
Abstract
The average mutation density of a mutant population is a major consideration when developing resources for the efficient, cost-effective implementation of reverse genetics methods such as Targeting Induced Local Lesions in Genomes (TILLING). Reliable estimates of mutation density can be achieved by the analysis of several selected loci from hundreds of individuals via mismatch cleavage of heteroduplexes or sequencing. A more rapid and less expensive alternative involves reduced representation sequencing of the genomes of a few individuals. Here we present a detailed protocol for the construction of restriction enzyme sequence comparative analysis (RESCAN) sequencing libraries using a combination of single restriction enzyme digestion and solid phase reversible immobilization-based size selection of restriction fragments. Indexing of the libraries using barcoded adapters enables cost-saving multiplexing prior to sequencing on an Illumina platform. Mutation density can be determined from the resulting sequence data with or without a reference genome.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Traditional mutagenesis has a rich history with regard to the generation of useful genetic variation for plant breeding (Micke et al. 1987; Ahloowalia et al. 2004). More recently, advances in genomic technologies have elevated the efficiency of mutation discovery, thus allowing populations generated by chemical mutagens or irradiation to be utilized for reverse genetics. Traditional mutagenesis holds several advantages over insertional approaches such as transposon and T-DNA tagging. With relative ease and broad applicability of chemical or irradiation mutagenesis, functional genomics can be pursued in plant species that are recalcitrant to tissue culture or genetic transformation. Mutagens such as ethyl methanesulfonate and gamma rays also produce a wider array of genetic lesions (i.e. point mutations, deletions and translocations) than is typically observed with insertional mutagenesis. Point mutations are of particular interest as missense mutations which result in varying degrees of functionality can provide significant insight into gene function in addition to the utility of nonsense mutations that abolish gene function (i.e. knockout mutations). Another advantage of traditional mutagenesis is the ability to produce high densities of randomly distributed mutations, thus enabling saturation mutagenesis to be achieved with much smaller populations than insertional mutagenesis would require (Henikoff and Comai 2003). While high mutation densities may complicate forward genetics screens and mutation breeding efforts, reverse genetics methods such as Targeting Induced Local Lesions in Genomes (TILLING) are predicated on the development of populations with sufficient numbers of mutations to enable cost-effective screening of DNA pools to identify those rare events (Comai and Henikoff 2006; Till et al. 2007).
The first step in establishing a reverse genetics pipeline based on traditional mutagenesis involves the identification or generation of populations with optimal mutation densities. Here a balance is struck between a sufficient mutation density to permit identifying rare mutations through screening of a reasonable number of M2 individuals (i.e. 2–4000) and adequate fertility of those individuals to permit the recovery of the mutations in the M3 generation. While mutation densities can be determined by screening amplicons of selected loci using sequencing or mismatch cleavage detection assays, hundreds of individuals must be examined to establish a reliable estimate for a given population (Comai and Henikoff 2006; Till et al. 2007; Monson-Miller et al. 2012). Such an approach is costly, time-consuming and impractical in the case where multiple populations are being evaluated or if a protocol for mutagenesis needs to be established. An alternative approach is to directly sequence a small subset of individuals from the population(s) of interest (Monson-Miller et al. 2012). This is now feasible given the advances in DNA sequencing which have greatly increased throughput while reducing costs.
Although whole-genome sequencing is becoming a more common approach in model and major economic species, the cost remains prohibitive for assessment of mutation densities in the context of developing reverse genetics resources in non-model species and specialty crops. A powerful alternative involves the reduction of genomic complexity. This can be achieved by targeting a subset of the genome prior to sequencing either by selective hybridization using specially designed oligonucleotide probes (e.g. exome capture) or through a combination of restriction enzyme digestion and fragment size selection (Monson-Miller et al. 2012). The selected DNA is then used to construct a reduced representation sequencing (RRS) library. This sampling approach increases the likelihood that a particular locus will be sequenced multiple times; sufficient sequencing coverage is necessary to reliably call mutations. During library construction, barcoded adapter sequences are used so that multiplexing of several libraries can be performed, thus increasing the numbers of individuals that can be sequenced concurrently.
