
Abstract
Here we give an overview of a worldwide effort, called the ENIGMA Consortium (http://enigma.ini.usc.edu), which unites scientists worldwide to determine how variants in our genetic code influence the brain, and how 12 major diseases affect the brain worldwide. At the time of writing, ENIGMA involves over 500 scientists from 185 institutions worldwide, working together on around 30 projects to discover factors that may help or harm the brain. By pooling genome-wide genomic data and brain imaging from over 33,000 people, ENIGMA has been able to identify single-nucleotide differences in the genome that are associated with differences in human brain structure and function. Given the broad interest in brain connectivity and the factors that affect it, we outline some tactics adopted by ENIGMA to discover specific genes that affect the brain; then we describe how ENIGMA is extending these methods to discover genetic influences on brain connectivity.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Background to ENIGMA
ENIGMA (Enhancing Neuroimaging Genetics through Meta-Analysis) is a worldwide network of researchers who work together to investigate various questions about the brain. The consortium pools brain imaging and genetic data from over 200 institutions around the world. The main goals of ENIGMA are to discover factors that help and harm the brain; the sheer size of the dataset is unprecedented, making it possible to see which effects on the brain are robust and consistent by pooling data worldwide. The idea for ENIGMA originated in late 2009 and the consortium has since published some of the largest brain imaging studies in the world—both in terms of the total number of individuals genotyped and scanned (now over 33,000) and in terms of the number of scientists collaborating [several hundred co-authors, in Stein et al. (2012), Thompson et al. (2014), and Hibar et al. (2015)]. Also, by pooling brain imaging and genomic data from tens of thousands of people, we were able to overcome several technical and sociological barriers; here we outline some of the strategies employed and the main findings and lessons learned. As befits a chapter in a book on brain connectivity, we also summarize the tactics that ENIGMA is beginning to employ to discover genetic influences on brain connectivity.
Genetic Influences on the Brain
By 2009, nearly 100 studies had been published showing that numerous measures of brain structure are heritable (Blokland et al. 2012). In other words, individual differences in our genetic code do affect specific features of the brain, such as the overall volume of the brain, the size of the hippocampus, and even measures of functional activity based on EEG or functional MRI. To establish this, researchers began by studying family-based cohorts or twins who were scanned with anatomical or functional MRI; when people with greater genetic similarity were compared, their brains were found to be more similar, on average, than were unrelated people of the same age and sex.
To formalize these ideas, the classical twin design has often been used to estimate the heritability of a behavioral trait by studying both identical and fraternal twins (siblings or other family members are often evaluated as well; Boomsma et al. 2002). Based on structural equation models, or even based on simpler approaches involving correlations, twin studies are able to estimate what fraction of the observed variability in a brain measure is due to genetics, that is, due to the genetic differences among individuals. Many measures of brain structure, such as the total amount of gray or white matter in the brain or the overall volume of the ventricles, were found to be heritable; that is to say, genetic factors are involved in determining their eventual values. Note that this type of genetic analysis does not require the direct examination of the DNA sequence, only the study of resemblances among family members with different degrees of familial relatedness (e.g., identical twins, siblings, etc.).
Soon afterwards, 3D “maps” of heritability began to be produced for a variety of brain measures, such as regional gray matter volumes in the cortex (Thompson et al. 2001), cortical thickness (Joshi et al. 2012), surface area (Chen et al. 2012), and fiber microstructure in diffusion-weighted MRI scans (Chiang et al. 2009). The proportion of variance due to genetic factors is not expected to be completely uniform across the brain. In general, genetic variation accounts for around half of the observed variance for many brain measures, in some cases more, making neuroimaging measures an attractive target for in-depth genetic analysis (Glahn et al. 2007).
The high heritability of brain structure is in line with many behavioral genetic studies showing substantial genetic effects on behavior and even risk for neurological and psychiatric illnesses, such as Alzheimer’s disease and schizophrenia . Genetic studies have shown that numerous traits relating to personality, cognition, and even risk for neurological or psychiatric disease are influenced by genetics to some degree. The influence of genetic versus environmental factors on cognition and intellectual performance was one of the most hotly debated scientific topics of the twentieth century (Jensen 1969; Lewontin et al. 1984). Of course, even if we concede that genes play some role in behavior, several caveats to heritability calculations apply: genetic variants do not influence the brain independently of other factors, and their effects may depend on a person’s age, sex, level of nutrition, education, or many other contextual factors in the population (Visscher et al. 2008). Although they are not necessarily heritable, epigenetic factors, such as methylation and acetylation, act on the genome to switch off or promote the action of certain parts of our genetic code. Also, an individual’s environment may be correlated to some degree with their genotype; for example, people with a natural aptitude for certain kinds of activity may seek out environments that promote those activities. This makes the effects of genes and environment difficult or impossible to disentangle. Gene x Environment interactions are also found, where a gene’s effect on the brain or behavior is accentuated or suppressed under certain conditions. In fact, much work in the fields of pharmacogenomics and personalized medicine depends on the notion that people with certain genetic risk factors may be less or more responsive to medication or other kinds of therapy. As such, the quest to identify genetic variants that relate to brain measures is likely to accelerate our genetic understanding of brain disease and mental illness. With this in mind, ENIGMA has several projects that relate brain measures to genomic variation and to disease, a topic that we will return to later.
Finding the Genes Involved
Knowing that a brain measure is heritable—or influenced by genetic factors—is the first step on the long road towards identifying specific differences in the genome that influence it. By 2009, genetic “sequencing” had become relatively inexpensive, and it was possible to reliably identify a person’s individual DNA sequence at each of over one million genetic locations, based on a person’s blood or saliva sample. Although well over 99 % of the genetic code is identical across healthy individuals, people do differ substantially in specific areas of the genetic code: there are deletions, expansions, and even single-nucleotide or single “letter” spelling differences in the base pair sequence. Some of these genetic differences do not affect the protein product, if the gene is expressed at all. Other genetic differences render the protein product dysfunctional or modify its activity, and they may influence brain function and behavior and our risk for disease.
