
Abstract
G-protein-coupled receptors (GPCRs) are the largest family of transmembrane receptors in fungi. These receptors have an important role in the transduction of extracellular signals into intracellular sites in response to diverse stimuli. They enable fungi to coordinate cell function and metabolism, thereby promoting their survival and propagation, and sense certain fundamentally conserved elements, such as nutrients, pheromones, and stress, for adaptation to their niches, environmental stresses, and host environment, causing disease and pathogen virulence. This chapter highlights the role of GPCRs in fungi in coordinating cell function and metabolism. Fungal cells sense the molecular interactions between extracellular signals. Their respective sensory systems are described here in detail.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
A wide range of bioactivemolecules, biotic or abiotic stimuli as diverse as light, visual, protons, taste stimuli, biogenic amines, Ca2+, odorants, amino acids, nucleotides, proteins, peptides, steroids, fatty acids, hormones, yeast mating factors, and even photons, transduce their extracellular signals to the intracellular environment by specific interaction with a class of G proteins coupled to receptors (GPCRs) (Maller 2003). Alfred Gilman and Martin Rodbell received the Nobel prize in 1994 for discovering GPCRs. The DNA and deduced amino acid sequences of more than 700 GPCRs are known, and all of these have stretches of 20–28 hydrophobic amino acids capable of forming transmembrane α-helices. Structural homology in the putative transmembrane regions between different members of the receptor superfamily have facilitated the molecular cloning of cDNAs encoding novel receptor sequences, and studies have highlighted the significance of discontinuous structural determinants in the definition of functional domains of these receptors (for more details, see Lismaa and Shine 1992). Josefsson (1999) suggested that the GPCRs can be classified into three superfamilies. The generation of multigene families by ectopic gene conversion (EGC) was first recognized as important for maintaining sequence identity between repeat copies of genes within the large rRNA gene cluster. The many multigene families include, for example, histones, GPCRs, ubiquitins, immunoglobulins, and major histocompatibility complex (MHC) genes. Functional analyses have demonstrated that the diversity of function elicited by individual neurotransmitters, hormones, etc. is at least partly derived from the existence of distinct structural receptor subtypes. Moreover, studies of the expression of cloned receptors in different cell lines have demonstrated that the functional response to receptor activation depends not only on which receptor subtype is involved, but also on the available repertoire of G proteins and intracellular effector systems. GPCRs consist of a single polypeptide that is folded into a globular shape and embedded in the plasma membrane of the cell. Seven segments of this molecule span the entire width of the membrane, which is the reason why GPCRs are sometimes called seven-transmembrane hydrophobic domain receptors (7TMs). The intervening portions loop both inside as well as outside the cell and have an extracellular N-terminus and a cytosolic C-terminus. GPCRs consist of a heterotrimer, possessing a predominantly hydrophilic guanine nucleotide-binding α-subunit (38–52 kDa), a β-subunit (35 kDa), and a γ-subunit (8–10 kDa) (Vauquelin and Von Mentzer 2007). The β- and γ-subunits are always closely associated, and the β–γ-heterodimers are presumed to be interchangeable from one G protein to another. These heterodimers are associated with the membrane via isoprenyl modifications of the γ-subunit and promote the predominantly hydrophilic α-subunit association with membranes and receptors. Generally, the α-subunits constitute the receptor-recognizing part of the G proteins and are largely involved in the recognition of effector components, which explains why the identity of a G protein is determined by the identity of its α-subunit. Based on the sequence of the α-subunits, G proteins have been grouped into four families: Gs, Gi/o, Gq/11, and G12/13, encoded by 16 genes (Vauquelin and Von Mentzer 2007). Repeated stimulation of a GPCR with its agonist over minutes results in a response that is decreased compared to the initial response. These effects that limit repeated GPCR activation are referred to as desensitization (Rajagopal and Shenoy 2018).
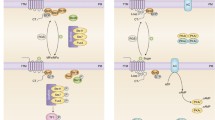
In higher plant genomes, only a single gene (or at most, a few genes) for the putative Gαs are found in contrast with the existence of a large number of genes for Gαs in mammalian genomes (23 Gα, 5 Gβ, 12 Gγ in humans). Members of each of the four families regulate key effectors (e.g., adenylate cyclase, phospholipase C, or directly regulate ion channel or kinase function) and generate secondary messengers (e.g., cAMP, Ca2+, IP3) that in turn trigger distinct signaling cascades. Therefore, GPCRs are likely to represent the most diverse signal transduction systems in eukaryotic cells. Signal perception occurs at the GPCRs, which act as guanine nucleotide exchange factors (GEFs) and facilitate the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on Gα (Fig. 3.1). The replacement of GDP bound to the α-subunit of the G protein by GTP after the activation of the receptor causes the dissociation of the GTP–Gα complex from the β–γ-dimer. In turn, GTP bound to the α-subunits of G protein initiates intracellular signaling responses by acting on effector molecules such as adenylate cyclases or phospholipases or by directly regulating ion channel or kinase function. The signal is turned off when GTP is hydrolyzed to GDP by the intrinsic GTPase activity of Gα, resulting in the reformation of the inactive heterotrimer GDP–Gαβγ. During the resting phase the Gα is guanosine diphosphate (GDP) bound and the three subunits form an inactive trimeric complex, GDP–Gαβγ. The rates of GTP hydrolysis of the Gα-subunit determine the intensity of the signal. Among many regulatory mechanisms, regulators of G-protein signaling (RGS proteins, COLD1, or phospholipases, Dα1) have a key function in the tight control of GPCR-G-protein-mediated signaling by accelerating the inherent GTPase activity of Gα, causing a faster turnover of the cycle. RGS proteins are pivotal in upstream regulation of fundamental biological processes in filamentous fungi, including vegetative growth, sporulation, mycotoxin/pigment production, pathogenicity, and mating. As an example, five distinct RGS proteins are found in the Aspergillus nidulans genome. Some genetic studies on yeast and studies on mammalian cells suggest that β–γ-subunits of G proteins may also regulate effector pathways. GPCRs are also expressed in proliferating cells, not only in fully differentiated cell functions. GPCRs have been implicated in embryogenesis, tissue regeneration, and growth stimulation. Many ligands acting via GPCRs are known to elicit a mitogenic response in a variety of cell types. Accumulated evidence indicates that GPCRs and their signaling molecules can harbor oncogenic potential. Plants possess hundreds of membrane-localized receptor-like kinases (RLKs). Interestingly, there is a surplus of receptor-like kinases (RLKs) that provide signal recognition at the plant cell surface. RLKs have conserved domain architecture, an N-terminal extracellular domain that is involved in signal perception, one to three transmembrane regions, and an intracellular protein kinase domain that transduces the signal downstream, typically by phosphorylating the effectors. There are multiple examples of interactions between plant G-protein components and RLKs (Choudhury and Pandey 2016).
|
|
|



3.2 Fungal GPCRs
In fungi, G proteins are integral for cell growth and division, mating, cell–cell fusion, morphogenesis, chemotaxis, virulence establishment, pathogenic development, and secondary metabolite production. Most filamentous fungi have three conserved Gα-subunits (I, II, III), one Gβ protein, and one Gγ protein. Several studies have identified bioinformatically the GPCRs encoded by various fungi: these include Aspergillus nidulans, Aspergillus fumigatus, Aspergillus oryzae, Magnaporthe grisea, Cryptococcus neoformans, Neurospora crassa, Verticillium spp., and Trichoderma spp. (Lafon et al. 2006). GPCRs have been divided into six families: A, B, C, D, E, and F. Among these families the following are related to fungi: family D is unique to fungi and comprises fungal pheromone receptors: fungal pheromone P-, α-factor receptors, and yeast GPR1 glucose receptors; and family E contains fungal pheromone A- and M-factor and cAMP receptors (Harmar 2001; Kulkarni et al. 2005). Han et al. (2004) identified nine GPCRs (GprA-I) in the A. nidulans genome, which are categorized into classes. Classes I and II include GprA (PreB) and GprB (PreA), which are similar to the yeast pheromone receptors Ste2 and Ste3, and function in self-fertilized sexual development (Seo et al. 2004). Class III includes GprC, GprD, and GprE receptors that might be involved in carbon source sensing on the basis of their high similarity to the Saccharomyces cerevisiae Gpr1 receptor (Xue et al. 1998; Kraakman et al. 1999). Class IV includes GprF and GprG, which are similar to the Schizosaccharomyces pombe Stm1 receptor, and the nutrient sensor Stm1-like proteins (Chung et al. 2001). The Stm1 receptor senses the cell nutritional state, thereby driving the cells to enter meiosis when encountering nutritionally deficient conditions. Class V includes GprH and GprI, which are similar to the Dictyostelium discoideum cAMP receptor cAR1 and thus have been proposed to be involved in cAMP sensing (Galagan et al. 2003). Later, Lafon et al. (2006) carried out an exhaustive comparative analysis of the genomes of three aspergilli: Aspergillus nidulans, A. fumigatus, and A. oryzae, and identified 7 additional GPCRs in A. nidulans: GprJ (class IV), GprK (class VI), GprM and GprN (class VII), GprO and GprP (class VIII), and NopA (class IX), totaling 16 potential GPCRs classified into nine classes (Lafon et al. 2006). Recently, a total of 10 GPCRs in N. crassa were divided into five classes: pheromone receptors (Pre-1 and Pre-2), cAMP receptor-like proteins (Gpr-1, Gpr-2, Gpr-3), carbon sensors (Gpr-4), putative nitrogen sensors (Gpr-5 and Gpr-6), and microbial opsins (Nop-1 and Orp-1) (Borkovich et al. 2004; Li et al. 2007). In the basidiomyceteCryptococcus neoformans, Xue et al. (2006) identified a large gene family of 7-TM proteins. Krishnan et al. (2012) provided the first evidence that four of the five main mammalian families of GPCRs, namely rhodopsin, adhesion, glutamate, and frizzled, are present in fungi. In the N. crassa genome a total of 10 receptors were predicted (Galagan et al. 2003). A recent report for A. nidulans identified GPCRs similar to the yeast pheromone receptors, the glucose-sensing receptor GPR1, the nitrogen-starvation sensing STM1, and the D. discoideum cAMP receptors (Han et al. 2004). In A. nidulans, the G-alpha subunit GanB and the G-protein-coupled receptor (GPCR) GprH have been shown to be involved in glucose sensing. GanB is involved in mediating activation of cAMP synthesis and subsequent PKA activation in the presence of glucose during early conidial germination events. The near-complete identification and characterization of both positive (GPCRs, G proteins, PhLPs, and effectors) and negative (RGS proteins) controllers of G-protein signaling in A. nidulans will provide us with insights into understanding the mechanisms underlying morphogenesis, pathogenicity, and toxigenesis in less genetically tractable but otherwise medically and agriculturally important fungi. Moreover, as many human diseases are associated with deleterious G-protein-mediated signals, understanding the molecular events resulting from dysfunctional regulation of G-protein signaling in A. nidulans may illuminate the nature of certain human diseases (Yu 2006). It is established that G proteins are involved in plant defense and suggested that they relay signals from defense-related receptor-like proteins (RLKS).
