
Abstract
Deep Convolutional Neural Networks (DCNN) has shown excellent performance in a variety of machine learning tasks. This paper presents Deep Convolutional Neural Fields (DeepCNF), an integration of DCNN with Conditional Random Field (CRF), for sequence labeling with an imbalanced label distribution. The widely-used training methods, such as maximum-likelihood and maximum labelwise accuracy, do not work well on imbalanced data. To handle this, we present a new training algorithm called maximum-AUC for DeepCNF. That is, we train DeepCNF by directly maximizing the empirical Area Under the ROC Curve (AUC), which is an unbiased measurement for imbalanced data. To fulfill this, we formulate AUC in a pairwise ranking framework, approximate it by a polynomial function and then apply a gradient-based procedure to optimize it. Our experimental results confirm that maximum-AUC greatly outperforms the other two training methods on 8-state secondary structure prediction and disorder prediction since their label distributions are highly imbalanced and also has similar performance as the other two training methods on solvent accessibility prediction, which has three equally-distributed labels. Furthermore, our experimental results show that our AUC-trained DeepCNF models greatly outperform existing popular predictors of these three tasks. The data and software related to this paper are available at https://github.com/realbigws/DeepCNF_AUC.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others
Keywords
- Sequence Labeling Problem
- Deep Convolutional Neural Networks (DCNN)
- Disorder Prediction (DISO)
- Label Distribution
- Conditional Neural Fields (CNF)
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Deep Convolutional Neural Networks (DCNN), originated by Yann LeCun at 1998 [30] for document recognition, is being widely used in a plethora of machine learning (ML) tasks ranging from speech recognition [22], to computer vision [27], and to computational biology [9]. DCNN is good at capturing medium- and/or long-range structured information in a hierarchical manner. To handle structured data, [5] has integrated DCNN with fully connected Conditional Random Fields (CRF) for semantic image segmentation. Here we present Deep Convolutional Neural Fields (DeepCNF), which is an integration of DCNN and linear-chain CRF, to address the task of sequence labeling and apply it to three important biology problems: solvent accessibility prediction (ACC), disorder prediction (DISO), and 8-state secondary structure prediction (SS8) [24, 34].
A protein sequence can be viewed as a string of amino acids (also called residues in the protein context) and we want to predict a label for each residue. In this paper we consider three types of labels: solvent accessibility, disorder state and 8-state secondary structure. These three structure properties are very important to the understanding of protein structure and function. The solvent accessibility is important for protein folding [10], the order/disorder state plays an important role in many biological processes [37], and protein secondary structure(SS) relates to local backbone conformation of a protein sequence [38]. The label distribution in these problems varies from almost uniform to highly imbalanced. For example, only \(\sim \)6 % of residues are shown to be disordered [19]. Some SS labels, such as 3–10 helix, beta-bridge, and pi-helix are extremely rare [46]. The widely-used training methods, such as maximum-likelihood [29] and maximum labelwise accuracy [16], perform well on data with balanced labels but not on highly-imbalanced data [8].
This paper presents a new maximum-AUC method to train DeepCNF for imbalanced sequence data. Specifically, we train DeepCNF by maximizing Area Under the ROC Curve (AUC), which is a good measure for class-imbalanced data [7]. Taking disorder prediction as an example, random guess can obtain \(\sim \)94 % per-residue accuracy, but its AUC is only \(\sim \)0.5. AUC is insensitive to changes in class distribution because the ROC curve specifies the relationship between false positive (FP) rate and true positive (TP) rate, which are independent of class distribution [7]. However, it is very challenging to directly optimize AUC. A few algorithms have been developed to maximize AUC on unstructured data [21, 23, 36], but to the best of our knowledge, there is no such an algorithm for imbalanced structured data (e.g., sequence data addressed here). To train DeepCNF by maximum-AUC, we formulate the AUC function in a ranking framework, approximate it by a polynomial Chebyshev function [3] and then use L-BFGS [31] to optimize it.
Our experimental results show that when the label distribution is almost uniform, there is no big difference between the three training methods. Otherwise, maximum-AUC results in better AUC and Mcc than the other two methods. Tested on several publicly available benchmark data, our AUC-trained DeepCNF model obtains the best performance on all the three protein sequence labeling tasks. In particular, at a similar specificity level, our method obtains better precision and sensitivity for those labels with a much smaller occurring frequency.
