
Abstract
Socioeconomic disadvantage (SED) has adverse impacts on physical (Adler and Rehkopf 2008; Blair and Raver 2012; Braverman and Egerter 2008; Cohen et al. 2010; Poulton et al. 2002) and psychological (Adler and Rehkopf 2008; Bradley and Corwyn 2002; Grant et al. 2003) health development. SED is similar to low socioeconomic status (SES) which is based on occupation, income, and education or a composite of more than one of these indicators (McLoyd 1998). However, we conceptualize SED more broadly than socioeconomic status to also include subjective perception of social position and contextual indicators of disadvantage, such as neighborhood deprivation. One of the most commonly used SED indicators is poverty, which is calculated using income that falls below an annually adjusted federal poverty line. Some studies suggest independent effects of different SED variables, but each variable tends to be highly correlated with the others; thus, it is often nearly impossible to disentangle independent effects. Therefore, in this chapter, we will review findings on different SED variables but discuss the impact of SED inclusively. In adulthood, SED is assessed based on factors in an individual’s own background such as income, occupation, and education level, whereas in childhood, SED is typically assessed based on these factors for the parents.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


1 Socioeconomic Disadvantages and Health Across the Lifespan
Socioeconomic disadvantage (SED) has adverse impacts on physical (Adler and Rehkopf 2008; Blair and Raver 2012; Braverman and Egerter 2008; Cohen et al. 2010; Poulton et al. 2002) and psychological (Adler and Rehkopf 2008; Bradley and Corwyn 2002; Grant et al. 2003) health development. SED is similar to low socioeconomic status (SES) which is based on occupation, income, and education or a composite of more than one of these indicators (McLoyd 1998). However, we conceptualize SED more broadly than socioeconomic status to also include subjective perception of social position and contextual indicators of disadvantage, such as neighborhood deprivation . One of the most commonly used SED indicators is poverty, which is calculated using income that falls below an annually adjusted federal poverty line. Some studies suggest independent effects of different SED variables, but each variable tends to be highly correlated with the others; thus, it is often nearly impossible to disentangle independent effects. Therefore, in this chapter, we will review findings on different SED variables but discuss the impact of SED inclusively. In adulthood, SED is assessed based on factors in an individual’s own background such as income, occupation, and education level, whereas in childhood, SED is typically assessed based on these factors for the parents.
SED is a widely recognized concern because long-lasting adverse effects on health have been robustly and consistently reported throughout the lifespan (Chen et al. 2007; Chen et al. 2002; Cohen et al. 2010; Evans and Kim 2010; Seeman et al. 2010) . With respect to psychological health development, exposure to SED has been associated with poor language, cognitive deficits, and behavioral problems during childhood (Blair and Raver 2012; Brooks-Gunn and Duncan 1997; Evans 2004; Evans and Kim 2013; McLoyd 1998). In adolescents, SED has been associated with higher rates of psychopathology including substance abuse, disruptive behaviors, and depression (Doan et al. 2012; Fauth et al. 2007; Goodman et al. 2005; Sariaslan et al. 2014). In adulthood, individuals with SED exposure are more likely to exhibit reduced productivity at work, increased involvement in crime, as well as higher rates of mental illness (Duncan et al. 2010; Lorant et al. 2003; Ross 2000).
SED has also been associated with physical health development. In infancy, SED is linked to higher infant mortality and negative birth outcomes (Brooks-Gunn and Duncan 1997; Metcalfe et al. 2011). In childhood and adolescence, SED is associated with higher risk for asthma, dental problems, and physical inactivity (Chen et al. 2004; Miller and Chen 2013). Individuals exposed to SED are more frequently sick, and they die early, with higher morbidity and mortality rates throughout adulthood. Adults exposed to SED have higher rates of cancer, cardiovascular disease, and diabetes (Seeman et al. 2001). Finally, childhood SED exposure has long-lasting impacts on both psychological and physical health development after accounting for adult exposure to SED (Cohen et al. 2010; Danese and McEwen 2012; Evans et al. 2012).
Elevated levels of stress have been identified as a primary mediating mechanism underlying the effects of SED on health development. Childhood SED such as family poverty is associated with greater exposure to stressors such as harsh parenting, exposure to violence, separation from the parent, lower school quality, negative peer relations, substandard housing, pollutants, noise, and crowding (Evans 2004). Neighborhood deprivation is associated with lack of access to physical and cultural resources such as fresh fruits and vegetables, open space and other recreational amenities, libraries, and transportation, as well as greater exposure to violence and crime (Meijer et al. 2012). Adulthood SED is also associated with financial hardship, perceived discrimination, lack of control and reward at work, and job insecurities (Ferrie et al. 2002; Matthews and Gallo 2011). Moreover, one of the distinct characteristics of being exposed to SED is that individuals with SED are far more likely to experience multiple stressors over long periods of time. Chronic and severe levels of stress exposure immobilize the individual’s ability to cope with stressors (Blair et al. 2011; Evans and Kim 2013; Wadsworth and Compas 2002).
2 Neurobiological Mechanisms of Stress Regulation
The next step in linking SED and health development is building an understanding of relevant neurobiological mechanisms. Chronic exposure to cumulative risk factors associated with SED can cause dysfunction in the brain and physiological systems, which in turn increases risks for psychological and physical illnesses. Neurobiological mechanisms of stress highlight three brain regions for their key roles in stress perception, appraisal, and regulation – the amygdala, hippocampus, and medial prefrontal cortex. These brain regions regulate the physiological stress systems, particularly the hypothalamus-pituitary-adrenal axis and autonomic nervous system , which lead to physical responses to stress such as “fight or flight” (Ulrich-Lai and Herman 2009).
2.1 Brain
The amygdala is a key region for detecting threats or potentially threatening information from the environment (Rodrigues et al. 2009). When an individual experiences actual or anticipated threats, the amygdala signals the paraventricular nucleus of the hypothalamus which then activates two primary physiological stress systems, the hypothalamus-pituitary-adrenal axis and the autonomic nervous system. Activation of the hypothalamus-pituitary-adrenal axis increases glucocorticoid (cortisol in humans) secretion and autonomic nervous system activation, including release of epinephrine and norepinephrine (Ulrich-Lai and Herman 2009). Long-term exposure to stressors perceived as stress can cause changes in microstructure and function of the amygdala, leading to neurogenesis and synaptogenesis (Davidson and McEwen 2012; Tottenham and Sheridan 2009). These changes may lead to hyperactivity of physiological stress regulation and increased behavioral reactivity to stress (Rodrigues et al. 2009).
The hippocampus is important for memory formation but also for stress regulation (Jacobson and Sapolsky 1991; McEwen 2001). While the amygdala rapidly activates the physiological stress system, the hippocampus plays a regulatory role by decreasing glucocorticoid secretion. This, in turn, leads to recovery of the hypothalamus-pituitary-adrenal axis activation to baseline. The hippocampus has a rich distribution of glucocorticoid receptors; therefore, its structure and function are particularly vulnerable to increased stress hormone level exposure. Exposure to chronic stress injures the microstructure and function of the hippocampus by retraction of apical dendrites and a reduction in spine density (McEwen 2001). The impact is further exacerbated because impaired hippocampal function reduces glucocorticoid negative feedback or regulatory functioning, which in turn leads to higher cortisol levels.
The prefrontal cortex is involved in excitatory and inhibitory physiological stress systems. The prefrontal cortex interacts with the amygdala and hippocampus, with the medial prefrontal cortex inhibiting both hypothalamus-pituitary-adrenal and autonomic stress responses by suppressing amygdala activation (Kalisch et al. 2006; Radley et al. 2006). On the other hand, the orbitofrontal cortex is involved in appraisal of emotional information and activates both hypothalamus-pituitary-adrenal and autonomic stress responses by increasing activity in the amygdala (Milad and Rauch 2007). Exposure to chronic stress impairs the medial prefrontal cortex through structural atrophy and suppression of neurogenesis while increasing dendritic connections of neurons in the orbitofrontal cortex (Davidson and McEwen 2012). Behaviorally, prefrontal cortex dysfunction contributes to difficulty in regulating emotional distress. Indeed, increased amygdala and orbitofrontal cortex activation and decreased hippocampal and medial prefrontal cortex activation are neural risk markers for many mental illnesses such as depression, anxiety, and antisocial behaviors (Coccaro et al. 2007; McEwen 2005; Mervaala et al. 2000; Shin et al. 2006). Similarly, abnormal brain morphometry typically assessed by gray matter volumes, cortical thickness, or white matter structure in the amygdala, hippocampus, and medial prefrontal cortex has also been associated with a range of psychiatric disorders (Phan et al. 2009; Price and Drevets 2012).
The three brain regions discussed above − amygdala, hippocampus, and prefrontal cortex – have been intensely examined in animal studies because of their role in stress regulation and vulnerability to chronic stress exposure. In humans, other brain regions also play an important role in responding to stress, and these too can be affected by chronic stress exposure. Rather than directly influencing human physiological stress responses, these brain areas communicate with and therefore influence the medial prefrontal cortex, hippocampus, and amygdala. The first of these neural networks is the lateral prefrontal cortex and anterior cingulate cortex, which are involved in executive function, decision-making, and self-regulation (Kalisch 2009; Kalisch et al. 2006; Ochsner et al. 2012). Activation in these regions suppresses amygdala activity by supporting cognitive reappraisal strategies and attentional control to help people cope with stressful situations. Chronic stress exposure has been shown to alter microstructure and function in this network, which contributes to difficulties in executive function (Holmes and Wellman 2009).
The second network of neural regions involved in human stress and copin g includes the insula, superior temporal gyrus, temporoparietal junction, and posterior cingulate cortex, brain areas which are centrally related to social cognition (Uddin et al. 2007). In humans, most everyday life experiences take place in social contexts, and these brain regions are involved in processing crucial social information such as others’ mental states, thoughts, and intentions. Language is also critical for communication during social interactions and likely contributes to coping processes such as cognitive appraisal. Therefore, brain regions involved in language production and comprehension, including Broca’s area, Wernicke’s area, and the angular gyrus, have also been examined by human stress researchers. Lack of cognitive and social stimulation has been associated with impaired functions in these networks (Adolphs 2001; Hackman and Farah 2009).
In sum, animal and human research consistently highlights the significant role of the amygdala, hippocampus, and medial prefrontal cortex in stress reactivity and regulation. In human research, these three key regions are interconnected with other neural networks integrally involved in higher-level thought, including the executive function network (the lateral prefrontal cortex and anterior cingulate cortex), the social cognition network (insula, superior temporal gyrus, and posterior cingulate cortex), and language regions (Broca’s area, Wernicke’s area, and the angular gyrus). The three key regions work together with these other networks to process information from positive and negative experience and decide appropriate responses.
2.2 Physiological Systems
When individuals are exposed to stress, information is initially processed and evaluated in the brain, which then activates the primary physiological stress systems, the hypothalamus-pituitary-adrenal axis, and autonomic systems. To activate the hypothalamus-pituitary-adrenal axis, signals from the amygdala cause the paraventricular nucleus of the hypothalamus to release corticotropin-releasing hormone and arginine vasopressin, which trigger secretion of adrenocorticotropic hormone from the pituitary gland. The adrenocorticotropic hormone then causes release of glucocorticoids, cortisol (stress hormone) in humans, by the adrenal cortex. Increased cortisol increases blood pressure and heart rate and stimulates anti-inflammatory and immunosuppressive actions.
The autonomic nervous system includes both the sympathetic and parasympathetic nervous systems . The sympathetic branch of the autonomic nervous system provokes immediate changes such as increased heart rate and blood pressure, representing a “fight-or-flight” response to threat. Neuroendocrine biomarkers for these systems include glucocorticoids (cortisol) for the hypothalamus-pituitary-adrenal axis and epinephrine and norepinephrine for the autonomic nervous system. Increased cortisol levels suppress inflammatory cytokines. Thus hypothalamus-pituitary-adrenal axis dysregulation can lead to inflammatory system dysregulation. Biomarkers of inflammatory system dysregulation include interleukin-6, tumor necrosis factor alpha, C-reactive protein, and insulin-like growth factor-1.
Dysregulation of hypothalamus-pituitary-adrenal axis and autonomic nervous system activation can also suppress and disrupt metabolic processes. Biomarkers of metabolic system dysfunction include low levels of high lipoprotein cholesterol and high levels of low-density lipoprotein cholesterol, triglycerides, glycosylated hemoglobin, glucose, and insulin. Additionally, adiposity as indicated by body mass index and waist-to-hip ratio are potentially downstream indicators of stress dysregulation. The primary stress response systems increase blood pressure and heart rates and thus elevate risk for cardiovascular disease. Biomarkers include systolic and diastolic blood pressure, as well as heart rate (Krantz and Falconer 1995).
The allostatic load model proposes that exposure to chronic stress may cause wear and tear in one or more primary stress regulatory systems (the hypothalamus-pituitary-adrenal axis and autonomic nervous system) and subsequently in secondary physiological stress systems (metabolic processes, inflammatory and immune responses, and cardiovascular responses). Compromised functioning across these multiple physiological systems from chronic environmental demands may lead to long-term damage. Dysregulation of multiple physiological systems, indexed by allostatic load, is a powerful predictor for health development outcomes and adults, including cardiovascular disease, diabetes, as well as cognitive impairment and premature mortality. SED has also been studied for links to allostatic load because SED increases exposure to repeated, severe, and chronic stressors, which elevate allostatic load over the lifespan (Gruenewald et al. 2012; Seeman et al. 2010).
3 Overview of SED and Neurobiological Impacts Over the Life Course
In this section of the chapter, we review current understanding of the effects of SED on neurobiological stress regulatory across the lifespan including (1) how childhood SED affects childhood neurobiological systems, (2) how adulthood SED affects adult neurobiological systems, and (3) how childhood SED affects adult neurobiological systems. Within each temporal focus, we review literature on the brain and physiological systems. Brain regions of interest include the hippocampus, amygdala, prefrontal cortex, and other related regions. We reviewed both structural and functional variations that have been associated with SED. Relevant physiological systems include the hypothalamus-pituitary-adrenal axis, sympathetic nervous system , cardiovascular responses, inflammatory and immune responses, and metabolic processes, as well as allostatic load . When available, the findings on the relations between the brain and physiological systems are also reviewed.
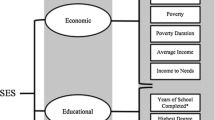
Following this overview of the literature on SED and neurobiological systems, we discuss the literature on life course health development principles (see Halfon and Forrest 2017). In particular, we discuss the multilevel and multidimensional interactions between neurobiological and environmental mechanisms underlying the associations between SED and health development. Finally, we discuss ways in which current scientific understanding of the effects of SED on human biology provides information for interventions and policy from a life course health development perspective (Figs. 1 and 2).

Conceptual mod el for chapter

A summary of socioeconomic disadvantages (SEDs) and neurobiological mechanisms across lifespan (SEDs socioeconomic disadvantages; PFC prefrontal cortex, SNS sympathetic nervous system; HPA hypothalamus-pituitary-adrenal)
4 Socioeconomic Disadvantages (SEDs) and Neurobiological Mechanisms During Childhood
4.1 Brain : Structure
There is a fast-growing body of literature on the effects of SED on the developing brain suggesting that childhood SED has negative impacts on brain development from infancy to late adolescence. We will discuss recent literature on the development of both brain structure and function in the context of SED exposure in the hippocampus, amygdala, and prefrontal cortex.
In the hippocampus, SED is associated with alterations in its structure across developmental stages (Hanson et al. 2011; Noble et al. 2015). Importantly, these effects have been documented not only in the USA but internationally as well (Jednorog et al. 2012). In a longitudinal study, family income was inversely associated with bilateral hippocampus gray matter volumes in children at age 9. This link was mediated by exposure to stress, both caregiving quality and stressful life events, assessed 3 years earlier (Luby et al. 2013). Another study suggests that reduced hippocampal volume is associated with increased externalizing of behavioral problems (Hanson et al. 2014). The reduced volumes in the hippocampus mediated the links between early life adversity and behavioral problems at school including problems with teachers and disruptive behaviors (Hanson et al. 2014).
Amygdala structure has been associated with major chronic stressors in early life such as maternal deprivation (Tottenham and Sheridan 2009). However, the patterns of the associations between early adversity and amygdala structure appear to be more complex compared to the patterns found in the hippocampus. Among children in middle childhood and early adolescence, SED was associated with reduced amygdala gray matter volumes (Hanson et al. 2014; Luby et al. 2013), whereas among children in middle childhood to late adolescence, SED was associated with increased amygdala gray matter volume (Noble et al. 2012). Others have failed to demonstrate the links between SED and amygdala structure (Hanson et al. 2011). More developmental research is needed to identify what SED factors may be associated with different developmental trajectories of the amygdala. Consistent with life course health development principles (see Halfon and Forrest 2017), work to date suggests that both timing and duration of SED exposure are critical for understanding SED impacts on amygdala structure and function.
The prefrontal cortex and other regions for language and social cognition are affected by exposure to SED. Only one study has examined the effects of family income on brain structure in the first years of life (Hanson et al. 2013). Longitudinal analyses revealed that infants from low-income families had lower gray matter volumes in the frontal and parietal lobes compared to their counterparts from mid- and high-income families. Moreover, the low-income infants had more dampened brain growth trajectories compared to mid- and high-income infants: although the brain volumes were comparable during the first year of life, by 3–4 years of age, low-income children showed significantly reduced volumes compared to high- and mid-income children. The reduced volume in the frontal lobe, a brain area involved in cognitive control, was associated with more externalizing behaviors, but not with internalizing behaviors. In another study with children aged 4–18, SED based on income and education was associated with cortical thinning in the prefrontal cortex, particularly in the right anterior cingulate cortex and left superior frontal gyrus, regions that are important for behavioral regulation and executive functions (Lawson et al. 2013). This study also provided evidence that changes due to stress exposure are not linear based on age. In the low SES group, from ages 5–17, gray matter volumes decreased in the inferior and superior temporal gyri, areas involved in language development and cognitive controls. On the other hand, in the high SES group, gray matter volumes in these areas increased over time (Noble et al. Noble et al. 2012b). Given that the pruning process occurs mid- to late adolescence (Giedd 2004), the rapid decline in gray matter volume in the low SES group may limit normal development in the prefrontal cortex. This may be further associated with compromised cognitive functions. In middle childhood, SED was linked to reduced gyrification in the medial prefrontal cortex, indicating delayed maturation (Jednorog et al. 2012).
