
Abstract
Recent advances in neuroscience have enabled increasingly detailed insight into the activity and structure of brain circuitry. In previous work, we have developed and applied methods for precisely controlling the activity of specific cells and projections within neural systems during behavior (optogenetics). Here I review distinct complementary technological approaches for observing natural activity patterns in these cells and projections during behavior (fiber photometry) and for obtaining anatomical insights into the wiring and molecular phenotype of these circuit elements within the intact mammalian brain (CLARITY-optimized lightsheet microscopy). Together these approaches may help further advance understanding of the circuit dynamics and wiring patterns that underlie adaptive and maladaptive behavior.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Neuronal Afferents
- Molecular Label
- Large Tissue Volume
- Light Sheet Microscopy
- Light Sheet Fluorescence Microscopy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
As we and others have noted (Tomer et al. 2014), a goal of modern neuroscience is to map neural circuits with wiring-level resolution, with brainwide perspective, and with knowledge of the natural and causal activity patterns occurring in these circuits in the context of behavior. Principles fundamental to the understanding of neural systems could result from such an integrative approach, but while progress has been made, many challenges and opportunities remain. Here I review our recent efforts to develop a recording technique sensitive enough to track the real-time dynamics of genetically and topologically specified subsets of neuronal projections in freely moving mice for direct in vivo measurement of a previously inaccessible variable: the coordinated activity of neuronal afferents projecting to a particular downstream target in the brain of a behaving animal (Gunaydin et al. 2014). Complementing this technology for the observation of natural behaviorally evoked activity patterns, I review rapid light-sheet microscopy methods for the efficient collection of high-resolution anatomical information within mammalian brains that have been made transparent to light (and permeable to macromolecular labels) with the CLARITY technology (Tomer et al. 2014). These two technologies can be employed alongside optogenetic tools that, in turn, can define the causal roles of these same projections in modulating behavior.
CLARITY
As we have described (Tomer et al. 2014), circuit wiring questions have attracted attention from generations of scientists, beginning with Cajal’s detailed representations of neurons visualized at high resolution with the Golgi staining technique while still embedded within semi-intact brain tissue. Over the last few decades, electron microscopy (EM) has emerged as a foundational method for deciphering details of neuronal circuit structure (Bock et al. 2011; Briggman et al. 2011). The key advantage of EM in this regard (relative to light microscopy) is identification of the presynaptic active zones containing neurotransmitter vesicles apposed to postsynaptic structures. In addition, EM facilitates visualization of some of the very finest branches of axons. However, EM tissue mapping requires relatively slow steps involving ultrathin sectioning/ablation and reconstruction; most importantly, the sample contrast preparation is largely incompatible with rich molecular phenotyping that could provide critical information on cell and synapse type. Ideally, datasets resulting from intact-brain mapping should be linkable to molecular information on the types of cells and synapses that are imaged structurally and even to dynamical information on natural activity pattern history (in these same circuits) known to be causally relevant to animal behavior. Suitable light-based imaging approaches, combined with specific genetic or histochemical molecular labeling methods, have emerged as important tools to visualize the structural, molecular and functional architecture of biological tissues, with a particularly vital role to play in emerging brainwide, high-resolution neuroanatomy.
Confocal methods revolutionized light microscopy by enabling optical sectioning in thick (tens of micrometers) fluorescently labeled samples, thereby allowing 3D reconstruction without the need for ultrathin physical sectioning (Conchello and Lichtman 2005). Two-photon microscopy further increased the accessible imaging depth (to hundreds of micrometers) even in living tissue samples (Helmchen and Denk 2005), and adaptive-optics approaches have improved imaging depth further (Tang et al. 2012). However, light microscopy remains limited for imaging throughout intact vertebrate nervous systems (for example, mouse brains span many millimeters even in the shortest spatial dimension and are opaque on this scale, due chiefly to light scattering). A common work-around to this limitation has been to slice brains into thin sections, in manual or automated fashion, followed by confocal or two-photon imaging (Micheva et al. 2010; Ragan et al. 2012); however, detailed labeling and reconstruction from thin sections have been (so far) limited to small volumes of tissue. An ideal integrative approach would be to label and image entirely intact vertebrate brains at high resolution.
