
Abstract
Epidemiological studies support a strong association between high-density lipoprotein (HDL) cholesterol levels and heart failure incidence. Experimental evidence from different angles supports the view that low HDL is unlikely an innocent bystander in the development of heart failure. HDL exerts direct cardioprotective effects, which are mediated via its interactions with the myocardium and more specifically with cardiomyocytes. HDL may improve cardiac function in several ways. Firstly, HDL may protect the heart against ischaemia/reperfusion injury resulting in a reduction of infarct size and thus in myocardial salvage. Secondly, HDL can improve cardiac function in the absence of ischaemic heart disease as illustrated by beneficial effects conferred by these lipoproteins in diabetic cardiomyopathy. Thirdly, HDL may improve cardiac function by reducing infarct expansion and by attenuating ventricular remodelling post-myocardial infarction. These different mechanisms are substantiated by in vitro, ex vivo, and in vivo intervention studies that applied treatment with native HDL, treatment with reconstituted HDL, or human apo A-I gene transfer. The effect of human apo A-I gene transfer on infarct expansion and ventricular remodelling post-myocardial infarction illustrates the beneficial effects of HDL on tissue repair. The role of HDL in tissue repair is further underpinned by the potent effects of these lipoproteins on endothelial progenitor cell number, function, and incorporation, which may in particular be relevant under conditions of high endothelial cell turnover. Furthermore, topical HDL therapy enhances cutaneous wound healing in different models. In conclusion, the development of HDL-targeted interventions in these strategically chosen therapeutic areas is supported by a strong clinical rationale and significant preclinical data.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- High-density lipoproteins
- Apolipoprotein A-I
- Heart failure
- Ventricular remodelling
- Drug development
- Ischaemia/reperfusion injury
- Tissue repair
- Wound healing
- Diabetic ulcer
1 Introduction
Heart failure is the pathophysiological state in which cardiac dysfunction is responsible for failure of the heart to pump blood at a rate commensurate with the requirements of metabolising tissues. This clinical syndrome is characterised by symptoms and signs of increased tissue and organ water and of decreased tissue and organ perfusion. Heart failure is a growing public health problem, the leading cause of hospitalisation and a major cause of mortality (Rathi and Deedwania 2012). The prevalence of heart failure can be estimated at 2 % in the Western world, and the incidence approaches 5–10 per 1,000 persons per year (Mosterd and Hoes 2007). However, the prevalence and incidence progressively increase with age. The prevalence of heart failure is 7 % in the age group 75–84 years and over 10 % in those older than 85 years (Mosterd et al. 1999). Projections show that by 2030, the prevalence of heart failure will increase 25 % from 2013 estimates (Go et al. 2013). The 5-year age-adjusted mortality rates after onset of heart failure are 50 % in men and 46 % in women (Roger et al. 2004).
Epidemiological studies support a strong association between high-density lipoprotein (HDL) cholesterol levels and heart failure incidence. In Framingham Heart Study participants free of coronary heart disease at baseline, low HDL cholesterol levels were independently associated with heart failure incidence after adjustment for interim myocardial infarction and clinical covariates (Velagaleti et al. 2009). These seminal prospective data are in agreement with earlier cross-sectional studies showing that post-infarct ejection fraction is lower in patients with low HDL cholesterol levels (Kempen et al. 1987; Wang et al. 1998). Low HDL cholesterol levels and low levels of apolipoprotein (apo) A-I, the main apo of HDL, indicate an unfavourable prognosis in patients with heart failure independent of the aetiology (Iwaoka et al. 2007; Mehra et al. 2009). An intriguing observation is that apo A-I is expressed in the human heart and in the heart of human apo A-I transgenic mice (Baroukh et al. 2004; Lowes et al. 2006).
The observed relationship between HDL and heart failure in epidemiological studies might be due to residual confounding. Low HDL may be an integrated biomarker of adverse metabolic processes including abnormal metabolism of triglyceride-rich lipoproteins, insulin resistance, and ongoing tissue inflammation. Crosstalk between inflammatory processes and metabolic dysregulation may be a critical player in the pathogenesis of heart failure (Palomer et al. 2013). Here, we will present experimental evidence from different angles, which support the view that low HDL is unlikely an innocent bystander in the development of heart failure. As will be discussed in the next sections, HDL exerts direct cardioprotective effects, which are mediated via their interactions with the myocardium and more specifically with cardiomyocytes (Sect. 2). HDL may improve cardiac function in several ways. Firstly, HDL may protect the heart against ischaemia/reperfusion injury resulting in a reduction of infarct size and thus in myocardial salvage (Sect. 3). Secondly, HDL can improve cardiac function in the absence of ischaemic heart disease as illustrated by its beneficial effects in diabetic cardiomyopathy (Sect. 4). Thirdly, HDL may improve cardiac function by reducing infarct expansion and by attenuating ventricular remodelling (Sect. 7). Since this latter effect is an example of the role of HDL in tissue repair, the reparative functions of HDL will be further dealt with in Sect. 5 on HDL and endothelial progenitor cells (EPCs) and in Sect. 6 on topical HDL therapy for cutaneous wound healing.
2 In Vitro Effects of HDL on Cardiomyocytes
HDL may exert an indirect protective influence on the heart derived from their ability to limit atherosclerotic plaque formation, thus preventing myocardial ischaemia and loss of myocardial tissue in acute coronary syndromes. Data are now accumulating to indicate that HDL have a more immediate and direct effect on cardiomyocytes. Discussion of these direct effects in this section will provide a more complete picture of the cardioprotective role of these lipoproteins and will also raise questions about the use of the HDL complex itself as a therapeutic agent.
Although HDL is found in plasma, they are also present in the interstitial space in amounts that correspond to approximately 25 % of their plasma concentration (Parini et al. 2006). It is therefore physiologically relevant to evaluate their direct impact on cells. The direct impact of HDL on cardiomyocytes has mainly been studied in two in vitro models: (1) rat neonatal cardiomyocytes and (2) adult mouse cardiomyocytes. These models allow the investigation of the specific actions of HDL on cardiomyocytes independent of the systemic parameters and of endothelial cells whose activity is regulated by HDL. Although studies investigating the direct role of HDL on cardiomyocytes are relatively limited, they all show a beneficial impact of HDL. These protective effects have been attributed to the activation of intracellular signalling cascades involved in pro-survival cell fate.
2.1 Effects of HDL in Rat Neonatal Cardiomyocytes
In cultured rat neonatal cardiomyocytes, HDL treatment inhibits the cytotoxic effects induced by oxidative stress from glucose and growth factor depletion or doxorubicin incubation. This protective effect has mainly been evaluated by its capacity to counteract the proapoptotic signals such as caspase 3 activation and DNA fragmentation (Frias et al. 2010; Theilmeier et al. 2006). HDL incubation induces the phosphorylation of phosphoinositide 3-kinase (PI3K)/Akt, extracellular signal-regulated kinase (ERK) 1/2, p38 mitogen-activated protein kinase (p38 MAPK), and the transcription factor signal transducer and activator of transcription 3 (STAT3) (Frias et al. 2009). Although the precise mechanism of action has not been completely elucidated, experiments using specific inhibitors have defined their role in counteracting apoptotic signals induced by doxorubicin. These studies suggest that the cardioprotective effect involves the activation of the pro-survival proteins ERK1/2 and STAT3, but not p38 MAPK. HDL also induces the phosphorylation of connexin 43 (Morel et al. 2012). This phosphorylation leads to a reduction in gap junction permeability, which may limit the spread of mediators implicated in death pathways. Connexin 43 phosphorylation requires the activation of protein kinase C (Morel et al. 2012). Although there is not yet direct proof of the implication of connexin 43 in the protective effects of HDL, connexin 43 has been shown to be implicated in ischaemic conditioning (Schulz et al. 2007; Schulz and Heusch 2004).
2.2 Adult Mouse Cardiomyocytes
In mouse adult cardiomyocytes, the protective actions of HDL have been demonstrated using a model of hypoxia and reperfusion. Incubation of cardiomyocytes with HDL before the hypoxia period (preconditioning) improved cell survival (Frias et al. 2013). This protection involves the preservation of mitochondrial integrity, as HDL treatment inhibits mitochondrial permeability transition pore opening. Similarly to the results obtained in neonatal cardiomyocytes, HDL induces that activation of intracellular signalling pathways such as PI3K/Akt, ERK1/2, and STAT3, which play a role in this protection. This protective action of HDL is significantly inhibited in cardiomyocytes from cardiomyocyte-restricted STAT3 knockout mice (Frias et al. 2013). Similarly, cardiomyocytes treated with HDL after hypoxia (at reperfusion) show improved cell survival compared to non-treated cells. In this context, the effects of HDL were attributed to PI3K/Akt and ERK1/2 (Tao et al. 2010). The possible role of STAT3 has not been investigated in this reperfusion protocol.
Taken together, HDL-induced activation of several target proteins in cardiomyocytes appears to be cardioprotective. These beneficial effects cannot be attributed to its role in cholesterol transport.
2.3 Apo A-I and Sphingosine-1-Phosphate Mediate the Cytoprotective Effects of HDL
As mentioned above, HDL can modulate the activation of signalling pathways in cardiac cells. HDL is a complex particle, which is composed of numerous proteins and bioactive lipids. Among these constituents, apo A-I and sphingosine-1-phosphate (S1P) are the two major effectors that have been involved in the activation of signalling pathways. Activation occurs via the binding of the HDL particle to scavenger receptor class B, type I (SR-BI), and of S1P to S1P receptors. S1P binds to five (S1P1–S1P5) high-affinity G protein-coupled receptors generating multiple downstream signals. Of the different S1P receptor subtypes, only S1P1, S1P2, and S1P3 receptors are expressed in the heart (Means and Brown 2009). S1P has been shown to be cytoprotective and to confer cardioprotection. For a more detailed discussion of the cardioprotective role of S1P, the interested reader is referred to recent reviews (Karliner 2013; Sattler and Levkau 2009).
In vitro, most of the beneficial effects of HDL mentioned above have been attributed to S1P. Data from experiments using pharmacological antagonists specific for S1P1, S1P2, and S1P3 receptors demonstrate an inhibition of the HDL actions. For example, several experiments suggest that S1P3 is involved in PI3K/Akt signalling and that S1P2 and/or S1P1 might be involved in ERK1/2 and STAT3 signalling (Frias et al. 2009; Tao et al. 2010). S1P2 was shown to play a key role in the HDL-induced protection against the apoptotic effects of doxorubicin (Frias et al. 2010). In the protection against hypoxia reperfusion injury, a predominant, mediatory role was indicated for the S1P1 and S1P3 receptors, via ERK1/2 and PI3K/Akt activation. No role was attributed to S1P2, but neither was it investigated (Tao et al. 2010). The evidence that reinforces the cytoprotective role of S1P comes from experiments, where addition of S1P to reconstituted HDL (rHDL) containing apo A-I improved the protection against the apoptotic action of doxorubicin (Frias et al. 2010).
It should be acknowledged that S1P cannot freely circulate in plasma and that the major plasma lipoprotein source of this lipid is HDL. Therefore, it is not surprising that the effects of HDL can be mediated by S1P. However, conflicting data suggest S1P-independent effects of apo A-I. One explanation proposed initially by Nofer and colleagues (2004) is that SR-BI can anchor the HDL particle at the cell membrane and allow S1P to interact with its specific receptors. In this context, Means and colleagues (Means et al. 2008) have shown that S1P1 is present in lipid raft structures from adult mouse cardiomyocytes. Lipid rafts are known to harbour SR-BI (Babitt et al. 1997). Although increasing evidence suggests a major role of S1P, the mechanism of signal transduction from HDL particle to cell is still under intensive investigation, and a possible role of SR-BI cannot be eliminated.
Crosstalk between intracellular signalling pathways increases the complexity of the regulation of HDL-induced cascades and impedes identification of the specific role of HDL constituents. Nevertheless, it is important to keep in mind that such complexity can result in differing and conflicting data. Further investigations will be necessary to elucidate the precise mechanism of action of HDL in cardiomyocytes in vitro.
3 HDL Confer Protection Against Ischaemia Reperfusion Injury
Whereas the previous section was dedicated to HDL-induced signalling in cardiomyocytes and to cytoprotective effects of HDL in vitro, the role of HDL in this section is extended to cardiac ischaemia/reperfusion injury (IRI).
Ischaemic heart disease is the leading cause of death in the world. After myocardial infarction, early reperfusion is the most effective strategy to limit cell death and subsequent complications that can lead to heart failure. However, re-establishing perfusion is paradoxically detrimental to the heart, giving rise to IRI. This phenomenon is termed lethal reperfusion injury and is defined as a myocardial injury caused by the restoration of coronary flow after an ischaemic episode. This injury culminates in the death of cardiomyocytes that were viable immediately before the myocardial reperfusion. Combating IRI is a notable focus of attempts to improve treatment strategies.
Experimental animal models of IRI are adequate to investigate the precise mechanisms of action and the potential targets of therapeutic drugs. Most investigations used the isolated rodent heart (ex vivo model) or transient ligature of the left anterior descending coronary artery (in vivo model). All studies that evaluated the effects of HDL on the heart submitted to the IRI protocol are unanimous and demonstrate a beneficial effect on cardiac function. Among these studies, a few demonstrated similar effects using rHDL, composed of apo A-I and phospholipids (palmitoyl-oleoyl-phosphatidylcholine). Some studies used rHDL containing the apo A-I Milano mutant.
3.1 IRI in the Isolated Heart Model
An early study using HDL perfusion in isolated hearts suggested that HDL significantly reduced post-ischaemic arrhythmias (Mochizuki et al. 1991). In contrast, perfusion with low-density lipoproteins (LDL) had no effects indicating HDL specificity. The mechanism was not defined, but it was hypothesised that the lipoprotein stabilised bioactive arachidonic acid metabolites (Mochizuki et al. 1991). These antiarrhythmogenic effects were later confirmed in vivo when treatment with rHDL decreased the incidence of post-ischaemic arrhythmias (Imaizumi et al. 2008). Surprisingly, treatment with apo A-I or phospholipids did not significantly reduce these arrhythmogenic responses. The effect of rHDL was mediated via the production of nitric oxide (NO), through an Akt/ERK/NO pathway in endothelial cells (Imaizumi et al. 2008). Unfortunately, the direct impact on cardiomyocytes was not investigated.
