
Abstract
Research is increasingly becoming data-driven, and natural sciences are not an exception. In both biology and medicine, we are observing an exponential growth of structured data collections from experiments and population studies, enabling us to gain novel insights that would otherwise not be possible. However, these growing data sets pose a challenge for existing compute infrastructures since data is outgrowing limits within compute. In this work, we present the application of a novel approach, Memory-Driven Computing (MDC), in the life sciences. MDC proposes a data-centric approach that has been designed for growing data sizes and provides a composable infrastructure for changing workloads. In particular, we show how a typical pipeline for genomics data processing can be accelerated, and application modifications required to exploit this novel architecture. Furthermore, we demonstrate how the isolated evaluation of individual tasks misses significant overheads of typical pipelines in genomics data processing.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Life and medical sciences are evolving towards a data-driven research model. The leading biological institutions face a new challenge in dealing with data. Interdisciplinary collaborations are aimed at accelerating this digitalization of research. The use of compute clusters and cloud services has become more common with growing data sets. High-performance computing (HPC) is relevant for many modeling and simulation aspects in the life sciences, as indicated by the study of genome-scale biochemical reaction networks [12].
This trend towards data-driven research challenges a community that is witnessing an explosion in accumulated data sizes. The European Bioinformatics Institute (EBI) has reported [10] a growth of data stored from 120 PB to 160PB from 2016 to 2018. The number of deposited high-throughput sequencing data sets at the US National Center for Bioinformatics (NCBI) shows exponential growth in the last years, as depicted in Fig. 1. This can be explained by the decline in sequencing costs, reduction in sequencing times, and availability of diverse sequencing platforms.
Another source of increasing data sets is comprehensive collections like population studies. These studies follow a large number of participants for a long time and acquire different modalities, like image data and multi-omics data. Storing and processing such data collections is challenging not only because of their sizes but also because the data needs to be available without delays for the researchers. While researching health-related questions, large time-series data sets can be helpful but pose a challenge for storage systems.

Number of high throughput sequencing (HTS) data sets uploaded to NCBI per year. The 2019 data is still incomplete, since most studies are only uploaded to NCBI once they are published.
In this work, we want to demonstrate how a novel architecture, named Memory-Driven Computing (MDC), can be used for processing genomics sequencing data.
2 Memory-Driven Computing
While the data sets used in research are continually growing, the computing power is not keeping up. The end of Moore’s law [31] seems to have finally been reached. Current approaches to scaling focus on distributing workloads among processing nodes in large clusters. However, these clusters come with growing costs, in particular from energy consumption that limits similar future scaling. Also, genomics tasks often cannot be easily distributed among many nodes, e.g., gene regulation network processing deals with densely connected graphs.
2.1 Novel Architecture Tailored for Data Science
To overcome these shortcomings, Hewlett Packard Enterprise (HPE) has proposed a novel architecture: Memory-Driven Computing. Unlike traditional, processor-centric systems, it puts the memory (and therefore the user data) at the center of the architecture [7]. This is realized by going from the traditional von Neumann architecture, as shown in Fig. 2a, established since 1953, towards a more memory-centric approach (Fig. 2b). To eliminate overheads, remote data access no longer has to traverse the host processor, but is handled by the memory fabric. Traditional local storage and memory are bundled into a persistent memory pool that can be realized through technologies such as the memristor [9]. Finally, the underlying fabric between all components, including processors, will be switching from electrical to optical connections. This is necessary to overcome the required energy for moving growing data sets.

(a) The traditional von Neumann architecture defines the processor as the central component. (b) MDC puts persistent memory at the center of the system. Components are linked through an optical fabric. (c) The connecting fabric, e.g., Gen-Z, puts different components from the processor-, memory- and accelerator class in a single shared address space. Other specialized devices like sequencers for data generation could also be integrated.