As a step in this direction, new methods have emerged to increase tissue transparency (Dodt et al. 2007; Hama et al. 2011; Ke et al. 2013) by chemically reducing the scattering of light travelling through the tissue sample. While intriguing and effective, these approaches are not generally suitable for detailed molecular phenotyping, since most tissues (such as the intact mature brain) remain largely impenetrable to macromolecular antibody or oligonucleotide labels (Kim et al. 2013). In cases where pieces of soft tissue such as mammary glands can be stained using hydrophobic clearing solutions that reduce lipid barriers to antibody labeling (Ertürk et al. 2012), fluorophores become highly unstable or quenched in the clearing process (a step that nevertheless must follow the antibody-staining phase, as transparency is otherwise lost; Ertürk et al. 2012). These limitations motivated the recent development of CLARITY (Chung et al. 2013; Kim et al. 2013), which involves removal of lipids in a stable hydrophilic chemical environment to achieve transparency of intact tissue, preservation of ultrastructure and fluorescence, and accessibility of native biomolecular content to antibody and nucleic acid probes. Subsequent screens for diverse hydrophilic lipid solubilization compounds have been productive and can be integrated with CLARITY (Susaki et al. 2014). The CLARITY technical platform enables multiple rounds of molecular, structural and activity-history interrogation throughout intact adult mammalian brains, which is relevant not only for neuroscience but also for research into any intact biological system.
Clarifying Large Tissue Volumes
CLARITY (Chung et al. 2013) builds upon chemical principles to grow hydrogel polymers from inside the tissue to provide a support framework for structural and biomolecular content (Fig. 1). This is achieved first by infusing a cold (4 °C) cocktail of hydrogel monomers (for example, acrylamide with bisacrylamide, but other types of monomers may also be used; Chung et al. 2013), formaldehyde, and thermally triggered initiators into the tissue, followed by polymerization of the hydrogel at 37 °C. Formaldehyde serves the dual purposes of cross-linking amine-containing tissue components to each other and covalently binding the hydrogel monomers to these native biomolecules, which include proteins, nucleic acids and other small molecules but not the vast majority of cellular membrane phospholipids. After the hydrogel polymerization is triggered, lipids (responsible for preventing access of both photons and molecular labels to deep structures) can then be readily removed without destroying or losing native tissue components using strong ionic detergent-based clearing solution (borate-buffered 4 % sodium-dodecyl-sulfate) at 37 °C, either passively with gentle recirculation or with active electrophoretic forcing (the latter greatly accelerates clearing but introduces some experimental complexity and risk). The resulting lipid-extracted and structurally stable tissue-hydrogel hybrid is immersed in a refractive index homogenization solution to render the intact brain transparent to light (Chung et al. 2013).

CLARITY pipeline overview. The tissue sample, e.g. an intact mouse brain, is perfused with cold hydrogel monomer solution that contains a cocktail of acrylamide, bisacrylamide, formaldehyde and thermal initiator. Formaldehyde mediates crosslinking of biomolecules to acrylamide monomers via amine groups; the presumptive chemistry of this process is shown. Hydrogel polymerization is initiated by incubating the perfused tissue at 37 °C, resulting in a meshwork of fibers that preserves biomolecules and the structural integrity of the tissue. Lipid membranes are removed by passive thermal clearing in SBC solution at 37 °C or by electrophoretic tissue clearing (ETC). The resulting intact tissue-hydrogel hybrid can undergo multiple rounds of molecular and structural interrogation using immunohistochemistry and light microscopy. A dedicated computational infrastructure is needed to analyze and store the data. Figure and text adapted from Tomer et al. (2014)
An additional feature of the stable hydrogel-tissue hybrid is that it can be subjected to multiple rounds of molecular interrogation (Chung et al. 2013). Typically, immunohistochemistry methods only allow investigation of two to three biomarkers at once in a tissue sample, but more simultaneous labels are required to define cells in terms of precise molecular/genetic identity, wiring, and activity history. This limitation is traditionally approached by combining information from multiple samples into a standard reference atlas. However, this strategy fails to fully phenotype individual cells, cannot capture the joint statistics among the different kinds of labels within a single preparation, and suffers from 3D alignment artefacts and variability among different individual tissue samples. By allowing multiple rounds of histochemical labeling and elution in the same tissue, CLARITY provides unusually rich access to molecular and structural information (Chung et al. 2013).