The direct cardioprotective impact of HDL on IRI has been studied by Calabresi and colleagues (Calabresi et al. 2003). They reported that treatment with rHDL improved post-ischaemic functional recovery and decreased creatine kinase release in the coronary effluent. Lipid-free apo A-I or phosphatidylcholine liposomes were not effective in protecting the heart from IRI. rHDL caused a dose-dependent reduction of ischaemia-induced cardiac tumour necrosis factor-α (TNF-α) expression and content, which correlated with the improved functional recovery (Calabresi et al. 2003). In agreement with this prior study, rHDL improved cardiac function (Rossoni et al. 2004) and reduced infarct size compared to non-treated animals (Gu et al. 2007). Recent studies showed that treatment with HDL significantly reduced the infarct size in rodents submitted to IRI compared to non-treated IRI animals (Frias et al. 2013; Morel et al. 2012). The protective impact of HDL or rHDL did not depend on the timing of injection. Indeed, the treatment at the moment of reperfusion was also effective, but to a lesser extent than the treatment before the ischaemic period.
The precise mechanism of HDL-induced cardioprotection is under intensive investigation, and the activation of intracellular signalling pathways plays an important role in such cardioprotection. In this context, the protective role of STAT3, previously shown in cardiomyocytes, has been confirmed in the isolated heart model. The reduction of the infarct size induced by HDL was significantly inhibited in STAT3 knockout mice (Frias et al. 2013). Mitochondria play an important role in cell survival and may be the key player in cardioprotection. Interestingly, HDL and rHDL containing apo A-I Milano have been shown to preserve mitochondrial integrity (Frias et al. 2013; Marchesi et al. 2008), consistent with a direct impact of HDL and rHDL on cardiac cells.
3.2 IRI in In Vivo Models
The data obtained ex vivo were extended to in vivo models. The first in vivo data on cardiac function after HDL injection demonstrated that HDL injection stimulates myocardial perfusion. This effect was mediated via the activation of endothelial nitric oxide synthase (eNOS) (Nofer et al. 2004). Although this study did not investigate IRI, it underlines the potential response of the heart to HDL in vivo. The effect of HDL was extended to an in vivo transient ligature of the left anterior descending coronary artery model (Theilmeier et al. 2006). One single injection of native HDL before ischaemia reduced infarct size, apoptotic cell death, and neutrophil infiltration. These protective effects were also significantly inhibited in the presence of the eNOS inhibitor L-NG-nitroarginine methyl ester (L-NAME) and were partially inhibited in S1P receptor subtype 3 KO mice (Theilmeier et al. 2006). Taken together, these results suggest that the HDL action is mediated via NO production and is dependent on the S1P content of HDL.
Similarly to the results obtained in the isolated heart, the protective role of rHDL containing apo A-I Milano during ischaemia or at reperfusion significantly reduced the infarct size induced by IRI in rabbits (Marchesi et al. 2004). Protective effects of apo A-I were also demonstrated (Gu et al. 2007). Injection of apo A-I 10 min before reperfusion significantly improved cardiac function, which was associated with a reduction of myocardial TNF-α and interleukin (IL)-6 levels. Histological analysis of cardiac tissue from rats treated with apo A-I showed a decrease in intercellular adhesion molecule (ICAM) expression and neutrophil infiltration compared to non-treated animals (Gu et al. 2007). It should be acknowledged that apo A-I and rHDL can absorb phospholipids, including S1P, in plasma. This could modulate their impact on IRI.
3.3 Apo A-I and S1P Confer Cardioprotective Effects on HDL
Based on all published reports, it is not easy to delineate the precise role of S1P or apo A-I in the cardioprotective effects of HDL. Both constituents have been investigated individually, and both have been shown to protect against IRI individually. However, their precise impact on the actions of HDL has not always been investigated in the same study or in the same model. Interestingly, levels of HDL-associated S1P were diminished in patients with coronary artery disease (Sattler et al. 2010). Very recent data demonstrate a key role of apolipoprotein M (apoM) in S1P binding to HDL particles (Christoffersen et al. 2011). Indeed, the amount of S1P contained in HDL was very low in apoM KO and higher in apoM overexpressing mice. Unfortunately, IRI in these mice has not yet been investigated.
With respect to S1P, given that its plasma concentration is many times higher than its affinity for S1P receptors, it has been suggested that HDL might play the role of neutralising and limiting the actions of circulating S1P (Murata et al. 2000). The impact of S1P association with HDL on the function of HDL still remains to be elucidated. It is probable that in the HDL particle, both are necessary to obtain the maximal positive response.
The mechanisms of HDL-induced cardioprotection against IRI are complex and cannot be attributed to the role of HDL in reverse cholesterol transport. More precise knowledge on the role of different HDL constituents in intracellular signalling and cardioprotection would help to design a therapeutic compound, which replicates all the beneficial aspects of HDL in the heart. Figure 1 illustrates conceptually the direct actions of HDL on cardiomyocytes.

Proposed protective intracellular signalling pathways induced by HDL in cardiomyocytes. HDL induces the activation of intracellular signalling pathways in cardiomyocytes. This activation leads to cardioprotection. Most of the activated signalling pathways induced by HDL involve its S1P constituent (HDL-S1P) and the participation of S1P receptor subtypes. Further investigations are necessary to elucidate the precise role of SR-BI in cardioprotection induced by HDL. The arrows indicate intracellular signalling as described in cardiomyocytes. Cx 43; connexin 43; PKC protein kinase C; MEK1/2: ERK (MAPK) kinase 1/2
4 Human Apo A-I Gene Transfer Attenuates Diabetic Cardiomyopathy
Diabetic cardiomyopathy is a cardiac disorder, which takes place in the absence of coronary artery disease and hypertension, and is characterised by impaired left ventricular function due to cardiac inflammation, oxidative stress, cardiomyocyte apoptosis, cardiac perivascular and interstitial fibrosis, intramyocardial microangiopathy, endothelial dysfunction, disturbed cardiac substrate metabolism, cardiomyocyte hypertrophy, abnormal intracellular Ca2+ handling, and impaired functionality of cardiac stem cells. This cardiac entity has first been recognised by Rubler et al. (1972), and its existence has been confirmed during the last three decades via epidemiological, clinical, and experimental studies. Hyperglycaemia, hyperinsulinaemia, and dyslipidaemia each contribute to the pathogenesis of diabetic cardiomyopathy via triggering cellular signalling and subsequent specific alterations in cardiac structure. However, hyperglycaemia-induced oxidative stress has been proposed to be the key determinant in the development of diabetic cardiomyopathy. Left ventricular diastolic dysfunction represents the earliest preclinical manifestation of diabetic cardiomyopathy, preceding systolic dysfunction, which can progress to symptomatic heart failure (Cosson and Kevorkian 2003; Freire et al. 2007; Raev 1994).
HDLs have antidiabetic properties. They reduce hyperglycaemia via their capacity to decrease pancreatic β-cell apoptosis (Abderrahmani et al. 2007; Fryirs et al. 2010; Rutti et al. 2009) and to stimulate glucose disposal in skeletal muscle (Han et al. 2007). They decrease dyslipidaemia via lowering adipocyte lipolysis (Van Linthout et al. 2010a) and induce insulin sensitivity (Berg et al. 2001) via upregulating the expression of adiponectin (Van Linthout et al. 2010a), which abrogates the development of hyperinsulinaemia. Consequently, increased HDL may, from a theoretical point of view, antagonise the main triggers of diabetic cardiomyopathy.
The main hallmarks of diabetic cardiomyopathy, including cardiac oxidative stress, interstitial inflammation, apoptosis, fibrosis, and endothelial dysfunction, on the one hand and the pleiotropic effects of HDL on the other hand further support the hypothesis that an increase in HDL may reduce the development of diabetic cardiomyopathy (Fig. 2). Via a human apo A-I gene transfer strategy, this hypothesis has been evaluated in an experimental rat model of streptozotocin-induced diabetes mellitus associated with severe hyperglycaemia and an LDL to HDL ratio of 1:1 (Van Linthout et al. 2007; Young et al. 1988).

HDL reduces the development of diabetic cardiomyopathy. Type 2 diabetes mellitus is associated with hyperglycaemia, dyslipidaemia, and hyperinsulinaemia. Hyperglycaemia is the main trigger that contributes to the development of diabetic cardiomyopathy via the induction of oxidative stress. Hallmarks of diabetic cardiomyopathy are, besides cardiac oxidative stress, cardiac fibrosis, inflammation, apoptosis, and endothelial dysfunction, which reciprocally affect one another (interconnected grey lines). HDLs have antidiabetic features (dotted lines): they reduce hyperglycaemia and dyslipidaemia via—among other mechanisms—their capacity to decrease pancreatic β-cell apoptosis and adipocyte lipolysis, respectively, and they increase insulin sensitivity via the induction of adiponectin, abrogating the development of hyperinsulinaemia. Besides interference with the triggers that lead to diabetic cardiomyopathy, HDLs reduce the features of diabetic cardiomyopathy via their pleiotropic effects, including their anti-oxidative, antifibrotic, anti-inflammatory, antiapoptotic, and endothelial-protective effects (full lines)
4.1 Human Apo A-I Gene Transfer Influences Metabolic Parameters in Streptozotocin-Induced Diabetes Mellitus
Apo A-I transfer resulted in a 60 % increase in HDL cholesterol, which was paralleled with a significant decline in very-low-density lipoprotein (VLDL) cholesterol, intermediate-density lipoprotein (IDL) cholesterol, and triglycerides, whereas LDL cholesterol was unaffected (Van Linthout et al. 2008). Despite the well-described antidiabetic effects of HDL involving their ability to reduce pancreatic β-cell apoptosis (Abderrahmani et al. 2007; Fryirs et al. 2010; Rutti et al. 2009), apo A-I transfer did not reduce blood glucose levels (Van Linthout et al. 2008), potentially due to the severity of the streptozotocin model, which is associated with a remaining insulin production below 1 % (Hughes et al. 2001). The decline in triglycerides and in the triglyceride-rich lipoproteins VLDL and IDL (Sztalryd and Kraemer 1995) after apo A-I transfer suggests an HDL-mediated decrease in lipolysis in adipose tissue, leading to less free fatty acids in the circulation, less triglyceride synthesis in the liver, and subsequent less VLDL and IDL synthesis (Tunaru et al. 2003). This hypothesis is corroborated by the ability of HDL to reduce the expression of hormone-sensitive lipase (Van Linthout et al. 2010a), the rate-limiting enzyme of adipocyte lipolysis in abdominal fat (Sztalryd and Kraemer 1995), and to increase the phosphorylation of the PI3K downstream target Akt in abdominal fat (Van Linthout et al. 2010a). Akt is involved in the anti-lipolytic and lipogenic effects of insulin in adipose tissue (Whiteman et al. 2002) and in the regulation of the expression of the adipokine adiponectin (Cong et al. 2007; Pereira and Draznin 2005), which is known to improve insulin sensitivity under diabetes (Peterson et al. 2008). However, an involvement of HDL in the hepatic expression of genes involved in triglyceride metabolism may not be excluded. The decrease in cardiac glycogen content (Van Linthout et al. 2008) suggests that apo A-I transfer in streptozotocin rats partly restored glucose metabolism as an energy source in the heart instead of nearly exclusive reliance on fatty acid metabolism for production of ATP (Kota et al. 2011).
4.2 Human Apo A-I Gene Transfer Attenuates Diabetes-Associated Oxidative Stress, Cardiac Fibrosis, and Endothelial Dysfunction
Hyperglycaemia induces oxidative stress via creating a disbalance between the generation of reactive oxygen species and their resolution by antioxidant enzymes, like superoxide dismutases, which convert O2 − anions into molecular oxygen and hydrogen peroxide (Nishikawa et al. 2000). Reactive oxygen species initiate diverse stress-signalling pathways including ERK, c-Jun N-terminal kinase (JNK), and p38 MAPK, alter cellular proteins, and induce lipid peroxidation. In diabetic cardiomyocytes, reactive oxygen species are predominantly generated by mitochondria, due to mitochondrial oxidation of fatty acids (Kota et al. 2011), and by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Li et al. 2010b). The pathological importance of p38 MAPK in the diabetic heart follows from the observation that p38 MAPK inhibition reduces cardiac inflammation and improves left ventricular dysfunction in streptozotocin-induced diabetic mice (Westermann et al. 2006). Besides decreasing the activity of p38 MAPK, apo A-I transfer reduced cardiac oxidative stress via upregulating the expression of the diabetes-downregulated mitochondrial superoxide dismutase-2 and extracellular-superoxide dismutase (Van Linthout et al. 2008). Both forms of superoxide dismutase are important for the heart as outlined in studies whereby cardiac overexpression of superoxide dismutase-2 protected the mitochondrial respiratory function and blocked apoptosis induction (Shen et al. 2006; Suzuki et al. 2002) and overexpression of extracellular-superoxide dismutase decreased macrophage infiltration and fibrosis and improved left ventricular dysfunction (Dewald et al. 2003). These studies postulate that the decrease in cardiac fibrosis after apo A-I transfer in streptozotocin-diabetic rats can be partly explained by the reduction in oxidative stress and inflammation, including downregulated expression of pro-fibrotic cytokines (inflammatory fibrosis) (Tschope et al. 2005), as well as by the decrease in cardiac apoptosis (cfr. supra) and subsequent replacement fibrosis, resulting in improved left ventricular function. The anti-inflammatory and antifibrotic effects of HDL are supported by findings in apo A-I knockout mice, which are associated with increased inflammatory cells and collagen deposition in the lung (Wang et al. 2010), and from observations with the mimetic apo A-I peptide 4F demonstrating an L-4F-mediated decreased endothelial cell matrix production (Ou et al. 2005). Since cardiac NADPH oxidases play a predominant role in the development of diabetic cardiomyopathy (Guo et al. 2007; Wold et al. 2006) and in diabetic cardiac remodelling (Li et al. 2010b), decreased cardiac NADPH oxidase activity following apo A-I transfer (Van Linthout et al. 2009) may be a critical mediator of reduced cardiac oxidative stress and subsequent fibrosis in the myocardium.
