribution [39], perhaps because this has been studied more accurately with the development of techniques such as fluorescence recovery after photo-bleaching (FRAP) microscopy and quantum dots (Q-dots) over the last years. This so-called lateral diffusion of NMDARs (i.e. rapid and seemingly Brownian movement of the receptor at the surface of the plasma membrane) was elegantly evidenced at the surface of young hippocampal neurons by Tovar and Westbrook in 2002 [36]. Using the NMDAR antagonist MK-801, an activity-dependent and irreversible open-channel blocker, to completely block synaptic NMDARs and abolish synaptic NMDAR-mediated currents, they were able to show a partial recovery of synaptic NMDAR-mediated responses within minutes, demonstrating that synapses had been refilled with unblocked NMDARs from extra-synaptic sites via lateral diffusion of receptors and that blocked synaptic receptors had diffused away from the PSD to the extra-synaptic compartment.
Today, we can directly image the trajectory of single-NMDARs in real-time with the use of Q-dotes (Fig. 2.2) and we know that approximately 30–40 % of surface NMDARs are mobile with an average diffusion coefficient of 0.05 μm2/s. In comparison, 50 % of surface AMPARs are mobile in basal conditions with an instantaneous diffusion coefficient in the range of 100–1000 μm2/s. It was shown that about a third of NMDARs traffic between the synaptic and extra-synaptic compartment, which means that NMDARs reside at the synapse for a matter of minutes, rather than days as previously thought [35, 40, 41]. The lateral diffusion of NMDARs is strongly influenced by the subunit composition of the receptor. In particular, GluN2B-containing NMDARs appear more mobile than GluN2A-containing NMDARs, in line with predictions that could be made from their interactions with distinct intracellular partners. Whether this mobility is affected by activity, in particular by the binding of glutamate, remains unclear. It was shown in vivo that the blockade of NMDARs leads to a rapid and striking redistribution of GluN2B subunits away from synapses [30]. On the other hand, inhibiting NMDARs does not affect diffusion properties in brain slices [13].

Illustration of the lateral diffusion of GluN2B- and GluN2A-countaining NMDARs at the surface of a dendritic spine of a cultured hippocampal neuron. The representative trajectory (over 50 ms) of a surface GluN2A-containing NMDAR is depicted in red, that of a GluN2B-containing NMDAR in blue. They were imaged using single Quantum-Dot (QD) tracking approach (inset, lower right). Each trajectory represents the diffusion of a single particle-receptor complex. The GluN2A–QD is confined in the PSD at the spine head while the GluN2B–QD diffuses from the head of the spine to the dendritic shaft. Scale bar = 200 nm. Adapted from [35]
2.2 NMDAR Intracellular Partners at Synapses and Extra-Synaptic Sites
Membrane-associated guanylate kinases (MAGUKs), such as PSD-95 and SAP-102, are direct intracellular partners that coordinate trafficking, anchoring, and signaling of NMDARs (among other receptors) by interacting with their cytoplasmic tail and by recruiting other signaling and scaffolding proteins at the PSD. It is still unclear to what extent particular NMDAR subtypes associate preferentially with specific MAGUKs but it is generally accepted that mature synapses are enriched in PSD-95, while immature/developing synapses contain SAP-102 [42–44]. Consequently, it is thought that NMDARs are anchored at mature synapses primarily via interactions with PSD-95 [45], while interactions with SAP-102 is responsible for their trafficking and anchoring at immature synapses. SAP-102 is also expressed in the adult but a strong competition with PSD-95 for insertion into the PSD causes its exclusion from the synapse. SAP-102 is thus thought to be enriched at peri-synaptic or extra-synaptic locations in adult. Importantly, it can still be found at synapses [42, 44], such that this view is only a gross approximation. According to this view, in the adult, interaction of NMDARs with SAP-102 would result in trafficking and addressing of the receptor to an extra-synaptic location. Because the spatial expression of SAP-102 seems less compartmentalized than that of PSD-95 this would also result in a higher mobility of the receptor. New data also support a role for SAP-102 in the synaptic clearance of NMDARs, indicating that it could play a role in regulating receptor content at synapses over time, which is very interesting in the context of activity-dependent reorganization of NMDARs that was observed at many occasions [30]. At extra-synaptic sites, it is unclear what MAGUK prevails, but consistent with the description above, the SAP-102/PSD-95 ratio is overall enriched in favor of a higher content of SAP-102. Petralia et al. found that (in cultures) SAP-102 is not enriched at synapses but rather expressed evenly in both extra-synaptic and synaptic compartments, in contrast with PSD-95 which they found to be five times more concentrated at synapses. The relative functional enrichment of SAP-102 at extra-synaptic sites would thus result solely from the preferential location of PSD-95 at synapses. Whether the nature of intracellular partners present at extra-synaptic locations varies according to the identity of the cellular processes that NMDARs are facing is unknown, but is an interesting possibility.
2.3 Subunit Composition at Synaptic and Extrasynaptic Sites
Details about the assembling of NMDARs subunits into a functional tetrameric receptor can be found in any of the many reviews from Dr. Paoletti. This section will focus on the functional properties that subunit composition confers to the receptor. Briefly, the assembling of NMDAR requires four subunits, two of which are necessarily the obligatory GluN1 subunit (any of its two splice variants). The two remaining subunits can be any of the 4 GluN2 subunits (A–D) and/or of the 2 GluN3 subunits (A and B). The most notorious of these combinations are the GluN2A-NMDARs (di-heteromers made of 2 GluN1 assembled with 2 GluN2B) and the GluN2B-NMDARs (GluN1-GluN2B di-heteromers) for which we have had very selective and efficient antagonists since the late 1980s. Like we already mentioned, the identity of the subunits that assemble confer a unique set of properties to the NMDAR they form together (Table 2.1). Owing to differences in their intercellular C-terminal domain (CTD), GluN2 subunits also interact with different scaffolding proteins, which strongly influences the surface localization and diffusion of NMDAR subtypes [3, 4], and this is thought to be the basis for location-dependent differences in NMDAR subunit composition. For instance, it is generally believed that GluN2A-NMDARs associate preferentially with PSD-95, and this interaction is responsible for trapping these receptors at synapses [35, 42–44, 46, 47]. GluN2B-NMDARs on the other hand tend to interact with SAP-102 [42–44]. According to the view described in Sect. 2.2 this would make GluN2B-NMDARs more mobile and addressed at extra-synaptic sites, which is remarkably consistent with existing data (see Sect. 2.1.2 and Fig. 2.2 above, and Sect. 2.3.2 below).
2.3.1 Subunit Composition of Synaptic NMDARs Is Highly Variable in Time and Space
NMDAR subunit composition changes over time throughout the CNS, as evidenced by their mRNA profile characterized with in situ hybridizations [48, 49]. This has also been confirmed at the protein level on multiple occasions using Western blotting and, to some extent, at the functional level using electrophysiology and pharmacology (Fig. 2.3). While the GluN1 subunit is ubiquitously expressed in the CNS during embryonic, postnatal development, and throughout adulthood, the GluN2 subunits, as well as GluN3 subunits, differ strikingly in their spatial and temporal expression profile. During early development, GluN2B has the highest level of expression of all GluN2 subunits throughout the CNS. This expression peaks during this second week of postnatal development, but then declines steadily and becomes restricted to the forebrain in adulthood. Similar to GluN2B, the expression of GluN2D peaks during the second week after birth, with a wide distribution at this time (including in the forebrain), before it declines again to weaker expression levels restricted to the brainstem and diencephalon in adulthood. In contrast, GluN2A and GluN2C are barely detectable before birth which drastically changes during the first 2 weeks of postnatal development. The levels of GluN2A rapidly increase during this period and it becomes the predominant GluN2 subunit in the entire CNS in adulthood. The GluN2C subunit appears later in postnatal development (~P10) as well but its expression remains restricted to the cerebellum and the olfactory bulbs throughout adulthood. Among the GluN3 subunits, GluN3A is expressed at low levels during embryonic development, peaks during the first week of postnatal development when its expression is surprisingly widespread, and decreases to low levels in adulthood. Finally, while little attention was dedicated to the GluN3B subunit until recently, its expression profile suggests that it could be central to the physiology of NMDARs in the CNS. Indeed, its expression increases slowly after birth to reach peak and ubiquitous expression in the adult CNS in a similar manner to that of the obligatory GluN1 subunit.

