
Abstract
Multiple Sclerosis (MS) is the most common demyelinating disease of man with over 400,000 cases in the United States and over 2.5 million cases worldwide. There are over 64,000 citations in Pubmed dating back as far as 1887. Much has been learned over the past 129 years with a recent burst in therapeutic options (mostly anti-inflammatory) with newer medications in development that are neuroprotective and/or neuroreparative. However, with all these advancements the cause of MS remains elusive. There is a clear interplay of genetic, immunologic, and environmental factors that influences both the development and progression of this disorder. This chapter will give a brief overview of the history and pathogenesis of MS with attention to how host immune responses in genetically susceptible individuals contribute to the MS disease process. In addition, we will explore the role of infectious agents in MS as potential “triggers” of disease. Models of virus-induced demyelination will be discussed, with an emphasis on the recent interest in human herpesviruses and the role they may play in MS disease pathogenesis. Although we remain circumspect as to the role of any microbial pathogen in MS, we suggest that only through well-controlled serological, cellular immune, molecular, and animal studies we will be able to identify candidate agents. Ultimately, clinical interventional trials that either target a specific pathogen or class of pathogens will be required to make definitive links between the suspected agent and MS.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
Multiple Sclerosis
History, Pathogenesis Epidemiology, and Etiology
History
Saint Lidwina of Schiedam (1380–1433), patron saint of the chronically ill and of ice skating, was afflicted with a debilitating illness at the age of 16; her symptoms included walking difficulties, headaches, unrelenting pain, a “split-face” with hanging lip, visual disturbances, and prolonged episodes of aphagia (Van Beiren 1427; Medaer 1979). Although historical texts suggest that Lidwina experienced periods of disease remission, her condition ultimately deteriorated, leaving her paralyzed with the exception of her left hand by the time of her death (Medaer 1979). The clinical picture that emerges from the fourteenth century descriptions of Lidwina’s suffering is consistent with the demyelinating disease now known as multiple sclerosis (MS) and it has been postulated that the description of her life constitutes the first documentation of this disorder (Medaer 1979).
In 1868, Dr. Jean-Martin Charcot classified MS as a discrete clinical entity that he called la sclerose en plaque disseminées. Charcot provided an accurate description of the clinical symptomatology and pathology of MS and had important insights into mechanisms of disease pathophysiology (Charcot 1868). Nearly 600 years after Lidwina’s death and more than 100 years after Charcot’s pioneering work in the field of neurology, many therapeutic interventions for MS have been developed. The establishment of a definitive cause or cure for this disease, however, remains elusive. In this chapter, we will review the complex amalgamation of genetic, immunologic, and environmental factors that influences the development of multiple sclerosis, with an emphasis on the association of virus infections with this debilitating neurologic disease.
Pathogenesis
Multiple sclerosis (MS ) is the most prevalent demyelinating disease of the central nervous system (CNS) and the most common disabling neurologic disease of young people, affecting an estimated 400,000 in the United States and 2–2.5 million people worldwide (Markowitz 2013). It is a chronic and incurable disease that significantly detracts from an individual’s quality of life and imposes substantial cost burdens on patients, their families, healthcare systems, and society (Battaglia et al. 2000; Parise et al. 2013). A diverse array of neurological signs and symptoms are associated with MS. These include limb weakness, impaired motor function, sensory symptoms, visual symptoms, eye movement disorders, bladder symptoms, sexual dysfunction, fatigue, ataxia, deafness, spasticity, dementia, and cognitive impairment (Rodgers et al. 1999). The life expectancy of a patient with MS is 5–10 years lower than that of the general population (Compston and Coles 2008).
Although the clinical progression of MS is variable and unpredictable, several disease phenotypes have been described; it is critical to distinguish disease phenotypes for both prognosis and decisions regarding treatment strategies. There are four distinct clinical courses in which the majority of MS patients can be classified: relapsing-remitting MS (RRMS) ; secondary-progressive MS (SPMS) ; primary-progressive MS (PPMS) ; and progressive-relapsing MS (PRMS) (Lublin and Reingold 1996; Confavreux and Vukusic 2014). About 80 % of people with MS are initially diagnosed with RRMS; this disease phenotype is characterized by disease exacerbations where new symptoms appear or existing symptoms become more severe (Markowitz 2013). In RRMS, disease exacerbations last for variable amounts of time and are followed by periods of total or partial recovery. Disease in RRMS patients may be inactive for months or years. Approximately 65 % of individuals who are initially diagnosed with RRMS will develop SPMS characterized by the accumulation of progressive disability with or without superimposed relapses. On average, the time between disease onset and conversion from RRMS to SPMS occurs in 19 years (Rovaris et al. 2006). The least common of the four major subtypes of MS, PRMS, describes those individuals who, from onset, have a steady neurologic decline but also have clear superimposed attacks (Lublin and Reingold 1996). These forms of progressive MS are distinct from the primary-progressive form (PPMS) , which is characterized by a lack of discernible attacks. Individuals with PPMS experience a gradual onset of disease with a steady worsening of symptoms (Confavreux and Vukusic 2014; Karussis 2014). In addition to the four major disease courses of MS, an international panel assembled in 2013 added clinically isolated syndrome (CIS ; attack of demyelination that does not fulfill all criteria of MS) and radiologically isolated syndrome (RIS; magnetic resonance imaging (MRI) findings suggestive of multiple sclerosis (MS) in asymptomatic/clinically silent patients) as MS phenotypes without changing the established framework four major disease courses (Lublin et al. 2014).
The onset of MS frequently occurs between the ages of 20 and 40 years and, like most autoimmune disorders, women are affected more often (1.5–2 times) than men (Karussis 2014). Multiple sclerosis (MS) is typically perceived as a disease of young adults and was previously believed to be rare in children. In recent years, however, there has been a marked increase in the number of children diagnosed with MS and it is now appreciated that pediatric-onset (defined here as disease onset prior to the age of 18) occurs in 2–5.6 % of all MS cases; indeed, this may have been the case for Lidwina of Schiedam (Krupp et al. 2007; Renoux et al. 2007; Chitnis et al. 2009; Ghezzi et al. 1997; Boiko et al. 2002; Banwell 2013). Pediatric-onset MS typically takes more time to reach the progressive stage (Compston and Coles 2008). However, these individuals still convert to a progressive disease phenotype at a lower average age than patients with adults-onset MS (Compston and Coles 2008).
Etiology: Genes, the Immune System, and the Environment
Genetic Factors
The etiology of MS is unknown. In part, this is owing to the variability of this disease. This suggests that many determinants many be involved in the range of clinical phenotypes that are defined as MS. Accordingly, the prevailing view of the etiology of MS is that it is multifactorial and that genetics, immunological, and environmental factors contribute collectively to MS susceptibility (Venkatesan and Johnson 2014). This view is consistent with an emerging paradigm shift in the field of immunology that suggests that our immune system is shaped as much, if not more, by our environment as our genes (Brodin et al. 2015).
A strong influence of genetic background on MS disease susceptibility is supported by epidemiological, familial, and molecular studies. The worldwide distribution of MS is skewed with areas of lower prevalence in Asia, Africa, and South America and areas of high prevalence in North America, Europe, New Zealand, and Australia (Kurtzke 1995). The incidence and prevalence of MS follows a north–south gradient in both hemispheres with rates of 30–80 cases per 100,000 in northern Europe, southern Canada, and the northern United States, in contrast with rates of 6–20 per 100,000 in most of southern Europe and the southern United States (Sadovnick and Ebers 1993; Compston 1994; Ebers et al. 1995; Kurtzke 2005). The north–south gradient observed in the New World is believed to reflect the tendency of individuals from Northern Europe to migrate to the northern regions of the United States and Canada and, conversely, those from regions of Europe with a lower incidence of MS to migrate to southern regions of the United States and South America. It is important to note that the latitudinal gradient of MS prevalence is not absolute; exceptions include the low prevalence of MS in Eskimo populations and the high prevalence of MS (150 per 100,000) in Sardinia (Acers and Acers-Warn 1994; Montomoli et al. 2002; Cocco et al. 2011). Furthermore, a recent comparison of 38 studies from Australia, Europe, and the United States suggests that the latitudinal gradient of MS disease prevalence may be diminishing (Alonso and Hernan 2008).
The influence of genetic background on susceptibility to MS is further supported by epidemiologic studies demonstrating different rates of MS among genetically distinct populations living in the same geographic area. For example, the frequency of MS is increased in Hungarians and Bulgarians of Caucasian decent compared to gypsy populations residing in the same regions (Kalman et al. 1991; Milanov et al. 1999). A similar situation exists in Australia and New Zealand where those of European descent have a far higher risk of developing MS than the indigenous populations (Hammond et al. 1988; Miller et al. 1986). In the United States, the prevalence of MS among people of Japanese ancestry living on the Pacific Coast (6.7/100,000) is considerably lower than that of Caucasians living in California (30/100,000) (Visscher et al. 1977). However, people of Japanese descent living on the West Coast of the United States have a slightly higher prevalence of MS than those living in Japan (2/100,000) suggesting that environmental factors influence disease susceptibility (Visscher et al. 1977). Collectively, the observed variations in MS prevalence among specific ethnic groups suggest that genetic traits that confer MS susceptibility may be enriched in populations that are at higher risk for MS and that these traits may be rare in populations at lower risk for MS. Moreover, there are other factors (both cultural and environmental) that differ between these groups (e.g., diet and lifestyle) that could confound the interpretation of these epidemiologic studies (Cree 2014).
Family and twin studies also indicate that there is a strong genetic component in the development of MS. Adoption studies have demonstrated that biological relatives of patients with MS have a greater likelihood of developing MS than adoptees and, conversely, that family members of adopted individuals with MS do not have an increased risk of developing the disorder (Ebers et al. 1995). The lifetime risk of MS increases with closer biological relationships. The risk is greatest for siblings of affected individuals, especially sisters, and decreases in half-siblings as well as second- and third-degree relatives (Sadovnick and Ebers 1993; Sadovnick et al. 1988, 1996). Additionally, the rate of MS concordance is greater in monozygotic (18–30 %) than in dizygotic twins (3–5 %). The incomplete penetrance of MS among monozygotic twins clearly suggests that a susceptible genetic background alone is not sufficient to cause disease (Bobowick et al. 1978; O’Gorman et al. 2013). Of interest, the concordance rate for fraternal twins is less than that of identical twins, but greater than that of other first-degree relatives of MS patients (Willer et al. 2003). Because twins share the same intrauterine environment and similar postnatal environments, the large difference in concordance between monozygotic and dizygotic twins must, at least in part, be attributable to genetic factors. Moreover, the increased concordance rate of MS between dizygotic twins compared with non-twin siblings reinforces the view that environmental factors are also in play.
The inheritance of MS susceptibility does not follow classic models of genetic inheritance. It is generally believed that MS is a complex heterogenous disorder, influenced by several genes, each exerting a relatively modest effect. These genes may act independently or epistatically on MS pathogenesis. Some genes may be involved in the induction of this complex disorder while others may be involved in disease progression, further complicating genetic studies in MS. Over the years, several genes, most of which are associated with immune function, myelin structure, or mitochondria have been tentatively associated with an increased risk of MS (He et al. 1998; Kellar-Wood et al. 1994; Reynier et al. 1999; Thompson et al. 1996). Many of these associations have not been demonstrated consistently in different studies. However, a strong association between MS and the major histocompatibility complex (MHC) was first identified in the 1970s (Jersild et al. 1975). Specifically, an increased risk of MS in individuals with MHC class II alleles DR2 and DQw1 has been found consistently in several populations (Cook 1997); in the preponderance of these studies the primary risk allele for MS is HLA-DRB1*15. Recently, additional high-risk MHC and non-MHC loci have been identified through genome-wide association studies (GWAS) comparing single nucleotide polymorphisms (SNPs) in affected and non-affected individuals (Australia and New Zealand Multiple Sclerosis Genetics Consortium 2009; De Jager et al. 2009; Sawcer and Hellenthal 2011; International Multiple Sclerosis Genetics Consortium et al. 2007). Interestingly, the majority of the loci associated with MS risk have known immunologic functions and many contribute to the risk of inheriting other autoimmune diseases (Cree 2014). This study was recently replicated with the association confirmed in 44 of the 58 genes initially implicated (Lin et al. 2015).
Immune Factors
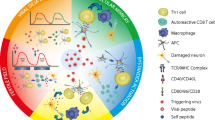
It is widely accepted that an aberrant immune response also plays an important role in the etiology of MS. This is based on the association of MS with genes involved with the immune response; the immunopathology of the disease; the clinical response of MS patients to immunomodulatory and immunosuppressive treatments; and similarities with experimental immune-mediated demyelinating diseases in animals. Historically, a T helper-1 (Th-1)-mediated autoimmune component driven by myelin protein-specific, pro-inflammatory cytokine secreting CD4+ T cells was believed to mediate the pathogenesis of this disorder. However, more recent evidence suggests that CD8+ T cells , macrophages, B cells, and the innate immune system also play important roles in the inflammatory cascade that results in demyelination and axonal loss in MS (Hoftberger et al. 2004; David and Lacroix 2003; Kotter et al. 2005; Prinz et al. 2006).
As described above, the MHC class II background of an individual is important in disease susceptibility. Many studies have concentrated on MHC-peptide interactions to determine how MHC class II alleles may confer diseases susceptibility. It has been suggested that the binding affinity of an antigenic peptide to an MHC allele determines T cell immunogenicity and encephalitogenecity (Greer et al. 1996). The vast majority of studies concerning MHC-peptide interactions relevant to MS have focused on myelin basic protein (MBP) as an antigenic peptide. MBP-specific T cells have been demonstrated in the peripheral blood of both MS patients and normal individuals. The frequencies of MBP-specific T cells tend to be higher in MS patients than controls. However, similar frequencies of MBP-specific T cells have been demonstrated in affected and unaffected family members, thereby suggesting that the frequency of MBP-specific T cells may be linked to the immunogenetic background of an individual and may be a prerequisite, rather than a consequence, of disease development (Joshi et al. 1993). Molecular mimicry, a phenomenon by which environmental antigens cross-react with normal host cell components, may induce an immune response against host proteins such as MBP. Therefore, individuals with higher frequencies of MBP reactive T cells may be more likely to develop autoreactive T cell responses as a result of environmental, non-self epitopes mimicking MBP.
Although the vast majority of studies have examined autoreactive CD4+ T cells , CD8+ T cells and B cells may also play a critical role in the neuropathogenesis of MS . Myelin antigen-specific CD8+ T cells have been identified and it has been demonstrated that severe experimental allergic encephalomyelitis (EAE) can be induced by adoptive transfer of anti-MBP CD8+ T cells (Hoftberger et al. 2004; Huseby et al. 2001). In addition, it is now widely accepted that B cells are important in the pathophysiology of MS. This newly appreciated role for B cells has largely arisen from clinical trials demonstrating a clear effect of monoclonal antibodies against the B-cell antigen CD20 (ocrelizumab and ofatumumab ) on clinical relapses and lesion formation in contrast with negative results obtained using a monoclonal antibody (anti-IL12/23 p40, ustekinumab ) that targets both Th1 and Th17 T cells (Hauser et al. 2008; Kappos et al. 2011; Segal et al. 2008).
A number of immune abnormalities are frequently observed in MS patients and a complex series of immunological mechanisms associated with events both in the peripheral blood system and in the CNS have been implicated in disease pathogenesis (Joshi et al. 1993; Martin et al. 1997). One of the hallmarks of MS is the intrathecal synthesis of oligoclonal antibodies. Oligoclonal bands (OCBs) and elevated IgG/albumin indices are found in the CSF of greater than 90 % of MS patients and can be used in the diagnosis of the disease . OCBs are not typically directed against a single antigen and antibody bands specific for viral bacterial, and self-antigens have been described (Sindic et al. 1994; Sriram et al. 1999; Virtanen et al. 2014; Franciotta et al. 2011). Therefore, it is unclear whether or not the intrathecal synthesis of immunoglobulins observed in MS results from the presence of cells that are actively recruited into the CNS after the pathogenetically relevant cells crossed the blood–brain barrier (BBB). In addition to the presence of OCBs , other immunological markers of disease activity have been described in MS. Several proinflammatory cytokines, including TNF-α, IFN-γ, and osteopontin, are upregulated in MS (Chabas et al. 2001). Treatment of MS patients with IFN-γ resulted in a marked increase in exacerbations, supporting the model of MS as an inflammatory autoimmune disease (Panitch et al. 1987). Furthermore, an increase in TNF-α expression precedes relapses and inflammatory activity as measured by MRI, while the mRNA levels of anti-inflammatory cytokines such as IL-10 and TGF-β β decline. The overexpression of these cytokines may be involved in disease pathogenesis by causing the upregulation of MHC and adhesion molecule expression on endothelial and glial cells, activating macrophages and recruiting TH1 cells, or by damaging oligodendroglial cells and myelin sheathes directly. The soluble adhesion molecules ICAM-1 and E-selectin are elevated in MS sera while soluble VCAM-1 and E-selectin are increased in the CSF of MS patients (Dore-Duffy et al. 1995).
The effectiveness of immunosuppressive and anti-inflammatory therapies in MS also supports an autoimmune component in disease pathogenesis. Although corticosteroid treatment does not alter the long-term course of MS, it is used effectively in the treatment of MS exacerbations. It has been demonstrated that the administration of high-dose steroids immediately stops BBB leakage as visualized by gadolinium-enhanced MRI (Burnham et al. 1991). A number of immunosuppressive and chemotherapeutic drugs including cyclophosphamide and methotrexate have been used in the treatment of MS with variable success. Over the past two decades, two immunomodulatory therapies, namely IFN-β and Copolymer-1 (Cop-1)/glatiramer acetate , have been used extensively in the treatment of MS with an excellent track record of efficacy and safety. IFN-β counteracts many of the effects of IFN-γ including the recruitment of inflammatory cells and the upregulation of MHC and adhesion molecules (Paty and Li 1993; Stone et al. 1995). Additionally, IFN-β has been shown to lower the exacerbation rate in MS patients with a relapsing remitting course and inflammatory activity as demonstrated by MRI. COP-1 is a synthetic polypeptide consisting of a random sequence of four amino acids, blocks peptides for the MHC binding groove. COP-1 has been demonstrated to be approximately as effective as IFN-β β in early RRMS (Johnson et al. 1995). More recently, monoclonal antibody-based therapies have been designed to act through various mechanisms, including blocking alpha-4 integrin interactions (natalizumab) or eliminating cells that express specific surface markers (e.g., CD52, alemtuzumab ; CD20 ocrelizumab and ofatumumab ; IL-2 receptor, dacluzimab ). These immunomodulatory therapies are highly effective, but have the potential for serious complications; for example, natalizumab is associated with progressive multifocal leukoencephalopathy (caused by JC virus) and alemtuzumab is associated with the development of new autoimmune disorders. Three new oral, immunomodulatory therapies have been approved for MS; fingolimod is an antagonist of normal endogenous sphingosine-1-phosphate (S-1-P) on naïve and activated T cells, teriflunomide interferes with lymphocyte proliferation by inhibiting the enzyme dihydroorotate dehydrogenase, and dimethyl fumarate counters oxidative stress (Cross and Naismith 2014). Fingolimod was recently linked to a reactivation of herpes simplex 1 and severe encephali tis in patients with MS (Pfender et al. 2015).
