
Abstract
Neutrophil granules contain serine proteases that are central components of the antimicrobial weapons of the innate immune system. Neutrophil proteases also contribute to the amplification and resolution of inflammatory responses through defined proteolytic cleavage of mediators, cell surface receptors, and extracellular matrix proteins. In the blood and at mucosal surfaces, neutrophil serine proteases are regulated by serpins found in plasma and by non-serpin secreted inhibitors. Distinct mechanisms leading to neutrophil cell death have been described for the granule serine proteases, neutrophil elastase, cathepsin G, and proteinase-3. Granule leakage in neutrophils triggers death pathways mediated by cathepsin G and proteinase-3, and both proteases are tightly regulated by their inhibitor SERPINB1 in a cell intrinsic manner. Although stored in the same types of granules, neutrophil elastase does not significantly contribute to cell death following intracellular release from granules into the cytoplasm. However, heterozygous mutations in ELANE, the gene encoding elastase, are the cause of severe congenital neutropenia, a life-threatening condition characterized by the death of neutrophils at an early precursor stage in the bone marrow. This chapter focuses on recent work exploring the biology of clade B intracellular serpins that inhibit neutrophil serine proteases and their functions in neutrophil homeostasis and serine protease control at sites of inflammation.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
5.1 Neutrophil Granules and Their Proteases
5.1.1 Neutrophil Biology and Neutrophil Serine Proteases
Neutrophils are white blood cells with a prominent function in the control of microbes at mucosal surfaces and in tissues. The containment and elimination of bacteria and fungi can be severely affected if too few neutrophils are available or if there is a defect in their ability to reach the infection site. Neutrophils carry a vast repertoire of antimicrobial molecules and enzymes that directly contribute to killing pathogens. While neutrophils are essential to fight bacterial and fungal infections, the presence and death of neutrophils at inflammatory sites has been associated with delayed healing, tissue damage, and pathogenesis of chronic diseases. For these reasons, neutrophils and their potentially harmful granule cargo have been considered a double-edged sword in inflammatory diseases. Neutrophils are now emerging as a central orchestrator of inflammatory responses and their resolution through continuous interactions with components of the innate and adaptive responses (Nathan 2006; Kruger et al. 2015). In this chapter, the function of the neutrophil granule serine proteases in neutrophil life cycle and their function in host defense and in inflammatory diseases will be reviewed with a special focus on their regulation by intracellular serpins.
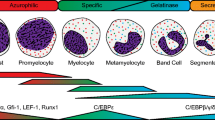
Neutrophils develop in the bone marrow from hematopoietic stem cells and differentiate progressively through progenitors with more restricted lineage potential known as the common myeloid progenitors and the granulocyte-macrophage progenitors (GMPs). The transcription factor PU.1 is essential for the generation of GMPs. At the GMP stage, the expression of the transcription factors, CCAAT/enhancer-binding protein-α (C/EBPα) and growth factor independent-1 (GFI-1), appears essential for commitment toward the neutrophil lineage, whereas PU.1 promotes monocyte differentiation (Cheng et al. 1996; Zhang et al. 1997; Dahl et al. 2003). C/EBP-α and GFI-1 promote proliferation of neutrophil precursors from myeloblasts, promyelocytes, and myelocytes, known as the mitotic pool of neutrophils in the bone marrow. These transcription factors are also key for the expression of neutrophil serine proteases and other primary granule proteins such as myeloperoxidase and defensins at the promyelocyte stage. As C/EPB-α and GFI-1 levels diminish, terminal granulocytic differentiation to mature neutrophils is under the transcriptional regulation of C/EBP-ε, which controls the expression of secondary and tertiary granule proteins. These stages are also known as the postmitotic pool of neutrophils. Mature neutrophils accumulate in the bone marrow as a large reserve pool that can be rapidly released to the circulation. During the last phase of differentiation, neutrophils start expressing receptors to transduce signals for mobilization to the blood and for effector functions. Once in the blood circulation, neutrophils have a relatively short life span of a few hours. They can sense tissue injury through cytokines, peptides, and lipid mediators secreted and/or presented by endothelial cells. Once firmly arrested on the vascular endothelium, neutrophils migrate into the tissue in a highly directional manner toward the injury site. Recruited neutrophils are highly efficient phagocytes and kill microbes using a broad range of antimicrobial peptides and enzymes. In the absence of inflammation, the neutrophil life cycle ends by efferocytosis, where neutrophils undergo apoptosis and are phagocytosed. Efferocytosis of neutrophils occurs in all tissues but, in steady-state conditions, it occurs principally in the spleen, the liver, and the bone marrow. This form of cell death is important for several reasons (Vandivier et al. 2006). First, it dampens the production of inflammatory mediators through the production of TGF-β, for example. Second, and in contrast to necrotic neutrophils, apoptotic neutrophils do not release their highly reactive contents, thus preventing damage to neighboring cells and extracellular matrix proteins. Third, impaired removal of apoptotic cells is associated with autoimmune disease. Last but not least, phagocytosis of apoptotic neutrophils reduces the production of IL-23 by the macrophages. IL-23 induces the production of IL-17 by αβ and γδ T cells. IL-17, in turn, upregulates G-CSF production by stromal cells leading to increased granulopoiesis (Stark et al. 2005). Therefore, the removal of apoptotic cells in the periphery sends a negative feedback loop that ultimately regulates neutrophil production in the bone marrow.
5.1.2 Granule Subsets and Protease Packaging
Neutrophil serine proteases (NSPs) contribute to many aspects of neutrophil biology in host defense and diseases associated with neutrophilic inflammation (Pham 2006). Human neutrophils contain four NSPs with an active catalytic triad: neutrophil elastase (NE), proteinase-3 (PR3), cathepsin G (CG), and the recently characterized neutrophil serine protease-4 (NSP4). NE and PR3 are elastinolytic proteases, and CG has chymotrypsin-like activity, whereas NSP4 is a trypsin-like protease. Azurocidin is encoded by a phylogenetically related serine protease gene, but its catalytic domain is mutated rendering the protease inactive. NSPs evolved from a common ancestor and are phylogenetically related to other serine proteases found in innate immune cells including granzymes and mast cell proteases (Ahmad et al. 2014). NSPs are conserved in mice, and in contrast to granzymes and mast cell proteases, there is a single mouse homolog for each of the human NSP genes.