In situ hybridizations showing the distribution of NMDAR GluN2 subunits mRNA in sagittal sections of rat brain over time (birth day (P1) to adulthood). Note the ubiquitous expression of GluN1 at any time in development and adulthood, consistent with the fact that it is an obligatory subunit required for the formation of a functional NMDAR. Adapted from Akazawa et al. [48]
At the functional level, the change in expression profile of NMDAR subunits throughout development is accompanied by changes in kinetics, magnesium-block properties, Ca2+ permeability and sensitivity to allosteric modulators of NMDAR-mediated currents (Table 2.1). A typical case of this spatiotemporal maturation of NMDAR subunit expression is the developmental ‘switch’ from GluN2B to GluN2A in the forebrain; that is, the expression of GluN2B subunits declines as that of GluN2A increases, which has led to the idea that GluN2B-containing NMDARs are replaced by GluN2A-containing NMDARs through postnatal development in the forebrain. This ‘GluN2B/GluN2A developmental switch’ is a well-accepted concept that prevails in the area CA1 of the hippocampus in particular at the canonical CA3-CA1 synapse. Surprisingly, it has not been fully characterized at the electrophysiological level. But it can still be validated by gathering data from individual studies that have assessed the contribution of GluN2B-homodimers to NMDAR-mediated synaptic currents at the CA3-CA1 synapses at different ages through the use of specific antagonists (Fig. 2.4). Overall it is established that, in adults, GluN2B-NMDARs are absent from CA3-CA1 synapses while GluN2A-homodimers are predominant, such that the population of NMDARs comprises of up to 75 % of GluN2A-NMDARs and about 25 % of GluN2A/GluN2B triheteromers (made of 2 GluN1, one GluN2A and one GluN2B). Therefore, at CA3-CA1 synapses, the replacement of GluN2B by GluN2A is almost total and GluN2B subunits only contribute to the formation of triheteromeric receptors. GluN2A-NMDARs were also found to be the predominant synaptic NMDAR subtype in neurons located in the substantia gelatinosa region of adult rat spinal cord, based on the decay time of synaptic currents, apparent K d for magnesium and ifenprodil insensitivity [25] .

Developmental profile of the sensitivity of NMDAR-mediated synaptic responses to the GluN2B-NMDAR specific antagonist Ro25-6981 at the canonical CA3-CA1 synapse in rat hippocampal acute slices, from data found in the literature. While GluN2B-NMDARs compose most of the population of synaptic NMDARs before P10, they are functionally absent from synapses past P40
This spatiotemporal shift in subunit expression is supported by the developmental profile of the intercellular partners involved in anchoring NMDARs at synapse, in particular SAP-102 and PSD95 ([42] and see Sect. 2.2 above). Indeed, SAP-102, which is thought to preferentially interact with the cytoplasmic tail of GluN2B subunits, is enriched at synapses during early stages of development. In the first weeks following birth, however, the expression of PSD-95, which preferentially interacts with GluN2A, increases. Eventually PSD-95 replaces SAP-102 at synapses due to a competition for insertion into the PSD [17, 44, 50, 51].
Unfortunately, biology does not comply with our need for simplicity and the replacement of GluN2B- with GluN2A-NMDARs at synapses is nothing but the exception of a few particular examples of synapses or brain regions and not a general rule. Indeed, GluN2B-NMDARs represent a major portion of synaptic NMDARs in many other areas of the adult CNS, including in the cortex [52] or in the spinal cord lamina I [53] where the contribution of GluN2B-NMDARs and GluN2D-NMDARs to the synaptic population far exceeds that of GluN2A-NMDARs. Additionally, the NMDAR-subtype content of synapses, in particular GluN2A- and GluN2B-NMDAR content, can be regulated by synaptic activity [3]. Indeed, it was shown that the GluN2B-subunit content is altered following long-term potentiation or depression of glutamatergic synapses in the hippocampus [54], indicating that GluN2B-NMDArs can contribute to synaptic NMDAR signaling in adult even at CA3-CA1 synapses.
2.3.2 Subunit Composition of Extra-Synaptic NMDARs Is Unresolved
Given that extra-synaptic NMDARs represent as high as ¾ of the population of NMDARs in the immature hippocampus [14] and an estimated 1/3 of the total NMDAR population on adult hippocampal neurons [12, 17], it is tempting to speculate from ‘global’ in situ hybridization and Western blotting studies that extra-synaptic receptors undergo developmental modifications of their subunit content as well. This, however, has never been addressed, probably because probing extra-synaptic NMDARs in brain slices has proven challenging due to the fact that there is no easy way to directly stimulate and record from this population of receptors.
Nonetheless, it is believed that extra-synaptic NMDARs are enriched in GluN2B-NMDARs in the adult hippocampus, in contrast with the GluN2A-NMDAR content at synapses. This fits with the idea that extra-synaptic NMDARs constitute a distinct population mediating a different function than synaptically located NMDARs. This idea is also quite satisfying when one considers that GluN2B-NMDARs have a higher affinity for glutamate, which is present at lower concentrations in the extra-synaptic space [3, 4, 55]. Additionally, this is consistent with the fact that GluN2B subunits, as well as SAP-102, while not predominant at synapses, are still abundantly expressed by hippocampal neurons in the adult [42–44]. These observations together strongly fuel the idea that GluN2B-NMDARs exist at extra-synaptic sites more abundantly than at synapses. Using the same kind of evidence and reasoning it is believed that GluN2D/C subunits are present at extra-synaptic sites on principal neurons in the cortex and spinal cord [25]. However, the subunit composition of extra-synaptic NMDARs remains a mystery in most brain regions.
Just like different NMDAR subtypes can be segregated at different synapses on a same neuron, it is certainly wise to hypothesize that extra-synaptic NMDARs do not form one homogeneous population at the surface of an entire neuron as far as subunit composition is concerned. It would be interesting to investigate whether, for example, extra-synaptic NMDARs located on apical dendrites share the same composition and properties as those located on distal dendrites, whether different subunit-compositions prevail at the different extra-synaptic specializations (i.e., neuron-glia, or axo-dendritic appositions), whether subunit composition at extra-synaptic sites can be regulated by activity, and whether subunit composition of extra-synaptic NMDARs undergoes a reorganization throughout postnatal development. Additionally, with the development of better subunit-specific antagonists it will soon be possible to reliably address the involvement or GluN2C-, GluN2D- and even GluN3-containing NMDARs in various processes, and it would be interesting to characterize better the extent to which these subtypes participate to extra-synaptic NMDAR signaling.
2.4 Gating of Synaptic and Extra-Synaptic NMDARs
2.4.1 Agonists of the Glutamate-Binding Site of Synaptic NMDARs
Glutamate is the endogenous agonist that binds to the agonist-binding site of synaptic NMDARs, which is located on the GluN2 subunit. It is estimated that glutamate concentration exceeds 3 mM within the first microseconds following a single synaptic release event [15] (Hamilton and Attwell). Considering that the dissociation constant of NMDARs for glutamate is in the micro-molar range, it can be calculated that 97 % of synaptic NMDARs will bind the two molecules of glutamate required for their activation during the first hundreds of microseconds following the release of a single vesicle of glutamate. This means that synaptic NMDARs are fully saturated during a synaptic transmission event. Therefore the rules that govern the activation of the glutamate-binding site of synaptic NMDARs follow a binary probability: 0 in the absence of synaptic activity, 1 during synaptic transmission. This simple fact, though often ignored, highlights the tremendous importance of (1) the existence of another agonist-binding site controlled by a distinct transmitter (D-serine or glycine, see Sects. 2.4.3 and 2.4.4 below) to regulate the gating of NMDARs and (2) the Mg2+ block of the pore of the ion channel that, independently the activation of the receptor per se, will dictate whether or not an ion flux is permitted through the channel.