Finally, a recent study demonstrated a robust association among SED, cortical structure, and cognitive function throughout childhood (age 3–20) (Noble et al. 2015). In the sample of 1099 participants, family income was associated with increased cortical surface area as well as thickness. The gradient relations between family income and cortical surface area were quadratic so that the children of the lower-income families were more affected by income changes compared to the children of the higher-income families. The differences in the cortical surface explained the lower neurocognitive abilities among low-income children. Similar findings that support the role of cortical structure differences in the income-achievement gap were recently reported in a smaller sample of adolescents (Mackey et al. 2015).
4.2 Brain: Function
Existing evidence suggests the effects of SED on whole-brain functioning can start very early in life. A longitudinal study demonstrated that both income and maternal education were positively associated with maturity of the sensorimotor network (primary sensorimotor/auditory) and default mode network at 6 months of age (Gao et al. 2014). At 6 months, babies can sit up and interact more extensively with their environment; therefore, these brain networks may support sensorimotor exploration and a sense of agency. Delayed development in these networks due to exposure to SED during the first year may portend adverse long-term impacts on child health development.
The impact of childhood SED on prefrontal cortex functioning and its role in cognitive development have been examined extensively in the literature, whereas the effects on hippocampal and amygdala functions are less understood. One study focused on the associations between SED and hippocampal function and included both objective measures of SES and maternal subjective ratings of social standing (Sheridan et al. 2013). Interestingly, perceived social standing, but not income or education, was associated with hippocampal function. Among children 8–12 years old, higher perceived social standing was associated with greater hippocampal activation during a memory encoding task. This finding highlights the importance of examining subjective and objective social standing in relation to brain development over the life course.
The role of SED exposure in amygdala functioning is not well understood. In adolescents, low SES was associated with increased amygdala responses to angry faces (Muscatell et al. 2012), which suggests greater neural sensitivity to negative emotional information in social contexts. In a longitudinal study, early adversity including low family income in infancy was associated with reduced functional connectivity between the amygdala and prefrontal cortex in female adolescents (Burghy et al. 2012). This reduced connectivity, which may be related to difficulties in emotion regulation, was further associated with higher anxiety symptoms.
The impact of SED exposure on the prefrontal cortex and other neural regions associated with cognitive functions has been detected during the first few years of life. Six- to eight-month-old infants from low-income families exhibited reduced gamma band power in the frontal lobe compared to infants from high-income families, after controlling for several covariates such as exposure to smoke (Tomalski et al. 2013). Reduced gamma power may be an early risk marker for poor attentional control and delayed language development, both of which are more prevalent in low SES children (Hackman and Farah 2009; Hoff 2006). This link between SES and frontal lobe development was found in older children as well. Low SES was associated with less mature EEG activity patterns in the frontal lobe across ages 4–6 (Otero et al. 2003; Otero 1997).
In middle childhood, SED exposure was associated with prefrontal cortex activity and executive functioning. Children 7–12 years old performed a target detection task designed to probe cognitive flexibility and working memory, which is related to lateral prefrontal cortex activation (Kishiyama et al. 2009). Lower SES children showed reduced prefrontal cortex activation compared to children of high SES families, indicating delayed prefrontal cortex development and increased risks for delay in executive function. Sheridan et al. (2012) examined the link between SED and neural activation for executive functioning among 8–12-year-old children (Sheridan et al. 2012). Interestingly, low SES children showed greater, rather than reduced, activation in right middle frontal gyrus compared to high SES children. Although there were no behavioral performance differences by SES, in low SES children, increased activation in the right middle frontal gyrus was associated with better accuracy. Therefore, low SES children may have to recruit this region more strongly than high SES children in order to perform at the same level. Additionally, low SED adolescent girls exhibited increased activation in the anterior cingulate cortex during an inhibition control task, which indicated ineffective recruitment of the brain region for behavioral performance (Spielberg et al. 2015).
Neural activations for attention control are also associated with SED exposure in children. In both early childhood (Stevens et al. 2009) and adolescence (D’Angiulli et al. 2008; D’Angiulli et al. 2012), low SES children showed reduced responses to target stimuli and increased responses to distractor stimuli. This pattern of neural activation may contribute to poor suppression of responses to distracting information and less filtering of irrelevant information. In adolescence, low SES was also associated with noisier and inefficient neural responses to auditory stimuli compared to mid-SES (Skoe et al. 2013). Such altered neural responses may require low SES children to exert more effort in order to perform at the same levels as their high SES counterparts, which suggests that SED reduces the overall adaptability and threatens the long-term heath development of these children.
Given consistent evidence of delayed language development in low SES children, the impact of SED exposure on brain regions involved in language development has been examined. Among kindergarteners, neural activations during a phonological awareness task of discriminating rhymed monosyllabic words versus nonwords were assessed (Raizada and Kishiyama 2010). Phonological processing is a critical, underlying process in reading acquisition. Children with high family SES exhibited left inferior frontal gyrus specialization, which is important for language development, while children of low family SES were less likely to exhibit such specialization. On the other hand, in middle childhood (ages 7–11), low SES background was associated with increased, not reduced, activations in left fusiform regions during a reading task, which was further associated with better phonological skills within the low SES group (Noble et al. 2006). Fusiform activation is thought to support visual word recognition, but it is not typically associated with effective phonological skills. Middle SES background was associated with increased activation in the perisylvian and superior frontal regions, typical activation regions for language skills. However, among low SES children, perisylvian and superior frontal region activation was less likely to be shown during a reading task. These results show complex associations between brain activations and development in the context of SED.
4.3 Physiological Systems
The sympathetic nervous system is commonly assessed by resting blood pressure, blood pressure reactivity, and blood pressure recovery in response to an acute stress exposure in children and adolescents. In children, SED is associated with dysregulation of the sympathetic nervous system marked by elevated resting blood pressure and heightened blood pressure reactivity to stress (Chen et al. 2004; Gump et al. 2007; Jackson et al. 1999; Kapuku et al. 2002).
On the other hand, in adolescence, the associations between SED and blood pressure are not consistent or in the opposite direction from childhood. Some researchers have failed to find a consistent association between SED and resting blood pressure during adolescence (Chen et al. 2002). A particularly interesting longitudinal study showed that maternal education was inversely related to childhood resting blood pressure at ages 7 and 9 years, but then had no association among 11- and 15-year-olds (Howe et al. 2013). When exposed to an acute stressor, SED adolescents exhibit dampened rather than elevated blood pressure reactivity (Evans and Kim 2007; Musante et al. 2000). Other studies have failed to reveal any association between SED and blood pressure reactivity in adolescents (Chen et al. 2002). One reason for less consistent findings with blood pressure in adolescence is an increased significance of peer and school environment relative to the family environment during adolescence. For example, neighborhood SED but not family SED was significantly associated with elevated ambulatory blood pressure in adolescents (McGrath et al. 2006). Although it is not consistently found, researchers identified slower blood pressure recovery – blood pressure taking longer to come back to baseline – after lower SES adolescents were exposed to acute stress (Evans et al. 2007; Walter and Hofman 1987). Epinephrine, a hormone that regulates the sympathetic nervous system , was elevated overnight among 9-year-olds exposed to SED (Evans and English 2002; Evans and Kim 2007). SED also reduced expressions of genes regulated by catecholamines in children with asthma, suggesting dysregulation of the sympathetic nervous system (Chen et al. 2009).
Cortisol , a hormone marker of the hypothalamus-pituitary-adrenal axis, is dysregulated in children and adolescents exposed to SED. SED is associated with elevated baseline levels of cortisol during childhood (Essex et al. 2002; Evans and English 2002; Flinn and England 1997; Gustafsson et al. 2006; Lupien et al. 2001; Lupien et al. 2000) and adolescence (Evans and Kim 2007) although Lupien et al. (2001) found that by age 12, SES is no longer related to baseline cortisol levels. The longer children experience poverty, the higher the cortisol level (Blair et al. 2013). In a particularly noteworthy study, Chen et al. (2010) examined salivary cortisol every 6 months for a 2-year period among a group of adolescents. Saliva readings were taken throughout the day. Overall levels were higher among youth from low-income families and, of particular interest, rose faster as the adolescent matured. Children exposed to SED in middle childhood also exhibited heightened reactivity to an acute stressor (Gump et al. 2009). Similar to the findings on blood pressure, neighborhood SES predicted higher cortisol reactivity in adolescent boys (Hackman et al. 2012). These same authors, however, also found a more rapid return of cortisol levels to baseline in relation to residence in more deprived neighborhoods: a result opposite to what one would expect.
Research also suggests that alleviation of SED can improve hypothalamus-pituitary-adrenal axis regulation in early childhood . Fernald and Gunnar (2009) evaluated the impacts of a conditional cash transfer program on assessments of cortisol during a visit to children’s homes. In this experimental program, low-income families are randomly assigned to receive cash in exchange for compliance with better health care for their child (e.g., nutrition supplements, physicals, child health education) plus more school attendance, or they are enrolled in a control group. The amount of cash transfer is considerable, averaging between 20 and 30% of a household’s annual income. The results showed that primary school-aged children in the income supplement program had lower overall levels of salivary cortisol if they also had a depressed mother. For low-income families in which the mother was not depressed, there were no impacts of additional income on cortisol levels.
Higher obesity rates are found in children exposed to SED. Obesity is associated with malfunctioning of the metabolism system. Metabolic dysregulation is most commonly indexed by high levels of lipids, low glucose tolerance, and high body mass index (BMI) which is an indicator of adiposity. Children exposed to SED show higher BMI (Shrewsbury and Wardle 2008; Sobal and Stunkard 1989). In an interesting study, Hargreaves et al. (2013) examined childhood obesity (≥ 95th% for age and gender) using four different UK national birth cohorts to illustrate secular trends from 1999 to 2009. Occupational level of the head of household was inversely related to obesity, and the degree of inequality rose 25% over the decade. Some researchers find that the link between SED and adiposity is stronger in younger children, females, and Caucasian children. It is also worth noting that when excess adiposity is defined in terms of obesity (typically >95th percentile for age and gender) rather than by actual BMI, the SES trends are stronger and more consistent.
When metabolic dysregulation was examined across childhood and adolescence, exposure to SED from birth to age 9 years was associated with high BMI at age 9 and also a steeper trajectory of weight gain over the next 8 years (Wells et al. 2010). Howe et al. (2013) tracked the same birth cohort over time and found that whereas there were no differences in BMI in relation to SES prior to age 4, around 60 months of age, clear SES differences emerged and continued to widen up to age 10, the end of their study.
A smaller number of studies have examined weight at different ages during development in relation to childhood SES. Kakinami et al. (2013b) took these early life course analyses a step further by generating poverty trajectory classes from birth to obesity assessments at ages 6, 8, 10, and 12 years among a large, representative sample of children from the province of Quebec. They found four classes: always poor, never poor, increased likelihood of poverty over time, and decreased likelihood of poverty over time. The largest differences occurred between the stable poor and the stable, nonpoor groups, but the effects took time to emerge. For age- and gender-adjusted BMI, the stable poverty group had higher BMI percentiles than the stable, nonpoor group at ages 10 and 12 years. Thus, at 6 and 8 years old, there was no link between duration of early poverty and BMI. For the likelihood of being overweight or obese (≥BMI 85th %), differences emerged by age 8 and continued through to age 12. Two other aspects of their data are noteworthy in thinking about health development. First, the older the child, the greater the impact on weight. Second, they also found elevated risk of greater weight in the group with decreased likelihood of poverty over time (i.e., started out poor but became nonpoor) relative to the stable nonpoor group, but did not find this for the opposite trajectory where children started out nonpoor but became poor. Thus, there is evidence that early poverty exposure has long-lasting impacts, regardless of subsequent upward mobility even among children. All of these effects remained with statistical controls for current income, birth weight, and parental weight.
The effects of SED may begin very early in life. Klebanov et al. (2014) showed that lower-income households have lower-weight infants at birth, a common finding in the literature on SES and birth weight (Spencer 2000). However, Klebanov and colleagues then showed that weight gain was faster in infants from low-income households in comparison to those from middle-income households. Low birth weight babies are at risk for more rapid weight gain because of maternal efforts to help the baby become healthier, leading to overfeeding as well as biological compensatory mechanisms within the baby herself that alter metabolism in order to gain weight.
Glucose metabolism is reduced in children exposed to SED, which contributes to the risk for obesity and metabolic system dysregulation. Several SED variables have been associated with dysregulated glucose metabolism. The National Health and Nutrition Examination Survey (NHANES) data, a nationally representative US dataset, revealed family income was inversely associated with glycosylated hemoglobin levels among children from age 1 to 19 (Seeman et al. 2010). Low parental education was also inversely associated with several metabolic markers including total cholesterol and the ratio of low- to high-density lipoproteins (Leino et al. 1996). Neighborhood deprivation was also associated with dysregulation of the metabolic system including higher low-density lipoproteins, triglycerides, as well as body fat among children aged 9–15 (Gliksman et al. 1990). Kakinami et al. (2013a) conducted a longitudinal study among children beginning at birth until age 10 years old. Poverty exposure was defined in terms of both timing (birth to age 2, 3–6, 7–10 years) and duration (number of age periods [0–3] with poverty exposure). Poverty timing and duration were each linked with elevated triglycerides and insulin with in-depth controls for a host of sociodemographic and physical variables, including family histories of pertinent diseases. Interestingly, for triglycerides, the duration of poverty exposure was most important, whereas for insulin, timing mattered, with exposure from birth to two most consequential. High-density lipids were also reduced by poverty exposure but once the covariates were included in the model, this effect was no longer significant.
Children exposed to SED exhibit elevated inflammatory responses. Chen and colleagues (Chen et al. 2003; Chen et al. 2006) examined the role of childhood SED exposure in inflammation and immune responses particularly among children with asthma. Asthma has a strong association with SED exposure, and the risk of asthma is related to dysregulation of inflammatory response. Among children and adolescents with asthma, low neighborhood SES was associated with increased cytokine responses to allergens (Chen et al. 2003; Chen et al. 2006). On the other hand, in healthy children, no association between neighborhood SES and inflammatory responses was found. Chronic exposure to low SES across ages 1–9 was associated with increased immune responses measured by interleukin-6 production, but a trajectory of initially low SES followed by upward mobility was associated with normal immune responses (Azad et al. 2012).
Inflammatory responses are regulated by gene expression of T cells . Among children with asthma, SED was associated with overexpression of T-cell genes that are involved in chemokine activity and responses to wounds (Chen et al. 2009). This was due to decreased activity of the transcription factors cyclic AMP-binding protein, AP1, and nuclear factor Y as well as increased activity of genes regulated by the proinflammatory transcription factor, nuclear factor kappa-B. Such findings suggest gene and inflammatory pathways linking SED and immune dysfunction among children. Childhood SES was further associated with the genetic mechanisms of inflammatory responses. Low family SES was associated with increased levels of messenger RNA for TLR4, a receptor involved in inflammatory response regulation in adolescence (ages 13–19) (Miller & Chen 2007b). The higher levels of TLR4 suggest leukocyte inflammatory responses to endotoxins, thus greater susceptibility to pathogens such as the cold virus. In addition to objective SED indicators, perceived level of SED was associated with higher proinflammatory biomarkers, IL-6, and TNF-α soluble receptor 2 (sTNFR2) in white, but not black, adolescents (Pietras and Goodman 2013).
When these different physiological systems are considered together in a cumulative fashion, SED is associated with higher allostatic load , indicating chronic wear and tear on physiological stress regulatory systems. Evans and colleagues demonstrated that exposure to chronic stressors including poverty is longitudinally associated with higher allostatic load in middle childhood and early adolescence (Evans 2003; Evans et al. 2007). Longer periods of living in poverty since birth was also associated with high allostatic load at age 13 (Evans 2003; Evans et al. 2007). Although literature on SED and allostatic load in childhood is limited, others have also uncovered significant links between SED and allostatic load in children and adolescents (Goodman et al. Goodman et al. 2005b). In adolescence, neighborhood poverty more significantly predicted higher allostatic load than family poverty (Brody et al. 2014). Using the nationally representative data of the NHANES, researchers also showed that the link between SED and allostatic load is greater in African Americans compared to white and Hispanic adolescents (Rainisch and Upchurch 2013). However, the relative advantages in the white and Hispanic adolescents became smaller as they grew older. The evidence on separate stress systems and allostatic load reveal mechanisms by which SED alters the physiological stress regulatory systems, which may further diminish the ability to cope with stress and increase risks for physical and mental illnesses.
5 SED and Neurobiological Mechanisms During Adulthood
5.1 Brain: Structure
The impact of socioeconomic disadvantage on adult brain structure has been researched less than in children. Existing studies suggest that disadvantages in adulthood are associated with aberrant structure in brain regions involved in language, executive functions, and emotion regulation. It should be noted, however, that while adults living in SED are more likely to have experienced similar disadvantages in childhood, most studies do not control for childhood SES or the trajectories of SED that span childhood, adolescence, and adulthood. Therefore, it is difficult to disentangle the timing effects of SED when understanding the effects on brain and physiological systems. Herein, noting this limitation, we review studies examining the cross-sectional association between adult SED and neurobiological dysfunctions in adulthood.
Exposure to disadvantages, particularly low educational attainment, has consistently been associated with reduced hippocampal volume. Among cognitively healthy seniors, low SES was associated with impaired neural anatomy including reduced white matter integrity in the hippocampus (Piras et al. 2011; Teipel et al. 2009). Among individuals from a wide age range (18–87 years), fewer years of education was also associated with a smaller hippocampus (Noble et al. Noble et al. 2012a). The association between SED and hippocampus structure starts at age 35, showing a decline in this area. Evidence of associations between adult SED and amygdala structure is limited, but SED was related to smaller amygdala structure in adulthood (Noble et al. Noble et al. 2012a), which may also be associated with mood dysregulation among adults exposed to SED.
SED exposure has also been linked to changes in regions involved with cognitive functioning. Among men aged 35–64, high neighborhood deprivation was associated with smaller cortical volume and thinning in the language-related Wernicke’s and Broca’s areas, as well as smaller cortical surface area in the frontoparietal regions, which are implicated in cognitive control (Krishnadas et al. Krishnadas et al. 2013b). In a sample of more than 300 brains of seniors (average age of 76), low educational attainment was associated with reduced gray matter volumes in the left temporoparietal area and in the bilateral orbitofrontal cortex (Foubert-Samier et al. 2012). In young adult participants, low subjective ratings of social standing using the MacArthur Scale of Subjective Social Status were associated with smaller gray matter volumes in the anterior cingulate cortex, but not in the hippocampus and amygdala (Gianaros et al. 2007). Standard SES, based on objective measures of income, education, and occupation, was not associated with gray matter volume in the anterior cingulate cortex.