Systemically, apo A-I transfer decreased the oxidative stress in rats with streptozotocin-induced diabetes as indicated by the decline in serum levels of thiobarbituric acid reactive substances (TBARS) (Van Linthout et al. 2008), a marker of lipid peroxidation (Tschope et al. 2005). This finding is corroborated by Mackness et al. (1991), who reported that HDLs decrease the formation of TBARS on oxidised LDL. This is likely mediated by an increase in the activity of paraoxonase or platelet-activating factor-acetyl hydrolase, 2 enzymes with anti-oxidative features, which are associated with HDL and which are known to be induced after apo A-I gene transfer (De Geest et al. 2000).
In the vasculature of diabetic rats, apo A-I transfer decreased oxidative stress as indicated by the reduction in diabetes-enhanced NADPH oxidase activity and eNOS uncoupling (Van Linthout et al. 2009), a phenomenon occurring when eNOS produces O2 − rather than NO. Uncoupling of eNOS is, besides NADPH oxidases (Cai 2005), an important source of reactive oxygen species in diseased, including diabetic, blood vessels (Hink et al. 2001). Consistent with the demonstrated role of the angiotensin II receptor, type 1 (AT1R) in mediating increased NADPD oxidase activity, and eNOS uncoupling in diabetes (Oak and Cai 2007), the downregulation in diabetes-induced AT1R (Hodroj et al. 2007; Nyby et al. 2007) after apo A-I transfer (Van Linthout et al. 2009) was postulated to be the predominant mediator of reduced NADPH oxidase activity and eNOS uncoupling. This hypothesis is further supported by in vitro findings showing that the HDL-mediated reduction in AT1R expression in human aortic endothelial cells was associated with a decline in hyperglycaemia-induced oxidative stress and a reduced responsiveness to angiotensin II (Van Linthout et al. 2009). These observations underline the finding of Tolle et al. (2008), who showed that HDL reduce NADPH oxidase-dependent reactive oxygen species generation via inhibition of the activation of Rac1, which is a downstream AT1R-dependent mediator of angiotensin II (Ohtsu et al. 2006). The exact mechanism by which HDL affect AT1R regulation under diabetes mellitus requires further fundamental studies. However, since oxidised LDL (Li et al. 2000) and reactive oxygen species (Gragasin et al. 2003) play a role in the induction of the AT1R in human aortic endothelial cells, one may speculate that HDL via intrinsic anti-oxidative features (cfr. supra, via paraoxonase and platelet-activating factor-acetyl hydrolase) may contribute to the downregulation of the AT1R under diabetes mellitus, which results in less NADPH oxidase activity and reactive oxygen formation and in turn may decrease AT1R expression. Concomitant with the reduced vascular oxidative stress, including decreased eNOS uncoupling, apo A-I transfer in streptozotocin rats resulted in an enhanced NO bioavailability (Chalupsky and Cai 2005) and consequently in a decrease in endothelial dysfunction (Van Linthout et al. 2009), which is another hallmark of diabetic cardiomyopathy.
4.3 Human Apo A-I Gene Transfer Reduces Diabetes-Induced Cardiac Inflammation
In agreement with the direct anti-inflammatory properties of HDL (Cockerill et al. 2001; Hyka et al. 2001), apo A-I transfer decreased the diabetes-induced intramyocardial inflammation (Westermann et al. 2007b, c) as indicated by the reduction in ICAM-1, vascular cell adhesion molecule (VCAM)-1, and TNF-α mRNA expression (Van Linthout et al. 2008) and VCAM-1 protein expression (Van Linthout et al. 2010b). Downregulation of VCAM-1/ICAM-1 expression suppresses monocyte-endothelial cell adhesion and subsequent transendothelial migration of inflammatory cells. This in turn reduced local expression of cytokines like TNF-α and consequently may attenuate the potentiation of the intramyocardial inflammatory reaction. Hyperglycaemia (Li et al. 2010a; Piga et al. 2007), oxidised LDL (Lee et al. 2010) (Hodgkinson et al. 2008; Peterson et al. 2007), and increased angiotensin II (Alvarez et al. 2004; Rius et al. 2010) under diabetes mellitus upregulate the expression of VCAM-1/ICAM-1. Since HDL did not affect blood glucose levels, the reduction in adhesion molecule expression was not an indirect consequence of an HDL-mediated antidiabetic effect. However, the reduced levels of TBARS, which are also retrieved on oxidised LDL, in diabetic rats treated with apo A-I transfer compared to diabetic rats treated with control vector suggest that apo A-I transfer decreases oxidised LDL and subsequent cardiac inflammation. Oxidised LDLs are agonists of Toll-like receptor 4 (TLR4), which is expressed on the cell surface of cardiac cells, including cardiomyocytes, smooth muscle cells, and endothelial cells. A role for TLR4 in the development of diabetic cardiomyopathy has recently been suggested (Zhang et al. 2010). The documented HDL-mediated reduction in endothelial TLR4 expression and subsequent decrease in NF-κB activity (Van Linthout et al. 2011) suggest that the reduction in cardiac inflammation after apo A-I transfer is further partly mediated via a decrease in endothelial TLR4 expression, limiting oxidised LDL-endothelial TLR4 interactions and subsequent activation of NF-κB. Furthermore, the activation of the AT1R, whose expression is upregulated under diabetes mellitus (Cohen 1993; Nyby et al. 2007), contributes to the development of diabetic cardiomyopathy. After all, AT1R antagonism under diabetes mellitus improves endothelial function (Cheetham et al. 2000) and reduces cardiac inflammation and fibrosis, resulting in an improvement in cardiac function (Westermann et al. 2007a). The observations that apo A-I transfer reduces aortic AT1R expression in streptozotocin-induced diabetic rats and that HDLs decrease the hyperglycaemia-induced AT1R expression in endothelial cells (Van Linthout et al. 2009) suggest that apo A-I transfer reduces cardiac endothelial AT1R and subsequent angiotensin II responsiveness including the induction of VCAM-1/ICAM-1 expression and subsequent monocyte-endothelial cell adhesion (Alvarez et al. 2004; Rius et al. 2010). Downregulation of the expression of the anti-inflammatory adipokine adiponectin (Ouchi et al. 2000, 2001; Ouedraogo et al. 2007) as observed in diabetic patients and in streptozotocin rats (Thule et al. 2006) has been implicated in chronic inflammatory phenotype in these subjects/rats. The positive correlation between apo A-I/HDL plasma levels and adiponectin concentrations (Verges et al. 2006) and the finding that apo A-I transfer increases the expression of adiponectin in an experimental model of extreme inflammation (Van Linthout et al. 2010a) further suggest that the effects of HDL on adiponectin expression may contribute to its anti-inflammatory effects and to the attenuation of cardiac inflammation in rats with streptozotocin-induced diabetes. Finally, since apo A-I induces regulatory T cells (Wilhelm et al. 2010), which protect against the pro-inflammatory status of endothelial cells (He et al. 2010), an induction in regulatory T cells following apo A-I transfer may also partly account for the reduced cardiac inflammation in diabetic rats.
Since inflammation triggers fibrosis (Ismahil et al. 2013; Savvatis et al. 2012; Westermann et al. 2011), the less pronounced diabetes-induced cardiac fibrosis (cfr. infra) following apo A-I transfer might partly be explained by the decrease in cardiac inflammation. In this context, the influence of a systemic HDL-mediated immunomodulation on cardiac inflammation and fibrosis needs further investigation.
4.4 Human Apo A-I Gene Transfer Reduces Diabetes-Associated Cardiac Apoptosis and Improves the Cardiac Endothelial Integrity
Cardiac apoptosis is another hallmark of diabetic cardiomyopathy. The incidence of cardiac apoptosis is higher in diabetic patients (Frustaci et al. 2000) and in diabetic animals (Cai et al. 2002) compared to non-diabetic controls and is directly linked to hyperglycaemia-induced oxidative stress (Cai et al. 2002). Mitochondria play an important role in oxidative stress-induced apoptosis. Caspase 3 and caspase 7 are essential mediators in the mitochondrial processes of apoptosis (Lakhani et al. 2006). In accordance with the reduction in oxidative stress, including the upregulation of cardiac mitochondrial superoxide dismutase-2 expression, apo A-I transfer decreased the induced caspase 3/7 activity in streptozotocin rats (Van Linthout et al. 2008). Concomitantly, apo A-I transfer raised the ratio of the antiapoptotic Bcl-2, a ‘guardian’ against mitochondrial initiation of caspase activation (Susin et al. 1996), towards the proapoptotic Bax. This ratio is a marker of increased cardiomyocyte survival probability (Condorelli et al. 1999). Furthermore, apo A-I transfer normalised the diabetes-reduced phosphorylation of the pro-survival protein kinase B Akt (Montanari et al. 2005; Uchiyama et al. 2004) and of its effector eNOS to levels found in non-diabetic hearts (Van Linthout et al. 2008). This finding was further corroborated by experiments in cardiomyocytes. HDL supplementation on cardiomyocytes in the presence of in vitro hyperglycaemia reduced apoptosis in a PI3K- and NO-dependent manner (Van Linthout et al. 2008). The antiapoptotic effects of apo A-I transfer were reflected at ultrastructural level by a reduced number of cardiomyocytes with swollen mitochondria and apoptotic bodies and a more intact endothelium and basement membrane (Van Linthout et al. 2008). These findings and the presence of activated Akt in cardiomyocytes, as well as in cardiac endothelial cells documented via immunofluorescence staining, underscore the importance of Akt activation after apo A-I transfer in the reduction of cardiomyocyte apoptosis as well as in the improvement in endothelial integrity. The ameliorated integrity of the endothelium suggests a potential restoration of the microvascular homeostasis, which has been demonstrated to reduce cardiomyocyte apoptosis and to result in the recovery of cardiac function in diabetic cardiomyopathy (Yoon et al. 2005). The significance of the cardiac endothelium on the contractile state and Ca2+ handling of subjacent cardiomyocytes (Brutsaert 2003; Nishida et al. 1993; Ramaciotti et al. 1992) suggests that part of the HDL-mediated improvement in left ventricular function in rats with streptozotocin-induced diabetes was indirectly due to their protective effect on the cardiac endothelium. This hypothesis is supported by the improvement in endothelial function found after apo A-I transfer in streptozotocin-induced diabetic rats (Van Linthout et al. 2009). In addition, HDL supplementation on isolated cardiomyocytes directly improved their contractility under hyperglycaemia-induced stress in a PI3K- and NO-dependent manner (Van Linthout et al. 2008).
Besides the direct HDL-mediated cardiomyocyte-protective effects and the endothelial-protective features, the decrease in cardiac apoptosis after apo A-I transfer in diabetic rats might be attributed to several other in parallel HDL-mediated triggered processes: (1) HDL may act as biological buffers capable of rapidly removing active proapoptotic TNF-α from the heart; (2) they have the potential to increase (Rossoni et al. 2004), stabilise, and activate prostaglandins (Aoyama et al. 1990). This enhanced prostanoid availability/activity may contribute to the HDL-mediated cardioprotection, by acting directly on cardiomyocytes (Zacharowski et al. 1999) and/or by inhibiting cardiac TNF-α production (Shinomiya et al. 2001); (3) HDL may reduce the expression of TLR 4 (Van Linthout et al. 2011), which has recently been shown to play an important role in cardiac apoptosis in diabetic cardiomyopathy.
In conclusion, apo A-I transfer attenuates the development of experimental diabetic cardiomyopathy via its anti-oxidative, anti-inflammatory, antifibrotic, and antiapoptotic actions (Fig. 2). Beyond beneficial vascular/cardioprotective long-term effects, direct myocardial effects of HDL may contribute to the improvement of cardiac function under severe streptozotocin-induced stress. Further studies are required to investigate the potential of apo A-I transfer to ameliorate established diabetic cardiomyopathy, especially in the context of type 2 diabetes mellitus, and to improve the function of HDL, which is impaired under diabetic conditions (Hedrick et al. 2000; Persegol et al. 2006; Sorrentino et al. 2009; Zheng et al. 2004). Finally, also the impact of the apo A-I/HDL-induced immunomodulatory effects on diabetes-associated endothelial dysfunction and cardiac fibrosis needs further clarification.
5 HDL and Tissue Repair: Modulation of EPC Biology via SR-BI
The term EPC is most often used in a very broad sense. There is no consensus definition of EPCs, which is a major source of confusion in the literature. EPCs encompass different categories of cells with different phenotypic and functional properties that affect neovascularisation and reendothelialisation. In a narrow, literally correct, and unambiguous sense, EPCs are immature precursor cells capable of differentiating into mature endothelial cells in vivo (Hagensen et al. 2010). Thus, an EPC sensu stricto corresponds to an in vivo category and is defined from a functional point of view. However, there are very significant methodological challenges to detect incorporation of circulating progenitor cells in the endothelium (Hagensen et al. 2010). Incorporation of EPCs is in general a relatively rare event (Feng et al. 2008, 2009a, b; Hagensen et al. 2010). High-resolution multichannel sequential confocal scanning microscopy provides a platform for colocalisation analysis with high specificity, which is required in lege artis cell tracking studies (De Geest 2009). Alternatively, EPCs may act in a paracrine way by releasing angiogenic factors and proteases to stimulate sprouting of local vessels or promote reendothelialisation indirectly (Rehman et al. 2003; Takakura et al. 2000; Urbich et al. 2005). To make a distinction with EPCs sensu stricto, these cells may be designated as proangiogenic haematopoietic progenitor cells (Fig. 3).

