
Abstract
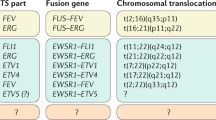
Sarcomas are a heterogeneous group of malignant tumors that are derived from mesenchymal tissues, including bone, muscle, and cartilage. Unlike carcinomas, which affect the elderly and are the final result of a long history of progressively accumulating preneoplastic lesions that allowed the molecular definition of multiple carcinogenic events, mostly sarcomas arise abruptly in infants, and their natural history of sarcomas is still mostly unknown. However, in the last decade, we have gained significant new insights into the genetic abnormalities that underlie the pathogenesis of these tumors. Specific molecular alterations have been associated with specific histological subtypes of sarcomas, leading to a new classification of many sarcomas. Conventionally grouped in either soft tissue or bone sarcomas according to the site of their origin, these tumors can now be genetically distinguished in two main groups: those carrying a tumor-specific recurrent chromosome aberrations that appear to be central to the pathogenesis of the tumor and are therefore included among diagnostic criteria and those with complex karyotypes and variable genetic alterations (Helman and Meltzer 2003; Wunder et al. 2007). Sarcomas with recurrent molecular changes include, among others, Ewing sarcoma family tumors, synovial sarcoma, alveolar rhabdomyosarcoma, myxoid liposarcoma, and myxoid chondrosarcoma. These tumors typically carry disease-specific chromosome translocations that frequently result in the expression of an oncogenic chimeric transcription factor, such as EWS-FLI1 in Ewing sarcoma. EWS-FLI1, which is present in around 85 % of Ewing sarcoma, derives specifically from a chromosomal translocation between chromosomes 11 and 22 and is referred to as t(11;22). While other translocations have also been described in Ewing sarcoma, including t(21;22) and t(7;22), all of the translocations involve the fusion of the EWS gene with an ETS family gene. Survival rates of patients with the different translocations appear to be the same (Le Deley et al. 2010; van Doorninck et al. 2010). Forced expression of EWS-FLI1 in normal cells can induce tumorigenesis. However, the effects of EWS-FLI1 expression are strongly dependent on cellular background (Kovar 2005). For example, EWS-FLI1 transforms immortalized murine NIH3T3 fibroblasts or mesenchymal stem cells and is required for the oncogenic phenotype of patient-derived EWS cells, but its introduction of into primary human or murine fibroblasts leads to growth arrest or cell death, respectively. These data suggest that oncogenic transformation by EWS-FLI requires a permissive cellular background. The critical factors in the permissive background are largely unknown, but may include disruption of the p53 and RB pathways and the presence of an intact IGF pathway as well as of CD99, a 32 kD integral membrane glycoprotein that is highly expressed in most cases of Ewing sarcoma. Recently it was clearly shown how EWS-FLI1 can induce upregulation of IGF1, inducing autocrine activation of IGF-1R and/or of CD99, thus sustaining its transforming activity in mesenchymal stem cells (Cironi et al. 2008; Riggi et al. 2005; Herrero-Martín et al. 2009; McKinsey et al. 2011).