3.3 GPCR and Yeast-Secreted Pheromones
Yeast, which was the first eukaryotic genome to be sequenced, provides an exemplary model system and tools for improving our knowledge of GPCRs and their signaling in multicellular eukaryotes (Xue et al. 2008). In Saccharomyces cerevisiae, two different haploid cells exist: mating type a (MATa, a-cell) and mating type α (MATα, α-cell) as a result of meiosis, defining what is termed a bipolar system. The two types of haploid (ascospores) are often called mating types because they describe the mating behavior: mating occurs only between an a-cell and an α-cell. The mating type of a haploid cell is determined by its genotype at the mating-type (MAT) locus on chromosome III. The two variants of the MAT locus, MATα and MATa, are referred to as idiomorphs rather than alleles because they differ in sequence, size, and gene content (Seraj Uddin et al. 2016; Singh et al. 1983). The MATα idiomorph contains two genes, MATα1 and MATα2, whereas the MATa idiomorph contains a single gene, MATa1, and those three genes code for transcription regulators (Hanson and Wolfe 2017). They determine the cell type of the haploid by activating or repressing the expression of a-specific genes (asg) and α-specific (αsg) genes (Haber 2012). In Saccharomyces cerevisiae, the pheromone genes MFa1 (a-mating factor, MFa, a-pheromone, which is a post-translationally modified peptide, its precursor protein does not enter the secretory pathway but is processed and matured in the cytoplasm where the generated peptide exits the cell via a transporter) and MFα1 (α-mating factor, MFα, α-pheromone produced from a long precursor that enters the secretory pathway and is processed by KEX1, KEX2, and STE13 proteases to finally release repeated peptides through exocytosis) are asg and αsg genes, respectively. The α-pheromone is produced from prepro-proteins, which display a signal peptide and are not post-translationally modified, a proregion, and four (MFα1) or two (MFα2) repetitions of nearly identical motifs, each of which is preceded by an easily recognizable protease cleavage site. This cleavage site is composed of the “KR” dipeptide followed by “EA” or “DA” dipeptides (e.g., KREA or KRDA). The KEX1 and KEX2 proteases, respectively, cut before the K and after the R of the KR dipeptide. STE13 then cleaves after the A of the EA or DA residues, thus releasing the repeated peptides. The motif recognized by STE13 is, however, more variable in sequence and in length and is often a repetition of XA or XP dipeptides (X, any amino acid). This processing occurs in the Golgi apparatus while the protein passes through the secretory pathway. The pheromone signaling pathway G-protein-subunit genes GPA1, STE4, and STE18 are haploid-specific genes (hsg) hsgs, and the mitogen-activated protein kinase “MAP kinase” FUS3 is a general pheromone-activated gene (Sorrells et al. 2015). In the haploid α-cells, the MATα1 gene codes for the HMG-domain transcription activator α1 (previously referred to as an “α-domain” protein but now recognized as a divergent HMG domain) (Martin et al. 2010) and the MATα2 gene code for the homeodomain-transcription repressor α2. The α1 and α2 proteins can both individually form complexes with the constitutively expressed Mcm1 (MADS domain) protein, which binds upstream of asgs and αsgs. In α-cells, the transcription of αsgs is activated because the α1–Mcm1 complex recruits the transcription factor Ste12 to their promoters, whereas the transcription of asgs is repressed because the α2–Mcm1 complex recruits the Tup1-Ssn6 corepressor. The MAT locus in a-haploid cells contains only the MATa1 gene coding for the homeodomain protein a1, but this protein is not required for a cell-type identity, which is instead defined by the absence of both α1 (the activator of αsgs) and α2 (the repressor of asgs). Instead of requiring an a-specific activator, asgs are activated by Mcm1 and Ste12, which are constitutively expressed in all cell types. Thus, in S. cerevisiae, the a-cell type is the default type, and yeast cells lacking a MAT locus will mate with α-haploid cells. In a/α diploid cells, αsgs, asgs, and hsgs are all repressed. These cells have MATα1 and MATα2 genes at the MAT locus on one chromosome, and MATa1 on the other, which results in the formation of a heterodimer a1–α2 of the two homeodomain proteins. There are approximately 5 to 12 asgs and αsgs, depending on the species (Sorrells et al. 2015). In addition to these, a shared set of haploid-specific genes (hsgs) (~12–16 in number) that facilitate mating is constitutively expressed in both a and α cells but not in a/α diploids (Booth et al. 2010), and a larger group of about 100 general pheromone-activated genes is induced in haploids of both types once a pheromone signal from the opposite type of haploid is detected (Sorrells et al. 2015). Because S. cerevisiae uses the formation of a heterodimer to sense heterozygosity of its MAT locus, and because this heterodimer is a repressor, there are no “diploid-specific” genes in S. cerevisiae (Galgoczy et al. 2004). Indeed, diploid-specific processes such as meiosis and sporulation are repressed in haploids. This repression is achieved via the hsg RME1, a haploid-specific activator that transcribes IRT1, a noncoding RNA which in turn represses IME1, the master inducer of meiosis. Thus, the combined action of RME1 and IRT1 inverts the output of the hsg regulatory logic to restrict IME1 expression to diploids. IME1 expression also requires the environmental signals of nitrogen and glucose depletion that initiate meiosis. No genes have constitutive diploid (a/α) specific expression in the same way that hsgs, αsgs, and asgs have constitutive cell type-specific expression in haploids. Mating of MATa and MATα cells produces diploid zygotes (MATa/MATα), which will not exhibit any mating type and therefore cease to secrete pheromones. Mating is initiated in response to the pheromone secreted by haploid cells. The lipopeptide a-pheromone released by a-cells is a C-terminally methylated and farnesylated 12-residue peptide that makes it very hydrophobic and acts on α-cells whereas the simple α-pheromone is an ordinary 13-residue peptide that acts on a-cells (Aksam et al. 2013). Their presence is communicated to the response machinery within the cell by means of a heterotrimeric G-protein complex that, when activated by the pheromone-bound receptor, serves in turn to activate a downstream mitogen-activated protein (MAP) kinase cascade module. The two loci MFAL1 and MFAL2 (MFαl and MFα2) are coding for the α-pheromone that arrests the a-cells at the G1 stage of the cell cycle as a method of synchronization of the two haploid cells before mating, thus ensuring that only unbudded and mononucleate haploids fuse to form diploid zygotes. Similarly, a-cells produce the peptide a-factor, which arrests the division cycle of α-cells at the Gl phase. In addition to regulating cell division, these factors induce formation of cell-surface agglutinins encoded by the alpha-agglutinin structural gene, AG alpha1. Alpha-agglutinin is a cell adhesion glycoprotein that promotes the aggregation of opposite cell types, and α-factor as well as a-factor elicit localized elongations “shmoos” of the target a-cell and α-cell, respectively, which may form the site for nuclear migration and fusion. Although α-cell mating requires only one of its two genes to be functional, typically both loci are transcribed, albeit at different rates (MFAL1 ⋙ MFAL2) (Kurjan 1985). The ability to respond to pheromones is controlled by at least eight additional genes: STE2, STE4, STE5, STE7, STE8, STE9, STE11, and STE12. Among these STE genes, STE2 and STE3 are receptors responsible for pheromone sensing. The a-cells express a unique GPCR, Ste2, which is the receptor for α-factor; α-cells express a different GPCR, Ste3, which is the receptor for a-factor. Mutations in any one of these genes prevent a-cells from arresting cell division, producing agglutinins, and altering cell morphology in response to α-factor. Similarly, pheromone sensitivity of α-cells requires the same genes with the exception of STE2. Cell-type specificity of the ste2 mutation raises the possibility that the STE2 gene encodes a cell-surface receptor that recognizes α-factor (Hartwell 1980; Jenness et al. 1983; MacKay and Manney 1974). The STE2 gene, which is necessary for stability of the binding activity, is likely to encode a structural component of the α-factor receptor, and it is likely to encode an integral part of the α-factor receptor because mutations in this gene affect the physical properties of the binding activity (Jenness et al. 1983). Their results were consistent with the view that a single type of receptor elicits different responses at different α-factor concentrations. Because of the large number of α-factor binding sites detected, they concluded that a-cells can potentially sense a wide range of α-factor concentrations. Physiological responses may be controlled by an intracellular “signal” generated by the binding of α-factor to its receptor. Di Segni et al. (2008, 2011) showed that a cryptic polyadenylation site is present inside the coding region of the a-specific STE2 gene, encoding the receptor for the α-factor. The two cell types (a- and α-cells) produce an incomplete STE2 transcript, but only a-cells generate full-length STE2 mRNA. The tRNA splicing endonuclease is able to produce trans-spliced mRNAs. During their work, Di Segni et al. discovered a previously unnoticed cryptic polyadenylation site early in the STE2 coding region. Cleavage and polyadenylation of pre-mRNAs are essential to ensure transcription termination. For this kind of regulation to be effective, the repression should be very tight. If α-cells produced even a small amount of the Ste2 receptor, they would undergo autocrine activation of the mating pathway by the α-factor that these cells secrete, leading to growth arrest. Therefore, the genes encoding the pheromone receptors should be very strictly regulated. The internal poly(A) site would eliminate rare transcripts of STE2 escaping repression. Conversely, the other mating-type receptor gene, STE3, is induced only in a-cells and not expressed at all in α-cells. The regulation of yeast mating genes is achieved through a concerted mechanism that involves transcriptional and posttranscriptional events. In a-cells, the STE2 gene is actively transcribed, the upstream poly(A) site is skipped as a result of the high transcription rate, and the canonical poly(A) site in the 3′-UTR is prevalently used. In contrast, in α-cells, STE2 is repressed and rare transcripts escaping the repression will abort as a result of polyadenylation at the cryptic site inside the coding region. The early cryptic polyadenylation site in STE2 contributes to its shutoff in α-cells, thus avoiding autocrine activation of the pheromone response pathway that could occur as a result of a leaky repression of transcription. Conversely, the STE3 gene, being always turned off except for where it should be expressed, does not need this further level of control, and indeed, no cryptic polyadenylation site is found in its coding region. The α-factor secreted by α-cells is bound by the Ste2 receptor and ectopically expressed; it induces pheromone response. Mating is elicited by the binding of α-factor and a-factor, respectively, to G-protein-coupled receptors Ste2 and Ste3, specifically expressed in a- or α-cells (Burkholder and Hartwell 1985; Hartig et al. 1986). Ste2 and Ste3 both activate the same Gα-subunit Gpa1 to facilitate the replacement of GDP by GTP, which dissociates the G-protein subunits Gα from the Gβγ (Ste4/Ste18) complex. Expression of all these genes (SCGI, STE4, STE78, STE2, STE3) is haploid specific (they are transcribed in both haploids a- and α-cells, but not in a/α diploids), as is the response to mating pheromones. Unusually, in the Saccharomyces cerevisiae pheromone-signaling pathway, it is the Gβγ complex that functions as the main driving force, not Gα, to induce the downstream pheromone-signaling responses, and cells lacking either subunit of the Gβγ complex are blocked for all mating responses (Whiteway et al. 1989). Free Gβγ then activates the signaling branch responsible for regulating cell division and the cell polarity branch responsible for polarized growth (Johnson et al. 2011). The Gβγ-subunit binds to a Ste5–Ste11 complex and to the Ste20 kinase (Leeuw et al. 1998). The scaffold protein