The structural integrity of the brain in the context of SED has also been examined. Healthy adults aged 30–50 years were examined for the effects of SED on the fractional anisotropy of white matter tracts, which represents the integrity of neural circuits across different regions (Gianaros et al. 2013). All three SED indicators – education, income, and neighborhood quality – were associated with reduced white matter integrity, assessed by the fractional anisotropy, reflecting axonal caliber, myelin thickness, or fiber coherence (Jung et al. 2010) throughout the brain. In another study, neighborhood deprivation alone was also associated with a less efficient and noisier neural network among men aged 35–64 (Krishnadas et al. 2013a). When both young adults and elders were compared, education attainment was not associated with brain structure in young adults but was positively associated with white matter integrity in seniors age 65 and above (Johnson et al. 2013). Reduced white matter integrity in the bilateral superior frontal gyrus was further associated with poor working memory performance in seniors with low educational attainment.
Several studies examined the role of physiological systems in the link between SED and the adult brain. In particular, the role of inflammation dysfunction has been highlighted as a mediator of the relations between SED and brain. The inflammation factor mediates the links between neighborhood deprivation and reduced cortical thickness in key language regions, including the left Wernicke’s area (Krishnadas et al. Krishnadas et al. 2013b). Inflammatory dysfunction (C-reactive protein) and metabolic dysregulation (adiposity) were also examined as a physiological risk marker of cardiovascular diseases. Inflammatory factors mediated the link between SED and adiposity, and adiposity was further associated with reduced integrity of neural circuits (Gianaros et al. 2013).
5.2 Physiological Systems
There is relatively robust evidence on sympathetic nervous system dysregulation in conjunction with adult SED exposure. Several studies have shown that SED is associated with higher resting blood pressure. High resting blood pressure contributes to greater risks for cardiac diseases from young to late adulthood. In developed countries like the USA, Canada, and the UK, evidence is stronger and more consistent than in developing countries (Colhoun et al. 1998; Kaplan and Keil 1993). Neighborhood SED is also associated with higher resting blood pressure among adults in many studies (Chaix et al. 2010; Chaix et al. 2008; Cozier et al. 2007; Cubbin et al. 2000; Diez-Roux et al. 2000; Harburg et al. 1973; McGrath et al. 2006).
Several other markers of sympathetic nervous system dysfunction have been found among adults exposed to SED. SED was associated with dampened blood pressure reactivity to stress (Steptoe et al. 2002a; Steptoe et al. 2003) and reduced heart rate variability (Hemingway et al. 2005; Sloan et al. 2005). Individuals exposed to SED are also less likely to exhibit the normal overnight dip in blood pressure, which is a risk marker for cardiovascular diseases (Spruill et al. 2009; Stepnowsky et al. 2004). One intriguing explanation for these nighttime effects could be sleep disturbance, which appears to be more common among lower SES adults (Buckhalt 2011). In addition, SED was associated with higher norepinephrine and epinephrine levels among adults (Cohen et al. Cohen et al. 2006a; Janicki-Deverts et al. 2007; Seeman et al. 2004).
Cortisol, a hormone index of hypothalamus-pituitary-adrenal axis activation, has a natural diurnal rhythm throughout the day. The level is highest immediately after awakening, and then it sharply decreases and continues to decrease until it gradually increases overnight. Hypothalamus-pituitary-adrenal axis dysregulation is indicated by lower levels of cortisol awakening response and dampened and slower decreases during the day. This has been demonstrated in several developed countries, across different ages of adulthood. Lower levels of awakening cortisol and slower decline during the day both result in overall elevated levels of cortisol in the system (Cohen et al. 2006; Cohen et al. 2006a; Li et al. 2007). SED-exposed adults are more likely to exhibit these circadian patterns (Adam and Kumari 2009; Brandtstädter et al. 1991), indicating dysregulation of the hypothalamus-pituitary-adrenal axis. In the context of low SES, compared to younger adults, older adults exhibited higher circadian levels overall (Karlamangla et al. 2013). In addition, when cortisol level was assessed using overnight urinary sampling, the accumulated level of cortisol was higher in SED-exposed adults compared to non-SED-exposed adults (Gruenewald et al. 2012).
Extensive evidence shows that SED is associated with elevated levels of inflammation among adults. Low socioeconomic status variables such as low education attainment, lower class occupation, and low income have been associated with higher levels of C-reactive protein, interleukin-6, and fibrinogen (Alley et al. 2006; Brunner et al. 1996; Brunner et al. 1997; De Boever et al. 1995; Gruenewald et al. 2009; Hemingway et al. 2003; Ishizaki et al. 2000; Jousilahti et al. 2003; Kivimaki et al. 2005; Koster et al. 2006; Loucks et al. 2006; Lubbock et al. 2005; McDade et al. 2006; Owen et al. 2003; Panagiotakos et al. 2004; Petersen et al. 2008; Pollitt et al. 2007, 2008; Rathmann et al. 2006; Steptoe et al. 2003; Steptoe et al. 2002; Wamala et al. 1999; Wilson et al. 1993). Neighborhood poverty is also associated with higher levels of C-reactive protein and interleukin-6 (Gallo and Matthews 2003). Markers for long-term upregulation of proinflammatory cytokines including tumor necrosis factor-α and interleukin-1 receptor antagonist and hemostatic factors such as factor VII, von Willebrand factor, and plasminogen activator inhibitor-1 have also been identified among SED individuals (Steptoe et al. Steptoe et al. 2002b; Wamala et al. 1999). Elevations in inflammatory markers suggest poor immune response regulation, which increases the risk for greater inflammatory reactions to pathogens and irritants and thus portends eventual morbidity (Gruenewald et al. 2012).
Consistent findings on SED and increased markers of metabolic malfunction, higher fasting/postload glucose, and higher fasting triglycerides were shown in adults with SED (Brunner et al. 1997; Matthews et al. 1989). Higher levels of low-density lipoproteins and lower levels of high-density lipoproteins have also been found in SED adults relative to their more advantaged peers (Matthews et al. 1989; Wamala et al. 1997). Not surprisingly, SED adults have higher rates of diabetes (Everson et al. 2002; Guize et al. 2008; Lidfeldt et al. 2007; Maty et al. 2005; Maty et al. 2008) and other metabolic illnesses including hypertension, obesity, dyslipidemia, and hyperglycemia (Brunner et al. 1997; Chichlowska et al. 2009; Loucks et al. 2007; Wamala et al. 1999).
Taken together, there is evidence for links between SED exposure and physiological stress dysregulation across the adult lifespan. Such evidence is reflected in the association between SED and allostatic load in adulthood. Both individual SED variables such as occupation, income, and education level as well as neighborhood SED exposure have been associated with elevated allostatic load (Bird et al. 2010; Crimmins et al. 2009; Geronimus et al. 2006; Karlamangla et al. 2005; Merkin et al. 2009; Schulz et al. 2012; Seeman et al. 2008; Singer and Ryff 1999). Higher allostatic load predicts elevated morbidity and mortality (Juster et al. 2011; Juster et al. 2010).
6 Childhood SED and Adult Neurobiological Mechanisms
6.1 Brain : Structure
When the effects of childhood SED exposure on adult hippocampus and amygdala were examined, mixed results were reported. It is important to note that in order to be confident that the relationship between childhood SED and adult neural alterations is not simply reflecting concurrent SED status, these life course studies should examine the association of childhood SED and adult brain independently of adult SED. In a sample of over 200 68-year-old adults in England, childhood exposure to SED at age 11 years was prospectively associated with reduced hippocampal volumes after controlling for adulthood exposure to SED at age 65 (Staff et al. 2012). However, another study provides evidence for the opposite result. Among 403 adults in Australia, aged 44–48 years, the impacts of self-reported childhood poverty exposure as well as financial hardship during the past 12 months on hippocampal and amygdalar volumes were examined (Butterworth et al. 2012). Analysis revealed that the current financial hardship, but not childhood poverty exposure, was associated with reduction in gray matter volumes of bilateral hippocampus and amygdala. However, it should be noted that the study sample was predominantly mid- and high-SES individuals and few individuals from low SES were included. The differences between the two studies may be related to the variance of childhood adversity included in the study samples.
Studies examining the prefrontal cortex and other cortical regions demonstrate that stronger evidence exists for the effects of childhood exposure, rather than adulthood exposure, to SED. In a longitudinal study in Germany, brain structure of 134 young adults was assessed. Early life poverty based on family income at 3 months of age was associated with reduced orbitofrontal gyrus gray matter volume at age 25 (Holz et al. 2015). The reduced orbitofrontal cortex volume further mediated the link between early life poverty and conduct disorders. Exposure to current poverty did not predict the orbitofrontal gyrus volume. Other studies have not directly tested the timing effect of childhood SED by controlling for adulthood SED; however, they suggest that childhood SED contributes to reduced structure in the prefrontal cortex (Cohen et al. Cohen et al. 2006b; Narita et al. 2010).
6.2 Brain: Function
The effects of childhood SED on the adult brain are better understood in relation to socio-emotional information processing as compared to cognitive processes. This is different from the literature on the effects of SED in adulthood, which is primarily focused on cognitive functions.
Adults who were exposed to childhood SED exhibited greater neural reactivity to negative emotional information in the amygdala, which may indicate elevated stress reactivity. Low subjective ratings of parental social standing, a retrospective report on childhood SED, were associated with greater amygdala response to angry faces, after controlling for adult SES (Gianaros et al. 2008). Adult SES did not predict amygdala response. Lower social status in childhood may be associated with more negative social interactions such as family conflicts and neighborhood adversity which may lead to greater amygdala sensitivity to angry faces. Childhood SED has also been prospectively associated with neural functions for emotion reactivity and regulation in adulthood. In one study, 49 young adults were asked to regulate negative emotions in response to negative images using cognitive strategies (Kim et al. 2013). Family income at age 9 significantly predicted reduced dorsolateral prefrontal cortex and ventrolateral prefrontal cortex activation during emotion regulation. These results were independent of concurrent adult income. Family income at age 9 years was also positively associated with amygdala activation during emotion regulation, which may suggest inefficient suppression of the amygdala for emotion regulation by dorsolateral prefrontal cortex and ventrolateral prefrontal cortex. Moreover, chronic exposure to stress such as family conflicts, violence, and substandard housing quality mediated the links between family income at age 9 and ventrolateral prefrontal cortex and dorsolateral prefrontal cortex activations.
Effects of childhood SED on brain functions for emotion regulation in a social context were also examined in adults. In young adults, low childhood SES, retrospectively reported, was associated with greater distress and reduced right ventrolateral prefrontal cortex activation in response to social exclusion (Yanagisawa et al. 2013). In another study with young adults, low neighborhood quality at age 13 was associated with greater activations in dorsomedial prefrontal cortex, superior frontal gyrus, and dorsal anterior cingulate cortex during social rejection (Gonzalez et al. 2014). These increased neural activations among individuals from SED backgrounds suggest a need for greater recruitment of these brain regions in order to regulate emotions during social rejection.
Individuals exposed to childhood SED showed neural markers of greater reactivity to negative information and difficulties in regulating emotional distress. They also showed neural indicators for difficulties processing positive information. In a study of African American young adults, individuals were divided into low- and high-SES groups based on both their childhood and current SES (Silverman et al. 2009). Low SES was associated with reduced brain responses to positive pictures (compared to neutral pictures) in insula, anterior cingulate cortex, and caudate compared to high SES. Such reduced neural responses to positive stimuli can be risk factors for depression (Epstein et al. 2006). Childhood exposure to SED was also associated with abnormal neural responses to social support. Low neighborhood quality at age 13 was associated with increased orbitofrontal cortex and insula activation to threats while holding a close friend’s hand (Coan et al. 2013), indicating that individuals with early SED experience did not benefit from social support when coping with stress and threats .
6.3 Physiological Syste ms
The long-lasting effects of childhood SED on the physiological stress systems have been shown in several studies. In the sympathetic nervous system function, low paternal education in childhood was associated with greater blood pressure reactivity to stress (Williams et al. 2008). Low childhood SES was associated with elevated blood pressure reactivity to stress in men but not in women (Taylor et al. 2004b), suggesting the link may be stronger in men. When adolescents experienced both low parental education and low neighborhood SES, SED exposure was also associated with greater reactivity among African Americans as compared to their white counterparts (Wilson et al. 2000). Finally, low SES in childhood is also associated with elevated resting blood pressure in adulthood (Hardy et al. 2003; Lehman et al. 2009). Using data from a nationally representative US longitudinal study, middle-aged adults who were from lower-income families measured during pregnancy through age 2 years had higher incidence of hypertension (Ziol-Guest et al. 2012) independently of concurrent income at middle age. Another important contribution of this study is the authors also had income data for ages 3–5 years and between 6 and 15 years. Only the earliest childhood period of poverty exposure made a difference for middle-aged hypertension. Both Hardy et al. (2003) and Lehman et al. (2009) also showed that increases in adult blood pressure with aging occurred faster among those who grew up in low SES households. In an unusual multigenerational study, adolescents whose parents grew up in a low SES household revealed a pattern of greater elevations in resting blood pressure over a period of one and a half years (Schreier and Chen 2010). Note that SED was measured in terms of their grandparents’ SES levels. Thus, not only is resting blood pressure in adults and adolescents elevated by early experiences of SED, but blood pressure rises faster as individuals from low SES backgrounds age. This is a potentially critical finding in thinking about health development and policies to mitigate the effects of childhood poverty.
Childhood SED also influences hypothalamus-pituitary-adrenal axis regulation in later life. Middle-aged men and women exhibited greater awakening cortisol levels when exposed to low SES in adolescence, independent from their adulthood SES (Gustafsson et al. 2010a). Among young adults, low childhood SES was associated with elevated baseline cortisol as well as greater reactivity to stress (Taylor et al. 2004a). One study suggests gender differences in the impact of timing and duration of SED exposure. In middle-aged women, continuous exposure to low SES from childhood to adulthood was associated with elevated cortisol, whereas in middle-aged men, low childhood SES was associated with elevated cortisol independent of adulthood SES (Li et al. 2007). The role of childhood SED has also been revealed in the exploration of genetic mechanisms of the hypothalamus-pituitary-adrenal axis. Lower parental occupational status between ages 0 and 5 was associated with downregulation of genes involved in glucocorticoid receptor expressions, which was further associated with higher levels of salivary cortisol in adults aged 25–40 (Miller & Chen 2007b; Miller et al. 2009b).
Inflammatory responses and immune function in later life are influenced by childhood SED exposure. In a large UK sample, controlling for adult SES, low SES at birth was associated with multiple markers of inflammation over-reactivity – fibrinogen, C-reactive protein, von Willebrand factor antigen, and a tissue plasminogen activator antigen (Tabassum et al. 2008). Similarly, in a large US sample, retrospectively reported low childhood SES was associated with multiple markers of inflammation over-reactivity – C-reactive protein, fibrinogen, white blood cell count, and von Willebrand factor – when adulthood SES was controlled for (Pollitt et al. 2007). Additional studies have identified significant associations between childhood SED, assessed by low parental educational attainment, and inflammation markers in adults (Brunner et al. 1996; Taylor et al. 2006). Similar to the hypothalamus-pituitary-adrenal axis, investigations of genetic mechanisms revealed that childhood SED predicted increased activity of genes regulated by the proinflammatory transcription factor, nuclear factor kappa-B, and increased activity of the transcription factor cyclic AMP-binding protein in adults (Miller et al. 2009). The upregulation of such genetic activity markers likely play a role in the links between childhood SED exposure and inflammation. Childhood poverty from the prenatal period to the second year of life significantly predicts rates of immune-mediated chronic diseases in adulthood including arthritis and hypertension, which are associated with reduced productivity at work (Ziol-Guest et al. 2012).
Similar to physiological systems, childhood SED is associated with adulthood metabolic processes. Controlling for SES in adulthood, low childhood SES was linked to higher BMI, (Laaksonen et al. 2004; Langenberg et al. 2003; Moore et al. 1962; Poulton et al. 2002; Power et al. 2005; Power et al. 2003), and low parental occupational status in childhood predicted lower high-density lipoprotein (HDL) levels (Brunner et al. 1999; Wanamethee et al. 1996). The associations between childhood SED and metabolic processes seem to be stronger in women than in men. Low SES at age 15 years was related to the development of obesity in women, but not men, from age 16–21 years (Lee et al. 2008). Additionally, low SES at age 4 years predicted metabolic syndrome as well as low levels of HDL and high body fat in women, but not men (Langenberg et al. 2006).
Given the impact of childhood SED across physiological systems, it is not surprising that childhood SED is associated with allostatic load , the cumulative index of dysregulation in multiple physiological stress systems, in later life. Indeed, studies suggest the long-lasting effects of childhood SED on allostatic load in adulthood (Gruenewald et al. 2012). This does not mean that childhood SED is independent of adulthood SED. Cumulative exposure to SED throughout childhood and adulthood appears to be the most significant predictor of adult allostatic load. For example, growing up in poverty was associated with greater allostatic load at age 60 (Singer and Ryff 1999). However, when both childhood and adulthood incomes were examined, the greatest risk for high allostatic load was related to the combination of childhood and adulthood poverty. The next highest risk was for individuals who did not grow up in poverty, but fell into poverty in adulthood. Adults who were never poor or who were poor in childhood but moved out of poverty as they aged showed the least elevated allostatic load. Another study points to the cumulative effects of SED across the lifespan. At age 42, individuals who were consistently exposed to low SES across ages 16, 21, 30, and 42 had the highest level of allostatic load compared to individuals who were exposed to low SES at one or more time points in life (Gustafsson et al. 2010b).
7 Links to the Life Course Health Development Principles
Some of what we are learning about how SED gets into human brain and biology is related to the seven life course health development principles presented by Halfon and Forrest (2017) and discussed in many other chapters of this handbook. Converging evidence from numerous studies on SED and neurobiological systems demonstrates that SED continuously influences the brain and physiological systems throughout the lifespan. Elder and others suggest that childhood is for developing and acquiring capacities and skills, while older adulthood is for managing losses (Baltes 1987; Elder 1998; Elder et al. 2003). We see from the evidence reviewed in this chapter that SED can disrupt or delay neurobiological development that underlies optimal cognitive and emotional functioning during childhood. In older adulthood, SED can accelerate the loss of neurobiological functions, which is in turn associated with memory loss and physical illness.
Neurobiological responses to SED exposure may support processes of adaptation and increased chances of survival from an evolutionary perspective (Gluckman and Hanson 2004; Worthman and Kuzara 2005). Increased amygdala or cardiovascular reactivity to negative environmental cues may help SED individuals more effectively detect threats in the environment. From a perspective such as the active calibration model (Ellis and Del Giudice 2014), the neurobiological changes due to SED reflect processes to prioritize adaptation to a threatening environment, thereby enabling individuals to maximize their chances of survival.