Contributions. 1. A novel training algorithm that directly maximizes the empirical AUC to learn DeepCNF model from imbalanced structured data. 2. Studying three training methods, i.e. maximum-likelihood, maximum labelwise accuracy, and maximum-AUC, for DeepCNF and testing them on three real-world protein sequence labeling problems, in which the label distribution varies from almost uniform to highly imbalanced. 3. Achieving the state-of-the-art performance on three important protein sequence labeling problems. 4. All benchmarks are public available, and the code is available online at https://github.com/realbigws/DeepCNF_AUC. A web server is also implemented and available at http://raptorx.uchicago.edu/StructurePropertyPred/predict/ [43].
1.1 Notations
Let L denote the sequence length, [L] denote the set {1, 2,..., L}. For a finite set S, let |S| denote its cardinality. Let \(X = (X_1, X_2, \dots , X_L), y=(y_1, y_2, \dots , y_L)\) denote the input features and labels respectively for position \(i, i\in [L]\). Denote \(\varSigma \) as the set of all possible labels, i.e., \(y_i \in \varSigma , \forall i\in [L]\).
2 Related Work
Class imbalance issue is a long-standing notorious problem. Early works have addressed this issue through data-level methods, which change the empirical distribution of the training data to create a new balanced dataset [20]. These methods include (a) under-sampling the majority class; (b) over-sampling the minority class; or (c) combining both under-sampling and over-sampling [4, 13, 32].
As AUC is an unbiased measurement for class-imbalanced data, a variety of approaches have been proposed to directly optimize the AUC value. In particular, (a) Cortes et al. [7] optimized AUC by RankBoost algorithm; (b) Ferri et al. [15] trained a decision tree by using AUC as splitting criteria; (c) Herschtal and Raskutti [21] trained a neural network by optimizing AUC; and (d) Joachims [23] proposed a generalized Support Vector Machines (SVM) that optimizes AUC.
However, all these approaches could only be applied on unstructured models. Recently, Rosenfeld et al. [40] have proposed a learning algorithm for structured models with AUC loss. However, there are three fundamental differences of our method with theirs: (a) our method targets at a sequence labelling problem (of course a structured model) with an imbalance label assignment, while their model is proposed for a ranking problem. Specifically, sequence labeling requires the prediction of the label (might not necessarily be binary) at each position, while the focus of structured ranking is on prediction of binary vectors \((y_1;\dots ; y_n)\) where it is hard (or unnecessary) to exactly predict which \(y_i\) have the value 1. Instead the goal of structured ranking is to rank the items \(1, \dots , n\) such that elements with \(y_i = 1\) are ranked high [40]; (b) our method is based on CRF, while they used structured SVM; and (c) we also studied deep learning extension of our method, while they did not. In summary, to the best of our knowledge, our work is the first sequence labelling study that aims to optimize the AUC value directly under a deep learning framework.
3 Method
3.1 DeepCNF Architecture
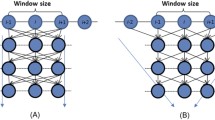
As shown in Fig. 1, DeepCNF has two modules: (i) the Conditional Random Fields (CRF) module consisting of the top layer and the label layer, and (ii) the deep convolutional neural network (DCNN) module covering the input to the top layer. When only one hidden layer is used, DeepCNF becomes Conditional Neural Fields (CNF), a probabilistic graphical model described in [39].

Illustration of a DeepCNF. Here i is the position index and \(X_i\) the associated input features, \(H^k\) represents the k-th hidden layer, and y is the output label. All the layers from the first to the K-th (i.e., top layer) form a DCNN with parameter \(W^k, k\in [K]\), where K is number of hidden layers. The K-th layer and the label layer form a CRF, in which the parameter U specifies the relationship between the output of the K-th layer and the label layer and T is the parameter for adjacent label correlation. Windows size is set to 3 only for illustration.