Interestingly, some evidence has recently revived the idea that aspartate (d- and l-) could be an endogenous ligand of NMDARs as well. Similar to glutamate, it is synthesized in the CNS, seemingly found in synaptic vesicles released by nerve terminals upon depolarization and taken up after transmission events [56]. Deletion or inhibition of the D-aspartate catabolism enzyme increases the levels of endogenous D-aspartate by 10–20-fold, and mice knocked out for this enzyme have elevated levels of the N-methyl derivative of d-aspartate (i.e., NMDA) and enhanced NMDAR-dependent functions such as LTP and spatial learning. While this is by no means a proof that d-, l- or N-methyl aspartate are endogenous ligands of NMDARs under normal conditions, this has generated a craze, over the past 15 years, for the idea that aspartate and glutamate could be stored and released from the same vesicle, or that aspartate could have its own vesicular loading and exocytotic pathway, and directly contribute to the activation of NMDARs at glutamatergic synapses. This is supported by the finding that a sialic acid transporter, closely related in sequence to the vesicular glutamate transporters (VGluTs), can transport both glutamate and aspartate [57, 58]. Interestingly, while many vesicular transporters can indeed package several transmitters in the same vesicle, VGluTs do not transport or even recognize aspartate [59, 60]. These observations predicted that, in the absence of VGluTs, excitatory synapses would only signal using synaptic vesicles that contain aspartate; which would permit the activation of NMDARs while AMPAR-mediated transmission would be suppressed. However, it was shown in 2015 by the group of Roger Nicoll that in VGluT1 knock-out mice, NMDAR-mediated synaptic responses were virtually absent and the AMPA/NMDA ratio (whenever measurable) was unaltered. This is consistent with the traditional view that glutamate fully accounts for the activation of synaptic NMDARs and suggested that if aspartate is released at synapses it is present at concentrations too low to have any physiological relevance. Interestingly, however, these data do not exclude the possibility that aspartate could signal to extra-synaptic NMDARs or pre-synaptic NMDARs.
2.4.2 Agonists of the Glutamate-Binding Site of Extra-Synaptic NMDARs
While synaptic NMDARs are only activated by brief and acute (i.e. phasic) release of glutamate from the presynaptic terminal, extra-synaptic NMDARs seem to be exposed to different sources of agonist and types of release. The existence of a tonic current mediated by NMDARs was reported in principal neurons of the hippocampus in 1989 [61]. This tonic current is mediated by receptors located outside of synapses and occurs due to sufficient concentrations of ambient agonist, of non-synaptic origin [61–64]. Indeed, this tonic current persists when synaptic activity is suppressed with TTX and, conversely, synaptic NMDAR activity remains intact after the NMDAR-mediated tonic current is blocked with MK-801 [64]. This establishes those receptors as distinct and necessarily distant from synaptic NMDARs. To date, the origin of the ambient glutamate that allows such tonic activation of extra-synaptic NMDARs is still unclear and whether this is indeed glutamate that allows this tonic activation has not, in fact, been demonstrated. Aside from the observation that it is of non-synaptic origin [62, 64] little evidence exist for the source or the nature of the agonist responsible for the NMDAR-mediated tonic current. It is known that the regulation of extra-synaptic glutamate concentration heavily relies on active uptake by astrocytes. Indeed, the glutamate transporter GLT1 is predominantly present on these glial cells and mediates 95 % of the glutamate uptake. Interestingly, the amplitude of NMDAR-mediated tonic current is strongly enhanced either by blocking GLT1 [65, 66], which enables glutamate spill over from synapses, or when slices are challenged with glial toxins [65]. Additionally, a recent study showed that the extent of NMDAR-mediated tonic current depends on glial coverage of synapses in the supra-optic nucleus of the hypothalamus [65]. Therefore, an interesting possibility is that astrocytes could be a key regulator of NMDAR-mediated tonic current by either confining synaptic space with glial processes, thus maintaining the synaptic/extra-synaptic compartmentation, or by modulating ambient glutamate levels.
The existence of an extra-synaptic NMDAR-mediated tonic current implies that a subset of extra-synaptic NMDARs is constitutively activated. However, extra-synaptic NMDARs can also be recruited experimentally by exogenous applications of NMDA or glutamate. This indicates that another subpopulation of extra-synaptic NMDARs exists that is normally silent and can be activated by phasic, non-synaptic release of glutamate. The main manifestation of such receptors are “slow inward currents” (SICs), characterized by a slow decay-time (on the order of seconds), low frequency of occurrence (~0.05 Hz) and a large amplitude (hundreds of pA). They occur spontaneously and have been recorded from principal neurons of the CA1 region of the hippocampus [67–69] and in the superficial layers of the dorsal horn of the spinal cord [70]. Like NMDAR-mediated tonic current, SICs persist in the absence of neuronal and/or synaptic activity [67]. They are also completely suppressed by glial inhibitors [70] and evidence suggests that they are caused by glutamate release from astrocytes onto extra-synaptic NMDARs [22, 55, 68, 71, 72]. It is easy to hypothesize that such currents could result from a “synaptic-like” release of glutamate from glial processes at specialized neuron–glia appositions such as those described above Sect. (2.1.3 and Fig. 2.1).
Challenging view: Beside their mechanistic relevance, these observations are interesting because they open the possibility that extra-synaptic NMDARs may exist as two functionally distinct pools (1), tonically activated receptors, which face supra-threshold amounts of glutamate and co-agonist and (2), overall silent extra-synaptic NMDARs, which can be acutely recruited by exogenous or endogenous agonist release (similar to synaptic NMDARs). If true, this would mean that glutamate availability is not homogenous outside of synapses and could be spatially regulated, giving rise to different sub-compartments within the extra-synaptic space, like suggested by the distribution of extra-synaptic NMDARs themselves. Although this is purely speculative, we propose that it could be relevant to consider the extra-synaptic space as being comprised of separate compartments instead of one large homogenous volume, at least from the point of view of glutamate and NMDARs.
2.4.3 Agonists of the Glycine-Binding Site of Synaptic NMDARs
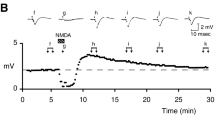
In 1987 a simple, yet revolutionary observation was published in a Nature article and described in those words: “In cultured neurons, the magnitude of the whole cell current produced by NMDA appeared to depend on the speed of perfusion of NMDA-containing solutions: slower movement of the perfusion solution resulted in larger response.” The authors hypothesized that “a substance that augments the response to NMDA is tonically released from the cultured cells (neurons or glia), and accumulates when the perfusion is slow” [73]. Johnson and Ascher had just made the discovery that NMDARs require two agonists for their activation. Their investigation led them to individually screen the effect of amino acids present in the cultured medium, and they made the discovery that “the effects of conditioned medium were reproduced by glycine.” Indeed, the main finding of this work Fig. (2.4) is that application of NMDA alone (or glutamate) on outside-out excised patches does not permit the opening of the NMDAR ion channel, and neither do applications of glycine alone. When both are co-applied however, NMDARs are activated and this results in sustained currents flowing through the receptor’s ion channel Fig. (2.5). While authors concluded that “glycine augmented the response to glutamate as well as that to NMDA” the correct conclusion of this experiment is that glycine allowed the response to glutamate as well as that to NMDA [74]; and this finding is the first demonstration that NMDARs harbor two distinct agonist-binding sites that recognize, and are activated by, two different ligands. As a concluding remark, authors hypothesized that “the glycine-binding site and the NMDA-binding site are on two distinct proteins [i.e. subunits]”, which was confirmed the following year in a Science publication by Kleckner and Dingledine who demonstrated that the glycine-binding site is on the GluN1 subunit while the glutamate-binding site is on the GluN2 subunit. Since then, the agonist binding domain of the GluN1 subunit of NMDARs has been coined the “glycine-binding site” and glycine was termed “co-agonist” of NMDARs. The idea that glycine, an abundant and ubiquitous amino acid present in culture medium and in the extracellular space of the brain parenchyma, is the endogenous agonist of the glycine-binding site remained unchallenged for nearly 20 years. It was supported by the existence of glycine transporters (GlyT1) on glial processes that surround synaptic contacts and mediate a powerful uptake of glycine. This was thought to regulate the concentration of glycine at excitatory synapses, providing a powerful means to modulate the degree of activation of the NMDAR glycine-binding site.