2.2 Composable Infrastructure
In this composable infrastructure, compute power can be attached to the memory as required to allow scaling. All components share the address space and are connected through a fabric like Gen-Z [13] that also controls data access and security, which are essential properties when working with sensitive medical data. Besides CPUs and memory, other components like accelerators and GPUs can be integrated. Since the fabric is well-specifiedFootnote 1, it is also possible to incorporate more specialized data sources, e.g., sequencing machines, to avoid data transfer to the fabric attached memory. Traditional HPC architectures typically offer large numbers of standardized nodes, grouped into different classes, like high-memory or GPU-nodes. Often these systems are specially tailored for simulation applications but do not fit well to other tasks like genomics data processing. With MDC, it is possible to group the fabric-connected components dynamically into task-tailored systems.
2.3 Transition Path Towards MDC
The idea behind MDC touches all components in computer systems. A hardware transition to MDC cannot realistically happen in a single step, but rather is a process. The first components like Gen-Z enabled devices, are expected in 2020, and other techniques like memristor-based storage for persistent fabric attached memory are still a few years out. Nonetheless, it is already possible to apply MDC principles. Existing large-memory machines can exploit an abundance of memory. Systems like the HPE Superdome Flex offer a shared address space between multiple nodes for applications. Software development can also use smaller servers or even laptops to emulate fabric attached memory using the Fabric Attached Memory Emulation (FAME)Footnote 2.
Software adapted for MDC has provided large performance gains [8] and allows us to think in new paradigms as memory pools enable new programming models [19]. The transition to MDC follows an iterative process of preparing and modifying an application. The preparation starts with the definition of goals and metrics, defining the optimization target, e.g., doubling the number of tasks per time. This is followed by a baseline performance measurement. The results are used to perform a cost/benefit analysis to identify the MDC modifications, e.g., modifications to exploit the abundance of memory through different data structures or the elimination of I/O. During the modification phase, the developers need to apply MDC principles and modify the application. Finally, the fine-tuning based on the initially defined metrics and goals can be performed. More details can be found in Sect. 5.
3 Related Work
Genomics data processing is a challenging task and, therefore, an excellent use case to evaluate MDC. In this section, we discuss prior work, beginning with in-memory genomics data processing approaches. Schapranow et al. [29] have employed an in-memory database to accelerate the alignment tool BWA on a cluster, a method similar to MapReduce. They have reported an improvement of 89% for a cluster of 25 machines. Firnkorn et al. [11] have followed a similar path and compare the use of in-memory databases (SAP HANA) to a traditional RDBMS (MySQL) for alignment focussed on direct matches. While they have observed an acceleration factor of 27, the comparison was made against a highly unusual tool that is not specialized in alignment. Finally, Li et al. [25] have improved the processing of genomic data using the SPARK framework by introducing improved compression of genomic data to optimize data transfer between the nodes. Hajj et al. [15] have demonstrated new address space concepts using samtools.
Other approaches use specialized hardware. Luo et al. [26] have used GPUs for alignment. Lavenier et al. [23] and Kim et al. [21] have explored the use of completely new hardware for genomics data processing. They propose to use processing within the memory to minimize data access time and maximize bandwidth. The pre-alignment steps of Kim et al. lead to an end-to-end improvement between 2\(\times \) and 3.5\(\times \). Alser et al. [5] propose another FPGA based tool for pre-alignment; they achieve a 10\(\times \) acceleration for this task using a Virtex-7 FPGA using Xilinx VC709 board running at 250 MHz. Kaplan et al. [18] propose a novel resistive approximate similarity search accelerator (RASSA) that exploits charge distribution and parallel in-memory processing to reflect a mismatch count between DNA sequences. Their pre-alignment software achieves 16–77\(\times \) improvements in long reads.
Finally, different programming languages have been explored to accelerate the preprocessing of genomics data. Herzeel et al. [16] have proposed a multithreaded framework for sequence analysis to leverage the concurrency features of the Go programming language. Tarasow et al. [30] have investigated the parallel processing features of the programming language D to improve genomics processing speed.