Role of bone marrow-derived proangiogenic haematopoietic progenitor cells and EPCs sensu stricto in endothelial repair and neovascularisation. Proangiogenic haematopoietic progenitor cells, the in vivo equivalent of ‘early EPCs’, contribute to vascularisation and endothelial repair indirectly by secreting paracrine factors such as VEGF, hepatocyte growth factor (HGF), granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), placental growth factor (PLGF), and IL-8. These factors stimulate local endothelial cell proliferation. On the other hand, EPCs sensu stricto enhance neovascularisation and endothelial repair directly by incorporation into the endothelium. EPCs sensu stricto can be considered to constitute the in vivo equivalent of ECFCs or BOECs (‘late outgrowth EPCs’). ECs endothelial cells; SMCs smooth muscle cells
Different protocols for short-term culture of blood mononuclear cells on fibronectin (with or without gelatin)-coated plates have been established by Vasa et al. (2001) and Hill et al. (2003). These cells are stimulated by in vitro culture conditions to mimic many features of endothelial cells (Hirschi et al. 2008). However, the putative EPCs identified by these assays do not give rise to a lineage of endothelial progeny, but cultured cells in these assays consist of monocytes or a population of haematopoietic cells with monocyte-macrophage or T-cell lineage commitment. Nevertheless, these so-called early EPCs or early outgrowth EPCs may regulate the angiogenic response in a paracrine way (Hirschi et al. 2008). In contrast, endothelial colony-forming cells (ECFCs) (also called ‘late outgrowth EPCs’ or ‘blood outgrowth endothelial cells’ (BOECs)) are immature precursor cells capable of differentiating into mature endothelial cells in vivo. They are derived from long-term culture of adherent peripheral blood mononuclear cells in endothelial conditions (‘late outgrowth EPCs’) and consist of colonies that display a cobblestone morphology (Ingram et al. 2004). ECFCs express cell surface antigens like primary endothelium, clonally propagate, can be replated into secondary and tertiary ECFCs, form capillary-like structures in vitro, and become endothelial cells in vivo (Ingram et al. 2004). ECFCs display postnatal vasculogenic activity upon transplantation in a matrix scaffold (Richardson and Yoder 2011). ECFCs could be considered to be similar to EPCs sensu stricto: they have a potential for postnatal vasculogenesis and endothelial repair at sites of endothelial damage. However, one should always consider that cell categories that are defined based on in vitro cell culture conditions have always a rather artificial character.
HDLs enhance EPC-mediated repair (Feng et al. 2008, 2009a, b; Tso et al. 2006). The effect of HDL on EPC number, function, and incorporation occurs via SR-BI (Feng et al. 2009b). Since HDLs have been demonstrated to increase vascular endothelial growth factor (VEGF) production by early EPCs in vitro, HDLs may improve reendothelialisation by its effects on these circulating proangiogenic cells (Feng et al. 2011). On the other hand, HDLs also improve ECFC function in vitro, and these effects are dependent on signalling via SR-BI, ERKs, and NO and on increased β1 integrin expression (Feng et al. 2011). Taken together, HDL exerts potent effects on different functional categories of EPCs via SR-BI.
The endothelial-protective and endothelial-reparative properties of HDL may be pathophysiologically and clinically relevant under conditions of high endothelial turnover. In healthy individuals, the endothelial layer is renewed at a low replication rate of 0–1 % per day (Erdbruegger et al. 2006). In pathological conditions of arteriosclerosis, endothelial turnover may be higher. Arteriosclerosis is a broad term that encompasses all diseases that lead to arterial hardening, including native atherosclerosis, post-injury neointima formation, cardiac allograft vasculopathy (CAV), and vein graft atherosclerosis. Areas of low shear stress in human arteries have an increased rate of endothelial turnover (Tricot et al. 2000). Augmented endothelial turnover is also observed in atherosclerosis-prone areas in apo E −/− mice (Foteinos et al. 2008). However, the highest degree of endothelial turnover is likely observed after arterial injury, in allografts (Rahmani et al. 2006), and in vein grafts. Although there is significant evidence that HDLs exert beneficial effects in models of angioplasty- or injury-induced arteriosclerosis (Ameli et al. 1994; De Geest et al. 1997; Shah and Amin 1992), we will focus here on allograft vasculopathy and vein graft atherosclerosis.
Orthotopic heart transplantation is a well-established therapy for selected patients with end-stage congestive heart failure. The long-term success of heart transplantation is limited by CAV. EPC-mediated repair may inhibit the progression of CAV. Although CAV is primarily the result of chronic rejection, non-immunological factors are modifiers of CAV progression. In the ‘response to injury’ concept of CAV, vascular lesions are considered to be the result of cumulative endothelial injury both by alloimmune responses and by non-alloimmune insults (Vassalli et al. 2003). T-cell alloimmunity, antibody-mediated immune attack, and non-immune factors induce endothelial cell death or endothelial dysfunction. Since the endothelium regulates vascular tone, inflammation, smooth muscle cell proliferation, and thrombosis (Behrendt and Ganz 2002), restoration of endothelial integrity and function is pivotal to attenuate the development of CAV (Pinney and Mancini 2004). Markers of endothelial injury have been shown to discriminate between CAV-positive and CAV-negative heart transplant recipients (Singh et al. 2012), which is consistent with a postulated pivotal role of endothelial injury in the pathogenesis of CAV.
Current strategies to prevent or treat CAV are clearly insufficient. Disruption of the endothelial lining due to endothelial cell apoptosis can be restored by proliferation of adjacent endothelial cells but also by incorporation of circulating bone marrow-derived EPCs (Friedrich et al. 2006; Walter et al. 2002; Werner et al. 2002, 2003). Hu et al. (2003) have demonstrated that EPCs contribute to endothelial regeneration in murine allografts. Moreover, increased HDL induced by hepatocyte-directed human apo A-I gene transfer in C57BL/6 apo E −/− mice enhanced the incorporation of bone marrow-derived EPCs into the injured endothelium of allografts from donor BALB/c mice and reduced neointima formation in these allografts (Feng et al. 2008). In subsequent experiments using BALB/c allografts in wild-type C57BL/6 mice, it was demonstrated that increased EPC incorporation and attenuation of allograft vasculopathy induced by hepatocyte-directed human apo A-I gene transfer are strictly dependent on SR-BI expression in bone marrow and bone marrow-derived cells. This conclusion was reached based on experiments in mice transplanted with wild-type bone marrow or alternatively with SR-BI−/− bone marrow. These data suggest that the effect of HDL on EPC incorporation and/or paracrine effects of EPCs are critical for the attenuation of allograft vasculopathy in this model. Nevertheless, the implications of these data for clinical CAV are unknown since murine experiments are performed in the absence of immunosuppressive therapies.
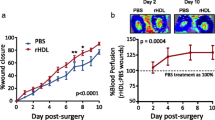
Surgical revascularisation using autologous vein grafts is limited by vein graft failure, which is frequently due to an aggressive form of atherosclerosis (Campeau et al. 1984). Vein graft failure is observed in 30–50 % of coronary bypass grafts within 10 years (Fitzgibbon et al. 1996). Hypercholesterolaemia is a risk factor for vein graft failure (Goldman et al. 2004), which may be related to adverse effects on endothelial integrity and function (Raja et al. 2004). Previously, it has been demonstrated that endothelial regeneration is retarded, and neointima formation is accelerated in vein grafts in hypercholesterolaemic apo E −/− mice compared to normocholesterolaemic controls (Dietrich et al. 2000; Xu et al. 2003). Topical HDL therapy reduced vein graft atherosclerosis in apo E −/− mice (Feng et al. 2011). Topical HDL application involves formulation of HDL in 20 % pluronic F-127 gel (pH 7.2) (Feng et al. 2011). Pluronic F-127 is a biocompatible and non-toxic substance and is characterised by thermoreversible gel formation at temperatures above 21 °C (Hu et al. 1999). One of the main advantages of topical HDL therapy is that the ‘distribution volume’ of the therapeutic agent is small compared to systemic administration. In addition, the extracellular protein concentration of HDL is 300–400 μg/ml (Parini et al. 2006; Sloop et al. 1987). This is approximately 20 % of the plasma protein concentration. Therefore, therapeutic effects of topical HDL can be expected at HDL concentrations that are substantially lower than the plasma concentration. Topical HDL administration on the adventitial side of vein grafts improved vein graft patency and function (Feng et al. 2011). Caval veins of C57BL/6 apo E −/− mice were grafted to the right carotid arteries of recipient C57BL/6 TIE2-LacZ/apo E −/− mice. HDL in 20 % pluronic F-127 gel was applied on the adventitial side of vein grafts. Topical HDL application reduced intimal area by 55 % (p < 0.001) at day 28 compared to control mice. Blood flow quantified by micro-magnetic resonance imaging at day 28 was 2.8-fold (p < 0.0001) higher in grafts of topical HDL-treated mice than in control mice. Topical HDL potently reduced intimal inflammation and resulted in enhanced endothelial regeneration as evidenced by an increase in the number of CD31-positive endothelial cells. As stated supra, HDL potently enhanced migration and adhesion of ECFCs in vitro, and these effects were dependent on signalling via SR-BI, ERK, and NO and on increased β1-integrin expression. Correspondingly, the number of CD31 β-galactosidase double-positive cells, reflecting incorporated circulating progenitor cells, was 3.9-fold (p < 0.01) higher in grafts of HDL-treated mice than in control grafts. Importantly, the effects of topical HDL therapy on vein graft atherosclerosis were similar compared to the effects of systemically increased HDL cholesterol after human apo A-I gene transfer. Taken together, topical HDL application is a new paradigm of HDL therapy.
Whereas this section was focused on HDL- and EPC-mediated endothelial repair, the next two sections will deal with HDL and tissue repair. In these therapeutic areas, the effect of HDL on EPCs may be an important mediator of the beneficial effects of HDL-targeted therapies.
6 Development of Topical HDL Therapy for Cutaneous Wound Healing
Wound healing results from complex interactions between extracellular matrix molecules, soluble mediators, resident skin cells, and infiltrating leukocytes as well as infiltrating EPCs. Rather artificially, wound healing can be divided in an inflammation phase, a phase of tissue formation with accumulation of granulation tissue and reepithelialisation, and finally a phase of tissue remodelling (Diegelmann and Evans 2004).
Granulation tissue contains tissue macrophages, fibroblasts, numerous new vessels, and extracellular matrix molecules. Macrophages stimulate fibroplasia and angiogenesis by secretion of various growth factors. Fibroblasts deposit and remodel the extracellular matrix of wounds. Blood vessels are needed to provide oxygen and nutrients, whereas extracellular matrix provides a conduit for cell migration and cell ingrowth. Formation of granulation tissue starts a few days after injury and is mainly triggered by growth factors such as platelet-derived growth factor and transforming growth factor-β, which stimulate fibroblast proliferation and migration (Singer and Clark 1999). Fibroblasts start to deposit and remodel extracellular matrix, which initially consists of fibronectin and hyaluronan but is later on replaced by proteoglycans and type I and III collagens (Toriseva and Kahari 2009). The formation of new blood vessels is a critical component of to granulation tissue formation. Angiogenesis is initially induced by tissue destruction and hypoxia and is subsequently stimulated by various molecules such as basic fibroblast growth factor, VEGF, and transforming growth factor-β, secreted by macrophages, keratinocytes, and endothelial cells. A role of EPCs in granulation tissue formation is directly suggested by experiments demonstrating that cell therapy with EPCs enhances wound healing in mice (Suh et al. 2005). The release of various growth factors such as VEGF and platelet-derived growth factor by EPCs appears to enhance wound healing (Suh et al. 2005). Reepithelialisation is the term used to describe the appearance of a new epithelial layer on top of a healing skin wound. It is dependent on the formation of a provisional wound bed matrix and involves the migration and proliferation of keratinocytes, the differentiation and stratification of new epithelium, and finally the reformation of the basement membrane.
Typical chronic cutaneous wounds are diabetic ulcers, ischaemic ulcers, and pressure ulcers. Diabetic ulcers occur in patients with type 1 and type 2 diabetes, whereas ischaemic ulcers are observed in patients with peripheral arterial diseases. Pressure ulcers occur in patients with paralysis or in other conditions that inhibit movement of body parts. Pressure ulcers or decubitus wounds typically occur on sacrum, shoulder blades, and heels. Several factors contribute to deficient wound healing in patients with diabetes: deficient growth factor production (Galkowska et al. 2006), impaired neovascularisation (Galiano et al. 2004b), attenuated keratinocyte and fibroblast proliferation and migration (Gibran et al. 2002), and altered balance between extracellular matrix accumulation and remodelling of the extracellular matrix by matrix metalloproteinases (Lobmann et al. 2002). Foot ulcers occur in a diabetic population with a prevalence of 5 % and lifetime incidence of 15 % (Abbott et al. 2002; Muller et al. 2002). Even with lege artis treatment, amputation is required in 14–24 % of cases. Every 30 s, a lower limb is lost due to diabetes. Approximately 85 % of these amputations are preceded by an ulcer. Mortality is up to 50 % in the first 3 years after amputation. Various studies show that costs to treat diabetic foot ulcers are extremely high, especially if hospitalisation is required (Matricali et al. 2007).
HDL may beneficially affect wound healing by accelerating resolution of inflammation, by enhancing granulation tissue formation involving increased EPC incorporation and increased paracrine effects of EPCs, and by accelerating reepithelialisation. Keratinocytes express SR-BI, and SR-BI expression is upregulated in dividing keratinocytes (Tsuruoka et al. 2002). Whereas in humans healing is primarily the result of reepithelialisation and granulation tissue formation, wound healing in mice occurs predominantly by wound contraction (Greenhalgh 2003). In the excisional wound healing model (Galiano et al. 2004a), a circular full-thickness wound is applied on the back of each mouse. Subsequently, a silicone splint is fixed around the wound with nylon sutures to counteract wound contraction. Consequently, wound healing in this model occurs by granulation tissue formation and reepithelialisation from the border. Granulation tissue formation and reepithelialisation were significantly delayed in C57BL/6 apo E−/− mice compared to C57BL/6 mice (Gordts et al. 2014). Topical administration of HDL (protein concentration 800 μg/ml; volume 80 μl) formulated in 20 % pluronic F-127 gel (pH 7.2) every 2 days on wounds in C57BL/6 apo E−/− mice significantly improved granulation tissue formation and reepithelialisation. Wound healing in mice treated with topical HDL therapy was very similar compared to C57BL/6 mice. Topical gel without HDL did not enhance wound healing. Improved wound healing induced by topical HDL therapy was also observed in C57BL/6 mice with streptozotocin-induced diabetes mellitus and in male type 2 diabetic leptin receptor-deficient mice (unpublished data).