Effect of glycine (1 μM) on the current induced by NMDA (10 μM) on outside-out excised patches containing NMDARs. Note that applications of NMDA or glycine alone do not elicit currents, consistent with the fact that the simultaneous binding of the two agonists on their respective agonist-binding site is required for the activation of NMDAR. From [73]
This canonical view was challenged by the groups of Salmon Snyder and Stephane Oliet, first in 1995 and then in 2006, respectively. After the discovery that d-amino acids exist in mammalian CNS [75–77], the observation was made by Schell et al. [78] that the localization of D-serine throughout the brain resembles that of NMDARs, and the binding pattern of (3H)D-serine revealed by autoradiography strongly resembled that of (3H)glycine binding. This strongly suggested that D-serine is an endogenous ligand for the glycine-binding site of NMDARs, an idea that received very little attention from most of the neuroscience community—with the notable exception of the glial community. Indeed, as the study of glial cells had started to thrive, the publication by Schell et al. showed outstanding potential contained in this statement: “We were surprised to observe under high magnification that D-serine immunoreactivity was associated exclusively with glia”.
The first demonstration that D-serine, and not glycine, is the endogenous ligand of the glycine-binding site of the NMDAR in brain tissue came from Panatier et al. [79], and echoed similar earlier demonstrations carried out in cultures by the Snyder group. Soon after, it was established that D-serine is the endogenous co-agonist of NMDARs at many, but not all, central synapses (reviewed in Papouin et al. [80]), such as in layer 5 of the cortex, in the retina, in the amygdala, in the nucleus accumbens, in the spinal cord and, notably, at the canonical CA3-CA1 synapses of the hippocampus [13]. The strength and impact of this finding are greatly underappreciated. Indeed, given that the gating of NMDARs by glutamate at synapses is binary (all or none, see Sect. 2.4.1), and given the importance of NMDARs in the development, physiology and pathology of the CNS, the notion that glial cells are responsible for the supply and regulation of the glycine-binding site’s agonist makes glial cells the primary regulator of the most investigated receptor in neuroscience. This also means that astrocytes are directly involved in processes such as synaptic plasticity and learning. Additionally, with its glutamate-binding site controlled by the pre-synaptic neuron, its glycine-binding site controlled by astrocytes, and its magnesium block reliant on the activity of the post-synaptic neuron, the NMDAR can be viewed as a molecular-scale model of the tripartite synapse and thus a remarkable tool to study it. The rules that govern the release of D-serine are still incompletely elucidated and subject to debates that spread beyond the scope of this chapter.
That D-serine is the endogenous ligand of the glycine-binding site of synaptic NMDARs is not true at every synapse in the CNS. Many instances have been found where the historical co-agonist glycine happens to be the endogenous ligand of the glycine-binding site. Such is the case in the spinal cord [81], on retinal ganglion cells [82] and in the nucleus of the tractus solitarius [79]. In structures where glycinergic innervation is abundant, such as in the retina and the spinal cord, it was shown that glycine that serves as an endogenous co-agonist of NMDARs at glutamatergic synapses [81, 82] originates from nearby inhibitory synapses. It is thought that following inhibitory synapses activity, glycine spills over and diffuses to bind to NMDARs at remote excitatory synapses. This highlights an interesting cross-talk between inhibitory and excitatory synapses in these regions along with the potential for inhibitory synapses to impact synaptic plasticity occurring at neighboring excitatory terminals.
Challenging view : When it was discovered in 1987 that NMDARs bind another ligand, and that this second-agonist is required to permit the activation of the receptor by glutamate, it was very naturally (but unfortunately) termed “co-agonist” of NMDARs. This misnomer has resulted in a multitude of inaccurate views and descriptions of NMDAR co-agonist function, ranging from “helper” for receptor activation to “allosteric modulator”. Not only is the terminology used in these examples erroneous, but the view they attempt to describe about the function of the co-agonist is wrong as well. In this case, like often in science, inspection of the original publication by Johnson and Ascher proves to be enlightening as it reveals that the so-called NMDAR co-agonist is nothing less than a full agonist at the receptor: (1) it binds to its own, separate, ligand-binding site on the GluN1 subunit, (2) it is absolutely required for the opening of the ion pore and (in the presence of glutamate) is sufficient for this activation, and (3) the efficacy of glycine and D-serine at the GluN1 agonist-binding site is ~100 %, which means they are full, not partial, agonists. In other words, NMDAR possesses two distinct agonist-binding sites, one harbored by each GluN1 subunit that binds glycine/D-serine, and one harbored by each GluN2 subunits that binds glutamate; in such a way that glutamate and glycine/D-serine play the exact same function on different subunits. The only reason why glycine was termed “co-agonist”, while glutamate is termed “agonist”, is because its role in controlling the activation of NMDARs was discovered after that of glutamate, but from a biological and molecular point of view, there is no such thing as a main or primary agonist of NMDARs. These considerations are also well illustrated by the simple fact that GluN3-NMDARs (made of two GluN1 and two GluN3 subunits that bind glycine/D-serine but not glutamate) lack a glutamate-binding site and are therefore only activated by glycine or D-serine while insensitive to glutamate.
2.4.4 Co-agonists of the Glycine-Binding Site of Extra-Synaptic NMDARs
The identity of the co-agonist of extra-synaptic NMDARs has only been addressed once, by our group [80]. We showed that, on apical dendrites of CA1 pyramidal neurons, extra-synaptic NMDARs are gated by glycine, and not D-serine. In striking contrast, NMDARs located at CA3-CA1 synapses on the same neurons are gated by D-serine, not glycine, like mentioned earlier. It is reasonable to assume that the availability of glycine and D-serine are regulated by distinct mechanisms. Therefore, on CA1 pyramidal neurons, the gating of the co-agonist binding site of NMDARs contributes to segregating synaptic and extra-synaptic NMDARs as two functionally distinct pools. Whether glycine is also the co-agonist of extra-synaptic NMDARs located elsewhere on CA1 neurons, and whether this applies to other brain areas, remains to be addressed. If the identity of the co-agonist is used as a way to more efficiently separate synaptic from extra-synaptic NMDARs as a general rule, then one might expect that wherever D-serine is the co-agonist of synaptic NMDARs, glycine would gate extra-synaptic NMDARs, and vice versa.