However, as reviewed above, such neurobiological changes are associated with worse long-term health consequences such as anxiety disorders and cardiovascular diseases, in part through elevated allostatic load . This trade-off in short-term survival strategies and long-term well-being may be related to the principle that optimal health development should enhance thriving and protect against diseases in individuals. Evidence presented in this chapter robustly shows that SED can undermine neurobiological development through the allostatic load; thus, individuals exposed to SED may be more likely to die early and develop illnesses across the lifespan.
Two of the life course health development principles merit particular attention with respect to the question of how SED influences health trajectories: (1) health development is highly sensitive to the timing and social structure of environmental exposures, and (2) health development is a complex, nonlinear process that results from person-environment interactions that are multidimensional, multidirectional, and multilevel.
7.1 Timing Versus Duration of SED Exposure on Neurobiological Systems
Different models have been used to explain how SED exerts its influence. The first model focuses on the timing of SED exposure. In this model, early exposure to SED causes stable and long-lasting adaptive changes in neurobiological systems that have inimical long-term effects. The second model focuses on the duration of SED exposure, with longer exposure generating the greatest impact on neurobiological systems. Empirical findings suggest that the two views are not mutually exclusive and both mechanisms may be operative. Early exposure to SED may have a long-lasting impact, but continuous exposure may further increase the gap between health development trajectories. Additionally, different neurobiological systems may be more influenced by one of the two models. Here, we review the two models and discuss how different neurobiological systems may be affected by SED.
The biological embedding model (Finch & Crimmins 2004; Hertzman 1999) highlights the role of timing of exposure to risks like SED. Critical and sensitive periods are key ideas in this model, meaning that the brain or some other physiological system has a period when it is particularly vulnerable to certain experiences or it requires certain experiences for normal development (Coe & Lubach 2005; Levine 2005; Newport et al. 2002). During a critical or sensitive period, certain experiences will produce adaptive responses that confer some short-term advantage to the individual but leave long-lasting negative influences on health developmental trajectories. In the case of SED, this model suggests that early exposure to SED may lead to epigenetic changes that alter the development of the brain and physiological systems in ways that increase vulnerability to disease across the lifespan. Some argue that the biological embedding model is too deterministic because it implies an individual is programmed to exhibit predicted outcomes when exposed to SED. While the biological embedding model is important for understanding different health developmental trajectories due to SED exposure, it is important to acknowledge considerable interindividual variation. This led some to suggest the term biological conditioning or priming might be more appropriate because it allows room for plasticity that may be influenced by future environments (Hanson and Gluckman 2014).
One example of the biological embedding model is the fetal origins theory (Barker 1990). During the critical prenatal period, a pregnant woman’s exposure to extreme physical or psychological stress such as malnutrition or wartime conditions can lead to long-lasting changes in the development of her child’s brain as well as the hypothalamus-pituitary-adrenal and sympathetic nervous system s. The changes affected by prenatal events can increase risks in offspring for physical illnesses like diabetes and cardiovascular disease or psychological illnesses like schizophrenia across the lifespan.
Another example of the biological embedding model is epigenetic changes due to early adversity. Animal studies demonstrate that through methylation of DNA or remodeling of the chromatin structure, early adversity such as low quality maternal care can increase expression of the glucocorticoid receptor gene in hippocampal tissue. Such upregulation of the hypothalamus-pituitary-adrenal axis and glucocorticoid levels in the brain then leads to long-term changes in other regions of the brain and in physiological systems (Szyf et al. 2008; Weaver et al. 2004). Animal studies suggest the critical period for early adversity in rodents is the first 8 days, which is equivalent to the first weeks of life in humans. Miller, Chen, and colleagues also have identified associations between early exposure to SED and signaling in leukocyte transcriptome that appear to persist into adulthood (Miller et al. 2009).
The human brain has sensitive periods that differ by region. The brain and biological processes are most plastic in the first few years of life (Lupien et al. 2009). During early childhood, the amygdala and hippocampus develop quickly and are therefore more vulnerable to stress. On the other hand, the prefrontal cortex develops gradually over the course of the entire childhood and young adulthood. Early normative changes include an increase in structure through neurogenesis (increase in number of neurons) and synaptogenesis (increase in dendritic spines of neurons). To promote effective communication within the brain, neurons and connections strengthen through myelination and other mechanisms when they are used and die if unused (Alexander-Bloch et al. 2013). Brain regions are thought to preserve plasticity in adulthood, but changes are more likely driven by gliogenesis (increase in number of glia, nonneuronal cells for supporting neurons) and vascular alterations rather than neurogenesis. Changes based on neurogenesis and synaptogenesis are longer lasting compared to changes driven by gliogenesis and other mechanisms. This explains how childhood SED exposure may have more significant and enduring impacts on the brain than adulthood SED exposure. While the prefrontal cortex and hippocampus seem to preserve plasticity into adulthood and may be able to recover from or reverse the impacts of SED, this does not appear to be true for the amygdala (Tottenham et al. 2010).
A biological embedding model also exists for development of the hypothalamus-pituitary-adrenal axis. SED exposure, indicated by a lack of family home ownership, in early childhood was associated with reduced levels of glucocorticoid receptor RNA in adolescence (Miller and Chen 2007a). When the timing of exposure was further examined, lack of home ownership before age 2 was a stronger predictor than between ages 5 and 9, while home ownership after age 10 had no effects on the outcome. The study demonstrates the role of childhood SED, especially early exposure, in the development of the hypothalamus-pituitary-adrenal axis regulatory system. Another study supports an embedding model for the immune system (Miller et al. 2011). When exposed to the cold virus, adults who were without home ownership from ages 0 to 9 were more likely to develop a cold for the next 5-day period (Cohen et al. 2004). Furthermore, there was a gradient relationship between the age when the parents first owned a home during the first 9 years and the likelihood of developing a cold (Cohen et al. 2004).
The biggest limitation of the biological embedding model is that it does not take later experience into account, which is not consistent with life course health development principles that posit continuous person⇔environment interactions across the life course. It is possible that early adversity leads to modifications in an individual’s later environment and behavior, which may further increase the risk for disease.
A second model focuses on the duration and accumulation of SED exposure. According to this model, early adversity can cause “chains of risks” (Kuh and Ben-Shlomo 2004), as individuals and their environments continuously and dynamically influence one another. For example, children living in poverty are more likely to be exposed to chronic stressors like family conflicts and harsh parenting, which may lead them to be more helpless. Less proactive cognitive and behavioral styles give rise to low academic performance and poor social skills, which then lowers the individual’s probability of finding and maintaining a good job. As a result, a person is likely to live in poverty as an adult and experience a range of individual and neighborhood deprivation and negative health outcomes. This is the idea that adversity begets negative health development, which begets more adversity. In addition, longer exposure to SED may contribute to an accumulation of chronic stress and lead to more severe damage in neurobiological systems. Indeed, in an earlier section, we explained that allostatic load is affected by SED in an additive fashion. The longer individuals were exposed to low SES, the higher their allostatic load. In addition, cumulative disadvantage is a powerful predictor of many biological markers of mortality and morbidity (Ackerman et al. 1975; Ader et al. 1960; Cameron et al. 2005; Chida et al. 2007; Fenoglio et al. 2006; Kruschinski et al. 2008; Lyons et al. 2009).
Both embedding and accumulative models play a role in predicting health outcomes in SED-exposed individuals. This is true for physiological systems. For example, among women at age 60, the associations among childhood and adulthood SED and metabolic processing including insulin resistance, HDL, and triglycerides were examined. Parental occupation status in childhood was a stronger predictor of metabolic processing than own occupational status in adulthood (Lawlor et al. 2002). However, women who were chronically exposed to low occupational status throughout childhood and adulthood exhibited the worst profile, including a 234% likelihood of being obese compared to women who were never exposed to low occupational status. The results suggest that childhood SED can be embedded in a physiological system to set the developmental trajectory, but accumulative processes may compound these earlier, poorer trajectories leading to worse outcomes throughout the lifespan.
The biggest challenge to understanding exact mechanisms of SED exposure (the embedding versus accumulative model) is that an individual tends to be exposed to poverty early and continuously. Thus, it may be most productive to consider a life course health development model, which examines the relational dynamic person⇔environment interactions, the timing and social patterning of exposures, and evolutionarily determined adaptive plastic responses that unfold during development. It is also difficult to tease apart the role of early exposure versus later exposure to SED because many studies are cross-sectional or measure early exposure to SED retrospectively. Thus, more longitudinal studies are needed to understand how health development is produced over time. Also, intervention studies such as cash transfers to families to improve SED provide opportunities to establish causal and temporal relations between SED and health outcomes.
7.2 Multilevel Neurobiology-Environment Interactions
Interactions between SED and neurobiological systems are highly complex not only because of the multilevel nature of neurobiological systems but also because SED is further associated with multilevel environmental factors (Sameroff 2010). SED is a major risk factor for a host of adverse outcomes because it serves as a proxy for many risk factors that result in psychological and physical illnesses (Chen et al. 2002; Evans and Kim 2010; Repetti et al. 2002). Understanding interactions between multilevel and multidimensional neurobiological and environmental systems is critical for developing effective clinical and community-level interventions to reduce the adverse impact of SED exposure on health.
First, many psychosocial factors have been suggested as mediators. In childhood, threat appraisal and family environment are well-known mediators for the links between SED and physical and psychological outcomes (Repetti et al. 2002). Retrospectively reported elements of the early family environment such as abusive and harsh parenting were associated with direct relationships between amygdala and ventrolateral prefrontal cortex activations, which suggests ineffective suppression of ventrolateral prefrontal cortex on the amygdala (Taylor et al. 2006). Maltreatment and abuse in childhood were also associated with weaker default mode network connectivity and increased connectivity between the amygdala and medial prefrontal cortex (Philip et al. 2013). Lack of maternal warmth in early childhood and early adolescence was associated with less activation in the reward circuits such as the medial prefrontal cortex during loss and winning, which may indicate altered reward processing and more difficulties in handling disappointment (Morgan et al. 2014).
Second, cumulative risk factors including physical environment – noise, crowding, and substandard housing quality – have been identified as strong mediators between SED and neurobiological outcomes across the lifespan (Evans and Cassells 2014; Evans and Kim 2010; Kim et al. 2013; Zalewski et al. 2012). SED exposure is also associated with chaotic home environment and poor quality in social environments outside the home, such as school and neighborhood (Evans et al. in press; Roy et al. 2014; Schreier et al. 2014). Along with these risk factors, it is important to consider factors that protect from the negative effects of SED exposure in childhood. Supportive parenting and support from others and adaptive coping skills have been suggested as protective factors. For example, active coping skills like distraction were associated with better psychological outcomes than avoidance and disengaging coping in children and adolescents (Wadsworth and Berger 2006; Wadsworth et al. in press).
Third, in adulthood, individuals who are exposed to SED are more likely to experience psychological stress, discrimination, negative emotions, and more limited resources such as social support (Cohen et al. 2006a, b; Kubzansky et al. 1999; Kunz-Ebrecht et al. 2004; Marmot et al. 1997). On the other hand, there are protective factors including optimism, mastery, self-esteem, and social support that can help to reduce the negative effects of SED exposure on the neurobiological systems (Kubzansky et al. 2001; Matthews et al. 2010; Taylor et al. 2008).
Fourth, sleep quality , an example of a personal factor, may also play a significant role in the neural and physiological systems throughout the lifespan. Sleep disturbance appears to be more common among lower SES adults (Buckalt 2011) and children (Jarrin et al. 2014). Adults with low income or education levels had significantly shorter sleep duration (Whinnery et al. 2013) and poorer sleep quality and higher levels of daytime sleepiness compared to adults with mid- to high income or education levels (Jarrin et al. 2013). The association between poverty and sleep quality was stronger in white adults than in African American adults (Patel et al. 2010). In children and adolescents, both low subjective social standing ratings and low objective SED indicators including family income and parental education were significantly associated with shorter sleep duration and poorer sleep quality (Jarrin et al. 2014). Sleep duration and quality are a concern because they can significantly influence cognitive functions (Dewald et al. 2010; Steenari et al. 2003; Walker 2008) and mood regulation (Dahl and Lewin 2002; Eidelman et al. 2012; Kahn et al. 2013) in children and adults. Poor sleep has also been associated with physiological stress dysregulation (Jarcho et al. 2013; Rao et al. 1996), brain structure including the hippocampus (Riemann et al. 2007), and brain functions such as risk-taking behaviors in adolescence (Telzer et al. 2013). Thus, examining the role of sleep in the associations between poverty and neurobiological stress regulation is an important future direction for health disparities research.
Furthermore, adding to the complexity of understanding how SED influences health development, there are multiple levels of neurobiological systems including genetic, neural, and physiological stress regulatory systems interacting with each other. While few studies exist, they have identified the significant role of the hypothalamus-pituitary-adrenal axis and immune processes in the associations among SED and brain structure and functions. For example, higher cortisol levels in children mediated the link between childhood SES and prefrontal cortex responses during cognitive tasks (Sheridan et al. 2012). Increased levels of inflammatory markers also mediated the associations between SED and white matter structure integrity in adulthood (Gianaros et al. 2013). Childhood poverty exposure prospectively predicted weaker default mode network activity, which was associated with higher levels of cortisol in response to social stress (Sripada et al. 2014).
In addition, it is important to understand how genes and environment interact in the context of SED across lifespan. Many studies have identified certain genetic variations that may lead individuals to be more susceptible or vulnerable to environmental influences (Ellis et al. 2011). Individuals with such susceptible genetic profiles would likely exhibit greater influence from SED exposure compared to individuals with insusceptible genetic profiles. Thus, the role of genetic profiles in neural and physiological system development throughout the lifespan in the context of SED will be important for refined identification of at-risk individuals (Kim-Cohen et al. 2004). Understanding gene expression, which is known to be affected by person ⇔environment interactions, will also be a fruitful avenue of future investigation.
Last, it is important to pursue understanding of the ways in which these multilevel, multidimensional interactions between the environment and neurobiology are linked to specific health development outcomes, in order to develop effective intervention approaches. Few studies have directly tested the mediating role of neurobiological systems in the relations between SED and health development, largely due to limitations in sample size. Testing mediation requires a large sample size for appropriate statistical power. Cumulative measures of physiological systems such as allostatic load are also useful in building a comprehensive understanding of the effects of SED exposure. While the research has been limited, studies that have included a large number of biological markers have provided evidence for allostatic load as a mediator for the links between SED and health outcomes (Seeman et al. 2004). More research involving large sets of biological markers is needed and will require population-based studies and collaboration across fields.
Population-based studies are also important for statistical power in testing interactions among individual differences such as gender and race/ethnicity. Differences across individuals are likely to be associated with different outcomes and vulnerabilities to SED exposure. For example, some evidence suggests that SED exposure has a greater impact on allostatic load in African American compared to white adults (Geronimus et al. 2006), while others did not find racial differences in the associations between SED exposure and allostatic load (Seeman et al. 2008).
8 Suggestions for Policy and Interventions
This discussion leads to the final part of the chapter on interventions. A greater benefit-cost ratio has been revealed for early intervention (Shonkoff et al. 2009; Heckman 2006). Evidence of the long-lasting effects of early experience also emphasizes the importance of early interventions. Future studies including those we suggested above may help to more clearly identify critical periods when investment would produce the biggest effects and long-term returns (Blair and Raver 2014; Shonkoff 2010).
There are several ways to intervene. The first is to directly intervene with SED, particularly family income. Cash transfer or income increases for a child’s parents early in childhood have been shown to improve the physiological stress regulation function as well as cognitive outcomes (Duncan et al. 2011; Fernald et al. 2008). Other ways to intervene would be to target the mediators we discussed above. Interventions for parenting skills, parental psychopathology, health-care quality, as well as a child’s cognitive functions have been shown to improve child health outcomes including cognitive and psychological functions and inflammatory regulation (Beeber et al. 2008; Miller et al. 2014; Olds et al. 1997). Third, interventions on protective factors such as coping have also been shown to improve physical and health outcomes in SED children and adults (Wadsworth et al. 2011).
Lastly, given the importance of parents in directly modifying home environments for children, a two-generation approach has received increasing attention (Kim and Watamura 2015). When both parents received a parenting intervention at the same time that children received attention training, children’s attentional control and brain functions improved more than when only children received the intervention (Neville et al. 2013). In particular, pregnancy and the first years of a child’s life can be an optimal time for the two-generation approach to intervention. Not only is this the period when a child’s brain and body are most rapidly developing, it is also a period when a parent’s brain undergoes plastic changes to support the parental role (Kim and Bianco 2014; Swain et al. 2014). For example, during the first few months of the postpartum period, both new mothers and fathers exhibit structural growth in brain regions including the striatum and prefrontal cortex that support warm and supportive parenting behaviors (Kim et al. 2010b; Kim et al. 2014). Sensitive neural activations, particularly for one’s own baby, have also been consistently identified among new mothers and fathers during these same periods, when parents and infants establish long-lasting emotional relationships (Barrett and Fleming 2011; Mascaro et al. 2013; Musser et al. 2012). At the same time, parents in poverty are far more vulnerable to development of postpartum depression (Boury et al. 2004; U.S. Department of Health and Human Services 2002; Walker et al. 2002), and it is possible that as the result of depression and the stress of living in poverty, they are also more likely to exhibit abusive or harsh parenting (Sedlak and Broadhurst 1996; Widom and Nikulina 2012). Parents who received poor quality of parental care in their own childhood or experienced chronic stress such as poverty are also more likely to show reduced biological and behavioral sensitivity to their own children (Champagne et al. 2003; Kim et al. 2015; Kim et al. 2010a) and to repeat harsh parenting (Belsky et al. 2005; Van Ijzendoorn 1992). Thus, interventions that pay more attention to the well-being of parents with SED backgrounds may ultimately promote optimal development of the next generations and reduce the intergenerational transmission of SED and harsh environments.
References
Ackerman, S. H., Hofer, M. A., & Weiner, H. (1975). Age at maternal separation and gastric erosion susceptibility in the rat. Psychosomatic Medicine, 37, 180–184.
Adam, E. K., & Kumari, M. (2009). Assessing salivary cortisol in large-scale, epidemiological research. Psychoneuroendocrinology, 34, 1423–1436.
Ader, R., Tatum, R., & Beels, C. C. (1960). Social factors affecting emotionality and resistance to disease in animals: I. Age of separation from the mother and susceptibility to gastric ulcers in the rat. Journal of Comparative and Physiological Psychology, 53, 446–454.
Adler, N. E., & Rehkopf, D. H. (2008). U.S. disparities in health: Descriptions, causes, and mechanisms. Annual Review of Public Health, 29, 235–252.
Adolphs, R. (2001). The neurobiology of social cognition. Current Opinion in Neurobiology, 11, 231–239.