This extensive lipid removal intrinsic to CLARITY appears to be essential not only for transparency but also for achieving efficient antibody penetration throughout intact brains; this stringent de-lipidation would normally be a destructive process causing extensive loss of biological molecules (Chung et al. 2013), but it is enabled by the hydrophilic hydrogel-tissue hybrid in a way that also preserves fine processes and ultrastructure even over multiple rounds of staining and elution (Chung et al. 2013). Many variations are possible and will continue to be explored; the “CUBIC” screen (Susaki et al. 2014) recently provided an intriguing diversity of additional hydrophilic lipid solubilization reagents, including aminoalcohols that can be used in CLARITY protocols (Susaki et al. 2014), though it was noted that the aminoalcohol incubation leaves behind significant lipid content in the tissue (Susaki et al. 2014), such that lipid-rich structures including white matter remain partially opaque. Also, the approach may not allow antibody penetration for molecular labeling and resolution of fine processes deeper than 0.5–0.8 mm into mature brain tissue (Susaki et al. 2014). The ETC process (though not essential even for whole-mouse-brain CLARITY , as in a typical 3-week passive-CLARITY approach that achieves full transparency and antibody access) still accelerates de-lipidation and might, therefore, help enhance an aminoalcohol approach to CLARITY, though this remains to be seen and will depend on the size and charge of the resulting lipid-containing particles.
CLARITY-Optimized Light Sheet Microscopy
Not only the speed of de-lipidation but also the speed of imaging is crucial for CLARITY. While confocal and two-photon microscopes have been the workhorse systems in volumetric imaging for the reasons described above, over the past two decades light sheet fluorescence microscopy has emerged as a powerful approach for high-speed volumetric imaging. Confocal and two-photon microscopies are point-scanning techniques, detecting optical signals point-by-point to construct an image. Confocal achieves optical sectioning by the use of a pinhole at the detection focal plane to reject out-of-focus light, whereas two-photon utilizes the fact that only simultaneous absorption of two photons results in fluorescence emission, an event much more likely to occur at the point of highest light intensity in the sample (the focal plane). Light sheet microscopy, in contrast, builds upon a 100-year-old idea to illuminate the sample from the side with a thin sheet of light and detect the emitted fluorescence signal with an in-focus orthogonally arranged objective (Siedentopf and Zsigmondy 1903; Huisken and Stainier 2009). The optical sectioning is achieved by the confinement of illumination to a selective plane, which allows use of fast CCD or sCMOS cameras to capture the whole image simultaneously, and results in an increase of 2–3 orders of magnitude in imaging speed compared to confocal and two-photon microscopy. Moreover, light sheet microscopy minimizes photo-bleaching by confining illumination to the plane of interest. Taken together, these properties of light sheet microscopy may be well suited for the imaging of large clarified samples, consistent with its previously demonstrated utility for minimizing unnecessary illumination.