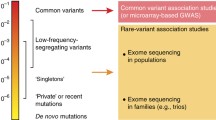
Genotyping companies began to offer genotyping services whereby over a million common genetic variants—or single nucleotide polymorphisms (SNPs)—could be assessed cheaply; in the United States, for example, some personalized genomics companies offered to send a person a million “letters,” or nucleotides, of their genetic code for $99 (in U.S. dollars). This ability to genotype common variants in the genome led to a surge in the popularity of genome-wide association studies (GWAS), efforts to identify markers or common variants in the human genome that are statistically associated with a certain trait, such as obesity, schizophrenia , depression, or Alzheimer’s disease . Many of these genomic screens were very successful. For instance, certain “risk genes,” such as APOE, CLU, and TREM2, have alternative sequences wherein one form is more commonly found in patients with Alzheimer’s disease (Harold et al. 2009; Jonsson et al. 2013). The quest to find these risk-associated genetic variants is motivated by finding new drug targets or, in the short term, evaluating a person’s risk for a specific disease, which can help in clinical trial design.
Again, several caveats apply. Common variants are not the only source of genetic variations that have an impact on the brain; in fact, rare variants—or even private variants found only within a single family or individual—have been found that associate with risk for autism or other disorders (Sanders et al. 2012; Purcell et al. 2014). When GWAS was first feasible on a large scale, studies of tens of thousands of individuals began to unearth common genetic differences associated with cholesterol levels in the blood and with bone density, obesity, or stroke, and a range of other common conditions. In each study, the genome was scanned for sequence variations associated with a single trait, such as a person’s height, body mass index, or a psychiatric diagnosis such as schizophrenia or bipolar illness, for example. Because of the high risk of false positives—searching millions of letters of the genetic code would likely detect many false associations—geneticists began to enforce a very high statistical threshold to implicate a genetic variant in a disorder, often requiring tens of thousands of subjects to find an association and replicate it.
GWAS of the Brain
Around 2009, GWAS began to be performed on brain measures [see supplementary information in Medland et al. (2014)], such as temporal lobe volume (Stein et al. 2010). Although some of the top “hits” in these studies seemed convincing from a mechanistic point of view, many geneticists argued that the power to detect common genetic variants that affect the brain was very limited, even in samples of approximately 1000 subjects. As brain imaging data are expensive and time-consuming to collect, only the largest national initiatives could even achieve sample sizes of 1000 subjects; the Alzheimer’s Disease Neuroimaging Initiative (ADNI; Jack et al. 2008), for example, was one of the largest studies ever attempted with neuroimaging. ADNI still took many years to recruit and scan a cohort of 800 people at 58 sites across North America. Power calculations suggest that cohorts of 10,000 or more subjects should be needed to zero in on genomic regions with reliable associations to brain measures, unless of course their effect sizes are extremely large. And so began a debate as to whether imaging would offer a more efficient way to detect influential genetic variants.
Counterarguments and Power
Two arguments were commonly advanced to suggest that large samples might not be required for successful genetic studies of brain images, but the evidence for each argument began to wane. The first was that some image-derived measures might be more highly reproducible than psychiatric diagnostic tests or cognitive scores; some measures from images (such as the density of connections between brain regions) might be closer to the biology of the gene action as well and therefore show a stronger effect. For example, a growth factor gene, such as BDNF, might influence the cell numbers or cell volumes in a specific structure of the brain, such as the hippocampus. If so, then the statistical association between common variants influencing the function of that gene and the size of the brain structure should be fairly easy to identify in a database of brain scans. As we shall see, this optimism had to be tempered; at least for the brain measures assessed so far, sample sizes needed for successful genetic association studies have been about the same as those needed to discover risk genes for clinical conditions such as Alzheimer’s disease or schizophrenia , though less than those for major depression, and have been on the order of tens of thousands. Even so, one should bear in mind that the large samples required to detect effects does not mean effects are trivial or unimportant. Rare variants with large effect, for example TREM2, appear to double a person’s risk for Alzheimer’s disease (Guerreiro et al. 2013; Jonsson et al. 2013) and cause brain tissue loss at twice the normal rate (Rajagopalan et al. 2013). Despite the fact that only 1 % of people carry this risk allele, the aggregate effect on society is no doubt substantial, perhaps similar to other mental disorders with similar prevalence but with devastating impact.
A second argument was that we should focus on candidate genes when looking for factors that affect the brain, rather than performing a completely open-ended, genome-wide search. Because certain growth factors in the brain—BDNF, and NGF, for example—have polymorphic variants within their genes, they could be natural candidates for affecting volumes of the brain and perhaps other more subtle features of brain function, such as functional activation or metabolism. Except for major risk genes such as APOE, a risk factor for late-onset Alzheimer’s disease, ENIGMA’s data would ultimately show that many of these candidate genes, long thought to affect brain measures, did not appear to do so in much larger sample sizes. This finding was confirmed in samples of 10,000 brain scans or more, samples large enough to detect effects accounting for as little as 1 % of the variance in a brain measure.
Between 2009 and 2012, over 20 cohorts worldwide came together to form ENIGMA. The initial study (called “ENIGMA1”; Stein et al. 2012) found common variants near the TESC gene that were associated with hippocampal volume measured in MRI scans of the brain. The SNPs involved also affected gene expression in living brain tissue, as confirmed by analysis of post-mortem brain tissue. Carrying one form of the gene was associated with a hippocampal volume that was smaller by an amount equivalent to about 3 years of brain aging, a small but substantial effect on a brain scan; the possible cognitive effects of this genetic change, and their effects on disease risk, are now the target of study. Other findings of ENIGMA1 included an association between intracranial volume in healthy subjects and a genetic variant in HMGA2, a gene that had formerly been associated with height and whose role in cell proliferation was beginning to be understood. ENIGMA would not have been able to demonstrate that these associations were robust without the help of another large consortium, CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology), whose GWAS studies of the aging brain in five large elderly cohorts were crucial in establishing the generality of the findings. In fact, when the two consortia exchanged their top findings for genetic variants associated with hippocampal and intracranial volumes, their top five hits were the same. The most associated SNPs in each consortium were the same ones, even though the studies assessed different individuals and were designed independently (Bis et al. 2012).