Environmental Factors
Over the years, many environmental factors, including vitamin D deficiency, diet, dog ownership, and smoking (Riccio et al. 2011; Suresh Kumar et al. 2013), have been associated with the pathophysiology of MS. In addition, infectious agents have been suspected in the etiology of MS for over a century (Johnson 1994). The first suggestion that infectious agents may be involved in the pathogenesis of MS came from Dr. Pierre Marie, a pupil of Charcot, in 1884. Marie posited that several organisms were involved in the pathogenesis of MS, either alone or in combination, based on the anecdotal association of acute infectious diseases (including typhoid, malaria, pneumonia, and childhood exanthemata) with the onset of disease. Marie hypothesized that MS was triggered by infection, which led to changes in blood vessels ultimately resulting in an inflammatory interstitial reaction of glia (Marie 1895). In addition Marie rather presciently emphasized the contribution of axonal degeneration in disease progression.
Evidence implicating infectious agents in the pathogenesis of MS includes: (1) epidemiological evidence of childhood exposure to infectious agents and an increase in disease exacerbations with viral infection (Johnson 1994; Weinshenker 1996); (2) geographic association of disease susceptibility with evidence of MS clustering (Kurtzke 1995; Haahr et al. 1997); (3) data suggesting that migration to and from high-risk areas before the age of 15 influences the likelihood of developing MS (Weinshenker 1996); (4) abnormal immune responses to a variety of microbes (Jacobson et al. 1985; Neighbour et al. 1981; Soldan et al. 2000); and (5) pathobiological similarities with animal viruses (including canine distemper virus (CDV), murine coronavirus, Theiler’s mouse encephalomyelitis virus , and visna virus) and human viruses (e.g., measles virus , JC virus , and HTLV-I ) that can cause disease with long incubation periods, a relapsing remitting course, and demyelination (Rodriguez et al. 1987; Wang et al. 1990; Appel et al. 1969; Berger et al. 1987; Filipowicz et al. 1983; Jacobson et al. 1990). Although many different viruses have been implicated in the pathogenesis of MS, either as etiologic agents, disease triggers, or cofactors in disease progression , none has been universally accepted as the causative agent of MS (Soldan and Jacobson 2004).
As described above, the distribution of MS follows a geographic distribution with an increased prevalence in northern latitudes, which may be the result of both genetic and environmental influences. Migration studies have supported an infectious etiology of MS (Alter et al. 1966, 1978; Alter and Okihiro 1971; Dean and Kurtzke 1971; Kurtzke et al. 1970). In general, individuals migrating from high-risk to low-risk areas after the age of 15 tend to take their risk of MS with them. However, individuals who migrate from high-risk to low-risk areas before the age of 15 acquire a lower risk. These data suggest that exposure to an environmental factor, perhaps an infectious agent, must occur before the age of 15 in order to influence MS susceptibility.
Reports of clusters or epidemics of MS also support a role for an infectious agent in MS. In the Faroe Islands off the coast of Denmark, no cases of MS were reported from 1929 to 1943. However, after the occupation of the Faroe Islands by British troops in 1940, 20 islanders developed MS between 1940 and the end of the war. The areas of the Faroe Island MS epidemic were found to correlate with the locations of British troop encampments after 1940 (Kurtzke et al. 1997; Rohowsky-Kochan et al. 1995). This initial “outbreak” of MS was followed by additional waves of the disease—each separated by 13 years. It has been proposed that the original cluster of MS in the Faroe Islands was initiated by an infectious agent that only affected the MS susceptibility of individuals between the ages of 11 and 12. The susceptible individuals harbored this microbe in a latent state through adolescence, thus accounting for the 13-year interval between MS clusters on the Faroe Islands (Kurtzke 1995). Other examples of MS epidemics have been described in the Shetland-Orkney Islands; Iceland; Key West, Florida; Mossyrock, Washington; and Mansfield, Massachusetts (Kurtzke et al. 1997; Kurtzke and Page 1997).
Infectious Agents in Multiple Sclerosis
Models of Virus-Induced Demyelination
Viruses have been implicated in a number of demyelinating diseases of the CNS in both animal and human subjects. The association of viruses in other demyelinating diseases further supports an infectious contribution to the development of MS. These examples of virus-mediated neurologic disorders demonstrate the capability of infectious agents to persist in the CNS and to induce demyelination years after the initial infection. Viruses involved in demyelinating diseases of nonhuman animals and man belong to disparate families including: Coronavirus (mouse hepatitis virus (MHV); Lentivirus (Visna virus and Caprine arthritis encephalitis virus); Orthoretrovirus (HTLV-I), Polyoma (SV40); Paramyxovirus (CDV and measles virus), Picornavirus (Theiler’s murine encephalomyelitis virus (TMEV) , encephalomyocarditis virus), Rhabdovirus (Chandipura virus , vesicular stomatitis virus ), and Togavirus (Ross River virus, Semliki Forest virus (SFV) , and Venezuelan equine encephalitis virus) (Rodriguez et al. 1987; Appel 1969; Lavi et al. 1984; Haase 1975). Demyelination from virus infection can occur through various mechanisms including direct infection and/or destruction of oligodendrocytes, indirect demyelination associated with the infection of neurons or astrocytes, or cytokine-mediated damage driven by infected monocytes/macrophages (Johnson 2004). The virus-mediated demyelinating disorders with greatest similarity to MS or that have been used effectively as disease models for MS are described herein (Procaccini et al. 2015) (Table 1).
TMEV is a mouse enterovirus that belongs to the family Picornaviridae and is typically found in the gut. Morphologically, it is similar to human poliovirus. Of interest, TMEV occasionally penetrates the CNS and causes an acute inflammation of the anterior horn cells that pathologically resembles poliomyelitis. One to five months after recovering from the acute paralytic disease, surviving mice develop a progressive inflammatory myelopathy characterized by extensive demyelination (Lipton 1975). TMEV is used as a model for MS because the pathological anomalies are limited to the CNS, infection is latent and persistent, demyelination is mediated by the immune system and occurs after a long incubation period, antibodies to myelin and proteolipid protein can be detected in diseased animals, and there are recurrences of demyelination and remyelination reminiscent of RRMS (Rodriguez et al. 1987). Moreover, there is a clear genetic component in the development of TMEV-mediated disease in mice; TMEV-infected disease-susceptible SJL/J mice develop a persistent CNS infection and later develop autoimmune demyelination, while disease-resistant C57BL/6 (B6) mice rapidly clear the infection and develop no autoimmune pathology (Chastain et al. 2015). The susceptibility of specific mouse strains to TMEV-mediated demyelinating disease is associated with MHC class I genes (Borrow et al. 1992; Thompkins et al. 2002). Of interest, it is the persistent, avirulent strain of TMEV that causes chronic CNS disease (Pevear et al. 1988). As an animal model of MS, TMEV has been particularly useful in the study of axonal damage and inflammatory-induced demyelination (Procaccini et al. 2015). The model also has relevance in investigations focused on the mechanisms of therapeutic drugs in MS. For example, dimethyl fumarate, a relatively new therapeutic option for MS, has been found to suppress TMEV-induced demyelinating disease via modulation of the Nrf2-Keap1 pathway (Kobayashi et al. 2015). In vivo depletion and neutralization studies implicate both CD4+ and CD8+ T cells in TMEV-induced demyelination (Tsunoda and Fujinami 2010; Mecha et al. 2013) and the deletion of virus-specific T cells enhances remyelination (Denic et al. 2014). In addition, deficient NK Dendritic responses and the innate immune system have been demonstrated to play a role in disease progression in persistently infected mice (Chastain et al. 2015; Gilli et al. 2016). Importantly, epitope spreading, in which the initial response is directed against Theiler’s virus and then broadens to myelin proteins, occurs in this model, providing a unique opportunity to investigate epitope spreading in the pathophysiology of a demyelinating disease.
Murine hepatitis virus (MHV ) is a coronavirus that, as the name suggests, primarily infects the liver. However, several strains, including the JHM strain, are neurotropic and cause encephalitis with subsequent CNS demyelination (Mecha et al. 2013). The virus readily infects oligodendrocytes and neurons and kills most animals. However, MHV can establish a persistent infection of astrocytes and those animals that survive acute infection develop a chronic progressive neurologic disease characterized by scattered demyelinating lesions and CNS infiltration of macrophages and lymphocytes (Kyuwa and Stohlman 1990). As the disease progresses, lymphocytic infiltration diminishes while demyelination and astrogliosis increase. These lesions resemble the chronic plaques of MS. The role of the immune system in the pathology of MHV-induced demyelination is complex; CNS disease can occur in the absence of B- and T cells, but does occur with the transfer of virus-specific antibodies or T cells (Matthews et al. 2002; Kim and Perlman 2005; Stohlman and Hinton 2001). B cells are necessary to prevent reactivation of virus in the CNS following clearance of acute infection (Bender and Weiss 2010). In addition, monocyte-derived macrophages and microglia are present in regions of demyelination (Bender and Weiss 2010). Recent evidence suggests that MHV directly activates microglia during acute inflammation, which leads to phagocytosis of the myelin sheath leading to chronic inflammatory demyelination (Chatterjee et al. 2013).
CDV is a member of the morbilliviruses and is related to measles and rinderpest viruses. CDV is a common infection of dogs and other canines. Acute infection with CDV may be followed by a demyelinating encephalomyelitis called subacute diffuse sclerosing encephalitis. This encephalitis is characterized by tremor, paralysis, and convulsions and my not appear until weeks or months after acute infection. Lesions observed show demyelination with sparing of axons and perivascular cuffs of lymphocytes and macrophages (Appel et al. 1969; Appel 1969). Antibodies to CDV and CNS myelin are detected in the serum and CSF of affected animals. Canine distemper demyelinating encephalomyelitis strongly resembles subacute sclerosing panencephalitis (SSPE) pathologically, virologically, and immunologically (Appel et al. 1969; Appel 1969). In this disorder, the initiation of demyelination may result from metabolic insufficiency of oligodendrocytes; macrophages and antiviral antibodies play a greater role in demyelination during relapses and in later stages of the disease (Sips et al. 2007; Vandevelde and Zurbriggen 2005). Of interest, a possible relationship between CDV and MS was investigated based on the description of increased CDV antibody titers in patients with MS (Rohowsky-Kochan et al. 1995). However, this association is not considered robust (Sips et al. 2007).
First recognized in Iceland, Visna is a progressive paralytic disease of sheep (Sigurdsson and Palsson 1958). Visna virus is a member of the lentiviruses, which includes human immunodeficiency viruses I and II. Infection of sheep with Visna virus results in gait abnormalities followed by paraplegia and total paralysis. The disease course is variable and neurologic signs typically correlated with elevations in CSF protein and pleocytosis. Visna virus has a primary tropism for monocytes and macrophages and is transported to the CNS by infected monocytes that release viral particles when they differentiate into macrophages (Narayan et al. 1983; Kennedy et al. 1985). Once released into the CNS, the virus infects microglia and leads to the recruitment and proliferation of cytotoxic T lymphocytes (CTL) . It is believed that the demyelinating lesions observed in this disease may be the result of damage by virus-specific CTL , autoantibodies, and the release of proinflammatory cytokines and chemokines (Peterson and Chesebro 2006).
Examples of viral-induced demyelinating diseases of man include progressive multifocal leukoencephalopathy (PML ), SSPE , and HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP). PML is a rare subacute demyelinating disease associated with JC virus, a polyoma virus that is widespread in human populations worldwide. Patients with PML present with variable symptoms, depending on the locations of CNS lesions (Johnson 1977). Approximately 50–70 % of adult humans are seropositive for JC virus, 65 % of which are infected by the age of 14. The virus typically remains latent in the kidneys, bone marrow, and B cells. In immunocompromised individuals or those with defective cellular immunity, however, the virus enters the brain via infected B cells where it attacks oligodendrocytes (Tornatore et al. 1992). It has been reported that 3–5 % of AIDS patients develop PML (Berger et al. 1987). With the widespread use of highly active antiretroviral therapy in HIV+ individuals, the incidence of PML has decreased, but remained above pre-epidemic levels in the HIV+ population (Christensen et al. 2010). New monoclonal antibody therapies for MS, including natalizubmab , have been linked to the development of PML (Yousry et al. 2006), bringing assays to detect anti-JC virus antibodies and investigations into the pathogenesis of PML to the forefront (Lamdhade et al. 2014).
SSPE is a CNS disease of children and young adults that develops as a rare consequence of measles virus infection. The clinical course of SSPE typically begins with subtle mental deterioration followed by lack of coordination and other motor abnormalities (Filipowicz et al. 1983). The clinical course of SSPE may last either months or years and ultimately results in coma and death. SSPE patients have high serum and CSF antibodies to all measles structural proteins with the exception of the membrane (M) protein. CNS lesions in SSPE are characterized by perivascular cuffing with infiltrates of lymphocytes and plasma cells in both gray and white matter. Extensive demyelination and an increase in hypertrophic astrocytes are also observed in this disease. Cowdry type A and B inclusion bodies containing measles virus-specific antigens are found in both neurons and glia (Souraud et al. 2009). However, intact measles virus particles are absent in brain material of SSPE patients and the mechanism by which measles virus particles are absent in brain material of SSPE patients and the mechanism by which measles virus enters the CNS is unknown. The development of SSPE has not been associated with particular strains of measles virus and, therefore, it is likely that other host factors are required for this rare disorder to occur (Moulin et al. 2011). Recently, a decrease in regulatory T cells, and CD8(+) T cells expressing the inhibitory NKG2A(+) receptor accompanied by elevated activating NK receptors on NK cells has been described in patients with SSPE, supporting a role for chronic stimulation by viral antigens and an over-activated immune response in SSPE pathogenesis {Yentur, 2014 #41}.
The human T-lymphotropic virus type I (HTLV-I ) is associated with a chronic, progressive neurologic disease known as HAM/TSP. The clinical hallmark of HAM/TSP is a gradual onset of lower extremity weakness, bowel and bladder dysfunction, fecal incontinence, Babinski sign, variable sensory loss (Osame 1990a, b; Osame et al. 1987). The onset of HAM/TSP is gradual in most patients and the disease is clinically indistinguishable from the chronic progressive form of MS. Cerebrospinal fluid (CSF) analysis in HAM/TSP is remarkable for OCBs, some of which are directed against HTLV-I (Jacobson et al. 1990). MRI has demonstrated demyelinating lesions in both the white matter and the periventricular regions of HAM/TSP brains and swelling or atrophy in the spinal cord. Although the distinct plaques characteristic of MS are not observed in HAM/TSP, loss of myelin with some preservation of axons has been described (Nakagawa et al. 1995). The incubation period between infection with HTLV-I and the development of HAM/TSP is typically long and only occurs in a minority of those infected.
The propensity for certain individuals to develop either HAM/TSP , ATL, or other HTLV-I associated inflammatory diseases while others remain clinically asymptomatic is not fully understood. It has been suggested that host genetics and immune abnormalities influence an individual’s predisposition to HAM/TSP, as they are believed to influence the likelihood of developing MS (Jacobson 1995). Therefore, the use of HAM/TSP as a “model” of a chronic progressive neurologic disease that occurs in only a small percentage of infected individuals is particularly germane in examining the possible involvement of a ubiquitous virus in the etiology of MS. The lifetime risk of an HTLV-I infected individual acquiring HAM/TSP over a lifetime is estimated to be 0.25 % (Osame 1990b). In Japan, associations have been made between the likelihood of developing either HAM/TSP or ATL and particular HLA haplotypes (Sonoda 1992; Sonoda et al. 1996). HAM/TSP patients of Japanese decent have an increased frequency of certain HLA-Cw7, B7, and DR1 alleles represented by the A26CwB16DR9DQ3 and A24Cw7BDR1DQ1 haplotypes. In contrast, Japanese ATL patients have an increased frequency of HLA-A26, B16, and DR19 and a decreased frequency of HLA A24 and Cw1 compared with controls. The HLA types DRB1*0901, DQN1*0303, and DRB1*1501 in ATL patients and HLA types DB1*0101, DRB1*0803, DRB1*1403, and DRB1* in HAM/TSP patients were found to be mutually exclusive (Sonoda et al. 1996).
The neuropathology of HAM/TSP indicates that immune-mediated mechanisms are involved in the progression of this disease. Furthermore, several lines of evidence indicate that the cellular and humoral immune responses of HAM/TSP patients are altered from that of HTLV-I asymptomatic carriers and uninfected controls. The immunologic hallmarks of HAM/TSP include an increase in ex vivo spontaneous lymphoproliferation in the absence of antigenic stimulation or IL-2 (Kramer et al. 1989), the presence of HTLV-I specific, CD8+ CTL in the PBL, and an increase in antibodies to HTLV-I in sera and CSF. In HAM/TSP, natural killer cells tend to be diminished in both number and activity. HTLV-1 is primarily found in the CD4(+)CD25(+) T cell subset (Regulatory T cells:Tregs ); these cells are critical for establishing peripheral immune tolerance and known to be dysfunctional in HAM/TSP. Recent work by our group suggests that epigenetic modification of the FOXP3 Treg-specific demethylated region decreases the functional suppression of Tregs and may play an important role in the neuropathogenesis of HAM/TSP (Anderson et al. 2014). Although the suggestion of disease-specific HTLV-I strains has been dismissed as a factor in the determination of disease susceptibility, increased viral load has been implicated in the pathogenesis of HAM/TSP . It has been suggested that increased proviral loads may be a predictor for the progression from the asymptomatic carrier state to HAM/TSP. It has also been suggested that the HLA class I allele A2*01 may confer a protective effect on the development of HAM/TSP by influencing the proviral load in infected individuals. Of interest, the HLA A2*01 haplotype has also been shown to decrease the overall risk of MS in an HLA allele comparison study from a cohort of Swedish and Norwegian MS patients and healthy controls using PCR-SSCP (Fogdell-Hahn et al. 2000).