4 Application in Bioinformatics
From an HPC perspective, bioinformatics applications often fall in either of two categories: I/O-bound and compute-bound. I/O-bound applications typically transform or annotate data. They operate on significant inputs and produce large outputs while the actual transformation task takes little to no computational effort. Compute-bound tasks are not limited by I/O, their primary work, e.g., assembly or modeling or cellular interaction, is the limiting factor. While many compute-bound applications operate on large data sets, the I/O part can often be neglected in comparison to the main functionality. In previous work, it has been shown that compute-bound applications still can benefit from the abundance of memory available in MDC [6].
4.1 Typical Preprocessing Tasks and Pipeline Structures
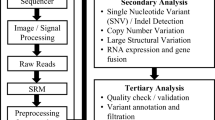
In this work, we will focus on optimizing an I/O-bound application since there is a lot of overhead that can be eliminated with MDC, and such applications are common in the typical structures of bioinformatic preprocessing tasks. The preprocessing of next-generation sequencing (NGS) genomics data consists of initial quality controls, demultiplexing of data from multiple experiments (which have been multiplexed to reduce costs), alignment to a reference genome and further quality controls as shown in Fig. 3. These steps can be performed through several competing tools that often specialize in certain types of experiments. Considering single-cell genomics, the number of involved tools increases, and the pipelines become more complex. This has lead to the development of specialized bioinformatics pipelines and dedicated workflow managers like Snakemake [22].

Classical bioinformatics pipelines are a series of tools that exchange data through large files on disk-based storage. With MDC, the data is kept in memory, and I/O can be avoided. Parallel processing with different tools does not impose a penalty for random data access that is known from existing storage systems that often are optimized for serial or streaming data access.
These pipelines deal with large input files, and many tools transform the data, annotate information, or change the order of the content. They take an input and produce an output of a similar size. This leads to a large number of intermediate files, and therefore these tools are often I/O-bound. The last step in the preprocessing typically performs a data reduction, going from alignments to gene expression information. Here, we will demonstrate the use case for the I/O-bound application samtools, which is a common tool for processing aligned reads or alignments.
4.2 Application: Samtools
Samtools [24] is a standard tool for processing alignments in Structured Alignment Map (SAM) and binary SAM (BAM) files [2]. These files contain tens to hundreds of millions of alignments in a single file and must be efficient to deal with growing data sets. An alignment consists of core information like genomic position, the actual sequence, the associated quality values, and auxiliary information, including tags to store key-value pairs of different data types.
We have evaluated the broad functionality of samtools (version 1.9) to select commands that are used most frequently in the community as well as those that could benefit most from eliminating I/O. In the following, we list these commands, briefly describe their functionality, and classify the output that they produce. A comprehensive overview of samtools and the details for each command can be found in the documentation [3].
View. Samtools provides the view command to convert between SAM and BAM format. Often tools only support one of these formats, or conversion to BAM is needed to reduce file sizes in storage. SAM files can be useful for visual inspection of the data since SAM is human readable. It is also possible to specify filter criteria, e.g., to filter for a specific chromosome.
Sort. After the alignment of input files to a reference genome, the output file (e.g., in BAM format) stores the alignments in random order. Many downstream steps such as read-count generation, variant calling, or visualization in IGV require the alignments in the file to be ordered. Alignments can be sorted by genomic order based on their coordinates on each chromosome or by read or query names to get a technical ordering.
Markdup. The markdup function can be used to identify duplicate alignments from a coordinate/position sorted file. The duplicate reads are referred to as the primary reads whose coordinates are matching. The highest quality of a duplicate is kept, and others are marked with the duplicate flag. Removal of duplicate reads, generated due to PCR amplification or sequencing, is an indispensable step for the processing of alignment data as it affects the overall quality and downstream steps of NGS analysis.
Fixmate. To prepare a file for the markdup command, it has to be modified with the fixmate command to add MS (mate score) and MC (CIGAR string for mate/next segment) tags. The MS tag is used by the markdup function to select the best reads to keep.