Non-biological Information Arising from ENIGMA
After ENIGMA’s first study, some hypotheses had to be revised about which genes might affect brain measures and how easy it would be to detect their effects. Some of the “hallowed” candidate genes in psychiatric genetics—COMT, for example—were initially hailed as explaining a fair proportion of the risk for psychiatric illness, only to be found less relevant or not well supported in follow-up studies [see Button et al. (2013) for an analysis of this “winner’s curse” effect]. Perhaps for the same reasons, many genes expected to influence brain structure were not found to do so, even in ENIGMA’s highly powered study. Only APOE had a convincing effect on hippocampal volume, with many growth factors and common psychiatric risk genes not showing demonstrable effects in much larger sample sizes than previously studied. Although it is not possible to rule out an effect that is undetected, the effects of these genes would likely be less than 1 % of the measured variance, much smaller than some originally thought.
On the bright side, the power to replicate findings across the whole diverse range of cohorts and populations in ENIGMA was surprising and encouraging. Most studies contributing to ENIGMA were designed with other goals in mind, on different scanners and some on different continents. As the data were pooled after the fact, substantial work went into showing that reproducible and accurate measures could be made of the same brain regions across sites and scanners [see Supplemental Materials in Stein et al. (2012)]. On the genomic side, ENIGMA’s use of reference panels such as HapMap3 and the 1000 Genomes datasets to “impute” genetic data collected from different genotyping chips also made it possible to pool data across sites, attaining a power not previously imagined for a brain imaging study.
But Do the ENIGMA Genes Affect Disease Risk?
Shortly after the initial study was published, a second initiative was started to screen the genome for common variants associated with volumes of seven other subcortical structures (the project was called “ENIGMA2”; Hibar et al. 2015) and 34 other cortical structures (ENIGMA3; in progress). In the course of these studies, a collaborative partnership began with the Psychiatric Genomics Consortium (PGC) to see if any of the brain-relevant genes were “enriched” in the PGC’s own screens for genes associated with psychiatric illnesses such as schizophrenia . ENIGMA studies of schizophrenia, epilepsy, obsessive compulsive disorder, and Alzheimer’s disease are currently underway. There is some optimism that these enrichment analyses may show that some of the same genes that affect the structure of the brain also create risk for disease. Several disease risk genes are known to be convincingly associated with brain differences: many of the top 20 or so Alzheimer’s risk genes (according to alzgene.org) are associated with differences in brain structure, metabolism, or pathology identifiable with brain imaging. Some of the logistics involved in looking up ENIGMA’s genes in other psychiatric GWAS involves performing “checksum” tests to exclude people who have taken part in both GWAS studies; such participants could cause spurious associations, making it important to screen out non-independent data.
In parallel, ENIGMA launched several working groups to identify brain measures that showed the greatest patient vs. control differences in cohorts of patients with schizophrenia (Turner et al. 2014; van Erp et al. 2015), bipolar illness (Hibar et al. 2014), depression (Schmaal et al. 2014, 2015), and ADHD (Hoogman et al. 2014). Some of these studies now number 4000–8000 subjects, making them the largest studies ever of their respective disorders. Clearly, the power to identify correlates of behavioral and cognitive dysfunction, and relevant modulators of illness such as medication effects, makes these efforts highly informative. All these studies are in their earlier phases now, but ultimately they may yield new sources of information to distinguish psychiatric profiles based on brain imaging and genetics and for differential diagnosis and even perhaps prognosis.
Searching Brain Images for Statistical Effects
In brain imaging studies more generally, it is common to align a group of subjects’ images to a standardized coordinate space and try to find parts of the brain with consistent activations or brain regions whose activity relates to modifiable parameters of the experimental design. One such approach, called statistical parametric mapping, or SPM, can identify brain regions where brain signals relate to some external predictor, such as a task performed in the scanner, or psychiatric diagnosis. To do this, often a regression model is fitted at thousands to millions of different locations in a 3D brain image and the significant regions are shown, after some suitable correction for the multiple statistical tests made in the image.
Brain-Wide Genome-Wide Scanning
Although it may seem a daunting task, Stein et al. (2010) proposed a method to screen every voxel (location) in the brain and every genotyped variant in a genomic screen to search both images and genomes at once for promising associations. The sheer number of computations can exceed one billion statistical tests. The first such efforts found no genuinely replicated associations and were computationally feasible only on a massively parallel computer cluster.
Due to the massive number of statistical tests, the significance threshold that needs to be achieved to control for false positives is around a billion to one (see Medland et al. 2014). Even so, this threshold was achievable and far exceeded by several “hits” (i.e., genetic associations) in ENIGMA2, making the approach feasible statistically as well. Although voxel-wise GWAS is a tour de force computationally, it can be combined with other techniques for dimension reduction to focus the search on promising signals. These methods can be statistical, based on genetic clustering or prioritizing brain measures with highest heritability , or they can be based on biology and known genetic pathways. Such efforts are reviewed in Thompson et al. (2013, 2014, 2015).
Genetic Screening of the Connectome
Based on the power that has been achieved so far through ENIGMA to discover common genetic influences on brain structure, it should now be clear that genome-wide analysis can also be extended to measures beyond that of individual neuroanatomical structures to discover factors that influence how regions of the brain are connected or work together, i.e., measures of brain connectivity. Brain connectivity can be modeled in terms of networks describing how different regions of the brain function together (functional connectivity) or how they are physically connected in terms of the strength, integrity, or pattern of the white matter fibers (structural connectivity) (Fig. 1).

Various forms of connectivity measures extracted from brain images; all these methods allow us to study the brain from a higher dimensional perspective and observe correlations and connections between regions. In the more classical approaches, voxelwise maps of activity or DTI-based integrity measures can be mapped out. In addition to MRI-based imaging, electrodes can be placed around the brain to obtain functional activation or electrophysiological signals. Structural or functional connections between different regions can be estimated. A broad search over all possible connections can lead to mapping the information in a matrix to form a mathematical graph representation. Global properties of this matrix can then be thought of as measures that describe the network as a whole. For example, one measure of interest examines the shortest path lengths in the network or the paths with the lowest numbers of connections between one region, or node, and all the others
Family and twin studies found that specific connections and global organizational measures are heritable in both functional and structural networks. Glahn et al. (2010) found that the resting state functional network, derived from blood oxygen level-dependent functional MRI imaging, is remarkably heritable; Smit et al. (2010) used EEG-based measures of connectivity to study the heritability of measures of network “clustering” and path length. Fornito et al. (2011) examined local and global measures of efficiency and connection distance, along with overall density for resting state networks. In a similar investigation of functional connectivity in children, however, van den Heuvel et al. (2013) did not detect significant heritability for certain local measures while robustly finding that more global measures of network organization were heritable. Structural connectivity and patterns of organization are also influenced by genetic factors. Jahanshad et al. (2013b) showed that a fraction of the total number of detected connections are indeed highly heritable, while Bohlken et al. (2014) studied the network’s topology to establish heritability for other global measures of fiber connections.