Several models for the immunopathogenesis of HAM/TSP have been proposed. All of these models are based on an HTLV-I-induced immune-mediated response in the CNS to either specific viral antigens or cross-reactive self peptides and none of which are mutually exclusive. The proposed models for the immunopathogenesis of HAM/TSP are similar to those suggested for MS. Therefore, it is hoped that insights into the pathogenesis of HAM/TSP will lead to a better understanding of MS and other neurologic disorders, such as neuro-AIDS, in which virus-mediated immunopathogenesis may occur in a subset of infected individuals.
Potential Mechanisms of Virus-Induced Demyelination in MS
There are a number of models of virus-induced demyelination in MS that seek to explain the complex series of events that ultimately result in the MS lesion. The molecular mimicry model suggests that an immune response against viral antigens that cross-reacts with normal host cell components may contribute to the pathogenesis of MS. Several viral sequences contain part of the human MBP sequence (Jahnke et al. 1985). Moreover, a number of viruses have been shown to share peptide motifs or other antigenic similarities with the components of myelin (Hogeboom 2015; Mameli et al. 2014; Zheng and Zhang 2014). Other relevant examples of molecular mimicry include the cross-reactivity of antibodies against proteins of herpes simplex and measles virus with human intermediate filaments (Fujinami et al. 1983; McCoy et al. 2006; Gabibov et al. 2011; Boucher et al. 2007). In vivo models have supported a role for molecular mimicry in inflammatory neurologic disease. For example, rabbits immunized with a synthetic peptide containing sequences of the hepatitis B virus polymerase develop experimental autoimmune encephalomyelitis (EAE) lesions . Rabbits that develop EAE as a consequence of immunizations with synthetic peptide generated a humoral and cell-mediated immune response to both myelin and hepatitis B polymerase (Fujinami and Oldstone 1985). Additionally, cross-reactivity between a monoclonal antibody to the VP1 protein of TMEV and oligodendrocytes has been demonstrated. Demyelinating disease was observed in mice who were administered the VPI monoclonal antibody. Subsequently, amino acid homologies between immunogenic epitopes of SFV and myelin autoantigens, MBP, myelin proteolipid protein (PLP) and myelin oligodendrocyte glycoprotein (MOG) were identified (Mokhtarian et al. 1999). Immunization of B6 mice with SFV proteins induced significant lymphocyte proliferation to the SFV E2 peptide as well as MOG peptide, 18–32, but not to MBP or PLP peptides. Immunization with both MOG 18–32 and E2 115–129 induced a later-onset chronic EAE-like disease (Mokhtarian et al. 1999). These examples of molecular mimicry support the possibility that immunological recognition of viral peptides of sufficient structural similarity to the immunodominant MBP peptide may lead to clonal expansion of MBP-reactive T cells in MS. Therefore, viruses which are known to cause latent or persistent infections such as herpes viruses may lead to a chronic antigenic stimulation of autoreactive T-cell clones (Allegretta et al. 1990).
A second possible mechanism of virus-induced demyelination is that of a “nonspecific bystander” effect resulting from the reaction of lymphocytes or macrophages to diverse antigens. In this case, oligodendrocytes or myelin sheaths could be damaged by lymphokines or proteases released by activated macrophages and immune cells in response to viral infection. The induction of inflammatory cytokines alone, such as TNF-α, has been shown to induce demyelination. This mechanism of viral stimulation of immunocompetent cells that nonspecifically attack the myelin sheath could explain demyelination in the number of infections with diverse viruses. The mechanism for virus-induced demyelination is also proposed in the pathogenesis of HAM/TSP . It has been suggested that the recognition of HTLV-I gene products in the CNS results in the lysis of glial cells and cytokine release (Ijichi et al. 1993). This model is based on the observation that HTLV-I-specific CTL restricted to immunodominant epitopes of HTLV-I gene products can be demonstrated in the PBL and CSF of HAM/TSP patients and that the frequency of HTLV-I-specific CTLs in the CNS could be either a resident glial cell (oligodendrocytes, astrocytes, or resident microglia) infected with HTLV-I or an infiltrating CD4+ cell. HLA class I and II are not normally expressed in the CNS which would prevent antigen presentation necessary for CTL activity. However, HLA class I and class II expression are upregulated by several cytokines including IFN-γ and TNF-α which can be induced by HTLV-I and are known to be upregulated in HAM/TSP patients. The release of cytokine and chemokine production by HTLV-I is potentially destructive to cells of the CNS. A similar mechanism could explain the virus induction of demyelinating disease in MS. Alternatively, it has been proposed that a combination of two mechanisms, molecular mimicry and bystander activation, induced by virus infection, can lead to CNS demyelinating diseases in MS. In this scenario, viral proteins having molecular mimicry with self-proteins in the CNS can prime genetically susceptible individuals. Once this priming has occurred, an immunologic challenge could result in disease through cytokine-mediated bystander activation (McCoy et al. 2006).
Demyelination may also develop as a consequence of virus-induced autoimmune reaction against brain antigens. Indirect evidence in support of this theory comes from EAE in animals and parainfectious encephalomyelitis in humans where virus-specific CD4+ lymphocytes proliferate in response to MBP (Liebert et al. 1988). It is unknown how viruses break immune tolerance and force the host to mount a strong cell-mediated immune response to brain antigens. As the virus replicates, it may incorporate host antigens into its envelope and inserts, modifies, or coats itself with cellular antigens on the cell surface. It is biologically possible that these newly exposed antigens may be recognized and treated by the host as foreign (Hirsch et al. 1975). For example, the presence of CD13 has been demonstrated in the envelope of human cytomegalovirus (HCMV) . CD13 becomes associated with HCMV upon budding in the Golgi-derived vacuoles during early egress (Soderberg et al. 1996). CD13-specific antibodies were detected in the majority of patients with HCMV viremia or disease after bone marrow transplantation (Soderberg et al. 1996; Naucler et al. 1996). These antibodies were found to cross-react with structures in normal skin biopsies (Soderberg et al. 1996; Naucler et al. 1996). Alternatively, lymphotropic viruses might interact with the immune regulatory system by destroying some populations of lymphocytes or stimulating the generation of autoreactive lymphocyte clones. Many lymphotropic viruses are capable of transforming infected cells and rendering them immortal. It has been demonstrated in vitro that cells immortalized by viruses, such as EBV, are capable of secreting autoantibodies (Rosen 1977). Further, it has been demonstrated that EBV infection transforms CD25+ B cells into antibody-secreting cells in rheumatoid arthritis. These CD25(+) B cells are enriched in peripheral blood and synovial fluid of RA patients, suggesting a relationship between EBV-dependent changes in B-cell phenotype and rheumatoid arthritis (Brisslert et al. 2013). Similar virus/B-cell interactions have been implicated in other models of autoimmunity {Sansonno, 2009 #7}.
Infectious Agents Historically Associated with Multiple Sclerosis
Infectious agents have been suspected in the etiology of MS since the late 1800s (Johnson 1994; Marie 1895). Over the years, several viruses and bacteria have been associated with MS based primarily on elevated antibody titers or the isolation of a particular virus/bacterium from MS material (Table 1). Despite the extensive efforts to associate MS with a particular pathogen, none of these viruses has been definitively associated with the disease. Elevated antibody titers to several viruses including influenza C, herpes simplex 1 and 2, measles, varicella-zoster, rubeola, vaccinia, Epstein-Barr virus (EBV), mumps, SV5, and human herpes virus 6 (HHV-6) have been reported in patients with MS in comparison with healthy controls (Henson et al. 1970; Alperovitch et al. 1991; Ito et al. 1975; Soldan et al. 1997; Sumaya et al. 1980). Most of these reported agents have been discounted from consideration in the pathogenesis of MS . However, a few remain candidate agents of interest.
Of the many viruses from disparate families (Table 2) that have been associated with MS, measles virus has received consideration. Measles virus can establish persistence, both in tissue culture and in vivo, as is the case in SSPE, a chronic progressive demyelinating disease of the CNS. In addition, both humoral and cellular immune responses to measles virus differs in MS patients compared to healthy controls (Jacobson et al. 1985). Intrathecal synthesis of measles-specific antibodies has been demonstrated in the CSF of MS patients and, paradoxically, decreased measles-specific CTL are found in MS patients compared with healthy individuals (Jacobson et al. 1985). Cytoplasmic tubular structures resembling measles nucleocapsids have been found in the astrocytes of one MS patient. Of interest, an explant of this patient’s brain tissue developed a cytopathic effect, which was blocked by pretreatment with an anti-measles serum (Field et al. 1972). Additionally, in one study, measles virus-specific RNA has been detected by in situ hybridization in brain material of patients. However, subsequent studies did not confirm these results.
Several bacteria have also been identified as potential etiologic agents in MS. More than 80 years ago, spirochetes were believed to cause MS based on the fact that syphilis can cause a relapsing/remitting inflammatory disease of the CNS with OCB present in the CSF, while Lyme disease can cause delayed neurologic symptoms with similarities to MS (Oksenberg and Barcellos 2000). Other associations of bacteria and MS have been suggested based on the observation of increased antibody titers in MS patients compared with controls (Sriram et al. 1999; Salmi et al. 1981, 1983). It is unknown whether elevated antibodies to infectious agents found in the CNS of MS patients represent local production of antibody in the CNS as a result of resident lymphocytes or if they are a consequence of “spill-over” of circulating serum antibodies as a consequence of a damaged BBB.
A potential association between Chlamydia pneumonia and MS has received a great deal of consideration. C. pneumonia is an obligate intracellular bacterium of the respiratory tract that causes community acquired pneumonia. While C. pneumonia primarily infects mucosal surfaces, systemic dissemination of the bacterium from the respiratory tract may occur via monocytes and macrophages (Hahn et al. 2002; Campbell and Kuo 2002; Moazed et al. 1998). Epidemiologic evidence suggests that C. pneumonia, like HHV-6, is a ubiquitous human pathogen (Leinonen 1993; Grayston et al. 1989). Since its identification as a unique chlamydial species in 1989, C. pneumonia has been controversially associated with several non-respiratory human pathologies of the cardiovascular system and CNS including artherosclerosis, giant cell arteritis, vasculitis, Alzheimer’s disease, encephalitis, HIV associated dementia, and MS (Balin et al. 1998; Contini et al. 2003; Helweg-Larsen et al. 2002; Koskiniemi et al. 1996; Rimenti et al. 2000; Rosenfeld et al. 2000; Sriram et al. 1998; Little et al. 2014; Grayston et al. 2015).
A relationship between C. pneumonia and MS was first suggested in a case report that demonstrated the presence of the bacterium in the CSF of an MS patient with rapidly progressive MS (Sriram et al. 1998). Of interest, antibiotic treatment of this patient resulted in marked clinical improvement (Sriram et al. 1998). This initial report was extended to a larger study that examined the presence of C. pneumonia in 17 RRMA , 20 and 27 OND controls (Sriram et al. 1999). IN this study approximately 47 % of RRMS and 80 % of SPMS patients were culture positive for C. pneumoniae, compared to only 11 % of OND controls (Sriram et al. 1999). In addition, CSF from all relapsing-remitting and 19/20 secondary progressive MS patients were PCR-positive for the C. pneumonia outer membrane gene compared to only 5/27 other neurologic disease controls (Sriram et al. 1999). Of interest, intrathecal antibodies specific for C. pneumoniae were significantly higher in MS patients than in controls with other neurologic diseases (Sriram et al. 1999). Subsequent studies from this group have demonstrated that OCB could be partially absorbed out of CSF from the majority of MS patients using C. pneumonia antigens and that the intraperitoneal inoculation of mice with C. pneumonia, after immunization with neural antigens, increased the severity of EAE (Du et al. 2002). PCR studies attempting to reproduce the PCR amplification of C. pneumonia from the CSF of MS patients have been inconsistent. Several studies reported no C. pneumonia positive samples by either culture or PCR (Boman et al. 2000; Numazaki and Chibar 2001; Saiz et al. 2001). Others have reported low positivity rates for C. pneumonia in MS (Layh-Schmitt et al. 2000; Aghaei et al. 2011), or high frequency of detection of C. pneumonia in other neurologic disease controls (Gieffers et al. 2001).
Viruses Currently Under Investigation for Their Putative Role in MS
Retroviruses
Although an association between the human retrovirus HTLV-I and MS has not been supported, the possibility of a retroviral etiology of MS has not been entirely excluded (Antel et al. 1990; Chen et al. 1990). In the absence of evidence for an exogenous retrovirus clearly associated with MS, it has been suggested that human endogenous retroviruses (HERV) could be involved (Rasmussen et al. 2000; Rudge 1991). HERV comprise up to 1 % of human DNA and have been implicated as “triggers” in a variety of autoimmune disorders (Krieg et al. 1992; Nakagawa et al. 1997). The proposed pathogenic role for HERV is based on the correlation of superantigen expression from the endogenous retrovirus termed IDDMK1,2,22 and insulin-dependent diabetes mellitus and the presence of autoantibodies that cross-react with HERV proteins in patients with systemic lupus erythematosus and Sjögren’s syndrome (Benoist and Mathis 1997; Conrad et al. 1997; Garry 1994). Endogenous retroviruses may influence immune regulation through direct effects on gene products due to integration sites, interactions with exogenous viruses, or as autoantigens (Rasmussen et al. 2000).
An endogenous retrovirus, known as the multiple sclerosis retrovirus (MSRV), has been associated with MS (Perron et al. 1997). An extensive characterization of this family of HERV, which has been designated as human endogenous retrovirus-W (HERV-W) , classifies MSRV as a member of the HERV-W family (Blond et al. 1999; Fujinami and Libbey 1999). MSRV pol (polymerase gene encoding retroviral reverse transcriptase) sequences were isolated from retroviral particles released by leptomeningeal cells (LM7) cultured from the CSF of an MS patient (Perron et al. 1997). Additionally, pol sequences were isolated from the serum of a significantly higher percentage of MS patients than controls (Garson et al. 1998). Of interest, proteins from HSV-I have been demonstrated to transactivate MSRV in vitro (Perron et al. 1997). This observation may be consistent with the correlation between MS exacerbations and viral infections (Sibley et al. 1985). Sequence analysis of the MSRV pol gene indicates that it is virtually identical to the pol gene of the endogenous retrovirus-9 family, which is expressed in MS and control human tissues (Brahic and Bureau 1997). Interestingly, it has been suggested that MSRV pol sequences are transcriptionally active and expressed in lymphocytes from both MS patients and controls (Nowak et al. 2003). Notably, in this initial study, MSRV Pol was expressed more frequently in lymphocytes and serum from MS patients compared with controls (Nowak et al. 2003). Subsequent neuropathological analysis has suggested that MSRV-Env protein is localized to MS plaques with higher expression of MSRV-Env in active plaques compared to inactive plaques (Mameli et al. 2007; Perron et al. 2012).
A pro-inflammatory mechanism for MSRV’s role in MS involving interactions between MSRV and Toll-like receptor 4 (TLR4) in PBMCs has been suggested (Rolland et al. 2006). In addition, MSRV-Env may interfere with remyelination via TLR4-mediated inhibition of oligodendrocyte differentiation on oligodendrocyte precursor cells (OPCs) (Kremer et al. 2013). Based on its direct toxicity to PBMCs and its ability to activate the innate immune system, MSRV-Env was deemed a relevant therapeutic target for MS. Accordingly, the mAB GNbAc1 was developed as an MSRV antagonist and has been demonstrated to abrogate HERV-W envelope protein-mediated oligodendroglial maturation blockade (Kremer et al. 2015). A phase IIa randomized clinical trial using two doses of GNbAc1 was recently concluded in 10 MS patients . The drug appears to be safe and well tolerated with a larger, proof-of-concept study pending (Derfuss et al. 2015).
Herpesviruses
HHV-6
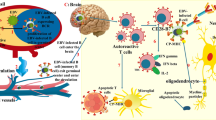
The human herpesvirus 6 (HHV-6 ) is a beta herpesvirus for which seroprevalence rates vary from 72 to 100 % in healthy adults worldwide (Asano et al. 1994). In spite of its universally high seroprevalence in MS patients and the unaffected population alike, HHV-6 has been one of the most serious contenders as an important infectious/environmental factor in the etiology of MS for the last 20 years. The virion architecture of HHV-6 is similar to that of other herpesviruses and consists of a core containing a linear double-stranded DNA, an icosadeltahedral capsid, a tegument surrounding the capsid, and an envelope spiked with viral glycoproteins on its surface. Although HHV-6 replicates primarily in T-lymphocytes , it is a pleiotropic virus that can either productively or nonproductively infect cells from several lineages including B cells, microglia, oligodendrocytes, and astrocytes (Ablashi et al. 1988; Albright et al. 1998; He et al. 1996; Takahashi et al. 1989).
In 1992, HHV-6 was subdivided into two similar, but unique variants (HHV-6A and HHV-6B ) based on genomic, antigenic, and biological differences (Ablashi et al. 1991). The genomes of HHV-6A and HHV-6B range in length from 160 to 170 kbp and encode approximately 100 proteins (Dewhurst et al. 1993). The overall nucleotide sequence identity between HHV-6A and HHV-6B is approximately 90 % (Dewhurst et al. 1993). Of the genes with less than 70 % identity between the two variants, all but one (U47) were found in the immediate-early region. Eighteen open reading frames are unique to either HHV-6A or HHV-6B (Dominguez et al. 1999). Because of the close sequence identities between the two viruses and the absence of serological assays that can easily discriminate between the two variants, HHV-6A and HHV-6B were historically treated as variants of the same virus species. However, it was argued that differences in the biology, cellular tropisms, growth profiles, and restriction endonuclease profiles of the two variants are sufficient to classify them as distinct herpesviruses rather than as variants of the same virus (Dewhurst et al. 1993; Dominguez et al. 1999). In 2012, the International Committee on Taxonomy of Viruses (ICTV ) formally recognized HHV-6A and HHV-6B as two distinct species; the genus Roseolovirus now comprises HHV-6A, HHV-6B, and HHV-7 and the other human beta herpesvirus, cytomegalovirus (CMV or HHV-5), is classified within the genus Cytomegalovirus, along with a number of beta herpesviruses found in nonhuman primates (Adams and Carstens 2012).