Neutrophils contain three types of granules (primary, secondary, and tertiary) and secretory vesicles. NSPs are principally stored in primary, also termed azurophil, granules. However, PR3 was also reported in other types of granules and secretory vesicles. Together with NSPs, primary granules carry myeloperoxidase, lysozyme, and antimicrobial peptides such as α-defensins and cathelicidin. How each cargo protein ends up in the correct granule appears largely controlled by transcriptional timing (Cowland and Borregaard 1999; Theilgaard-Mönch et al. 2005). As described above, different transcription factors dominate at different phases of neutrophil differentiation and mRNA expression of NSPs is highest in promyelocytes, which is the time where primary granules are formed.
NSPs are synthesized as pre-pro-enzymes and require posttranslational cleavage at the N-terminus and C-terminus to gain their fully mature active form. First, the signal peptide is cleaved by a signal peptidase. The resulting pro-enzymes are then further processed by dipeptidyl peptidase I (DPPI), also known as cathepsin C, which cleaves the N-terminal dipeptide in the endoplasmic reticulum (ER) and Golgi apparatus before packaging into granules. Cleavage of the pro-dipeptide by DPPI is essential for the enzymatic activity of the NSPs. Neutrophils of dppi −/− mice have no detectable NSP activity, and CG protein is not detectable in neutrophil lysates (Adkison et al. 2002). The C-terminus of the pro-enzymes is also cleaved before packaging into granules, but the protease responsible for this process has not yet been identified. Mutants of NE and CG lacking the C-terminal peptide and expressed in RBL cell line are correctly routed to granules and are enzymatically active indicating that the C-terminal domain is not required for these functions (Gullberg et al. 1995).
The mechanisms of targeting and retention of NSPs in primary granules are not fully elucidated. Cleavage of the C-terminus of NE appears to promote the routing of NE to granules by two mechanisms: first, the C-terminus contains a putative transmembrane domain that leads to membrane anchoring and leads to targeting to the plasma membrane; second, the cleavage of the C-terminus leads to the uncovering of a binding domain for adaptor protein complex 3 (AP3), which may favor protein distribution to granules (Benson et al. 2003). The proteoglycan serglycin contributes to the retention of proteases and inflammatory mediators in leukocytes and endothelial cells (Kolset and Tveit 2008). In neutrophils of serglycin-deficient mice, only NE appeared to require serglycin for proper localization into granules, whereas sorting of CG and PR3 was not affected (Niemann et al. 2007). While early studies showed that PR3 localized principally in primary granules (Borregaard and Cowland 1997), PR3 has also been reported in secretory vesicles and other types of granules, which may explain the more rapid mobilization of PR3 to the plasma membrane (Witko-Sarsat et al. 1999; Loison et al. 2014).
5.2 NSP Inhibitors
5.2.1 Secreted Clade A Serpins
Several inhibitors of the clade A and the clade B serpins, as well as non-serpin inhibitors of the macroglobulin and chelonianin families, contribute to the regulation of the activity of NSPs in different compartments of the cell, tissues, and the whole organism.
The clade A serpins α1-antitrypsin (AAT , SERPINA1) and α1-antichymotrypsin (ACT, SERPINA3) are the two principal serpins found in plasma that inhibit NSPs. AAT inhibits NE, CG, and PR3 by the classical suicide-substrate mechanism of serpins (Huntington et al. 2000). The three NSPs cleave the reactive center loop (RCL) of AAT at the same P1 site Met-358. ACT, in contrast, inhibits only CG. AAT and ACT are produced by the liver and respiratory airway epithelial cells and function by regulating NSPs in blood and extracellular compartments when NSPs are released from neutrophils following degranulation or necrosis.
AAT is also expressed in neutrophils during the late stages of granulopoiesis in the bone marrow and is found at relatively higher levels in mature differentiated cells (Missen et al. 2006). AAT protein is found within neutrophil granules and can be secreted upon stimulation (Mason et al. 1991; Clemmensen et al. 2011). In circulating neutrophils, AAT levels in neutrophils are increased as the secretory vesicles are formed by engulfing plasma proteins that can later be released in response to inflammatory stimuli (Borregaard et al. 1992). The relative importance and function of AAT within the different granules of neutrophils in homeostatic and inflammatory conditions remain to be defined. There is also growing evidence that AAT functions as an anti-inflammatory mediator independently of NSP inhibition (Jonigk et al. 2013). One mechanism may be through interference with TNF-α signaling pathways (Bergin et al. 2014).
In mice and rats, AAT and ACT genes have been duplicated multiple times. The number of AAT genes and plasma concentrations appear to vary between different laboratory mouse strains. Modeling AAT deficiency by deleting the mouse genes has proved to be even more challenging because of embryonic lethality (Wang et al. 2011).
5.2.2 Intracellular Clade B Serpins
Clade B serpins regroup phylogenetically related proteins with nuclear and/or cytoplasmic localization. Vertebrate clade B serpins evolved from a single serpinB1-like gene that has remained conserved in fish, birds, and mammals (Kaiserman and Bird 2005; Benarafa and Remold-O’Donnell 2005). In man, there are 13 clade B serpins found on two loci. SERPINB1, SERPINB6, and SERPINB9 genes are found on chromosome 6p25, and the ten other clade B serpin genes are clustered on chromosome 18q21. Serpins encoded in the 6p25 cluster inhibit granule serine proteases of leukocytes, and high expression of these serpins is found in cells that also carry target proteases.