The genetic influences on these brain measures have also been established by exploring the effect of known disease risk genes on the connectome. Candidate gene analyses have even suggested that connectome properties may be associated with genetic risk factors for diseases and disorders such as autism (Scott-Van Zeeland et al. 2010; Dennis et al. 2011), schizophrenia (Braskie et al. 2012), and dementia (Brown et al. 2011; Jahanshad et al. 2012); given the history of candidate gene associations in psychiatric genetics, these findings will need to be replicated and assessed in larger samples. There is clear potential for using connectivity measures as targets for genetic analysis or perhaps even for successfully discovering disease risk genes through a genome-wide search.
While functional connectivity measures also appear to be promising targets for genetic study, here we focus our discussion on expanding structural connectivity analyses for large-scale genetic analyses in ENIGMA. Figure 2 shows the structural connectivity matrix from an individual: it stores information on the proportion of detected fibers connecting each pair of brain regions. Jahanshad et al. (2013a, b) proposed a method to map structural connectivity based on diffusion-weighted MRI and prioritize the resulting connections for a genome-wide screen to identify common variants that affect brain connectivity. Not all possible connections are found in all individuals and not all parts of the brain are directly connected to all the others, so the connectivity matrices are relatively sparse (see Fig. 2). As such, a matrix that represents some measure of the quality or density of connections between all pairs of regions on the cortex may represent a number of possible connections that is equal to the square of the number of regions, in theory. For example, breaking up the cortex into 70 regions (Desikan et al. 2006) would lead to a connectivity matrix of almost 5000 elements, but only around 1 % of these might show high reproducibility and heritability in a population.

The structural connectivity matrix. Using standard anatomical MRI and a variant called diffusion-weighted MRI for fiber tracking, we can map out the structural connectivity network of the brain. To do this, we combine a cortical parcellation (top left) with a set of fiber pathways computed using tractography algorithms (bottom left). The resulting connections between all pairs of cortical regions are organized into a connectivity matrix (right). Its rows and columns correspond to the cortical regions and the magnitudes of the elements represent properties of the connections detected between them, such as fiber integrity or density
Using a classical twin model based on identical and fraternal twins, Jahanshad et al. (2013a, b) identified the heritable connections within structural connectivity matrices of several thousand elements and carried forward only the approximately 50 heritable connections into a genome-wide screen. The gene showing a genome-wide and connectome-wide level association with a particular connection within the connectome, SPON1, was subsequently also associated with cognitive decline in an independent study, albeit at a different locus (Sherva et al. 2014). This gene is also implicated in amyloid processing (Hafez et al. 2012), a key component of Alzheimer’s disease pathology.
Clearly, the ability to pursue such an approach on a large scale, within ENIGMA, depends on several factors: a working group, ENIGMA-DTI, was set up to assess its feasibility. First, unless diffusion-weighted MRI measures show greater genetic effect sizes than other traits assessed so far, there must be tens of thousands of DTI scans available from people with GWAS for such a study to be well powered. Second, the format of the connectivity matrix must be sufficiently standardized and agreed on in advance, to allow the exchange and pooling of brain connectivity data across sites.
Encouragingly, by mid-2014, the ENIGMA-DTI working group had amassed around 10,000 DTI scans. Pilot studies showed that the data could be analyzed in a consistent way (Jahanshad et al. 2013a; Kochunov et al. 2014). As the ENIGMA3 project involves a cortical volumetric analysis, the current plan is for ENIGMA to use those cortical regions as the basis for a structural connectivity analysis, using the same voxel-wise analysis of the connections as advocated in Stein et al. (2010) and Jahanshad et al. (2013b). It will be interesting to see if similar sample sizes, tens of thousands, are needed to find and replicate genetic associations with measures of structural brain connectivity. It could be that mathematical tactics for dimension reduction, or network-based measures, are also attractive targets for genetic analysis; so far the relative merits of each of these measures remains to be seen.
Caveats for Multi-site Genomic Analysis of the Connectome
In addition to the caveats noted for pooling multi-site structural MRI data, several additional caveats make the analysis of connectivity challenging. First, the choice of tractography methods can result in different matrices; the method only detects fibers that the algorithm can identify, so many true connections may be missed and some “false positive” connections will also be detected. As with standard MRI, these factors are largely influenced by the signal to noise ratio and resolutions of the images. Often, an arbitrary threshold is implemented to remove the false positive connections, but short fibers can be filtered out. If a connection appears to be weak or inconsistent across subjects, this connection may also be removed. Interestingly, Fornito et al. (2011) found that, for resting state networks at different thresholds, the degree of heritability varied for different global measures, and heritability was not uniform across all nodes; there were various levels of genetic influence for each measure. Lastly, while seemingly intuitive, the results do depend on the parcellation of the cortex, the way the cortical surface is split up into regions of interest. Depending on the goals of the study, parcellation schemes can be improved to maximize power.
Before embarking on large-scale collaborative efforts combining connectivity matrices and network metrics, confounding factors such as these should be properly investigated as is currently being done in ENIGMA’s working groups, such as the ENIGMA-DTI and EEG working groups, among others. For example, in single site studies, Buchanan et al. (2014) performed test-retest reliability analyses to explore the reliability of measures after exploring a variety of commonly used approaches. Dennis et al. (2012) and Zhan et al. (2013) explored the consequences of altering the thresholds used to define networks as well as different methods of tractography, respectively.