HHV-6B has been identified as the causative agent of exanthema subitum and accounts for the majority of symptomatic HHV-6 infections in infants; seropositivity for HHV-6B has been reported to be up to 100 %. In contrast, HHV-6A is less prevalent in the USA, Europe, and Japan (Braun et al. 1997). However, an increased neurovirulence of HHV-6A compared to HHV-6B has been suggested based on a greater detection of the HHV-6A than the HHV-6B in the CSF of children and adults (Hall et al. 1998). Additionally, HHV-6A has been isolated from the CNS of AIDS patients with areas of demyelination (Knox and Carrigan 1995).
Among the multitude of viruses investigated in association with MS, measles virus and a number of herpesviruses have been given the most serious consideration. HHV-6 is an attractive candidate as a possible etiologic agent in MS for several reasons. (1) Primary infection with HHV-6 usually occurs during the first few years of life and the involvement of HHV-6 with MS is consistent with epidemiological evidence in MS suggesting that exposure to an etiologic agent before puberty (Kurtzke 1995). (2) HHV-6, particularly the HHV-6A variant, is highly neurotropic (Hall et al. 1998). (3) Primary infection with HHV-6 occasionally results in neurologic complications including meningitis and meningo-encephalitis and febrile seizures (Asano et al. 1992). HHV-6 has been demonstrated to cause fatal encephalitis in AIDS patients and in individuals immunosuppressed as a consequence of bone marrow transplantation (Knox and Carrigan 1995). Furthermore, a neuropathogenic role for HHV-6 has been suggested based on the development of a variety of disorders associated with active HHV-6 infection including fulminant demyelinating encephalomyelitis, subacute leukoencephalitis, necrotizing encephalitis, PML, and chronic myelopathy (Carrigan et al. 1996; Novoa et al. 1997; Mackenzie et al. 1995). One of the fundamental properties of herpesviruses is their tendency to reactivate. The same factors that often lead to herpesvirus reactivation, such as stress and infection with another agent, have also been associated with MS exacerbations. Unfortunately, the mechanisms by which HHV-6 achieves latency and reactivation are poorly understood. It believed that the virus reactivates frequently as it is readily found in the saliva and peripheral blood mononuclear cells of healthy adults and reactivation in transplant recipients is not uncommon and can cause severe clinical manifestations such as encephalitis, and bone marrow suppression (Yoshikawa 2004). (4) Herpesviruses are typically latent in nervous tissue and cannot be structurally identified in a latent state. Therefore, herpesviruses are not likely to be found by electron microscopy. (5) HHV-6 is pleiotrophic and infects cells of both lymphoid and nonlymphoid origin. The pleiotropism of HHV-6 could explain abnormalities observed in both the immune and nervous systems of patients with MS.
In 1995, Challoner and colleagues were the first to suggest a potential role for HHV-6 in MS based on an unbiased search for nonhuman DNA by representational difference analysis (RDA) (Challoner et al. 1995). This technique is based on successive rounds of subtractive hybridization and PCR amplification, enriched for DNA sequences present in DNA preparations from MS disease material and control PBMCs. In this study, over 70 DNA fragments were analyzed. One of these fragments was found to be homologous to the MDBP gene of the HHV-6B variant Z29. HHV-6 DNA was found in 78 % of MS brains and 74 % of control brains. However, monoclonal antibodies against the HHV-6 101 K protein and the DNA binding protein p41 were detected in the brain tissues of MS patients and not in controls. In MS brains , nuclear staining was found in oligodendrocytes surrounding MS plaques more frequently than in uninvolved white matter (Challoner et al. 1995). While this study did not establish a causal link between HHV-6 and MS, it was the first study to suggest an association between a virus and MS using an unbiased technology.
Soon after the Challoner report , the association of HHV-6 with MS was supported by immunological and molecular studies from our group and others (Soldan et al. 1997). Significantly higher antibody titers against HHV-6 whole virus preparations in MS patients compared with normal controls and increase in HHV-6 DNA in the PBMCs of MS patients by PCR were reported (Sola et al. 1993). While these preliminary studies were intriguing, they were based on methodologies that do not discriminate between latent and active infection of HHV-6. Therefore, to distinguish between these stages of HHV-infection, early antibody responses to the HHV-6 p41/38 early antigen and the presence of HHV-6 serum DNA by nested PCR were examined (Soldan et al. 1997). It was demonstrated that HHV-6 serum DNA correlates with active HHV-6 infection and is not found in healthy individuals (Secchiero et al. 1995). In this original report, a significant increase in IgM response to the p41/38 early antigen was demonstrated in patients with the relapsing remitting form of MS in comparison to healthy controls and individuals with other neurologic disease. An increased IgM response to the p41/38 early antigen was also observed in a group of patients with other inflammatory diseases. Additional studies have confirmed the presence of increased IgM responses to HHV-6 in patients with MS (Ablashi et al. 1998; Friedman et al. 1999; Ramroodi et al. 2013; Khaki et al. 2011), while no correlation has been found in other reports (Enbom et al. 1999; Gustafsson et al. 2014; Simpson et al. 2014). A recent study including 2163 serum samples from 596 MS patients has supported the increase in HHV-6 antibody titers in MS patients and further demonstrated that levels of anti HHV-6 A/B IgG and IgM titers correlate with relapses and progression in MS. Interestingly, IgG and IgM titers reached their highest value 2 weeks 1 month, respectively, before relapse, suggesting that anti-HHV-6 antibody responses have potential as a biomarkers for disease monitoring and modifying therapies (Ortega-Madueno et al. 2014).
In addition to examining the antibody response to HHV-6 in MS, we used a highly sensitive nested PCR assay to amplify HHV-6 DNA from the serum of MS patients and controls as a marker of HHV-6 infection (Soldan et al. 1997). HHV-6 serum DNA was detected in 30 % of MS patients and in 0 % of controls (0/47) consisting of healthy individuals, patients with other inflammatory diseases and patients with other neurologic diseases (Soldan et al. 1997). This NIH cohort was expanded to include a total of 167 MS patients and 70 controls (Berti et al. 2002). Our group has continued to demonstrate the presence of HHV-6 DNA in the serum of approximately 23 % of MS patients and 0 % of controls (Soldan et al. 1997; Berti et al. 2002; Akhyani et al. 2000). Subsequent studies by a number of other groups have reexamined the presence of HHV-6 serum DNA in MS patients. Overall, the results from these studies were equivocal (Soldan et al. 1997; Akhyani et al. 2000; Locatelli et al. 2000; Tejada-Simon et al. 2002) and it was suggested that discrepancies in these reports may be attributable to differences in patient selection, timing of sample acquisition, techniques, and reagents used (Fillet et al. 1998; Jacobson 1998).
To extend the observation of cell free serum HHV-6 DNA in MS patients, a longitudinal study consisting of 215 samples obtained from 59 MS patients followed prospectively for a 5 month period of time was conducted (Berti et al. 2002). Serum HHV-6 DNA was detected from significantly fewer sera obtained during periods of clinical remission. While data from previous studies represent single time points, this study analyzed a large group of MS patients over time and suggests that there is a statistically greater likelihood of detecting HHV-6 DNA in the serum of an MS patient during an exacerbation. Therefore, the time of serum sampling is likely to affect the detection of HHV-6 DNA in the serum of MS patients. Additionally, this report further supports a role for HHV-6 in the pathogenesis of MS by suggesting that the presence of serum HHV-6 DNA, similar to the presence of gadolinium enhancing lesions, coincides with clinical worsening in a subset of patients (Berti et al. 2002). Later studies measuring the presence of active HHV-6 in PBMC and CSF found that high levels of HHV-6A were frequently present in the early stages of MS and suggested that viral replication takes place within the CNS (Rotola et al. 2004). It was further confirmed that HHV-6 viral loads are also elevated during relapses/exacerbations of MS (Alvarez-Lafuente et al. 2006a, b). Moreover, using a novel digital droplet PCR assay, Leibovitch et al. have demonstrated that both the presence of DNA from the more neurotropic HHV-6A and the simultaneous detection of HHV-6A and HHV-6B (representative of co-infection) occur more frequently in saliva from MS patients than controls (Leibovitch et al. 2014).
Cellular immune responses to HHV-6 have been compared in MS patients and controls (Soldan et al. 2000; Tejada-Simon et al. 2002, 2003). In a report examining the T-cell lymphoproliferative responses of healthy controls and patients with MS to both variants of HHV-6 and HHV-7 using whole virus lysates it was demonstrated that there was no difference in either the frequency or magnitude of proliferative responses between healthy controls and patients with MS to either HHV-6B or HHV-7 (Soldan et al. 2000). However, a significantly higher percentage of patients with MS had proliferative responses to HHV-6A (66 %) compared to healthy controls (33 %). At present it is unknown whether the increased frequency of lymphoproliferative responses to the HHV-6A lysate in patients with MS is the result of a higher seroprevalence of the HHV-6A in MS patients or in an altered host immune response (Soldan et al. 2000). Moreover, subsequent studies have successfully amplified HHV-6A in PBMC, serum, and urine of MS patients, but not in controls (Akhyani et al. 2000; Kim et al. 2000). Collectively, the description of an increased lymphoproliferative response to HHV-6A and the unique amplification of HHV-6A DNA in the PBMC, serum, and urine of MS patients further support the association of HHV-6 with MS. Moreover, these studies suggest that the highly neurotropic A variant rather than the B variant may play a role in this disease (Soldan et al. 2000). These studies underscore the importance of considering HHV-6A and HHV-6B separately and not as a single entity.
In a study examining cellular immune responses to HHV-6, T-cell responses to recombinant HHV-6 101 K protein were described in MS patients and controls (Tejada-Simon et al. 2002). The 101 K protein used for this study was cloned from the HHV-6B variant and has 81 % homology with HHV-6A. A lower precursor frequency of 10 K specific T cells was observed in MS patients compared with controls (Tejada-Simon et al. 2002). The impaired T-cell response to the 101 K protein of HHV-6 was associated with increased HHV-6-specific IgM responses. Of interest, HHV-6-specific T-cell lines derived from MS patients demonstrated Th-1 biased cytokine profiles marked by the inability to produce Il-4 and IL-10. In a report from Tejada Simon et al., cross-reactivity between MBP (residues 96–102) and HHV-6 U24 (residues 4–10) were examined. Increased precursor frequencies of MBP/HHV-6 cross-reactive T cells were found in MS patients compared with healthy controls (Tejada-Simon et al. 2003). Importantly, this study demonstrates a relationship between HHV-6 and autoreactive immune responses to MBP. In 2012, these observations were confirmed in a cohort of Chinese MS patients (Cheng et al. 2012). These studies further support an association between HHV-6 and MS and suggest a potential role for HHV-6-specific T cells in the pathogenesis of MS via molecular mimicry.
Because MS is a CNS disorder , detailed analysis of brain material is paramount in assessing the potential association between HHV-6 and MS. HHV-6 is a commensal pathogen of the CNS and HHV-6 DNA can be amplified from 20 to 70 % of brains of MS patients and controls (Challoner et al. 1995; Luppi et al. 1995; Sanders et al. 1996). A number of studies have addressed the frequency of HHV-6 in control and MS brains and identified infected cell phenotypes in pathologically defined tissue (Cermelli et al. 2003; Goodman et al. 2003). Goodman et al. used in situ-PCR (ISPCR ) to identify individual cells containing the HHV-6 genome from surgical biopsy specimens from MS patients presenting with acute disease who had not received immunomodulatory therapy. This study demonstrated that high frequencies of HHV-6 genome positive neuroglial and inflammatory cells are present in acute-phase lesion tissue and that oligodendrocytes are the predominant cell infected in the acute MS lesion (Cermelli et al. 2003; Goodman et al. 2003). In a companion study, pathologically defined material from brain autopsies were isolated by laser-assisted microdissection prior to HHV-6 DNA amplification ; representing a significant advance over PCR amplification of virus DNA from pathologically undefined bulk brain tissue HHV-6 DNA was amplified from brains of both MS patients and controls, HHV-6 DNA was detected at a significantly higher frequency in MS plaques compared to brain tissue from non-MS neurologic disorders, non-MS inflammatory, and normal appearing white matter from MS brains. Collectively, these studies suggest that HHV-6 is present early in the evolution of the MS lesion and may play a significant role in the demyelinative pathogenesis of MS .
Perhaps the most compelling evidence implicating HHV-6 in MS is the presence of HHV-6-specific OCBs in the CSF of 20–25 % of MS patients (Virtanen et al. 2011, 2014; Derfuss et al. 2005). As discussed above, OCBs are common among CNS disorders with an infectious component. Moreover, when the causative agent is known, OCBs are typically specific to the agent of interest; in SSPE, for example, OCBs primarily target measles virus; in HAM/TSP, the OCBs are specific for HTLV-I, etc. Therefore, the identification of HHV-6-specific bands in a subset of MS patients has strengthened the association of HHV-6 and MS and indicates that HHV-6 infection of the CNS is involved in the pathophysiology of the disorder. In 2014, Pietläinen-Nicklén and colleagues analyzed patients with demyelinating diseases, including MS, and HHV-6-reactive CSF OCB, and found that patients with HHV-6 OCB appear to form a separate group that skews younger than patients without HHV-6 OCB, suggesting that antibody reactivity to HHV-6 may be a key, early event in the neuropathogenesis of MS (Pietilainen-Nicklen et al. 2014). In addition, it has demonstrated that herpesvirus-specific CSF OCB inversely correlated with the detection of CSF viral DNA, and that MS patients with CSF viral DNA have significantly more contrast enhancing lesions compared to those without detectable CSF viral DNA. The reduction in antibodies correlates with both the detection of virus in the CSF and MRI activity, which is highly suggestive of an active inflammatory process involving the intrathecal humoral immune response to HHV-6 (Virtanen et al. 2014). Although the majority of recent immune studies have focused on OCBs and CNS B-cell reactivity to HHV-6 in MS, a recent report demonstrating an enrichment of HHV-6 and EBV-specific T cell responses in CSF compared to peripheral blood of MS patients (progressive and relapsing-remitting subtypes) supports a role for intrathecal T-cell responses as well (Wuest et al. 2014).
EBV and Multiple Sclerosis
Arguably no other candidate infectious agent matches the epidemiology, risk profile, and pathobiology of MS as well as EBV. EBV is a near ubiquitous gamma-herpesvirus that causes infectious mononucleosis. Latent infection with EBV results in the expression of viral genes that promote the survival of the host cell as well as the virus. These genes can drive cellular proliferation and oncogenic properties in host cells. Therefore, EBV is considered to be a causative agent in several malignancies including Burkitt’s lymphoma, Hodgkin’s lymphoma, gastric cancers, and nasopharyngeal carcinoma (Chen et al. 2015; Houldcroft and Kellam 2015). EBV is also involved in hairy leukoplakia and CNS lymphomas in the context of HIV infection {Riedel, 2015 #64}. It is estimated that 1 % of all human cancers or 200,000 cancer cases per year are attributable to EBV (Smets and Sokal 2014). In addition, EBV is associated with a variety of autoimmune disorders including Sjögren’s syndrome, dermatomyelitis, rheumatoid arthritis, systemic lupus erythematosus, and MS (Pender 2012; Ascherio and Munger 2010).
There are two main strains of EBV, denoted as types I and II, with each strain comprising several variants and both strains have been isolated from patients with MS (Santon et al. 2011). The virus is spread through salivary contact, establishing infection in the mucosal epithelium of the oropharynx. From this initial site of infection, EBV is transmitted to locally infiltrating B cells, where it establishes a stable, latent infection in memory B cells and can also be found in the epithelial cells of the nasopharynx and gastro-intestinal tract for the duration of a person’s lifetime (Rickinson and Kieff 1996). Occasionally, latent EBV switches to the replicative lytic cycle, which may result in the production and release of virions into the saliva. A strong host immune response ensures that the EBV lytic cycles are brief. Primary infection is generally asymptomatic in children. However, if EBV is contracted during adolescence or early adulthood, it is more often symptomatic and 40 % of primary infections in adolescents and adults results in infectious mononucleosis.
In North America and Western Europe, roughly 50 % of children are infected with EBV by the age of five with seroprevalence rates increasing to up to 95 % after the age of 13; this is in contrast to the developing world where >90 % of individuals are infected in early childhood (Niederman and Evans 1997). A history of infectious mononucleosis is more common in MS patients and in areas where MS is prevalent and there are high standards of hygiene (Warner and Carp 1988). The overlapping epidemiology of infectious mononucleosis and MS is consistent with the “hygiene hypothesis,” which suggests that decreased exposure to childhood infections predisposes individuals to proinflammatory or autoimmune responses and increases MS risk and may explain the lower incidence of MS in areas where there is an increased prevalence of childhood infections (Leibowitz et al. 1966; Bach 2002). The “hygiene hypothesis ” suggests that either (1) MS and infectious mononucleosis arise independently as a consequence of living in conditions of high hygiene in childhood or (2) high hygiene in childhood delays exposure to EBV infection, which subsequently results in an increased risk of MS (Ascherio and Munger 2010).
An association between EBV and MS is supported by numerous studies demonstrating: MS patients have higher EBV-specific antibody titers 15–20 years before onset of neurological symptoms; the presence of EBV infection in brain-infiltrating B cells and plasma cells of 95 % of autopsy cases examined; a history of infectious mononucleosis is more common in MS patients and in areas where MS is prevalent; and a reduced risk of MS among individuals who are EBV seronegative, which increases sharply if the same individuals seroconvert (Warner and Carp 1988; DeLorenze et al. 2006; Ascherio et al. 2001; Serafini et al. 2007; Levin et al. 2010).
In addition to epidemiologic consistency between the risk of MS and the acquisition of EBV infection, a preponderance of laboratory studies have provided additional support for a role of EBV in both adult and pediatric-onset MS (Ascherio and Munger 2010). Of interest, it has been demonstrated that there is an increased seroprevalence of EBV for an increased rate of remote EBV infection in patients with pediatric MS compared with controls (Alotaibi et al. 2004; Banwell et al. 2007; Yea et al. 2013; Pohl et al. 2006). Moreover, among seropositive children tested for EBV DNA in saliva, increased oral shedding was found for EBV, but not for other herpesviruses, in pediatric MS patients compared with controls. Of the virus detected in the saliva of these pediatric patients, it was found that type I EBV was detected more frequently than type II (Yea et al. 2013). In a recent prospective study following children with acquired demyelinating syndromes (ADS), remote EBV infection (defined by the presence of anti-EBNA1 and anti-VCA IgG antibodies were present) was found to be more common in children with MS than those with monophasic ADS (Makhani et al. 2016). By contrast, CMV infection was more common in children with monophasic ADS (Makhani et al. 2016). This study suggests that children with evidence of remote EBV infection without CMV infection were at highest risk of subsequent MS diagnosis and may support previous reports demonstrating a protective role for CMV infection in MS (Sundqvist et al. 2014; Waubant et al. 2011).