Given \(X=(X_1, \dots , X_L)\) and \(y=(y_1, \dots , y_L)\), DeepCNF calculates the conditional probability of y on the input X with parameter \(\theta \) as follows,
where \(f_\theta (y, X, i)\) is the binary potential function specifying correlation among adjacent labels at position i, \(g_\theta (y, X, i)\) is the unary potential function modeling relationship between \(y_i\) and input features for position i, and Z(X) is the partition function. Formally, \(f_\theta (\cdot )\) and \(g_\theta (\cdot )\) are defined as follows:

where a and b represent two specific labels for prediction, \(\delta (\cdot )\) is an indicator function, \(A_{a,h}(X,i,W)\) is a deep neural network function for the h-th neuron at position i of the top layer for label a, and W, U and T are the model parameters to be trained. Specifically, W is the parameter for the neural network, U is the parameter connecting the top layer to the label layer, and T is for label correlation. The two potential functions can be merged into a single binary potential function \(f_\theta (y,X,i) = f_\theta (y_{i-1},y_i,X,i) = \sum _{a,b,h} T_{a,b,h} A_{a,b,h}(X,i,W)\delta (y_{i-1}=a) \delta (y_i=b)\). Note that these deep neural network functions for different labels could be shared to \(A_h(X, i, W)\). To control model complexity and avoid over-fitting, we add a \(L_2\)-norm penalty term as the regularization factor.
Figure 1 shows two adjacent layers of DCNN. Let \(M_k\) be the number of neurons for a single position at the k-th layer. Let \(X_i(h)\) be the h-th feature at the input layer for residue i and \(H_i^k(h)\) denote the output value of the h-th neuron of position i at layer k. When \(k=1\), \(H^k\) is actually the input feature X. Otherwise, \(H^k\) is a matrix of dimension \(L\times M_k\). Let \(2N_k+1\) be the window size at the k-th layer. Mathematically, \(H_i^k(h)\) is defined as follows:
Meanwhile, \(\pi (\cdot )\) is the activation function, either the sigmoid (i.e. \(1/(1+\exp (-x))\)) or the tanh (i.e. \((1-\exp (-2x))/(1+\exp (-2x))\)) function. \(W_n^k (-N_k \le n \le N_k)\) is a 2D weight matrix for the connections between the neurons of position \(i+n\) at layer k and the neurons of position i at layer \(k+1\). \(W_n^k(h, h')\) is shared by all the positions in the same layer, so it is position-independent. Here \(h'\) and h index two neurons at the k-th and \((k+1)\)-th layers, respectively. See Appendix about how to calculate the gradient of DCNN by back propagation.
3.2 Objective Functions
Let T be the number of training sequences and \(L_t\) denote the length of sequence t. We study three different training methods: maximum-likelihood, maximum labelwise accuracy, and proposed maximum-AUC.
Maximum-Likelihood. The log-likelihood is a widely-used objective function for training CRF [29]. Mathematically, the log-likelihood is defined as follows:
where \(P_\theta (y|X)\) is defined in Eq. (1).
Maximum Labelwise Accuracy. Gross et al. [16] proposed an objective function that could directly maximize the labelwise accuracy defined as
where \(y_i^{(\tau )}\) denotes the real label at position i, \(P_\theta (y_i^{(\tau )})\) is the predicted probability of the real label at position i. It could be represented by the marginal probability
where \(F_{l_1:l_2}(y,X,\theta ) = \sum _{i=l_1}^{l_2} f_\theta (y, X, i)\).
To obtain a smooth approximation to this objective function, [16] replaces the indicator function with a sigmoid function \(Q_\lambda (x) = 1/(1+\exp (-\lambda x))\) where the parameter \(\lambda \) is set to 15 by default. Then it becomes the following form:
where \(\tilde{y}_i^{(\tau )}\) denote the label other than \(y_i^{(\tau )}\) that has the maximum posterior probability at position i.
Maximum-AUC. The AUC of a predictor function \(P_\theta \) on label \(\tau \) is defined as:
where \(P(\cdot )\) is the probability over all pairs of positive and negative examples, \(D^\tau \) is a set of positive examples with true label \(\tau \), and \(D^{!\tau }\) is a set of negative examples with true label not being \(\tau \). Note that the union of \(D^\tau \) and \(D^{!\tau }\) contains all the training sequence positions, i.e., \(D^\tau =\cup _{t=1}^T \cup _{i=1}^{L_t} \delta _{i,t}^\tau \) where \(\delta _{i,t}^\tau \) is an indicator function. If the true label of the i-th position from sequence t equals to \(\tau \), then \(\delta _{i,t}^\tau \) is equal to 1; otherwise 0. Again, \(P_\theta (y_i^\tau )\) could be represented by the marginal probability \(P_\theta ( y_i^\tau | X^t)\) from the training sequence t. Since it is hard to calculate the derivatives of Eq. (2), we use the following Wilcoxon-Mann-Whitney statistic [18], which is an unbiased estimator of \(AUC(P_\theta , \tau )\):
Finally, by summing over labels, the overall AUC objective function is
\(\sum _\tau AUC^{WMW}(P_\theta , \tau )\).