SERPINB1 was the first cytoplasmic inhibitor of elastase to be identified in neutrophils and monocytes/macrophages (Remold-O’Donnell 1985, 1989; Potempa et al. 1988). It was thus named monocyte/neutrophil elastase inhibitor (MNEI) and leukocyte elastase inhibitor (LEI). SERPINB1 also inhibits CG and PR3 through the classical serpin complex inhibition mechanism. NE and PR3 target the RCL of SERPINB1 at Cys-344 at the classical P1 position, corresponding to Met-358 of AAT . In contrast, CG and other chymotrypsin-like proteases cleave the reactive center loop (RCL) of SERPINB1 at Phe-343 (Cooley et al. 2001). SERPINB1 is expressed in most tissues with higher levels in lymphoid organs such as the bone marrow and the spleen as well as in the pancreas and the lungs. In hematopoietic cells, SERPINB1 is expressed in stem cells, all leukocyte lineages, and platelets. SERPINB1 levels are the highest in the neutrophil lineage, where highest mRNA expression is found in the early stages of granulopoiesis and protein levels remain high in bone marrow and blood neutrophils (Benarafa et al. 2002; Missen et al. 2006). Its role in neutrophil homeostasis is further described below.
SERPINB6, previously described as proteinase inhibitor-6 (PI-6), inhibits CG but not NE and PR3 (Scott et al. 1999). SERPINB6 is also broadly expressed in various tissues and cells with high levels in all myeloid cells with relatively higher levels in monocytic/dendritic cells (Scarff et al. 2003; Missen et al. 2006).
First identified as squamous cell carcinoma antigen (SCCA), SERPINB4 (SCCA2) is also an inhibitor of CG but with slower association rate constants than SERPINB6 and SERPINB1 (Schick et al. 1997). Its closest homolog in mice, serpinb3a, also inhibits CG but is not expressed in hematopoietic cells (Al-Khunaizi et al. 2002; Askew et al. 2004).
5.2.3 Non-serpin Inhibitors of NSPs
α2-Macroglobulin (α2M) is a very large (725 kDa) protease inhibitor found in plasma. It inhibits NSPs as well as many other serine proteases by a cleavage-induced conformational change that traps the proteases into a cavity within α2M (Barrett and Starkey 1973). In contrast to serpin-protease complexes, proteases trapped by α2M can still cleave small molecular weight substrates that can reach the proteolytic domain of the protease. Because of its large size, the diffusion of α2M into tissues during inflammation was considered to be limited and its realm of activity is thus likely limited to the blood circulation, where it regulates fibrinolysis, coagulation, complement, and NSPs (de Boer et al. 1993). However, α2M is found in the lung epithelial lining fluid in adult respiratory distress syndrome (ARDS), where it traps elastase and prevents cleavage of large substrates (Wewers et al. 1988).
Secretory leukocyte protease inhibitor (SLPI) and elafin are two chelonianins that are secreted by epithelial cells and contribute to inhibition of extracellular NSPs at mucosal surfaces and in tissues (Sallenave 2010). They use a reversible keyhole type of inhibition, where the small approximately 10 kDa inhibitors bind the catalytic pocket of the protease with high affinity. SLPI inhibits both NE and CG, while elafin inhibits NE and PR3.
The relative importance of each inhibitor depends on expression levels, compartmentalization, and posttranslational modifications. In addition, because NSPs are usually released together, inhibitors that are inactivated by proteolysis by one NSP may be disarmed before inhibiting their target. While redundancy and compensatory mechanisms are expected and observed between the different inhibitors, essential physiological functions have also been attributed to individual NSP inhibitors.
5.3 Neutrophil Death in Steady-State Conditions
SERPINB1 has emerged as an important regulator of neutrophil survival. A cytoprotective function for intracellular serpins was postulated based on the presence of these inhibitors in cells carrying cell death -inducing granule proteases, such as granzymes (Bird 1999). In the case of SERPINB1, expression profiling during granulopoiesis supported this hypothesis since mRNA and protein levels of SERPINB1 peaked in promyelocyte and myelocyte stages, which coincided with the high but transient transcription of NSPs and other primary granule proteins (Theilgaard-Mönch et al. 2005; Benarafa et al. 2011). The evidence that SERPINB1 is cytoprotective for neutrophil has been elucidated in a series of studies of mice with a targeted deletion of serpinb1a (Benarafa et al. 2007), the mouse ortholog of the human gene (Benarafa et al. 2002). In these mice, neutrophil numbers in the bone marrow are approximately 50 % of the levels of wild-type mice. The phenotype is highly penetrant as it is observed in 129S6 and C57BL/6J backgrounds. Circulating blood neutrophil numbers in serpinb1a −/− mice are only marginally lower than in wild-type mice in normal conditions, but a faster decline in circulating neutrophils is observed after myeloablation with cyclophosphamide (Benarafa et al. 2011). In mice, reduction of neutrophil numbers in the bone marrow is physiologically relevant as it holds over 90 % of mature neutrophils. Indeed, laboratory strains of mice have on average 1 × 106 neutrophils/ml of blood with a total volume of 2 ml; and a mouse femur represents approximately 6 % of the total bone marrow and contains 4–6 × 106 neutrophils depending on the mouse strain.
Neutropenia in serpinb1a −/− mice is due to a cell intrinsic survival defect affecting differentiated neutrophils, while the development of mitotic neutrophil precursors is normal (Benarafa et al. 2011). The mechanism of cell death in the absence of serpinb1a is dependent on CG in steady state in vivo. Indeed, neutropenia in serpinb1a −/− mice was fully rescued by deficiency in CG (Ctsg −/−). In contrast, NE deficiency had no effect on neutrophil survival in serpinb1a −/− mice (Baumann et al. 2013). Competitive stem cell transplantation of irradiated mice showed a survival advantage of wild-type neutrophils over serpinb1a −/− neutrophils but not over Ctsg −/−.serpinb1a −/− double-deficient neutrophils. These mixed bone marrow chimera experiments demonstrated that neutrophil death was cell intrinsic in vivo and support intracellular leakage of granule proteins in mature neutrophils as a mechanism of cell death in the absence of SERPINB1. Importantly, mice lacking one or all three NSPs, as in dppi −/− mice, do not present increased neutrophil numbers in bone marrow and blood. These findings indicate that in homeostatic conditions, neutrophil homeostasis is not critically regulated by NSPs unless the intracellular anti-protease shield is deficient.
SERPINB6 may also contribute to neutrophil homeostasis in steady state as it is expressed in the cytoplasm of neutrophils and inhibits CG. However, mice deficient in serpinb6a, the ortholog of the human gene (Kaiserman et al. 2002), do not present an obvious defect in neutrophil numbers in vivo (Scarff et al. 2004). This somewhat surprising result may be explained by a compensatory increase in serpinb1a expression in bone marrow cells of serpinb6a −/− mice. The characterization of mice deficient in the two serpins may reveal complementary functions.