The origin of glycine available to extra-synaptic NMDARs is unclear, especially in the hippocampus. In this structure, the presence of glycinergic terminals has never been established (the inhibitory transmission is entirely abolished by GABA receptor antagonists) and the expression of functional glycine receptors (GlyRs) is thought to stop after birth [83–85], even though the existence of extra-synaptic GlyRs has been documented in adults [86, 87]. Yet, in vivo microdialysis reported amounts of free glycine as high as 10 μM in the hippocampus [88, 89]. In vivo, a major source of extracellular glycine in the CNS could be the blood flow since approximately 200 μM of glycine are found in the blood [90] and since glycine is able to cross the endothelial wall of capillaries by means of glycine transporters [91, 92]. In slices, however, blood vessels are emptied of their initial content, indicating that a source of glycine exists in the brain parenchyma itself. With an average intracellular glycine concentration of 3–6 mM [93], glial cells could be a major source of glycine in brain slices. In the hippocampus, it was also proposed that glycine could be co-released with GABA at inhibitory synapses by interneurons [94] similar to what occurs in the thalamus, the brainstem, the spinal cord and the cortex [95, 96]. Notably, that glycine is present at 10 μM in brain parenchyma would suggest that the amounts of glycine available to extra-synaptic NMDARs are high enough to be saturating [88, 89], especially if those receptors are GluN2B-NMDARs (K d < 1 μM). This would leave little room for the modulation of NMDAR activity through the co-agonist-binding site at extra-synaptic locations. Although surprising, this idea is supported by observations that exogenous applications of co-agonist do not enhance the amplitude of NMDAR-mediated tonic current [64].
2.4.5 Endogenous and Exogenous NMDARs Allosteric Modulators
Several competitive antagonists, binding directly onto the agonist-binding site of either the GluN1 subunit or the GluN2 subunit where they compete with the endogenous agonist and prevent or reduce its conformational activation of the site, are available, efficient and specific for NMDARs if used at appropriate concentrations. This includes the glycine-binding site antagonist 5,7-Dichlorokynurenic acid (DCKA) and the glutamate binding-site antagonist d(–)-2-Amino-5-phosphonopentanoic acid (D-AP5). Blockers of the ion channel such as Dizocilpine (MK-801) also exist that act as non-competitive antagonists since they alter the permeability of the channel without changing the activation rules of the receptor. Beside these competitive and non-competitive antagonists, a number of endogenous and exogenous compounds can modulate the activity of NMDARs by binding on specific sites that are distinct from the agonist binding sites or the ion channel. They act by changing the conformation of the receptor which usually modifies the open channel probability (Po) but, contrary to agonists and competitive antagonists, they do not interfere with the activation of the receptor. Among the endogenous allosteric modulators, the most unacknowledged of them all is also the simplest and the most important: H+ ions (protons). Protons bind to the NMDAR at an unknown location thought to be closely associated with the channel gate. Proton binding impacts the N-terminal domain (NTD) of the GluN2 subunit and trigger a “closed” conformation of this domain which favors the closure of the ion pore of NMDAR channel. Therefore, H+ binding reduces the Po of the channel [97]. This is of the uttermost importance mainly for two reasons: (1) The EC50 of the proton-binding site for protons is in the range of physiological pH (~6.9 for GluN2A and 7.5 for GluN2B subunit) which means that protons exert a tonic and basal inhibition of NMDAR channel Po and that this inhibition can differentially affect particular NMDAR-subtypes for subtle pH changes. (2) The inhibitory effect of H+ is directly responsible for the subunit-selectivity and efficacy of other allosteric modulators such as zinc (Zn2+) and Ifenpodil/Ro25-6981. Indeed these competitive antagonists act by causing conformational changes in the receptor that increase the sensitivity of the proton-binding site and thus enhance the inhibition by H+, therefore further reducing the Po of the channel. Zinc, like protons, is an endogenous allosteric modulator of NMDARs that binds in the NTD of GluN2A subunits with a remarkable selectivity since its affinity for GluN2A-NTD is 1000 times better than that for any other subunit. Under physiological pH, zinc only produces a ~70 % inhibition of GluN2A-NMDAR mediated current. Interestingly, zinc is co-stored in vesicles and co-released with glutamate. While, ambient extracellular zinc levels are too low to inhibit synaptic GluN2A-NMDARs in a tonic manner, sustained synaptic transmission elicits a surge of zinc concentration at excitatory synapses that causes an endogenous subunit-specific inhibition of NMDAR transmission that is thought to mediate an activity-dependent regulation of neuronal circuits [98].
Surprisingly, 40 years after the discovery of the first NMDAR antagonist, the number of pharmacological reagents available to discriminate between NMDAR-subtypes is still very limited as reviewed in Neyton and Paoletti [99]. In particular, antagonists selective for GluN2C-, GluN2D- and GluN3-containing receptors as well as specific antagonists for NMDAR tiheteromers, such as GluN2A/GluN2B-NMDARs, are still lacking. Yet, a large number of compounds are available online from various chemical suppliers that are described as subunit-selective based on their tendency to prefer a particular NMDAR-subtype. Unfortunately, while some of them indeed have a higher affinity for a particular NMDAR subunit or subtype, their affinity is not high enough to allow a full inhibition of this subtype/subunit without impacting other subtypes/subunits. This is typically the case of PPDA, often presented as a GluN2C- and GluN2D-specific antagonist based on its preferential binding to these subunits (Ki ~ 0.1 μM). Unfortunately, it Ki in the very same order of magnitude for the other subunits (GluN2B: 0.3 μM; GluN2A: 0.6 μM), such that PPDA is not a subunit-selective antagonist specific for GluN2C/GluN2D-containing NMDARs. The same has been established for NVP-AAM077 [100], a putative GluN2A-NMDAR specific antagonist. As a general rule, it is safe to consider that an antagonist is only selective for a NMDAR subtype/subunit if its affinity for the latter is at least an order of magnitude higher than that for other subtypes/subunits [99].
2.5 Functions of NMDARs in Relation to Their Location
While the general role of NMDARs in various CNS functions is clearly establishes, the precise contribution of synaptic vs. extra-synaptic receptors to each of them remains mostly elusive. Some studies have reported a role for non-synaptic NMDARs in extra-synaptic inhibition [101] and dendritic dynamic range compression [102], but the main functions in which extra-synaptic NMDARs have been involved and studied are synaptic plasticity, excitotoxicity/ischemia, excitability, and neuron-glia interactions.
2.5.1 Synaptic Plasticity, a Matter of Subunits or Location?
Using genetic and pharmacological approaches, there has been intense speculation about the role of specific NMDAR subtypes in the selective induction of LTP and LTD [103–108]. It was proposed that GluN2A-NMDARs preferentially trigger LTP, whereas GluN2B-NMDARs are preferentially associated with LTD. Given the very complex and diversified expression pattern of NMDAR subunits throughout the CNS and over time, this tempting proposal seemed improbable. While it might be true in specific cases, this dichotomy has indeed been significantly and repeatedly challenged [104, 106] and the idea that a particular subtype of NMDAR is specifically involved in inducing potentiation or depression at glutamatergic synapses seems over-simplifying, as reviewed in great detail in [3]. Since synaptic and extra-synaptic NMDARs often seem to have distinct subunit compositions, this controversy raised the question of whether NMDAR location, rather than subunit composition per se, could constitute a determining factor to the direction of synaptic plasticity and be the biological reason for discrepancies between studies [109]. While this interesting idea was formulated 10 years ago, the role of extra-synaptic NMDARs in LTP and LTD has rarely been characterized directly, i.e. independently of subunit composition. Because there is no simple rule linking the cellular location of NMDARs to their subunit composition, and because the subunit content of receptors located outside of synapses is unresolved, it is difficult to interpret results involving extra-synaptic NMDARs in synaptic plasticity based on subtype-specific pharmacological experiments which, unfortunately, is the vast majority of studies on that matter. Activating or silencing specifically synaptic or extra-synaptic NMDARs thus requires different approaches that do not rely on subunit composition. The main alternative is the use of the open-channel blocker MK-801, which allows inactivation of NMDARs that are recruited while leaving the silent/not recruited receptors intact. When combined with low-frequency stimulation of afferent fibers, this method allows the selective blockade of synaptic NMDARs and leaves most of their extra-synaptic counterparts intact [12, 13, 64]. This method presents the advantage that it only relies on whether receptors are active or not during MK-801 application, and therefore circumvents caveats associated with the use of subunit-selective reagents. Interestingly, using such an approach, Xu et al. [110] found that selective stimulation of extra-synaptic NMDARs triggers LTD in CA1 neurons. Another approach is based on the discovery that D-serine and glycine gate synaptic and extra-synaptic NMDARs respectively [13]. Taking advantage of this segregation, we showed in adult rat hippocampal slices that synaptic NMDARs, but not extra-synaptic ones, are required for LTP induction, whereas activation of receptors at both locations is required for LTD. These findings thus confirm the idea proposed by Dr. Rusakov [109] that what matters in LTP and LTD induction may be the location, rather than the subtype, of NMDARs recruited. If the compartmentalization of NMDAR by their co-agonist gating was to be generalized to other brain regions, this might be a very powerful and convenient way, in addition to the MK-801 approach, to study the role of synaptic and extra-synaptic NMDARs in various functions independently of their composition.