For a large dataset, the computational cost of AUC by Eq. (3) is high. Recently, Calders and Jaroszewicz [3] proposed a polynomial approximation of AUC which can be computed in linear time. The key idea is to approximate the indicator function \(\delta (x>0)\), where x represents \(P_\theta (y_i^\tau | X) - P_\theta (y_j^\tau |X)\) by a polynomial Chebyshev approximation. That is, we approximate \(\delta (x>0)\) by \(\sum _{\mu \in [d]} c_\mu x^\mu \) where d is the degree and \(c_\mu \) the coefficient of the polynomial [3]. Let \(n_1 = |D^\tau |\) and \(n_0=|D^{!\tau }|\). Using the polynomial Chebyshev approximation, we can approximate Eq. (3) as follows:
where \(\mathcal {Y}_{\mu l} = c_\mu \left( {\begin{array}{c}\mu \\ l\end{array}}\right) (-1)^{\mu -l}\), \(s(P^l, D^\tau ) = \sum _{i\in D^\tau } P(y_i^\tau )^l\) and \(v(P^l, D^{!\tau }) = \sum _{j\in D^{!\tau }} P(y_j^\tau )^l\). Note that we have \(s(P^l, D^\tau ) = \sum _{t\in [T]} \sum _{i\in [L_t]} \delta _{i,t}^\tau P(y_i^\tau )^l\) and a similar structure for \(v(P^l, D^{!\tau })\).
4 Results
In this section presents our experimental results of the AUC-trained DeepCNF models on three protein sequencing problems, which are summarize as follows:
ACC. We used DSSP [26] to calculate the absolute accessible surface area for each residue in a protein and then normalize it by the maximum solvent accessibility to obtain the relative solvent accessibility (RSA) [6]. Solvent accessibility of one residue is classified into 3 labels: buried (B) for RSA from 0 to 10), intermediate (I) for RSA from 10 to 40 and exposed (E) for RSA from 40 to 100. The ratio of these three labels is around 1:1:1 [33].
DISO. Following the definition in [35], we label a residue as disordered (label 1) if it is in a segment of more than three residues missing atomic coordinates in the X-ray structure. Otherwise it is labeled as ordered (label 0). The distribution of these two labels (ordered vs. disordered) is 94:6 [45].
SS8. The 8-state protein secondary structure is calculated by DSSP [26]. In particular, DSSP assigns 3 types for helix (G for 310 helix, H for alpha-helix, and I for pi-helix), 2 types for strand (E for beta-strand and B for beta-bridge), and 3 types for coil (T for beta-turn, S for high curvature loop, and L for irregular) [44]. The distribution of these 8 labels (H,E,L,T,S,G,B,I) is 34:21:20:11:9:4:1:0 [43].
4.1 Dataset
To use a set of non-redundant protein sequences for training and test, we pick one representative sequence from each protein superfamily defined in CATH [42] or SCOP [1]. The test proteins are in different superfamilies than the training proteins, so we can reduce the bias incurred by the sequence profile similarity between the training and test proteins. The publicly available JPRED [11] dataset (http://www.compbio.dundee.ac.uk/jpred4/about.shtml) satisfies such a condition, which has 1338 training and 149 test proteins, respectively, each belonging to a different superfamily. We train the DeepCNF model using the JPRED training set and conduct 7-fold cross validation to determine the model hyper-parameters for each training method.
We also evaluate the predictive performance of our DeepCNF models on the CASP10 [28] and CASP11 [25] test targets (merged to a single CASP dataset) and the recent CAMEO [17] hard test targets. To remove redundancy, we filter the CASP and CAMEO datasets by removing those targets sharing >25 % sequence identity with the JPRED training set. This result in 126 CASP and 147 CAMEO test targets, respectively. See Appendix for their test results.
4.2 Evaluation Criteria
We use Qx to measure the accuracy of sequence labeling where x is the number of different labels for a prediction task. Qx is defined as the percentage of residues for which the predicted labels are correct. In particular, we use Q3 accuracy for ACC prediction, Q8 accuracy for SS8 prediction and Q2 accuracy for disorder prediction.