5.4 Spontaneous Neutrophil Death In Vitro
Mechanisms regulating neutrophil cell death are often investigated in vitro, where isolated neutrophils rapidly die by caspase-dependent apoptosis. This process is often referred to as spontaneous apoptosis. The survival of isolated neutrophils can be extended by addition of synthetic caspase inhibitors or by acting on upstream survival pathways. Serpinb1a −/− neutrophils also demonstrate reduced survival in vitro that is only partly inhibited by caspase inhibitors. In contrast, the survival defect is fully rescued if neutrophils are concomitantly deficient in CG, indicating that in the absence of serpinb1 , CG induce neutrophil death through both caspase-dependent and caspase-independent pathways. While CG has been shown to activate caspase-7 (Zhou and Salvesen 1997), the targets of CG leading to caspase-independent cell death are not yet know.
Interestingly, isolated neutrophils of mice deficient in PR3 (prtn3 −/−) show improved survival in vitro compared to WT neutrophils and PR3 is found in the cytoplasm of aging neutrophils in vitro. Inhibitory complexes between PR3 and serpinb1 are found in the cytoplasm of aging neutrophils, supporting the notion that granule leakage promotes cell death . In contrast to CG or NE, PR3 directly activates pro-caspase-3 leading to apoptosis (Loison et al. 2014). Therefore, serpinb1 appears to act at the intersection of death pathways mediated by CG and PR3. It is conceivable that the two proteases function in a cascade because CG deletion alone rescues the cell intrinsic death pathway in serpinb1a −/− mice in vitro and in steady-state conditions in vivo. This hypothesis could be tested in prtn3 −/−.serpinb1a −/− double knockout mice. Alternatively, CG and PR3 could induce neutrophil death through parallel pathways that would enhance each other by depletion of the serpin shield.
5.5 Neutrophil Elastase and Severe Congenital Neutropenia
5.5.1 Genetic Causes of Severe Congenital Neutropenia
Severe congenital neutropenia (SCN) is a rare inherited disease characterized by persistently low neutrophil counts (<500/μl) in blood and a maturation arrest of neutrophil precursors in the bone marrow. Infants with SCN consequently develop mouth ulcers and pneumonia that can be fatal. Mutations in the neutrophil elastase gene (ELANE or ELA2) are the cause of most cases of SCN and of all cases of cyclic neutropenia (CyN) (Dale et al. 2000; Dale and Link 2009). CyN is a milder form of SCN where blood neutrophil counts oscillate between normal and very low counts with a cyclic period of 21 days (Horwitz et al. 1999).
SCN was first described in the 1950s by Dr. Rolf Kostmann in a family in northern Sweden. The genetic cause of the original cases of Kostmann’s syndrome has recently been attributed to a homozygous mutation in the HAX1 (HCLS1-associated protein X-1) gene (Klein et al. 2007). Other infrequent causes of SCN include mutations in adenylate kinase 2 (AK2), the transcriptional repressor (GFI1), and the glucose-6-phosphatase G6PC3 genes (Klein 2011). X-linked neutropenia has also been described in a subset of patients with Wiskott-Aldrich syndrome (WAS) carrying activating mutations in the WAS gene (Albert et al. 2011). Finally, a significant proportion of SCN cases remain genetically undefined, and with the advances in sequencing technology, new genetic defects leading to SCN will certainly be discovered (Boztug and Klein 2011). Because it is genetically heterogeneous, several molecular pathways are likely leading to SCN.
5.5.2 ELANE Mutations in SCN and CyN
Close to two hundred distinct mutations in ELANE have been identified in patients with SCN and CyN (Germeshausen et al. 2013; Makaryan et al. 2015). How mutant elastase leads to neutropenia is not completely elucidated, but the current working paradigm is that the mutated NE protein induces apoptosis in developing neutrophils in the bone marrow via the initiation of the unfolded protein response (UPR) (Köllner et al. 2006; Grenda et al. 2007). NE mutants are predicted to have defective folding as the protein is synthesized in the ER. Misfolded proteins are detected by ER sensors, which trigger the UPR that can ultimately lead to apoptosis. As very large amounts of mutant misfolded NE are produced at the promyelocyte stage, the UPR triggers apoptosis resulting in the absence of differentiated neutrophils in the bone marrow and blood. Genotype-phenotype analysis studies suggest that the pattern of mutations in ELANE is different in patients that develop SCN or CyN (Makaryan et al. 2015). However, some mutations can lead to both SCN and CyN suggesting that additional disease modifiers may affect the severity of the phenotype (Germeshausen et al. 2013). Furthermore, about 25 % of mutations leading to SCN, but none to CyN, are associated with the development of myelodysplasia (MDS) and acute myeloid leukemia (AML). However, only a fraction of the patients with these mutations also develop MDS/AML, indicating again that other genetic, epigenetic, and environmental factors contribute to disease initiation and progression.
5.5.3 A Role for Serpins in SCN?
Whether mutations in intracellular serpin genes contribute to the severity of SCN or to the subsequent development of MDS/AML has yet to be demonstrated in humans. Studies presented above suggest that CG and PR3 can contribute to neutrophil death if the serpin shield is deficient as in serpinb1a −/− mice. In addition, serpinb1a −/− mice are more susceptible and succumb to lung infections with Pseudomonas aeruginosa (Benarafa et al. 2007). However, the level of neutropenia observed in serpinb1a −/− mice is milder than that observed in SCN and no maturation arrest is observed in the bone marrow (Benarafa et al. 2011).