Further preclinical studies are required to evaluate the robustness of this strategy in other models of delayed wound healing. However, murine models of diabetic wound healing differ in many respects to clinical diabetic wound healing. They do not take into account that there is a significant degree of heterogeneity of ulcers in patients with diabetes. Three categories can be discerned: ‘purely neurotrophic’ diabetic ulcers, ‘purely ischaemic’ ulcers in patients with diabetes, and poorly healing ulcers in diabetic patients with microangiopathy. The ‘purely neurotrophic’ diabetic ulcers heal well after off-loading. The ‘purely ischaemic’ ulcers should be treated with revascularisation strategies. Typically, diabetic ulcers in patients with more pronounced microangiopathy and macroangiopathy are characterised by defects in granulation tissue formation and constitute a potential therapeutic area for HDL-targeted therapies. However, no murine model can adequately mimic these chronic diabetic ulcers. Decubitus wounds constitute another target for topical HDL therapy but lack of adequate animal models hinders preclinical development.
7 Beneficial Effects of Selective HDL-Raising Gene Transfer on Cardiac Remodelling and Cardiac Function After Myocardial Infarction in Mice
Loss of myocardial tissue following acute myocardial infarction results in a decreased systolic ejection and increased left ventricular end-diastolic volume and pressure. The Frank-Starling mechanism, implying that an increased end-diastolic volume results in an increased pressure developed during systole, may help to restore cardiac output. However, the concomitant increased wall stress may induce regional hypertrophy in the non-infarcted segment, whereas in the infarcted area expansion and thinning may occur. Experimental animal studies show that the infarcted ventricle hypertrophies and that the degree of hypertrophy is dependent on the infarct size (Anversa and Sonnenblick 1990). Taken together, architectural remodelling is characterised by the formation of a discrete collagen scar, ventricular dilatation, and ventricular hypertrophy. This process may continue for weeks or months until the distending forces are counterbalanced by the tensile strength of the collagen scar. Remodelling post-myocardial infarction is complex since it involves the infarct area, the infarct border zone, and the non-infarcted myocardium (Sutton and Sharpe 2000). Following myocardial infarction, dyskinetic bulging may occur in infarct area. The myocardial fibres contiguous to this segment in the infarct border zone become exposed to a more pronounced increase in wall stress because of the more prominent change in the radius of curvature induced by the regional dilatation. Cardiomyocytes in the border zone of the infarct become larger than cardiomyocytes in the remote area of the ventricle. This is consistent with the view that the infarct-induced stress on the chamber walls is an important determinant of cardiomyocyte hypertrophy (Cohn 1993).
Post-infarct remodelling occurs in the setting of volume overload, since the stretched and dilated infarcted tissue increases the left ventricular volume. An increased ventricular volume not only implies increased preload (passive ventricular wall stress at the end of diastole) but also increased afterload (total myocardial wall stress during systolic ejection). Afterload is increased since the systolic radius is increased. Since both systolic and diastolic wall stress are increased, remodelling and hypertrophy post-myocardial infarction are characterised by mixed features of pressure overload and volume overload.
The myocardium consists of 3 integrated components: cardiomyocytes, extracellular matrix, and capillary microcirculation. All 3 components are involved in the remodelling process. The role of the extracellular matrix is distinct in the early phase of remodelling (within 72 h) and the late phase (beyond 72 h). Neutrophil infiltration of matrix metalloproteinases induces degradation of intermyocyte collagen struts and cardiomyocyte slippage. This leads to infarct expansion characterised by the disproportionate thinning and dilatation of the infarct segment (Erlebacher et al. 1984). Infarct expansion predisposes to myocardial rupture and congestive heart failure (Eaton et al. 1979; Erlebacher et al. 1982; Jugdutt and Michorowski 1987; Schuster and Bulkley 1979). Increased wall stress as a result of infarct expansion leads to mechanical stretch-elicited local angiotensin II release and activation of a fetal gene programme (Sutton and Sharpe 2000). Local angiotensin II release promotes cardiomyocyte hypertrophy. In later stages of remodelling, interstitial fibrosis is induced. Transforming growth factor-β1 transforms fibroblasts into myofibroblasts and induces activation of tissue inhibitors of metalloproteinases and production of type I and type III collagen. The resultant interstitial fibrosis negatively affects the diastolic properties of the heart.
Post-infarct ejection fraction is lower in patients with low HDL cholesterol levels (Kempen et al. 1987; Wang et al. 1998). Although this could be due to differences in atherosclerosis or in the microvasculature or could be related to a decrease in infarct size due to beneficial effects of HDL in IRI, the possibility that HDL beneficially affects infarct expansion and ventricular remodelling should be considered. This can be investigated in models of permanent ligation of, e.g. the left anterior descending coronary artery. An attenuation of post-infarct left ventricular remodelling and improved infract healing was induced by infusion of rHDL once a week for 4 weeks in rats following ligation of the proximal left coronary artery (Kiya et al. 2009). Interestingly, rHDL-treated rats also showed an increase of phosphorylation of ERK1/2 in the left ventricular tissue, but not of p38 MAPK or JNK (Kiya et al. 2009). Gordts et al. (2013) have recently shown that human apo A-I gene transfer in C57BL/6 LDL receptor-deficient mice increases survival, decreases infarct expansion, attenuates left ventricular dilatation, and improves cardiac function following permanent ligation of the left anterior descending coronary artery. Gene transfer in C57BL/6 LDL receptor-deficient mice was performed with the E1E3E4-deleted adenoviral vector AdA-I, inducing hepatocyte-specific expression of human apo A-I, or with the control vector Adnull. A ligation of the left anterior descending coronary artery was performed 2 weeks after transfer or saline injection. Permanent ligation of the left anterior descending coronary artery excludes salutary coronary effects of HDL on myocardial salvage as observed in models of IRI (Theilmeier et al. 2006). Consequently, the infarct area and the area at risk were nearly identical at 24 h after ligation implying that the initial increase in loading conditions was not different between human apo A-I gene transfer myocardial infarction mice and control myocardial infarction mice. HDL cholesterol levels were persistently 1.5 times (p < 0.0001) higher in AdA-I mice compared to controls. Survival was increased (p < 0.01) in AdA-I myocardial infarction mice compared to control myocardial infarction mice during the 28-day follow-up period (hazard ratio for mortality 0.42; 95 % CI 0.24–0.76). Longitudinal morphometric analysis demonstrated attenuated infarct expansion (reduced infract length and increased infarct thickness of the infarct in AdA-I myocardial infarction mice) and inhibition of left ventricular dilatation in AdA-I myocardial infarction mice compared to controls. AdA-I transfer exerted immunomodulatory effects and increased neovascularisation in the infarct zone. Increased HDL after AdA-I transfer significantly improved systolic and diastolic cardiac function post-MI and led to a preservation of peripheral blood pressure.
The effects of AdA-I transfer on remodelling have significant consequences for the progressive development of heart failure. Ventricular dilatation increases the loading conditions of the heart. After all, ‘preload’ at the organ level corresponds to passive ventricular wall stress at the end of diastole (preloadLV = (EDPLV) (EDRLV)/2WTLV where EDP is end-diastolic pressure, EDR is end-diastolic radius, and WT is wall thickness). ‘Afterload’ at the organ level reflects myocardial wall stress during systolic ejection (afterloadLV = (SPLV) (SRLV)/2WTLV where SP is systolic pressure, SR is systolic radius, and WT is wall thickness). Thus, the attenuation of infarct expansion and ventricular dilatation induced by increased HDL is critical for long-term preservation of left ventricular function. Although an increase of end-diastolic volume may be physiologically beneficial in terms of the Frank–Starling mechanism, the increase in afterload in a larger ventricle is clearly not beneficial. The increase in afterload corresponds at the cellular level to an increase in tension that each muscle fibre must develop, and correspondingly, oxygen consumption increases. In addition, ventricular hypertrophy may be associated with an imbalance between the vascular and cardiomyocyte compartment in the myocardium (Shimizu et al. 2010; Tirziu et al. 2007). Increased oxygen consumption together with a decrease in relative vascularity may lead to tissue hypoxia, cardiomyocyte dysfunction, and late cardiomyocyte death. There was a trend for a higher capillary density and a higher relative vascularity at day 28 in AdA-I myocardial infarction mice compared to control myocardial infarction mice. Taken together, ventricular dilatation initially produced by an external stimulus (ligation of the left anterior descending coronary artery) induces a vicious cycle where an increase of loading conditions leads to an intrinsic pathological heart muscle (cardiomyopathy) characterised by cardiomyocyte death and dysfunction (reduced intrinsic myocardial contractility and relaxation), collagen deposition, and progressive dilatation of the heart. Although no follow-up was performed in the current study after 28 days, we speculate that differences in ventricular function between control MI mice and human apo A-I gene transfer MI mice will be more marked after longer follow-up.
Neovascularisation may play a role in scar formation and scar tissue remodelling (Wang et al. 2005). Increased neovascularisation was observed in the infarcts of AdA-I myocardial infarction mice at day 28 after gene transfer. HDL exerts potent effects on the endothelium. These effects occur via enhanced endothelial survival (Nofer et al. 2001), endothelial cell migration (Seetharam et al. 2006), and EPC-mediated repair (Feng et al. 2009a, b; Tso et al. 2006). Beneficial effects on EPCs may have contributed to improved infarct healing.
Taken together, attenuation of remodelling following human apo A-I gene transfer may be considered to constitute the cardiac equivalent of enhanced wound healing and tissue repair induced by HDL.
Conclusions
One of the pitfalls in the field of HDL-targeted interventions is the lack of selective HDL-raising drugs. The development of gene transfer technologies with a sufficiently high therapeutic index may pave the road for a selective and effective HDL-targeted therapeutic intervention (Jacobs et al. 2012; Van Craeyveld et al. 2010). Nathwani et al. (2011) performed a landmark study that demonstrated for the first time long-term expression of a transgene product at therapeutic levels following systemic hepatocyte-directed gene transfer in humans. In this study (Nathwani et al. 2011), haemophilia B was successfully treated with an adeno-associated viral serotype 8 human FIX expressing vector. These seminal data highlight that hepatocyte-directed adeno-associated viral serotype 8 gene transfer may become a clinical reality in the next decades. The recent positive assessment by the European Medicines Agency’s Committee for Medicinal Products for Human Use of the marketing authorisation for the first gene transfer product in Europe (alipogene tiparvovec (Glybera®)) highlights an important paradigm shift by regulatory agencies, as well as the biotechnological entrepreneurs and investors. Nevertheless, there is no short-term perspective for widespread clinical use of gene transfer technologies.
Three modalities discussed in this review are selective HDL-targeted therapies: infusion of rHDL, human apo A-I gene transfer, and topical HDL therapy. The main strategic questions with regard to HDL-targeted therapies are related to the choice of therapeutic areas in which a real clinical difference could be made. These questions are inextricably linked to the current state of the art of evidence-based medicine. Since a significant part of this review is focused on heart failure, we will illustrate this point on the basis of a very significant dichotomous classification of these patients.
Among patients with hospitalised heart failure, approximately 50 % have heart failure with reduced ejection fraction (HFrEF), and 50 % have heart failure with preserved ejection fraction (HFpEF) with the proportion of patients with HFpEF increasing with time (Liu et al. 2013; Vaduganathan and Fonarow 2013). The cutoff value for preserved ejection fraction is 0.5. Most clinical heart failure trials have been focused on patients with HFrEF. Inhibition of the renin-angiotensin-aldosterone and sympathetic nervous systems improves survival and decreases hospitalisations in patients with HFrEF (Reed et al. 2014). In contrast to these significant advances in treatment and reduction in mortality in patients with HFrEF, drug strategies with strong evidence in HFrEF have proved unsuccessful in HFpEF, and the mortality in patients with HFpEF has remained unchanged (Liu et al. 2013). HFpEF is a complex clinical syndrome that is characterised by classical heart failure symptoms with increased left ventricular filling pressures and preserved left ventricular ejection fraction. Increased end-diastolic ventricular stiffness is observed in HFpEF, but the exact mechanisms that induce HFpEF are unknown. This heart failure subtype disproportionately affects women and the elderly and is commonly associated with other cardiovascular comorbidities, such as hypertension and diabetes. Most therapeutic gains can likely be made in the field HFpEF. Interestingly, human apo A-I gene transfer has been shown to improve diastolic function in C57BL/6 LDL receptor-deficient mice (Gordts et al. 2012). The same strategic questions should also be raised in relation to the role of HDL in prevention and treatment of coronary artery disease. One might speculate that perspectives for an HDL-targeted therapy in patients with stable coronary artery disease are limited, whereas beneficial coronary and myocardial effects in the setting of acute coronary syndromes may still constitute a window of opportunity.