For high-speed collection of imaging data from large clarified volumes, COLM is 100–1000 times faster than conventional scanning methods, leading to vastly decreased photo-bleaching (Tomer et al. 2014). The properties of COLM are not only useful for mouse brains but also will be particularly relevant for maintaining this high cellular and subcellular resolution at practical speeds in brains from larger-brained organisms. The fast COLM approach described here for clarified intact mouse brains maintains high resolution even 5–6 mm deep in tissue using the 0.95 NA objective; ultimate resolution in any light microscope, of course, remains limited by the laws of diffraction (λ/2NA = ~180 nm), but the emergence of super-resolution (or “diffraction unlimited”) imaging methods, such as STED/RESOLFT and PALM/STORM, could in the future allow a further four- to fivefold improvement in achievable resolution.
We assess the compatibility of clarified samples with light sheet, observing greater than two orders of magnitude faster imaging speed (Tomer et al. 2014) with minimal photo-bleaching results (Fig. 2). For example, it was possible to image an entire mouse brain in about 4 h using a 10× magnification objective and in about 1.5 days using a 25× objective, as opposed to many days and months, respectively, with a confocal microscope. COLM is especially well suited for interrogation of large tissue samples labeled with transgenic or histochemical approaches. The increased speed of acquisition and higher quality of data generated via CLARITY using new microscopy methods, combined with high-speed CLARITY processing itself enabled by efficient tissue transformation protocols, define a versatile and efficient platform for structural and molecular interrogation of large and fully assembled tissues (Tomer et al. 2014).

Fast high-resolution imaging of clarified brain using COLM. A 3.15 mm × 3.15 mm × 5.3 mm volume acquired from an intact clarified Thy1-eYFP mouse brain using COLM with ×25 magnification; the brain had been perfused with 0.5 % acrylamide monomer solution. The complete image dataset was acquired in ~1.5 h; for optimal contrast, the LUT of zoomed-in images was linearly adjusted between panels (a) and (b), magnified views from the panel (c) region defined by yellow squares (d–i), maximum-intensity projections of over a 50 μm-thick volume, as shown by the progression of cyan and yellow boxes and arrows. A camera exposure time of 20 ms was used; refractive index liquid 1.454 was used as the immersion medium. All scale bars: 100 μm. Figure and text adapted from Tomer et al. (2014)
As a final comment for future work, we note that very large datasets result from this new capability for high-speed imaging of large tissue volumes at high resolution, and extensive innovation will be needed in image analysis and data management (for example, if the intact 0.3 cm3 mouse brain is represented by 0.5 × 0.5 × 0.5 cubic-micron 16-bit voxels, at least 4.8 terabytes of raw data result). Fortunately, big-data and high-performance computing have led to advanced image-compression technologies such as JPEG 2000 3D, increased computational capacity with GPU parallel computation technology, and cloud infrastructures (such as Amazon S3) for data storage and sharing. We expect that the integration and application of these methods to CLARITY (Tomer et al. 2014) will allow increasingly complete access to, and understanding of, the molecular and structural organization of large intact tissues.
Tracking Activity in Deep Genetically Targeted Neurons of Behaving Animals
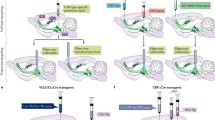
To observe not only the structure but also the real-time activity of specified neural cells and projections, we developed a method termed fiber photometry, with a simple design (only a single multimode optical fiber), suitable for recording from deep brain structures and sensitive enough to detect activity changes not only in cell bodies but also in axons during behavior, where signals are considerably smaller (Gunaydin et al. 2014). This fiber photometry (light measurement with a single-fiberoptic device sensitive enough to detect activity in axonal fibers) relies on a lock-in amplifier and a high-sensitivity photoreceiver along with custom software to record (through an implanted 400 μm optical fiber) the population activity of neural circuit elements expressing a genetically encoded Ca2+ indicator (Fig. 3). The single fiber allows chronic, stable, minimally disruptive access to deep brain regions and interfaces with a flexible patchcord on the skull surface (Gunaydin et al. 2014).