2.5.2 Is Excitotoxicity Caused by Extra-Synaptic NMDARs?
Because NMDARs are highly permeable to calcium, they not only relay a physiological signal to neurons, but can also trigger intracellular signaling cascades leading to cell death. In fact, the link between excitotoxicity [111] and excessive glutamate release [112] were described years before synaptic plasticity. NMDARs were demonstrated to be the main source of calcium responsible for glutamate-induced excitotoxicity in the late 1980s [113–115]. Ever since, NMDARs have been renowned for their dual roles in physiology and pathophysiology, and the mechanisms of NMDAR-induced cell death is one of the most investigated aspects of this receptor.
In 2002, Hardingham et al. published the first compelling evidence that synaptic and extra-synaptic NMDAR activation have opposing effects on cell fate. Ever since, the prevailing theory is that the activity of synaptic NMDARs favors neuronal survival, via the phosphorylation of intracellular factors such as CREB or Erk1/2. In contrast, cell death is mainly mediated by the activation of extra-synaptic NMDARs (mostly GluN2B-NMDARs in this study) which inhibits the CREB and Erh1/2 pathways in addition to directly promoting pro-death signaling via caspase-3 [16]. This view gained additional attention as the use of memantine developed. Memantine is thought to be an extra-synaptic NMDARs blocker and was shown to reduce neuronal death in models for neurodegenerative disorders such as Huntington’s [116] or Alzheimer’s disease [117–119]. However, numerous observations now point to the limitation of the theory that synaptic NMDARs favor survival while extra-synaptic NMDARs promote cell-death. Indeed, this finding, obtained in neuronal cultures, has hardly been replicated in slices or in vivo. In fact, several experiments point to a role for synaptic NMDARs in cell death during NMDA application in acute slices or oxygen glucose deprivation (OGD) in vivo [13, 80, 120, 121]. Amadoro et al. [122] demonstrated that synaptic NMDARs mediate hypoxia-induced neurotoxicity almost entirely, whereas blocking extra-synaptic NMDARs provides no protection. Similarly, we showed that NMDA-induced excitotoxicity in slices is entirely mediated by synaptic NMDARs in the area CA1 of the hippocampus since it can be prevented by silencing receptors at this location [13]. On the contrary, silencing extra-synaptic NMDARs had no effect on NMDA-induced cell-death. Other recent work [120, 123], following the protocols established by Hardingham et al. [124], failed to replicate the findings that extra-synaptic NMDARs, but not synaptic ones, promote cell-death. This controversy, questioning the role of extra-synaptic NMDARs in excitotoxicity, is also fueled by recent and thorough insights into the mechanism of the use-dependent NMDAR blocker memantine [125]. Memantine was proposed to preferentially block extra-synaptic receptors over synaptic ones, owing to its fast off-rate, low affinity, voltage-dependent binding and uncompetitive nature [16, 125, 126]. The fact that memantine can successfully prevent neuronal death in vitro or in pathological conditions [127, 128] has thus strongly contributed to the extra-synaptic hypothesis of excitotoxicity. However, the group of Steven Mennerick demonstrated, in two studies, that memantine has a strong and fast inhibitory effect on synaptic NMDARs (80 % in 5 min) under basal low-frequency stimulation. Additionally, they show that the neuroprotective effect of memantine is mediated by the blockade of synaptic, not extra-synaptic, NMDARs [125] (Katsuki et al.). This body of evidence strongly questions the prevalent role of extra-synaptic NMDARs in triggering neuronal death during excitotoxic conditions in culture, and suggests that this theory does not apply to more intact preparations.
2.5.3 Neuron-Glia Interactions and Excitability
The mechanisms and roles of SICs and tonic currents, and how astrocytes play a role in these forms of signaling have been recently reviewed by Balázs Pál (Front Cell Neuro [129]).
It is accepted that the tonic current mediated by extra-synaptic NMDARs plays a role in the excitability of principal neurons and in modulating dendritic inputs [130]. This might be particularly true for interneurons [131] which excitability is crucial in setting synchronized neuronal population dynamics. It also becomes increasingly clear that NMDAR-mediated tonic current can be upregulated in pathological conditions, ranging from cocaine addiction [132] to Alzheimer’s disease [117]. However, the relevance of such tonic current to neuronal physiology and the impact of its pathological disturbance need further investigation. Notably, even though its exact function is unknown, NMDAR-mediated tonic current certainly plays a major role in neuronal and network physiology because it has been observed in virtually every brain region thus far.
SICs were shown to result from the release of glutamate by astrocytes onto neuronal extra-synaptic NMDARs, and to occur simultaneously in distinct neurons within 100 μm of each other [67], which fits with the idea that an astrocyte’s anatomical territory spans an area approximately 50–100 μm in diameter. Based on these observations, SICs have been proposed to play a role in neuronal synchrony and network excitability [67, 81] but this aspect still remains poorly understood and would require more investigation. In addition, since they have only been observed in slices so far, often under non-physiological conditions (low magnesium, GLT1 blockers), the question of whether SICs represent an important feature of neuron–glia signaling that also occurs in vivo remains to be established. Based on the observation that extra-synaptic NMDARs are often located at seemingly specialized neuron-astrocyte contacts, it might be relevant to investigate SICs as the manifestation of an extra-synaptic, yet synaptic-like, astrocyte-to-neuron form of signaling.
2.6 Concluding Remarks
In conclusion we would like to draw readers’ attention to the fact that the subcellular location of the NMDAR has emerged as a key determinant of NMDAR-mediated physiological functions as well as NMDAR-related pathological conditions. Such location appears to be equally or potentially more important than NMDAR subunit-composition. Furthermore, extreme methodological and intellectual caution should be used when employing subunit-based pharmacological approaches to decipher the role of a particular subset of NMDAR in a given context. Not only are some of the available reagents not selective enough, but also the subunit composition of NMDARs is not conclusively telling of their location since there seems to be no clear or absolute subunit-hallmarks that differentiate synaptic from extra-synaptic NMDARs. Finally, many aspects of extra-synaptic NMDARs suggest that they could be involved in a complex and specific type of extra-synaptic cell-to-cell communication. That this facet of NMDAR physiology has remained largely unexplored promises new exiting lines research and future paradigm-shifting discoveries. As a starting point, we propose that the availability of glutamate and co-agonists, and the distribution of NMDARs, are not homogenous outside of synapses and could be spatially regulated, thus delineating distinct functional and morphological sub-compartments within the extra-synaptic space.
References
Biscoe TJ et al. D-alpha-aminoadipate as a selective antagonist of amino acid-induced and synaptic excitation of mammalian spinal neurones. Nature. 1977;270:743.