Alexander-Bloch, A., Raznahan, A., Bullmore, E., & Giedd, J. (2013). The convergence of maturational change and structural covariance in human cortical networks. Journal of Neuroscience, 33, 2889–2899.
Alley, D. E., Seeman, T. E., Ki Kim, J., Karlamangla, A., Hu, P., & Crimmins, E. M. (2006). Socioeconomic status and C-reactive protein levels in the US population: NHANES IV. Brain, Behavior, and Immunity, 20, 498–504.
Azad, M. B., Lissitsyn, Y., Miller, G. E., Becker, A. B., HayGlass, K. T., & Kozyrskyj, A. L. (2012). Influence of socioeconomic status trajectories on innate immune responsiveness in children. PloS One, 7, e38669.
Baltes, P. B. (1987). Theoretical propositions of life-span developmental psychology: On the dynamics between growth and decline. Developmental Psychology, 23(5), 611–626.
Barker, D. J. (1990). The fetal and infant origins of adult disease. BMJ: British Medical Journal, 301(6761), 1111.
Barrett, J., & Fleming, A. S. (2011). Annual research review: All mothers are not created equal: Neural and psychobiological perspectives on mothering and the importance of individual differences. Journal of Child Psychology and Psychiatry, 52(4), 368–397. doi:10.1111/j.1469-7610.2010.02306.x.
Beeber, L. S., Perreira, K. M., & Schwartz, T. (2008). Supporting the mental health of mothers raising children in poverty - how do we target them for intervention studies? Reducing the Impact of Poverty on Health and Human Development: Scientific Approaches, 1136, 86–100.
Belsky, J., Jaffee, S. R., Sligo, J., Woodward, L., & Silva, P. A. (2005). Intergenerational transmission of warm-sensitive-stimulating parenting: A prospective study of mothers and fathers of 3-year-olds. Child Development, 76, 384–396.
Bird, C. E., Seeman, T., Escarce, J. J., Basurto-Dávila, R., Finch, B. K., Dubowitz, T., et al. (2010). Neighbourhood socioeconomic status and biological ‘wear and tear’in a nationally representative sample of US adults. Journal of Epidemiology and Community Health, 64, 860–865.
Blair, C., & Raver, C. C. (2012). Child development in the context of adversity: Experiential canalization of brain and behavior. American Psychologist, 67, 309–318.
Blair, C., & Raver, C. C. (2014). Closing the achievement gap through modification of neurocognitive and neuroendocrine function: Results from a cluster randomized controlled trial of an innovative approach to the education of children in kindergarten. PloS One, 9, e112393.
Blair, C., Raver, C. C., Granger, D., Mills-Koonce, R., & Hibel, L. (2011). Allostasis and allostatic load in the context of poverty in early childhood. Development and Psychopathology, 23, 845–857.
Blair, C., Berry, D., Mills-Koonce, R., & Granger, D. (2013). Cumulative effects of early poverty on cortisol in young children: Moderation by autonomic nervous system activity. Psychoneuroendocrinology, 38, 2666–2675.
Boury, J. M., Larkin, K. T., & Krummel, D. A. (2004). Factors related to postpartum depressive symptoms in low-income women. Women and Health, 39(3), 19–34. doi:10.1300/J013v39n03_02.
Bradley, R. H., & Corwyn, R. F. (2002). Socioeconomic status and child development. Annual Review of Psychology, 53, 371–399.
Brandtstädter, J., Baltes-Götz, B., Kirschbaum, C., & Hellhammer, D. (1991). Developmental and personality correlates of adrenocortical activity as indexed by salivary cortisol: Observations in the age range of 35 to 65 years. Journal of Psychosomatic Research, 35, 173–185.
Braverman, P., & Egerter, S. (2008). Overcoming obstacles to health: Robert Wood Johnson Foundation Commission to build a healthier America. Princeton: Robert Wood Johnson Foundation.
Brody, G. H., Lei, M.-K., Chen, E., & Miller, G. E. (2014). Neighborhood poverty and allostatic load in African American youth. Pediatrics, 134, e1362–e1368.
Brooks-Gunn, J., & Duncan, G. J. (1997). The effects of poverty on children. The Future of Children, 7, 55–71.
Brunner, E., Davey Smith, G., Marmot, M., Canner, R., Beksinska, M., & O’Brien, J. (1996). Childhood social circumstances and psychosocial and behavioural factors as determinants of plasma fibrinogen. Lancet, 347, 1008–1013.
Brunner, E., Marmot, M., Nanchahal, K., Shipley, M., Stansfeld, S., Juneja, M., & Alberti, K. (1997). Social inequality in coronary risk: Central obesity and the metabolic syndrome. Evidence from the Whitehall II study. Diabetologia, 40, 1341–1349.
Brunner, E., Shipley, M. J., Blane, D., Davey-Smith, G., & Marmot, M. (1999). When does cardiovascular risk start? Past and present socioeconomic circumstances and risk factors in adulthood. Journal of Epidemiology and Community Health, 53, 757–764.
Buckhalt, J. A. (2011). Insufficient sleep and the socioeconomic status achievement gap. Child Development Perspectives, 5, 59–65.
Burghy, C. A., Stodola, D. E., Ruttle, P. L., Molloy, E. K., Armstrong, J. M., Oler, J. A., et al. (2012). Developmental pathways to amygdala-prefrontal function and internalizing symptoms in adolescence. Nature Neuroscience, 15, 1736–1741.
Butterworth, P., Cherbuin, N., Sachdev, P., & Anstey, K. J. (2012). The association between financial hardship and amygdala and hippocampal volumes: Results from the PATH through life project. Social Cognitive and Affective Neuroscience, 7, 548–556.
Cameron, N. M., Champagne, F. A., Parent, C., Fish, E. W., Ozaki-Kuroda, K., & Meaney, M. J. (2005). The programming of individual differences in defensive responses and reproductive strategies in the rat through variations in maternal care. Neuroscience and Biobehavioral Reviews, 29, 843–865.
Chaix, B., Ducimetiere, P., Lang, T., Haas, B., Montaye, M., Ruidavets, J.-B., et al. (2008). Residential environment and blood pressure in the PRIME study: Is the association mediated by body mass index and waist circumference? Journal of Hypertension, 26, 1078–1084.
Chaix, B., Bean, K., Leal, C., Thomas, F., Havard, S., Evans, D., et al. (2010). Individual/neighborhood social factors and blood pressure in the RECORD cohort study which risk factors explain the associations? Hypertension, 55, 769–775.
Champagne, F. A., Weaver, I. C., Diorio, J., Sharma, S., & Meaney, M. J. (2003). Natural variations in maternal care are associated with estrogen receptor alpha expression and estrogen sensitivity in the medial preoptic area. Endocrinology, 144(11), 4720–4724.
Chen, E., Matthews, K. A., & Boyce, W. T. (2002). Socioeconomic differences in children’s health: How and why do these relationships change with age? Psychological Bulletin, 128, 295–329.
Chen, E., Fisher, E. B., Bacharier, L. B., & Strunk, R. C. (2003). Socioeconomic status, stress, and immune markers in adolescents with asthma. Psychosomatic Medicine, 65, 984–992.
Chen, E., Langer, D. A., Raphaelson, Y. E., & Matthews, K. A. (2004). Socioeconomic status and health in adolescents: The role of stress interpretations. Child Development, 75, 1039–1052.
Chen, E., Hanson, M. D., Paterson, L. Q., Griffin, M. J., Walker, H. A., & Miller, G. E. (2006). Socioeconomic status and inflammatory processes in childhood asthma: The role of psychological stress. The Journal of Allergy and Clinical Immunology, 117, 1014–1020.
Chen, E., Chim, L. S., Strunk, R. C., & Miller, G. E. (2007). The role of the social environment in children and adolescents with asthma. American Journal of Respiratory and Critical Care Medicine, 176, 644–649.
Chen, E., Miller, G. E., Walker, H. A., Arevalo, J. M., Sung, C. Y., & Cole, S. W. (2009). Genome-wide transcriptional profiling linked to social class in asthma. Thorax, 64, 38–43.
Chen, E., Cohen, S., & Miller, G. W. (2010). How low socioeconomic status effects 2-year hormonal trajectories in children. Psychological Science, 21, 31–37.
Chichlowska, K. L., Rose, K. M., Diez-Roux, A. V., Golden, S. H., McNeill, A. M., & Heiss, G. (2009). Life course socioeconomic conditions and metabolic syndrome in adults: The atherosclerosis risk in communities (ARIC) study. Annals of Epidemiology, 19, 875–883.
Chida, Y., Sudo, N., Sonoda, J., Hiramoto, T., & Kubo, C. (2007). Early-life psychological stress exacerbates adult mouse asthma via the hypothalamus-pituitary-adrenal axis. American Journal of Respiratory and Critical Care Medicine, 175, 316–322.
Coan, J. A., Beckes, L., & Allen, J. P. (2013). Childhood maternal support and social capital moderate the regulatory impact of social relationships in adulthood. International Journal of Psychophysiology, 88, 224–231.
Coccaro, E. F., McCloskey, M. S., Fitzgerald, D. A., & Phan, K. L. (2007). Amygdala and orbitofrontal reactivity to social threat in individuals with impulsive aggression. Biological Psychiatry, 62, 168–178.
Coe, C. L., & Lubach, G. R. (2005). Prenatal origins of individual variation in behavior and immunity. Neuroscience and Biobehavioral Reviews, 29, 39–49.
Cohen, S., Doyle, W. J., Turner, R. J., Alper, C. M., & Skoner, D. P. (2004). Childhood socioeconomic status and host resistance to infectious illness in adulthood. Psychosomatic Medicine, 66, 553–558.
Cohen, S., Schwartz, J. E., Epel, E., Kirschbaum, C., Sidney, S., & Seeman, T. (2006a). Socioeconomic status, race, and diurnal cortisol decline in the coronary Artery risk development in young adults (CARDIA) study. Psychosomatic Medicine, 68, 41–50.
Cohen, R. A., Grieve, S., Hoth, K. F., Paul, R. H., Sweet, L., Tate, D., et al. (2006b). Early life stress and morphometry of the adult anterior cingulate cortex and caudate nuclei. Biological Psychiatry, 59, 975–982.
Cohen, S., Doyle, W. J., & Baum, A. (2006c). Socioeconomic status is associated with stress hormones. Psychosomatic Medicine, 68, 414–420.
Cohen, S., Janicki-Deverts, D., Chen, E., & Matthews, K. A. (2010). Childhood socioeconomic status and adult health. Annals of the New York Academy of Sciences, 1186, 37–55.
Colhoun, H. M., Hemingway, H., & Poulter, N. R. (1998). Socioeconomic status and blood pressure: An overview analysis. Journal of Hypertension, 12, 91–110.
Cozier, Y. C., Palmer, J. R., Horton, N. J., Fredman, L., Wise, L. A., & Rosenberg, L. (2007). Relation between neighborhood median housing value and hypertension risk among black women in the United States. American Journal of Public Health, 97, 718–724.
Crimmins, E. M., Kim, J. K., & Seeman, T. E. (2009). Poverty and biological risk: The earlier “aging” of the poor. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, 64, 286–292.
Cubbin, C., Hadden, W. C., & Winkleby, M. A. (2000). Neighborhood context and cardiovascular disease risk factors: The contribution of material deprivation. Ethnicity & Disease, 11, 687–700.
Dahl, R. E., & Lewin, D. S. (2002). Pathways to adolescent health sleep regulation and behavior. Journal of Adolescent Health, 31, 175–184.
Danese, A., & McEwen, B. S. (2012). Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiology & Behavior, 106, 29–39.
D’Angiulli, A., Herdman, A., Stapells, D., & Hertzman, C. (2008). Children’s event-related potentials of auditory selective attention vary with their socioeconomic status. Neuropsychology, 22, 293–300.
D’Angiulli, A., Van Roon, P. M., Weinberg, J., Oberlander, T. F., Grunau, R. E., Hertzman, C., & Maggi, S. (2012). Frontal EEG/ERP correlates of attentional processes, cortisol and motivational states in adolescents from lower and higher socioeconomic status. Frontiers in Human Neuroscience, 6, 306.
Davidson, R. J., & McEwen, B. S. (2012). Social influences on neuroplasticity: Stress and interventions to promote well-being. Nature Neuroscience, 15, 689–695.
De Boever, E., Braeckman, L., Baele, G., Rosseneu, M., & De Backer, G. (1995). Relation of fibrinogen to lifestyles and to cardiovascular risk factors in a working population. International Journal of Epidemiology, 24, 915–921.
Dewald, J. F., Meijer, A. M., Oort, F. J., Kerkhof, G. A., & Bögels, S. M. (2010). The influence of sleep quality, sleep duration and sleepiness on school performance in children and adolescents: A meta-analytic review. Sleep Medicine Reviews, 14, 179–189.
Diez-Roux, A. V., Link, B. G., & Northridge, M. E. (2000). A multilevel analysis of income inequality and cardiovascular disease risk factors. Social Science & Medicine, 50, 673–687.
Doan, S. N., Fuller-Rowell, T. E., & Evans, G. W. (2012). Cumulative risk and adolescent’s internalizing and externalizing problems: The mediating roles of maternal responsiveness and self-regulation. Developmental Psychology, 48, 1529–1539.
Duncan, G. J., Ziol-Guest, K. M., & Kalil, A. (2010). Early-childhood poverty and adult attainment, behavior, and health. Child Development, 81, 306–325.
Duncan, G. J., Morris, P. A., & Rodrigues, C. (2011). Does money really matter? Estimating impacts of family income on young children’s achievement with data from random-assignment experiments. Developmental Psychology, 47, 1263.
Eidelman, P., Gershon, A., McGlinchey, E., & Harvey, A. G. (2012). Sleep and psychopathology. In C. M. Morin & C. A. Espie (Eds.), The Oxford handbook of sleep and sleep disorders (pp. 172–189). New York: Oxford University Press.
Elder, G. H. (1998). The life course as developmental theory. Child Development, 69(1), 1–12.
Elder, G. H., Jr., Johnson, M. K., & Crosnoe, R. (2003). The emergence and development of life course theory. In Handbook of the life course (pp. 3–19). New York: Springer.
Ellis, B. J., & Del Giudice, M. (2014). Beyond allostatic load: Rethinking the role of stress in regulating human development. Development and Psychopathology, 26(1), 1–20. doi:10.1017/S0954579413000849.
Ellis, B. J., Boyce, W. T., Belsky, J., Bakermans-Kranenburg, M. J., & van Ijzendoorn, M. H. (2011). Differential susceptibility to the environment: An evolutionary-neurodevelopmental theory. Development and Psychopathology, 23, 7–28.
Epstein, J., Pan, H., Kocsis, J., Yang, Y., Butler, T., Chusid, J., et al. (2006). Lack of ventral striatal response to positive stimuli in depressed versus normal subjects. American Journal of Psychiatry, 163, 1784–1790.
Essex, M. J., Klein, M. H., Cho, E., & Kalin, N. H. (2002). Maternal stress beginning in infancy may sensitize children to later stress exposure: Effects on cortisol and behavior. Biological Psychiatry, 52, 776–784.
Evans, G. W. (2003). A multimethodological analysis of cumulative risk and allostatic load among rural children. Developmental Psychology, 39, 924–933.
Evans, G. W. (2004). The environment of childhood poverty. American Psychologist, 59, 77–92.
Evans, G. W., & Cassells, R. C. (2014). Childhood poverty, cumulative risk exposure, and mental health in emerging adults. Clinical Psychological Science, 2(3), 287–296.
Evans, G. W., & English, K. (2002). The environment of poverty: Multiple stressor exposure, psychophysiological stress, and socioemotional adjustment. Child Development, 73, 1238–1248.
Evans, G. W., & Kim, P. (2007). Childhood poverty and health: Cumulative risk exposure and stress dysregulation. Psychological Science, 18, 953–957.
Evans, G. W., & Kim, P. (2010). Multiple risk exposure as a potential explanatory mechanism for the socioeconomic status-health gradient. Annals of the New York Academy of Sciences, 1186, 174–189.
Evans, G. W., & Kim, P. (2012). Childhood poverty and young adults’ allostatic load: The mediating role of childhood cumulative risk exposure. Psychological Science, 23, 979–983.
Evans, G. W., & Kim, P. (2013). Childhood poverty, chronic stress, self-regulation, and coping. Child Development Perspectives, 7, 43–48.
Evans, G.W., & Schamberg, M.A. (2009). Childhood poverty, chronic stress, and adult working memory. Proceedings of the National Academy of Sciences, 106, 6545–6549.
Evans, G. W., Kim, P. K., Ting, A. H., Tesser, H. B., & Shanis, D. (2007). Cumulative risk, maternal responsiveness, and allostatic load among young adolescents. Developmental Psychology, 43, 341–351.
Evans, G.W., Wells, N.M. & Schamberg, M.A. (2010). The ecological context of SES and obesity. In L. Dube, A. Bechara, A. Dagher, D. Drewnowski, J. LeBel, J., P. James, D. Richard, R.Y. Yada (Eds.). Obesity prevention: The role of society and brain on individual behavior (pp. 713–725). New York: Elsevier.
Evans, G. W., Chen, E., Miller, G. E., & Seeman, T. E. (2012). How poverty gets under the skin: A lifecourse perspective. In V. Maholmes & R. King (Eds.), The Oxford handbook of poverty and child development (pp. 13–36). New York: Oxford University Press.
Evans, G. W., Exner-Cortens, D., Kim, P., & Bartholomew, D. (2013). Childhood poverty and late adolescents’ blood pressure reactivity and recovery to an acute stressor: The mediating role of family conflict. Psychosomatic Medicine, 75, 691–700.
Evans, G. W., Eckenrode, J., & Marcynyszyn, L. A. (in press). Poverty and chaos. In G. W. Evans & T. D. Wachs (Eds.), Chaos and its influence on children’s development: An ecological perspective. Washington, DC: American Psychological Association.
Everson, S. A., Maty, S. C., Lynch, J. W., & Kaplan, G. A. (2002). Epidemiologic evidence for the relation between socioeconomic status and depression, obesity, and diabetes. Journal of Psychosomatic Research, 53, 891–895.
Fauth, R. C., Leventhal, T., & Brooks-Gunn, J. (2007). Welcome to the neighborhood? Long-term impacts of moving to low-poverty neighborhoods on poor children’s and adolescents’ outcomes. Journal of Research on Adolescence, 17, 249–284.
Fenoglio, K. A., Brunson, K. L., & Baram, T. Z. (2006). Hippocampal neuroplasticity induced by early-life stress: Functional and molecular aspects. Frontiers in Neuroendocrinology, 27, 180–192.
Fernald, L. C., Gertler, P. J., & Neufeld, L. M. (2008). Role of cash in conditional cash transfer programmes for child health, growth, and development: An analysis of Mexico’s Oportunidades. The Lancet, 371, 828–837.