Several studies have investigated NE activity in the context of ELANE mutations, and the combined evidence does not support a role for altered NE activity as the direct cause of neutrophil precursor death in the bone marrow. In transfection studies, recombinant NE mutants showed variable activity on peptide substrates ranging from the absence of activity, reduction in activity, to a higher activity than wild-type NE. In addition, AAT inhibited all active mutants in vitro (Li and Horwitz 2001). Because SCN patients have heterozygous mutations in ELANE, it was hypothesized that mutant NE might interfere with wild-type NE. The overall NE activity was tested in blood neutrophils of SCN patients with or without ELANE mutations. In single cell assays by flow cytometry, which are associated with technical caveats such as dye loading standardization, average NE activity was similar in neutrophils of SCN patients and those of normal subjects, but the variation between individuals was greater in the SCN patient group (Germeshausen et al. 2013). In the same study, NE activity in lysates of blood neutrophils of a small subset of SCN patients with mutated NE was significantly lower than the activity of neutrophil lysates of SCN patients without ELANE mutation and than that of normal donors. Therefore, ELANE mutations do not appear to be linked with consistently lower or higher NE activity. In addition, elane −/− mice have normal granulopoiesis and neutrophil counts (Belaaouaj et al. 1998), further indicating that reduced or absent NE activity is not required for neutrophil survival and differentiation. Measurements of CG and PR3 activity and levels of SERPINB1 and SERPINB6 in neutrophils of SCN patients with or without ELANE mutations have not been investigated and may provide additional mechanistic clues on pathways leading to neutropenia.
5.5.4 Therapy and Mouse Models for SCN
G-CSF therapy is the standard treatment for almost all types of neutropenia, whether congenital or induced by chemotherapy. Long-term G-CSF therapy has considerably improved the quality of life and reduced mortality from infections in SCN patients (Rosenberg et al. 2008). Yet, as in other severe inherited diseases with defects in hematopoiesis, there is a high long-term risk of developing malignancy. Therefore, better understanding of disease mechanisms may help design better targeted therapies for SCN. In serpinb1a −/− mice, G-CSF treatment increased precursor proliferation and accumulation of mature neutrophils in the bone marrow to levels comparable to those of untreated wild-type mice. However, G-CSF treatment failed to accumulate neutrophils in blood of serpinb1a −/− mice indicating that G-CSF therapy does not fully block NSP-mediated death in circulating cells (Baumann et al. 2013).
Attempts at modeling SCN in mice expressing ELANE mutants found in SCN patients unfortunately did not live up to expectations. Knock-in mice expressing the human V72M mutant of NE instead of mouse NE did not develop SCN and had normal granulopoiesis in steady-state and in stress conditions (Grenda et al. 2002). Another knock-in mouse expressing the G193X mutation that leads to the truncation of the carboxy-terminal 27 amino acids of the mature NE protein was also generated but similarly showed normal granulopoiesis in steady-state conditions (Nanua et al. 2011). However, neutrophil precursors of mice carrying the G193X mutation showed reduced proliferation and survival after tunicamycin and bortezomib treatment, which block N-linked glycosylation and proteasome activity, respectively. These effects were associated with endoplasmic reticulum stress and markers of unfolded protein responses suggesting that modulating the unfolded protein response may be of therapeutic benefit. Finally, the overall lack of effect of these mutants may be due to the significantly lower expression levels of NE in mice compared to human neutrophils (Nanua et al. 2011). Expressing ELANE mutants under the control of a stronger promoter may provide a model.
5.6 NSPs and Intracellular Serpins Beyond Neutrophils
5.6.1 Inflammatory Lung Disease
AAT , SLPI, and elafin have been long established as part of the antiprotease shield against NSPs in the lungs (Greene and McElvaney 2009). Most prominently, patients with AAT deficiency develop lung emphysema at a young age (Laurell and Eriksson 1963). This finding is one of the pillars of protease-antiprotease paradigm of chronic lung disease, where NSPs destroy the lung elastin fibers and other matrix proteins leading to emphysema. Single nucleotide polymorphism that alters the protein sequence of ACT has also been associated with chronic obstructive pulmonary disease (Lomas and Silverman 2001), suggesting that CG may contribute to pathogenesis.
SERPINB1 is also found in airway fluids during lung inflammatory disease (Cooley et al. 2011; Davies et al. 2010; Yasumatsu et al. 2006). Indeed, SERPINB1 can be secreted by an alternative mechanism used by other leaderless cytoplasmic proteins such as IL-1 family members (Keller et al. 2008). However, the relative importance of this secreting pathway and the release of SERPINB1 in the extracellular milieu after cell death remain to be established. Because AAT and SERPINB1 are both fast inhibitors of all three NSPs, a role for SERPINB1 in preventing the development and the severity of pulmonary emphysema in aging mice and following cigarette smoke exposure was tested in serpinb1a −/− mice. Serpinb1a was expressed in the lungs of control mice and expression was higher after cigarette smoke exposure. However, stereological analysis of the lungs and lung function tests revealed that serpinb1a −/− mice did not develop early onset emphysema as they aged. In addition, they developed cigarette smoke-induced emphysema to a comparable extent as wild-type mice after 6-month exposure (Cremona et al. 2013). These findings suggest distinct functions for the intracellular and extracellular serpins in emphysema development.
5.6.2 Lung Infection Models
Excessive inflammatory host response increases morbidity and mortality associated with seasonal respiratory influenza, and highly pathogenic virus strains are characterized by massive infiltration of leukocytes that produce a storm of injurious cytokines. Following up on studies showing increased production of inflammatory cytokines in P. aeruginosa infection of serpinb1a −/− mice (Benarafa et al. 2007), a role for SERPINB1 in influenza A virus infection was investigated. After challenge with a high-dose influenza A/Philadelphia/82 (H3N2), the survival of serpinb1a −/− mice was significantly reduced. Sublethally infected animals suffered increased morbidity, delayed resolution of lung injury, and increased immune cell death (Gong et al. 2011). Importantly, early virus-induced cytokine and chemokine burst and influx of PMNs and monocytes were also normal, and these responses were associated with normal viral clearance in serpinb1a −/− mice compared to wild-type. Whereas initial cytokines and chemokines rapidly decreased in WT mice, TNF-α, IL-6, KC/CXCL1, G-CSF, IL-17A, and MCP-1/CCL2 remained elevated in serpinb1a −/− mice. Monocyte-derived cells were the dominant immune cells in influenza-infected lungs, and those from serpinb1a −/− mice produced more IL-6 and TNF-α when tested ex vivo (Gong et al. 2011). Because viral clearance was unimpaired, the study highlights the critical role of serpinB1 in mitigating tissue injury, restricting inflammatory cytokine production, and reducing morbidity.