The original RRS method was described by Altshuler et al. (2000) well over a decade ago and involved pooling of a modest number of human DNA samples that had been digested with various restriction enzymes followed by size selection using agarose gel fractionation. Size-selected fragments were then cloned and subjected to Sanger sequencing to identify single nucleotide polymorphisms. A number of variations on this approach have been developed for ultrahigh-throughput sequencing platforms (Monson-Miller et al. 2012; Baird et al. 2008; van Orsouw et al. 2007; Van Tassell et al. 2008; Andolfatto et al. 2011; Elshire et al. 2011), and comparisons of these methods have been reviewed recently (Davey et al. 2011; Poland and Rife 2012). Restriction enzyme sequence comparative analysis (RESCAN), our version of RRS, involves the simple construction of barcoded Illumina sequencing libraries from DNA samples that have been digested using a restriction enzyme with a 4 bp recognition site (e.g. NlaIII, MseI) and subjected to solid phase reversible immobilization (DeAngelis et al. 1995) bead-based size selection. Libraries can be constructed from as little as 100 ng of DNA and barcoding facilitates multiplexing of up to 96 libraries/individuals per sequencing lane. The RESCAN method has been successfully employed to assess mutation density in rice mutants derived from sodium azide mutagenesis with and without the use of the Nipponbare reference genome sequence (Monson-Miller et al. 2012). RESCAN has also been used in various genotyping applications including QTL mapping (Seymour et al. 2012), evaluation of genetic diversity (Kim and Tai 2013a) and identifying introgressed regions in advanced backcross lines (Kim and Tai 2013b).
Since the original report of the RESCAN method (Monson-Miller et al. 2012), minor modifications have been made in the library construction protocol to improve the efficiency of adapter ligation and the subsequent library enrichment step. In our current version of RESCAN, restriction enzyme-digested DNAs are subjected to end repair and A-tailing prior to adapter ligation rather than directly ligating digested DNAs to adapters with compatible ends. As a result, commercially available next-generation sequencing (NGS) library kits may be used for ease, efficiency and robustness. Integration of the standard NGS library construction procedures within the RESCAN protocol has improved ligation efficiency, leading to more consistent and better enrichment of libraries (i.e. fewer cycles of PCR amplification) prior to sequencing. In addition to a detailed protocol for construction of RESCAN libraries, considerations for sequencing and an overview of the computational pipeline for identifying mutations from RESCAN sequence data are described.
2 Materials
2.1 DNA Restriction Digestion
-
1.
Genomic DNA (see Note 1).
-
2.
NlaIII restriction enzyme (New England Biolabs). Store at −80 °C (see Note 2).
-
3.
10x NlaII digestion buffer (New England Biolabs) supplied with NlaIII. Store at −80 °C.
-
4.
Thermocycler or other incubator set at 37 °C.
2.2 Digest Clean-Up and Size Fractionation
-
1.
1× Tris-acetate EDTA (TAE) buffer and 1 % agarose/1× TAE gel (see Note 3).
-
2.
DNA size standards such 1 kb ladder or λ DNA-HindIII. Store at 4 °C or −20 °C depending on frequency of use (see Note 4).
-
3.
DNA stain (e.g. ethidium bromide) and gel documentation equipment.
-
4.
Agencourt AMPure XP beads (Beckman Coulter). Store at 4 °C (see Note 5).
-
5.
Magnet stand (see Note 6).
2.3 RESCAN Library Preparation
-
1.
Agencourt AMPure XP beads.
-
2.
Indexed adapters (NEXTflex DNA Barcodes, Bioo Scientific) for multiplexing of DNA sequencing libraries (see Note 7).
-
3.
Thermocycler.
-
4.
Magnet stand.
-
5.
NGS library preparation kit (KAPA Biosystems). Store at −20 °C (see Note 8).
2.4 PCR Enrichment
-
1.
Phusion High-Fidelity DNA polymerase (New England Biolabs). Stored at −20 °C.
-
2.
5× Phusion HF Buffer (New England Biolabs) supplied with Phusion polymerase. Store at −20 °C.
-
3.
Adapter primers (5 μM), complementary to indexed adapters, for library amplification (see Note 9). Store at −20 °C.
-
4.
2 % agarose/1× TAE gel and 1× TAE electrophoresis buffer.
-
5.
100 bp DNA ladder size standard (New England Biolabs). Store at −20 °C.
3 Methods
3.1 NlaIII Restriction Digestion
-
1.