Fernald, L. C., & Gunnar, M. R. (2009). Poverty-alleviation program participation and salivary cortisol in very lowincome children. Social Science and Medicine, 68, 2180–2189.
Ferrie, J. E., Shipley, M. J., Stansfeld, S. A., & Marmot, M. G. (2002). Effects of chronic job insecurity and change in job security on self reported health, minor psychiatric morbidity, physiological measures, and health related behaviours in British civil servants: The Whitehall II study. Journal of Epidemiology and Community Health, 56, 450–454.
Finch, C. E., & Crimmins, E. M. (2004). Inflammatory exposure and historical changes in human life-spans. Science, 305, 1736–1739.
Flinn, M. V., & England, B. (1997). Social economics of childhood glucocorticoid stress response and health. American Journal of Physical Anthropology, 102, 33–53.
Foubert-Samier, A., Catheline, G., Amieva, H., Dilharreguy, B., Helmer, C., Allard, M., & Dartigues, J.-F. (2012). Education, occupation, leisure activities, and brain reserve: A population-based study. Neurobiology of Aging, 33(2), 423.e15–423.e25.
Gallo, L. C., & Matthews, K. A. (2003). Understanding the association between socioeconomic status and physical health: Do negative emotions play a role? Psychological Bulletin, 129, 10–31.
Gao, W., Alcauter, S., Elton, A., Hernandez-Castillo, C. R., Smith, J. K., Ramirez, J., & Lin, W. (2014). Functional network development during the first year: Relative sequence and socioeconomic correlations. Cerebral Cortex, 25(9), 2919–2928.
Geronimus, A. T., Hicken, M., Keene, D., & Bound, J. (2006). “weathering” and age patterns of allostatic load scores among blacks and whites in the United States. American Journal of Public Health, 96(5), 826–833.
Gianaros, P. J., Horenstein, J. A., Cohen, S., Matthews, K. A., Brown, S. M., Flory, J. D., et al. (2007). Perigenual anterior cingulate morphology covaries with perceived social standing. Social Cognitive and Affective Neurosciences, 2, 161–173.
Gianaros, P. J., Horenstein, J. A., Hariri, A. R., Sheu, L. K., Manuck, S. B., Matthews, K. A., & Cohen, S. (2008). Potential neural embedding of parental social standing. Social Cognitive and Affective Neurosciences, 3, 91–96.
Gianaros, P. J., Marsland, A. L., Sheu, L. K., Erickson, K. I., & Verstynen, T. D. (2013). Inflammatory pathways link socioeconomic inequalities to white matter architecture. Cerebral Cortex, 23, 2058–2071.
Giedd, J. N. (2004). Structural magnetic resonance imaging of the adolescent brain. Annals of the New York Academy of Sciences, 1021, 77–85.
Gliksman, M. D., Dwyer, T., & Wlodarczyk, J. (1990). Differences in cardiovascular disease risk factors in Australian schoolchildren. Preventive Medicine, 19, 291–304.
Gluckman, P. D., & Hanson, M. A. (2004). Living with the past: Evolution, development, and patterns of disease. Science, 305(5691), 1733–1736. doi:10.1126/science.1095292.
Gonzalez, M. Z., Beckes, L., Chango, J., Allen, J. P., & Coan, J. A. (2014). Adolescent neighborhood quality predicts adult dACC response to social exclusion. Social Cognitive and Affective Neuroscience. doi:10.1093/scan/nsu137.
Goodman, E., McEwen, B. S., Dolan, L. M., Schafer-Kalkhoff, T., & Adler, N. E. (2005a). Social disadvantage and adolescent stress. Journal of Adolescent Health, 37, 484–492.
Goodman, E., McEwen, B. S., Huang, B., Dolan, L. M., & Adler, N. E. (2005b). Social inequalities in biomarkers of cardiovascular risk in adolescence. Psychosomatic Medicine, 67, 9–15.
Grant, K. E., Compas, B. E., Stuhlmacher, A. F., Thurm, A. E., McMahon, S. D., & Halpert, J. A. (2003). Stressors and child and adolescent psychopathology: Moving from markers to mechanisms of risk. Psychological Bulletin, 129, 447–466.
Gruenewald, T. L., Cohen, S., Matthews, K. A., Tracy, R., & Seeman, T. E. (2009). Association of socioeconomic status with inflammation markers in black and white men and women in the coronary Artery risk development in young adults (CARDIA) study. Social Science & Medicine, 69, 451–459.
Gruenewald, T. L., Karlamangla, A. S., Hu, P., Stein-Merkin, S., Crandall, C., Koretz, B., & Seeman, T. E. (2012). History of socioeconomic disadvantage and allostatic load in later life. Social Science & Medicine, 74, 75–83.
Guize, L., Jaffiol, C., Gueniot, M., Bringer, J., Giudicelli, C., Tramoni, M., et al. (2008). Diabetes and socio-economic deprivation. A study in a large French population. Bulletin de l’Académie Nationale de Médecine, 192, 1707–1723.
Gump, B. B., Reihman, J., Stewart, P., Lonky, E., Darvill, T., & Matthews, K. A. (2007). Blood lead (Pb) levels: A potential environmental mechanism explaining the relation between socioeconomic status and cardiovascular reactivity in children. Health Psychology, 26, 296–304.
Gump, B. B., Reihman, J., Stewart, P., Lonky, E., & Matthews, K. A. (2009). Blood lead (Pb) levels: Further evidence for an environmental mechanism explaining the association between socioeconomic status and psychophysiological dysregulation in children. Health Psychology, 28, 614–620.
Gustafsson, P. E., Gustafsson, P. A., & Nelson, N. (2006). Cortisol levels and psychosocial factors in preadolescent children. Stress and Health, 22, 3–9.
Gustafsson, P. E., Janlert, U., Theorell, T., & Hammarstrom, A. (2010a). Life-course socioeconomic trajectories and diurnal cortisol regulation in adulthood. Psychoneuroendocrinology, 35, 613–623.
Gustafsson, P. E., Janlert, U., Theorell, T., Westerlund, H., Hammarström, A. (2010b). Socioeconomic status over the life course and allostatic load in adulthood: Results from the Northern Swedish Cohort. Journal of Epidemiology & Community Health, jech-2010.
Haber, S. N., & Knutson, B. (2009). The reward circuit: Linking primate anatomy and human imaging. Neuropsychopharmacology, 35, 4–26.
Hackman, D. A., & Farah, M. J. (2009). Socioeconomic status and the developing brain. Trends in Cognitive Sciences, 13, 65–73.
Hackman, D. A., Betancourt, L. M., Brodsky, N. L., Hurt, H., & Farah, M. J. (2012). Neighborhood disadvantage and adolescent stress reactivity. Frontiers in Human Neuroscience, 6, 277.
Halfon, N., & Forrest, C. B. (2017). The emerging theoretical framework of life course health development. In N. Halfon, C. B. Forrest, R. M. Lerner, & E. Faustman (Eds.), Handbook of life course health-development science. Cham: Springer.
Hanson, M. A., & Gluckman, P. D. (2014). Early developmental conditioning of later health and disease: Physiology or pathophysiology? Physiological Reviews, 94(4), 1027–1076. doi:10.1152/physrev.00029.2013.
Hanson, J. L., Chandra, A., Wolfe, B. L., & Pollak, S. D. (2011). Association between income and the hippocampus. PloS One, 6, e18712.
Hanson, J. L., Hair, N., Shen, D. G., Shi, F., Gilmore, J. H., Wolfe, B. L., & Pollak, S. D. (2013). Family poverty affects the rate of human infant brain growth. PloS One, 8, e80954.
Hanson, J. L., Nacewicz, B. M., Sutterer, M. J., Cayo, A. A., Schaefer, S. M., Rudolph, K. D., et al. (2014). Behavior problems after early life stress: Contributions of the hippocampus and amygdala. Biological Psychiatry, 77(4), 314–323.
Harburg, E., Erfurt, J. C., Chape, C., Hauenstein, L. S., Schull, W. J., & Schork, M. A. (1973). Socioecological stressor areas and black-white blood pressure: Detroit. Journal of Chronic Diseases, 26, 595–611.
Hardy, R., Kuh, D., Langenberg, C., & Wadsworth, M. E. (2003). Birthweight, childhood social class, and change in adult blood pressure in the 1946 British birth cohort. The Lancet, 362, 1178–1183.
Hargreaves, D. S., Marbini, A. D., & Viner, R. M. (2013). Inequality trends in health and future health risk among English children and young people, 1999–2009. Archives of Disease in Childhood, 98, 850–855.
Heatherton, T. F. (2011). Neuroscience of self and self-regulation. Annual Review of Psychology, 62, 363–390.
Heckman, J. J. (2006). Skill formation and the economics of investing in disadvantaged children. Science, 312, 1900–1902.
Hemingway, H., Shipley, M., Mullen, M. J., Kumari, M., Brunner, E., Taylor, M., et al. (2003). Social and psychosocial influences on inflammatory markers and vascular function in civil servants (the Whitehall II study). American Journal of Cardiology, 92, 984–987.
Hemingway, H., Shipley, M., Brunner, E., Britton, A., Malik, M., & Marmot, M. (2005). Does autonomic function link social position to coronary risk? The Whitehall II study. Circulation, 111, 3071–3077.
Hertzman, C. (1999). The biological embedding of early experience and its effects on health in adulthood. Annals of the New York Academy of Sciences, 896, 85–95.
Hoff, E. (2006). How social contexts support and shape language development. Developmental Review, 26, 55–85.
Holmes, A., & Wellman, C. L. (2009). Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neuroscience & Biobehavioral Reviews, 33, 773–783.
Holz, N. E., Boecker, R., Hohm, E., Zohsel, K., Buchmann, A. F., Blomeyer, D., & Plichta, M. M. (2015). The longterm impact of early life poverty on orbitofrontal cortex volume in adulthood: results from a prospective study over 25 years. Neuropsychopharmacology, 40(4), 996–1004.
Howe, L. D., Lawlor, D. A., & Propper, C. (2013). Trajectories of socioeconomic inequalities in health, behaviors and academic achievement across childhood and adolescence. Journal of Epidemiology and Community Health, 67, 358–364.
Ishizaki, M., Martikainen, P., Nakagawa, H., & Marmot, M. (2000). The relationship between employment grade and plasma fibrinogen level among Japanese male employees. YKKJ Research Group. Atherosclerosis, 151, 415–421.
Jackson, R. W., Treiber, F. A., Turner, J. R., Davis, H., & Strong, W. B. (1999). Effects of race, sex, and socioeconomic status upon cardiovascular stress responsivity and recovery in youth. International Journal of Psychophysiology, 31, 111–119.
Jacobson, L., & Sapolsky, R. (1991). The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocrine Reviews, 12, 118–134.
Janicki-Deverts, D., Cohen, S., Adler, N. E., Schwartz, J. E., Matthews, K. A., & Seeman, T. E. (2007). Socioeconomic status is related to urinary catecholamines in the coronary Artery risk development in young adults (CARDIA) study. Psychosomatic Medicine, 69, 514–520.
Jarcho, M. R., Slavich, G. M., Tylova-Stein, H., Wolkowitz, O. M., & Burke, H. M. (2013). Dysregulated diurnal cortisol pattern is associated with glucocorticoid resistance in women with major depressive disorder. Biological Psychology, 93, 150–158.
Jarrin, D. C., McGrath, J. J., Silverstein, J. E., & Drake, C. (2013). Objective and subjective socioeconomic gradients exist for sleep quality, sleep latency, sleep duration, weekend oversleep, and daytime sleepiness in adults. Behavioral Sleep Medicine, 11, 144–158.
Jarrin, D. C., McGrath, J. J., & Quon, E. C. (2014). Objective and subjective socioeconomic gradients exist for sleep in children and adolescents. Health Psychology, 33, 301.
Jednorog, K., Altarelli, I., Monzalvo, K., Fluss, J., Dubois, J., Billard, C., et al. (2012). The influence of socioeconomic status on children’s brain structure. PloS One, 7, e42486.
Johnson, N. F., Kim, C., & Gold, B. T. (2013). Socioeconomic status is positively correlated with frontal white matter integrity in aging. Age, 35, 2045–2056.
Jousilahti, P., Salomaa, V., Rasi, V., Vahtera, E., & Palosuo, T. (2003). Association of markers of systemic inflammation, C reactive protein, serum amyloid a, and fibrinogen, with socioeconomic status. Journal of Epidemiology & Community Health, 57, 730–733.
Jung, R. E., Grazioplene, R., Caprihan, A., Chavez, R. S., & Haier, R. J. (2010). White matter integrity, creativity, and psychopathology: Disentangling constructs with diffusion tensor imaging. PloS One, 53, e9818.
Juster, R.-P., McEwen, B. S., & Lupien, S. J. (2010). Allostatic load biomarkers of chronic stress and impact on health and cognition. Neuroscience & Biobehavioral Reviews, 35, 2–16.
Juster, R.-P., Bizik, G., Picard, M., Arsenault-Lapierre, G., Sindi, S., Trepanier, L., et al. (2011). A transdisciplinary perspective of chronic stress in relation to psychopathology throughout life span development. Development and Psychopathology, 23, 725–776.
Kahn, M., Sheppes, G., & Sadeh, A. (2013). Sleep and emotions: Bidirectional links and underlying mechanisms. International Journal of Psychophysiology, 89, 218–228.
Kakinami, L., Seguin, L., Lambert, M., Gauvin, L., Nikiema, B., & Paradis, G. (2013a). Comparison of three life course models of poverty in predicting cardiovascular disease risk in youth. Annals of Epidemiology, 23, 485–491.
Kakinami, L., Séguin, L., Lambert, M., Gauvin, L., Nikiema, B., Paradis, G. (2013b). Poverty’s latent effect on adiposity during childhood: Evidence from a Québec birth cohort. Journal of epidemiology and community health, jech-2012.
Kalisch, R. (2009). The functional neuroanatomy of reappraisal: Time matters. Neuroscience & Biobehavioral Reviews, 33, 1215–1226.
Kalisch, R., Wiech, K., Critchley, H. D., & Dolan, R. J. (2006). Levels of appraisal: A medial prefrontal role in high-level appraisal of emotional material. NeuroImage, 30, 1458–1466.
Kaplan, G., & Keil, J. (1993). Sociodemographic factors and cardiovascular disease. Circulation, 88, 1973–1998.
Kapuku, G. K., Treiber, F. A., & Davis, H. C. (2002). Relationships among socioeconomic status, stress induced changes in cortisol, and blood pressure in African American males. Annals of Behavioral Medicine, 24, 320–325.
Karlamangla, A. S., Singer, B. H., Williams, D. R., Schwartz, J. E., Matthews, K. A., Kiefe, C. I., & Seeman, T. E. (2005). Impact of socioeconomic status on longitudinal accumulation of cardiovascular risk in young adults: The CARDIA study (USA). Social Science & Medicine, 60, 999–1015.
Karlamangla, A. S., Friedman, E. M., Seeman, T. E., Stawksi, R. S., & Almeida, D. M. (2013). Daytime trajectories of cortisol: Demographic and socioeconomic differences—Findings from the National Study of daily experiences. Psychoneuroendocrinology, 38, 2585–2597.
Kim, P., & Bianco, H. (2014). How motherhood and poverty change the brain. Zero to Three, 34(4), 29–36.
Kim, P., & Watamura, S. E. (2015). Two open windows: Infant and parent neurobiologic change. Retrieved from http://ascend.aspeninstitute.org/pages/two-open- windows-infant-and-parent-neurobiologic-change.
Kim, P., Leckman, J. F., Mayes, L. C., Newman, M.-A., Feldman, R., & Swain, J. E. (2010a). Perceived quality of maternal care in childhood and structure and function of mothers’ brain. Developmental Science, 13(4), 662–673.
Kim, P., Leckman, J. F., Mayes, L. C., Feldman, R., Wang, X., & Swain, J. E. (2010b). The plasticity of human maternal brain: Longitudinal changes in brain anatomy during the early postpartum period. Behavioral Neuroscience, 124(5), 695–700. doi:10.1037/a0020884.
Kim, P., Evans, G. W., Angstadt, M., Ho, S. S., Sripada, C. S., Swain, J. E., et al. (2013). Effects of childhood poverty and chronic stress on emotion regulatory brain function in adulthood. The Proceedings of the National Academy of Sciences, 110, 18442–18447.
Kim, P., Rigo, P., Mayes, L. C., Feldman, R., Leckman, J. F., & Swain, J. E. (2014). Neural plasticity in fathers of human infants. Social Neuroscience, 9(5), 522–535. doi:10.1080/17470919.2014.933713.
Kim, P., Ho, S. S., Evans, G. W., Liberzon, I., & Swain, J. E. (2015). Childhood social inequalities influences neural processes in young adult caregiving. Developmental Psychobiology, 57(8), 948–960.
Kim-Cohen, J., Moffitt, T. E., Caspi, A., & Taylor, A. (2004). Genetic and environmental processes in young children’s resilience and vulnerability to socioeconomic deprivation. Child Development, 75, 651–668.
Kishiyama, M. M., Boyce, W. T., Jimenez, A. M., Perry, L. M., & Knight, R. T. (2009). Socioeconomic disparities affect prefrontal function in children. Journal of Cognitive Neuroscience, 21, 1106–1115.
Kivimaki, M., Lawlor, D. A., Juonala, M., Smith, G. D., Elovainio, M., Keltikangas-Jarvinen, L., et al. (2005). Lifecourse socioeconomic position, C-reactive protein, and carotid intima-media thickness in young adults: The cardiovascular risk in young Finns study. Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 2197–2202.
Klebanov, P. K., Evans, G. W., & Brooks-Gunn, J. (2014). Poverty, ethnicity, and risk of obesity among low birth weight infants. Journal of Applied Developmental Psychology, 35, 245–253.
Koster, A., Bosma, H., Penninx, B. W., Newman, A. B., Harris, T. B., van Eijk, J. T., et al. (2006). Association of inflammatory markers with socioeconomic status. Journal of Gerontology: Medical Sciences, 61A(3), 284–290.
Krantz, D. S., & Falconer, J. (1995). Measurement of cardiovascular responses. In S. Cohen, R. C. Kessler, & L. Gordon (Eds.), Measuring stress (pp. 193–212). New York: Oxford University Press.
Krishnadas, R., Kim, J., McLean, J., Batty, G. D., McLean, J. S., Millar, K., et al. (2013a). The envirome and the connectome: Exploring the structural noise in the human brain associated with socioeconomic deprivation. Frontiers in Human Neuroscience, 7, 722.