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Sarcomas are a heterogeneous group of malignant tumors that are derived from mesenchymal tissues, including bone, muscle, and cartilage. Unlike carcinomas, which affect the elderly and are the final result of a long history of progressively accumulating preneoplastic lesions that allowed the molecular definition of multiple carcinogenic events, mostly sarcomas arise abruptly in infants, and their natural history of sarcomas is still mostly unknown. However, in the last decade, we have gained significant new insights into the genetic abnormalities that underlie the pathogenesis of these tumors. Specific molecular alterations have been associated with specific histological subtypes of sarcomas, leading to a new classification of many sarcomas. Conventionally grouped in either soft tissue or bone sarcomas according to the site of their origin, these tumors can now be genetically distinguished in two main groups: those carrying a tumor-specific recurrent chromosome aberrations that appear to be central to the pathogenesis of the tumor and are therefore included among diagnostic criteria and those with complex karyotypes and variable genetic alterations (Helman and Meltzer 2003; Wunder et al. 2007). Sarcomas with recurrent molecular changes include, among others, Ewing sarcoma family tumors, synovial sarcoma, alveolar rhabdomyosarcoma, myxoid liposarcoma, and myxoid chondrosarcoma. These tumors typically carry disease-specific chromosome translocations that frequently result in the expression of an oncogenic chimeric transcription factor, such as EWS-FLI1 in Ewing sarcoma. EWS-FLI1, which is present in around 85 % of Ewing sarcoma, derives specifically from a chromosomal translocation between chromosomes 11 and 22 and is referred to as t(11;22). While other translocations have also been described in Ewing sarcoma, including t(21;22) and t(7;22), all of the translocations involve the fusion of the EWS gene with an ETS family gene. Survival rates of patients with the different translocations appear to be the same (Le Deley et al. 2010; van Doorninck et al. 2010). Forced expression of EWS-FLI1 in normal cells can induce tumorigenesis. However, the effects of EWS-FLI1 expression are strongly dependent on cellular background (Kovar 2005). For example, EWS-FLI1 transforms immortalized murine NIH3T3 fibroblasts or mesenchymal stem cells and is required for the oncogenic phenotype of patient-derived EWS cells, but its introduction into primary human or murine fibroblasts leads to growth arrest or cell death, respectively. These data suggest that oncogenic transformation by EWS-FLI requires a permissive cellular background. The critical factors in the permissive background are largely unknown, but may include disruption of the p53 and RB pathways and the presence of an intact IGF pathway as well as of CD99, a 32 kD integral membrane glycoprotein that is highly expressed in most cases of Ewing sarcoma. Recently it was clearly shown how EWS-FLI1 can induce upregulation of IGF1, inducing autocrine activation of IGF-1R and/or of CD99, thus sustaining its transforming activity in mesenchymal stem cells (Cironi et al. 2008; Riggi et al. 2005; Herrero-Martín et al. 2009; McKinsey et al. 2011).
The presence of specific chimeric product is very attractive from a therapeutic point of view. Unfortunately the chimeric transcription factors that give rise to Ewing sarcoma are not druggable at the best of our current knowledge. Thus, the most interesting therapeutic options are druggable pathways regulated by EWS-FLI1, such as the IGF-1R-mediated signaling pathway. Antibodies or tyrosine-kinase inhibitors directed against the IGF-1 receptor protein have also been studied as a potential treatment for advanced Ewing sarcoma (Manara et al. 2007) and have implications for therapy. However, phase I–III clinical studies with anti-IGF-IR drugs have clearly indicated modest toxic effects, with mild and reversible hyperglycemia as the most common toxicity, but limited effectiveness. Particularly in Ewing’s sarcoma (EWS), despite the presence of the target in all tumors and ample preclinical evidence supporting the potential value of anti-IGF-IR agents, less than 10 % of cases extraordinarily responded to this therapy (Olmos et al. 2010; Pappo et al. 2011). Evidences for a compensatory role of IR-A when IGF-1R is disrupted (Garofalo et al. 2011, 2012) have been provided, indicating the relationship between these two receptors as one mechanism responsible for acquired and intrinsic resistance to selective anti-IGF-IR therapy. However, further studies are clearly necessary to better define patients that may really benefit from an anti-IGF-IR therapy as well as to rationalize the use of this targeted therapy in combination treatments.