Turn on thermocycler or incubator and set to 37 °C (hold).
-
2.
Measure DNA concentration of genomic DNA and calculate the volume containing 500 ng (see Note 10).
-
3.
On ice combine the following in a 0.2 ml tube:
-
44.5 μl DNA (500 ng; add ultrapure water to final volume as needed)
-
5.0 μl 10× NlaIII digestion buffer
-
0.5 μl NlaIII enzyme (10 units/μl)
-
-
4.
Mix well by gently pipetting up and down and spin down contents for a few seconds. Incubate restriction digest(s) at 37 °C for >6 h (see Note 11).
3.2 NlaIII Digest Check and Clean-Up
-
1.
Check that DNA digest has proceeded to completion by evaluating 2 μl (out of 50 μl) of the digest on a 1 % agarose/1× TAE gel. Include appropriate DNA size standards (e.g. 0.5–1 μl of 1 kb DNA ladder). DNA should appear as a smear with no distinct bands.
-
2.
During the gel electrophoresis, place AMPure XP beads at room temperature for 20 min to allow for equilibration.
-
3.
Following confirmation of complete digestion of the DNA, prepare a fresh solution of 80 % ethanol.
-
4.
Thoroughly mix remaining digest with 50 μl of equilibrated AMPure XP beads. Let stand at room temperature 5–15 min to allow the DNA to bind to beads.
-
5.
Place beads on magnet and wait until liquid has cleared (approximately 2 min). Discard supernatant.
-
6.
With the tube still on the magnet, wash beads two times with 200 μl ethanol (80 %). Work quickly so that the AMPure beads do not dry. Discard final ethanol wash (pipetting off all excess) and let beads dry on the magnet (~2–10 min; see Note 12).
-
7.
Remove dry beads from magnet and resuspend in 51 μl elution buffer (EB; 10 mM Tris–HCl, pH = 8.0) by pipetting up and down at least ten times. Let beads stand at room temperature for 5 min to elute DNA from beads.
-
8.
Return sample to the magnet and once clear, carefully pipette 50 μl cleared liquid (no beads!) into a new tube. Discard the tube with the beads. Sample is now ready for end repair or storage in the freezer (see Note 13).
3.3 RESCAN Library Preparation (End Repair, A-Tailing, Adapter Ligation and Size Selection)
-
1.
Place AMPure XP beads and PEG/NaCl SPRI solution (supplied with KAPA NGS library kit) at room temperature for 20 min (see Note 14).
-
2.
Programme a thermocycler with the following temperature profiles:
-
20 °C, 30 min
-
30 °C, 30 min
-
20 °C, 15 min
-
-
3.
On ice, prepare the KAPA end repair master mix as follows (see Note 15):
-
Water, 8 μl
-
10× end repair buffer, 7 μl
-
End repair enzyme mix, 5 μl
-
-
4.
For end repair, combine 20 μl master mix with 50 μl NlaIII-digested DNA in a 0.2 ml PCR tube, mix well, place in thermocycler, and incubate at 20 °C for 30 min. Proceed immediately to next step (end repair clean-up).
-
5.
To each end repair reaction, add 120 μl AMPure XP beads and pipette up and down to thoroughly mix. Let stand at room temperature 5–15 min to allow the DNA to bind to beads.
-
6.
During binding period, prepare A-tailing master mix as follows (see Note 15):
-
Water, 42 μl
-
A-tailing buffer, 5 μl
-
A-tailing enzyme mix, 3 μl
-
-
7.
Place beads on magnet and wait until liquid has cleared (approximately 2 min). Remove liquid by pipetting and discard (do not remove tube from magnet).
-
8.
With the tube still on the magnet, wash two times with 200 μl ethanol (80 %). Work quickly so that the AMPure XP beads do not dry. Discard final ethanol wash (pipetting off all excess) and let beads dry on the magnet stand (~2–10 min; see Note 12).
-
9.
Remove beads from magnet and resuspend beads in 50 μl of master mix by pipetting up and down.
-
10.
Incubate at 30 °C for 30 min and then proceed immediately to clean-up.
-
11.
To each A-tailing reaction with beads, add 90 μl PEG/NaCl SPRI solution and thoroughly mix by pipetting. Total volume is now 140 μl. Let stand at room temperature for 5–15 min to allow the DNA to bind to beads.