Q3 accuracy, mean Mcc and AUC of solvent accessibility (ACC) prediction with respect to the DCNN architecture: (left) the number of neurons, (middle) window size, and (right) the number of hidden layers. Training methods: maximum likelihood (black), maximum labelwise accuracy (red) and maximum AUC (green). (Color figure online)
From TP (true positives), TN (true negatives), FP (false positives) and FN (false negatives), we may also calculate sensitivity (sens), specificity (spec), precision (prec) and Matthews correlation coefficient (Mcc) as \(\frac{TP}{TP + FN}, \frac{TN}{TN+FP}, \frac{TP}{TP+FP}\) and \(\frac{TP\times TN - FP\times FN}{\sqrt{(TP+FP)(TN+FP)(TP+FN)(TN+FN)}}\), respectively. We also use AUC as a measure. Mcc and AUC are generally regarded as balanced measures which can be used on class-imbalanced data. Mcc ranges from \(-1\) to +1, with +1 representing a perfect prediction, 0 random prediction and \(-1\) total disagreement between prediction and ground truth. AUC has a minimum value 0 and the best value 1.0. When there are more 2 different labels in a labeling problem, we may also use mean Mcc (denoted as \(\bar{Mcc}\)) and mean AUC (denoted as \(\bar{AUC}\)), which are averaged over all the different labels.
4.3 Performance Comparison on Objective Functions
The architecture of the DCNN in DeepCNF model is mainly determined by the following 3 factors (see Fig. 1): (i) the number of hidden layers; (ii) the number of different neurons at each layer; and (iii) the window size at each layer. We compared three different methods for training the DeepCNF model: maximum likelihood, maximum labelwise accuracy, and maximum AUC for the prediction of three-label solvent accessibility (ACC), two-label order/disorder (DISO), and eight-label secondary structure element (SS8), respectively.

Q2 accuracy, mean Mcc and AUC of disorder (DISO) prediction with respect to the DCNN architecture: (left) the number of neurons, (middle) window size, and (right) the number of hidden layers. Training methods: maximum likelihood (black), maximum labelwise accuracy (red) and maximum AUC (green). (Color figure online)

Q8 accuracy, mean Mcc and AUC of 8-state secondary structure (SS8) prediction with respect to the DCNN architecture: (left) the number of neurons, (middle) window size, and (right) the number of hidden layers. Training methods: maximum likelihood (black), maximum labelwise accuracy (red) and maximum AUC (green). (Color figure online)
We conduct 7-fold cross-validation for each possible DCNN architecture, each training method, and each labeling problem using the JPRED dataset. To simplify the analysis, we use the same number of neurons and the same windows size for all hidden layers. By default we use 5 hidden layers, each with 50 different hidden neurons and windows size 11.
Overall, as shown in Figs. 2, 3 to 4, Our DeepCNF model reaches peak performance when it has 4 to 5 hidden layers, 50 to 100 different hidden neurons at each layer, and windows size 11. Further increasing the number of layers, the number of different hidden neurons, and the windows size does not result in significant improvement in Qx accuracy, mean Mcc and AUC, regardless of the training method.
For ACC prediction, as shown in Fig. 2, since the three labels are equally distributed, no matter what training methods are used, the best Q3 accuracy, the best mean Mcc and the best mean AUC are 0.69, 0.45, 0.82, respectively; For DISO prediction, since the two labels are highly imbalanced, as shown in Fig. 3, although all three training methods have similar Q2 accuracy 0.94, maximum-AUC obtains mean Mcc and AUC at 0.51 and 0.89, respectively, greatly outperforming the other two; For SS8 prediction, as shown in Fig. 4, since there are three rare labels (i.e., G for 3–10 helix, B for beta-bridge, and I for pi-helix), maximum-AUC has the overall mean Mcc at 0.44 and mean AUC at 0.86, respectively, much better than maximum labelwise accuracy, which has mean Mcc at 0.41 and mean AUC less than 0.8, respectively.
4.4 Performance Comparison with State-of-the-art
Programs to Compare. Since our method is ab initio, we do not compare it with consensus-based or template-based methods. Instead, we compare our method with the following ab initio predictors: (i) for ACC prediction, we compare to SPINE-X [14] and ACCpro5-ab [34]. SPINE-X uses neural networks (NN) while ACCpro5-ab uses bidirectional recurrent neural network (RNN); (ii) for DISO prediction, we compare to DNdisorder [12] and DisoPred3-ab [24]. DNdisorder uses deep belief network (DBN) while DisoPred3-ab uses support vector machine (SVM) and NN for prediction; (iii) for SS8 prediction, we compare our method with SSpro5-ab [34] and RaptorX-SS8 [46]. SSpro5-ab is based on RNN while RaptorX-SS8 uses conditional neural field (CNF) [39]. We cannot evaluate Zhous method [48] since it is not publicly available.