CD99 is another molecule being studied as a potential immunotherapy target for the treatment of Ewing sarcoma. Engagement by anti-CD99 monoclonal antibodies induces massive apoptosis and reduces malignant potential of Ewing sarcoma cells (Scotlandi et al. 2000; Cerisano et al. 2004). An increased antitumor effectiveness of the anti-CD99 monoclonal antibody has recently been demonstrated both in vitro and in vivo when combined with doxorubicin (Scotlandi et al. 2006). In addition recent research suggests that CD99 plays a role in preventing the normal neural differentiation of Ewing cells (Rocchi et al. 2010). Apoptosis also occurs following activation of cell surface receptors by the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and the use of agonistic monoclonal antibodies activating the TRAIL receptors is one of the emerging targeted strategies in cancer management, either as monotherapy or in association with other treatment modalities. There is evidence that Ewing’s sarcoma cells are exquisitely sensitive to TRAIL-mediated apoptosis. A preclinical study showing efficacy of TRAIL in animals was recently reported (Picarda et al. 2010). These drugs are still purely investigational at this point.
Finally EWS-FLI1 fusion genes were recently found to act in a positive feedback loop to maintain the expression of PARP1, which was required for EWS-FLI-mediated transcription, thereby enforcing oncogene-dependent sensitivity to PARP-1 inhibition (Brenner et al. 2012). Ewing sarcoma cells, primary tumor xenografts, and tumor metastases were all highly sensitive to PARP1 inhibition. Addition of a PARP1 inhibitor to the second-line chemotherapeutic agent temozolomide resulted in complete responses of all treated tumors in an EWS-FLI1-driven mouse xenograft model of Ewing sarcoma. These findings offer a strong preclinical rationale to target the EWS-FLI1:PARP1 intersection as a therapeutic strategy to improve the treatment of Ewing sarcoma.
Bibliography
Brenner JC, Feng FY, Han S, Patel S, Goyal SV, Bou-Maroun LM, Liu M, Lonigro R, Prensner JR, Tomlins SA, Chinnaiyan AM (2012) PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res 72(7):1608–1613. doi:10.1158/0008-5472.CAN-11-3648. Epub 2012 Jan 27
Cerisano V, Aalto Y, Perdichizzi S, Bernard G, Manara MC, Benini S, Cenacchi G, Preda P, Lattanzi G, Nagy B, Knuutila S, Colombo MP, Bernard A, Picci P, Scotlandi K (2004) Molecular mechanisms of CD99-induced caspase-independent cell death and cell-cell adhesion in Ewing’s sarcoma cells: actin and zyxin as key intracellular mediators. Oncogene 23(33):5664–5674
Cironi L, Riggi N, Provero P, Wolf N, Suvà ML, Suvà D, Kindler V, Stamenkovic I (2008) IGF1 is a common target gene of Ewing’s sarcoma fusion proteins in mesenchymal progenitor cells. PLoS One 3(7):e2634
Garofalo C, Manara MC, Nicoletti G, Marino MT, Lollini PL, Astolfi A, Pandini G, López-Guerrero JA, Schaefer KL, Belfiore A, Picci P, Scotlandi K (2011) Efficacy of and resistance to anti-IGF-1R therapies in Ewing’s sarcoma is dependent on insulin receptor signaling. Oncogene 30(24):2730–2740. doi:10.1038/onc.2010.640. Epub 2011 Jan 31
Garofalo C, Mancarella C, Grilli A, Manara MC, Astolfi A, Marino MT, Conte A, Sigismund S, Carè A, Belfiore A, Picci P, Scotlandi K (2012) Identification of common and distinctive mechanisms of resistance to different anti-IGF-IR agents in Ewing’s sarcoma. Mol Endocrinol 26(9):1603–1616. doi:10.1210/me.2012-1142. Epub 2012 Jul 13
Helman LJ, Meltzer P (2003) Mechanisms of sarcoma development. Nat Rev Cancer 3(9):685–694