-
12.
During the binding period, prepare adapter ligation master mix as follows (see Note 15):
-
Water, 34 μl
-
5× KAPA ligation buffer, 10 μl
-
KAPA T4 DNA ligase, 5 μl
-
-
13.
Place beads on magnet stand and wait until liquid has cleared (approximately 2 min). With tube still on the magnet, remove liquid by pipetting and discard. Wash beads two times with 200 μl of 80 % ethanol. Work quickly so that the AMPure XP beads do not dry between washes. Discard final ethanol wash (pipetting off all excess) and let beads dry on the magnet (~2–10 min; see Note 12). Immediately proceed to next step (i.e. adapter ligation).
-
14.
Remove beads from magnet and suspend thoroughly in 49 μl adapter ligation master mix by pipetting up and down. Carefully pipette 1 μl of 0.5–5 μM premixed adapter (Table 19.1), mix well, and incubate at 20 °C for 15 min (see Note 16).
-
15.
Following incubation, immediately add 50 μl of the PEG/NaCl SPRI solution to the ligation and thoroughly mix by pipetting. Reaction volume should be 100 μl. Incubate at room temperature for 5–15 min to allow DNA to binds to the beads.
-
16.
Place beads on magnet and wait until liquid has cleared (approximately 2 min). Remove liquid by pipetting and discard (do not remove tube from magnet).
-
17.
With the tube still on the magnet stand, wash two times with 200 μl ethanol (80 %). Work quickly so that the AMPure XP beads do not dry. Discard final ethanol wash (pipetting off all excess) and let beads dry on the magnet (~2–10 min; see Note 12).
-
18.
Remove beads from magnet and suspend in 100 μl elution buffer (10 mM Tris–HCl, pH = 8.0). Incubate at room temperature for 2 min to elute the DNA. Proceed to the next step (i.e. size selection) or store sample at 4 °C (see Note 17).
-
19.
For size selection, add 60 μl PEG/NaCl SPRI solution to the 100 μl of eluted DNA (buffer and beads) and mix thoroughly by pipetting. Total volume is now 160 μl. Incubate at room temperature for 5–15 min to allow DNA fragments larger than 450 bp to bind to the beads.
-
20.
Place beads on magnet stand and wait until liquid has cleared completely. Do not remove tube from the magnet stand. Transfer 155 μl of the supernatant to a clean tube. The supernatant should contain DNA fragments ≤450 bp (see Note 18).
-
21.
Add 20 μl of room temperature-equilibrated AMPure XP beads and mix thoroughly by pipetting. Incubate at room temperature for 5–15 min to bind DNA fragments greater than 250 bp to the beads.
-
22.
Place beads on magnet and wait until liquid has cleared (approximately 2 min). Remove liquid by pipetting and discard (do not remove tube from magnet).
-
23.
With the tube still on the magnet stand, wash two times with 200 μl freshly prepared 80 % ethanol. Work quickly so that the AMPure XP beads do not dry between washes. Discard final wash and make sure to remove all traces of ethanol. Let beads dry on the magnet for 2–10 min (see Note 12).
-
24.
Remove beads from magnet and resuspend in 25 μL elution buffer (10 mM Tris–HCl, pH = 8.0). Incubate beads at room temperature for 2 min to elute DNA. Place back on magnet stand to clear beads from the liquid and transfer the supernatant to a new tube. Sample is now ready for library enrichment (see Note 19).
3.4 PCR Enrichment of RESCAN Libraries
-
1.
Programme thermocycler with the following amplification profile for enrichment (see Note 20):
-
98 °C for 45 s (initial denaturation)
-
10 cycles of | 98 °C for 15 s |
60 °C for 30 s | |
72 °C for 30 s |
-
72 °C for 1 min (final extension)
-
4 °C hold
-
2.
Thaw 2× KAPA mix and primers
-
3.
For each sample (i.e. library) combine the following in a 0.2 ml PCR tube:
-
Size-selected DNA, 20 μl
-
2× KAPA mix, 25 μl
-
Primers (10 μM), 5 μl
-
-
4.
Load samples in thermocycler and run enrichment profile.
-
5.
Verify enrichment by removing 4 μl from the 50 μl PCR reaction and running this sample on a 2 % agarose/1× TAE gel along with a 100 bp DNA ladder (see Note 21). Store remainder of the reaction at 4 °C.