In lung infection models, serpinb1a −/− deficiency is characterized by defective microbial clearance and increased production of inflammatory cytokines (Benarafa et al. 2007; Gong et al. 2011). Neutrophil extracellular traps (NETs) are web of nuclear DNA, histones, and antimicrobial molecules released by neutrophils following stimulation by endogenous and pathogen-associated inflammatory mediators. The generation of NETs, or NETosis, was initially described as the ultimate effort by neutrophils to contain microbes and prevent their dissemination by executing this form of programmed cell death (Brinkmann et al. 2004; Fuchs et al. 2007). However, some agonists, such as Staphylococcus aureus, may not require neutrophil death for NET release (Pilsczek et al. 2010); and GM-CSF-primed neutrophils release NETs composed of mitochondrial DNA following stimulation with complement C5a (Yousefi et al. 2009). In vitro, serpinb1a −/− neutrophils release more NETs than wild-type neutrophils in response to agonists that induce NETs via reactive oxygen species-dependent and species-independent routes (Farley et al. 2012). These findings suggest that SERPINB1 regulates a conserved portion of the NET release pathway. SERPINB1 was also shown to regulate DNA release from activated neutrophils in vivo following Pseudomonas infection. Yet, despite producing more NETs, neutropenic serpinb1a −/− mice fail to control Pseudomonas infection in the lung and cannot prevent systemic bacterial spreading (Benarafa et al. 2007; Farley et al. 2012). On the contrary, increased NET generation in these mice may contribute to increased inflammation and tissue injury as observed during influenza infection (Gong et al. 2011).
Several questions thus remain on the potential molecular partners of SERPINB1 in DNA release and associated inflammation. Potential mechanistic pathways include inhibition of NE activity, which is required for NET generation (Papayannopoulos et al. 2010). Moreover, prtn3 −/− mice show reduced inflammation and increased survival in a peritonitis model (Loison et al. 2014). Because SERPINB1 translocates to the nucleus during NET generation and is associated with chromatin, it was also hypothesized that SERPINB1 is involved in chromatin decondensation (Popova et al. 2006; Farley et al. 2012).
5.6.3 SERPINB6 and Deafness
A homozygous truncating mutation in SERPINB6 is associated with non-syndromic sensorineural hearing loss in humans (Sirmaci et al. 2010). Most strikingly, progressive age-related hearing loss is also observed in serpinb6a −/− mice (Tan et al. 2013). Serpinb6a is expressed in cells of the cochlea and its absence leads to degeneration of the organ of Corti, which is composed of hair cells required for transmitting auditory signals. Whether inhibition of proteases, and CG in particular, is involved in this process remains to be determined.
References
Adkison AM, Raptis SZ, Kelley DG, Pham CTN (2002) Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 109:363–371. doi:10.1172/JCI13462
Ahmad J, Bird PI, Kaiserman D (2014) Analysis of the evolution of granule associated serine proteases of immune defence (GASPIDs) suggests a revised nomenclature. Biol Chem 395:1253–1262. doi:10.1515/hsz-2014-0174
Albert MH, Notarangelo LD, Ochs HD (2011) Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Curr Opin Hematol 18:42–48. doi:10.1097/MOH.0b013e32834114bc
Al-Khunaizi M, Luke CJ, Askew YS et al (2002) The serpin SQN-5 is a dual mechanistic-class inhibitor of serine and cysteine proteinases. Biochemistry 41:3189–3199
Askew DJ, Askew YS, Kato Y et al (2004) Comparative genomic analysis of the clade B serpin cluster at human chromosome 18q21: amplification within the mouse squamous cell carcinoma antigen gene locus. Genomics 84:176–184. doi:10.1016/j.ygeno.2004.01.015
Barrett AJ, Starkey PM (1973) The interaction of alpha 2-macroglobulin with proteinases. Characteristics and specificity of the reaction, and a hypothesis concerning its molecular mechanism. Biochem J 133:709–724
Baumann M, Pham CTN, Benarafa C (2013) SerpinB1 is critical for neutrophil survival through cell-autonomous inhibition of cathepsin G. Blood 121:3900–3907. doi:10.1182/blood-2012-09-455022
Belaaouaj AA, McCarthy R, Baumann M et al (1998) Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med 4:615–618
Benarafa C, Remold-O’Donnell E (2005) The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proc Natl Acad Sci USA 102:11367–11372. doi:10.1073/pnas.0502934102
Benarafa C, Cooley J, Zeng W et al (2002) Characterization of four murine homologs of the human ov-serpin monocyte neutrophil elastase inhibitor MNEI (SERPINB1). J Biol Chem 277:42028–42033. doi:10.1074/jbc.M207080200
Benarafa C, Priebe GP, Remold-O’Donnell E (2007) The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J Exp Med 204:1901–1909. doi:10.1084/jem.20070494
Benarafa C, LeCuyer TE, Baumann M et al (2011) SerpinB1 protects the mature neutrophil reserve in the bone marrow. J Leukoc Biol 90:21–29. doi:10.1189/jlb.0810461
Benson KF, Li F-Q, Person RE et al (2003) Mutations associated with neutropenia in dogs and humans disrupt intracellular transport of neutrophil elastase. Nat Genet 35:90–96. doi:10.1038/ng1224
Bergin DA, Reeves EP, Hurley K et al (2014) The circulating proteinase inhibitor α-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med 6:217ra1. doi:10.1126/scitranslmed.3007116
Bird PI (1999) Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins. Immunol Cell Biol 77:47–57. doi:10.1046/j.1440-1711.1999.00787.x
Borregaard N, Cowland JB (1997) Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89:3503–3521
Borregaard N, Kjeldsen L, Rygaard K et al (1992) Stimulus-dependent secretion of plasma proteins from human neutrophils. J Clin Invest 90:86–96. doi:10.1172/JCI115860
Boztug K, Klein C (2011) Genetic etiologies of severe congenital neutropenia. Curr Opin Pediatr 23:21–26. doi:10.1097/MOP.0b013e32834262f8