Ste5, the PAK kinase Ste20, and the Cdc24/Far1 complex are three main downstream targets of the Gβγ complex. When bound to Ste5, the Ste4–Ste18 complex facilitates its membrane recruitment and places the scaffold protein, the entire mitogen-activated protein kinase (MAPK) module and Ste20, into close proximity to enable signaling circuit activation (Leeuw et al. 1998; Pryciak and Huntress 1998). The MAPK module is a three-tiered phospho-relay system composed of Ste11 (MAPKKK), Ste7 (MAPKK), and Fus3 (MAPK). Upon signal activation, the phosphor-activated Fus3 releases the downstream transcription factor Ste12 from inhibition by Dig1/Dig2, which induces the expression of several mating-specific genes (Bardwell et al. 1994). Haploid yeast cells use a prototypic cell signaling system to transmit information about the extracellular concentration of mating pheromone secreted by potential mating partners (Yu et al. 2008). Recent studies on the yeast pheromone response have shown how positive feedback generates switches, negative feedback enables gradient detection, and coherent feedforward regulation underlies cellular memory (Atay and Skotheim 2017). The ability of cells to respond distinguishably to different pheromone concentrations depends on how much information about pheromone concentration the system can transmit. They showed that the MAPK Fus3 mediates fast-acting negative feedback that adjusts the dose response of the downstream system response to match that of receptor–ligand binding. This “dose–response alignment,” defined by a linear relationship between receptor occupancy and downstream response, can improve the fidelity of information transmission by making downstream responses corresponding to different receptor occupancies more distinguishable and reducing amplification of stochastic noise during signal transmission. They also showed that one target of the feedback is a novel signal-promoting function of the RGS protein Sst2. Negative feedback is a general mechanism used in signaling systems to align dose responses and thereby increase the fidelity of information transmission (Yu et al. 2008). When cells sense pheromones, the interaction between a pheromone and its receptor in either haploid cell type triggers a MAP-kinase signaling cascade resulting in G1-phase arrest of mitotic proliferation. Then, the mating pathway is activated, the transcriptional profile changes, and they exhibit a chemotactic response by the formation of a mating projection (shmooing) toward the mating partner polarized toward the pheromone source, and finally mating by cell and nuclear fusion to generate a diploid zygote (Fig. 3.2). Components of the pheromone-signaling pathway, from the upstream receptor–G-protein complex to the downstream transcription factor, are all required for these mating responses (Herskowitz 1995).

The mating pathway is activated, the transcriptional profile changes, and they exhibit a chemotactic response by the formation of a mating projection (shmooing) toward the mating partner polarized toward the pheromone source. Blue balls refer to a pheromone while red square refer to α pheromone and each of which are bound to their respective receptors
For haploid cells (both a and α), cell type-specific processes include the induction of competence to mate and the repression of sporulation, whereas diploid cells require repression of mating and the ability to initiate meiosis and sporulation. The mating process of budding yeast (S. cerevisiae) is, to date, the best studied example of chemotropism. In mating mixtures, haploid yeast cells can interpret a shallow pheromone gradient, the chemotrope, toward the closest mating partner, and fuse to form a diploid zygote. How yeast cells accurately position the polarity machinery toward the source of pheromone is unclear. It is well known that the pheromone receptor and its cognate G protein are uniformly distributed on the plasma membrane of vegetative cells and that they polarize in response to pheromone (Wang and Stone 2017). Gradient sensing, inhibition of receptor phosphorylation by Gβγ, results in differential phosphorylation of the receptor across the cell surface, and, consequently, lesser internalization of the receptor and G protein on the up-gradient side of the cell. A key question is how the uniformly distributed surface receptor competes for a limited amount of G protein. Wang and Stone (2017) showed that in mating cells the initially uniform receptor and G protein first localize as polarized crescents at the default polarity site. The receptor and G-protein crescents then track along the plasma membrane until they reach the region of highest pheromone concentration, centered around the position at which the cell ultimately shmoos toward its partner. They also showed that polarization of Gβ to the default polarity site is independent of receptor phosphorylation and polarization, whereas Gβ tracking from the default site to the eventual chemotropic site does not occur if receptor phosphorylation and redistribution are blocked. These observations suggest a new mechanism that localizes the receptor with its much less abundant G protein. In their revised model, they proposed that mating cells that are arrested in G1 cell-cycle phase concentrate Gβg at the default polarity site, likely through its interaction with Far1-Bem1-Cdc24-Cdc42. The polarized Gβg then protects the receptor from being phosphorylated and internalized, thereby triggering local accumulation of the receptor and G protein. Because the pheromone gradient is mirrored by a gradient of signaling activation within the receptor/G-protein crescent, there are higher proportions of active unphosphorylated receptors and active G protein closer to the pheromone source. The peak of signaling activity incrementally moves up the pheromone gradient, as unprotected receptors are phosphorylated and co-internalized with G proteins at the back, while vesicles containing nascent receptors and G proteins preferentially dock where the receptor is most abundant.
The phenomenon that the two haploid cell types of yeast (a and α) are able to interconvert in a reversible manner by means of a programmed DNA rearrangement process is called mating-type switching. Mating-type switching is the process by which a haploid a-cell can become a haploid α-cell, by changing its genotype at the mating-type (MAT) locus from MATa to MATα, or vice versa. Unicellular organisms that do not contain separate germline and somatic DNA cannot make permanent changes to their genomes during development, as these will be transmitted to offspring. Instead, programmed DNA rearrangements underlying cell-type specification in these organisms must be reversible (Nieuwenhuis and Immler 2016). In unicellular organisms every cell must retain the capacity to produce every other type of cell. Mating-type switching was the subject of early studies in S. cerevisiae genetics and molecular biology (Oshima 1993; Barnett 2007; Klar 2010). Its mechanism of switching is complex and involves multiple components as well as multiple levels of regulation. The dissection of how cell-type specification and mating-type switching is controlled in S. cerevisiae led to breakthroughs in our understanding of many other fundamental cellular processes including homologous recombination, cell signaling pathways, gene silencing, and mechanisms of transcriptional regulation (Haber 2012). In fact, the idea of using arrows and T-bar symbols in network diagrams to symbolize gene activation and repression, respectively, is attributable to Ira Herskowitz (Botstein 2004), whose laboratory discovered the cassette mechanism of switching in S. cerevisiae (Fig. 3.3a and b). Switching seemed to appear abruptly within the family Saccharomycetaceae (Butler et al. 2004). The characterization of homothallic and heterothallic strains of S. cerevisiae led to the discovery of genetic loci controlling homothallism and ultimately to the cassette model of mating-type switching. Switching mating types in S. cerevisiae involves a unidirectional DNA replacement. The cassette model states that a haploid cell can switch its genotype at the MAT locus (from MATa to MATα idiomorph or vice versa) by a gene conversion process. Although mating-type switching in S. cerevisiae is often called gene conversion, it is more accurately described as a synthesis-dependent strand annealing (SDSA) process because of the nonhomology of the Y-regions (Fig. 3.3a) between the outgoing and incoming alleles (Ira et al. 2006). The current active gene content at the MAT locus of a haploid cell is replaced by copying a reserve version of the MAT gene of the opposite allele, stored at a transcriptionally silent location (Lee and Haber 2015). This process requires the genome to have three copies of mating-type sequence information, all of which are on chromosome III in S. cerevisiae: the active MAT locus (either MATa or MATα), two silent loci termed HML (containing the reserve copy of MATα sequence information) and HMR (containing the reserve copy of MATa sequence information) (Fig. 3.3b). All three loci are flanked by identical sequence regions called X and Z (Fig. 3.3a). The Y-region in the center comes in two forms, Ya and Yα, that are allelic but completely different in sequence. During switching, the actively expressed MAT locus is cleaved by the endonuclease HO encoded by the HO gene on chromosome IV, at a site that marks the boundary between the Y-sequences unique to the MATa or MATα idiomorphs and the shared Z-sequence flanking them, and eliminates the allele at the active MAT locus. Switching is initiated when the HO endonuclease makes a dsDNA break at the Y–Z junction of MAT. The 3′-end of a DNA strand from the Z-region beside MAT then invades the donor (HMR or HML) locus and is extended by a DNA polymerase through the donor Y-region and into the X-region, after which it reinvades the MAT locus. Finally, the second strand of the new Y-region at MAT is synthesized in the direction from X to Z. Switching is slow, taking approximately 70 min, and is more than 100 times more error prone than normal DNA replication (Hicks et al. 2011; Hanson and Wolfe 2017). In other words, the gap is subsequently filled in through gene conversion guided by homology at the X- and Z-regions; the cleaved MAT locus uses HML or HMR as a template for DNA repair with a strong preference for the silent locus containing the mating-type information opposite to the current MAT genotype (Haber 2012). After MATa is cleaved by HO endonuclease, it is normally repaired by copying HML-alpha, creating a new MAT-alpha cell; this occurs in about 95% of the cells. In the other 5% of cells, the cleaved MATa is repaired by copying HMRa, creating MATa again. This MATa to MATa switching is called “futile switching” because it does not make any difference to the cell. Similarly, when a MAT-alpha cell tries to switch, about 5% are futile MAT-alpha to MAT-alpha switches, and 95% are productive MAT-alpha to MATa switches. The futile switches occur because the recombination enhancer (RE) element is not perfect; that is, the RE makes the direction switching strongly biased but not completely biased. It is possible that during meiosis DNA double-strand breaks are induced similarly at high frequency in the MAT flanking recombination hot spots and the intra-MAT gene conversion hot spot. In S. cerevisiae, cells typically switch their mating types every other generation and only mother cells that have divided twice can switch (Sun and Heitman 2016). HO is expressed only in cells that have budded once, which means that only mother cells switch mating type and can mate their second daughter neighboring cells. Hence, most natural isolates of S. cerevisiae are diploid and phenotypically homothallic. The question of whether a mating-type system similar to that of S. cerevisiae is found in other hemiascomycetes has become pertinent recently because of the discovery of mating-type-like (MTL) loci in Candida species that had been regarded as asexual. The Candida albicans genome sequence includes an MTL locus but neither silent cassettes nor a HO endonuclease gene (Butler et al. 2004), although C. albicans seems to have homologues of all the elements of a functional pheromone response pathway involved in mating in S. cerevisiae (STE2, STE3, Gα(Gpa1), Gβ(Ste4), Gγ(Ste18), (MAP) STE20, STE11, STE7, FUS3, and Ste12p) but lacks many homologues of S. cerevisiae genes for meiosis (Tzung et al. 2001).