Krishnadas, R., McLean, J., Batty, G. D., Burns, H., Deans, K. A., Ford, I., et al. (2013b). Socioeconomic deprivation and cortical morphology: Psychological, social, and biological determinants of ill health study. Psychosomatic Medicine, 75, 616–623.
Kruschinski, C., Skripuletz, T., Bedoui, S., Raber, K., Straub, R. H., Hoffmann, T., et al. (2008). Postnatal life events affect the severity of asthmatic airway inflammation in the adult rat. Journal of Immunology, 180, 3919–3925.
Kubzansky, L. D., Kawachi, I., & Sparrow, D. (1999). Socioeconomic status, hostility, and risk factor clustering in the Normative Aging Study: Any help from the concept of allostatic load? Annals of Behavioral Medicine, 21(4), 330–338.
Kubzansky, L. D., Sparrow, D., Vokonas, P., & Kawachi, I. (2001). Is the glass half empty or half full? A prospective study of optimism and coronary heart disease in the normative aging study. Psychosomatic Medicine, 63, 910–916.
Kuh, D., & Ben-Shlomo, Y. (2004). A life course approach to chronic disease epidemiology. New York: Oxford University Press.
Kunz-Ebrecht, S. R., Kirschbaum, C., & Steptoe, A. (2004). Work stress, socioeconomic status and neuroendocrine activation over the working day. Social Science and Medicine, 58, 1523–1530.
Laaksonen, M., Sarlio-Lahteenkorva, S., & Lahelma, E. (2004). Multiple dimensions of socioeconomic position and obesity among employees: The Helsinki health study. Obesity Research, 12, 1851–1858.
Langenberg, C., Hardy, R., Kuh, D., Brunner, E., & Wadsworth, M. E. (2003). Central and total obesity in middle aged men and women in relation to lifetime socioeconomic status: evidence from a national birth cohort. Journal of Epidemiology and Community Health, 57, 816–822.
Langenberg, C., Kuh, D., Wadsworth, E. J., Brunner, E., & Hardy, R. (2006). Social circumstances and education: Life course origins of social inequalities in metabolic risk in a prospective national birth cohort. American Journal of Public Health, 96, 2216–2221.
Lawlor, D. A., Ebrahim, S., & Smith, G. D. (2002). Socioeconomic position in childhood and adulthood and insulin resistance: Cross sectional survey using data from British women’s heart and health study. BMJ, 325, 805.
Lawson, G. M., Duda, J. T., Avants, B. B., Wu, J., & Farah, M. J. (2013). Associations between children’s socioeconomic status and prefrontal cortical thickness. Developmental Science, 16, 641–652.
Lee, J., Harris, K. M., & Gordon-Larsen, P. (2008). Life course perspectives on the links between poverty and obesity during the transition to adulthood. Population Research and Policy Review, 28, 505–532.
Lehman, B. J., Taylor, S. E., Kiefe, C. I., & Seeman, T. E. (2009). Relationship of early life stress and psychological functioning to blood pressure in the CARDIA study. Health Psychology, 28, 338.
Leino, M., Porkka, K. V. K., Raitakari, O. T., Laitinen, S., Taimela, S., & Viikari, J. S. (1996). Influence of parental occupation on coronary heart disease risk factors in children. The cardiovascular risk in young Finns study. International Journal of Epidemiology, 25, 1189–1195.
Levine, S. (2005). Developmental determinants of sensitivity and resistance to stress. Psychoneuroendocrinology, 30, 939–946.
Li, L., Power, C., Kelly, S., Kirschbaum, C., & Hertzman, C. (2007). Life-time socio-economic position and cortisol patterns in mid-life. Psychoneuroendocrinology, 32, 824–833.
Lidfeldt, J., Li, T. Y., Hu, F. B., Manson, J. E., & Kawachi, I. (2007). A prospective study of childhood and adult socioeconomic status and incidence of type 2 diabetes in women. American Journal of Epidemiology, 165, 882–889.
Lorant, V., Deliege, D., Eaton, W., Robert, A., Philippot, P., & Ansseau, M. (2003). Socioeconomic inequalities in depression: A meta-analysis. American Journal of Epidemiology, 157, 98–112.
Loucks, E. B., Sullivan, L. M., Hayes, L. J., D’Agostino, R. B., Larson, M. G., Vasan, R. S., et al. (2006). Association of educational level with inflammatory markers in the Framingham offspring study. American Journal of Epidemiology, 163, 622–628.
Loucks, E. B., Magnusson, K. T., Cook, S., Rehkopf, D. H., Ford, E. S., & Berkman, L. F. (2007). Socioeconomic position and the metabolic syndrome in early, middle, and late life: Evidence from NHANES 1999–2002. Annals of Epidemiology, 17, 782–790.
Love, J. M., Kisker, E. E., Ross, C. M., Schochet, P. Z., Brooks-Gunn, J., Paulsell, D., et al. (2002). Making a difference in the lives of infants and toddlers and their families: the impacts of Early Head Start. U.S. Department of Health and Human Services. Princeton: Mathematica policy research.
Lubbock, L. A., Goh, A., Ali, S., Ritchie, J., & Whooley, M. A. (2005). Relation of low socioeconomic status to C-reactive protein in patients with coronary heart disease (from the heart and soul study). American Journal of Cardiology, 96, 1506–1511.
Luby, J., Belden, A., Botteron, K., Marrus, N., Harms, M. P., Babb, C., et al. (2013). The effects of poverty on childhood brain development: The mediating effect of caregiving and stressful life events. Journal of the American Medical Association Pediatrics, 167(12), 1135–1142.
Lupien, S. J., King, S., Meaney, M. J., & McEwen, B. S. (2000). Child’s stress hormone levels correlate with mother’s socioeconomic status and depressive state. Biological Psychiatry, 48, 976–980.
Lupien, S. J., King, S., Meaney, M., & McEwen, B. S. (2001). Can poverty can under your skin? Basal cortisol levels and cognitive function in children from low and high socioeconomic status. Development and Psychopathology, 13, 653–676.
Lupien, S. J., McEwen, B. S., Gunnar, M. R., & Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nature Reviews Neuroscience, 10, 434–445.
Lyons, D. M., Parker, K. J., Katz, M., & Schatzberg, A. F. (2009). Developmental cascades linking stress inoculation, arousal regulation, and resilience. Frontiers in Behavioral Neuroscience, 3, 32.
Mackey, A. P., Finn, A. S., Leonard, J. A., Jacoby-Senghor, D. S., West, M. R., Gabrieli, C. F., & Gabrieli, J. D. (2015). Neuroanatomical correlates of the income- achievement gap. Psychological Science, 26(6), 925–933. doi:10.1177/0956797615572233.
Marmot, M. G., Bosma, H., Hemingway, H., Brunner, E., & Stansfeld, S. (1997). Contribution of job control and other risk factors to social variations in coronary heart disease incidence. Lancet, 350, 235–239.
Mascaro, J. S., Hackett, P. D., & Rilling, J. K. (2013). Testicular volume is inversely correlated with nurturing-related brain activity in human fathers. Proceedings of the National Academy of Sciences of the United States of America, 110(39), 15746–15751. doi:10.1073/pnas.1305579110.
Matthews, K. A., & Gallo, L. C. (2011). Psychological perspectives on pathways linking socioeconomic status and physical health. Annual Review of Psychology, 62, 501–530.
Matthews, K. A., Kelsey, S. F., Meilahn, E. N., Kuller, L. H., & Wing, R. R. (1989). Educational attainment and behavioral and biological risk factors for coronary heart disease in middle-aged women. American Journal of Epidemiology, 129, 1132–1144.
Matthews, K. A., Gallo, L. C., & Taylor, S. E. (2010). Are psychosocial factors mediators of socioeconomic status and health connections? Annals of the New York Academy of Sciences, 1186, 146–173.
Maty, S. C., Everson-Rose, S. A., Haan, M. N., Raghunathan, T. E., & Kaplan, G. A. (2005). Education, income, occupation, and the 34-year incidence (1965-99) of type 2 diabetes in the Alameda County study. International Journal of Epidemiology, 34, 1274–1281.
Maty, S. C., Lynch, J. W., Raghunathan, T. E., & Kaplan, G. A. (2008). Childhood socioeconomic position, gender, adult body mass index, and incidence of type 2 diabetes mellitus over 34 years in the Alameda County study. American Journal of Public Health, 98, 1486–1494.
McDade, T. W., Hawkley, L. C., & Cacioppo, J. T. (2006). Psychosocial and behavioral predictors of inflammation in middle-aged and older adults: The Chicago health, aging, and social relations study. Psychosomatic Medicine, 68, 376–381.
McEwen, B. S. (2001). Plasticity of the hippocampus: Adaptation to chronic stress and allostatic load. Annals of the New York Academy of Sciences, 933, 265–277.
McEwen, B. S. (2002). The end of stress as we know it. Washington, DC: John Henry Press.
McEwen, B. S. (2005). Glucocorticoids, depression, and mood disorders: Structural remodeling in the brain. Metabolism, 54, 20–23.
McGrath, J. J., Matthews, K. A., & Brady, S. S. (2006). Individual versus neighborhood socioeconomic status and race as predictors of adolescent ambulatory blood pressure and heart rate. Social Science and Medicine, 63, 1442–1453.
McLoyd, V. C. (1998). Socioeconomic disadvantage and child development. American Psychologist, 53, 185–204.
Meijer, M., Röhl, J., Bloomfield, K., & Grittner, U. (2012). Do neighborhoods affect individual mortality? A systematic review and meta-analysis of multilevel studies. Social Science & Medicine, 74, 1204–1212.
Merkin, S. S., Basurto-Dávila, R., Karlamangla, A., Bird, C. E., Lurie, N., Escarce, J., & Seeman, T. (2009). Neighborhoods and cumulative biological risk profiles by race/ethnicity in a national sample of US adults: NHANES III. Annals of Epidemiology, 19, 194–201.
Mervaala, E., Föhr, J., Könönen, M., Valkonen-Korhonen, M., Vainio, P., Partanen, K., et al. (2000). Quantitative MRI of the hippocampus and amygdala in severe depression. Psychological Medicine, 30, 117–125.
Metcalfe, A., Lail, P., Ghali, W. A., & Sauve, R. S. (2011). The association between neighbourhoods and adverse birth outcomes: A systematic review and meta-analysis of multi-level studies. Paediatric and Perinatal Epidemiology, 25, 236–245.
Milad, M. R., & Rauch, S. L. (2007). The role of the orbitofrontal cortex in anxiety disorders. Annals of the New York Academy of Sciences, 1121, 546–561.
Miller, G., & Chen, E. (2007a). Unfavorable socioeconomic conditions in early life presage expression of proinflammatory phenotype in adolescence. Psychosomatic Medicine, 69, 402–409.
Miller, G. E., & Chen, E. (2013). The biological residue of childhood poverty. Child Development Perspectives, 7, 67–73.
Miller, G. E., Chen, E., Fok, A. K., Walker, H., Lim, A., Nicholls, S., et al. (2009). Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. Proceedings of the National Academy of Sciences, 106, 14716–14721.
Miller, G. E., Chen, E., & Parker, K. J. (2011). Psychological stress in childhood and susceptibility to the chronic diseases of aging: Moving toward a model of behavioral and biological mechanisms. Psychological Bulletin, 137(6), 959–997.
Miller, G. E., Brody, G. H., Yu, T., & Chen, E. (2014). A family-oriented psychosocial intervention reduces inflammation in low-SES African American youth. Proceedings of the National Academy of Sciences, 111, 11287–11292.
Moore, M. E., Stunkard, A., & Srole, L. (1962). Obesity, social class, and mental illness. Journal of the American Medical Association, 181, 138–142.
Morgan, J. K., Shaw, D. S., & Forbes, E. E. (2014). Maternal depression and warmth during childhood predict age 20 neural response to reward. Journal of the American Academy of Child and Adolescent Psychiatry, 53(108–117), e101.
Musante, L., Treiber, F. A., Kapuku, G. K., Moore, D., Davis, H., & Strong, W. B. (2000). The effects of life events on cardiovascular reactivity to behavioral stressors as a function of socioeconomic status, ethnicity, and sex. Psychosomatic Medicine, 62, 760–767.
Muscatell, K. A., Morelli, S. A., Falk, E. B., Way, B. M., Pfeifer, J. H., Galinsky, A. D., et al. (2012). Social status modulates neural activity in the mentalizing network. NeuroImage, 60, 1771–1777.
Musser, E. D., Kaiser-Laurent, H., & Ablow, J. C. (2012). The neural correlates of maternal sensitivity: An fMRI study. Developmental Cognitive Neuroscience, 2(4), 428–436. doi:10.1016/j.dcn.2012.04.003.
Narita, K., Takei, Y., Suda, M., Aoyama, Y., Uehara, T., Kosaka, H., et al. (2010). Relationship of parental bonding styles with gray matter volume of dorsolateral prefrontal cortex in young adults. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 34, 624–631.
Neville, H. J., Stevens, C., Pakulak, E., Bell, T. A., Fanning, J., Klein, S., & Isbell, E. (2013). Family-based training program improves brain function, cognition, and behavior in lower socioeconomic status preschoolers. Proceedings of the National Academy of Sciences, 110, 12138–12143.
Newport, D. J., Stowe, Z. N., & Nemeroff, C. B. (2002). Parental depression: Animal models of an adverse life event. American Journal of Psychiatry, 159, 1265–1283.
Noble, K. G., Wolmetz, M. E., Ochs, L. G., Farah, M. J., & McCandliss, B. D. (2006). Brain–behavior relationships in reading acquisition are modulated by socioeconomic factors. Developmental Science, 9, 642–654.
Noble, K. G., Grieve, S. M., Korgaonkar, M. S., Engelhardt, L. E., Griffith, E. Y., Williams, L. M., & Brickman, A. M. (2012a). Hippocampal volume varies with educational attainment across the life-span. Frontiers in Human Neuroscience, 6, 307.
Noble, K. G., Houston, S. M., Kan, E., & Sowell, E. R. (2012b). Neural correlates of socioeconomic status in the developing human brain. Developmental Science, 15, 516–527.
Noble, K. G., Houston, S. M., Brito, N. H., Bartsch, H., Kan, E., Kuperman, J. M., et al. (2015). Family income, parental education and brain structure in children and adolescents. Nature Neuroscience, 18(5), 773–778. doi:10.1038/nn.3983.
Ochsner, K. N., Silvers, J. A., & Buhle, J. T. (2012). Functional imaging studies of emotion regulation: A synthetic review and evolving model of the cognitive control of emotion. Annals of the New York Academy of Sciences, 1251, E1–E24.
Olds, D. L., Eckenrode, J., Henderson, C. R., Jr., Kitzman, H., Powers, J., Cole, R., et al. (1997). Long-term effects of home visitation on maternal life course and child abuse and neglect. Fifteen-year follow-up of a randomized trial. The Journal of the American Medical Association, 278, 637–643.
Otero, G. A. (1997). Poverty, cultural disadvantage and brain development: A study of pre-school children in Mexico. Electroencephalography and Clinical Neurophysiology, 102, 512–516.
Otero, G., Pliego-Rivero, F., Fernández, T., & Ricardo, J. (2003). EEG development in children with sociocultural disadvantages: A follow-up study. Clinical Neurophysiology, 114, 1918–1925.
Owen, N., Poulton, T., Hay, F. C., Mohamed-Ali, V., & Steptoe, A. (2003). Socioeconomic status, C-reactive protein, immune factors, and responses to acute mental stress. Brain, Behavior, and Immunity, 17, 286–295.
Panagiotakos, D. B., Pitsavos, C. E., Chrysohoou, C. A., Skoumas, J., Toutouza, M., Belegrinos, D., et al. (2004). The association between educational status and risk factors related to cardiovascular disease in healthy individuals: The ATTICA study. Annals of Epidemiology, 14, 188–194.
Patel, N. P., Grandner, M. A., Xie, D., Branas, C. C., & Gooneratne, N. (2010). "sleep disparity" in the population: Poor sleep quality is strongly associated with poverty and ethnicity. BMC Public Health, 10, 475.
Petersen, K. L., Marsland, A. L., Flory, J., Votruba-Drzal, E., Muldoon, M. F., & Manuck, S. B. (2008). Community socioeconomic status is associated with circulating interleukin-6 and C-reactive protein. Psychosomatic Medicine, 70, 646–652.
Phan, K. L., Orlichenko, A., Boyd, E., Angstadt, M., Coccaro, E. F., Liberzon, I., & Arfanakis, K. (2009). Preliminary evidence of white matter abnormality in the uncinate fasciculus in generalized social anxiety disorder. Biological Psychiatry, 66, 691–694.
Philip, N. S., Sweet, L. H., Tyrka, A. R., Price, L. H., Bloom, R. F., & Carpenter, L. L. (2013). Decreased default network connectivity is associated with early life stress in medication-free healthy adults. European Neuropsychopharmacology, 23, 24–32.
Pietras, S. A., & Goodman, E. (2013). Socioeconomic status gradients in inflammation in adolescence. Psychosomatic Medicine, 75, 442–448.
Piras, F., Cherubini, A., Caltagirone, C., & Spalletta, G. (2011). Education mediates microstructural changes in bilateral hippocampus. Human Brain Mapping, 32, 282–289.
Pollitt, R. A., Kaufman, J. S., Rose, K. M., Diez-Roux, A. V., Zeng, D., & Heiss, G. (2007). Early-life and adult socioeconomic status and inflammatory risk markers in adulthood. European Journal of Epidemiology, 22, 55–66.
Pollitt, R. A., Kaufman, J. S., Rose, K. M., Diez-Roux, A. V., Zeng, D., & Heiss, G. (2008). Cumulative life course and adult socioeconomic status and markers of inflammation in adulthood. Journal of Epidemiology & Community Health, 62, 484–491.
Poulton, R., Caspi, A., Milne, B. J., Thomson, W. M., Taylor, A., Sears, M. R., & Moffitt, T. E. (2002). Association between children’s experience of socioeconomic disadvantage and adult health: A life-course study. The Lancet, 360, 1640–1645.
Power, C., Manor, O., & Matthews, S. (2003). Child to adult socioeconomic conditions and obesity in a national cohort. International Journal of Obesity, 27, 1081–1086.
Power, C., Graham, H., Due, P., Hallqvist, J., Joung, I., Kuhn, D., & Lynch, J. W. (2005). The contribution of childhood and adult socioeconomic position to adult obesity and smoking behavior: An international comparison. International Journal of Epidemiology, 34, 335–344.
Price, J. L., & Drevets, W. C. (2012). Neural circuits underlying the pathophysiology of mood disorders. Trends in Cognitive Sciences, 16, 61–71.
Radley, J. J., Arias, C. M., & Sawchenko, P. E. (2006). Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. The Journal of Neuroscience, 26, 12967–12976.