Future Directions: Adaptive Connectomics and EPIC
In Prasad et al. (2014), we introduced a method called “EPIC” (Evolving Partitions in Connectomics) to compute brain connectivity in such a way as to be optimally sensitive to statistical effects in a population, such as the effect of Alzheimer’s disease or depression. Clearly, the brain can be divided into regions in many different ways, such as spectral clustering (Craddock et al. 2012), hierarchical clustering (Blumensath et al. 2013), or even genetic clustering (Chen et al. 2012). Each one leads to a different definition of brain connectivity between the resulting regions. Although the set of possible partitions is truly astronomical in number, EPIC offers a principled approach to identify the optimal set of brain regions to find specific statistical effects on the connectivity of the resulting regions. Put another way, if we are seeking brain regions whose connectivity is disrupted in Alzheimer’s disease, the algorithm will merge and split parts of the brain until it reaches a set of connections that best differentiates Alzheimer’s disease patients from controls.
With this adaptive method in mind, it is easy to see how the brain could be partitioned in such a way to maximize the heritability of the connections, automatically de-selecting unfavorable measures before performing a genome-wide screen. If that were done, genomic screens of the connectome might be more efficient, allowing a two-way interplay between discovered genes and the search for connections they might affect.
Still further potential is available once a genome-wide hit is detected; in that case, it should be possible to merge and split cortical sectors so that the genetic effect of a SNP or set of SNPs is more powerfully detected. In other words, one could adjust the cortical partition to maximize the proportion of variance that can be attributed to SNPs or common genetic variants. These high-dimensional searches of the connectome and genome at once will draw upon the full breadth of ingenuity of mathematicians and geneticists alike.
With the scale of ENIGMA and other consortia now planned, it seems likely that we may crack the “Enigma code” of the brain’s connectivity network, using intelligent algorithms and the concerted efforts of the worldwide scientific community. Identifying the genetic influences on the structure and function of the human brain can allow us to understand what makes us human and help uncover the mechanisms causing psychiatric illness.
References
Bis JC, DeCarli C, Smith AV, van der Lijn F, Crivello F, Fornage M, Debette S, Shulman JM, Schmidt H, Srikanth V, Schuur M, Yu L, Choi SH, Sigurdsson S, Verhaaren BF, DeStefano AL, Lambert JC, Jack CR Jr, Struchalin M, Stankovich J, Ibrahim-Verbaas CA, Fleischman D, Zijdenbos A, den Heijer T, Mazoyer B, Coker LH, Enzinger C, Danoy P, Amin N, Arfanakis K, van Buchem MA, de Bruijn RF, Beiser A, Dufouil C, Huang J, Cavalieri M, Thomson R, Niessen WJ, Chibnik LB, Gislason GK, Hofman A, Pikula A, Amouyel P, Freeman KB, Phan TG, Oostra BA, Stein JL, Medland SE, Vasquez AA, Hibar DP, Wright MJ, Franke B, Martin NG, Thompson PM, Enhancing Neuro Imaging Genetics through Meta-Analysis Consortium, Nalls MA, Uitterlinden AG, Au R, Elbaz A, Beare RJ, van Swieten JC, Lopez OL, Harris TB, Chouraki V, Breteler MM, De Jager PL, Becker JT, Vernooij MW, Knopman D, Fazekas F, Wolf PA, van der Lugt A, Gudnason V, Longstreth WT Jr, Brown MA, Bennett DA, van Duijn CM, Mosley TH, Schmidt R, Tzourio C, Launer LJ, Ikram MA, Seshadri S (2012) Cohorts for heart and aging research in genomic epidemiology consortium. Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat Genet 44:545–551
Blokland GA, de Zubicaray GI, McMahon KL, Wright MJ (2012) Genetic and environmental influences on neuroimaging phenotypes: a meta-analytical perspective on twin imaging studies. Twin Res Hum Genet 15:351–371
Blumensath T, Jbabdi S, Glasser MF, Van Essen DC, Ugurbil K, Behrens TE, Smith SM (2013) Spatially constrained hierarchical parcellation of the brain with resting-state fMRI. Neuroimage 76:313–324
Bohlken MM, Mandl RC, Brouwer RM, van den Heuvel MP, Hedman AM, Kahn RS, Hulshoff Pol HE (2014) Heritability of structural brain network topology: a DTI study of 156 twins. Hum Brain Mapp 35:5295–5305
Boomsma D, Busjahn A, Peltonen L (2002) Classical twin studies and beyond. Nat Rev Genet 3:872–882
Braskie MN, Jahanshad N, Stein JL, Barysheva M, Johnson K, McMahon KL, de Zubicaray GI, Martin NG, Wright MJ, Ringman JM, Toga AW, Thompson PM (2012) Relationship of a variant in the NTRK1 gene to white matter microstructure in young adults. J Neurosci 32(17):5964–72. doi:10.1523/JNEUROSCI.5561-11.2012
Brown JA, Terashima KH, Burggren AC, Ercoli LM, Miller KJ, Small GW, Bookheimer SY (2011) Brain network local interconnectivity loss in aging APOE-4 allele carriers. Proc Natl Acad Sci USA 108(51):20760–20765
Buchanan CR, Pernet CR, Gorgolewski KJ, Storkey AJ, Bastin ME (2014) Test-retest reliability of structural brain networks from diffusion MRI. Neuroimage 86:231–243
Button KS, Ioannidis JPA, Mokrysz C, Nosek BA, Flint J, Robinson ESJ, Munafò MR (2013) Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci 14:365–376