Overall Evaluation. Here we only compare our AUC-trained DeepCNF model (trained by the JPRED data) to the other state-of-the-art methods on the CASP and CAMEO datasets. As shown in Tables 1, 2 to 3, our AUC-trained DeepCNF model outperforms thPlease refer to appendix for a more detailed review for those problems and existing state-of-the art algorithms.e other predictors on all the three sequence labeling problems, in terms of the Qx accuracy, Mcc and AUC. When the label distribution is highly imbalanced, our method greatly exceeds the others in terms of Mcc and AUC. Specifically, for DISO prediction on the CASP data, our method achieves 0.55 Mcc and 0.89 AUC, respectively, greatly outperforming DNdisorder (0.37 Mcc and 0.81 AUC) and DisoPred3_ab (0.47 Mcc and 0.84 AUC). For SS8 prediction on the CAMEO data, our method obtains 0.42 Mcc and 0.83 AUC, respectively, much better than SSpro5_ab (0.37 Mcc and 0.78 AUC) and RaptorX-SS8 (0.38 Mcc and 0.79 AUC).
Sensitivity, Specificity, and Precision. Tables 4 and 5 list the sensitivity, specificity, and precision on each label obtained by our method and the other competing methods evaluated on the merged CASP and CAMEO data. Overall, at a high specificity level, our method obtains compatible or better precision and sensitivity for each label, especially for those rare labels such as G, I, B, S, T for SS8, and disorder state for DISO. Taking SS8 prediction as an example, for pi-helix (I), our method has sensitivity and precision 0.18 and 0.33 respectively, while the second best method obtains 0.03 and 0.12, respectively. For beta-bridge (B), our method obtains sensitivity and precision 0.13 and 0.42, respectively, while the second best method obtains 0.07 and 0.34, respectively (Table 6).
5 Discussions
We have presented a novel training algorithm that directly maximizes the empirical AUC to learn DeepCNF model (DCNN+CRF) from imbalanced structured data. We also studied the behavior of three training methods: maximum-likelihood, maximum labelwise accuracy, and maximum-AUC, on three real-world protein sequence labeling problems, in which the label distribution varies from equally distributed to highly imbalanced. Evaluated by AUC and Mcc, our maximum-AUC training method achieves the state-of-the-art performance in predicting solvent accessibility, disordered regions, and 8-state secondary structure.
Instead of using a linear-chain CRF, we may model a protein by Markov Random Fields (MRF) to capture long-range residue interactions [47]. As suggested in [41], the predicted residue-residue contact information could further contribute to disorder prediction under the MRF model. In addition to the three protein sequence labeling problems tested in this work, our maximum-AUC training algorithm could be applied to many sequence labeling problems with imbalanced label distributions [20]. For example, in post-translation modification (PTM) site prediction, the phosphorylation and methylation sites occur much less frequently than normal residues [2].