VTA-NAc projection activity encodes social interaction. Area plots, smoothed behavioral score: %total Ca2+ peaks representing specific social target-related and novel-object behaviors during VTA cell body (top) and VTA-NAc projection (bottom) fiber photometry (5 min; n = 10 and n = 11 mice, respectively). Arrows: target introduction. Note encoding of social interaction by VTA cell body and VTA-NAc projection activity. Figure and text adapted from Gunaydin et al. (2014)
For cell type-specific recording of Ca2+ transients—a proxy for certain neural activity—we injected a Cre-dependent adeno-associated virus (AAV) carrying the GCaMP5g gene into VTA of transgenic TH::Cre mice and implanted an optical fiber in VTA for simultaneous delivery of 473 nm excitation light and collection of GCaMP5g fluorescence emission (Gunaydin et al. 2014). Activity-dependent fluorescence emitted by cells in the volume was collected simultaneously; after propagating back through the same patchcord used to deliver excitation light, this fluorescence was spectrally separated using a dichroic, passed through a single band filter, and focused onto a photodetector. To first test if this system would be capable of detecting VTA activity in a temporally precise manner, we recorded Ca2+ signals in VTA neurons of TH-GCaMP mice given access to sucrose solution, an established natural reward. Sucrose consumption was assessed using a contact “lickometer,” which registered an event every time the mouse completed a circuit from a metal spout to a metal operant chamber floor, time-locked to the Ca2+ recording. This setup enabled readout of the VTA response with temporal precision on the order of milliseconds. VTA signals were tightly correlated in time with onset of licking bouts and habituated over recording epochs (Gunaydin et al. 2014).
Neural Activity that Encodes and Predicts Social Interaction
Next we applied fiber photometry during same-sex social interaction. We recorded from the VTA of female mice during home-cage social interaction, in which a novel social target mouse was introduced into the test mouse cage for a 5-min epoch, and video time-locked to the VTA GCaMP signal was collected (Gunaydin et al. 2014). Upon introduction of the social target, we observed a marked increase in activity of the targeted VTA neurons during interaction with this novel mouse. Such activity was absent in the eYFP control, indicating that observed transients were Ca2+ signals and not motion artifacts (Gunaydin et al. 2014). In separate trials, we exposed test mice to a novel object placed in the home cage (counterbalanced with novel-mouse exposure). Nevertheless, VTA activity in response to the novel object resembled peak VTA activity during social interaction, with similar amplitude (mean peak dF/F during interaction: 16.4 % ± 2.1 % SEM for social, 13.7 % ± 1.4 % SEM for novel object, n = 10; Wilcoxon signed-rank test, p = 0.5).
While these data were consistent with hypothesized importance of the VTA-NAc projection in social behavior, direct observation of endogeneous activity in the projection during social behavior remained lacking. This would require measuring a previously inaccessible but fundamental neural circuit quantity: native activity in a specific projection during behavior. Fiber photometry was designed for this technical challenge, and we next tested real-time tracking of Ca2+ transients in genetically specified VTA inputs to NAc. Using TH-GCaMP mice, we implanted an optical fiber in medial NAc to detect activity specifically in axon fibers corresponding to the projection in question during home cage social interaction and novel object investigation (Gunaydin et al. 2014).
We observed robust GCaMP signals across many social interaction bouts, demonstrating that fiber photometry could be used to selectively record from neuronal projections during behavior (Gunaydin et al. 2014). We observed smaller projection activation to novel object (n = 11, Wilcoxon signed rank test, mean peak dF/F: 6.9 % ± 1.4 % for social, 3.5 % ± 0.7 % for novel object, p = 0.016). Stronger encoding of social than of object interactions by the VTA-NAc projection (not seen at the cell bodies) supports the hypothesis that there are distinctly wired relevant subpopulations of VTA neurons. We sought to capitalize on this ability to track projection activity during behavior by probing in greater detail the encoding of specific behaviors by the VTA projection to NAc (in comparison with activity in VTA cell bodies) using multifactorial high-resolution quantitative behavioral assessment. We first employed an automated peak-finding algorithm to detect all Ca2+ peaks throughout the 5-min testing period, blind to mouse behaviors, for social and novel object conditions during both VTA cell body and VTA-NAc projection recordings. Next we automatically segmented video clips centered (±1 s) around the time of each Ca2+ peak and scored video segments for interaction, approach, withdrawal, ambulation, grooming, rest, burrowing, rearing, and head extension (Gunaydin et al. 2014).