Although EBV is a leading candidate infectious agent for multiple sclerosis (MS) and the increased risk of MS among individuals who have been infected with EBV is increasingly well accepted, the mechanism by which EBV may either cause or contribute to the pathogenesis of MS has not been established, though several possible hypotheses have been proposed. It has been suggested that a disruption in the normal sequence of common viral infections, owing to a late primary infection by EBV, may lead to immune dysregulation and ultimately MS in genetically susceptible individuals. Higher frequencies of CD4+ T cells specific for EBNA-1 and enhanced IFN-γ duction have been demonstrated in MS patients relative to healthy, seropositive individuals (Lunemann et al. 2006). In addition, virus-specific T cells may recognize EBV-infected cells in the brain (Serafini et al. 2007; Angelini et al. 2013) or cross-react with myelin antigens potentially contribute to the development of MS by cross-recognition of myelin antigens (Lang et al. 2002; Holmoy et al. 2004; Lunemann et al. 2008) or other autoantigens like αB-crystallin which is induced by B cells upon EBV infection (van Sechel et al. 1999). It has also been suggested that deficient CTL control of EBV infected B cells in MS may be involved in the pathogeneisis of MS; this theory is supported by increased spontaneous immortalization of EBV infected B cells of MS patients in culture (Fraser et al. 1979). In humanized mice, it has been shown that adoptive transfer of EBV-specific CD8+ T-cell clones are capable of transiently controlling infection {Antsiferova, 2014 #120}.
While some studies have demonstrated that the CD8+ T-cell response to EBV infected B cells is deficient in MS and decreases with age (Pender 2012), others have reported increased EBV-specific CD8+ responses in MS. Studies using HLA class I pentamers have shown that lytic antigen-specific CD8+ T cell responses are detected in fewer inactive MS patients than in active MS patients and controls, while the frequency of CD8+ T cells specific for EBV lytic and latent antigens is higher in both active and inactive MS patients (Angelini et al. 2013). In addition, analysis of postmortem MS brain samples showed expression of the EBV lytic protein BZLF-1 and interactions between cytotoxic CD8+ T cells and EBV lytically infected plasma cells in inflammatory white matter lesions and meninges (Angelini et al. 2013). The authors suggest that that inability to control EBV infection during inactive MS could set the stage for intracerebral viral reactivation and disease relapse (Angelini et al. 2013). A large study comprising 221 MS patients and 218 control subjects from Switzerland used HLA-A2, HLA-B7, and HLA-B8 restricted EBV and CMV-specific tetramers to determine the percentage of EBV-specific CD8+ T cells (Jilek et al. 2012). This study determined that there is a higher prevalence of MS patients with HLA HLA-B*0702/EBVRPP-specific CD8+ T cells ex vivo. However, the magnitude of the HLA-B*0702/EBVRPP-specific CD8+ T cell response was lower in MS patients. Upon stimulation with HLA-B7/EBVRPP peptide, MS patients had decreased CD8+ cytotoxic activity compared to controls (Jilek et al. 2012).
Because there is limited access to brain tissue, studies on CNS immune responses in MS patients typically focus on CSF samples. As mentioned previously, we reported the presence of EBV and HHV-6 reactive OCBs in MS CSF (Virtanen et al. 2014). EBV reactive OCBs were rarer than HHV-6 OCBs and, interestingly, mutually exclusive (i.e., OCB reactivity in individual MS patients was shown to either HHV-6 or EBV, but not to both) (Virtanen et al. 2014). In addition, Pfuhl et al. have correlated the presence of serum antibodies to Epstein–Barr nuclear antigen-1, but not to EBV viral capsid antigen, rubella, or varicella zoster virus, with an elevated ithrathecal IgGs in patients with early MS (Pfuhl et al. 2015), suggesting that EBV infection has a key role in early events in the pathogenesis of MS. In addition to elevated EBV-specific antibodies in the CSF, the presence of CTLs recognizing EBV lytic proteins in the CSF of MS patients was reported (van Nierop et al. 2016) complementing earlier studies demonstrating high EBV EBNA-1-specific CTLs in MS patients, particularly at the onset of disease (Jaquiery et al. 2010).
Concluding Thoughts
Epidemiologic evidence suggests that MS is a multifactorial disease, which develops at the intersection of genes, the immune response, and the environment. Several lines of evidence support a role for viruses as a key environmental component or, perhaps, a trigger in the etiology of MS. Although a number of infectious agents have been associated with MS, none of these viruses have been firmly determined to be “THE” causative agent. Additionally, mechanism(s) by which virus–host interactions may lead to demyelination are complex, diverse, and incompletely understood.
The involvement of multiple infectious agents in MS, as suggested originally by Dr. Pierre Marie over 100 years ago, could explain the difficulty in identifying a single agent responsible for this highly variable and chronic disease (Marie 1895). Accordingly, rather than a single, unique causative agent, we suggest that multiple viruses, including ubiquitous and often commensal pathogens like HHV-6 and EBV, may be involved in the etiology of MS that trigger disease in different subsets of individuals through common mechanisms via a virus-specific and/or a cross-reactive autoimmune process, resulting in clinical diseases in a subset of genetically susceptible individuals.
As we posit that that several viruses may be involved in the etiology of MS and that particular viruses trigger disease in different subsets of individuals, we therefore suggest that there may be common mechanism(s) or, perhaps, interactions with a common receptor(s). An insight into how this may be possible may come from further interrogation of the interactions between HHV-6 and its receptor. Santoro and colleagues demonstrated that CD46, also known as the membrane cofactor protein (MCP) , is the cellular receptor for HHV-6 (Santoro et al. 1999). CD46 is a member of family of glycoprotein that function as regulators of complement activation (RCA) and prevent spontaneous activation of complement on autologous cells. CD46 is expressed on all human nucleated cells and soluble forms can be found in plasma tears and seminal fluid of normal individuals (Hara et al. 1992). The use of a virtually ubiquitous human molecule as a surface receptor helps to explain the pleiotropism of HHV-6. Notably, HHV-6 (a large, double stranded DNA virus) and measles virus (a negative-sense, single stranded RNA virus) both use CD46 as a receptor and both viruses have been strongly associated with MS (Dorig et al. 1993; Oldstone et al. 1999). Additionally, other viruses use various members of the RCA family as cellular receptors; EBV uses CD21 while CD55 is used by several echoviruses and coxsakieviruses (Bergelson et al. 1994, 1995).
Could viruses that share a family of receptors in common, such as HHV-6, EBV, and measles virus cause MS by a similar mechanism? It is theoretically possible that the engagement of an RCA receptor by one or many of these viruses may result in increased activation of the complement cascade on autologous cells through downregulation of the receptor. This abnormal increase in complement could lead to widespread tissue damage through cytokine dysregulation, cytolysis, and nitric oxide production (Ghali and Schneider-Schaulies 1998; Karp et al. 1996). Increased levels of the complement regulatory protein CD46 have been demonstrated in both the serum and CSF of MS patients compared with healthy controls and other neurologic disease patients (Soldan et al. 2001). Elevated levels of soluble serum CD46 were also found in other inflammatory disease cohort, indicating that an increase in soluble serum CD46 may be a common phenomenon in autoimmune disorders (Soldan et al. 2001; Cuida et al. 1997; Kawano et al. 1999). Therefore, the increased levels of CD46 demonstrated in the serum and CSF of MS patients may be indicative of an increased activation of the complement system in MS, both peripherally and intrathecally. However, a significant correlation was observed between elevated levels of serum soluble CD46 and the detection of serum HHV-6 DNA in serum from MS patients, while no serum HHV-6 DNA was detected in other inflammatory disease controls (Soldan et al. 2001). Therefore, while an increase in serum soluble CD46 is common in inflammatory diseases in general, immune-pathogenic mechanisms involving both HHV-6 and its receptor are likely to be unique to MS.
Alternatively, these viruses and their common receptor could play a role as an MS “trigger’” either directly or indirectly through an autoimmune mechanism. As viruses replicate, they frequently incorporate host antigens, including their own receptors, into their envelopes as has been demonstrated for CMV, HIV-1, SIV, and HTLV-I. Incorporation of the receptor into the virus envelope could cause the antigen to be recognized and treated by the host as foreign (Soderberg et al. 1996; Marschang et al. 1995; Montefiori et al. 1994). This phenomenon could account for the increase in CD46-specific autoantibodies observed in MS (Pinter et al. 2000). Moreover, complement components, including CD46, play an important role in regulating activation of both B cells and T cells, presenting another possible mechanism for these viruses to influence the immune response and, potentially, the expression and activation of other viruses (Jabara et al. 2011; Fuchs et al. 2009; Ni Choileain and Astier 2011; Astier et al. 2003; Le Friec et al. 2012).
Currently, HHV-6 and EBV are receiving considerable attention as potential MS “triggers.” Although there is a preponderance of evidence supporting relationships between EBV and HHV-6 and MS, it can not be definitively asserted that either EBV or HHV-6 causes or drives the pathogenesis of MS. Additional serological, cellular immune, molecular, animal, and clinical studies, ideally including clinical trials with specific antiviral interventions, are necessary to elucidate the role, if any, of these herpesviruses in the pathogenesis of MS.
References
Ablashi DV, Josephs SF, Buchbinder A, Hellman K, Nakamura S, Llana T, Lusso P, Kaplan M, Dahlberg J, Memon S et al (1988) Human B-lymphotropic virus (human herpesvirus-6). J Virol Methods 21:29–48
Ablashi DV, Balachandran N, Josephs SF, Hung CL, Krueger GR, Kramarsky B, Salahuddin SZ, Gallo RC (1991) Genomic polymorphism, growth properties, and immunologic variations in human herpesvirus-6 isolates. Virology 184:545–552
Ablashi DV, Lapps W, Kaplan M, Whitman JE, Richert JR, Pearson GR (1998) Human Herpesvirus-6 (HHV-6) infection in multiple sclerosis: a preliminary report. Mult Scler 4:490–496
Acers TE, Acers-Warn A (1994) Incidence patterns of immunogenetic diseases in the North American Indians. J Okla State Med Assoc 87:309–314
Adams MJ, Carstens EB (2012) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch Virol 157:1411–1422
Aghaei M, Ashtari F, Bahar M, Falahian MR (2011) Chlamydia pneumoniae seropositivity in Iranian patients with multiple sclerosis: a pilot study. Neurol Neurochir Pol 45:128–131
Akhyani N, Berti R, Brennan MB, Soldan SS, Eaton JM, McFarland HF, Jacobson S (2000) Tissue distribution and variant characterization of human herpesvirus (HHV)-6: increased prevalence of HHV-6A in patients with multiple sclerosis. J Infect Dis 182:1321–1325
Albright AV, Lavi E, Black JB, Goldberg S, O’Connor MJ, Gonzalez-Scarano F (1998) The effect of human herpesvirus-6 (HHV-6) on cultured human neural cells: oligodendrocytes and microglia. J Neurovirol 4:486–494
Allegretta M, Nicklas JA, Sriram S, Albertini RJ (1990) T cells responsive to myelin basic protein in patients with multiple sclerosis. Science 247:718–721
Alonso A, Hernan MA (2008) Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology 71:129–135
Alotaibi S, Kennedy J, Tellier R, Stephens D, Banwell B (2004) Epstein-Barr virus in pediatric multiple sclerosis. JAMA 291:1875–1879
Alperovitch A, Berr C, Cambon-Thomsen A, Puel J, Dugoujon JM, Ruidavets JB, Clanet M (1991) Viral antibody titers, immunogenetic markers, and their interrelations in multiple sclerosis patients and controls. Hum Immunol 31:94–99
Alter M, Okihiro M (1971) When is multiple sclerosis acquired? Neurology 21:1030–1036
Alter M, Leibowitz U, Speer J (1966) Risk of multiple sclerosis related to age at immigration to Israel. Arch Neurol 15:234–237
Alter M, Kahana E, Loewenson R (1978) Migration and risk of multiple sclerosis. Neurology 28:1089–1093
Alvarez-Lafuente R, De Las HV, Bartolome M, Garcia-Montojo M, Arroyo R (2006a) Human herpesvirus 6 and multiple sclerosis: a one-year follow-up study. Brain Pathol 16:20–27
Alvarez-Lafuente R, Garcia-Montojo M, De las Heras V, Bartolome M, Arroyo R (2006b) Clinical parameters and HHV-6 active replication in relapsing-remitting multiple sclerosis patients. J Clin Virol 37(Suppl 1):S24–S26
Anderson R, Lundqvist A, Bergstrom T (1999) Successful treatment of generalized primary herpes simplex type 2 infection during pregnancy. Scand J Infect Dis 31:201–202
Anderson MR, Enose-Akahata Y, Massoud R, Ngouth N, Tanaka Y, Oh U, Jacobson S (2014) Epigenetic modification of the FoxP3 TSDR in HAM/TSP decreases the functional suppression of Tregs. J Neuroimmune Pharmacol 9:522–532
Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, Ruggieri S, Gasperini C, Buttari F, Centonze D, Mechelli R, Salvetti M, Borsellino G, Aloisi F, Battistini L (2013) Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog 9, e1003220
Antel J, Bangham C, Reingold SC (1990) Human T-lymphotrophic virus type I and multiple sclerosis. Ann Neurol 27:689–690
Appel MJ (1969) Pathogenesis of canine distemper. Am J Vet Res 30:1167–1182
Appel MJ, Menegus M, Parsonson IM, Carmichael LE (1969) Pathogenesis of canine herpesvirus in specific-pathogen-free dogs: 5- to 12-week-old pups. Am J Vet Res 30:2067–2073
Asano Y, Yoshikawa T, Kajita Y, Ogura R, Suga S, Yazaki T, Nakashima T, Yamada A, Kurata T (1992) Fatal encephalitis/encephalopathy in primary human herpesvirus-6 infection. Arch Dis Child 67:1484–1485
Asano Y, Yoshikawa T, Suga S, Kobayashi I, Nakashima T, Yazaki T, Kajita Y, Ozaki T (1994) Clinical features of infants with primary human herpesvirus 6 infection (exanthem subitum, roseola infantum). Pediatrics 93:104–108
Ascherio A, Munger KL (2010) Epstein-barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol 5:271–277
Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ, Hankinson SE, Hunter DJ (2001) Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA 286:3083–3088
Astier AL, Xu R, Svoboda M, Hinds E, Munoz O, de Beaumont R, Crean CD, Gabig T, Freedman AS (2003) Temporal gene expression profile of human precursor B leukemia cells induced by adhesion receptor: identification of pathways regulating B-cell survival. Blood 101:1118–1127
Australia and New Zealand Multiple Sclerosis Genetics Consortium (2009) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet 41:824–828
Bach JF (2002) The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347:911–920
Balin BJ, Gerard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, Whittum-Hudson JA, Hudson AP (1998) Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol 187:23–42
Banwell BL (2013) Pediatric multiple sclerosis. Handb Clin Neurol 112:1263–1274
Banwell B, Krupp L, Kennedy J, Tellier R, Tenembaum S, Ness J, Belman A, Boiko A, Bykova O, Waubant E, Mah JK, Stoian C, Kremenchutzky M, Bardini MR, Ruggieri M, Rensel M, Hahn J, Weinstock-Guttman B, Yeh EA, Farrell K, Freedman M, Iivanainen M, Sevon M, Bhan V, Dilenge ME, Stephens D, Bar-Or A (2007) Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol 6:773–781
Barbosa LH, Hamilton R (1973) Virological studies with multiple-sclerosis brain tissues. Lancet 1:1415–1417
Battaglia MA, Zagami P, Uccelli MM (2000) A cost evaluation of multiple sclerosis. J Neurovirol 6(Suppl 2):S191–S193
Bender SJ, Weiss SR (2010) Pathogenesis of murine coronavirus in the central nervous system. J Neuroimmune Pharmacol 5:336–354
Benoist C, Mathis D (1997) Autoimmune diabetes. Retrovirus as trigger, precipitator or marker? Nature 388:833–834
Bergelson JM, Chan M, Solomon KR, St John NF, Lin H, Finberg RW (1994) Decay-accelerating factor (CD55), a glycosylphosphatidylinositol-anchored complement regulatory protein, is a receptor for several echoviruses. Proc Natl Acad Sci U S A 91:6245–6248
Bergelson JM, Mohanty JG, Crowell RL, St John NF, Lublin DM, Finberg RW (1995) Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J Virol 69:1903–1906
Berger JR, Kaszovitz B, Post MJ, Dickinson G (1987) Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann Intern Med 107:78–87
Berti R, Brennan MB, Soldan SS, Ohayon JM, Casareto L, McFarland HF, Jacobson S (2002) Increased detection of serum HHV-6 DNA sequences during multiple sclerosis (MS) exacerbations and correlation with parameters of MS disease progression. J Neurovirol 8:250–256