Abbreviations
- Apo:
-
Apolipoprotein
- AT1R:
-
Angiotensin II receptor, type 1
- BOEC:
-
Blood outgrowth endothelial cells
- CAV:
-
Cardiac allograft vasculopathy
- ECFCs:
-
Endothelial colony-forming cells
- eNOS:
-
Endothelial nitric oxide synthase
- EPC:
-
Endothelial progenitor cell
- ERK:
-
Extracellular signal-regulated kinase
- HDL:
-
High-density lipoprotein
- ICAM:
-
Intercellular adhesion molecule
- IDL:
-
Intermediate-density lipoprotein
- HFpEF:
-
Heart failure with preserved ejection fraction
- HFrEF:
-
Heart failure with reduced ejection fraction
- IL:
-
Interleukin
- IRI:
-
Ischaemia/reperfusion injury
- JNK:
-
c-Jun N-terminal kinase
- LDL:
-
Low-density lipoprotein
- L-NAME:
-
L-NG-nitroarginine methyl ester
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NO:
-
Nitric oxide
- p38 MAPK:
-
p38 mitogen-activated protein kinase
- PI3K:
-
Phosphoinositide 3-kinase
- S1P:
-
Sphingosine-1-phosphate
- STAT3:
-
Signal transducer and activator of transcription 3
- SR-BI:
-
Scavenger receptor class B, type I
- TBARS:
-
Thiobarbituric acid reactive substances
- TLR4:
-
Toll-like receptor 4
- TNF-α:
-
Tumour necrosis factor-α
- VCAM:
-
Vascular cell adhesion molecule
- VEGF:
-
Vascular endothelial growth factor
- VLDL:
-
Very-low-density lipoprotein
References
Abbott CA, Carrington AL, Ashe H, Bath S, Every LC, Griffiths J, Hann AW, Hussein A, Jackson N, Johnson KE, Ryder CH, Torkington R, Van Ross ER, Whalley AM, Widdows P, Williamson S, Boulton AJ (2002) The North-West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based patient cohort. Diabet Med 19:377–384
Abderrahmani A, Niederhauser G, Favre D, Abdelli S, Ferdaoussi M, Yang JY, Regazzi R, Widmann C, Waeber G (2007) Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 50:1304–1314
Alvarez A, Cerda-Nicolas M, Naim Abu Nabah Y, Mata M, Issekutz AC, Panes J, Lobb RR, Sanz MJ (2004) Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood 104:402–408
Ameli S, Hultgardh-Nilsson A, Cercek B, Shah PK, Forrester JS, Ageland H, Nilsson J (1994) Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation 90:1935–1941
Anversa P, Sonnenblick EH (1990) Ischemic cardiomyopathy: pathophysiologic mechanisms. Prog Cardiovasc Dis 33:49–70
Aoyama T, Yui Y, Morishita H, Kawai C (1990) Prostaglandin I2 half-life regulated by high density lipoprotein is decreased in acute myocardial infarction and unstable angina pectoris. Circulation 81:1784–1791
Babitt J, Trigatti B, Rigotti A, Smart EJ, Anderson RG, Xu S, Krieger M (1997) Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J Biol Chem 272:13242–13249
Baroukh N, Lopez CE, Saleh MC, Recalde D, Vergnes L, Ostos MA, Fiette L, Fruchart JC, Castro G, Zakin MM, Ochoa A (2004) Expression and secretion of human apolipoprotein A-I in the heart. FEBS Lett 557:39–44
Behrendt D, Ganz P (2002) Endothelial function. From vascular biology to clinical applications. Am J Cardiol 90:40L–48L
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953
Brutsaert DL (2003) Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev 83:59–115
Cai H (2005) NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res 96:818–822
Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ (2002) Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51:1938–1948
Calabresi L, Rossoni G, Gomaraschi M, Sisto F, Berti F, Franceschini G (2003) High-density lipoproteins protect isolated rat hearts from ischemia-reperfusion injury by reducing cardiac tumor necrosis factor-alpha content and enhancing prostaglandin release. Circ Res 92:330–337
Campeau L, Enjalbert M, Lesperance J, Bourassa MG, Kwiterovich P Jr, Wacholder S, Sniderman A (1984) The relation of risk factors to the development of atherosclerosis in saphenous-vein bypass grafts and the progression of disease in the native circulation. A study 10 years after aortocoronary bypass surgery. N Engl J Med 311:1329–1332
Chalupsky K, Cai H (2005) Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA 102:9056–9061
Cheetham C, Collis J, O'Driscoll G, Stanton K, Taylor R, Green D (2000) Losartan, an angiotensin type 1 receptor antagonist, improves endothelial function in non-insulin-dependent diabetes. J Am Coll Cardiol 36:1461–1466
Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, Dahlback B (2011) Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA 108:9613–9618
Cockerill GW, Huehns TY, Weerasinghe A, Stocker C, Lerch PG, Miller NE, Haskard DO (2001) Elevation of plasma high-density lipoprotein concentration reduces interleukin-1-induced expression of E-selectin in an in vivo model of acute inflammation. Circulation 103:108–112
Cohen RA (1993) Dysfunction of vascular endothelium in diabetes mellitus. Circulation 87:V67–V76
Cohn JN (1993) Post-MI remodeling. Clin Cardiol 16:II21–II24
Condorelli G, Morisco C, Stassi G, Notte A, Farina F, Sgaramella G, de Rienzo A, Roncarati R, Trimarco B, Lembo G (1999) Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation 99:3071–3078
Cong L, Chen K, Li J, Gao P, Li Q, Mi S, Wu X, Zhao AZ (2007) Regulation of adiponectin and leptin secretion and expression by insulin through a PI3K-PDE3B dependent mechanism in rat primary adipocytes. Biochem J 403:519–525
Cosson S, Kevorkian JP (2003) Left ventricular diastolic dysfunction: an early sign of diabetic cardiomyopathy? Diabetes Metab 29:455–466
De Geest B (2009) The origin of intimal smooth muscle cells: are we on a steady road back to the past? Cardiovasc Res 81:7–8
De Geest B, Zhao Z, Collen D, Holvoet P (1997) Effects of adenovirus-mediated human apo A-I gene transfer on neointima formation after endothelial denudation in apo E-deficient mice. Circulation 96:4349–4356
De Geest B, Stengel D, Landeloos M, Lox M, Le Gat L, Collen D, Holvoet P, Ninio E (2000) Effect of overexpression of human apo A-I in C57BL/6 and C57BL/6 apo E-deficient mice on 2 lipoprotein-associated enzymes, platelet-activating factor acetylhydrolase and paraoxonase. Comparison of adenovirus-mediated human apo A-I gene transfer and human apo A-I transgenesis. Arterioscler Thromb Vasc Biol 20:E68–E75
Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML (2003) Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci USA 100:2700–2705
Diegelmann RF, Evans MC (2004) Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 9:283–289
Dietrich H, Hu Y, Zou Y, Huemer U, Metzler B, Li C, Mayr M, Xu Q (2000) Rapid development of vein graft atheroma in ApoE-deficient mice. Am J Pathol 157:659–669
Eaton LW, Weiss JL, Bulkley BH, Garrison JB, Weisfeldt ML (1979) Regional cardiac dilatation after acute myocardial infarction: recognition by two-dimensional echocardiography. N Engl J Med 300:57–62
Erdbruegger U, Haubitz M, Woywodt A (2006) Circulating endothelial cells: a novel marker of endothelial damage. Clin Chim Acta 373:17–26
Erlebacher JA, Weiss JL, Eaton LW, Kallman C, Weisfeldt ML, Bulkley BH (1982) Late effects of acute infarct dilation on heart size: a two dimensional echocardiographic study. Am J Cardiol 49:1120–1126
Erlebacher JA, Weiss JL, Weisfeldt ML, Bulkley BH (1984) Early dilation of the infarcted segment in acute transmural myocardial infarction: role of infarct expansion in acute left ventricular enlargement. J Am Coll Cardiol 4:201–208
Feng Y, Jacobs F, Van Craeyveld E, Brunaud C, Snoeys J, Tjwa M, Van Linthout S, De Geest B (2008) Human ApoA-I transfer attenuates transplant arteriosclerosis via enhanced incorporation of bone marrow-derived endothelial progenitor cells. Arterioscler Thromb Vasc Biol 28:278–283
Feng Y, Van Craeyveld E, Jacobs F, Lievens J, Snoeys J, De Geest B (2009a) Wild-type apo A-I and apo A-I(Milano) gene transfer reduce native and transplant arteriosclerosis to a similar extent. J Mol Med 87:287–297
Feng Y, van Eck M, Van Craeyveld E, Jacobs F, Carlier V, Van Linthout S, Erdel M, Tjwa M, De Geest B (2009b) Critical role of scavenger receptor-BI-expressing bone marrow-derived endothelial progenitor cells in the attenuation of allograft vasculopathy after human apo A-I transfer. Blood 113:755–764
Feng Y, Gordts SC, Chen F, Hu Y, Van Craeyveld E, Jacobs F, Carlier V, Zhang Z, Xu Q, Ni Y, De Geest B (2011) Topical HDL administration reduces vein graft atherosclerosis in apo E deficient mice. Atherosclerosis 214:271–278
Fitzgibbon GM, Kafka HP, Leach AJ, Keon WJ, Hooper GD, Burton JR (1996) Coronary bypass graft fate and patient outcome: angiographic follow-up of 5,065 grafts related to survival and reoperation in 1,388 patients during 25 years. J Am Coll Cardiol 28:616–626
Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q (2008) Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein E-deficient mice. Circulation 117:1856–1863
Freire CM, Moura AL, Barbosa Mde M, Machado LJ, Nogueira AI, Ribeiro-Oliveira A Jr (2007) Left ventricle diastolic dysfunction in diabetes: an update. Arq Bras Endocrinol Metabol 51:168–175
Frias MA, James RW, Gerber-Wicht C, Lang U (2009) Native and reconstituted HDL activate Stat3 in ventricular cardiomyocytes via ERK1/2: role of sphingosine-1-phosphate. Cardiovasc Res 82:313–323
Frias MA, Lang U, Gerber-Wicht C, James RW (2010) Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc Res 85:118–126
Frias MA, Pedretti S, Hacking D, Somers S, Lacerda L, Opie LH, James RW, Lecour S (2013) HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis 228:110–116
Friedrich EB, Walenta K, Scharlau J, Nickenig G, Werner N (2006) CD34-/CD133+/VEGFR-2+ endothelial progenitor cell subpopulation with potent vasoregenerative capacities. Circ Res 98:e20–e25
Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P (2000) Myocardial cell death in human diabetes. Circ Res 87:1123–1132
Fryirs MA, Barter PJ, Appavoo M, Tuch BE, Tabet F, Heather AK, Rye KA (2010) Effects of high-density lipoproteins on pancreatic beta-cell insulin secretion. Arterioscler Thromb Vasc Biol 30:1642–1648
Galiano RD, Jt M, Dobryansky M, Levine JP, Gurtner GC (2004a) Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 12:485–492
Galiano RD, Tepper OM, Pelo CR, Bhatt KA, Callaghan M, Bastidas N, Bunting S, Steinmetz HG, Gurtner GC (2004b) Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am J Pathol 164:1935–1947
Galkowska H, Wojewodzka U, Olszewski WL (2006) Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen 14:558–565
Gibran NS, Jang YC, Isik FF, Greenhalgh DG, Muffley LA, Underwood RA, Usui ML, Larsen J, Smith DG, Bunnett N, Ansel JC, Olerud JE (2002) Diminished neuropeptide levels contribute to the impaired cutaneous healing response associated with diabetes mellitus. J Surg Res 108:122–128
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB (2013) Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127:e6–e245
Goldman S, Zadina K, Moritz T, Ovitt T, Sethi G, Copeland JG, Thottapurathu L, Krasnicka B, Ellis N, Anderson RJ, Henderson W (2004) Long-term patency of saphenous vein and left internal mammary artery grafts after coronary artery bypass surgery: results from a Department of Veterans Affairs Cooperative Study. J Am Coll Cardiol 44:2149–2156
Gordts SC, Van Craeyveld E, Muthuramu I, Singh N, Jacobs F, De Geest B (2012) Lipid lowering and HDL raising gene transfer increase endothelial progenitor cells, enhance myocardial vascularity, and improve diastolic function. PLoS ONE 7:e46849
Gordts SC, Muthuramu I, Nefyodova E, Jacobs F, Van Craeyveld E, De Geest B (2013) Beneficial effects of selective HDL raising gene transfer on survival, cardiac remodelling, and cardiac function after myocardial infarction in mice. Gene Ther 20:1053–1061
Gordts SC, Muthuramu I, Amin R, Jacobs F, De Geest B (2014) The impact of lipoproteins on wound healing: topical HDL therapy corrects delayed wound healing in apolipoprotein E deficient mice. Pharmaceuticals 7(4):419–432
Gragasin FS, Xu Y, Arenas IA, Kainth N, Davidge ST (2003) Estrogen reduces angiotensin II-induced nitric oxide synthase and NAD(P)H oxidase expression in endothelial cells. Arterioscler Thromb Vasc Biol 23:38–44
Greenhalgh DG (2003) Wound healing and diabetes mellitus. Clin Plast Surg 30:37–45
Gu SS, Shi N, Wu MP (2007) The protective effect of ApolipoproteinA-I on myocardial ischemia-reperfusion injury in rats. Life Sci 81:702–709
Guo Z, Xia Z, Jiang J, McNeill JH (2007) Downregulation of NADPH oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by N-acetyl-L-cysteine. Am J Physiol Heart Circ Physiol 292:H1728–H1736
Hagensen MK, Shim J, Thim T, Falk E, Bentzon JF (2010) Circulating endothelial progenitor cells do not contribute to plaque endothelium in murine atherosclerosis. Circulation 121:898–905
Han R, Lai R, Ding Q, Wang Z, Luo X, Zhang Y, Cui G, He J, Liu W, Chen Y (2007) Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia 50:1960–1968
He S, Li M, Ma X, Lin J, Li D (2010) CD4+CD25+Foxp3+ regulatory T cells protect the proinflammatory activation of human umbilical vein endothelial cells. Arterioscler Thromb Vasc Biol 30:2621–2630
Hedrick CC, Thorpe SR, Fu MX, Harper CM, Yoo J, Kim SM, Wong H, Peters AL (2000) Glycation impairs high-density lipoprotein function. Diabetologia 43:312–320
Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T (2003) Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med 348:593–600
Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T (2001) Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88:E14–E22
Hirschi KK, Ingram DA, Yoder MC (2008) Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol 28:1584–1595
Hodgkinson CP, Laxton RC, Patel K, Ye S (2008) Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler Thromb Vasc Biol 28:2275–2281