|
|
Fig. 3.3 (a) Organization of repeat sequences flanking the MAT loci in four species (Klar 2007; Hanson et al. 2014). In Komagataella phaffii, the region that becomes inverted during mating-type switching is 138 kb long and was recently discovered to contain a centromere at its approximate center (Coughlan et al. 2016). CEN, centromere; TEL, telomere. (Source: By courtesy and permission of Kenneth H. Wolfe and Genetics Society of America) (https://doi.org/10.1534/genetics.117.202036) | Fig. 3.3 (b) Gene organization in the MAT, HML, and HMR loci on Sacchormyces cerevisiae chromosome III. Shading indicates genes whose transcription is repressed. (Source: By courtesy and permission of Kenneth H. Wolfe and Genetics Society of America) (https://doi.org/10.1534/genetics.117.202036) |


The ambiguity of the meiotic occurrence in Candida albicans to date does not let us know much about its switching mating-type mechanism. Chlamydospore formation is a characteristic of many fungal species, among them the closely related human-pathogenic dimorphic yeasts C. albicans and Candida dubliniensis. Although function and regulation of filamentation are well studied in these species, the basis of chlamydospore formation (although chlamydospores are non-meiotic, but are asexual spores) is mostly unknown (Böttcher et al. 2016; Navarathna et al. 2016). Ho-mediated switching between an active MAT locus and silent cassettes exists only in the Saccharomyces sensu stricto group and their closest relatives: Candida glabrata, Kluyveromyces delphensis, and Saccharomyces castellii. Butler et al. (2004) identified, in C. glabrata, an MTL1 as the ortholog of the MAT locus of K. delphensis and showed that switching between C. glabrata MTL1a and MTL1α genotypes occurs in vivo. The more distantly related species Kluyveromyces lactis has silent cassettes but switches mating type without the aid of Ho endonuclease. Very distantly related species such as C. albicans and Yarrowia lipolytica do not have silent cassettes. The Naumovozyma castellii mating-type system is organized as in S. cerevisiae, and in many other Saccharomycetaceae members, such as C. glabrata and Nakaseomyces delphensis (syn. Kluyveromyces delphensis) (Butler et al. 2004). The system includes a MAT locus, an HO endonuclease gene, silent HMR and HML cassettes, and the MATα1, MATα2, and MATa1 genes that express and regulate mating-type specific features (Butler et al. 2004). In N. castellii the MAT locus and HMRa and HMLα silent cassette are located on chromosome II. The sexual state of the cell is defined by expressing the genetic information present at the MAT locus. Thus, a mating-type switch involves replacement of this information with a copy of the opposite genotype that resides in the silent locus, that is, by a gene conversion process. The mating-type switch in N. castellii initiates with the introduction of a double-stranded break at the MAT locus, performed by Ho endonuclease encoded by the HO gene. The mating-type information is then copied from the silent HMRa and HMLα cassettes that store a- and α-specific sequence information, respectively. Overall, N. castellii therefore conforms to the conventional mating-type system as known for the very well studied pathway in S. cerevisiae, although with some deviations in the details (Andersson and Cohn 2017). The work by Le Marquer et al. (2019) presents the largest analysis of fungal secreted proteins predicted to release repeated peptides through KEX2 activity. The best characterized representatives of these proteins are the α sexual pheromones of Ascomycota. Cyclic peptides produced in Ascomycota from KEX2-processed precursor proteins belong to the family of ribosomally synthesized and post-translationally modified peptides (RiPPs), and they act as mycotoxins. Le Marquer et al. hypothesized that many proteins with characteristics similar to α sexual pheromones would be present in many if not all fungal species. A few species, scattered all along the fungal kingdom, were shown to possess such proteins. They reported that sexual pheromone genes are present in the following presumed asexual fungi: Cladosporium fulvum, Aspergillus niger, Verticillium dahliae, Fusarium oxysporum, and Trichoderma reesei. It is also important to consider that sexual pheromones have other important sex-independent roles such as biofilm production or conglutination of hyphae. These KEX2-processed repeat proteins (KEPs) present a clear STE13 signature, identical to that of α-type pheromone protein precursors of Ascomycota. GPCRs are also present in these fungi to perceive these peptides. The investigators speculated that KEPs with clear STE13 signatures may act as hormones or pheromones regulating distinct cellular programs. KEPs may be precursors of peptides, regulating some of the hyphal polar growth, septation, branching, fusion or healing, hyphal network coordination, and production of sexual and/or vegetative reproduction bodies and spores. KEPs displaying peptides with a STE13 signature are therefore good candidates for the release of peptidic hormones that may have distinct roles from sexuality and may regulate other aspects of fungal biology.
3.4 Mating in the Basidiomycota Ustilago maydis and Sporisorium scitamineum
Although Basidiomycota do not use α-type pheromones for their sexual reproduction, Le Marquer et al. (2019) identified several proteins with α-type pheromone features in their secretomes. Basidiomycota have conserved proteins with striking similarities to α-sexual pheromones so far only described in Ascomycota. The close relationship between Ustilago maydis and Sporisorium scitamineum (subphylum Ustilaginomycotina) can be evidenced by the misclassification of S. scitamineum as U. scitaminea. Generally, Ustilago infects all aerial parts of the corn plant and rapidly forms galls or tumors filled with spores. In contrast, Sporisorium infects young sugarcane seedlings, remains asymptomatic, and grows systemically until the emergence of a mass of sooty spores. The whole genome character of S. scitamineum is most similar to U. maydis, which belongs to a separate genus. In the phylum Basidiomycota, a wide variety of lifestyles are represented, ranging from well-known and conspicuous wood-decaying mushrooms, plant growth-promoting and mutualistic mycorrhizae, and crop-destroying smut and rust fungi, to yeast-like human pathogens. Lifestyle differences have consequences for the mating and breeding systems of these fungi. Basidiomycetes have been recognized as having diverse breeding systems, from homothallism to heterothallism. The study of breeding systems, for example, led to the discovery of the astounding variability in mating-type alleles among mushrooms, with thousands of different mating types in some species, and to the realization that in many fungal pathogens the process of sexual reproduction is closely linked to infection and pathogenicity. In basidiomycetes, the sexual cycle typically involves the fusion of genetically distinct homokaryotic hyphae or haploid yeast cells to produce a dikaryon in which the two haploid parental nuclei are replicated in a coordinated fashion without fusion during hyphal elongation. Nuclear fusion (karyogamy) then takes place in the basidia or in other specialized structures (e.g., teliospores), after which the diploid nucleus undergoes meiosis to generate haploid basidiospores (meiospores) and complete the life cycle. One common underlying feature shared by most fungi is the lack of genetically determined anisogamy [i.e., a situation in which a species has two classes of gamete sizes: one class of large gametes (female function) and one class of small gametes (male function) and a single genotype can produce both types of gametes in most anisogamous fungi]. When the two MAT loci are unlinked, four mating types can be generated by meiosis among the haploid progeny, defining this as a tetrapolar breeding system. Other basidiomycetes have instead a bipolar system controlled by a single MAT locus, either because the pheromones and pheromone receptors P/R and the heterodimeric or homeodomain-type HD loci are linked or because one has lost its function in mating-type determinism (Coelho et al. 2017). In mushroom-forming species (Agaricomycetes), there has been a generalized diversification of alleles at both MAT loci, in some cases yielding species with hundreds or thousands of possible mating types (Coelho et al. 2017). Mating is regulated by two genetic mating-type MAT loci: one locus (a) encodes two, a1 and a2, tightly linked genes encoding P/R (Rowell and DeVay 1954; Holliday 1961; Puhalla 1968), whereas the b locus encodes two subunits that harbor conserved genes of a HD transcription factor determining the viability following syngamy (i.e., haploid cell fusion during mating) (Bakkeren et al. 2008). Different a-alleles are necessary, together with different b-alleles, for the development of the filamentous form (Banuett and Herskowitz 1989). Schulz et al. (1990) reported that the fungal pathogen of corn, U. maydis, has two forms: one is yeast like and nonpathogenic, and the other is a dikaryotic filamentous and pathogenic form. The b mating-type genes encode two subunits consisting of an HD1 class and an HD2-class protein that are not related to each other in primary sequence; however, they share a common domain organization. The N-terminal regions of the HD proteins contain the highest degree of variation when different alleles are compared and are thus designated as the variable regions, whereas the C-terminal regions of the proteins, including the homeodomains, are highly conserved (Gillissen et al. 1992; Kronstad and