References
Andreeva, A., Howorth, D., Chothia, C., Kulesha, E., Murzin, A.G.: Scop2 prototype: a new approach to protein structure mining. Nucleic Acids Res. 42(D1), D310–D314 (2014)
Blom, N., Sicheritz-Pontén, T., Gupta, R., Gammeltoft, S., Brunak, S.: Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4(6), 1633–1649 (2004)
Calders, T., Jaroszewicz, S.: Efficient AUC optimization for classification. In: Kok, J.N., Koronacki, J., Lopez de Mantaras, R., Matwin, S., Mladenič, D., Skowron, A. (eds.) PKDD 2007. LNCS(LNAI), vol. 4702, pp. 42–53. Springer, Heidelberg (2007). doi:10.1007/978-3-540-74976-9_8
Chawla, N.V., Bowyer, K.W., Hall, L.O., Kegelmeyer, W.P.: Smote: synthetic minority over-sampling technique. J. Artif. Intell. Res. 16, 321–357 (2002)
Chen, L.-C., Papandreou, G., Kokkinos, I., Murphy, K., Yuille, A.L., Semantic image segmentation with deep convolutional nets, fully connected CRFs. arXiv preprint arXiv: 1412.7062 (2014)
Chothia, C.: The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 105(1), 1–12 (1976)
Cortes, C., Mohri, M.: Auc optimization vs. error rate minimization. Adv. Neural Inf. Process. Syst. 16(16), 313–320 (2004)
De Lannoy, G., François, D., Delbeke, J., Verleysen, M.: Weighted conditional random fields for supervised interpatient heartbeat classification. IEEE Trans. Biomed. Eng. 59(1), 241–247 (2012)
Di Lena, P., Nagata, K., Baldi, P.: Deep architectures for protein contact map prediction. Bioinformatics 28(19), 2449–2457 (2012)
Dill, K.A.: Dominant forces in protein folding. Biochemistry 29(31), 7133–7155 (1990)
Drozdetskiy, A., Cole, C., Procter, J., Barton, G.J.: Jpred4: a protein secondary structure prediction server. Nucleic Acids Res., gkv332 (2015)
Eickholt, J., Cheng, J.: Dndisorder: predicting protein disorder using boosting and deep networks. BMC Bioinf. 14(1), 88 (2013)
Estabrooks, A., Jo, T., Japkowicz, N.: A multiple resampling method for learning from imbalanced data sets. Comput. Intell. 20(1), 18–36 (2004)
Faraggi, E., Xue, B., Zhou, Y.: Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by guided-learning through a two-layer neural network. Proteins: Struct., Funct., Bioinf. 74(4), 847–856 (2009)
Ferri, C., Flach, P., Hernández-Orallo, J.: Learning decision trees using the area under the ROC curve. In: ICML, vol. 2, pp. 139–146 (2002)
Gross, S.S., Russakovsky, O., Do, C.B., Batzoglou, S.: Training conditional random fields for maximum labelwise accuracy. In: Advances in Neural Information Processing Systems, pp. 529–536 (2006)
Haas, J., Roth, S., Arnold, K., Kiefer, F., Schmidt, T., Bordoli, L., Schwede, T.: The protein model portala comprehensive resource for protein structure and model information. Database 2013, bat031 (2013)
Hanley, J.A., McNeil, B.J.: The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143(1), 29–36 (1982)
He, B., Wang, K., Liu, Y., Xue, B., Uversky, V.N., Dunker, A.K.: Predicting intrinsic disorder in proteins: an overview. Cell Res. 19(8), 929–949 (2009)
He, H., Garcia, E., et al.: Learning from imbalanced data. IEEE Trans. Knowl. Data Eng. 21(9), 1263–1284 (2009)
Herschtal, A., Raskutti, B.: Optimising area under the ROC curve using gradient descent. In: Proceedings of the Twenty-first International Conference on Machine Learning, p. 49. ACM (2004)
Hinton, G., Deng, L., Dong, Y., Dahl, G.E., Mohamed, A., Jaitly, N., Senior, A., Vanhoucke, V., Nguyen, P., Sainath, T.N., et al.: Deep neural networks for acoustic modeling in speech recognition: the shared views of four research groups. IEEE Sig. Process. Mag. 29(6), 82–97 (2012)
Joachims, T.: A support vector method for multivariate performance measures. In: Proceedings of the 22nd International Conference on Machine Learning, pp. 377–384. ACM (2005)
Jones, D.T., Cozzetto, D.: Disopred3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics 31(6), 857–863 (2015)
Joo, K., Joung, I., Lee, S.Y., Kim, J.Y., Cheng, Q., Manavalan, B., Joung, J.Y., Heo, S., Lee, J., Nam, M., et al.: Template based protein structure modeling by global optimization in CASP11. Proteins: Struct., Funct., Bioinform. (2015)
Kabsch, W., Sander, C.: Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22(12), 2577–2637 (1983)
Krizhevsky, A., Sutskever, I., Hinton, G.E.: Imagenet classification with deep convolutional neural networks. In: Advances in Neural Information Processing Systems, pp. 1097–1105 (2012)