-
6.
If enrichment is not observed, return the remaining 46 μl of the PCR reaction to the thermocycler and perform another two cycles of amplification using an abbreviated profile. Run another 4 μl on an agarose gel to check enrichment (see Note 20).
-
7.
To each PCR reaction, add 50 μl AMPure XP beads and mix thoroughly by pipetting. Let stand at room temperature 5–15 min to allow the DNA to bind to beads.
-
8.
Place beads on magnet stand and wait until liquid has cleared (approximately 2 min).
-
9.
Discard liquid and with the tube still on the magnet stand, wash beads two times with 200 μl of 80 % ethanol. Work quickly so that the AMPure XP beads do not dry between washes. Discard final ethanol wash (pipetting off all excess) and let beads dry on the magnet (~2–10 min; see Note 12).
-
10.
Remove beads from magnet stand and suspend beads in 21 μl EB (10 mM Tris–HCl, pH = 8.0). Incubate at room temperature for 2 min to elute DNA.
-
11.
Return sample to magnet stand and incubate beads until the liquid is clear. With the sample still on the magnet stand, carefully remove 20 μl of eluted DNA into clean 1.5 ml microfuge tube. Do not disturb the beads.
-
12.
Measure the concentration of 1 μl of eluted DNA. Run another 1–2 μl of the DNA on a 2 % agarose/1× TAE gel to ensure adapter dimers are not present prior to sequencing. Store sample(s) at −20 °C.
-
13.
Prior to sequencing, prepare multiplexed sample by combining an equal amount of DNA from each library to be sequence. We recommend pooling a maximum of 20 libraries per lane (see Note 22).
3.5 Sequencing
-
1.
Sequencing of RESCAN libraries is typically performed by dedicated sequencing core facilities or commercial sequencing labs. The Illumina sequencing platforms available (e.g. HiSeq 2500, MiSeq) and the protocols for submission, handling and analysis of samples are dependent on the facility. For RESCAN, we currently employ single-read 50 bp (SR50) or paired end read 100 bp (PE100) read lengths using a HiSeq 2500 (or higher) platform. Sequencing facilities will provide an option of determining the DNA concentration of the sample you submit using either qPCR or a bioanalyser. It is recommended that this quality control step is performed.
-
2.
At the time RESCAN libraries are submitted for sequencing, it should be specified that the samples are indexed and the size of the barcode (e.g. 8 bp) should be indicated.
3.6 Computational Analysis of Sequence Data
-
1.
Illumina raw sequence reads are provided as compressed fastq file (see Note 23), containing both reads that passed quality filters and those that did not. Reads are often divided across multiple files (e.g. each file might contain four million reads) for more efficient acquisition. Because of large file size, it is recommended that processing be performed on a computer cluster as memory requirements may exceed file size, especially during reference-dependent alignment and pile-up generation (see Note 24).
-
2.
Sort sequence reads by their barcodes and quality trim using ‘Allprep’, a custom python script available for download at https://github.com/Comai-Lab/allprep. The following parameters are used for both 50 and 100 bp runs: mean PHRED quality score over a sliding window of 5 bp ≥ 20 and sequence length ≥ 35 bp. Reads containing N bases can be discarded and index sequences are trimmed.
-
3.
Align processed reads reference genome using BWA version 0.7.5a (Li and Durbin 2009) and default mismatch parameters. Sam/bam files are automatically generated using Samtools (Li et al. 2009) and an additional python script (http://comailab.genomecenter.ucdavis.edu/index.php/ Bwa-doall; see Note 25).
-
4.
Create BWA Samtools-derived mpileup (mapping quality ≥20) and parsed mpileup files, using scripts available here: (http://comailab.genomecenter.ucdavis.edu/index.php/Mpileup).
-
5.