Blond JL, Beseme F, Duret L, Bouton O, Bedin F, Perron H, Mandrand B, Mallet F (1999) Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J Virol 73:1175–1185
Bobowick AR, Kurtzke JF, Brody JA, Hrubec Z, Gillespie M (1978) Twin study of multiple sclerosis: an epidemiologic inquiry. Neurology 28:978–987
Boiko A, Vorobeychik G, Paty D, Devonshire V, Sadovnick D, University of British Columbia MS Clinic Neurologists (2002) Early onset multiple sclerosis: a longitudinal study. Neurology 59:1006–1010
Boman J, Roblin PM, Sundstrom P, Sandstrom M, Hammerschlag MR (2000) Failure to detect Chlamydia pneumoniae in the central nervous system of patients with MS. Neurology 54:265
Borrow P, Tonks P, Welsh CJ, Nash AA (1992) The role of CD8+ T cells in the acute and chronic phases of Theiler’s murine encephalomyelitis virus-induced disease in mice. J Gen Virol 73(Pt 7):1861–1865
Boucher A, Desforges M, Duquette P, Talbot PJ (2007) Long-term human coronavirus-myelin cross-reactive T-cell clones derived from multiple sclerosis patients. Clin Immunol 123:258–267
Brahic M, Bureau JF (1997) Multiple sclerosis and retroviruses. Ann Neurol 42:984–985
Braun DK, Dominguez G, Pellett PE (1997) Human herpesvirus 6. Clin Microbiol Rev 10:521–567
Bray PF, Bloomer LC, Salmon VC, Bagley MH, Larsen PD (1983) Epstein-Barr virus infection and antibody synthesis in patients with multiple sclerosis. Arch Neurol 40:406–408
Brisslert M, Rehnberg M, Bokarewa MI (2013) Epstein-Barr virus infection transforms CD25+ B cells into antibody-secreting cells in rheumatoid arthritis patients. Immunology 140:421–429
Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, Maecker HT, Davis MM (2015) Variation in the human immune system is largely driven by non-heritable influences. Cell 160:37–47
Burks JS, DeVald BL, Jankovsky LD, Gerdes JC (1980) Two coronaviruses isolated from central nervous system tissue of two multiple sclerosis patients. Science 209:933–934
Burnham JA, Wright RR, Dreisbach J, Murray RS (1991) The effect of high-dose steroids on MRI gadolinium enhancement in acute demyelinating lesions. Neurology 41:1349–1354
Campbell LA, Kuo CC (2002) Chlamydia pneumoniae pathogenesis. J Med Microbiol 51:623–625
Carrigan DR, Harrington D, Knox KK (1996) Subacute leukoencephalitis caused by CNS infection with human herpesvirus-6 manifesting as acute multiple sclerosis. Neurology 47:145–148
Cermelli C, Berti R, Soldan SS, Mayne M, D’Ambrosia JM, Ludwin SK, Jacobson S (2003) High frequency of human herpesvirus 6 DNA in multiple sclerosis plaques isolated by laser microdissection. J Infect Dis 187:1377–1387
Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT, Sobel RA, Lock C, Karpuj M, Pedotti R, Heller R, Oksenberg JR, Steinman L (2001) The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 294:1731–1735
Challoner PB, Smith KT, Parker JD, MacLeod DL, Coulter SN, Rose TM, Schultz ER, Bennett JL, Garber RL, Chang M et al (1995) Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A 92:7440–7444
Charcot JM (1868) Histologie de la sclerose en plaques. Gazette des Hopitaux 41:554–558
Chastain EM, Getts DR, Miller SD (2015) Deficient natural killer dendritic cell responses underlay the induction of Theiler’s virus-induced autoimmunity. MBio 6, e01175
Chatterjee D, Biswas K, Nag S, Ramachandra SG, Das SJ (2013) Microglia play a major role in direct viral-induced demyelination. Clin Dev Immunol 2013:510396
Chen IS, Haislip AM, Myers LW, Ellison GW, Merrill JE (1990) Failure to detect human T-cell leukemia virus-related sequences in multiple sclerosis blood. Arch Neurol 47:1064–1065
Chen XZ, Chen H, Castro FA, Hu JK, Brenner H (2015) Epstein-Barr virus infection and gastric cancer: a systematic review. Medicine (Baltimore) 94, e792
Cheng W, Ma Y, Gong F, Hu C, Qian L, Huang Q, Yu Q, Zhang J, Chen S, Liu Z, Chen X, Zhou T, Zhang D (2012) Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front Biosci (Landmark Ed) 17:1648–1658
Chitnis T, Glanz B, Jaffin S, Healy B (2009) Demographics of pediatric-onset multiple sclerosis in an MS center population from the Northeastern United States. Mult Scler 15:627–631
Christensen KL, Holman RC, Hammett TA, Belay ED, Schonberger LB (2010) Progressive multifocal leukoencephalopathy deaths in the USA, 1979–2005. Neuroepidemiology 35:178–184
Cocco E, Sardu C, Massa R, Mamusa E, Musu L, Ferrigno P, Melis M, Montomoli C, Ferretti V, Coghe G, Fenu G, Frau J, Lorefice L, Carboni N, Contu P, Marrosu MG (2011) Epidemiology of multiple sclerosis in south-western Sardinia. Mult Scler 17:1282–1289
Compston A (1994) The epidemiology of multiple sclerosis: principles, achievements, and recommendations. Ann Neurol 36(Suppl 2):S211–S217
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372:1502–1517
Confavreux C, Vukusic S (2014) The clinical course of multiple sclerosis. Handb Clin Neurol 122:343–369
Conrad B, Weissmahr RN, Boni J, Arcari R, Schupbach J, Mach B (1997) A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell 90:303–313
Contini C, Fainardi E, Seraceni S, Granieri E, Castellazzi M, Cultrera R (2003) Molecular identification and antibody testing of Chlamydophila pneumoniae in a subgroup of patients with HIV-associated dementia complex. Preliminary results. J Neuroimmunol 136:172–177
Cook SD (1997) Multiple sclerosis and viruses. Mult Scler 3:388–389
Coulter-Mackie M, Adler R, Wilson G, Dales S (1985) In vivo and in vitro models of demyelinating diseases. XII. Persistence and expression of corona JHM virus functions in RN2-2 Schwannoma cells during latency. Virus Res 3:245–261
Cree BA (2014) Multiple sclerosis genetics. Handb Clin Neurol 122:193–209
Cross AH, Naismith RT (2014) Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med 275(4):350–363. doi:10.1111/joim.12203
Cuida M, Legler DW, Eidsheim M, Jonsson R (1997) Complement regulatory proteins in the salivary glands and saliva of Sjogren’s syndrome patients and healthy subjects. Clin Exp Rheumatol 15:615–623
Dal Canto MC, Rabinowitz SG (1982) Experimental models of virus-induced demyelination of the central nervous system. Ann Neurol 11:109–127
David S, Lacroix S (2003) Molecular approaches to spinal cord repair. Annu Rev Neurosci 26:411–440
De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, International MSGC, Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, Kappos L, Polman C, McArdle WL, Strachan DP, Evans D, Cross AH, Daly MJ, Compston A, Sawcer SJ, Weiner HL, Hauser SL, Hafler DA, Oksenberg JR (2009) Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 41:776–782
Dean G, Kurtzke JF (1971) On the risk of multiple sclerosis according to age at immigration to South Africa. Br Med J 3:725–729
DeLorenze GN, Munger KL, Lennette ET, Orentreich N, Vogelman JH, Ascherio A (2006) Epstein-Barr virus and multiple sclerosis: evidence of association from a prospective study with long-term follow-up. Arch Neurol 63:839–844
Denic A, Wootla B, Zoecklein L, Rodriguez M (2014) Deletion of virus-specific T-cells enhances remyelination in a model of multiple sclerosis. J Neurol Transl Neurosci 2
Derfuss T, Hohlfeld R, Meinl E (2005) Intrathecal antibody (IgG) production against human herpesvirus type 6 occurs in about 20 % of multiple sclerosis patients and might be linked to a polyspecific B-cell response. J Neurol 252:968–971
Derfuss T, Curtin F, Guebelin C, Bridel C, Rasenack M, Matthey A, Du Pasquier R, Schluep M, Desmeules J, Lang AB, Perron H, Faucard R, Porchet H, Hartung HP, Kappos L, Lalive PH (2015) A phase IIa randomized clinical study testing GNbAC1, a humanized monoclonal antibody against the envelope protein of multiple sclerosis associated endogenous retrovirus in multiple sclerosis patients—a twelve month follow-up. J Neuroimmunol 285:68–70
Dewhurst S, Dollard SC, Pellett PE, Dambaugh TR (1993) Identification of a lytic-phase origin of DNA replication in human herpesvirus 6B strain Z29. J Virol 67:7680–7683
Dominguez G, Dambaugh TR, Stamey FR, Dewhurst S, Inoue N, Pellett PE (1999) Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J Virol 73:8040–8052
Dore-Duffy P, Newman W, Balabanov R, Lisak RP, Mainolfi E, Rothlein R, Peterson M (1995) Circulating, soluble adhesion proteins in cerebrospinal fluid and serum of patients with multiple sclerosis: correlation with clinical activity. Ann Neurol 37:55–62
Dorig RE, Marcil A, Chopra A, Richardson CD (1993) The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305
Du C, Yao SY, Ljunggren-Rose A, Sriram S (2002) Chlamydia pneumoniae infection of the central nervous system worsens experimental allergic encephalitis. J Exp Med 196:1639–1644
Ebers GC, Sadovnick AD, Risch NJ (1995) A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature 377:150–151
Enbom M, Wang FZ, Fredrikson S, Martin C, Dahl H, Linde A (1999) Similar humoral and cellular immunological reactivities to human herpesvirus 6 in patients with multiple sclerosis and controls. Clin Diagn Lab Immunol 6:545–549
Field EJ, Cowshall S, Narang HK, Bell TM (1972) Viruses in multiple sclerosis? Lancet 2:280–281
Filipowicz A, Motta E, Piekarska-Konstanty A, Kazibutowska Z (1983) Subacute sclerosing panencephalitis—clinical observations. Pediatr Pol 58:471–475
Fillet AM, Lozeron P, Agut H, Lyon-Caen O, Liblau R (1998) HHV-6 and multiple sclerosis. Nat Med 4:537; author reply 538
Fogdell-Hahn A, Ligers A, Gronning M, Hillert J, Olerup O (2000) Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens 55:140–148
Franciotta D, Di Stefano AL, Jarius S, Zardini E, Tavazzi E, Ballerini C, Marchioni E, Bergamaschi R, Ceroni M (2011) Cerebrospinal BAFF and Epstein-Barr virus-specific oligoclonal bands in multiple sclerosis and other inflammatory demyelinating neurological diseases. J Neuroimmunol 230:160–163
Fraser KB, Haire M, Millar JH, McCrea S (1979) Increased tendency to spontaneous in-vitro lymphocyte transformation in clinically active multiple sclerosis. Lancet 2:175–176
Friedman JE, Lyons MJ, Cu G, Ablashl DV, Whitman JE, Edgar M, Koskiniemi M, Vaheri A, Zabriskie JB (1999) The association of the human herpesvirus-6 and MS. Mult Scler 5:355–362
Fuchs A, Atkinson JP, Fremeaux-Bacchi V, Kemper C (2009) CD46-induced human Treg enhance B-cell responses. Eur J Immunol 39:3097–3109
Fujinami RS, Libbey JE (1999) Endogenous retroviruses: are they the cause of multiple sclerosis? Trends Microbiol 7:263–264
Fujinami RS, Oldstone MB (1985) Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science 230:1043–1045
Fujinami RS, Oldstone MB, Wroblewska Z, Frankel ME, Koprowski H (1983) Molecular mimicry in virus infection: crossreaction of measles virus phosphoprotein or of herpes simplex virus protein with human intermediate filaments. Proc Natl Acad Sci U S A 80:2346–2350
Gabibov AG, Belogurov AA Jr, Lomakin YA, Zakharova MY, Avakyan ME, Dubrovskaya VV, Smirnov IV, Ivanov AS, Molnar AA, Gurtsevitch VE, Diduk SV, Smirnova KV, Avalle B, Sharanova SN, Tramontano A, Friboulet A, Boyko AN, Ponomarenko NA, Tikunova NV (2011) Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J 25:4211–4221
Garry RF (1994) New evidence for involvement of retroviruses in Sjogren’s syndrome and other autoimmune diseases. Arthritis Rheum 37:465–469
Garson JA, Tuke PW, Giraud P, Paranhos-Baccala G, Perron H (1998) Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis. Lancet 351:33
Ghali M, Schneider-Schaulies J (1998) Receptor (CD46)- and replication-mediated interleukin-6 induction by measles virus in human astrocytoma cells. J Neurovirol 4:521–530
Ghezzi A, Deplano V, Faroni J, Grasso MG, Liguori M, Marrosu G, Pozzilli C, Simone IL, Zaffaroni M (1997) Multiple sclerosis in childhood: clinical features of 149 cases. Mult Scler 3:43–46
Gieffers J, Pohl D, Treib J, Dittmann R, Stephan C, Klotz K, Hanefeld F, Solbach W, Haass A, Maass M (2001) Presence of Chlamydia pneumoniae DNA in the cerebral spinal fluid is a common phenomenon in a variety of neurological diseases and not restricted to multiple sclerosis. Ann Neurol 49:585–589
Gilli F, Li L, Pachner AR (2016) The immune response in the CNS in Theiler’s virus induced demyelinating disease switches from an early adaptive response to a chronic innate-like response. J Neurovirol 22(1):66–79. doi:10.1007/s13365-015-0369-4
Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM (2003) Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J Infect Dis 187:1365–1376
Goswami KK, Randall RE, Lange LS, Russell WC (1987) Antibodies against the paramyxovirus SV5 in the cerebrospinal fluids of some multiple sclerosis patients. Nature 327:244–247
Grayston JT, Wang SP, Kuo CC, Campbell LA (1989) Current knowledge on Chlamydia pneumoniae, strain TWAR, an important cause of pneumonia and other acute respiratory diseases. Eur J Clin Microbiol Infect Dis 8:191–202
Grayston JT, Belland RJ, Byrne GI, Kuo CC, Schachter J, Stamm WE, Zhong G (2015) Infection with Chlamydia pneumoniae as a cause of coronary heart disease: the hypothesis is still untested. Pathog Dis 73:1–9
Greer JM, Sobel RA, Sette A, Southwood S, Lees MB, Kuchroo VK (1996) Immunogenic and encephalitogenic epitope clusters of myelin proteolipid protein. J Immunol 156:371–379
Gudnadottir M, Helgadottir H, Bjarnason O, Jonsdottir K (1964) Virus isolated from the brain of a patient with multiple sclerosis. Exp Neurol 9:85–95
Gustafsson R, Reitsma R, Stralfors A, Lindholm A, Press R, Fogdell-Hahn A (2014) Incidence of human herpesvirus 6 in clinical samples from Swedish patients with demyelinating diseases. J Microbiol Immunol Infect 47:418–421
Haahr S, Munch M, Christensen T, Moller-Larsen A, Hvas J (1997) Cluster of multiple sclerosis patients from Danish community. Lancet 349:923
Haase AT (1975) The slow infection caused by visna virus. Curr Top Microbiol Immunol 72:101–156
Hahn DL, Azenabor AA, Beatty WL, Byrne GI (2002) Chlamydia pneumoniae as a respiratory pathogen. Front Biosci 7:e66–e76
Hall CB, Caserta MT, Schnabel KC, Long C, Epstein LG, Insel RA, Dewhurst S (1998) Persistence of human herpesvirus 6 according to site and variant: possible greater neurotropism of variant A. Clin Infect Dis 26:132–137
Hammond SR, McLeod JG, Millingen KS, Stewart-Wynne EG, English D, Holland JT, McCall MG (1988) The epidemiology of multiple sclerosis in three Australian cities: Perth, Newcastle and Hobart. Brain 111(Pt 1):1–25
Hara T, Kuriyama S, Kiyohara H, Nagase Y, Matsumoto M, Seya T (1992) Soluble forms of membrane cofactor protein (CD46, MCP) are present in plasma, tears, and seminal fluid in normal subjects. Clin Exp Immunol 89:490–494
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, HERMES Trial Group (2008) B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358:676–688
He J, McCarthy M, Zhou Y, Chandran B, Wood C (1996) Infection of primary human fetal astrocytes by human herpesvirus 6. J Virol 70:1296–1300
He B, Yang B, Lundahl J, Fredrikson S, Hillert J (1998) The myelin basic protein gene in multiple sclerosis: identification of discrete alleles of a 1.3 kb tetranucleotide repeat sequence. Acta Neurol Scand 97:46–51
Helweg-Larsen J, Tarp B, Obel N, Baslund B (2002) No evidence of parvovirus B19, Chlamydia pneumoniae or human herpes virus infection in temporal artery biopsies in patients with giant cell arteritis. Rheumatology (Oxford) 41:445–449
Henson TE, Brody JA, Sever JL, Dyken ML, Cannon J (1970) Measles antibody titers in multiple sclerosis patients, siblings, and controls. JAMA 211:1985–1988
Hirsch ME, Kelly AP, Proffitt MR, Black PH (1975) Cell-mediated immunity to antigens associated with endogenous murine C-type leukemia viruses. Science 187:959–961
Hoftberger R, Aboul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, Jellinger K, Lassmann H (2004) Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol 14:43–50
Hogeboom C (2015) Peptide motif analysis predicts lymphocytic choriomeningitis virus as trigger for multiple sclerosis. Mol Immunol 67(2 Pt B):625–635. doi:10.1016/j.molimm.2015.07.041
Holmoy T, Kvale EO, Vartdal F (2004) Cerebrospinal fluid CD4+ T cells from a multiple sclerosis patient cross-recognize Epstein-Barr virus and myelin basic protein. J Neurovirol 10:278–283
Houldcroft CJ, Kellam P (2015) Host genetics of Epstein-Barr virus infection, latency and disease. Rev Med Virol 25:71–84
Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J (2001) A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med 194:669–676
Ijichi S, Izumo S, Eiraku N, Machigashira K, Kubota R, Nagai M, Ikegami N, Kashio N, Umehara F, Maruyama I et al (1993) An autoaggressive process against bystander tissues in HTLV-I-infected individuals: a possible pathomechanism of HAM/TSP. Med Hypotheses 41:542–547
International Multiple Sclerosis Genetics Consortium, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL (2007) Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357:851–862
Ito M, Barron AL, Olszewski WA, Milgrom F (1975) Antibody titers by mixed agglutination to varicella-zoster, herpes simplex and vaccinia viruses in patients with multiple sclerosis. Proc Soc Exp Biol Med 149:835–839