Hodroj W, Legedz L, Foudi N, Cerutti C, Bourdillon MC, Feugier P, Beylot M, Randon J, Bricca G (2007) Increased insulin-stimulated expression of arterial angiotensinogen and angiotensin type 1 receptor in patients with type 2 diabetes mellitus and atheroma. Arterioscler Thromb Vasc Biol 27:525–531
Hu Y, Zou Y, Dietrich H, Wick G, Xu Q (1999) Inhibition of neointima hyperplasia of mouse vein grafts by locally applied suramin. Circulation 100:861–868
Hu Y, Davison F, Zhang Z, Xu Q (2003) Endothelial replacement and angiogenesis in arteriosclerotic lesions of allografts are contributed by circulating progenitor cells. Circulation 108:3122–3127
Hughes SJ, Powis SH, Press M (2001) Surviving native beta-cells determine outcome of syngeneic intraportal islet transplantation. Cell Transplant 10:145–151
Hyka N, Dayer JM, Modoux C, Kohno T, Edwards CK 3rd, Roux-Lombard P, Burger D (2001) Apolipoprotein A-I inhibits the production of interleukin-1beta and tumor necrosis factor-alpha by blocking contact-mediated activation of monocytes by T lymphocytes. Blood 97:2381–2389
Imaizumi S, Miura S, Nakamura K, Kiya Y, Uehara Y, Zhang B, Matsuo Y, Urata H, Ideishi M, Rye KA, Sata M, Saku K (2008) Antiarrhythmogenic effect of reconstituted high-density lipoprotein against ischemia/reperfusion in rats. J Am Coll Cardiol 51:1604–1612
Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, Pollok K, Ferkowicz MJ, Gilley D, Yoder MC (2004) Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 104:2752–2760
Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD (2013) Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 114(2):266–282
Iwaoka M, Obata JE, Abe M, Nakamura T, Kitta Y, Kodama Y, Kawabata K, Takano H, Fujioka D, Saito Y, Kobayashi T, Hasebe H, Kugiyama K (2007) Association of low serum levels of apolipoprotein A-I with adverse outcomes in patients with nonischemic heart failure. J Card Fail 13:247–253
Jacobs F, Gordts S, Muthuramu I, De Geest B (2012) The liver as a target organ for gene therapy: state of the art, challenges, and future perspectives. Pharmaceuticals 5:1372–1392
Jugdutt BI, Michorowski BL (1987) Role of infarct expansion in rupture of the ventricular septum after acute myocardial infarction: a two-dimensional echocardiographic study. Clin Cardiol 10:641–652
Karliner JS (2013) Sphingosine kinase and sphingosine 1-phosphate in the heart: a decade of progress. Biochim Biophys Acta 1831:203–212
Kempen HJ, van Gent CM, Buytenhek R, Buis B (1987) Association of cholesterol concentrations in low-density lipoprotein, high-density lipoprotein, and high-density lipoprotein subfractions, and of apolipoproteins AI and AII, with coronary stenosis and left ventricular function. J Lab Clin Med 109:19–26
Kiya Y, Miura S, Imaizumi S, Uehara Y, Matsuo Y, Abe S, Jimi S, Urata H, Rye KA, Saku K (2009) Reconstituted high-density lipoprotein attenuates postinfarction left ventricular remodeling in rats. Atherosclerosis 203:137–144
Kota SK, Kota SK, Jammula S, Panda S, Modi KD (2011) Effect of diabetes on alteration of metabolism in cardiac myocytes: therapeutic implications. Diabetes Technol Ther 13:1155–1160
Lakhani SA, Masud A, Kuida K, Porter GA Jr, Booth CJ, Mehal WZ, Inayat I, Flavell RA (2006) Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311:847–851
Lee WJ, Ou HC, Hsu WC, Chou MM, Tseng JJ, Hsu SL, Tsai KL, Sheu WH (2010) Ellagic acid inhibits oxidized LDL-mediated LOX-1 expression, ROS generation, and inflammation in human endothelial cells. J Vasc Surg 52:1290–1300
Li D, Saldeen T, Romeo F, Mehta JL (2000) Oxidized LDL upregulates angiotensin II type 1 receptor expression in cultured human coronary artery endothelial cells: the potential role of transcription factor NF-kappaB. Circulation 102:1970–1976
Li J, Jin HB, Sun YM, Su Y, Wang LF (2010a) KB-R7943 inhibits high glucose-induced endothelial ICAM-1 expression and monocyte-endothelial adhesion. Biochem Biophys Res Commun 392:516–519
Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T (2010b) Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59:2033–2042
Liu Y, Haddad T, Dwivedi G (2013) Heart failure with preserved ejection fraction: current understanding and emerging concepts. Curr Opin Cardiol 28:187–196
Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H (2002) Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia 45:1011–1016
Lowes BD, Zolty R, Minobe WA, Robertson AD, Leach S, Hunter L, Bristow MR (2006) Serial gene expression profiling in the intact human heart. J Heart Lung Transplant 25:579–588
Mackness MI, Arrol S, Durrington PN (1991) Paraoxonase prevents accumulation of lipoperoxides in low-density lipoprotein. FEBS Lett 286:152–154
Marchesi M, Booth EA, Davis T, Bisgaier CL, Lucchesi BR (2004) Apolipoprotein A-IMilano and 1-palmitoyl-2-oleoyl phosphatidylcholine complex (ETC-216) protects the in vivo rabbit heart from regional ischemia-reperfusion injury. J Pharmacol Exp Ther 311:1023–1031
Marchesi M, Booth EA, Rossoni G, Garcia RA, Hill KR, Sirtori CR, Bisgaier CL, Lucchesi BR (2008) Apolipoprotein A-IMilano/POPC complex attenuates post-ischemic ventricular dysfunction in the isolated rabbit heart. Atherosclerosis 197:572–578
Matricali GA, Dereymaeker G, Muls E, Flour M, Mathieu C (2007) Economic aspects of diabetic foot care in a multidisciplinary setting: a review. Diabetes Metab Res Rev 23:339–347
Means CK, Brown JH (2009) Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc Res 82:193–200
Means CK, Miyamoto S, Chun J, Brown JH (2008) S1P1 receptor localization confers selectivity for Gi-mediated cAMP and contractile responses. J Biol Chem 283:11954–11963
Mehra MR, Uber PA, Lavie CJ, Milani RV, Park MH, Ventura HO (2009) High-density lipoprotein cholesterol levels and prognosis in advanced heart failure. J Heart Lung Transplant 28:876–880
Mochizuki S, Okumura M, Tanaka F, Sato T, Kagami A, Tada N, Nagano M (1991) Ischemia-reperfusion arrhythmias and lipids: effect of human high- and low-density lipoproteins on reperfusion arrhythmias. Cardiovasc Drugs Ther 5(Suppl 2):269–276
Montanari D, Yin H, Dobrzynski E, Agata J, Yoshida H, Chao J, Chao L (2005) Kallikrein gene delivery improves serum glucose and lipid profiles and cardiac function in streptozotocin-induced diabetic rats. Diabetes 54:1573–1580
Morel S, Frias MA, Rosker C, James RW, Rohr S, Kwak BR (2012) The natural cardioprotective particle HDL modulates connexin43 gap junction channels. Cardiovasc Res 93:41–49
Mosterd A, Hoes AW (2007) Clinical epidemiology of heart failure. Heart 93:1137–1146
Mosterd A, Hoes AW, de Bruyne MC, Deckers JW, Linker DT, Hofman A, Grobbee DE (1999) Prevalence of heart failure and left ventricular dysfunction in the general population. The Rotterdam study. Eur Heart J 20:447–455
Muller IS, de Grauw WJ, van Gerwen WH, Bartelink ML, van Den Hoogen HJ, Rutten GE (2002) Foot ulceration and lower limb amputation in type 2 diabetic patients in dutch primary health care. Diabetes Care 25:570–574
Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, Ui M, Okajima F (2000) Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. Biochem J 352(Pt 3):809–815
Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, O'Beirne J, Smith K, Pasi J, Glader B, Rustagi P, Ng CY, Kay MA, Zhou J, Spence Y, Morton CL, Allay J, Coleman J, Sleep S, Cunningham JM, Srivastava D, Basner-Tschakarjan E, Mingozzi F, High KA, Gray JT, Reiss UM, Nienhuis AW, Davidoff AM (2011) Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365:2357–2365
Nishida M, Springhorn JP, Kelly RA, Smith TW (1993) Cell-cell signaling between adult rat ventricular myocytes and cardiac microvascular endothelial cells in heterotypic primary culture. J Clin Invest 91:1934–1941
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Nofer JR, Levkau B, Wolinska I, Junker R, Fobker M, von Eckardstein A, Seedorf U, Assmann G (2001) Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem 276:34480–34485
Nofer JR, van der Giet M, Tolle M, Wolinska I, von Wnuck LK, Baba HA, Tietge UJ, Godecke A, Ishii I, Kleuser B, Schafers M, Fobker M, Zidek W, Assmann G, Chun J, Levkau B (2004) HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest 113:569–581
Nyby MD, Abedi K, Smutko V, Eslami P, Tuck ML (2007) Vascular Angiotensin type 1 receptor expression is associated with vascular dysfunction, oxidative stress and inflammation in fructose-fed rats. Hypertens Res 30:451–457
Oak JH, Cai H (2007) Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 56:118–126
Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank G, Motley E, Eguchi S (2006) Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension 48:534–540
Ou J, Wang J, Xu H, Ou Z, Sorci-Thomas MG, Jones DW, Signorino P, Densmore JC, Kaul S, Oldham KT, Pritchard KA Jr (2005) Effects of D-4F on vasodilation and vessel wall thickness in hypercholesterolemic LDL receptor-null and LDL receptor/apolipoprotein A-I double-knockout mice on Western diet. Circ Res 97:1190–1197
Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, Ohmoto Y, Nakamura T, Yamashita S, Funahashi T, Matsuzawa Y (2000) Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 102:1296–1301
Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, Ishigami M, Kuriyama H, Kishida K, Nishizawa H, Hotta K, Muraguchi M, Ohmoto Y, Yamashita S, Funahashi T, Matsuzawa Y (2001) Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 103:1057–1063
Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, Chan L, Goldstein BJ, Scalia R (2007) Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest 117:1718–1726
Palomer X, Salvado L, Barroso E, Vazquez-Carrera M (2013) An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol 168:3160–3172
Parini P, Johansson L, Broijersen A, Angelin B, Rudling M (2006) Lipoprotein profiles in plasma and interstitial fluid analyzed with an automated gel-filtration system. Eur J Clin Invest 36:98–104
Pereira RI, Draznin B (2005) Inhibition of the phosphatidylinositol 3′-kinase signaling pathway leads to decreased insulin-stimulated adiponectin secretion from 3T3-L1 adipocytes. Metabolism 54:1636–1643
Persegol L, Verges B, Foissac M, Gambert P, Duvillard L (2006) Inability of HDL from type 2 diabetic patients to counteract the inhibitory effect of oxidised LDL on endothelium-dependent vasorelaxation. Diabetologia 49:1380–1386
Peterson SJ, Husney D, Kruger AL, Olszanecki R, Ricci F, Rodella LF, Stacchiotti A, Rezzani R, McClung JA, Aronow WS, Ikehara S, Abraham NG (2007) Long-term treatment with the apolipoprotein A1 mimetic peptide increases antioxidants and vascular repair in type I diabetic rats. J Pharmacol Exp Ther 322:514–520
Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, Abraham NG (2008) L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res 49:1658–1669
Piga R, Naito Y, Kokura S, Handa O, Yoshikawa T (2007) Short-term high glucose exposure induces monocyte-endothelial cells adhesion and transmigration by increasing VCAM-1 and MCP-1 expression in human aortic endothelial cells. Atherosclerosis 193:328–334
Pinney SP, Mancini D (2004) Cardiac allograft vasculopathy: advances in understanding its pathophysiology, prevention, and treatment. Curr Opin Cardiol 19:170–176
Raev DC (1994) Which left ventricular function is impaired earlier in the evolution of diabetic cardiomyopathy? An echocardiographic study of young type I diabetic patients. Diabetes Care 17:633–639
Rahmani M, Cruz RP, Granville DJ, McManus BM (2006) Allograft vasculopathy versus atherosclerosis. Circ Res 99:801–815
Raja SG, Haider Z, Ahmad M, Zaman H (2004) Saphenous vein grafts: to use or not to use? Heart Lung Circ 13:403–409
Ramaciotti C, Sharkey A, McClellan G, Winegrad S (1992) Endothelial cells regulate cardiac contractility. Proc Natl Acad Sci USA 89:4033–4036
Rathi S, Deedwania PC (2012) The epidemiology and pathophysiology of heart failure. Med Clin North Am 96:881–890
Reed BN, Rodgers JE, Sueta CA (2014) Polypharmacy in heart failure: drugs to use and avoid. Heart Fail Clin 10(4):577–590
Rehman J, Li J, Orschell CM, March KL (2003) Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 107:1164–1169
Richardson MR, Yoder MC (2011) Endothelial progenitor cells: quo vadis? J Mol Cell Cardiol 50(2):266–272
Rius C, Abu-Taha M, Hermenegildo C, Piqueras L, Cerda-Nicolas JM, Issekutz AC, Estan L, Cortijo J, Morcillo EJ, Orallo F, Sanz MJ (2010) Trans- but not cis-resveratrol impairs angiotensin-II-mediated vascular inflammation through inhibition of NF-kappaB activation and peroxisome proliferator-activated receptor-gamma upregulation. J Immunol 185:3718–3727
Roger VL, Weston SA, Redfield MM, Hellermann-Homan JP, Killian J, Yawn BP, Jacobsen SJ (2004) Trends in heart failure incidence and survival in a community-based population. JAMA 292:344–350
Rossoni G, Gomaraschi M, Berti F, Sirtori CR, Franceschini G, Calabresi L (2004) Synthetic high-density lipoproteins exert cardioprotective effects in myocardial ischemia/reperfusion injury. J Pharmacol Exp Ther 308:79–84
Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A (1972) New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 30:595–602
Rutti S, Ehses JA, Sibler RA, Prazak R, Rohrer L, Georgopoulos S, Meier DT, Niclauss N, Berney T, Donath MY, von Eckardstein A (2009) Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 150:4521–4530
Sattler K, Levkau B (2009) Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc Res 82:201–211
Sattler KJ, Elbasan S, Keul P, Elter-Schulz M, Bode C, Graler MH, Brocker-Preuss M, Budde T, Erbel R, Heusch G, Levkau B (2010) Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol 105:821–832