Leong 1990; Schulz et al. 1990). The b locus, with 25 different alleles and most probably more than that, regulates this dimorphism: any combination of 2 different alleles triggers pathogenic development, whereas the presence of identical alleles results in the yeast-like form. Syngamy is governed by, in its simplest form, small 10- to 15-amino acid–lipopeptide pheromones derived from 35 to 40 amino acid precursors through post-translation modifications at both the N- and C-termini. These diffusible pheromones are received by seven transmembrane-domain pheromone receptors, coupled to a G protein for downstream signal transduction. This molecular determination of mating fusion is similar in part to the a- and α-factor P/R system in the Ascomycetes. Therefore, instead of the a-factor/Ste3 and α-factor/Ste2 coupled sensing system characteristic of Saccharomyces cerevisiae, “chemo-sensing” specificity in basidiomycetes is mediated by allelic variants of the same type of genes. Mating is often initiated by a reciprocal exchange of pheromones recognized by matching pheromone receptor variants in both mating types, and thus two strains are needed to carry different alleles of the pheromone and receptor genes at the P/R locus. Gillissen et al. (1992), Kronstad and Leong (1990), and Schulz et al. (1990) have cloned four b alleles (bi, b2, b3, b4) and showed that the b locus contains a single open reading frame (ORF) of 410 amino acids with a variable N-terminal region and a highly conserved C-terminal region (60% and 93% identity, respectively). Mutational analysis confirms that this ORF is responsible for b activity. The b polypeptides appear to be DNA-binding proteins because they contain a motif related to the homeodomain in their constant region. The investigators proposed that combinatorial interactions between b polypeptides generate regulatory proteins that determine the developmental program of the fungus. They discussed also the association of b polypeptides. Snetselaar and McCann (2017) suggested revising the U. maydis life cycle based upon their recent findings. Their microscopic examination of both living and fixed tumor material showed that nuclei fuse long before sporulation begins and that tumors are filled with uninucleate cells undergoing mitosis. Quantification of DNA in the nuclei confirmed these observations. Additionally, fungal cells from tumor material placed on nutrient agar produced colonies of diploid budding cells. Time-lapse observations showed that at least some of these colonies arose from thin-walled fungal cells rather than from immature spores. Ultrastructural examination of developing teliospores from tumors confirmed that these were uninucleate. Condensed chromatin and other structures characteristic of nuclei in prophase I of meiosis were observed. The bipolar species Sporisorium scitamineum and the tetrapolar species Ustilago maydis possess one divergently transcribed gene pair that encodes the homeodomain proteins bE (HD1) and bW (HD2). The MAT-1 locus, gene order, and orientation, as well as the genomic context, are conserved in the b mating-type genes. Interestingly, both bE and bW mating-type genes are present in the genomes of Ustilaginaceae, including the two genera of Ustilago and Sporisorium. The a and b loci are linked, and the mating-type locus (MAT) segregates independently in the tetrapolar Sporisorium reilianum and U. maydis. S. scitamineum is a bipolar mating fungus; the a and b loci are linked, and the mating-type locus segregates as a single locus. Only the dikaryotic hyphae formed by fusion of compatible sporidia can infect the host (Feldbrügge et al. 2004). Que et al. (2014) explored the genomes of 12 fungi, S. scitamineum plus 11 other fungi, and identified members of the G-protein-coupled receptor family from the entire deduced proteomes. They demonstrated that S. scitamineum possesses only 6 GPCRs, which are grouped into five classes that are responsible for transducing extracellular signals into intracellular responses; however, the genome is without any PTH11-like GPCR. This total set of analyses also resulted in the identification of 7 and 5 putative GPCRs in S. reilianum and U. maydis, respectively; this was the first high-quality genome sequence of S. scitamineum and was also the first reported genome of sugarcane fungi. There are 192 virulence-associated genes in the genome of S. scitamineum, among which 31 are expressed in all the stages, which mainly encode for energy metabolism and redox of short-chain compound-related enzymes. Sixty-eight candidates for secreted effector proteins (CSEPs) were found in the genome of S. scitamineum, and 32 of them expressed in the different stages of sugarcane infection, which are probably involved in infection or triggering defense responses (Que et al. 2014).
The basidiomycete U. maydis is a member of the smut fungi, a large group of biotrophic parasites that infect mostly grasses. Genome-wide analysis demonstrated that U. maydis is more closely related to humans than to budding yeasts (Steinberg and Perez-Martin 2008). With the advent of molecular genetics, a wide range of tools was developed allowing precise genome modifications. Research in U. maydis initially followed three main directions: (i) the characterization of genes involved in DNA recombination, (ii) the study of mating-type loci, and (iii) the so-called killer phenomenon, related to the presence of virus-like particles. Subsequently, researchers became interested in different aspects of the biology ranging from gene regulation, signaling, and virulence to cell biology. Community efforts led to a high-quality genome sequence that now serves as a blueprint for comparative studies (Kämper et al. 2006). Under appropriate conditions, such as ambient temperature and humidity, the diploid spores germinate, undergo meiosis, and form a promycelium into which the four haploid nuclei migrate. Septation then produces compartments that contain one haploid nucleus. Following mitosis, haploid cells bud off from the promycelium and enter the vegetative life cycle, during which they proliferate by budding. On the leaf surface, haploid cells of different mating types fuse and form a filamentous cell cycle-arrested dikaryon, which is the pathogenic form (Snetselaar and Mims 1992; García-Muse et al. 2003). These filaments differentiate into infection structures known as appressoria (for more details, see Matei and Doehlemann 2016; Lanver et al. 2017). Cells differing in the a mating-type locus recognize each other via lipopeptide pheromones, the cell cycle arrests in the G2 phase, budding is stopped, and conjugation tubes are formed. These structures develop at one cell tip and grow toward each other guided through the pheromone gradient until they merge (Brefort et al. 2009). The resulting dikaryon switches to polar growth if the two mating partners carry different alleles of the two unlinked mating type loci, a and b. The a locus, which exists in two alleles, a1 and a2, controls the fusion of haploid cells and is, together with the b locus, responsible for maintenance of the filamentous form (Banuett and Herskowitz 1989). The a locus has been shown to encode an a-pheromone-based recognition system (Bölker et al. 1992). The b locus is multiallelic; each allele codes for a pair of homeodomain proteins termed bE and bW, which are assumed to act as transcriptional regulators when appropriately combined. This assertion is based on data which show that development cannot be initiated in the absence of b gene products (Gillissen et al. 1992). The gene products exist in inactive combinations in haploid strains and in active combinations in the dikaryon. Recent experimental evidence has indicated that inactive bE-bW combinations differ from active combinations in their ability to form the heterodimer. The b locus codes for a pair of homeodomain transcription factors that dimerize when derived from different alleles and control subsequent sexual and pathogenic development. In response to both chemical and physical signals of the plant surface, the dikaryotic filament forms poorly differentiated appressoria that penetrate the cuticle, probably via the action of lytic enzymes (Brefort et al. 2009). After penetration, the cell cycle is reactivated concomitantly with the development of clamp-like structures that allow the correct sorting of nuclei to maintain the dikaryotic status (see Brefort et al. 2009). In this way, the fungus proliferates, forms a massive network of hyphae, and induces plant tumors. Hyphal growth inside the tumors is followed by sporogenesis, a poorly understood process that includes karyogamy, hyphal fragmentation, and differentiation into melanized diploid teliospores. Eventually the tumors dry up, rupture, and release the diploid spores, which are dispersed by air, and this closes the life cycle. Here the author covered two members of the Basidiomycetes. For more details about mating-type genes in Pucciniomycotina, Ustilaginomycotina, and Agaricomycotina, see Kües et al. (2011), Coelho et al. (2017), and Snetselaar and McCann (2017).
Fungus-growing termites (Macrotermitinae, Isoptera) have practiced farming with their fungal symbionts of the basidiomycete genus Termitomyces. Surprisingly, unlike other basidiomycetes in which where sex is largely restricted to basidia, Termitomyces maximizes sexuality at the somatic stage, resulting in an ever-changing genotype composed of a myriad of coexisting heterogeneous nuclei in a heterokaryon. Somatic meiotic-like recombination may endow Termitomyces with the agility to cope with termite consumption by maximized genetic variability (Hsieh et al. 2017).