Kryshtafovych, A., Barbato, A., Fidelis, K., Monastyrskyy, B., Schwede, T., Tramontano, A.: Assessment of the assessment: evaluation of the model quality estimates in CASP10. Proteins: Struct., Funct., Bioinform. 82(S2), 112–126 (2014)
Lafferty, J., McCallum, A., Pereira, F.C.: Conditional random fields: probabilistic models for segmenting and labeling sequence data (2001)
LeCun, Y., Bottou, L., Bengio, Y., Haffner, P.: Gradient-based learning applied to document recognition. Proc. IEEE 86(11), 2278–2324 (1998)
Liu, D.C., Nocedal, J.: On the limited memory BFGS method for large scale optimization. Math. Program. 45(1–3), 503–528 (1989)
Liu, X.-Y., Jianxin, W., Zhou, Z.-H.: Exploratory undersampling for class-imbalance learning. IEEE Trans. Syst. Man, Cybern. Part B (Cybern.) 39(2), 539–550 (2009)
Ma, J., Wang, S.: Acconpred: Predicting solvent accessibility and contact number simultaneously by a multitask learning framework under the conditional neural fields model. BioMed Res. Int. 2015 (2015)
Magnan, C.N., Baldi, P.: SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 30(18), 2592–2597 (2014)
Monastyrskyy, B., Fidelis, K., Moult, J., Tramontano, A., Kryshtafovych, A.: Evaluation of disorder predictions in CASP9. Proteins: Struct., Funct., Bioinform. 79(S10), 107–118 (2011)
Narasimhan, H., Agarwal, S.: A structural SVM based approach for optimizing partial AUC. In: Proceedings of the 30th International Conference on Machine Learning, pp. 516–524 (2013)
Oldfield, C.J., Dunker, A.K.: Intrinsically disordered proteins and intrinsically disordered protein regions. Ann. Rev. Biochem. 83, 553–584 (2014)
Pauling, L., Corey, R.B., Branson, H.R.: The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Nat. Acad. Sci. 37(4), 205–211 (1951)
Peng, J., Bo, L., Xu, J.: Conditional neural fields. In: Advances in Neural Information Processing Systems, pp. 1419–1427 (2009)
Rosenfeld, N., Meshi, O., Globerson, A., Tarlow, D.: Learning structured models with the AUC loss and its generalizations. In: Proceedings of the Seventeenth International Conference on Artificial Intelligence and Statistics, pp. 841–849 (2014)
Schlessinger, A., Punta, M., Rost, B.: Natively unstructured regions in proteins identified from contact predictions. Bioinformatics 23(18), 2376–2384 (2007)
Sillitoe, I., Lewis, T.E., Cuff, A., Das, S., Ashford, P., Dawson, N.L., Furnham, N., Laskowski, R.A., Lee, D., Lees, J.G., et al.: Cath: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 43(D1), D376–D381 (2015)
Wang, S., Li, W., Liu, S., Jinbo, X.: Raptorx-property: a web server for protein structure property prediction. Nucleic Acids Res., gkw306 (2016)
Wang, S., Peng, J., Ma, J., Jinbo, X.: Protein secondary structure prediction using deep convolutional neural fields. Sci. Rep. 6 (2016)
Wang, S., Weng, S., Ma, J., Tang, Q.: Deepcnf-d: predicting protein order/disorder regions by weighted deep convolutional neural fields. Int. J. Mol. Sci. 16(8), 17315–17330 (2015)
Wang, Z., Zhao, F., Peng, J., Jinbo, X.: Protein 8-class secondary structure prediction using conditional neural fields. Proteomics 11(19), 3786–3792 (2011)
Jinbo, X., Wang, S., Ma, J.: Protein Homology Detection Through Alignment of Markov Random Fields: Using MRFalign. SpringerBriefs in Computer Science. Springer, Heidelberg (2015)
Zhou, J., Troyanskaya, O.G.: Deep supervised, convolutional generative stochastic network for protein secondary structure prediction. arXiv preprint arXiv:1403.1347 (2014)
Acknowledgments
The authors are grateful to the computing power provided by the UChicago Beagle and RCC allocations. The authors are also grateful to the National Institutes of Health [R01GM0897532 to J.X.] and National Science Foundation [DBI-0960390 to J.X.].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this paper
Cite this paper
Wang, S., Sun, S., Xu, J. (2016). AUC-Maximized Deep Convolutional Neural Fields for Protein Sequence Labeling. In: Frasconi, P., Landwehr, N., Manco, G., Vreeken, J. (eds) Machine Learning and Knowledge Discovery in Databases. ECML PKDD 2016. Lecture Notes in Computer Science(), vol 9852. Springer, Cham. https://doi.org/10.1007/978-3-319-46227-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-46227-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46226-4
Online ISBN: 978-3-319-46227-1
eBook Packages: Computer ScienceComputer Science (R0)