Chen CH, Gutierrez ED, Thompson W, Panizzon MS, Jernigan TL, Eyler LT, Fennema-Notestine C, Jak AJ, Neale MC, Franz CE, Lyons MJ, Grant MD, Fischl B, Seidman LJ, Tsuang MT, Kremen WS, Dale AM (2012) Hierarchical genetic organization of human cortical surface area. Science 335(6076):1634–1636
Chiang MC, Barysheva M, Shattuck DW, Lee AD, Madsen SK, Avedissian C, Klunder AD, Toga AW, McMahon KL, de Zubicaray GI, Wright MJ, Srivastava A, Balov N, Thompson PM (2009) Genetics of brain fiber architecture and intellectual performance. J Neurosci 29:2212–2224
Craddock RC, James GA, Holtzheimer PE 3rd, Hu XP, Mayberg HS (2012) A whole brain fMRI atlas generated via spatially constrained spectral clustering. Hum Brain Mapp 33:1914–1928
Dennis EL, Jahanshad N, Rudie JD, Brown JA, Johnson K, McMahon KL, de Zubicaray GI, Montgomery G, Martin NG, Wright MJ (2011) Altered structural brain connectivity in healthy carriers of the autism risk gene, CNTNAP2. Brain Connect 1(6):447–459
Dennis EL, Jahanshad N, Toga AW, McMahon KL, de Zubicaray GI, Martin NG, Wright MJ, Thompson PM (2012) Test-retest reliability of graph theory measures of structural brain connectivity. In: Ayache N, Delingette H, Golland P, Mori S (eds) Medical image computing and computer-assisted intervention‚ MICCAI 2012. Springer, Heidelberg, pp 305–312
Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ (2006) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31(3):968–980
Fornito A, Zalesky A, Bassett DS, Meunier D, Ellison-Wright I, Yucel M, Wood SJ, Shaw K, O’Connor J, Nertney D, Mowry BJ, Pantelis C, Bullmore ET (2011) Genetic influences on cost-efficient organization of human cortical functional networks. J Neurosci 31(9):3261–3270
Glahn DC, Thompson PM, Blangero J (2007) Neuroimaging endophenotypes: strategies for finding genes influencing brain structure and function. Hum Brain Mapp 28(6):488–501
Glahn DC, Winkler AM, Kochunov P, Almasy L, Duggirala R, Carless MA, Curran JC, Olvera RL, Laird AR, Smith SM, Beckmann CF, Fox PT, Blangero J (2010) Genetic control over the resting brain. Proc Natl Acad Sci USA 107(3):1223–1228
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127
Hafez DM, Huang JY, Richardson JC, Masliah E, Peterson DA, Marr RA (2012) F-spondin gene transfer improves memory performance and reduces amyloid-beta levels in mice. Neuroscience 223:465–472
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, Heun R, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41:1088–1093
Hibar DP, Westlye L, Thompson PM, Andreassen O, ENIGMA Bipolar Disorder Working Group (2014) ENIGMA bipolar disorder working group findings from 1,747 cases and 2,615 controls. Organization for Human Brain Mapping Conference, Hamburg, Germany
Hibar DP et al (2015) Common genetic variants influence human subcortical brain structures. Nature 520(7546):224–229
Hoogman M, Zwiers M, Mennes M, Franke B, ENIGMA ADHD Working Group (2014) Brain structure and ADHD across the life span: an ENIGMA collaboration. Organization for Human Brain Mapping Conference, Hamburg, Germany
Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, L Whitwell J, Ward C, Dale AM, Felmlee JP, Gunter JL, Hill DL, Killiany R, Schuff N, Fox-Bosetti S, Lin C, Studholme C, DeCarli CS, Krueger G, Ward HA, Metzger GJ, Scott KT, Mallozzi R, Blezek D, Levy J, Debbins JP, Fleisher AS, Albert M, Green R, Bartzokis G, Glover G, Mugler J, Weiner MW (2008) The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J Magn Reson Imaging 27(4):685–691
Jahanshad N, Valcour VG, Nir TM, Kohannim O, Busovaca E, Nicolas K, Thompson PM (2012) Disrupted brain networks in the aging HIV+ population. Brain Connect 2(6):335–344
Jahanshad N, Kochunov PV, Sprooten E, Mandl RC, Nichols TE, Almasy L, Blangero J, Brouwer RM, Curran JE, de Zubicaray GI, Duggirala R, Fox PT, Hong LE, Landman BA, Martin NG, McMahon KL, Medland SE, Mitchell BD, Olvera RL, Peterson CP, Starr JM, Sussmann JE, Toga AW, Wardlaw JM, Wright MJ, Hulshoff Pol HE, Bastin ME, McIntosh AM, Deary IJ, Thompson PM, Glahn DC (2013a) Multi-site genetic analysis of diffusion images and voxelwise heritability analysis: a pilot project of the ENIGMA-DTI working group. Neuroimage 81:455–469
Jahanshad N, Rajagopalan P, Hua X, Hibar DP, Nir TM, Toga AW, Jack CR Jr, Saykin AJ, Green RC, Weiner MW, Medland SE, Montgomery GW, Hansell NK, McMahon KL, de Zubicaray GI, Martin NG, Wright MJ, Thompson PM (2013b) Genome-wide scan of healthy human connectome discovers SPON1 gene variant influencing dementia severity. Proc Natl Acad Sci USA 110:4768–4773
Jensen AR (1969) How much can we boost IQ and scholastic achievement? Harvard Educ Rev 39:1–123
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–116
Joshi AA, Leporé N, Joshi SH, Lee AD, Barysheva M, Stein JL, McMahon KL, Johnson K, de Zubicaray GI, Martin NG, Wright MJ, Toga AW, Thompson PM (2012) The contribution of genes to cortical thickness and volume. Neuroreport 22:101–105