Combined Commands. Often, the functionality of samtools is not used independently, but multiple commands interact. An example is the marking of duplicate reads in a data set. This task consists of four consecutive steps: sorting by query name, the fixmate command, sorting by genomic position, and finally, actual marking of the duplicates. Often these steps are connected through files (see Fig. 4). It is also possible to connect the tools with a pipe; this avoids I/O but still requires serialization and deserialisation of the data between the individual steps. The serialization and deserialization steps convert between in-memory and on-disk representation of SAM records.

Samtools commands can be concatenated with multiple approaches: Data exchange through files (top) or a pipeline (middle). We propose to share data in memory (bottom) to remove unnecessary overhead.
5 Modifications to Exploit Memory-Driven Computing
Running an unmodified application in an MDC environment already yields the benefit of accelerated data access, since no storage systems and drivers are involved. However, to fully exploit this environment, memory-mapping the data is recommended and can often be easily added. Besides this, two significant areas for benefiting from MDC exist: First, we can eliminate all I/O operations for data input and output. This also applies to temporary data and data exchange between applications. Existing storage systems often are optimized for serial or streaming data access. Random access from a single application or multiple applications reduces data throughput significantly. Since the central memory pool does not require traditional access patterns for performance reasons (e.g., linear reading), parallel processing of data becomes feasible. This speeds up quality control tools and preprocessing in parallel or similar use cases, where I/O bottlenecks are common. In a second step, the internal data structures of an application can be modified to benefit from the abundance of memory in MDC environments. This could be, for example, extensive pre-calculation of intermediate results to replace standard computations with a simple look-up.
We have applied these principles to samtools to benefit from I/O elimination and improved data passing between different functionalities of the application.
5.1 Samtools
Samtools stores data in the Sequence Alignment Map (SAM)-format [2], which is text-based. A binary version of this format (BAM) exists as well. Finally, a text-based column-oriented version (CRAM) exists but is of no practical relevance. In this work, we present four contributions to the main functionality of samtools in the areas of I/O reduction that include input and output parsing, removal of intermediate files, and the better integration of multiple commands. Samtools is available as open source and written in C.
Parallel Input Parsing. We have modified the input loading and parsing for the two most common formats, SAM and BAM. In both formats, we first need to parse the header information that contains reference information about the alignment targets (e.g., chromosomes) and their sizes. Afterward, we split the file into chunks and parse them in parallel. To fully exploit the capabilities of MDC, we have modified all I/O to use memory-mapped files, which are based on the posix mmap() function that establishes a mapping between an address space of a process and a memory object. MDC already gives us the shared memory pool for storing our data, therefore, we can avoid the introduction of additional frameworks like MPI-IO which introduce additional layers.
Sequence Alignment Map (SAM) files are a line-based format where each line contains a single alignment. Alignments require header information, and it needs to be loaded first. Next, we split the file into byte-regions and process them in parallel. Through MDC, the data is already in memory, and random access bears no extra costs. The start of the regions does not necessarily coincide with the beginning of a line, hence parsing only starts after the next newline. Correspondingly, the last line is parsed beyond the end of the region to capture the full alignment information.
Binary SAM (BAM) files consist of a series of gzip-compressed blocks that contain the header and alignment information. Again, the header needs to be parsed first, just like for SAM files. Next, the compressed blocks containing the alignments can be processed. A single block can hold up to 64 KB of alignment information. Since there is no reliable indicator for the beginning of a compressed block, it is not possible to simply split the file into equally sized regions. Therefore, we first create an index of all blocks. This task takes very little time since the file is already in memory. The index is then used to distribute the contents for parallel decompression and alignment parsing, as shown in Fig. 5a.

Parallel input and output. The BAM file is initially scanned for blocks and then parsed in parallel. The output works by writing into sufficiently large regions of the file, followed by a later removal intermediate unused spaces.
Parallel Output Writing.
The process of writing alignment data starts with storing the header information. Afterward, we estimate the average size of an alignment to assess the overall file size. This allows us to allocate sufficient space in a memory-mapped file for the output. We split the output into parts that are saved into different regions of the output file. Unused space remains in each region because the estimation of the expected size is designed to overestimate so that all alignments will fit. In the last step, we move the file parts to eliminate the unused space, and finally, we truncate the file to the actual size. These steps are shown in Fig. 5b. For BAM files, we estimate the required size for uncompressed data. However, usually, good compression can be achieved, and therefore the final file size is much smaller.