Jabara HH, Angelini F, Brodeur SR, Geha RS (2011) Ligation of CD46 to CD40 inhibits CD40 signaling in B cells. Int Immunol 23:215–221
Jacobson S (1995) Human T lymphotropic virus, type-I myelopathy: an immunopathologically mediated chronic progressive disease of the central nervous system. Curr Opin Neurol 8:179–183
Jacobson S (1998) Association of human herpesvirus-6 and multiple sclerosis: here we go again? J Neurovirol 4:471–473
Jacobson S, Flerlage ML, McFarland HF (1985) Impaired measles virus-specific cytotoxic T cell responses in multiple sclerosis. J Exp Med 162:839–850
Jacobson S, Gupta A, Mattson D, Mingioli E, McFarlin DE (1990) Immunological studies in tropical spastic paraparesis. Ann Neurol 27:149–156
Jahnke U, Fischer EH, Alvord EC Jr (1985) Sequence homology between certain viral proteins and proteins related to encephalomyelitis and neuritis. Science 229:282–284
Jaquiery E, Jilek S, Schluep M, Meylan P, Lysandropoulos A, Pantaleo G, Du Pasquier RA (2010) Intrathecal immune responses to EBV in early MS. Eur J Immunol 40:878–887
Jersild C, Dupont B, Fog T, Platz PJ, Svejgaard A (1975) Histocompatibility determinants in multiple sclerosis. Transplant Rev 22:148–163
Jilek S, Schluep M, Harari A, Canales M, Lysandropoulos A, Zekeridou A, Pantaleo G, Du Pasquier RA (2012) HLA-B7-restricted EBV-specific CD8+ T cells are dysregulated in multiple sclerosis. J Immunol 188:4671–4680
Johnson RT (1977) Slow viral diseases of the central nervous system: transmissibility vs. communicability. Clin Neurosurg 24:590–599
Johnson RT (1994) The virology of demyelinating diseases. Ann Neurol 36(Suppl):S54–S60
Johnson RT (2004) Demyelinating diseases. In: Knobler SL, O’Connor S, Lemon SM et al (eds) Institute of medicine (US) forum on microbial threats. National Academies Press, Washington, DC
Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, Myers LW, Panitch HS, Rose JW, Schiffer RB (1995) Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 45:1268–1276
Joshi N, Usuku K, Hauser SL (1993) The T-cell response to myelin basic protein in familial multiple sclerosis: diversity of fine specificity, restricting elements, and T-cell receptor usage. Ann Neurol 34:385–393
Kalman B, Takacs K, Gyodi E, Kramer J, Fust G, Tauszik T, Guseo A, Kuntar L, Komoly S, Nagy C et al (1991) Sclerosis multiplex in gypsies. Acta Neurol Scand 84:181–185
Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, Yin M, Leppert D, Glanzman R, Tinbergen J, Hauser SL (2011) Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 378:1779–1787
Karp CL, Wysocka M, Wahl LM, Ahearn JM, Cuomo PJ, Sherry B, Trinchieri G, Griffin DE (1996) Mechanism of suppression of cell-mediated immunity by measles virus. Science 273:228–231
Karussis D (2014) The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J Autoimmun 48–49:134–142. doi:10.1016/j.jaut.2014.01.022
Kawano M, Seya T, Koni I, Mabuchi H (1999) Elevated serum levels of soluble membrane cofactor protein (CD46, MCP) in patients with systemic lupus erythematosus (SLE). Clin Exp Immunol 116:542–546
Kellar-Wood H, Robertson N, Govan GG, Compston DA, Harding AE (1994) Leber’s hereditary optic neuropathy mitochondrial DNA mutations in multiple sclerosis. Ann Neurol 36:109–112
Kennedy PG, Narayan O, Ghotbi Z, Hopkins J, Gendelman HE, Clements JE (1985) Persistent expression of Ia antigen and viral genome in visna-maedi virus-induced inflammatory cells. Possible role of lentivirus-induced interferon. J Exp Med 162:1970–1982
Khaki M, Ghazavi A, Ghasami K, Rafiei M, Payani MA, Ghaznavi-Rad E, Mosayebi G (2011) Evaluation of viral antibodies in Iranian multiple sclerosis patients. Neurosciences (Riyadh) 16:224–228
Kim TS, Perlman S (2005) Virus-specific antibody, in the absence of T cells, mediates demyelination in mice infected with a neurotropic coronavirus. Am J Pathol 166:801–809
Kim JS, Lee KS, Park JH, Kim MY, Shin WS (2000) Detection of human herpesvirus 6 variant A in peripheral blood mononuclear cells from multiple sclerosis patients. Eur Neurol 43:170–173
Knox KK, Carrigan DR (1995) Active human herpesvirus (HHV-6) infection of the central nervous system in patients with AIDS. J Acquir Immune Defic Syndr Hum Retrovirol 9:69–73
Kobayashi K, Tomiki H, Inaba Y, Ichikawa M, Kim BS, Koh CS (2015) Dimethyl fumarate suppresses Theiler’s murine encephalomyelitis virus-induced demyelinating disease by modifying the Nrf2-Keap1 pathway. Int Immunol 27:333–344
Koskiniemi M, Gencay M, Salonen O, Puolakkainen M, Farkkila M, Saikku P, Vaheri A (1996) Chlamydia pneumoniae associated with central nervous system infections. Eur Neurol 36:160–163
Kotter MR, Zhao C, van Rooijen N, Franklin RJ (2005) Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis 18:166–175
Kramer A, Jacobson S, Reuben JF, Murphy EL, Wiktor SZ, Cranston B, Figueroa JP, Hanchard B, McFarlin D, Blattner WA (1989) Spontaneous lymphocyte proliferation in symptom-free HTLV-I positive Jamaicans. Lancet 2:923–924
Kremer D, Schichel T, Forster M, Tzekova N, Bernard C, van der Valk P, van Horssen J, Hartung HP, Perron H, Kury P (2013) Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation. Ann Neurol 74:721–732
Kremer D, Forster M, Schichel T, Gottle P, Hartung HP, Perron H, Kury P (2015) The neutralizing antibody GNbAC1 abrogates HERV-W envelope protein-mediated oligodendroglial maturation blockade. Mult Scler 21:1200–1203
Krieg AM, Gourley MF, Perl A (1992) Endogenous retroviruses: potential etiologic agents in autoimmunity. FASEB J 6:2537–2544
Krupp LB, Banwell B, Tenembaum S, International Pediatric MS Study Group (2007) Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 68:S7–S12
Kurtzke JF (1995) MS epidemiology world wide. One view of current status. Acta Neurol Scand Suppl 161:23–33
Kurtzke JF (2005) Epidemiology and etiology of multiple sclerosis. Phys Med Rehabil Clin N Am 16:327–349
Kurtzke JF, Page WF (1997) Epidemiology of multiple sclerosis in US veterans: VII. Risk factors for MS. Neurology 48:204–213
Kurtzke JF, Dean G, Botha DP (1970) A method for estimating the age at immigration of white immigrants to South Africa, with an example of its importance. S Afr Med J 44:663–669
Kurtzke JF, Hyllested K, Arbuckle JD, Bronnum-Hansen H, Wallin MT, Heltberg A, Jacobsen H, Olsen A, Eriksen LS (1997) Multiple sclerosis in the Faroe Islands. 7. Results of a case control questionnaire with multiple controls. Acta Neurol Scand 96:149–157
Kyuwa S, Stohlman S (1990) Background paper. Advances in the study of MHV infection of mice. Adv Exp Med Biol 276:555–556
Lamdhade S, Ashkanani A, Alroughani R (2014) Prevalence of anti-JC virus antibody in multiple sclerosis patients in Kuwait. ISRN Neurol 2014:861091
Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, Hjorth P, Sondergaard L, Svejgaard A, Wucherpfennig K, Stuart DI, Bell JI, Jones EY, Fugger L (2002) A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol 3:940–943
Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR (1984) Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597–603
Layh-Schmitt G, Bendl C, Hildt U, Dong-Si T, Juttler E, Schnitzler P, Grond-Ginsbach C, Grau AJ (2000) Evidence for infection with Chlamydia pneumoniae in a subgroup of patients with multiple sclerosis. Ann Neurol 47:652–655
Le Friec G, Sheppard D, Whiteman P, Karsten CM, Shamoun SA, Laing A, Bugeon L, Dallman MJ, Melchionna T, Chillakuri C, Smith RA, Drouet C, Couzi L, Fremeaux-Bacchi V, Kohl J, Waddington SN, McDonnell JM, Baker A, Handford PA, Lea SM, Kemper C (2012) The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol 13:1213–1221
Leibovitch EC, Brunetto GS, Caruso B, Fenton K, Ohayon J, Reich DS, Jacobson S (2014) Coinfection of human herpesviruses 6A (HHV-6A) and HHV-6B as demonstrated by novel digital droplet PCR assay. PLoS One 9, e92328
Leibowitz U, Antonovsky A, Medalie JM, Smith HA, Halpern L, Alter M (1966) Epidemiological study of multiple sclerosis in Israel. II. Multiple sclerosis and level of sanitation. J Neurol Neurosurg Psychiatry 29:60–68
Leinonen M (1993) Pathogenetic mechanisms and epidemiology of Chlamydia pneumoniae. Eur Heart J 14(Suppl K):57–61
Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A (2010) Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol 67:824–830
Liebert UG, Linington C, ter Meulen V (1988) Induction of autoimmune reactions to myelin basic protein in measles virus encephalitis in Lewis rats. J Neuroimmunol 17:103–118
Lin X, Deng FY, Lu X, Lei SF (2015) Susceptibility genes for multiple sclerosis identified in a gene-based genome-wide association study. J Clin Neurol 11(4):311–318
Lipton HL (1975) Theiler’s virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun 11:1147–1155
Little CS, Joyce TA, Hammond CJ, Matta H, Cahn D, Appelt DM, Balin BJ (2014) Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front Aging Neurosci 6:304
Locatelli G, Santoro F, Veglia F, Gobbi A, Lusso P, Malnati MS (2000) Real-time quantitative PCR for human herpesvirus 6 DNA. J Clin Microbiol 38:4042–4048
Lublin FD, Reingold SC (1996) Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46:907–911
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sorensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, Bebo B Jr, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O’Connor PW, Petkau J, Pozzilli C, Rudick RA, Sormani MP, Stuve O, Waubant E, Polman CH (2014) Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 83:278–286
Lunemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Munz C, Martin R (2006) Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain 129:1493–1506
Lunemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, Munz C (2008) EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med 205:1763–1773
Luppi M, Barozzi P, Maiorana A, Marasca R, Trovato R, Fano R, Ceccherini-Nelli L, Torelli G (1995) Human herpesvirus-6: a survey of presence and distribution of genomic sequences in normal brain and neuroglial tumors. J Med Virol 47:105–111
Mackenzie IR, Carrigan DR, Wiley CA (1995) Chronic myelopathy associated with human herpesvirus-6. Neurology 45:2015–2017
Makhani N, Banwell B, Tellier R, Yea C, McGovern S, O’Mahony J, Ahorro JM, Arnold D, Sadovnick AD, Marrie RA, Bar-Or A, Canadian Pediatric Demyelinating Disease Network (2016) Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult Scler 22(3):385–388. doi:10.1177/1352458515595876
Mameli G, Astone V, Arru G, Marconi S, Lovato L, Serra C, Sotgiu S, Bonetti B, Dolei A (2007) Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not Human herpesvirus 6. J Gen Virol 88:264–274
Mameli G, Cossu D, Cocco E, Masala S, Frau J, Marrosu MG, Sechi LA (2014) Epstein-Barr virus and Mycobacterium avium subsp. paratuberculosis peptides are cross recognized by anti-myelin basic protein antibodies in multiple sclerosis patients. J Neuroimmunol 270:51–55
Marie P (1895) Lectures on diseases of the spinal cord. New Sydenham Society, London, pp 102–153
Markowitz CE (2013) Multiple sclerosis update. Am J Manag Care 19:s294–s300
Marschang P, Sodroski J, Wurzner R, Dierich MP (1995) Decay-accelerating factor (CD55) protects human immunodeficiency virus type 1 from inactivation by human complement. Eur J Immunol 25:285–290
Martin C, Enbom M, Soderstrom M, Fredrikson S, Dahl H, Lycke J, Bergstrom T, Linde A (1997) Absence of seven human herpesviruses, including HHV-6, by polymerase chain reaction in CSF and blood from patients with multiple sclerosis and optic neuritis. Acta Neurol Scand 95:280–283
Matthews AE, Lavi E, Weiss SR, Paterson Y (2002) Neither B cells nor T cells are required for CNS demyelination in mice persistently infected with MHV-A59. J Neurovirol 8:257–264
McCoy L, Tsunoda I, Fujinami RS (2006) Multiple sclerosis and virus induced immune responses: autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 39:9–19
McStreet GH, Elkunk RB, Latiwonk QI (1992) Investigations of environmental conditions during cluster indicate probable vectors of unknown exogenous agent(s) of multiple sclerosis. Comp Immunol Microbiol Infect Dis 15:75–77
Mecha M, Carrillo-Salinas FJ, Mestre L, Feliu A, Guaza C (2013) Viral models of multiple sclerosis: neurodegeneration and demyelination in mice infected with Theiler’s virus. Prog Neurobiol 101–102:46–64
Medaer R (1979) Does the history of multiple sclerosis go back as far as the 14th century? Acta Neurol Scand 60:189–192
Milanov I, Topalov N, Kmetski T (1999) Prevalence of multiple sclerosis in Gypsies and Bulgarians. Neuroepidemiology 18:218–222
Miller DH, Hornabrook RW, Dagger J, Fong R (1986) Ethnic and HLA patterns related to multiple sclerosis in Wellington, New Zealand. J Neurol Neurosurg Psychiatry 49:43–46
Moazed TC, Kuo CC, Grayston JT, Campbell LA (1998) Evidence of systemic dissemination of Chlamydia pneumoniae via macrophages in the mouse. J Infect Dis 177:1322–1325
Mokhtarian F, Zhang Z, Shi Y, Gonzales E, Sobel RA (1999) Molecular mimicry between a viral peptide and a myelin oligodendrocyte glycoprotein peptide induces autoimmune demyelinating disease in mice. J Neuroimmunol 95:43–54
Montefiori DC, Cornell RJ, Zhou JY, Zhou JT, Hirsch VM, Johnson PR (1994) Complement control proteins, CD46, CD55, and CD59, as common surface constituents of human and simian immunodeficiency viruses and possible targets for vaccine protection. Virology 205:82–92
Montomoli C, Allemani C, Solinas G, Motta G, Bernardinelli L, Clemente S, Murgia BS, Ticca AF, Musu L, Piras ML, Ferrai R, Caria A, Sanna S, Porcu O (2002) An ecologic study of geographical variation in multiple sclerosis risk in central Sardinia, Italy. Neuroepidemiology 21:187–193
Moulin E, Beal V, Jeantet D, Horvat B, Wild TF, Waku-Kouomou D (2011) Molecular characterization of measles virus strains causing subacute sclerosing panencephalitis in France in 1977 and 2007. J Med Virol 83:1614–1623
Nakagawa M, Izumo S, Ijichi S, Kubota H, Arimura K, Kawabata M, Osame M (1995) HTLV-I-associated myelopathy: analysis of 213 patients based on clinical features and laboratory findings. J Neurovirol 1:50–61
Nakagawa K, Brusic V, McColl G, Harrison LC (1997) Direct evidence for the expression of multiple endogenous retroviruses in the synovial compartment in rheumatoid arthritis. Arthritis Rheum 40:627–638
Narayan O, Kennedy-Stoskopf S, Sheffer D, Griffin DE, Clements JE (1983) Activation of caprine arthritis-encephalitis virus expression during maturation of monocytes to macrophages. Infect Immun 41:67–73
Naucler CS, Larsson S, Moller E (1996) A novel mechanism for virus-induced autoimmunity in humans. Immunol Rev 152:175–192
Neighbour PA, Miller AE, Bloom BR (1981) Interferon responses of leukocytes in multiple sclerosis. Neurology 31:561–566
Ni Choileain S, Astier AL (2011) CD46 plasticity and its inflammatory bias in multiple sclerosis. Arch Immunol Ther Exp (Warsz) 59:49–59
Niederman JC, Evans AS (1997) Epstein-Barr virus. In: Evans AS, Kaslow RA (eds) Viral infections of humans: epidemiolog and Control. Plenum Medical Book Company, New York, pp 253–283
Novoa LJ, Nagra RM, Nakawatase T, Edwards-Lee T, Tourtellotte WW, Cornford ME (1997) Fulminant demyelinating encephalomyelitis associated with productive HHV-6 infection in an immunocompetent adult. J Med Virol 52:301–308
Nowak J, Januszkiewicz D, Pernak M, Liwen I, Zawada M, Rembowska J, Nowicka K, Lewandowski K, Hertmanowska H, Wender M (2003) Multiple sclerosis-associated virus-related pol sequences found both in multiple sclerosis and healthy donors are more frequently expressed in multiple sclerosis patients. J Neurovirol 9:112–117
Numazaki K, Chibar S (2001) Failure to detect Chlamydia pneumoniae in the central nervous system of patients with MS. Neurology 57:746
O’Gorman C, Lin R, Stankovich J, Broadley SA (2013) Modelling genetic susceptibility to multiple sclerosis with family data. Neuroepidemiology 40:1–12
Oksenberg JR, Barcellos LF (2000) The complex genetic aetiology of multiple sclerosis. J Neurovirol 6(Suppl 2):S10–S14
Oldstone MB, von Herrath M, Lewicki H, Hudrisier D, Whitton JL, Gairin JE (1999) Use of a high-affinity peptide that aborts MHC-restricted cytotoxic T lymphocyte activity against multiple viruses in vitro and virus-induced immunopathologic disease in vivo. Virology 256:246–257
Ortega-Madueno I, Garcia-Montojo M, Dominguez-Mozo MI, Garcia-Martinez A, Arias-Leal AM, Casanova I, Arroyo R, Alvarez-Lafuente R (2014) Anti-human herpesvirus 6A/B IgG correlates with relapses and progression in multiple sclerosis. PLoS One 9, e104836
Osame M (1990a) Diagnosis and treatment of HTLV-I associated myelopathy. Nippon Naika Gakkai Zasshi 79:261–265
Osame M (1990b) HTLV-I-associated myelopathy (HAM). Tanpakushitsu Kakusan Koso 35:1320–1326
Osame M, Igata A, Matsumoto M, Tara M (1987) HTLV-I-associated myelopathy. Gan To Kagaku Ryoho 14:2411–2416
Panitch HS, Hirsch RL, Schindler J, Johnson KP (1987) Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology 37:1097–1102
Parise H, Laliberte F, Lefebvre P, Duh MS, Kim E, Agashivala N, Abouzaid S, Weinstock-Guttman B (2013) Direct and indirect cost burden associated with multiple sclerosis relapses: excess costs of persons with MS and their spouse caregivers. J Neurol Sci 330:71–77