Perform mutation detection using the mutations and polymorphisms surveyor (MAPS) bioinformatics pipeline (Henry et al. 2014) in mutation mode (available at http://comailab.genomecenter.ucdavis.edu/index.php/MAPS). In short, the MAPS pipeline is based on the assumption that mutations occur randomly and should not be shared by multiple individuals. It is therefore designed to detect mutations in single samples, using all other samples as controls. The minimum number of samples that can be analysed is three although we strongly recommend using higher number of samples (see Note 24). The size of the genome that is assayed for the presence of mutations is also important (see Note 24). Although reads are initially mapped to a reference sequence, wild-type alleles are not assumed to be the reference allele at that location. Instead, the wild-type allele is determined from the data as the allele shared by all samples except from the mutant sample. Most of the time, the wild-type and reference sequence alleles are identical but not always, especially when the samples originate from a different variety or species of rice than the reference sequence used. The MAPS pipeline is divided into two steps. Thresholds for mutation detection vary based on each experiment, but guidelines can be found in Henry et al. (2014). In our experience, the mutant allele coverage (MACov), i.e. the number of times that a mutant allele is observed in the mutant sample, is the most important criteria for robust mutation detection. For homozygous mutations, a MACov >3 can be sufficient. For heterozygous mutations, a MACov >5 is usually required. For EMS populations, the percentage of false positive mutations can be assessed by calculating the percentage of canonical mutations observed (CG>TA) (Henry et al. 2014).
4 Notes
-
1.
Any extraction protocol that yields DNA of high molecular weight (≥15 kb) and good purity (A260/A280 ratio of ≥1.8). We have used both home-made reagents (e.g. potassium acetate-sodium dodecyl sulphate method) and commercial kits (e.g. Qiagen DNeasy). Total genomic DNA (including chloroplast and mitochondrial genomes) is routinely used.
-
2.
Restriction enzymes with 4 bp recognition sites are typically used. We have had success with NlaIII and MseI although others, including those with longer recognition motifs, may work well. For species with high-quality reference genome sequence information, in silico restriction digests may be performed to estimate the number of restriction fragments in a given size range (Monson-Miller et al. 2012).
-
3.
TAE buffer is typically made at 50× (1 L contains 242 g Tris base, 57.1 ml glacial acetic acid and 100 ml of 0.5 mM ethylenediaminetetraacetic acid, pH 8.0). Dilute 1:50 with deionized water to make 1× TAE electrophoresis buffer. For 1 % agarose/1× TAE gel, add 1 g agarose (electrophoresis grade) per 100 ml of 1× TAE buffer and heat to boiling to completely dissolve agarose (stir as needed). Cool to 50–55 °C before pouring into gel mould.
-
4.
DNA size standards may be purchased ready to use or require the addition of standard gel loading dye solution. Comparable size standards (i.e. covering similar size ranges) can be purchased from several commercial sources. Home-made size standards may also be used.
-
5.
Solid phase reversible immobilization (SPRI) beads are sold commercially as AMPure XP beads. SPRI beads are paramagnetic (i.e. only magnetic in the presence of a magnetic field) and consist of a polystyrene core surrounded by a layer of magnetite which is coated with carboxyl molecules. In the presence of polyethylene glycol (PEG) and salt, DNA will reversibly bind to these carboxyl molecules. Depending on the concentration of the PEG/salt, different size DNA will preferentially bind to the carboxyl molecules, thus permitting size fractionation. Currently sold in quantities of 5 ml, 60 ml and 450 ml, AMPure beads should be well mixed and equilibrated to room temperature before use. We typically transfer small ‘working’ aliquots of the AMPure beads (0.5–1 ml) to 1.5 ml tubes. It is advised that the instructions provided by the manufacturer on the handling and storage of the beads are carefully reviewed. The greatest source of error (and loss of DNA) during the RESCAN library construction is due to the mishandling of the AMPure beads.
-
6.
Use of paramagnetic bead separation technology has become a ubiquitous tool in molecular biology. There are numerous manufacturers and suppliers of magnetic stands for various tube sizes and formats. In this protocol, we use a magnetic stand that can hold single 0.2 ml tubes as well as 0.2 ml tube strips (8, 12, and) or 96 well PCR-style plates for flexible throughput. We recommend stands in which the magnets are located in such a way that the beads collect along the side of the tubes so that pipette tips can touch the bottom of the tubes without disturbing the pellets.
-
7.
Indexed adapters that are compatible with Illumina sequencing platforms can be purchased with unique six or eight nucleotide barcodes.
-
8.
Next-generation sequencing (NGS) library kits are available from many commercial sources. Home-made kits consisting of components obtained from various sources may also be used. We have found the convenience and reliability of commercial kits to outweigh the possible cost savings of assembling individual reagents.