Intermediate Data Storage. The data processing commands of samtools often require some temporary storage. The sorting command pre-sorts data in blocks and finally merges them into the final file. For smaller files, these temporary blocks are held in memory, but this no longer applies to growing data sets. Similarly, the markdup and fixmate commands use temporary storage for intermediate data. In our approach, we load the full data set into memory to avoid additional I/O. We have modified samtools to remove temporary files. With all data in memory, sorting can be easily performed without temporary files using the C++ extensions for parallelism [17]. The temporary data storage of the fixmate and markdup commands can be resolved by instead keeping a list of references to the alignments that otherwise would have been written to disk.
Pipelining Commands. We have found that certain samtools commands are often run together. We have chosen the marking of duplicates pipeline for investigation and optimization. Since data parsing and I/O consume a large part of the runtime, we have modified the commands to take a list of alignments in memory and to work on that data. Therefore, we have modified the sorting, fixmate, and markdup commands to work with a list of alignments that we obtain from the input parsing. Although fixmate and markdup are sequential tasks, we still can remove the input parsing or deserialisation.
Memory-Management. A growing number of processing threads, in particular during the input parsing, allocate many small memory parts. With massive input files, tens to hundreds of million allocations are required during input parsing. With the default allocator (from glibc), the central lock for the memory-list is a bottleneck that significantly slows down the input parsing. The core information for an alignment is of fixed size, and it is accompanied by a variable-length string for storing sequence and auxiliary information. This results in two allocations per alignment. For the core information, the memory can be pre-allocated in larger batches due to its known size, the sequence and auxiliary information requires custom allocations. We have considered specialized allocators like jemalloc [1] and tcmalloc [14] that use per-thread pools as well as a non-freeing custom allocation solution, that acquires large portions of memory per thread and uses them for allocations. This custom allocator is experimental only to understand the difficulties of many small allocations from a large number of threads and should not be used in outside of experiments, since it does not track the allocated memory, and therefore, is not able to free memory again.
These modifications are designed to be reusable and are available on Github: https://github.com/schultzelab/samtools-mdc. This includes the parallel reading and writing functions for SAM and BAM files. Samtools-mdc is designed to be a drop-in replacement for samtools. SAM/BAM reading and writing has been separated from the data modification (e.g. sorting) to allow re-use of components.
6 Evaluation and Results
We have used two systems to evaluate our MDC-modifications. We took a typical Dell blade server, as it is common among bioinformatics groups, with two sockets, 32 cores, 64 threads, and a total of 768 GB memory. As a second system, we have used an HPE Superdome Flex, with two nodes, 16 sockets, 144 cores, and 288 threads and a total of 6 TB memory. The abundance of memory in this system is close to our expectation in an MDC environment, although larger systems (up to 48 TB) exist. The Superdome Flex implements MDC principles and provides a single address space across the nodes through a custom interconnect and firmware. Future systems are expected to include Gen-Z hardware. We have collected data points for different numbers of threads, with 1, 16, 32 and 64 collected on the smaller blade system and 144 and 288 gathered on the Superdome Flex system.
We have selected a range of samplesFootnote 3 from the National Institutes of Health Sequence Read Archive [4] to get coverage of typical input files sizes. These ten samples cover different studies and multiple diseases. An overview of the samples is shown in Table 1. We have grouped the samples by size into three categories: small, medium, and large.
6.1 Memory Allocation
Our initial experiments show that memory allocation is a crucial factor in processing SAM and BAM files. The loading process requires multiple allocations per read, and this becomes an increasing challenge with a growing number of parallel reading threads.