3.5 GPCR and Fungal Response to Stress
When used in traditional technologies such as baking, brewing, and distillers’ fermentations, yeasts are exposed to numerous environmental stresses that can be encountered in concert and sequentially. Yeasts exhibit a complex array of stress responses when under conditions that are less than physiologically ideal. These responses involve aspects of cell sensing, signal transduction, transcriptional and post-translational control, protein targeting to organelles, accumulation of protectants, and activity of repair functions. Attfield (1997) reviewed the aspects of stress and stress response in the context of baker’s yeast manufacturing and applications, and discussed the potential of improving the general robustness of the industrial baker’s yeast strains in relationship to physiological and genetic manipulations. Yun et al. (1997) suggested that Gpr1p is a G-protein-coupled receptor localized at the plasma membrane of Saccharomyces cerevisiae that is likely to monitor an extracellular signal such as nutrition and transduce it via Gpa2p, a possible positive regulator of cAMP level. The dual regulation of gene expression by two different stress conditions is observed in the regulation of the CUP1 gene, encoding a yeast metallothionein. If an essential nutrient is missing from the medium, cells are arrested, enter the stationary phase, and display phenotypes associated with low pKa activity, including high expression of stress-related genes and the production of stored carbohydrates. The protein phosphatase calcineurin is activated by heat shock. CUP1 expression is activated in response to heat shock and glucose starvation through the action of heat-shock factor (Hsf1) and heat-shock element (HSE) located within the CUP1 promoter. It has been observed that in sucrose nonfermenting 1 (snf1# or snf4#) mutants, induction of CUP1 by carbon starvation is abolished, without affecting heat-shock-induced regulation. Glucose starvation induces the activation of CUP1 by a process dependent on the phosphorylation of Hsf1 by Snf1 kinase. This phosphorylation causes trimerization and activation of Hsf1 (Hahn et al. 2001); this is probably the reason why snf1# mutants are more sensitive to heat stress because Hsf1 activity may not be appropriately regulated (Thompson-Jaeger et al. 1991). Snf1 protein kinase participates in the regulation of different cellular responses to different forms of stress, which could be an indication of the key role of the kinase as a sensor of the fuel and stress status of the cell. Sanz (2003) concluded that sucrose nonfermenting 1 (Snf1) protein kinase has a main role in transcriptional activation, repression of gene expression, the regulation of different cellular responses to stress status of yeast cell:nutrient limitation stress, glycogen synthesis stimulated by nutrient limitation, glucose depletion, salt stress, and heat shock. In contrast to the pheromone GPCRs, which are expressed in only haploid cells and in a mating-type-specific fashion, the Gpr1 receptor is expressed in both diploid and haploid cells of either mating type. Broach (2012) showed that we have a detailed understanding of some of the circuitry underlying nutritional sensing in yeast, but we are still somewhat vague on others. The availability of key nutrients, such as sugars, amino acids, and nitrogen compounds, dictates the developmental programs and the growth rates of yeast cells. A number of overlapping signaling networks, those centered on Ras/protein kinase A, AMP-activated kinase, and target of rapamycin complex I, for example, inform cells on nutrient availability and influence the cell transcriptional, translational, post-translational, and metabolic profiles as well as their developmental decisions. For instance, the interplay of positive and negative regulators and the various feedforward and feedback loops in regulating expression of glucose transporters is so well described that modeling efforts have yielded highly predictive dynamic descriptions of its behavior. In S. cerevisiae, Rgt2p and Snf3p are two sensors that upon glucose detection increase the expression of transporters, therefore enhancing glucose uptake. In the absence of glucose, Rgt1p forms a repressor complex with Mth1p, Std1p, and Ssn6p-Tup1p, inhibiting the expression of HXT transporter-encoding genes. When glucose is available, Mth1 and Std1 are phosphorylated by casein kinase I and ubiquitinated by the SCF (Skp1-Cullin-F-box protein) E3 ubiquitin ligase Grr1, which leads to their degradation by the 26S proteasome. Depletion of the corepressors dissociates Rgt1 and relieves repression of hexose transporter gene transcription. On the other hand, we still have no clear understanding of the upstream components of glucose signaling that regulate protein kinase A or the interplay between the various small G proteins in that process. Even more poorly described are the pathways sensing and responding to nitrogen levels, especially that nitrogen regulates pathogenicity and filamentation. In addition, cell growth and cell-cycle progression are dependent on the availability of nitrogen and other nutrients and subsequent signaling via the target of rapamycin complex1. Although many of the components of the target of rapamycin complex TORC1, which controls G2/M progression in yeast, and the signaling network have been identified and their interactions defined, we have less understanding of the pathways emanating from TORC1, particularly through protein phosphatases. Moreover, we can infer the existence of a second nitrogen-sensing pathway from the limits of TORC1 effects, but this pathway is poorly defined. Finally, we appreciate that significant crosstalk exists between and among the various nutrient signaling pathways, for instance, glucose sparing in nitrogen- or phosphate-starved cells, but the nature of that interplay is undefined. Thus, we have a number of important details to fill in regarding the structure of the nutrient-sensing networks. In addition, several fundamental questions regarding the interplay of nutrient availability and growth have yet to be solved. Roelants et al. (2017) reported that for eukaryotic cell growth, they must expand by inserting glycerolipids, sphingolipids, sterols, and proteins into their plasma membrane and maintain the proper levels and bilayer distribution. When it was found that the antibiotic rapamycin was a potent inhibitor of the proliferation of virtually every eukaryotic cell type examined (e.g., fungi, T cells, and tumor cells), it became clear that the molecular target of rapamycin must be highly conserved and its function is critical for cell survival. Indeed, ever since the authentic target of rapamycin (TOR) was first discovered using an elegant genetic approach in the budding yeast Saccharomyces cerevisiae, TOR has emerged as a universal, centrally important sensor, integrator, and controller of eukaryotic cell growth. TOR is a serine/threonine kinase of the phosphatidylinositol kinase-related kinase family, which shares conserved motifs (such as HEAT repeats, FAT, and FATC domains), and is structurally and functionally conserved in eukaryotes. The TOR pathway orchestrates the growth of cells in response to nutrients. Tor proteins sense nutrient signals, including amino acids, and regulate a broad range of cell developmental and signaling processes, including ribosome biosynthesis, protein translation, starvation-related transcriptional regulation, and autophagy. A fungal cell must coordinate growth with enlargement of its cell wall. In S. cerevisiae, a plasma membrane-localized protein kinase complex, TORC2, serves as a sensor and master regulator of these plasma membrane- and cell wall-associated events by directly phosphorylating and thereby stimulating the activity of two types of effector protein kinases: Ypk1, along with a paralog (Ypk2), and Pkc1. Ypk1 is a central regulator of pathways and processes required for plasma membrane lipid as well as protein homeostasis requires phosphorylation on its T-loop by eisosome-associated protein kinase Pkh1 and a paralog (Pkh2). For cell survival under various stresses, Ypk1 function requires TORC2-mediated phosphorylation at multiple sites near its C-terminus. Pkc1 controls diverse processes, especially cell wall synthesis and integrity. Pkc1 is also regulated by Pkh1- and TORC2-dependent phosphorylation, but in addition, by interaction with Rho1-GTP and lipids phosphatidylserine (PtdSer) and diacylglycerol (DAG).
There are an increasing number of studies on 14-3-3 proteins and GTPase families that are defined as functionally conserved eukaryotic proteins which participate in many important cellular processes in fungi. These studies reveal that 14-3-3 proteins are related to fungal growth, which is a complex phenomenon related to nutrient assimilation and development, such as dimorphic transition and mycelial growth, cell program regulation, development, autophagy, apoptosis, that might be a universal stress response signaling cascades and virulence. Fungi have genetic interactions with TOR signaling transduction pathways, which are also important during nutrient stress and could regulate metabolic processes, regulation of carbon and nitrogen metabolism, by directly interacting with and modifying the functions of target enzymes, with numerous roles in the regulation of biological processes as adapters, activators, or repressors in the regulation of signal transduction pathways. The BMH1 gene from Candida albicans is essential, and both wild-type alleles are necessary for the optimal growth and morphogenesis of this organism. They could control filamentation and the dimorphic transition in some dimorphic fungi by responding to different environmental factors, especially in C. albicans. In basidiomycetous fungus, it was reported that Pdc1 (a homolog of 14-3-3 in Ustilago maydis), had an important role in the regulation of the dimorphic switch in U. maydis. In S. cerevisiae, 14-3-3 proteins are required for the G1/S transition; in contrast, in U. maydis these are essential during the G2/M transition and have an important function in its sexual reproduction. To date, some 14-3-3 interacting proteins that regulate growth have been discovered in fungi; however, numerous biological processes (especially in mycelial development and secondary metabolism in filamentous fungi) and the regulatory mechanisms of 14-3-3 proteins are still unclear (Shi et al. 2019). The reprogramming of gene expression during stress typically involves initial global repression of protein synthesis, accompanied by the activation of stress-responsive mRNAs through both translational and transcriptional responses. Crawford and Pavitt (2018) summarized the major repressive mechanisms and discussed the mechanisms of translational activation in response to different stresses in S. cerevisiae. Taken together, a wide range of studies indicated that multiple elements act in concert to bring about appropriate translational responses: these include regulatory elements within mRNAs, altered mRNA interactions with RNA-binding proteins, and the specialization of ribosomes that each contribute toward regulating protein expression to suit the changing environmental conditions.
In budding yeast, an important part of extracellular glucose sensing and signaling is mediated by the cAMP-PKA pathway. A dual glucose-sensing system is involved in the activation of the cAMP-PKA pathway: on the one hand, extracellular glucose sensing occurs through the GPCR system composed of Gpr1 and its associated Gα protein, Gpa2, and on the other hand, an intracellular system dependent on glucose uptake and hexokinase-mediated phosphorylation that activates in some unknown way the Ras proteins (Rutherford et al. 2019). All Ras proteins are members of a eukaryotic subfamily of small GTPases involved mainly in cellular signal transduction. The structurally and functionally conserved TOR pathway has for a long time been suggested to contribute to the regulation of cell growth and many related properties by nutrient availability. However, no clear mechanisms have been identified by which the TOR pathway would detect extracellular nutrients, and more recent work suggests that the TOR proteins rather sense intracellular nitrogen availability, in particular mobilization of nitrogen reserves from the vacuole/lysosome. S. cerevisiae possesses two TOR genes: Tor1 and Tor2. The discovery of several types of plasma membrane nutrient sensors, including a GPCR, several transporter-like sensors, and multiple transceptors, has firmly established yeast as the leading model organism in the field of cellular nutrient sensing. A favored hypothesis is that the PKA-regulating transceptors act in a way that is analogous to GPCRs. A major challenge for the future is the elucidation of the molecular mechanisms involved in nutrient responses that at least partially depend on the metabolism of the nutrient. These mechanisms are much more difficult to identify because of the complex nature of metabolism and the many side effects caused by genetic modification of metabolic pathways. It can be predicted easily that there must be many more allosteric interactions between metabolic intermediates and components of signaling pathways than are currently known. A major mechanism likely to be identified soon is that involved in activation of the Ras proteins by one or more intermediates of glucose catabolism. Because of the importance of Ras in cancer induction in mammalian cells and the well-known overactive glycolysis in cancer cells, that is, the Warburg effect, elucidation of this mechanism may have major consequences for our general understanding of the connection between glycolysis and control of cellular proliferation. The TOR pathway has long been considered as the main integrator of multiple nutrient signals for cellular growth control. However, more and more evidence indicates that the TOR pathway is primarily a specific nitrogen-sensing pathway, with a main role in coordinating the availability of extracellular nitrogen with that of intracellular nitrogen reserves and with its effect on cellular growth being one of the multiple outcomes of this function. Hence, a major challenge for the future remains the identification of the nutrient sensors that regulate cellular growth. In this respect, it is important to realize that all essential nutrients, the macronutrients providing carbon, nitrogen, phosphorus, and sulfur, as well as the micronutrients such as metal ions and vitamins, have a decisive effect on cellular growth control and hence should all be sensed in some way to exert this function. A specific mechanism may exist for the regulation of cellular growth by each nutrient, but alternatively, a common principle may be involved in sensing all essential nutrients for cellular growth control, and these nutrient sensors may interact much more directly with the protein synthesis machinery than previously anticipated. At present, nutrient control of bulk protein synthesis remains vague and the true relevance of the few controls identified remains poorly defined. Another gap in our understanding is the link between initial nutrient responses and long-term adaptation to the same nutrient. At present, we know that in the nutrient responses for which it has been investigated, the two processes have different requirements, but how the rapid response proceeds to the long-term response at the molecular level is unknown. The powerful genomic and proteomic technologies currently available have led to rapid progress in identifying the scope of signal transduction pathway targets. Most of this information, however, has been obtained with gene deletion or overexpression strains, or using small-molecule inhibitors that completely inactivate the target protein. This point raises the question to what extent the very many changes in target genes or proteins usually detected are physiologically relevant. Another outcome of these studies has been that the signaling pathways investigated affect targets in other parts of metabolism than previously considered. Here too, the true physiological relevance of the widened scope remains to be determined (Conrad et al. 2014). It is well established that the protein kinases PKA and Sch9, and the tor complex 1 (TORC1), have a central role in the nutrient-induced signaling network that controls growth, survival, and longevity by maintaining a tight balance between proliferation and stress defense. PKA activity is regulated by the Ras–cAMP pathway and activation of adenylate cyclase. The latter requires extracellular sensing of glucose via the Gpr1–Gpa2 GPCR system as well as intracellular glucose sensing via the hexokinases Hxk1/2 and glucokinase Glk1, which in turn stimulate the small GTPases Ras1 and Ras2. Nitrogen sources activate TORC1 at the vacuolar membrane. Depending on their quality as a nitrogen source, amino acids act through an evolutionarily conserved mechanism comprising EGOC, a complex containing the Rag-like GTPases Gtr1 and Gtr2. Sch9 is a well-known TORC1 effector and shuttles between the cytoplasm and the vacuole in a glucose-dependent manner. Besides TORC1, Sch9 activity is also regulated by the sphingolipid-dependent PDK1 orthologs Pkh1-3 and the protein kinase Snf1, a key factor in glucose repression. The pathways controlled by PKA, TORC1, and Sch9 converge on the protein kinase Rim15, which ensures proper entry into the stationary phase by activating the expression of the so-called STRE- and PDS-controlled genes during the diauxic shift. When the stress is halted or when nutrients are again plentiful, trehalose is rapidly degraded and, here, the availability of Pi dictates the PKA-dependent activation of Nth1, the neutral trehalase that hydrolyzes the disaccharide in the cytoplasm.