Area plots of all VTA-DA Ca2+ peak times subdivided by behavioral category (Fig. 3) allowed direct comparison of total peak activity over time attributable to each category, including as a percent of total overall Ca2+ peak activity (Gunaydin et al. 2014). In the social case, a larger proportion of total Ca2+ peak activity occurred during interaction for VTA-to-NAc projections than for cell bodies, supporting the conclusion that this projection more selectively encodes social interaction than does the cell body signal. For novel object behavior, both cell bodies and projections poorly encoded approach or interaction; interestingly, while the VTA cell bodies seemed to strongly encode withdrawal from the object, the VTA-NAc projection only weakly encoded this specific behavior (Fig. 3). Across the entire 5-min testing period, VTA-NAc projections showed a decreased proportion of Ca2+ peak activity (compared with VTA cell-body data) occurring during target-relevant behavior (accounted for by withdrawal) in the case of novel object but not social behavior (Fig. 3). These data together support the conclusion that VTA-NAc projection activity represents a signal with specific importance to social behavior relative to object interactions (Gunaydin et al. 2014). Not only is this projection-specific activity parameter especially predictive in behavior (Fig. 3) but projection-specific activity is also in general particularly important for causal elicitation of complex behaviors (Deisseroth 2014). Therefore, this new ability to directly measure the activity of projections between brain regions provides a potentially relevant source of data on the behaviorally significant dynamics of information flow (Deisseroth 2014).
Outlook
We have developed and applied two new methods, fiber photometry and COLM, for direct measurement of the activity and structure of specified neuronal afferents projecting to a particular downstream target. Together, these results demonstrate the integrative value of complementary optical techniques in causally mapping specific projections and postsynaptic targets within neural circuitry. Projection-specific optogenetic manipulations complement the specificity of fiber photometry and COLM by enabling control of the corresponding projection dynamics. This approach may suggest circuit-based targets for further research into physiological or neuropsychiatric disease-related symptoms and may be generally applicable for investigation of specific circuit elements in mammalian behavior.
References
Bock DD, Lee WC, Kerlin AM, Andermann ML, Hood G, Wetzel AW, Yurgenson S, Soucy ER, Kim HS, Reid RC (2011) Network anatomy and in vivo physiology of visual cortical neurons. Nature 471:177–182. doi:10.1038/nature09802
Briggman KL, Helmstaedter M, Denk W (2011) Wiring specificity in the direction-selectivity circuit of the retina. Nature 471:183–188. doi:10.1038/nature09818
Chung K, Wallace J, Kim SY, Kalyanasundaram S, Andalman AS, Davidson TJ, Mirzabekov JJ, Zalocusky KA, Mattis J, Denisin AK, Pak S, Bernstein H, Ramakrishnan C, Grosenick L, Gradinaru V, Deisseroth K (2013) Structural and molecular interrogation of intact biological systems. Nature 497:332–337. doi:10.1038/nature12107
Conchello JA, Lichtman JW (2005) Optical sectioning microscopy. Nat Methods 2:920–931. doi:10.1038/nmeth815
Deisseroth K (2014) Circuit dynamics of adaptive and maladaptive behaviour. Nature 505:309–317
Dodt HU, Leischner U, Schierloh A, Jährling N, Mauch CP, Deininger K, Deussing JM, Eder M, Zieglgänsberger W, Becker K (2007) Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat Methods 4:331–336. doi:10.1038/nmeth1036