Paty DW, Li DK (1993) Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurology 43:662–667
Pender MP (2012) CD8+ T-cell deficiency, Epstein-Barr virus infection, vitamin D deficiency, and steps to autoimmunity: a unifying hypothesis. Autoimmune Dis 2012:189096
Perron H, Garson JA, Bedin F, Beseme F, Paranhos-Baccala G, Komurian-Pradel F, Mallet F, Tuke PW, Voisset C, Blond JL, Lalande B, Seigneurin JM, Mandrand B (1997) Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. The Collaborative Research Group on Multiple Sclerosis. Proc Natl Acad Sci U S A 94:7583–7588
Perron H, Germi R, Bernard C, Garcia-Montojo M, Deluen C, Farinelli L, Faucard R, Veas F, Stefas I, Fabriek BO, Van-Horssen J, Van-der-Valk P, Gerdil C, Mancuso R, Saresella M, Clerici M, Marcel S, Creange A, Cavaretta R, Caputo D, Arru G, Morand P, Lang AB, Sotgiu S, Ruprecht K, Rieckmann P, Villoslada P, Chofflon M, Boucraut J, Pelletier J, Hartung HP (2012) Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult Scler 18:1721–1736
Peterson KE, Chesebro B (2006) Influence of proinflammatory cytokines and chemokines on the neuropathogenesis of oncornavirus and immunosuppressive lentivirus infections. Curr Top Microbiol Immunol 303:67–95
Pevear DC, Borkowski J, Calenoff M, Oh CK, Ostrowski B, Lipton HL (1988) Insights into Theiler’s virus neurovirulence based on a genomic comparison of the neurovirulent GDVII and less virulent BeAn strains. Virology 165:1–12
Pfender N, Jelcic I, Linnebank M, Schwarz U, Martin R (2015) Reactivation of herpesvirus under fingolimod: a case of severe herpes simplex encephalitis. Neurology 84:2377–2378
Pfuhl C, Oechtering J, Rasche L, Giess RM, Behrens JR, Wakonig K, Freitag E, Pache FC, Otto C, Hofmann J, Eberspacher B, Bellmann-Strobl J, Paul F, Ruprecht K (2015) Association of serum Epstein-Barr nuclear antigen-1 antibodies and intrathecal immunoglobulin synthesis in early multiple sclerosis. J Neuroimmunol 285:156–160
Pietilainen-Nicklen J, Virtanen JO, Uotila L, Salonen O, Farkkila M, Koskiniemi M (2014) HHV-6-positivity in diseases with demyelination. J Clin Virol 61:216–219
Pinter C, Beltrami S, Caputo D, Ferrante P, Clivio A (2000) Presence of autoantibodies against complement regulatory proteins in relapsing-remitting multiple sclerosis. J Neurovirol 6(Suppl 2):S42–S46
Pohl D, Krone B, Rostasy K, Kahler E, Brunner E, Lehnert M, Wagner HJ, Gartner J, Hanefeld F (2006) High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology 67:2063–2065
Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, Rochford CD, Bruck W, Becher B (2006) Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest 116:456–464
Procaccini C, De Rosa V, Pucino V, Formisano L, Matarese G (2015) Animal models of multiple sclerosis. Eur J Pharmacol 759:182–191
Ramroodi N, Sanadgol N, Ganjali Z, Niazi AA, Sarabandi V, Moghtaderi A (2013) Monitoring of active human herpes virus 6 infection in Iranian patients with different subtypes of multiple sclerosis. J Pathog 2013:194932
Rasmussen HB, Lucotte G, Clausen J (2000) Endogenous retroviruses and multiple sclerosis. J Neurovirol 6(Suppl 2):S80–S84
Renoux C, Vukusic S, Mikaeloff Y, Edan G, Clanet M, Dubois B, Debouverie M, Brochet B, Lebrun-Frenay C, Pelletier J, Moreau T, Lubetzki C, Vermersch P, Roullet E, Magy L, Tardieu M, Suissa S, Confavreux C, Adult Neurology Departments KIDMUS Study Group (2007) Natural history of multiple sclerosis with childhood onset. N Engl J Med 356:2603–2613
Reynier P, Penisson-Besnier I, Moreau C, Savagner F, Vielle B, Emile J, Dubas F, Malthiery Y (1999) mtDNA haplogroup J: a contributing factor of optic neuritis. Eur J Hum Genet 7:404–406
Riccio P, Rossano R, Liuzzi GM (2011) May diet and dietary supplements improve the wellness of multiple sclerosis patients? A molecular approach. Autoimmune Dis 2010:249842
Rickinson AB, Kieff E (1996) Epstein-Barr virus. In: Fields BNKD, Howley PM (eds) Fields Virology, vol 2, 3rd edn. Lippincott-Raven, Philadelphia, pp 2397–2446
Rimenti G, Blasi F, Cosentini R, Moling O, Pristera R, Tarsia P, Vedovelli C, Mian P (2000) Temporal arteritis associated with Chlamydia pneumoniae DNA detected in an artery specimen. J Rheumatol 27:2718–2720
Rodgers MM, Mulcare JA, King DL, Mathews T, Gupta SC, Glaser RM (1999) Gait characteristics of individuals with multiple sclerosis before and after a 6-month aerobic training program. J Rehabil Res Dev 36:183–188
Rodriguez M, Oleszak E, Leibowitz J (1987) Theiler’s murine encephalomyelitis: a model of demyelination and persistence of virus. Crit Rev Immunol 7:325–365
Rohowsky-Kochan C, Dowling PC, Cook SD (1995) Canine distemper virus-specific antibodies in multiple sclerosis. Neurology 45:1554–1560
Rolland A, Jouvin-Marche E, Viret C, Faure M, Perron H, Marche PN (2006) The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J Immunol 176:7636–7644
Rosen FS (1977) Lymphoma, immunodeficiency and the Epstein-Barr virus. N Engl J Med 297:1120–1121
Rosenfeld ME, Blessing E, Lin TM, Moazed TC, Campbell LA, Kuo C (2000) Chlamydia, inflammation, and atherogenesis. J Infect Dis 181(Suppl 3):S492–S497
Ross RT, Nicolle LE, Cheang M (1995) Varicella zoster virus and multiple sclerosis in a Hutterite population. J Clin Epidemiol 48:1319–1324
Rotola A, Merlotti I, Caniatti L, Caselli E, Granieri E, Tola MR, Di Luca D, Cassai E (2004) Human herpesvirus 6 infects the central nervous system of multiple sclerosis patients in the early stages of the disease. Mult Scler 10:348–354
Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M (2006) Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol 5:343–354
Rudge P (1991) Does a retrovirally encoded superantigen cause multiple sclerosis? J Neurol Neurosurg Psychiatry 54:853–855
Sadovnick AD, Ebers GC (1993) Epidemiology of multiple sclerosis: a critical overview. Can J Neurol Sci 20:17–29
Sadovnick AD, Baird PA, Ward RH (1988) Multiple sclerosis: updated risks for relatives. Am J Med Genet 29:533–541
Sadovnick AD, Ebers GC, Dyment DA, Risch NJ (1996) Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet 347:1728–1730
Saiz A, Marcos MA, Graus F, Vidal J, Jimenez de Anta MT (2001) No evidence of CNS infection with Chlamydia pneumoniae in patients with multiple sclerosis. J Neurol 248:617–618
Salmi A, Viljanen M, Reunanen M (1981) Intrathecal synthesis of antibodies to diphtheria and tetanus toxoids in multiple sclerosis patients. J Neuroimmunol 1:333–341
Salmi A, Reunanen M, Ilonen J, Panelius M (1983) Intrathecal antibody synthesis to virus antigens in multiple sclerosis. Clin Exp Immunol 52:241–249
Sanders VJ, Felisan S, Waddell A, Tourtellotte WW (1996) Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol 2:249–258
Santon A, Cristobal E, Aparicio M, Royuela A, Villar LM, Alvarez-Cermeno JC (2011) High frequency of co-infection by Epstein-Barr virus types 1 and 2 in patients with multiple sclerosis. Mult Scler 17:1295–1300
Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P (1999) CD46 is a cellular receptor for human herpesvirus 6. Cell 99:817–827
Sawcer S, Hellenthal G (2011) The major histocompatibility complex and multiple sclerosis: a smoking gun? Brain 134:638–640
Schuller E, Ruzicka E, Allinquant B, Lebon P, Govaerts A, Degos JD (1988) Intrathecal antimeasles immunity in 140 patients with multiple sclerosis. Ann N Y Acad Sci 540:286–289
Seay AR, Wolinsky JS (1982) Ross River virus-induced demyelination: I. Pathogenesis and histopathology. Ann Neurol 12:380–389
Secchiero P, Carrigan DR, Asano Y, Benedetti L, Crowley RW, Komaroff AL, Gallo RC, Lusso P (1995) Detection of human herpesvirus 6 in plasma of children with primary infection and immunosuppressed patients by polymerase chain reaction. J Infect Dis 171:273–280
Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH, Ustekinumab MSI (2008) Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol 7:796–804
Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, Andreoni L, Trivedi P, Salvetti M, Faggioni A, Aloisi F (2007) Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med 204:2899–2912
Sibley WA, Bamford CR, Clark K (1985) Clinical viral infections and multiple sclerosis. Lancet 1:1313–1315
Sigurdsson B, Palsson PA (1958) Visna of sheep; a slow, demyelinating infection. Br J Exp Pathol 39:519–528
Simpson S Jr, Taylor B, Burrows J, Burrows S, Dwyer DE, Taylor J, Ponsonby AL, Blizzard L, Dwyer T, Pittas F, van der Mei I (2014) EBV & HHV6 reactivation is infrequent and not associated with MS clinical course. Acta Neurol Scand 130:328–337
Sindic CJ, Monteyne P, Laterre EC (1994) The intrathecal synthesis of virus-specific oligoclonal IgG in multiple sclerosis. J Neuroimmunol 54:75–80
Sips GJ, Chesik D, Glazenburg L, Wilschut J, De Keyser J, Wilczak N (2007) Involvement of morbilliviruses in the pathogenesis of demyelinating disease. Rev Med Virol 17:223–244
Smets F, Sokal EM (2014) Prevention and treatment for Epstein-Barr virus infection and related cancers. Recent Results Cancer Res 193:173–190
Soderberg C, Larsson S, Rozell BL, Sumitran-Karuppan S, Ljungman P, Moller E (1996) Cytomegalovirus-induced CD13-specific autoimmunity—a possible cause of chronic graft-vs-host disease. Transplantation 61:600–609
Sola P, Merelli E, Marasca R, Poggi M, Luppi M, Montorsi M, Torelli G (1993) Human herpesvirus 6 and multiple sclerosis: survey of anti-HHV-6 antibodies by immunofluorescence analysis and of viral sequences by polymerase chain reaction. J Neurol Neurosurg Psychiatry 56:917–919
Soldan SS, Jacobson S (2004) Infection and multiple sclerosis. In: Shoenfeld Y, Rose N (eds) Infections and autoimmunity. Elsevier, Philadelphia
Soldan SS, Berti R, Salem N, Secchiero P, Flamand L, Calabresi PA, Brennan MB, Maloni HW, McFarland HF, Lin HC, Patnaik M, Jacobson S (1997) Association of human herpes virus 6 (HHV-6) with multiple sclerosis: increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA. Nat Med 3:1394–1397
Soldan SS, Leist TP, Juhng KN, McFarland HF, Jacobson S (2000) Increased lymphoproliferative response to human herpesvirus type 6A variant in multiple sclerosis patients. Ann Neurol 47:306–313
Soldan SS, Fogdell-Hahn A, Brennan MB, Mittleman BB, Ballerini C, Massacesi L, Seya T, McFarland HF, Jacobson S (2001) Elevated serum and cerebrospinal fluid levels of soluble human herpesvirus type 6 cellular receptor, membrane cofactor protein, in patients with multiple sclerosis. Ann Neurol 50:486–493
Sonoda S (1992) Immune response to HTLV-I infection. Uirusu 42:29–39
Sonoda S, Fujiyoshi T, Yashiki S (1996) Immunogenetics of HTLV-I/II and associated diseases. J Acquir Immune Defic Syndr Hum Retrovirol 13(Suppl 1):S119–S123
Souraud JB, Faivre A, Waku-Kouomou D, Gaillard T, Aouad N, Meaudre E, Wild FT, Fouet B, Soulard R (2009) Adult fulminant subacute sclerosing panencephalitis: pathological and molecular studies—a case report. Clin Neuropathol 28:213–218
Sriram S, Mitchell W, Stratton C (1998) Multiple sclerosis associated with Chlamydia pneumoniae infection of the CNS. Neurology 50:571–572
Sriram S, Stratton CW, Yao S, Tharp A, Ding L, Bannan JD, Mitchell WM (1999) Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann Neurol 46:6–14
Stohlman SA, Hinton DR (2001) Viral induced demyelination. Brain Pathol 11:92–106
Stone LA, Frank JA, Albert PS, Bash C, Smith ME, Maloni H, McFarland HF (1995) The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann Neurol 37:611–619
Sumaya CV, Myers LW, Ellison GW (1980) Epstein-Barr virus antibodies in multiple sclerosis. Arch Neurol 37:94–96
Sundqvist E, Bergstrom T, Daialhosein H, Nystrom M, Sundstrom P, Hillert J, Alfredsson L, Kockum I, Olsson T (2014) Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult Scler 20:165–173
Suresh Kumar R, Syed S, Anand Kumar A, Subha Kumari KN, Sajitha K (2013) Serum vitamin d levels in Indian patients with multiple sclerosis. Indian J Clin Biochem 28:255–258
Takahashi K, Sonoda S, Higashi K, Kondo T, Takahashi H, Takahashi M, Yamanishi K (1989) Predominant CD4 T-lymphocyte tropism of human herpesvirus 6-related virus. J Virol 63:3161–3163
Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Killian JM, Zhang JZ (2002) Detection of viral DNA and immune responses to the human herpesvirus 6 101-kilodalton virion protein in patients with multiple sclerosis and in controls. J Virol 76:6147–6154
Tejada-Simon MV, Zang YC, Hong J, Rivera VM, Zhang JZ (2003) Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol 53:189–197
ter Meulen V, Koprowski H, Iwasaki Y, Kackell YM, Muller D (1972) Fusion of cultured multiple-sclerosis brain cells with indicator cells: presence of nucleocapsids and virions and isolation of parainfluenza-type virus. Lancet 2:1–5
Thompkins SM, Fuller KG, Miller SD (2002) Theiler’s virus-mediated autoimmunity: local presentation of CNS antigens and epitope spreading. Ann N Y AcadSci 958:26–38
Thompson RJ, Mason CR, Douglas AJ, Hinks LJ, Dwarswaard A, Price SE (1996) Analysis of polymorphisms of the 2′,3′-cyclic nucleotide-3′-phosphodiesterase gene in patients with multiple sclerosis. Mult Scler 2:215–221
Tornatore C, Berger JR, Houff SA, Curfman B, Meyers K, Winfield D, Major EO (1992) Detection of JC virus DNA in peripheral lymphocytes from patients with and without progressive multifocal leukoencephalopathy. Ann Neurol 31:454–462
Tsunoda I, Fujinami RS (2010) Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol 5:355–369
Van Beiren J (1427) Memoriale Johannis Duces de Bavaria. Rijksarchief, Den Haag
van Nierop GP, Mautner J, Mitterreiter JG, Hintzen RQ, Verjans GM (2016) Intrathecal CD8 T-cells of multiple sclerosis patients recognize lytic Epstein-Barr virus proteins. Mult Scler 22(3):279–291. doi:10.1177/1352458515588581
van Sechel AC, Bajramovic JJ, van Stipdonk MJ, Persoon-Deen C, Geutskens SB, van Noort JM (1999) EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol 162:129–135
Vandevelde M, Zurbriggen A (2005) Demyelination in canine distemper virus infection: a review. Acta Neuropathol 109:56–68
Venkatesan A, Johnson RT (2014) Infections and multiple sclerosis. Handb Clin Neurol 122:151–171
Virtanen JO, Pietilainen-Nicklen J, Uotila L, Farkkila M, Vaheri A, Koskiniemi M (2011) Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J Neuroimmunol 237:93–97
Virtanen JO, Wohler J, Fenton K, Reich DS, Jacobson S (2014) Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings. Mult Scler 20:27–34
Visscher BR, Detels R, Coulson AH, Malmgren RM, Dudley JP (1977) Latitude, migration, and the prevalence of multiple sclerosis. Am J Epidemiol 106:470–475
Wang FI, Stohlman SA, Fleming JO (1990) Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J Neuroimmunol 30:31–41
Warner HB, Carp RI (1988) Multiple sclerosis etiology—an Epstein-Barr virus hypothesis. Med Hypotheses 25:93–97
Waubant E, Mowry EM, Krupp L, Chitnis T, Yeh EA, Kuntz N, Ness J, Chabas D, Strober J, McDonald J, Belman A, Milazzo M, Gorman M, Weinstock-Guttman B, Rodriguez M, Oksenberg JR, James JA, US Pediatric MS Network (2011) Common viruses associated with lower pediatric multiple sclerosis risk. Neurology 76:1989–1995
Weinshenker BG (1996) Epidemiology of multiple sclerosis. Neurol Clin 14:291–308
Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC, Canadian Collaborative Study Group (2003) Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci U S A 100:12877–12882
Wuest SC, Mexhitaj I, Chai NR, Romm E, Scheffel J, Xu B, Lane K, Wu T, Bielekova B (2014) A complex role of herpes viruses in the disease process of multiple sclerosis. PLoS One 9, e105434
Yea C, Tellier R, Chong P, Westmacott G, Marrie RA, Bar-Or A, Banwell B, Canadian Pediatric Demyelinating Disease Network (2013) Epstein-Barr virus in oral shedding of children with multiple sclerosis. Neurology 81:1392–1399
Yoshikawa T (2004) Human herpesvirus 6 infection in hematopoietic stem cell transplant patients. Br J Haematol 124:421–432
Yousry TA, Major EO, Ryschkewitsch C, Fahle G, Fischer S, Hou J, Curfman B, Miszkiel K, Mueller-Lenke N, Sanchez E, Barkhof F, Radue EW, Jager HR, Clifford DB (2006) Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med 354:924–933
Zheng MM, Zhang XH (2014) Cross-reactivity between human cytomegalovirus peptide 981–1003 and myelin oligodendroglia glycoprotein peptide 35–55 in experimental autoimmune encephalomyelitis in Lewis rats. Biochem Biophys Res Commun 443:1118–1123
Zinchenko AP (1965) On the role of the rabies virus in the etiology of multiple sclerosis and encephalomyelitis. Zh Nevropatol Psikhiatr Im S S Korsakova 65:1634–1640
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Soldan, S.S., Jacobson, S. (2016). Virus-Induced Demyelination: The Case for Virus(es) in Multiple Sclerosis. In: Reiss, C. (eds) Neurotropic Viral Infections. Springer, Cham. https://doi.org/10.1007/978-3-319-33189-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-33189-8_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-33188-1
Online ISBN: 978-3-319-33189-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)