To sense the changes in nutrient availability, the yeast S. cerevisiae has three different classes of nutrient-sensing proteins acting at the plasma membrane: GPCRs, which detect the presence of certain nutrients and activate signal transduction in association with a G protein; non-transporting transceptors, that is, nutrient carrier homologs with only a receptor function, previously called nutrient sensors; and transporting transceptors (transport and receptor), active nutrient carriers that combine the functions of a nutrient transporter and receptor. Rubio-Texeira et al. (2010) provided an updated overview of the proteins involved in sensing nutrients for rapid activation of the protein kinase A pathway, which belong to the first and the third category, and they also provided a comparison with the best-known examples of the second category, the non-transporting transceptors, which control the expression of the regular transporters for the nutrient sensed by these proteins. Three well-known types of molecular machines employed for the sensing of the environment (receptors, transporters, and channels) are polytopic transmembrane proteins, monomeric or oligomeric, located in the plasma membrane of all types of cells. Strikingly, the transmembrane receptors involved in nutrient sensing (e.g., Ssy1, Mep2, Snf3, and Rgt2) are structurally homologous to nutrient transporters. In contrast to bona fide receptors, transporters and channels mediate the uptake of solutes, metabolites, drugs, or ions, which themselves can act as molecular signals once intracellularly accumulated (Diallinas 2017). Snf3 is used by S. cerevisiae to sense glucose, fructose, and mannose.
Microorganisms have multiple sensing systems to sense extracellular and intracellular nutrient signals to adapt to the environment and their own metabolic state. Fungal pathogens prefer certain carbon sources for rapid uptake and metabolism to provide energy for growth and host colonization. The GPCR gene family represents one important sensor system that has been found to have important roles in nutrient sensing in many fungal species (Xue et al. 1998, 2006; Lorenz et al. 2000; Bardwell 2004; Han et al. 2004; Lemaire et al. 2004; Miwa et al. 2004; Maidan et al. 2005a, b). In the yeast S. cerevisiae, Ca2+ signaling mediated by the Ca2+/calmodulin-dependent phosphatase, calcineurin, is required for survival during environmental stress. The phosphatase activates gene expression through its regulation of the Crz1p (“crazy”) transcription factor. Calcineurin dephosphorylates Crz1p and causes its rapid translocation from the cytosol to the nucleus, then Crz1p activates the transcription of genes whose products promote cell survival (Cyert 2003). Permeases and their homologs function as sensors for nutrients, including homologs of permeases for sugars, amino acids, ammonia, and phosphate (Bahn et al. 2007). Starved yeast cells anticipate exposure to glucose by activating the Hxt5p (hexose transporter 5) glucose transporter, providing an advantage during early phases after glucose resupply. When cAMP and glucose fluorescence resonance energy transfer (FRET) sensors were used to identify three signaling pathways that cooperate in the anticipatory Hxt5p activity in glucose-starved cells, it was found as expected that Snf1 and the AMP kinase pathway do cooperate, but surprisingly also are dependent on an extracellular G-protein-coupled Gpr1 (G-protein-coupled receptor1)/cAMP/PKA (protein kinase A) pathway and the Pho85 (phosphate metabolism85)/Plc (phospholipase C) 6/7 pathway. Gpr1/cAMP/PKA are key elements of a G-protein-coupled sugar response pathway that produces a transient cAMP peak to induce growth-related genes. A novel function of the Gpr1/cAMP/PKA pathway was identified in glucose-starved cells: during starvation, the Gpr1/cAMP/PKA pathway is required to maintain Hxt5p activity in the absence of glucose-induced cAMP spiking. During starvation, cAMP levels remain low, triggering expression of HXT5, whereas cAMP spiking leads to a shift to the high-capacity Hxt isoforms (Bermejo et al. 2013). Further work is needed specifically whether the pathway is constitutively active in the absence of glucose, whether PKA exists in different phosphorylation states, and whether the two states regulate different sets of genes. An interesting question to address will also be how the two pathways cooperate to trigger the ability to accumulate glucose and whether they measure distinct signals, such as lack of extracellular glucose or reduced energy status. A putative seven-transmembrane protein gene, stm1+, which is required for proper recognition of nitrogen starvation signals, was isolated as a multicopy suppressor of a ras1 synthetic lethal mutant in Schizosaccharomyces pombe (Chung et al. 2001). Under nitrogen-deficient conditions, transcription of the stm1 gene was induced. Under nutritionally sufficient conditions, overexpression of Stm1 inhibited vegetative cell growth, resulting in decreased intracellular cAMP levels, increased the expression of the meiosis-specific genes ste11, mei2, and mam2, and facilitated sexual development in homothallic cells. Induction of ste11, a meiosis-specific gene transcription factor, by Stm1 overexpression was enhanced in gpa2-deleted cells but was absent in a deletion mutant of sty1, a key protein kinase that links mitotic control with environmental signals and induces stress-responsive genes. Moreover, deletion of both stm1 and ras1 caused delayed entry into G1 arrest in S. pombe when the cells were grown on a nitrogen-deficient medium. Thus, it is considered that the stm1 gene can function through Gpa2-dependent and/or -independent pathways and may have a role in providing the prerequisite state for entering the pheromone-dependent differentiation cycle in which heterotrimeric Gα1 protein, Gpa1, and Ras1 are major factors. Stm1 could function as a sentinel molecule sensing the nutritional state of the cells, stopping the proliferative cell cycle, and preparing the cell to enter meiosis under nutritionally deficient conditions.
In several fungi, responses to contact with a surface are promoted by GPCR family members. As some GPCRs have been shown to be mechanosensitive, the mechanical forces that result from contact might activate fungal GPCRs and contribute to contact-dependent differentiation. The details of how mechanical forces influence GPCR function await clarification (Kumamoto 2008). A modified form of the human B2 bradykinin GPCR, which was fused to two fluorescent proteins to allow fluorescence resonance energy transfer (FRET) between the two fluorescent proteins, was shown to undergo conformational changes that reduced FRET in response to shear stress (Chachisvilis et al. 2006). Thus, GPCRs are mechanosensitive and undergo conformational changes in response to mechanical forces, such as stretching. The fungal cell wall is essential for its viability and an important target of antifungal drugs. In fungi, a conserved MAPK signaling module is responsible for maintaining cellular integrity, shape, and resilience to environmental stresses (Lesage and Bussey 2006). The plant pathogen Magnaporthe grisea, the causative agent of rice blast, undergoes appressorium formation following contact with a surface. Contact with a hard surface is required for appressorium formation, and neither soft surfaces nor liquids are inductive for appressorium development. Members of the GPCR family also regulate morphogenesis in Candida albicans and in the model organism S. cerevisiae. In C. albicans, the formation of filamentous hyphae is promoted by numerous cues from the environment, including contact. Contact sensing results in the formation of hyphae that penetrate into the substratum below the C. albicans cells. Filamentation in response to contact is defective in C. albicans mutants that lack the GPCR Gpr1p, whereas filamentation of this mutant in response to other environmental cues is normal. S. cerevisiae forms pseudohyphae when grown in contact with agar medium of the appropriate composition (for example, low nitrogen content), and S. cerevisiae GPR1 is important for pseudohyphal development. S. cerevisiae Gpr1p binds sugars such as glucose and sucrose directly and is thought to be a nutrient sensor. In the yeast S. cerevisiae, rapid activation of the cAMP pathway by glucose and sucrose requires the GPCR Gpr1. Lemaire et al. (2004) obtained results by cysteine scanning mutagenesis and the substituted cysteine accessibility method (SCAM) of residues in TMD VI that provided strong evidence that glucose and sucrose directly interact as ligands with Gpr1, with the affinity for sucrose being much higher (Rutherford et al. 2019). Structurally, mannose acts as an antagonist for both sucrose and glucose. Although the mechanosensitivity of these fungal GPCRs has not been directly investigated, the mechanosensitivity of other members of this protein family suggests that fungal GPCRs might respond to physical forces which act on the membrane. In summary, GPCR signaling is a conserved feature of contact-dependent morphogenesis in several fungal species. The observation that activation of some GPCRs is influenced by mechanical forces that act on the membrane suggests that contact-dependent perturbation of the membrane could potentiate or activate GPCRs in fungal contact sensing (Kumamoto 2008).