Ertürk A, Becker K, Jährling N, Mauch CP, Hojer CD, Egen JG, Hellal F, Bradke F, Sheng M, Dodt HU (2012) Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc 7:1983–1995. doi:10.1038/nprot.2012.119
Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RA, Tye KM, Anikeeva P, Malenka RC, Deisseroth K (2014) Natural neural projection dynamics underlying social behavior modulation. Cell 157:1535–1551
Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, Fukami K, Sakaue-Sawano A, Miyawaki A (2011) Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci 14:1481–1488. doi:10.1038/nn.2928
Helmchen F, Denk W (2005) Deep tissue two-photon microscopy. Nat Methods 2:932–940. doi:10.1038/nmeth818
Huisken J, Stainier DY (2009) Selective plane illumination microscopy techniques in developmental biology. Development 136:1963–1975. doi:10.1242/dev.022426
Ke MT, Fujimoto S, Imai T (2013) SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci 16:1154–1161. doi:10.1038/nn.3447
Kim SY, Chung K, Deisseroth K (2013) Light microscopy mapping of connections in the intact brain. Trends Cogn Sci 17:596–599. doi:10.1016/j.tics.2013.10.005
Micheva KD, Busse B, Weiler NC, O’Rourke N, Smith SJ (2010) Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron 68:639–653. doi:10.1016/j.neuron.2010.09.024
Ragan T et al (2012) Serial two-photon tomography for automated ex vivo mouse brain imaging. Nat Methods 9:255–258. doi:10.1038/nmeth.1854
Siedentopf H, Zsigmondy R (1903) Uber Sichtbarmachung und Grössenbestimmung ultramikroskopischer Teilchen, mit besonderer Anwendung auf Goldrubingläser. Ann Phys 10:1–39
Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, Yokoyama C, Onoe H, Eguchi M, Yamaguchi S, Abe T, Kiyonari H, Shimizu Y, Miyawaki A, Yokota H, Ueda HR (2014) Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157:726–739
Tang J, Germain RN, Cui M (2012) Superpenetration optical microscopy by iterative multiphoton adaptive compensation technique. Proc Natl Acad Sci USA 109:8434–8439. doi:10.1073/pnas.1119590109
Tomer R, Ye L, Hsueh B, Deisseroth K (2014) Advanced CLARITY methods for rapid and high-resolution imaging of large intact tissues. Nat Protoc 9:1682–1697
Acknowledgments
We thank the entire Deisseroth lab for helpful discussions. K.D. is supported by the DARPA Neuro-FAST program, NIMH, NSF, NIDA, the Simons Foundation, and the Wiegers Family Fund. All tools and methods described are distributed and supported freely (optogenetics.org, clarityresourcecenter.org, http://wiki.claritytechniques.org) and discussed in an open forum (forum.claritytechniques.org). Text and figures were taken from our in-press papers (Gunaydin et al. 2014; Tomer et al. 2014), and I am grateful to my collaborators and co-authors on these papers: Lisa Gunaydin, Logan Grosenick, Joel Finkelstein, Isaac Kauvar, Lief Fenno, Avishek Adhikari, Stephan Lammel, Julie Mirzabekov, Raag Airan, Kay Tye, Polina Anikeeva, Rob Malenka, Raju Tomer, Li Ye, and Brian Hsueh.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Deisseroth, K. (2016). Form Meets Function in the Brain: Observing the Activity and Structure of Specific Neural Connections. In: Kennedy, H., Van Essen, D., Christen, Y. (eds) Micro-, Meso- and Macro-Connectomics of the Brain. Research and Perspectives in Neurosciences. Springer, Cham. https://doi.org/10.1007/978-3-319-27777-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-27777-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27776-9
Online ISBN: 978-3-319-27777-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)