To test the allocations strategies, we read the three sample groups with different allocators and multiple thread numbers, ranging from 1 to 288, and report the cumulated time per group. The results, differentiated between bam and sam files, are shown in Fig. 6. It can be seen immediately that the default allocator consistently shows the worst performance, both with just a single thread as well as with large numbers of threads (144, 288). In most cases, tcmalloc performs equal or worse in comparison to jemalloc. Our custom allocator shows the best performance for most cases; however, we discarded it since it is not a full implementation but rather a proof-of-principle.

Cumulative times to fully load the small, medium and large samples in SAM and BAM format into memory using different allocators (see Sect. 5.1, Memory-Management) and number of threads.
6.2 Samtools Commands
Next, we evaluated different samtools commands. The results present a broad range of behavior, and we show examples in Fig. 7. All examples use the large bam sample group. The view command shows consistently better performance for the MDC-optimised version of samtools (mdc) when compared to the original (unmodified) samtools (orig). The fixmate use case contains program logic that is not parallelized, and the streaming architecture of the original samtools shows better performance than the modified version. Samtools (mdc) loads the complete data set first, performs the fixmate operation, and then writes the complete data set; samtools (orig) uses a streaming approach that is more suited for this specific task. Finally, sorting by query name shows slightly better results of the MDC-optimised version for smaller numbers of threads and similar performance for larger thread numbers (64, 144, 288). With growing thread numbers, samtools (orig) is configured to retain growing parts of the data set in memory, similar to our approach.

Comparison of original and mdc-modified samtools for three commands: view, fixmate and sorting by query name. Each command uses the large bam samples and threads ranging from 16 to 288.
6.3 Marking Duplicates Pipeline
Finally, we evaluated different choices to realize a duplicate marking pipeline: individual calls with intermediate files, a pipeline with serialization and deserialization, and our MDC approach with all data stored in memory. The results are presented in Fig. 8. We have evaluated all pipelines with sam and bam samples from all three groups. The MDC pipeline is always faster than the other options. For small files and large numbers of threads, the pipeline with serialization and deserialization can be slower than individual tools. The measurements for the individual tools show that the majority of processing time is spent in the fixmate functionality. Further inspection revealed that this step is not parallelized and it would be an opportunity for future work.

Evaluation of duplicate marking using individual commands (stacked results), a pipeline, and our MDC approach. We have tested the bam (left) and sam (right) files of different sizes and report cumulative results per size group. Single-threaded MDC results for sam files had to be omitted to technical problems. Sort/sort -n denotes sorting by query name, sort -p is sorting by genomic position.
7 Discussion
Samtools is already optimized for single commands; the streaming approach of samtools shows satisfactory performance. Some commands are not parallelized, and the streaming might prevent this. Especially the fixmate functionality consumes a significant amount of time. If we analyze not just single commands but rather pipelines that combine multiple commands, we can see that data exchange between the individual commands limits the throughput. Using a pipe instead of intermediate files shows an acceleration but still comes with overhead from serialization and deserialization.
With MDC, we can remove I/O from intermediate files, and we can share data in native data types. This gives an additional acceleration over the pipeline approach, and we believe this to be true of other pipelines. However, the input and output of the current MDC version are still files and need to be parsed and written - an overhead that could be avoided by using shared memory for data exchange between multiple tools.
Furthermore, we have found that selecting a proper allocation strategy is crucial for parsing genomics data. A single read requires at least two allocations, and with a growing number of threads, the synchronization overhead becomes a bottleneck. Some of the required memory objects are already known in size so that the pre-allocation of larger amounts is possible.
8 Outlook
A next step will be to expand the MDC optimized version of samtools to cover more of its functionality. After focusing on the most common commands, we aim to study the remaining commands as well. Since samtools is a central component in many pipelines, we want to improve data exchange between tools to remove the need for expensive serialization and deserialization and to establish a common way of memory-based data exchange.
Besides these steps, we want to expand from genomics data preprocessing to bioinformatics analysis, especially for the growing number of large single-cell data sets from consortia like the Human Cell Atlas [27] which try to catalog all cell types. For many biological questions, a large number of analysis methods already exist. Still, for many, the performance is increasingly becoming an issue, this has been shown for trajectory inference by Saelens et al. [28].