Brinkmann V, Reichard U, Goosmann C et al (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. doi:10.1126/science.1092385
Cheng T, Shen H, Giokas D et al (1996) Temporal mapping of gene expression levels during the differentiation of individual primary hematopoietic cells. Proc Natl Acad Sci USA 93:13158–13163
Clemmensen SN, Jacobsen LC, Rørvig S et al (2011) Alpha-1-antitrypsin is produced by human neutrophil granulocytes and their precursors and liberated during granule exocytosis. Eur J Haematol 86:517–530. doi:10.1111/j.1600-0609.2011.01601.x
Cooley J, Takayama TK, Shapiro SD et al (2001) The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry 40:15762–15770
Cooley J, Sontag MK, Accurso FJ, Remold-O’Donnell E (2011) SerpinB1 in cystic fibrosis airway fluids: quantity, molecular form and mechanism of elastase inhibition. Eur Respir J 37:1083–1090. doi:10.1183/09031936.00073710
Cowland JB, Borregaard N (1999) The individual regulation of granule protein mRNA levels during neutrophil maturation explains the heterogeneity of neutrophil granules. J Leukoc Biol 66:989–995
Cremona TP, Tschanz SA, von Garnier C, Benarafa C (2013) SerpinB1 deficiency is not associated with increased susceptibility to pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol 305:L981–9. doi:10.1152/ajplung.00181.2013
Dahl R, Walsh JC, Lancki D et al (2003) Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol 4:1029–1036. doi:10.1038/ni973
Dale DC, Link DC (2009) The many causes of severe congenital neutropenia. N Engl J Med 360:3–5. doi:10.1056/NEJMp0806821
Dale DC, Person RE, Bolyard AA et al (2000) Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 96:2317–2322
Davies PL, Spiller OB, Beeton ML et al (2010) Relationship of proteinases and proteinase inhibitors with microbial presence in chronic lung disease of prematurity. Thorax 65:246–251. doi:10.1136/thx.2009.116061
de Boer JP, Creasey AA, Chang A et al (1993) Alpha-2-macroglobulin functions as an inhibitor of fibrinolytic, clotting, and neutrophilic proteinases in sepsis: studies using a baboon model. Infect Immun 61:5035–5043
Farley K, Stolley JM, Zhao P et al (2012) A SerpinB1 regulatory mechanism is essential for restricting neutrophil extracellular trap generation. J Immunol 189:4574–4581. doi:10.4049/jimmunol.1201167
Fuchs TA, Abed U, Goosmann C et al (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241. doi:10.1083/jcb.200606027
Germeshausen M, Deerberg S, Peter Y et al (2013) The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum Mutat 34:905–914. doi:10.1002/humu.22308
Gong D, Farley K, White M et al (2011) Critical role of serpinB1 in regulating inflammatory responses in pulmonary influenza infection. J Infect Dis 204:592–600. doi:10.1093/infdis/jir352
Greene CM, McElvaney NG (2009) Proteases and antiproteases in chronic neutrophilic lung disease – relevance to drug discovery. Br J Pharmacol 158:1048–1058. doi:10.1111/j.1476-5381.2009.00448.x
Grenda DS, Johnson SE, Mayer JR et al (2002) Mice expressing a neutrophil elastase mutation derived from patients with severe congenital neutropenia have normal granulopoiesis. Blood 100:3221–3228. doi:10.1182/blood-2002-05-1372
Grenda DS, Murakami M, Ghatak J et al (2007) Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood 110:4179–4187. doi:10.1182/blood-2006-11-057299
Gullberg U, Lindmark A, Lindgren G et al (1995) Carboxyl-terminal prodomain-deleted human leukocyte elastase and cathepsin G are efficiently targeted to granules and enzymatically activated in the rat basophilic/mast cell line RBL. J Biol Chem 270:12912–12918
Horwitz M, Benson KF, Person RE et al (1999) Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet 23:433–436. doi:10.1038/70544
Huntington JA, Read RJ, Carrell RW (2000) Structure of a serpin-protease complex shows inhibition by deformation. Nature 407:923–926. doi:10.1038/35038119
Jonigk D, Al-Omari M, Maegel L et al (2013) Anti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci USA 110:15007–15012. doi:10.1073/pnas.1309648110
Kaiserman D, Bird PI (2005) Analysis of vertebrate genomes suggests a new model for clade B serpin evolution. BMC Genomics 6:167. doi:10.1186/1471-2164-6-167
Kaiserman D, Knaggs S, Scarff KL et al (2002) Comparison of human chromosome 6p25 with mouse chromosome 13 reveals a greatly expanded ov-serpin gene repertoire in the mouse. Genomics 79:349–362. doi:10.1006/geno.2002.6716
Keller M, Rüegg A, Werner S, Beer H-D (2008) Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818–831. doi:10.1016/j.cell.2007.12.040
Klein C (2011) Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol 29:399–413. doi:10.1146/annurev-immunol-030409-101259
Klein C, Grudzien M, Appaswamy G et al (2007) HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet 39:86–92. doi:10.1038/ng1940
Köllner I, Sodeik B, Schreek S et al (2006) Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood 108:493–500. doi:10.1182/blood-2005-11-4689
Kolset SO, Tveit H (2008) Serglycin--structure and biology. Cell Mol Life Sci 65:1073–1085. doi:10.1007/s00018-007-7455-6
Kruger P, Saffarzadeh M, Weber ANR et al (2015) Neutrophils: between host defence, immune modulation, and tissue injury. PLoS Pathog 11, e1004651. doi:10.1371/journal.ppat.1004651
Laurell CB, Eriksson S (1963) The electrophoretic α; 1-globulin pattern of serum in α; 1-antitrypsin deficiency. Scand J Clin Lab Invest 15:132–140. doi:10.1080/00365516309051324
Li FQ, Horwitz M (2001) Characterization of mutant neutrophil elastase in severe congenital neutropenia. J Biol Chem 276:14230–14241. doi:10.1074/jbc.M010279200
Loison F, Zhu H, Karatepe K et al (2014) Proteinase 3-dependant caspase-3 cleavage modulates neutrophil death and inflammation. J Clin Invest. doi:10.1172/JCI76246
Lomas DA, Silverman EK (2001) The genetics of chronic obstructive pulmonary disease. Respir Res 2:20–26