-
9.
Adapter-ligated DNA is usually very low in concentration; PCR amplification is necessary for proper cluster formation on the Illumina analyser. Primers for amplifying libraries are complementary to sequences in the indexed adapters. Both primers and indexed adapters can be purchased from Illumina or may be purchased separately from suppliers of DNA oligonucleotides.
-
10.
Any standard method for quantifying DNA is acceptable. Concentration of DNA should be at least 12 ng/μl, preferably much higher (100–200 ng/μl) to minimize the amount of possible contaminants added to the reaction mixture that might inhibit the restriction enzyme. Total library input can be as little as 100 ng, but higher (500 ng to 1 μg) inputs yield more consistent library preparation.
-
11.
It is important that the DNA is completely digested prior to size selection and ligation. It may be necessary to adjust enzyme amount based on concentration of input DNA. This can be determined empirically, but a good metric is 1 U of enzyme per 1 μg of DNA. Overnight incubation is preferred for complete digestion. Most thermocyclers have heated lids, which should be set to prevent evaporation of the reaction mixture. When performing digests in an incubator, place tubes in a sealed plastic bag with a wet paper towel covering the caps.
-
12.
Be careful not to overdry the AMPure beads. Overdrying can negatively impact yields. The surface of the beads (i.e. the pellet, Fig. 19.1) should not appear cracked. Optimal drying time is 3–5 min, but additional time (up to 10 min) may be needed if ethanol wash is not completely removed.
-
13.
This is a safe stopping point in the protocol. DNA can be stored in the elution buffer at −20 °C for 2 weeks.
-
14.
The PEG/NaCl solution is light sensitive so keep covered with aluminium foil.
-
15.
Volumes shown are for a single reaction. To process additional samples, prepare a master mix with 5 % excess to account for losses during pipetting. New reagent volumes = volumes shown × number of samples × 1.05.
-
16.
Each indexed forward ‘B’ adapter should be mixed with the universal ‘A’ adapter to the desired concentration prior to ligation. Recommendations for the adapter concentration can be found in the KAPA LTP Library Preparation Kit Technical Data Sheet, p. 5, KAPA Biosystems.
-
17.
This is a safe stopping point. Eluted DNA and beads can be stored for 24 h at 4 °C.
-
18.
Be careful not to transfer any beads when the supernatant is removed as DNA fragments greater than 450 bp are bound to them at this stage.
-
19.
This is a safe stopping point. Ligations can be kept at 4 °C for 24 h or −20 °C for up to 72 h.
-
20.
This amplification profile takes a total of about 30 min to complete. Amplification for more than 12 cycles is not recommended as this will significantly increase the percentage of clonal reads.
-
21.
A strong smear of DNA between 300 and 600 bp should be visible if good enrichment has been achieved. This is a safe stopping point. Enrichment can be kept at 4 °C for 24 h or −20 °C for up to 72 h.
-
22.
The throughput of the Illumina HiSeq platform depends on the read lengths, run mode and the number of lanes on which your samples will be run. These details are provided by the sequencing facility you use (additional information can be found at http://www.illumina.com/systems/hiseq_2500_1500/performance_specifications.html).
-
23.
In addition to sequence data, each file contains basic information such as Illumina instrument ID, run and lane number, read quality scores and index information.
-
24.
Mutation rate varies from population to population and within populations. It is therefore advised to use multiple individuals when assessing mutation rate. We recommend assessing a mutation rate in at least ten randomly selected individuals to obtain a mean rate for the population. Next, a sufficient number of bps should be assayed in order to reliably assess mutation rate. This will depend on the number of RESCAN fragments selected and the average number of reads obtained from each library (Table 19.2). How much of the genome is covered depends on what restriction enzyme is used and the size fraction selected (Table 19.3). The conditions used in this protocol result in the majority of the selected fragments being in the range of 250–450 bp. In species such as rice, the availability of a reference genome sequence enables in silico restriction digests to be performed to estimate the number of fragments generated by a restriction enzyme which are within a given size range (Monson-Miller et al. 2012) (Tables 19.2 and 19.3). Although the number of larger fragments generated by restriction enzymes with 4 bp recognition sites is smaller (in theory allowing for better coverage and greater multiplexing), in practice, size selection of larger fragments (>600 or 700 bp) is not very efficient; thus library construction is similarly inefficient (Monson-Miller et al. 2012) (Table 19.2).