Rainisch, B. K. W., & Upchurch, D. M. (2013). Sociodemographic correlates of allostatic load among a national sample of adolescents: Findings from the National Health and nutrition examination survey, 1999–2008. Journal of Adolescent Health, 53, 506–511.
Raizada, R. D., & Kishiyama, M. M. (2010). Effects of socioeconomic status on brain development, and how cognitive neuroscience may contribute to levelling the playing field. Frontiers in Human Neuroscience, 4, 3.
Rao, U., Dahl, R. E., Ryan, N. D., Birmaher, B., Williamson, D. E., Giles, D. E., et al. (1996). The relationship between longitudinal clinical course and sleep and cortisol changes in adolescent depression. Biological Psychiatry, 40, 474–484.
Rathmann, W., Haastert, B., Giani, G., Koenig, W., Imhof, A., Herder, C., et al. (2006). Is inflammation a causal chain between low socioeconomic status and type 2 diabetes? Results from the KORA survey 2000. European Journal of Epidemiology, 21, 55–60.
Repetti, R. L., Taylor, S. E., & Seeman, T. E. (2002). Risky families: Family social environments and the mental and physical health of offspring. Psychological Bulletin, 128, 330–366.
Riemann, D., Voderholzer, U., Spiegelhalder, K., Hornyak, M., Buysse, D. J., Nissen, C., et al. (2007). Chronic insomnia and MRI-measured hippocampal volumes: A pilot study. Sleep, 30, 955.
Rodrigues, S. M., LeDoux, J. E., & Sapolsky, R. M. (2009). The influence of stress hormones on fear circuitry. Annual Review of Neuroscience, 32, 289–313.
Ross, C. E. (2000). Neighborhood disadvantage and adult depression. Journal of Health and Social Behavior, 41(2), 177–187.
Roy, A. L., McCoy, D. C., & Raver, C. C. (2014). Instability versus quality: Residential mobility, neighborhood poverty, and children’s self-regulation. Developmental Psychology, 50(7), 1891–1896.
Sameroff, A. (2010). A unified theory of development: A dialectic integration of nature and nurture. Child Development, 81(1), 6–22. doi:10.1111/j.1467-8624.2009.01378.x.
Sariaslan, A., Larsson, H., D’Onofrio, B., Langstrom, N., & Lichtenstein, P. (2014). Childhood family income, adolescent violent criminality and substance misuse: Quasi-experimental total population study. British Journal of Psychiatry, 205(4), 286–290.
Schreier, H. M. C., & Chen, E. (2010). Socioeconomic status in one’s childhood predicts offspring cardiovascular risk. Brain, Behavior, and Immunity, 24, 1324–1331.
Schreier, H. M. C., & Chen, E. (2013). Socioeconomic status and the health of youth: A multilevel, multidomain approach to conceptualizing pathways. Psychological Bulletin, 139, 606–654.
Schreier, H. M., Roy, L. B., Frimer, L. T., & Chen, E. (2014). Family chaos and adolescent inflammatory profiles: The moderating role of socioeconomic status. Psychosomatic Medicine, 76, 460–467.
Schulz, A. J., Mentz, G., Lachance, L., Johnson, J., Gaines, C., & Israel, B. A. (2012). Associations between socioeconomic status and allostatic load: Effects of neighborhood poverty and tests of mediating pathways. American Journal of Public Health, 102, 1706–1714.
Sedlak, A. J., & Broadhurst, D. D. (1996). Third National Incidence Study of child abuse and neglect. Washington, DC: US Department of Health and Human Services.
Seeman, T. E., McEwen, B. S., Rowe, J. W., & Singer, B. H. (2001). Allostatic load as a marker of cumulative biological risk: MacArthur studies of successful aging. Proceedings of the National Academy of Sciences, 98, 4770–4775.
Seeman, T. E., Crimmins, E., Huang, M., Singer, B. H., Bucur, A., Gruenewald, T., et al. (2004). Cumulative biological risk and socioeconomic differences in mortality: MacArthur studies of successful aging. Social Science & Medicine, 58, 1958–1997.
Seeman, T., Merkin, S. S., Crimmins, E., Koretz, B., Charette, S., & Karlamangla, A. (2008). Education, income and ethnic differences in cumulative biological risk profiles in a national sample of US adults: NHANES III (1988–1994). Social Science & Medicine, 66, 72–87.
Seeman, T., Epel, E., Gruenewald, T., Karlamangla, A., & Mc Ewen, B. S. (2010a). Socio-economic differentials in peripheral biology: Cumulative allostatic load. Annals of the New York Academy of Sciences, 1186, 223–239.
Sheridan, M. A., Sarsour, K., Jutte, D., D’Esposito, M., & Boyce, W. T. (2012). The impact of social disparity on prefrontal function in childhood. PloS One, 7, e35744.
Sheridan, M. A., How, J., Araujo, M., Schamberg, M. A., & Nelson, C. A. (2013). What are the links between maternal social status, hippocampal function, and HPA axis function in children? Developmental Science, 16, 665–675.
Shin, L. M., Rauch, S. L., & Pitman, R. K. (2006). Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Annals of the New York Academy of Sciences, 1071, 67–79.
Shonkoff, J. P. (2010). Building a new biodevelopmental framework to guide the future of early childhood policy. Child Development, 81, 357–367.
Shonkoff, J. P. (2012). Leveraging the biology of adversity to address the roots of disparities in health and development. Proceedings of the National Academy of Sciences, 109, 17302–17307.
Shrewsbury, V., & Wardle, J. (2008). Socioeconomic status and adiposity in childhood. Obesity, 16, 275–284.
Silverman, M. E., Muennig, P., Liu, X., Rosen, Z., & Goldstein, M. A. (2009). The impact of socioeconomic status on the neural substrates associated with pleasure. The Open Neuroimaging Journal, 3, 58–63.
Singer, B. H., & Ryff, C. D. (1999). Hierarchies of life histories and associated health risks. In N. E. Adler, M. Marmot, B. S. McEwen, & J. Stewart (Eds.), Socioeconomic status and health in industrial nations (pp. 96–115). New York: New York Academy of Sciences.
Skoe, E., Krizman, J., & Kraus, N. (2013). The impoverished brain: Disparities in maternal education affect the neural response to sound. The Journal of Neuroscience, 33, 17221–17231.
Sloan, R. P., Huang, M.-H., Sidney, S., Liu, K., Williams, O. D., & Seeman, T. (2005). Socioeconomic status and health: Is parasympathetic nervous system activity an intervening mechanism? International Journal of Epidemiology, 34, 309–315.
Sobal, J., & Stunkard, A. J. (1989). Socioeconomic status and obesity: A review of the literature. Psychological Bulletin, 105, 260–275.
Spencer, N. (2000). Poverty and child health. Abingdon: Radcliffe Medical Press.
Spielberg, J. M., Galarce, E. M., Ladouceur, C. D., McMakin, D. L., Olino, T. M., Forbes, E. E., et al. (2015). Adolescent development of inhibition as a function of SES and gender: Converging evidence from behavior and fMRI. Human Brain Mapping. doi:10.1002/hbm.22838.
Spruill, T. M., Gerin, W., Ogedegbe, G., Burg, M., Schwartz, J. E., & Pickering, T. G. (2009). Socioeconomic and psychosocial factors mediate race differences in nocturnal blood pressure dipping. American Journal of Hypertension, 22, 637–642.
Sripada, R. K., Swain, J. E., Evans, G. W., Welsh, R. C., & Liberzon, I. (2014). Childhood poverty and stress reactivity are associated with aberrant functional connectivity in default mode network. Neuropsychopharmacology, 39(9), 2244–2251.
Staff, R. T., Murray, A. D., Ahearn, T. S., Mustafa, N., Fox, H. C., & Whalley, L. J. (2012). Childhood socioeconomic status and adult brain size: Childhood socioeconomic status influences adult hippocampal size. Annals of Neurology, 71, 653–660.
Steenari, M.-R., Vuontela, V., Paavonen, E. J., Carlson, S., Fjällberg, M., & Aronen, E. T. (2003). Working memory and sleep in 6-to 13-year-old schoolchildren. Journal of the American Academy of Child & Adolescent Psychiatry, 42, 85–92.
Stepnowsky, C. J., Jr., Nelesen, R. A., DeJardin, D., & Dimsdale, J. E. (2004). Socioeconomic status is associated with nocturnal blood pressure dipping. Psychosomatic Medicine, 66, 651–655.
Steptoe, A., Feldman, P. J., Kunz, S., Owen, N., Willemsen, G., & Marmot, M. (2002a). Stress responsivity and socioeconomic status: A mechanism for increased cardiovascular disease risk? European Heart Journal, 23, 1757–1763.
Steptoe, A., Owen, N., Kunz-Ebrecht, S., & Mohamed-Ali, V. (2002b). Inflammatory cytokines, socioeconomic status, and acute stress responsivity. Brain, Behavior, and Immunity, 16, 774–784.
Steptoe, A., Kunz-Ebrecht, S., Owen, N., Feldman, P. J., Rumley, A., Lowe, G. D., & Marmot, M. (2003). Influence of socioeconomic status and job control on plasma fibrinogen responses to acute mental stress. Psychosomatic Medicine, 65, 137–144.
Stevens, C., Lauinger, B., & Neville, H. (2009). Differences in the neural mechanisms of selective attention in children from different socioeconomic backgrounds: An event-related brain potential study. Developmental Science, 12, 634–646.
Swain, J. E., Kim, P., Spicer, J., Ho, S. S., Dayton, C. J., Elmadih, A., & Abel, K. M. (2014). Approaching the biology of human parental attachment: Brain imaging, oxytocin and coordinated assessments of mothers and fathers. Brain Research, 1580, 78–101. doi:10.1016/j.brainres.2014.03.007.
Szyf, M., McGowan, P., & Meaney, M. J. (2008). The social environment and the epigenome. Environmental and Molecular Mutagenesis, 49, 46–60.
Tabassum, F., Kumari, M., Rumley, A., Lowe, G., Power, C., & Strachen, D. (2008). Effects of socioeconomic position on inflammatory and hemostatic markers: A life-course analysis in the 1958 British birth cohort. American Journal of Epidemiology, 167, 1332–1341.
Taylor, S., Lerner, J., Sage, R., Lehman, B., & Seeman, T. (2004a). Early environments, emotions, responses to stress and health. Journal of Personality, 72, 1365–1393.
Taylor, S. E., Lerner, J. S., Sage, R. M., Lehman, B. J., & Seeman, T. E. (2004b). Early environment, emotions, responses to stress, and health. Journal of Personality, 72, 1365–1394.
Taylor, S. E., Eisenberger, N. I., Saxbe, D., Lehman, B. J., & Lieberman, M. D. (2006a). Neural responses to emotional stimuli are associated with childhood family stress. Biological Psychiatry, 60, 296–301.
Taylor, S. E., Lehman, B. J., Kiefe, C. I., & Seeman, T. E. (2006b). Relationship of early life stress and psychological functioning to adult C-reactive protein in the coronary Artery risk development in young adults study. Biological Psychiatry, 60, 819–824.
Taylor, S. E., Burklund, L. J., Eisenberger, N. I., Lehman, B. J., Hilmert, C. J., & Lieberman, M. D. (2008). Neural bases of moderation of cortisol stress responses by psychosocial resources. Journal of Personality and Social Psychology, 95, 197–211.
Teipel, S. J., Meindl, T., Wagner, M., Kohl, T., Bürger, K., Reiser, M. F., et al. (2009). White matter microstructure in relation to education in aging and Alzheimer’s disease. Journal of Alzheimer’s Disease, 17, 571–583.
Telzer, E. H., Fuligni, A. J., Lieberman, M. D., & Galván, A. (2013). The effects of poor quality sleep on brain function and risk taking in adolescence. NeuroImage, 71, 275–283.
Tomalski, P., Moore, D. G., Ribeiro, H., Axelsson, E. L., Murphy, E., Karmiloff-Smith, A., et al. (2013). Socioeconomic status and functional brain development – Associations in early infancy. Developmental Science, 16, 676–687.
Tottenham, N., & Sheridan, M. A. (2009). A review of adversity, the amygdala and the hippocampus: A consideration of developmental timing. Frontiers in Human Neuroscience, 3, 68.
Tottenham, N., Hare, T. A., Quinn, B. T., McCarry, T. W., Nurse, M., Gilhooly, T., et al. (2010). Prolonged institutional rearing is associated with atypically large amygdala volume and difficulties in emotion regulation. Developmental Science, 13, 46–61.
U.S. Department of Health and Human Services Administration for Children and Families. (2002). Making a difference in the lives of infants and toddlers and their families: The impacts of early head star. Mathematica Policy Research, Inc. Princeton, NJ Under Contract DHHS-105-95-1936.
Uddin, L. Q., Iacoboni, M., Lange, C., & Keenan, J. P. (2007). The self and social cognition: The role of cortical midline structures and mirror neurons. Trends in Cognitive Sciences, 11, 153–157.
Ulrich-Lai, Y. M., & Herman, J. P. (2009). Neural regulation of endocrine and autonomic stress responses. Nature Reviews Neuroscience, 10, 397–409.
Van Ijzendoorn, M. H. (1992). Intergenerational transmission of parenting: A review of studies in nonclinical populations. Developmental Review, 12(1), 76–99.
Wadsworth, M. E., & Berger, L. E. (2006). Adolescents coping with poverty-related family stress: Prospective predictors of coping and psychological symptoms. Journal of Youth and Adolescence, 35, 57–70.
Wadsworth, M. E., & Compas, B. E. (2002). Coping with family conflict and economic strain: The adolescent perspective. Journal of Research on Adolescence, 12, 243–274.
Wadsworth, M. E., Santiago, C. D., Einhorn, L., Etter, E. M., Rienks, S., & Markman, H. (2011). Preliminary efficacy of an intervention to reduce psychosocial stress and improve coping in low-income families. American Journal of Community Psychology, 48, 257–271.
Wadsworth, M. E., Evans, G. W., Grant, K. E., Carter, J. S., & Duffy, S. (in press). Poverty and the development of psychopathology. In D. Chicchetti (Ed.), Developmental psychopathology (3rd ed.). New York: Wiley.
Walker, M. P. (2008). Cognitive consequences of sleep and sleep loss. Sleep Medicine, 9, S29–S34.
Walker, L., Timmerman, G. M., Kim, M., & Sterling, B. (2002). Relationships between body image and depressive symptoms during postpartum in ethnically diverse, low income women. Women and Health, 36(3), 101–121. doi:10.1300/J013v36n03_07.
Walter, H. J., & Hofman, A. (1987). Socioeconomic status, ethnic origin, and risk factors for coronary heart disease in children. American Heart Journal, 113, 812–818.
Wamala, S. P., Wolk, A., Schenck-Gustafsson, K., & Orth-Gomér, K. (1997). Lipid profile and socioeconomic status in healthy middle aged women in Sweden. Journal of Epidemiology and Community Health, 51, 400–407.
Wamala, S. P., Murray, M. A., Horsten, M., Eriksson, M., Schenck-Gustafsson, K., Hamsten, A., et al. (1999). Socioeconomic status and determinants of hemostatic function in healthy women. Arteriosclerosis, Thrombosis, and Vascular Biology, 19, 485–492.
Wanamethee, G., Whincup, P. H., Shaper, G., & Walker, M. (1996). Influence of father’s social class on cardiovascular disease in middle-aged men. The Lancet, 348, 1254–1255.
Weaver, I. C. G., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004). Epigenetic programming by maternal behavior. Nature Neuroscience, 7, 847–854.
Wells, N. M., Evans, G. W., Beavis, A., & Ong, A. D. (2010). Early childhood poverty, cumulative risk exposure, and body mass index trajectories through young adulthood. American Journal of Public Health, 100(12), 2507–2512.
Whinnery, J., Jackson, N., Rattanaumpawan, P., & Grandner, M. A. (2013). Short and long sleep duration associated with race/ethnicity, sociodemographics, and socioeconomic position. Sleep, 37, 601–611.
Widom, C. S., & Nikulina, V. (2012). Long-term consequences of child neglect in low- income families. In V. Maholmes & R. B. King (Eds.), The Oxford handbook of poverty and child development (pp. 68–85). New York: Oxford University Press.
Williams, R. B., Marchuk, D. A., Seigler, I. C., Barefoot, J. C., Helms, M. J., Brummett, B. H., et al. (2008). Childhood socioeconomic status and serotonin transporter gene polymorphism enhance cardiovascular reactivity to mental stress. Psychosomatic Medicine, 70, 32–39.
Wilson, T. W., Kaplan, G. A., Kauhanen, J., Cohen, R. D., Wu, M., Salonen, R., & Salonen, J. T. (1993). Association between plasma fibrinogen concentration and five socioeconomic indices in the Kuopio ischemic heart disease risk factor study. American Journal of Epidemiology, 137, 292–300.
Wilson, D. K., Kliewer, W., Plybon, L., & Sica, D. A. (2000). Socioeconomic status and blood pressure reactivity in healthy black adolescents. Hypertension, 35, 496–500.
Worthman, C. M., & Kuzara, J. (2005). Life history and the early origins of health differentials. American Journal of Human Biology, 17(1), 95–112. doi:10.1002/ajhb.20096.
Yanagisawa, K., Masui, K., Furutani, K., Nomura, M., Yoshida, H., & Ura, M. (2013). Family socioeconomic status modulates the coping-related neural response of offspring. Social Cognitive and Affective Neuroscience, 8, 617–622.
Zalewski, M., Lengua, L. J., Kiff, C. J., & Fisher, P. A. (2012). Understanding the relation of low income to HPA-axis functioning in preschool children: Cumulative family risk and parenting as pathways to disruptions in cortisol. Child Psychiatry & Human Development, 43, 924–942.
Ziol-Guest, K. M., Duncan, G. J., Kalil, A., & Boyce, W. T. (2012). Early childhood poverty, immune-mediated disease processes, and adult productivity. Proceedings of the National Academy of Sciences, 109(Suppl 2), 17289–17293.
Acknowledgments
The authors wish to acknowledge Christian Capistrano for literature research assistance and Christina Congleton for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2018 The Author(s)
About this chapter

Cite this chapter
Kim, P., Evans, G.W., Chen, E., Miller, G., Seeman, T. (2018). How Socioeconomic Disadvantages Get Under the Skin and into the Brain to Influence Health Development Across the Lifespan. In: Halfon, N., Forrest, C., Lerner, R., Faustman, E. (eds) Handbook of Life Course Health Development . Springer, Cham. https://doi.org/10.1007/978-3-319-47143-3_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-47143-3_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-47141-9
Online ISBN: 978-3-319-47143-3
eBook Packages: MedicineMedicine (R0)