Makaryan V, Zeidler C, Bolyard AA et al (2015) The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol 22:3–11. doi:10.1097/MOH.0000000000000105
Mason DY, Cramer EM, Massé JM et al (1991) Alpha 1-antitrypsin is present within the primary granules of human polymorphonuclear leukocytes. Am J Pathol 139:623–628
Missen MA, Haylock D, Whitty G et al (2006) Stage specific gene expression of serpins and their cognate proteases during myeloid differentiation. Br J Haematol 135:715–724. doi:10.1111/j.1365-2141.2006.06360.x
Nanua S, Murakami M, Xia J et al (2011) Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood 117:3539–3547. doi:10.1182/blood-2010-10-311704
Nathan C (2006) Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6:173–182. doi:10.1038/nri1785
Niemann CU, Åbrink M, Pejler G et al (2007) Neutrophil elastase depends on serglycin proteoglycan for localization in granules. Blood 109:4478–4486. doi:10.1182/blood-2006-02-001719
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191:677–691. doi:10.1083/jcb.201006052
Pham CTN (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol 6:541–550. doi:10.1038/nri1841
Pilsczek FH, Salina D, Poon KKH et al (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185:7413–7425. doi:10.4049/jimmunol.1000675
Popova EY, Claxton DF, Lukasova E et al (2006) Epigenetic heterochromatin markers distinguish terminally differentiated leukocytes from incompletely differentiated leukemia cells in human blood. Exp Hematol 34:453–462. doi:10.1016/j.exphem.2006.01.003
Potempa J, Dubin A, Watorek W, Travis J (1988) An elastase inhibitor from equine leukocyte cytosol belongs to the serpin superfamily. Further characterization and amino acid sequence of the reactive center. J Biol Chem 263:7364–7369
Remold-O’Donnell E (1985) A fast-acting elastase inhibitor in human monocytes. J Exp Med 162:2142–2155
Remold-O’Donnell E, Nixon JC, Rose RM (1989) Elastase inhibitor. Characterization of the human elastase inhibitor molecule associated with monocytes, macrophages, and neutrophils. J Exp Med 169:1071–1086
Rosenberg PS, Alter BP, Link DC et al (2008) Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol 140:210–213. doi:10.1111/j.1365-2141.2007.06897.x
Sallenave J-M (2010) Secretory leukocyte protease inhibitor and elafin/trappin-2: versatile mucosal antimicrobials and regulators of immunity. Am J Respir Cell Mol Biol 42:635–643. doi:10.1165/rcmb.2010-0095RT
Scarff KL, Ung KS, Sun J, Bird PI (2003) A retained selection cassette increases reporter gene expression without affecting tissue distribution in SPI3 knockout/GFP knock-in mice. Genesis 36:149–157. doi:10.1002/gene.10210
Scarff KL, Ung KS, Nandurkar H et al (2004) Targeted disruption of SPI3/Serpinb6 does not result in developmental or growth defects, leukocyte dysfunction, or susceptibility to stroke. Mol Cell Biol 24:4075–4082
Schick C, Kamachi Y, Bartuski AJ et al (1997) Squamous cell carcinoma antigen 2 is a novel serpin that inhibits the chymotrypsin-like proteinases cathepsin G and mast cell chymase. J Biol Chem 272:1849–1855
Scott FL, Hirst CE, Sun J et al (1999) The intracellular serpin proteinase inhibitor 6 is expressed in monocytes and granulocytes and is a potent inhibitor of the azurophilic granule protease, cathepsin G. Blood 93:2089–2097
Sirmaci A, Erbek S, Price J et al (2010) A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet 86:797–804. doi:10.1016/j.ajhg.2010.04.004
Stark MA, Huo Y, Burcin TL et al (2005) Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22:285–294. doi:10.1016/j.immuni.2005.01.011
Tan J, Prakash MD, Kaiserman D, Bird PI (2013) Absence of SERPINB6A causes sensorineural hearing loss with multiple histopathologies in the mouse inner ear. Am J Pathol 183:49–59. doi:10.1016/j.ajpath.2013.03.009
Theilgaard-Mönch K, Jacobsen LC, Borup R et al (2005) The transcriptional program of terminal granulocytic differentiation. Blood 105:1785–1796. doi:10.1182/blood-2004-08-3346
Vandivier RW, Henson PM, Douglas IS (2006) Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 129:1673–1682. doi:10.1378/chest.129.6.1673
Wang D, Wang W, Dawkins P et al (2011) Deletion of Serpina1a, a murine α1-antitrypsin ortholog, results in embryonic lethality. Exp Lung Res 37:291–300. doi:10.3109/01902148.2011.554599
Wewers MD, Herzyk DJ, Gadek JE (1988) Alveolar fluid neutrophil elastase activity in the adult respiratory distress syndrome is complexed to alpha-2-macroglobulin. J Clin Invest 82:1260–1267. doi:10.1172/JCI113724
Witko-Sarsat V, Cramer EM, Hieblot C et al (1999) Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood 94:2487–2496
Yasumatsu R, Altiok O, Benarafa C et al (2006) SERPINB1 upregulation is associated with in vivo complex formation with neutrophil elastase and cathepsin G in a baboon model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 291:L619–L627. doi:10.1152/ajplung.00507.2005
Yousefi S, Mihalache C, Kozlowski E et al (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16:1438–1444. doi:10.1038/cdd.2009.96
Zhang DE, Zhang P, Wang ND et al (1997) Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA 94:569–574
Zhou Q, Salvesen GS (1997) Activation of pro-caspase-7 by serine proteases includes a non-canonical specificity. Biochem J 324(Pt 2):361–364
Acknowledgments
Charaf Benarafa is funded by the Swiss National Science Foundation (Grant number 310030_149790).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Benarafa, C. (2015). Regulation of Neutrophil Serine Proteases by Intracellular Serpins. In: Geiger, M., Wahlmüller, F., Furtmüller, M. (eds) The Serpin Family. Springer, Cham. https://doi.org/10.1007/978-3-319-22711-5_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-22711-5_5
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-22710-8
Online ISBN: 978-3-319-22711-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)