Furthermore, the integration of multiple data sources increases computational needs. Multi-omics data sets that combine approaches like genomics, lipidomics, and proteomics allow gaining novel insights into biological processes. Another domain that produces growing data sets for analysis is spatial approaches that combine the capture of omics data with spatial and image information. This also provides a link to population studies whose growing data production poses an increasing challenge.
Change history
15 June 2020
The original version of chapters 17 and 24 were previously published non-open access. They have now been made open access under a CC BY 4.0 license and the copyright holder has been changed to ‘The Author(s).’ The book has also been updated with the change.
The chapters 19 and 25 were inadvertently published open access. This has been corrected and the chapters are now non-open access.
Notes
References
jeMalloc. http://jemalloc.net
SAM specification (2019). http://samtools.github.io/hts-specs/SAMv1.pdf
SAMtools 1.9 documentation (2019)
The National Institutes of Health (NIH) Sequence Read Archive (SRA) (2019). https://www.ncbi.nlm.nih.gov/sra/
Alser, M., Hassan, H., Xin, H., Ergin, O., Mutlu, O., Alkan, C.: GateKeeper: a new hardware architecture for accelerating pre-alignment in DNA short read mapping. Bioinform. 33(21), 3355–3363 (2017). https://doi.org/10.1093/bioinformatics/btx342. (Oxford England)
Becker, M., et al.: Accelerated genomics data processing using memory-driven computing (accepted). In: Proceedings of the 6th International Workshop on High Performance Computing on Bioinformatics (HPCB 2019) in conjunction with the IEEE International Conference on Bioinformatics and Biomedicine (BIBM 2019), San Diego, USA (2019)
Bresniker, K.M., Singhal, S., Williams, R.S.: Adapting to thrive in a new economy of memory abundance. Computer 48(12), 44–53 (2015). https://doi.org/10.1109/JSTQE.2012.2236080
Chen, F., et al.: Billion node graph inference: iterative processing on the machine. Tech. rep. (2016). https://www.labs.hpe.com/publications/HPE-2016-101
Chua, L.: Memristor-the missing circuit element. IEEE Trans. Circuit Theory 18(5), 507–519 (1971). https://doi.org/10.1109/TCT.1971.1083337
Cook, C.E., et al.: The European Bioinformatics Institute in 2018: tools, infrastructure and training. Nucl. Acids Res. (2019). https://doi.org/10.1093/nar/gky1124
Firnkorn, D., Knaup-Gregori, P., Lorenzo Bermejo, J., Ganzinger, M.: Alignment of high-throughput sequencing data inside in-memory databases. Stud. Health Technol. Inform. 205, 476–480 (2014). https://doi.org/10.3233/978-1-61499-432-9-476
Fröhlich, F., Kaltenbacher, B., Theis, F.J., Hasenauer, J.: Scalable parameter estimation for genome-scale biochemical reaction networks. PLoS Comput. Biol. (2017). https://doi.org/10.1371/journal.pcbi.1005331
Gen-Z Consortium: Gen-Z core specification 1.0 (2018). https://genzconsortium.org/specification/core-specification-1-0/
Ghemawat, S., Menage, P.: Tcmalloc: thread-caching malloc (2007). http://goog-perftools.sourceforge.net/doc/tcmalloc.html
Hajj, I.E., et al.: SpaceJMP : programming with multiple virtual address spaces. In: ASPLOS, pp. 353–368, No. Section 3 (2016). https://doi.org/10.1145/2872362.2872366
Herzeel, C., Costanza, P., Decap, D., Fostier, J., Verachtert, W.: elPrep 4: a multithreaded framework for sequence analysis. PLoS ONE 14(2), 1–16 (2019). https://doi.org/10.1371/journal.pone.0209523
Programming Languages – Technical Specification for C++ Extensions for Parallelism. ISO/IEC TS 19570:2018. Standard (November 2018)