Davies J, Watkins JC. Actions of D and L forms of 2-amino-5-phosphonovalerate and 2-amino-4-phosphonobutyrate in the cat spinal cord. Brain Res. 1982;235:378–86.
Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400.
Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011;33:1351–65.
Monaghan DT, Jane DE. In: Van Dongen AM, editorBiology of the NMDA receptorBoca Raton, FL: CRC Press/Taylor & Francis; 2009.
LeMaistre JL, Sanders SA, Stobart MJ, Lu L, Knox JD, Anderson HD, Anderson CM. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J Cereb Blood Flow Metab. 2012;32:537–47.
Stobart JL, Lu L, Anderson HD, Mori H, Anderson CM. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2013;110:3149–54.
Lee MC, Ting KK, Adams S, Brew BJ, Chung R, Guillemin GJ. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One. 2010;5:14123.
Cao N, Yao ZX. Oligodendrocyte N-methyl-D-aspartate receptor signaling: insights into its functions. Mol Neurobiol. 2013;47:845–56.
Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci. 2002;22:2165–73.
Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–36.
Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–19.
Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–46.
Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–8.
Clements JD. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–71.
Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–96.
Petralia RS, Wang YX, Hua F, Yi Z, Zhou A, Ge L, Stephenson FA, Wenthold RJ. Organization of NMDA receptors at extrasynaptic locations. Neuroscience. 2010;167:68–87.
Zhang J, Diamond JS. Subunit- and pathway-specific localization of NMDA receptors and scaffolding proteins at ganglion cell synapses in rat retina. J Neurosci. 2009;29:4274–86.
Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. 2007;10:331–9.
Kunz PA, Roberts AC, Philpot BD. Presynaptic NMDA receptor mechanisms for enhancing spontaneous neurotransmitter release. J Neurosci. 2013;33:7762–9.
Aoki C, Rhee J, Lubin M, Dawson TM. NMDA-R1 subunit of the cerebral cortex co-localizes with neuronal nitric oxide synthase at pre- and postsynaptic sites and in spines. Brain Res. 1997;750:25–40.
Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhäuser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–20.
Clark BA, Farrant M, Cull-Candy SG. A direct comparison of the single-channel properties of synaptic and extrasynaptic NMDA receptors. J Neurosci. 1997;17:107–16.
Kharazia VN, Weinberg RJ. Immunogold localization of AMPA and NMDA receptors in somatic sensory cortex of albino rat. J Comp Neurol. 1999;412:292–302.
Momiyama A. Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. J Physiol. 2000;523:621–8.
Petralia RS, Wang YX, Wenthold RJ. Internalization at glutamatergic synapses during development. Eur J Neurosci. 2003;18:3207–17.
Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–12.
Valtschanoff JG, Burette A, Wenthold RJ, Weinberg RJ. Expression of NR2 receptor subunit in rat somatic sensory cortex: synaptic distribution and colocalization with NR1 and PSD-95. J Comp Neurol. 1999;410:599–611.
Petralia RS, Wang YX, Wenthold RJ. NMDA receptors and PSD-95 are found in attachment plaques in cerebellar granular layer glomeruli. Eur J Neurosci. 2002;15:583–7.
Fujisawa S, Aoki C. In vivo blockade of N-methyl-D-aspartate receptors induces rapid trafficking of NR2B subunits away from synapses and out of spines and terminals in adult cortex. Neuroscience. 2003;121:51–63.
Rosenmund C, Feltz A, Westbrook GL. Synaptic NMDA receptor channels have a low open probability. J Neurosci. 1995;15:2788–95.
Tao-Cheng JH, Gallant PE, Brightman MW, Dosemeci A, Reese TS. Structural changes at synapses after delayed perfusion fixation in different regions of the mouse brain. J Comp Neurol. 2007;501:731–40.
Takumi Y, Ramírez-León V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999a;2:618–24.
Groc L, Bard L, Choquet D. Surface trafficking of N-methyl-D-aspartate receptors: physiological and pathological perspectives. Neuroscience. 2009;158:4–18.
Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc Natl Acad Sci U S A. 2006;103:18769–74.
Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–64.
Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. 1976;16:1055–69.
Poo M. Rapid lateral diffusion of functional A Ch receptors in embryonic muscle cell membrane. Nature. 1982;295:332–4.
Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron. 2008;59:359–74.
Bard L, Groc L. Glutamate receptor dynamics and protein interaction: lessons from the NMDA receptor. Mol Cell Neurosci. 2011;48:298–307.
Bard L, Sainlos M, Bouchet D, Cousins S, Mikasova L, Breillat C, Stephenson FA, Imperiali B, Choquet D, Groc L. Dynamic and specific interaction between synaptic NR2-NMDA receptor and PDZ proteins. Proc Natl Acad Sci U S A. 2010;107:19561–6.
Yoshii A, Sheng MH, Constantine-Paton M. Eye opening induces a rapid dendritic localization of PSD-95 in central visual neurons. Proc Natl Acad Sci U S A. 2003;100:1334–9.
Sans N, Petralia RS, Wang YX, Blahos II J, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–71.
van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–37.
Valtschanoff JG, Weinberg RJ. J Neurosci. 2001;21:1211–7.
Pérez-Otaño I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–38.
Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Köhr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–81.
Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994;347:150–60.
Laurie DJ, Seeburg PH. Regional and developmental heterogeneity in splicing of the rat brain NMDAR1 mRNA. J Neurosci. 1994;14:3180–94.
Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–53.
Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–20.
Thomazeau A, Lassalle O, Iafrati J, Souchet B, Guedj F, Janel N, Chavis P, Delabar J, Manzoni OJ. Prefrontal deficits in a murine model overexpressing the down syndrome candidate gene dyrk1a. J Neurosci. 2014;34:1138–47.
Hildebrand ME, Pitcher GM, Harding EK, Li H, Beggs S, Salter MW. GluN2B and GluN2D NMDARs dominate synaptic responses in the adult spinal cord. Sci Rep. 2014;4:4094.
Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66:859–70.
Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–38.
Morland C, Nordengen K, Larsson M, Prolo LM, Farzampour Z, Reimer RJ, Gundersen V. Vesicular uptake and exocytosis of L-aspartate is independent of sialin. FASEB J. 2013;27:1264–74.
Miyaji T, Echigo N, Hiasa M, Senoh S, Omote H, Moriyama Y. Identification of a vesicular aspartate transporter. Proc Natl Acad Sci U S A. 2008;105:11720–4.
Miyaji T, Omote H, Moriyama Y. Functional characterization of vesicular excitatory amino acid transport by human sialin. J Neurochem. 2011;119:1–5.
Herring BE, Silm K, Edwards RH, Nicoll RA. Is Aspartate an Excitatory Neurotransmitter? J Neurosci. 2015;35:10168–71.
Reimer RJ, Edwards RH. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch. 2004;447:629–35.
Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science. 1989;246:815–8.
Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564:397–410.
Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–41.
Le Meur K, Galante M, Angulo MC, Audinat E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol. 2007;580:373–83.
Fleming TM, Scott V, Naskar K, Joe N, Brown CH, Stern JE. State-dependent changes in astrocyte regulation of extrasynaptic NMDA receptor signalling in neurosecretory neurons. J Physiol. 2011;589:3929–41.
Povysheva NV, Johnson JW. Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol. 2012;107:2232–43.
Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–7.
Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–43.
Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–63.
Nie H, Zhang H, Weng HR. Bidirectional neuron-glia interactions triggered by deficiency of glutamate uptake at spinal sensory synapses. J Neurophysiol. 2010;104:713–25.
Bergersen LH, Gundersen V. Morphological evidence for vesicular glutamate release from astrocytes. Neuroscience. 2009;158:260–5.
Bergersen LH, Morland C, Ormel L, Rinholm JE, Larsson M, Wold JF, Røe AT, Stranna A, Santello M, Bouvier D, et al. Immunogold detection of L-glutamate and D-serine in small synaptic-like microvesicles in adult hippocampal astrocytes. Cereb Cortex. 2012;22:1690–7.
Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–31.
Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–7.
Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T. Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert.-butyloxycarbonyl-L-cysteine and o-phthaldialdehyde. J Chromatogr. 1992;582:41–8.
Hashimoto A, Oka T, Nishikawa T. Anatomical distribution and postnatal changes in endogenous free D-aspartate and D-serine in rat brain and periphery. Eur J Neurosci. 1995;7:1657–63.
Hashimoto A, Oka T. Free D-aspartate and D-serine in the mammalian brain and periphery. Prog Neurobiol. 1997;52:325–53.
Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci U S A. 1995;92:3948–52.
Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–84.
Papouin T, Oliet SH. Organization, control and function of extrasynaptic NMDA receptors. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130601.
Ahmadi S, Muth-Selbach U, Lauterbach A, Lipfert P, Neuhuber WL, Zeilhofer HU. Facilitation of spinal NMDA receptor currents by spillover of synaptically released glycine. Science. 2003;300:2094–7.
Kalbaugh TL, Zhang J, Diamond JS. Coagonist release modulates NMDA receptor subtype contributions at synaptic inputs to retinal ganglion cells. J Neurosci. 2009;29:1469–79.
Flint AC, Liu X, Kriegstein AR. Nonsynaptic glycine receptor activation during early neocortical development. Neuron. 1998;20:43–53.
Ito S, Cherubini E. Strychnine-sensitive glycine responses of neonatal rat hippocampal neurones. J Physiol. 1991;440:67–83.
Xu TL, Gong N. Glycine and glycine receptor signaling in hippocampal neurons: diversity, function and regulation. Prog Neurobiol. 2010;91:349–61.
Chattipakorn SC, McMahon LL. Pharmacological characterization of glycine-gated chloride currents recorded in rat hippocampal slices. J Neurophysiol. 2002;87:1515–25.
Chattipakorn SC, McMahon LL. Strychnine-sensitive glycine receptors depress hyperexcitability in rat dentate gyrus. J Neurophysiol. 2003;89:1339–42.
Horio M, Kohno M, Fujita Y, Ishima T, Inoue R, Mori H, Hashimoto K. Levels of D-serine in the brain and peripheral organs of serine racemase (Srr) knock-out mice. Neurochem Int. 2011;59:853–9.
Yamamoto S, Morinobu S, Iwamoto Y, Ueda Y, Takei S, Fujita Y, Yamawaki S. Alterations in the hippocampal glycinergic system in an animal model of posttraumatic stress disorder. J Psychiatr Res. 2010;44:1069–74.
Holecek M, Kovarik M. Alterations in protein metabolism and amino acid concentrations in rats fed by a high-protein (casein-enriched) diet—effect of starvation. Food Chem Toxicol. 2011;49:3336–42.
Okamoto M, Akanuma S, Tachikawa M, Hosoya K. Characteristics of glycine transport across the inner blood-retinal barrier. Neurochem Int. 2009;55:789–95.
Zafra F, Aragón C, Olivares L, Danbolt NC, Giménez C, Storm-Mathisen J. Glycine transporters are differentially expressed among CNS cells. J Neurosci. 1995;15:3952–69.
Verleysdonk S, Martin H, Willker W, Leibfritz D, Hamprecht B. Rapid uptake and degradation of glycine by astroglial cells in culture: synthesis and release of serine and lactate. Glia. 1999;27:239–48.
Song W, Chattipakorn SC, McMahon LL. Glycine-gated chloride channels depress synaptic transmission in rat hippocampus. J Neurophysiol. 2006;95:2366–79.
Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–24.
O’Brien JA, Berger AJ. Cotransmission of GABA and glycine to brain stem motoneurons. J Neurophysiol. 1999;82:1638–41.
Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P. Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature. 2009;459:703–7.
Vergnano AM, Rebola N, Savtchenko LP, Pinheiro PS, Casado M, Kieffer BL, Rusakov DA, Mulle C, Paoletti P. Zinc dynamics and action at excitatory synapses. Neuron. 2014;82:1101–14.
Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Curr Opin Pharmacol. 2007;7:39–47.
Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–3.
Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron. 2001;31:1027–34.
Oikonomou KD, Short SM, Rich MT, Antic SD. Extrasynaptic glutamate receptor activation as cellular bases for dynamic range compression in pyramidal neurons. Front Physiol. 2012;3:334.
Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–4.
Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301.
Duffy S, Labrie V, Roder JC. D-serine augments NMDA-NR2B receptor-dependent hippocampal long-term depression and spatial reversal learning. Neuropsychopharmacology. 2008;33:1004–18.
Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–6.
Zhao JP, Constantine-Paton M. NR2A-/- mice lack long-term potentiation but retain NMDA receptor and L-type Ca2+ channel-dependent long-term depression in the juvenile superior colliculus. J Neurosci. 2007;27:13649–54.
Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–8.
Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–57.
Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, Baum ML, Bibb JA, Lombroso PJ. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29:9330–43.
Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–21.
Lucas D, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58:193–201.
Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture:attenuation by NMDA antagonists. J Neurosci. 1988;8:185–96.
Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cellculture. J Neurosci. 1987;7:357–68.
Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–79.
Wroge CM, Hogins J, Eisenman L, Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci. 2012;32:6732–42.
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:2518–27.
Zhou X, Ding Q, Chen Z, Yun H, Wang H. Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem. 2013;288:24151–9.
Zhou X, Hollern D, Liao J, Andrechek E, Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013;4:560.
Katsuki H, Nonaka M, Shirakawa H, Kume T, Akaike A. Endogenous D-serine is involved in induction of neuronal death by N-methyl-D-aspartate and simulated ischemia in rat cerebrocortical slices. J Pharmacol Exp Ther. 2004;311:836–44.
Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33.
Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A. 2006;103:2892–7.
Shleper M, Kartvelishvily E, Wolosker H. D-serine is the dominant endogenous coagonist for NMDA receptor neurotoxicity in organotypic hippocampal slices. J Neurosci. 2005;25:9413–7.
Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–14.
Emnett CM, Eisenman LN, Taylor AM, Izumi Y, Zorumski CF, Mennerick S. Indistinguishable synaptic pharmacodynamics of the N-methyl-D-aspartate receptor channel blockers memantine and ketamine. Mol Pharmacol. 2013;84:935–47.
Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–50.
Ditzler K. Efficacy and tolerability of memantine in patients with dementia syndrome. A double-blind, placebo controlled trial. Arzneimittelforschung. 1991;41:773–80.
Lipton SA. The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res. 2005;2:155–65.
Pál B. Astrocytic actions on extrasynaptic neuronal currents. Front Cell Neurosci. 2015;9:474.
Wu YW, Grebenyuk S, McHugh TJ, Rusakov DA, Semyanov A. Backpropagating action potentials enable detection of extrasynaptic glutamate by NMDA receptors. Cell Rep. 2012;1:495–505.
Riebe I, Seth H, Culley G, Dósa Z, Radi S, Strand K, Fröjd V, Hanse E. Tonically active NMDA receptors—a signalling mechanism critical for interneuronal excitability in the CA1 stratum radiatum. Eur J Neurosci. 2016;43:169–78.
Ortinski PI, Turner JR, Pierce RC. Extrasynaptic targeting of NMDA receptors following D1 dopamine receptor activation and cocaine self-administration. J Neurosci. 2013;33:9451–61.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Papouin, T., Oliet, S.H.R. (2017). Synaptic and Extra-Synaptic NMDA Receptors in the CNS. In: Hashimoto, K. (eds) The NMDA Receptors. The Receptors, vol 30. Humana Press, Cham. https://doi.org/10.1007/978-3-319-49795-2_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-49795-2_2
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-49793-8
Online ISBN: 978-3-319-49795-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)