Kochunov P, Jahanshad N, Sprooten E, Nichols TE, Mandl RC, Almasy L, Booth T, Brouwer RM, Curran JE, de Zubicaray GI, Dimitrova R, Duggirala R, Fox PT, Hong LE, Landman BA, Lemaitre H, Lopez L, Martin NG, McMahon KL, Mitchell BD, Olvera RL, Peterson CP, Starr JM, Sussmann JE, Toga AW, Wardlaw JM, Wright MJ, Wright SN, Bastin ME, McIntosh AM, Boomsma DI, Kahn RS, den Braber A, de Geus EJ, Deary IJ, Pol HEH, Williamson D, Blangero J, van ’t Ent D, Thompson PM, Glahn DC (2014) Multi-site study of additive genetic effects on fractional anisotropy of cerebral white matter: comparing meta and mega analytical approaches for data pooling. Neuroimage 95:136–150
Lewontin RC, Rose SPR, Kamin LJ (1984) Not in our genes: biology, ideology, and human nature. Pantheon Books, New York
Medland SE, Jahanshad N, Neale BM, Thompson PM (2014) Whole-genome analyses of whole-brain data: working within an expanded search space. Nat Neurosci 17:791–800
Prasad G, Joshi SH, Thompson PM (2014) Optimizing brain connectivity networks for disease classification using EPIC. IEEE 11th International Symposium on Biomedical Imaging, Beijing, China
Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190
Rajagopalan P, Hibar DP, Thompson PM (2013) TREM2 Alzheimer risk gene carriers lose brain tissue faster. N Engl J Med 369:1565–1567
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485:237–241
Schmaal L, Veltman DJ, Hibar DP (2014) Subcortical brain volume abnormalities in major depressive disorder: prospective meta-analytic findings from the Enigma Major Depressive Disorder Working Group. Society for Neuroscience Conference in Washington DC, USA, abstract 614.05
Schmaal L (2015) Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. doi:10.1038/mp.2015.69
Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, Bookheimer SY (2010) Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med 2(56):56–80
Sherva R, Tripodis Y, Bennett DA, Chibnik LB, Crane PK, de Jager PL, Farrer LA, Saykin AJ, Shulman JM, Naj A, Green RC, GENAROAD Consortium, Alzheimer’s Disease Neuroimaging Initiative, Alzheimer’s Disease Genetics Consortium (2014) Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement 10:45–52
Smit DJ, Boersma M, van Beijsterveldt CE, Posthuma D, Boomsma DI, Stam CJ, de Geus EJ (2010) Endophenotypes in a dynamically connected brain. Behav Genet 40(2):167–177
Stein JL, Hua X, Lee S, Ho AJ, Leow AD, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Dechairo BM, Potkin SG, Weiner MW, Thompson P (2010) Voxelwise genome-wide association study (vGWAS). Neuroimage 53:1160–1174
Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, Toro R, Appel K, Bartecek R, Bergmann O, Bernard M, Brown AA, Cannon DM, Chakravarty MM, Christoforou A, Domin M, Grimm O, Hollinshead M, Holmes AJ, Homuth G, Hottenga JJ, Langan C, Lopez LM, Hansell NK, Hwang KS, Kim S, Laje G, Lee PH, Liu X, Loth E, Lourdusamy A, Mattingsdal M, Mohnke S, Maniega SM, Nho K, Nugent AC, O’Brien C, Papmeyer M, Putz B, Ramasamy A, Rasmussen J, Rijpkema M, Risacher SL, Roddey JC, Rose EJ, Ryten M, Shen L, Sprooten E, Strengman E, Teumer A, Trabzuni D, Turner J, van Eijk K, van Erp TG, van Tol MJ, Wittfeld K, Wolf C, Woudstra S, Aleman A, Alhusaini S, Almasy L, Binder EB, Brohawn DG, Cantor RM, Carless MA, Corvin A, Czisch M, Curran JE, Davies G, de Almeida MA, Delanty N, Depondt C, Duggirala R, Dyer TD, Erk S, Fagerness J, Fox PT, Freimer NB, Gill M, Goring HH, Hagler DJ, Hoehn D, Holsboer F, Hoogman M, Hosten N, Jahanshad N, Johnson MP, Kasperaviciute D, Kent JW Jr, Kochunov P, Lancaster JL, Lawrie SM, Liewald DC, Mandl R, Matarin M, Mattheisen M, Meisenzahl E, Melle I, Moses EK, Muhleisen TW, Nauck M, Nothen MM, Olvera RL, Pandolfo M, Pike GB, Puls R, Reinvang I, Renteria ME, Rietschel M, Roffman JL, Royle NA, Rujescu D, Savitz J, Schnack HG, Schnell K, Seiferth N, Smith C, Steen VM, Valdes Hernandez MC, Van den Heuvel M, van der Wee NJ, Van Haren NE, Veltman JA, Volzke H, Walker R, Westlye LT, Whelan CD, Agartz I, Boomsma DI, Cavalleri GL, Dale AM, Djurovic S, Drevets WC, Hagoort P, Hall J, Heinz A, Jack CR Jr, Foroud TM, Le Hellard S, Macciardi F, Montgomery GW, Poline JB, Porteous DJ, Sisodiya SM, Starr JM, Sussmann J, Toga AW, Veltman DJ, Walter H, Weiner MW, Alzheimer’s Disease Neuroimaging Initiative, EPIGEN Consortium, IMAGEN Consortium, Saguenay Youth Study Group, Bis JC, Ikram MA, Smith AV, Gudnason V, Tzourio C, Vernooij MW, Launer LJ, DeCarli C, Seshadri S, Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium, Andreassen OA, Apostolova LG, Bastin ME, Blangero J, Brunner HG, Buckner RL, Cichon S, Coppola G, de Zubicaray GI, Deary IJ, Donohoe G, de Geus EJ, Espeseth T, Fernández G, Glahn DC, Grabe HJ, Hardy J, Hulshoff Pol HE, Jenkinson M, Kahn RS, McDonald C, McIntosh AM, McMahon FJ, McMahon KL, Meyer-Lindenberg A, Morris DW, Müller-Myhsok B, Nichols TE, Ophoff RA, Paus T, Pausova Z, Penninx BW, Potkin SG, Sämann PG, Saykin AJ, Schumann G, Smoller JW, Wardlaw JM, Weale ME, Martin NG, Franke B, Wright MJ, Thompson PM, Enhancing Neuro Imaging Genetics through Meta-Analysis Consortium (2012) Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet 44:552–561
Thompson PM, Cannon TD, Narr KL, van Erp T, Poutanen VP, Huttunen M, Lönnqvist J, Standertskjöld-Nordenstam CG, Kaprio J, Khaledy M, Dail R, Zoumalan CI, Toga AW (2001) Genetic influences on brain structure. Nat Neurosci 4(12):1253–1258
Thompson PM, Ge T, Glahn DC, Jahanshad N, Nichols TE (2013) Genetics of the connectome. Neuroimage 80:475–488
Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME, Toro R, Jahanshad N, Schumann G, Franke B, Wright MJ, Martin NG, Agartz I, Alda M, Alhusaini S, Almasy L, Almeida J, Alpert K, Andreasen NC, Andreassen OA, Apostolova LG, Appel K, Armstrong NJ, Aribisala B, Bastin ME, Bauer M, Bearden CE, Bergmann O, Binder EB, Blangero J, Bockholt HJ, Boen E, Bois C, Boomsma DI, Booth T, Bowman IJ, Bralten J, Brouwer RM, Brunner HG, Brohawn DG, Buckner RL, Buitelaar J, Bulayeva K, Bustillo JR, Calhoun VD, Cannon DM, Cantor RM, Carless MA, Caseras X, Cavalleri GL, Chakravarty MM, Chang KD, Ching CR, Christoforou A, Cichon S, Clark VP, Conrod P, Coppola G, Crespo-Facorro B, Curran JE, Czisch M, Deary IJ, de Geus EJ, den Braber A, Delvecchio G, Depondt C, de Haan L, de Zubicaray GI, Dima D, Dimitrova R, Djurovic S, Dong H, Donohoe G, Duggirala R, Dyer TD, Ehrlich S, Ekman CJ, Elvsashagen T, Emsell L, Erk S, Espeseth T, Fagerness J, Fears S, Fedko I, Fernandez G, Fisher SE, Foroud T, Fox PT, Francks C, Frangou S, Frey EM, Frodl T, Frouin V, Garavan H, Giddaluru S, Glahn DC, Godlewska B, Goldstein RZ, Gollub RL, Grabe HJ, Grimm O, Gruber O, Guadalupe T, Gur RE, Gur RC, Goring HH, Hagenaars S, Hajek T, Hall GB, Hall J, Hardy J, Hartman CA, Hass J, Hatton SN, Haukvik UK, Hegenscheid K, Heinz A, Hickie IB, Ho BC, Hoehn D, Hoekstra PJ, Hollinshead M, Holmes AJ, Homuth G, Hoogman M, Hong LE, Hosten N, Hottenga JJ, Hulshoff Pol HE, Hwang KS, Jack CR Jr, Jenkinson M, Johnston C, Jonsson EG, Kahn RS, Kasperaviciute D, Kelly S, Kim S, Kochunov P, Koenders L, Kramer B, Kwok JB, Lagopoulos J, Laje G, Landen M, Landman BA, Lauriello J, Lawrie SM, Lee PH, Le Hellard S, Lemaitre H, Leonardo CD, Li CS, Liberg B, Liewald DC, Liu X, Lopez LM, Loth E, Lourdusamy A, Luciano M, Macciardi F, Machielsen MW, Macqueen GM, Malt UF, Mandl R, Manoach DS, Martinot JL, Matarin M, Mather KA, Mattheisen M, Mattingsdal M, Meyer-Lindenberg A, McDonald C, McIntosh AM, McMahon FJ, McMahon KL, Meisenzahl E, Melle I, Milaneschi Y, Mohnke S, Montgomery GW, Morris DW, Moses EK, Mueller BA, Munoz Maniega S, Muhleisen TW, Muller-Myhsok B, Mwangi B, Nauck M, Nho K, Nichols TE, Nilsson LG, Nugent AC, Nyberg L, Olvera RL, Oosterlaan J, Ophoff RA, Pandolfo M, Papalampropoulou-Tsiridou M, Papmeyer M, Paus T, Pausova Z, Pearlson GD, Penninx BW, Peterson CP, Pfennig A, Phillips M, Pike GB, Poline JB, Potkin SG, Putz B, Ramasamy A, Rasmussen J, Rietschel M, Rijpkema M, Risacher SL, Roffman JL, Roiz-Santianez R, Romanczuk-Seiferth N, Rose EJ, Royle NA, Rujescu D, Ryten M, Sachdev PS, Salami A, Satterthwaite TD, Savitz J, Saykin AJ, Scanlon C, Schmaal L, Schnack HG, Schork AJ, Schulz SC, Schur R, Seidman L, Shen L, Shoemaker JM, Simmons A, Sisodiya SM, Smith C, Smoller JW, Soares JC, Sponheim SR, Sprooten E, Starr JM, Steen VM, Strakowski S, Strike L, Sussmann J, Samann PG, Teumer A, Toga AW, Tordesillas-Gutierrez D, Trabzuni D, Trost S, Turner J, Van den Heuvel M, van der Wee NJ, van Eijk K, van Erp TG, van Haren NE, van ’t Ent D, van Tol MJ, Valdes Hernandez MC, Veltman DJ, Versace A, Volzke H, Walker R, Walter H, Wang L, Wardlaw JM, Weale ME, Weiner MW, Wen W, Westlye LT, Whalley HC, Whelan CD, White T, Winkler AM, Wittfeld K, Woldehawariat G, Wolf C, Zilles D, Zwiers MP, Thalamuthu A, Schofield PR, Freimer NB, Lawrence NS, Drevets W, The Alzheimer’s Disease Neuroimaging Initiative (2014) The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav 8:153–182
Thompson PM et al (2015) ENIGMA and the individual: predicting factors that affect the brain in 35 countries worldwide. NeuroImage. doi:10.1016/j.neuroimage.2015.11.057
Turner J, van Erp T, Hibar D, Thompson PM, and the ENIGMA Schizophrenia Working Group (2014) Subcortical and cortical variations in schizophrenia: the ENIGMA SZ Working Group. Organization for Human Brain Mapping Conference, Hamburg, Germany
van den Heuvel MP, van Soelen IL, Stam CJ, Kahn RS, Boomsma DI, Hulshoff Pol HE (2013) Genetic control of functional brain network efficiency in children. Eur Neuropsychopharmacol 23:19–23
van Erp TG et al (2015) Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. doi:10.1038/mp.2015.118
Visscher PM, Hill WG, Wray NR (2008) Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet 9:255–266
Zhan L, Mueller BA, Jahanshad N, Jin Y, Lenglet C, Yacoub E, Sapiro G, Ugurbil K, Harel N, Toga AW (2013) Magnetic resonance field strength effects on diffusion measures and brain connectivity networks. Brain Connect 3(1):72–86
Acknowledgments
All authors are funded by the National Institutes of Health, including support from the NIH “Big Data to Knowledge” (BD2K) program.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2016 The Author(s)
About this chapter
Cite this chapter
Thompson, P.M., Hibar, D.P., Stein, J.L., Prasad, G., Jahanshad, N. (2016). Genetics of the Connectome and the ENIGMA Project. In: Kennedy, H., Van Essen, D., Christen, Y. (eds) Micro-, Meso- and Macro-Connectomics of the Brain. Research and Perspectives in Neurosciences. Springer, Cham. https://doi.org/10.1007/978-3-319-27777-6_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-27777-6_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27776-9
Online ISBN: 978-3-319-27777-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)