Kaplan, R., Yavits, L., Ginosar, R.: RASSA: resistive pre-alignment accelerator for approximate DNA long read mapping. IEEE Micro 39, 44–54 (2018). https://doi.org/10.1109/MM.2018.2890253
Keeton, K.: The machine : an architecture for memory-centric computing. In: Workshop on Runtime and Operating Systems for Supercomputers (ROSS), p. 2768406 (June 2015)
Kim, D., Paggi, J.M., Park, C., Bennett, C., Salzberg, S.L.: Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. (2019). https://doi.org/10.1038/s41587-019-0201-4
Kim, J.S., et al.: GRIM-filter: fast seed location filtering in DNA read mapping using processing-in-memory technologies. BMC Genomics 19(Suppl 2) (2018). https://doi.org/10.1186/s12864-018-4460-0
Köster, J., Rahmann, S.: Snakemake-a scalable bioinformatics workflow engine. Bioinformatics (2012). https://doi.org/10.1093/bioinformatics/bts480
Lavenier, D., Roy, J.F., Furodet, D.: DNA mapping using processor-in-memory architecture. In: Proceedings - 2016 IEEE International Conference on Bioinformatics and Biomedicine, BIBM 2016, pp. 1429–1435 (2017). https://doi.org/10.1109/BIBM.2016.7822732
Li, H., et al.: The sequence alignment/map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009). https://doi.org/10.1093/bioinformatics/btp352
Li, X., Tan, G., Wang, B., Sun, N.: High-performance genomic analysis framework with in-memory computing. ACM SIGPLAN Not. 53(1), 317–328 (2018). https://doi.org/10.1145/3200691.3178511
Luo, R., et al.: SOAP3-dp: fast, accurate and sensitive GPU-based short read aligner. PLoS ONE 8(5) (2013). https://doi.org/10.1371/journal.pone.0065632
Regev, A., et al.: The Human Cell Atlas White Paper (October 2018). http://arxiv.org/abs/1810.05192
Saelens, W., Cannoodt, R., Todorov, H., Saeys, Y.: A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37(5), 547–554 (2019). https://doi.org/10.1038/s41587-019-0071-9
Schapranow, M.P., Plattner, H.: HIG - an in-memory database platform enabling real-time analyses of genome data. In: Proceedings - 2013 IEEE International Conference on Big Data, Big Data 2013, pp. 691–696 (2013). https://doi.org/10.1109/BigData.2013.6691638
Tarasov, A., Vilella, A.J., Cuppen, E., Nijman, I.J., Prins, P.: Genome analysis Sambamba : fast processing of NGS alignment formats. Bioinformatics 31(November), 2032–2034 (2017). https://doi.org/10.5281/zenodo.13200.Contact
Theis, T.N., Philip Wong, H.S.: The end of Moore’s Law: a new beginning for information technology. Comput. Sci. Eng. 19(2), 41–50 (2017). https://doi.org/10.1109/MCSE.2017.29
Acknowledgment
This work was funded in part by the HGF grant sparse2big, the FASTGenomics grant of the German Federal Ministry for Economic Affairs and Energy, and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC2151/1—390873048. We want to thank Milind Chabbi, Bill Hayes, Keith Packard, Patrick Demichel, Binoy Arnold, Robert Peter Haddad, Eric Wu, Chris Kirby, Rocky Craig from Hewlett Packard Enterprise and Hewlett Packard Labs and the bioinformatics group at the AG Schultze at the Life and Medical Sciences Institute at the University of Bonn.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Becker, M. et al. (2020). Scaling Genomics Data Processing with Memory-Driven Computing to Accelerate Computational Biology. In: Sadayappan, P., Chamberlain, B.L., Juckeland, G., Ltaief, H. (eds) High Performance Computing. ISC High Performance 2020. Lecture Notes in Computer Science(), vol 12151. Springer, Cham. https://doi.org/10.1007/978-3-030-50743-5_17
Download citation
DOI: https://doi.org/10.1007/978-3-030-50743-5_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50742-8
Online ISBN: 978-3-030-50743-5
eBook Packages: Computer ScienceComputer Science (R0)