Savvatis K, van Linthout S, Miteva K, Pappritz K, Westermann D, Schefold JC, Fusch G, Weithauser A, Rauch U, Becher PM, Klingel K, Ringe J, Kurtz A, Schultheiss HP, Tschope C (2012) Mesenchymal stromal cells but not cardiac fibroblasts exert beneficial systemic immunomodulatory effects in experimental myocarditis. PLoS ONE 7:e41047
Schulz R, Heusch G (2004) Connexin 43 and ischemic preconditioning. Cardiovasc Res 62:335–344
Schulz R, Boengler K, Totzeck A, Luo Y, Garcia-Dorado D, Heusch G (2007) Connexin 43 in ischemic pre- and postconditioning. Heart Fail Rev 12:261–266
Schuster EH, Bulkley BH (1979) Expansion of transmural myocardial infarction: a pathophysiologic factor in cardiac rupture. Circulation 60:1532–1538
Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, Cummings ML, Kitchens RL, Marcel YL, Rader DJ, Shaul PW (2006) High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res 98:63–72
Shah PK, Amin J (1992) Low high density lipoprotein level is associated with increased restenosis rate after coronary angioplasty. Circulation 85:1279–1285
Shen X, Zheng S, Metreveli NS, Epstein PN (2006) Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 55:798–805
Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, Tateno K, Moriya J, Yokoyama M, Nojima A, Koh GY, Akazawa H, Shiojima I, Kahn CR, Abel ED, Komuro I (2010) Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest 120:1506–1514
Shinomiya S, Naraba H, Ueno A, Utsunomiya I, Maruyama T, Ohuchida S, Ushikubi F, Yuki K, Narumiya S, Sugimoto Y, Ichikawa A, Oh-ishi S (2001) Regulation of TNFalpha and interleukin-10 production by prostaglandins I(2) and E(2): studies with prostaglandin receptor-deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem Pharmacol 61:1153–1160
Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341:738–746
Singh N, Van Craeyveld E, Tjwa M, Ciarka A, Emmerechts J, Droogne W, Gordts SC, Carlier V, Jacobs F, Fieuws S, Vanhaecke J, Van Cleemput J, De Geest B (2012) Circulating apoptotic endothelial cells and apoptotic endothelial microparticles independently predict the presence of cardiac allograft vasculopathy. J Am Coll Cardiol 60:324–331
Sloop CH, Dory L, Roheim PS (1987) Interstitial fluid lipoproteins. J Lipid Res 28:225–237
Sorrentino SA, Besler C, Rohrer L, Meyer M, Heinrich K, Bahlmann FH, Mueller M, Horvath T, Doerries C, Heinemann M, Flemmer S, Markowski A, Manes C, Bahr MJ, Haller H, von Eckardstein A, Drexler H, Landmesser U (2009) Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 121(1):110–122
Suh W, Kim KL, Kim JM, Shin IS, Lee YS, Lee JY, Jang HS, Lee JS, Byun J, Choi JH, Jeon ES, Kim DK (2005) Transplantation of endothelial progenitor cells accelerates dermal wound healing with increased recruitment of monocytes/macrophages and neovascularization. Stem Cells 23:1571–1578
Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G (1996) Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med 184:1331–1341
Sutton MG, Sharpe N (2000) Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101:2981–2988
Suzuki K, Murtuza B, Sammut IA, Latif N, Jayakumar J, Smolenski RT, Kaneda Y, Sawa Y, Matsuda H, Yacoub MH (2002) Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia-reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation 106:I270–I276
Sztalryd C, Kraemer FB (1995) Regulation of hormone-sensitive lipase in streptozotocin-induced diabetic rats. Metabolism 44:1391–1396
Takakura N, Watanabe T, Suenobu S, Yamada Y, Noda T, Ito Y, Satake M, Suda T (2000) A role for hematopoietic stem cells in promoting angiogenesis. Cell 102:199–209
Tao R, Hoover HE, Honbo N, Kalinowski M, Alano CC, Karliner JS, Raffai R (2010) High-density lipoprotein determines adult mouse cardiomyocyte fate after hypoxia-reoxygenation through lipoprotein-associated sphingosine 1-phosphate. Am J Physiol Heart Circ Physiol 298:H1022–H1028
Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, Schober O, Hildebrand R, Schulz R, Heusch G, Haude M, von Wnuck LK, Herzog C, Schmitz M, Erbel R, Chun J, Levkau B (2006) High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114:1403–1409
Thule PM, Campbell AG, Kleinhenz DJ, Olson DE, Boutwell JJ, Sutliff RL, Hart CM (2006) Hepatic insulin gene therapy prevents deterioration of vascular function and improves adipocytokine profile in STZ-diabetic rats. Am J Physiol Endocrinol Metab 290:E114–E122
Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, Roncal C, Eriksson U, Fu Q, Elfenbein A, Hall AE, Carmeliet P, Moons L, Simons M (2007) Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest 117:3188–3197
Tolle M, Pawlak A, Schuchardt M, Kawamura A, Tietge UJ, Lorkowski S, Keul P, Assmann G, Chun J, Levkau B, van der Giet M, Nofer JR (2008) HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler Thromb Vasc Biol 28:1542–1548
Toriseva M, Kahari VM (2009) Proteinases in cutaneous wound healing. Cell Mol Life Sci 66:203–224
Tricot O, Mallat Z, Heymes C, Belmin J, Leseche G, Tedgui A (2000) Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation 101:2450–2453
Tschope C, Walther T, Escher F, Spillmann F, Du J, Altmann C, Schimke I, Bader M, Sanchez-Ferrer CF, Schultheiss HP, Noutsias M (2005) Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. Faseb J 19:2057–2059
Tso C, Martinic G, Fan WH, Rogers C, Rye KA, Barter PJ (2006) High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler Thromb Vasc Biol 26:1144–1149
Tsuruoka H, Khovidhunkit W, Brown BE, Fluhr JW, Elias PM, Feingold KR (2002) Scavenger receptor class B type I is expressed in cultured keratinocytes and epidermis. Regulation in response to changes in cholesterol homeostasis and barrier requirements. J Biol Chem 277:2916–2922
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S (2003) PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 9:352–355
Uchiyama T, Engelman RM, Maulik N, Das DK (2004) Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 109:3042–3049
Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S (2005) Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol 39:733–742
Vaduganathan M, Fonarow GC (2013) Epidemiology of hospitalized heart failure: differences and similarities between patients with reduced versus preserved ejection fraction. Heart Fail Clin 9:271–276, v
Van Craeyveld E, Gordts S, Jacobs F, De Geest B (2010) Gene therapy to improve high-density lipoprotein metabolism and function. Curr Pharm Des 16:1531–1544
Van Linthout S, Riad A, Dhayat N, Spillmann F, Du J, Dhayat S, Westermann D, Hilfiker-Kleiner D, Noutsias M, Laufs U, Schultheiss HP, Tschope C (2007) Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia 50:1977–1986
Van Linthout S, Spillmann F, Riad A, Trimpert C, Lievens J, Meloni M, Escher F, Filenberg E, Demir O, Li J, Shakibaei M, Schimke I, Staudt A, Felix SB, Schultheiss HP, De Geest B, Tschope C (2008) Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation 117:1563–1573
Van Linthout S, Spillmann F, Lorenz M, Meloni M, Jacobs F, Egorova M, Stangl V, De Geest B, Schultheiss HP, Tschope C (2009) Vascular-protective effects of high-density lipoprotein include the downregulation of the angiotensin II type 1 receptor. Hypertension 53:682–687
Van Linthout S, Foryst-Ludwig A, Spillmann F, Peng J, Feng Y, Meloni M, Van Craeyveld E, Kintscher U, Schultheiss HP, De Geest B, Tschope C (2010a) Impact of HDL on adipose tissue metabolism and adiponectin expression. Atherosclerosis 210:438–444
Van Linthout S, Spillmann F, Schultheiss HP, Tschope C (2010b) High-density lipoprotein at the interface of type 2 diabetes mellitus and cardiovascular disorders. Curr Pharm Des 16:1504–1516
Van Linthout S, Spillmann F, Graiani G, Miteva K, Peng J, Van Craeyveld E, Meloni M, Tolle M, Escher F, Subasiguller A, Doehner W, Quaini F, De Geest B, Schultheiss HP, Tschope C (2011) Down-regulation of endothelial TLR4 signalling after apo A-I gene transfer contributes to improved survival in an experimental model of lipopolysaccharide-induced inflammation. J Mol Med (Berl) 89:151–160
Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, Dimmeler S (2001) Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation 103:2885–2890
Vassalli G, Gallino A, Weis M, von Scheidt W, Kappenberger L, von Segesser LK, Goy JJ (2003) Alloimmunity and nonimmunologic risk factors in cardiac allograft vasculopathy. Eur Heart J 24:1180–1188
Velagaleti RS, Massaro J, Vasan RS, Robins SJ, Kannel WB, Levy D (2009) Relations of lipid concentrations to heart failure incidence: the Framingham Heart Study. Circulation 120:2345–2351
Verges B, Petit JM, Duvillard L, Dautin G, Florentin E, Galland F, Gambert P (2006) Adiponectin is an important determinant of apoA-I catabolism. Arterioscler Thromb Vasc Biol 26:1364–1369
Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM (2002) Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation 105:3017–3024
Wang TD, Wu CC, Chen WJ, Lee CM, Chen MF, Liau CS, Sung FC, Lee YT (1998) Dyslipidemias have a detrimental effect on left ventricular systolic function in patients with a first acute myocardial infarction. Am J Cardiol 81:531–537
Wang B, Ansari R, Sun Y, Postlethwaite AE, Weber KT, Kiani MF (2005) The scar neovasculature after myocardial infarction in rats. Am J Physiol Heart Circ Physiol 289:H108–H113
Wang W, Xu H, Shi Y, Nandedkar S, Zhang H, Gao H, Feroah T, Weihrauch D, Schulte ML, Jones DW, Jarzembowski J, Sorci-Thomas M, Pritchard KA Jr (2010) Genetic deletion of apolipoprotein A-I increases airway hyperresponsiveness, inflammation, and collagen deposition in the lung. J Lipid Res 51:2560–2570
Werner N, Priller J, Laufs U, Endres M, Bohm M, Dirnagl U, Nickenig G (2002) Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition. Arterioscler Thromb Vasc Biol 22:1567–1572
Werner N, Junk S, Laufs U, Link A, Walenta K, Bohm M, Nickenig G (2003) Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res 93:e17–e24
Westermann D, Rutschow S, Van Linthout S, Linderer A, Bucker-Gartner C, Sobirey M, Riad A, Pauschinger M, Schultheiss HP, Tschope C (2006) Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 49:2507–2513
Westermann D, Rutschow S, Jager S, Linderer A, Anker S, Riad A, Unger T, Schultheiss HP, Pauschinger M, Tschope C (2007a) Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes 56:641–646
Westermann D, Van Linthout S, Dhayat S, Dhayat N, Escher F, Bucker-Gartner C, Spillmann F, Noutsias M, Riad A, Schultheiss HP, Tschope C (2007b) Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes 56:1834–1841
Westermann D, Van Linthout S, Dhayat S, Dhayat N, Schmidt A, Noutsias M, Song XY, Spillmann F, Riad A, Schultheiss HP, Tschope C (2007c) Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 102:500–507
Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C (2011) Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 4:44–52
Whiteman EL, Cho H, Birnbaum MJ (2002) Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab 13:444–451
Wilhelm AJ, Zabalawi M, Owen JS, Shah D, Grayson JM, Major AS, Bhat S, Gibbs DP Jr, Thomas MJ, Sorci-Thomas MG (2010) Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr-/-, ApoA-I-/- mice. J Biol Chem 285(46):36158–36169
Wold LE, Ceylan-Isik AF, Fang CX, Yang X, Li SY, Sreejayan N, Privratsky JR, Ren J (2006) Metallothionein alleviates cardiac dysfunction in streptozotocin-induced diabetes: role of Ca2+ cycling proteins, NADPH oxidase, poly(ADP-Ribose) polymerase and myosin heavy chain isozyme. Free Radic Biol Med 40:1419–1429
Xu Q, Zhang Z, Davison F, Hu Y (2003) Circulating progenitor cells regenerate endothelium of vein graft atherosclerosis, which is diminished in ApoE-deficient mice. Circ Res 93:e76–e86
Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, Hanley A, Isner JM, Losordo DW (2005) Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 111:2073–2085
Young NL, Lopez DR, McNamara DJ (1988) Contributions of absorbed dietary cholesterol and cholesterol synthesized in small intestine to hypercholesterolemia in diabetic rats. Diabetes 37:1151–1156
Zacharowski K, Olbrich A, Piper J, Hafner G, Kondo K, Thiemermann C (1999) Selective activation of the prostanoid EP(3) receptor reduces myocardial infarct size in rodents. Arterioscler Thromb Vasc Biol 19:2141–2147
Zhang Y, Peng T, Zhu H, Zheng X, Zhang X, Jiang N, Cheng X, Lai X, Shunnar A, Singh M, Riordan N, Bogin V, Tong N, Min WP (2010) Prevention of hyperglycemia-induced myocardial apoptosis by gene silencing of Toll-like receptor-4. J Transl Med 8:133
Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL (2004) Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest 114:529–541
Acknowledgments
Dr. Sophie Van Linthout received grant support from the European Foundation for the Study of Diabetes (EFSD). Research in the labs of Dr. Frias was supported by grants from the Fondation Gustave et Simone Prévot, Wolfermann Nageli Stiftung, Novartis Consumable Foundation, Jubiläumsstiftung, and Fondation pour la luttre contre le cancer et investigations médico-biologiques. Dr. Bart De Geest received grant support of the FWO-Vlaanderen (grants G.0529.10N and G0A3114N) and of the KU Leuven (grant OT/13/090). Dr. Frias and Dr. De Geest participated in and received financial support from COST Action BM0904 ‘HDL—from biological understanding to clinical exploitation’.
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Van Linthout, S., Frias, M., Singh, N., De Geest, B. (2015). Therapeutic Potential of HDL in Cardioprotection and Tissue Repair. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)