Herrero-Martín D, Osuna D, Ordóñez JL, Sevillano V, Martins AS, Mackintosh C, Campos M, Madoz-Gúrpide J, Otero-Motta AP, Caballero G, Amaral AT, Wai DH, Braun Y, Eisenacher M, Schaefer KL, Poremba C, de Alava E (2009) Stable interference of EWS-FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target. Br J Cancer 101(1):80–90. Epub 2009 Jun 2
Kovar H (2005) Context matters: the hen or egg problem in Ewing’s sarcoma. Semin Cancer Biol 15(3):189–196
Le Deley MC, Delattre O, Schaefer KL, Burchill SA, Koehler G, Hogendoorn PC, Lion T, Poremba C, Marandet J, Ballet S, Pierron G, Brownhill SC, Nesslböck M, Ranft A, Dirksen U, Oberlin O, Lewis IJ, Craft AW, Jürgens H, Kovar H (2010) Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28(12):1982–1988
Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D, Lollini PL, Nanni C, Hofmann F, García-Echeverría C, Picci P, Scotlandi K (2007) Preclinical in vivo study of new insulin-like growth factor-I receptor–specific inhibitor in Ewing’s sarcoma. Clin Cancer Res 13(4):1322–1330
McKinsey EL, Parrish JK, Irwin AE, Niemeyer BF, Kern HB, Birks DK, Jedlicka P (2011) A novel oncogenic mechanism in Ewing sarcoma involving IGF pathway targeting by EWS/Fli1-regulated microRNAs. Oncogene 30(49):4910–4920
Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I, Worden FP, Gualberto A, Scurr M, de Bono JS, Haluska P (2010) Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol 11(2):129–135. Epub 2009 Dec 23
Pappo AS, Patel SR, Crowley J et al (2011) R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II sarcoma alliance for research through collaboration study. J Clin Oncol 29:4541–4547
Picarda G, Lamoureux F, Geffroy L, Delepine P, Montier T, Laud K, Tirode F, Delattre O, Heymann D, Rédini F (2010) Preclinical evidence that use of TRAIL in Ewing’s sarcoma and osteosarcoma therapy inhibits tumor growth, prevents osteolysis, and increases animal survival. Clin Cancer Res 16(8):2363–2374. Epub 2010 Apr 6
Riggi N, Cironi L, Provero P, Suvà ML, Kaloulis K, Garcia-Echeverria C, Hoffmann F, Trumpp A, Stamenkovic I (2005) Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res 65(24):11459–11468
Rocchi A, Manara MC, Sciandra M, Zambelli D, Nardi F, Nicoletti G, Garofalo C, Meschini S, Astolfi A, Colombo MP, Lessnick SL, Picci P, Scotlandi K (2010) CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J Clin Invest 120(3):668–680. doi:10.1172/JCI36667. Epub 2010 Feb 8
Scotlandi K, Baldini N, Cerisano V, Manara MC, Benini S, Serra M, Lollini PL, Nanni P, Nicoletti G, Bernard G, Bernard A, Picci P (2000) CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res 60(18):5134–5142
Scotlandi K, Perdichizzi S, Bernard G, Nicoletti G, Nanni P, Lollini PL, Curti A, Manara MC, Benini S, Bernard A, Picci P (2006) Targeting CD99 in association with doxorubicin: an effective combined treatment for Ewing’s sarcoma. Eur J Cancer 42(1):91–96. Epub 2005 Dec 2
van Doorninck JA, Ji L, Schaub B, Shimada H, Wing MR, Krailo MD, Lessnick SL, Marina N, Triche TJ, Sposto R, Womer RB, Lawlor ER (2010) Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol 28(12):1989–1994. Epub 2010 Mar 22
Wunder JS, Nielsen TO, Maki RG, O’Sullivan B, Alman BA (2007) Opportunities for improving the therapeutic ratio for patients with sarcoma. Lancet Oncol 8(6):513–524
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Scotlandi, K. (2014). Biology of Ewing Sarcoma. In: Picci, P., Manfrini, M., Fabbri, N., Gambarotti, M., Vanel, D. (eds) Atlas of Musculoskeletal Tumors and Tumorlike Lesions. Springer, Cham. https://doi.org/10.1007/978-3-319-01748-8_48
Download citation
DOI: https://doi.org/10.1007/978-3-319-01748-8_48
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-01747-1
Online ISBN: 978-3-319-01748-8
eBook Packages: MedicineMedicine (R0)