-
25.
By default, Bwa-DoAll performs a step that filters reads for clonal reads, i.e. reads that are the result of PCR amplification post-library construction, by identifying read pairs for which both the forward and reverse reads map at the exact same positions. Because of the repetitive nature of RESCAN libraries, i.e. reads are located adjacent to NlaIII cut sites and therefore have the same start positions, this step is omitted. Instead, the reads are filtered for chimeric sequences by searching read sequences for internal restriction site (ATGC). Assuming full digestion of the input DNA, restriction sites should only be found at the start and end of the DNA fragment but not in the middle. If an internal restriction site is found, the read is trimmed at the beginning of the internal site.

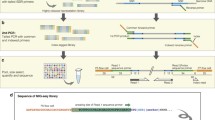
Size selection of restriction fragments using solid phase reversible immobilization (SPRI) bead technology. (a) AMPure XP beads in 8-tube strip on a 96-well magnet stand. (b) Pellets of AMPure beads. The position of the magnet results in the formation of a pellet on the side wall, allowing complete removal of the supernatant from the bottom of the tubes
References
Ahloowalia BS, Maluszynski M, Nichterlein K (2004) Global impact of mutation-derived varieties. Euphytica 135:187–204
Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES (2000) An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407:513–516
Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, Stern DL (2011) Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res 21:610–617
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376
Comai L, Henikoff S (2006) TILLING: practical single-nucleotide mutation discovery. Plant J 45:684–694
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
DeAngelis MM, Wang DG, Hawkins TL (1995) Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res 23:4742–4743
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS One 6:e19379
Henikoff S, Comai L (2003) Single-nucleotide mutations for plant functional genomics. Annu Rev Plant Biol 54:375–401
Henry IM, Nagalakshmi U, Lieberman MC, Ngo KJ, Krasileva KV, Vasquez-Gross H, Akhunova A, Akhunov E, Dubcovsky J, Tai TH, Comai L (2014) Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 26:1382–1397
Kim SI, Tai TH (2013a) Identification of SNPs in closely related temperate japonica rice cultivars using restriction enzyme-phased sequencing. PLoS One 8:e60176
Kim SI, Tai TH (2013b) High resolution genotyping by restriction enzyme-phased sequencing of advanced backcross lines of rice exhibiting differential cold stress recovery. Euphytica 192:107–115
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The Sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Micke A, Donini B, Maluszynski M (1987) Induced mutations for crop improvement—a review. Trop Agric 64:259–278
Monson-Miller J, Sanchez Mendez DC, Fass J, Henry IM, Tai TH, Comai L (2012) Reference genome-independent assessment of mutation density using restriction enzyme-phased sequencing. BMC Genomics 13:72
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5:92–102
Seymour DK, Filiault DL, Henry IH, Monson-Miller J, Ravi M, Pang A, Comai L, Chan SWL, Maloof JN (2012) Rapid creation of Arabidopsis doubled haploid lines for quantitative trait locus mapping. Proc Natl Acad Sci USA 109:4227–4232
Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L (2007) Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol 7:19
van Orsouw NJ, Hogers RC, Janssen A, Yalcin F, Snoeijers S, Verstege E, Schneiders H, van der Poel H, van Oeveren J, Verstegen H, van Eijk MJ (2007) Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes. PLoS One 2:e1172
Van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS (2008) SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods 5:247–252
Acknowledgments
The methods described here were developed with the support from the National Science Foundation Plant Genome Program (Plant Genome award no. DBI–0822383) to L.C. and the U.S. Department of Agriculture, Agricultural Research Service CRIS Project 5306-21000-017/021-00D to T.H.T.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2017 International Atomic Energy Agency
About this chapter
Cite this chapter
Burkart-Waco, D., Henry, I.M., Ngo, K., Comai, L., Tai, T.H. (2017). Determining Mutation Density Using Restriction Enzyme Sequence Comparative Analysis (RESCAN). In: Jankowicz-Cieslak, J., Tai, T., Kumlehn, J., Till, B. (eds) Biotechnologies for Plant Mutation Breeding. Springer, Cham. https://doi.org/10.1007/978-3-319-45021-6_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-45021-6_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45019-3
Online ISBN: 978-3-319-45021-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)