Although much is known about the mechanisms of stress-dependent signaling, less is known about coordination between the stress response and other cell signaling processes. Cells routinely experience changing and often unfavorable conditions in their environment. All cells respond to osmotic stress by implementing molecular signaling events to protect the organism. Many are transmitted by GPCRs or the high osmolarity glycerol (HOG) pathway. Salt and hyperosmotic stress trigger a complex adaptive response in yeast cells, which is coordinated by the HOG MAP kinase pathway. MAPKs are activated in response to osmotic stress, as well as by signals acting through GPCRs. For proper adaptation, the action of these kinases must be coordinated. To identify second messengers of stress adaptation. Shellhammer et al. (2017) conducted a mass spectrometry-based global metabolomics profiling analysis, quantifying nearly 300 metabolites in the yeast S. cerevisiae. They showed that three branched-chain amino acid (BCAA) metabolites increase in response to osmotic stress and require the MAPK Hog1. Ectopic addition of these BCAA derivatives promotes phosphorylation of the G protein α-subunit and dampens G-protein-dependent transcription, similar to that seen in response to osmotic stress. Conversely, genetic ablation of Hog1 activity or the BCAA regulatory enzymes leads to diminished phosphorylation of Gα and increased transcription. Taken together, their results define a new class of candidate second messengers that mediate crosstalk between osmotic stress and GPCR signaling pathways.
The molecular mechanisms that enable yeast cells to detect and transmit cold signals and their physiological significance in the adaptive response to low temperatures are unknown. To overcome environmental stresses, fungi, as do other eukaryotes, rely on the rapid transduction of signals through mitogen-activated protein kinase (MAPK) pathways. Panadero et al. (2006) demonstrated that the MAPK Hog1p is specifically activated in response to cold. Phosphorylation of Hog1p was dependent on Pbs2p, the MAPK kinase (MAPKK) of the high osmolarity glycerol (HOG) pathway, and Ssk1p, the response regulator of the two-component system Sln1p-Ypd1p. Although C. albicans Sln1 can functionally replace S. cerevisiae Sln1 and is indeed involved in osmosensing, Sln1 is not essential for viability of C. albicans. However, the HOG pathway is activated by heat and cold stress in S. cerevisiae, but these stressors do not activate the HOG pathway in C. albicans, in which mitochondrial function appears to be required for activation of CaHog1 in response to oxidative stress. Interestingly, phosphorylation of Hog1p was stimulated at 30°C in cells exposed to the membrane rigidifier agent dimethyl sulfoxide. Moreover, Hog1p activation occurred specifically through the Sln1 branch, which suggests that Sln1p monitors changes in membrane fluidity caused by cold. Quite remarkably, the activation of Hog1p at low temperatures affected the transcriptional response to cold shock. Indeed, the absence of Hog1p impaired the cold-instigated expression of genes for trehalose- and glycerol-synthesizing enzymes and small chaperones. Moreover, a downward transfer to 12° or 4°C stimulated the overproduction of glycerol in a Hog1p-dependent manner. However, hog1Δ mutant cells showed no growth defects at 12°C as compared with the wild type. On the contrary, deletion of HOG1 or GPD1 decreased tolerance to freezing of wild-type cells preincubated at a low temperature, whereas no differences could be detected in cells shifted directly from 30° to −20°C. Thus, exposure to low temperatures triggered a Hog1p-dependent accumulation of glycerol, which is essential for freeze protection. Steroids are perceived as stress by yeast cells that triggers general stress response leading to activation of heat-shock proteins, cell-cycle regulators, MDR transporters, etc. (Prasad et al. 2012).
Flammulina velutipes (Curt. ex Fr.) Sing, a white-rot fungus, is known as golden-needle mushroom, winter mushroom, or enokitake. Because of its high nutritional and medicinal value, F. velutipes is cultivated on a significant scale worldwide, which has both ecological and commercial importance. The fruit-body quality of F. velutipes is determined by both genotype and environment. Both yield and quality of F. velutipes fruit-bodies differ largely depending on their primordium number, and the initiation of primordium is prone to environmental factors. Knowledge of chronological protein expression patterns in F. velutipes mycelia in response to cold stress is an important metabolic engineering strategy that considers the impact of novel genes and pathways on cold adaptation in fungi. Proteomics is a broad instrument-intensive research area that has advanced rapidly since its inception. Liu et al. (2017) undertook to investigate chronological changes of protein expression during F. velutipes mycelia in response to cold stress. The proteomics data revealed that some interesting proteins upregulated in some central metabolic pathways might be important during mycelia response to cold stress in F. velutipes. More than four functional categories selected to evaluate the data sets were mainly involved in energy metabolic processes, amino acid biosynthesis and metabolism, signaling pathway, transport, and translation. They found that the global expressed and differentially expressed proteins participated in energy metabolism pathways including the citrate cycle, pentose phosphate pathway, glyoxylate and dicarboxylate metabolism, and sucrose metabolism. They showed complex protein abundance change patterns in acute normal culture to cold stress transfer in mycelia of F. velutipes at the molecular level. The four proteins that had higher expression levels after cold stress in mycelia and involved in energy metabolism were catalase, glucose-6-phosphate isomerase, trehalase, and beta-glucosidase. Catalase, universal in many fungi, a very important enzyme in protecting the cell from oxidative damage by reactive oxygen species (ROS), was significantly upregulated during exposure of mycelia to short-term cold stress, but its expression levels returned to normal after long-term cold stress. Worth noting, S. cerevisiae bZip transcription factor ScYap1 and its C. albicans homolog CaCap1 are also involved in oxidative stress response. To prevent damage to cells and tissues, catalase is frequently used by cells to rapidly catalyze the decomposition of hydrogen peroxide, a harmful by-product of many normal metabolic processes, into less reactive gaseous oxygen and water molecules. Glucose-6-phosphate isomerase (GPI), a dimeric enzyme universally distributed in eukaryotes, catalyzes the reversible isomerization of glucose-6-phosphate and fructose-6-phosphate, trehalase, and beta-glucosidase expression, and dramatically increased after long-term cold stress. In the cytoplasm, it is involved in both glycolysis and gluconeogenesis, as well as the pentose phosphate pathway. Outside the cell, GPI is also known to function as a maturation factor. Cloning experiments revealed that GPI is identical to the protein known as autocrine motility factor (AMF) produced and secreted by cancer cells that is thought to have a key role in regulating endoplasmic reticulum (ER) calcium release to protect against apoptosis in response to ER stress, and stimulates cell growth and motility as a growth factor by activating the MAPK/ERK or PI3K/AKT pathways. Such pathways are signal transduction pathways that promote survival and growth in response to extracellular signals. Beta-glucosidase, a heterogeneous group of exo-type glycosyl hydrolases, catalyzes the hydrolysis of β-glucosidic linkages in β-d-glucosides and oligosaccharides, with release of glucose monomers by breaking the β-1,4 glucosidic bonds of cellobiose. Beta-glucosidases are important in fundamental biological processes, such as nutrient uptake and developmental regulation or chemical defense against pathogen attack. Beta-glucosidase catalyzes the hydrolysis of β-glucosidic linkages in β-d-glucosides and oligosaccharides, with release of glucose monomers by breaking the β-1,4 glucosidic bonds of cellobiose. Trehalase, one of the important carbohydrate storages in fungi, is an important physiological process for fungal spore germination and the resumption of mycelium growth, and catalyzes the conversion of trehalose to glucose: this suggests that trehalase may be important in energy metabolism during fungal mycelia to fruit-bodies transfer and regulating fruit-body formation. The sugar is thought to form a gel phase as cells dehydrate, which prevents disruption of internal cell organelles, by effectively splinting them in position. Knowing the proteins associated with energy metabolic pathways responsible for the development of the mycelia is helpful to better understanding of the molecular mechanisms of mycelium resistance to cold stress and fruiting-body formation in F. velutipes. Amino acids serve as major nitrogen carriers in the long-distance transport systems, precursors to important metabolites, stress response molecules, and signaling molecules. Eleven DPSs were shown to be involved in the biosynthesis and metabolism of nine amino acids. Argininosuccinate synthase and class V chitinase ChiB1 were significantly upregulated in mycelia under short-term cold stress, but its expression levels were dramatically decreased during long-term cold stress. The metabolic pathways in which argininosuccinate synthase participates are linked to the varied uses of the amino acid arginine, such as arginine synthesis, urea synthesis, nitric oxide synthesis, polyamine synthesis, and creatine synthesis. Arginine is the main precursor for urea formation whereas accumulation of large quantities in fruit-bodies is a known feature of larger basidiomycetes. Arginine is a significant nutrient molecule for the transport from the mycelium during early primordium development. The class V chitinase expression level increased in mycelia under short-term cold stress, and it may increase energy supply to promote resistance to cold stress. An increased abundance of signaling pathways and other process-related proteins was shown after mycelia were exposed to cold stress. A total of 1198 proteins were identified and quantified in three different mycelium growth stages of F. velutipes, demonstrating the applicability of the iTRAQ approach to multiplexed proteomic profiling was successful. A broader comparison of the proteomes of the three mycelium developments revealed that changes in some central metabolic pathways may be involved in the cold stress responses of F. velutipes mycelia.
Recent advances on the control of pH homeostasis revealed interesting new aspects underpinning the crosstalk within the nutrient signaling network (Eskes et al. 2017). In the model ascomycete and occasional pathogen Aspergillus nidulans, the PacC transcription factor governs gene expression in response to extracellular pH and is vital for mammalian pathogenicity (Díez et al. 2002). Under alkaline conditions, a signaling cascade involving seven proteins is involved in activation of PacC. A putative pH sensor, PalH, has 7TMDs and a cytoplasmic C-terminus that interacts with a cognate arrestin encoded by palF. Unlike canonical GPCR receptors, PalH is not thought to act via interaction with G-protein subunits. When an alkaline response is triggered, PalF is phosphorylated and subsequently ubiquitinated in a PalH-dependent manner, leading to PalB-mediated, signal-dependent, proteolytic cleavage of the pH-responsive transcription factor PacC. Subsequent translocation of the truncated PacC protein, from cytoplasm to nucleus, permits alkaline adaptation via differential expression of genes required to enable growth under alkaline extracellular conditions (Espeso et al. 2000). In Aspergillus fumigatus the amino acid residues crucial for PalH and PalF interaction are conserved, and in split-ubiquitin analyses the proteins enter into close proximity.
Peroxisomes are ubiquitous, DNA-free organelles present in most eukaryotic cells. Yeast peroxisomes readily adapt their number and physiological function in response to changes in the metabolic state of the cell. Manzanares-Estreder et al. (2017) quantified the growth efficiency of yeast cultures upon NaCl stress while continuously lowering the glucose content. Growth on high-salinity media was dramatically affected by low sugar availability. They suggested that a possible explanation for this dramatic effect was that salt stress caused glucose starvation, which in turn would make necessary the use of alternative energy sources on stress. They suggest that during the defense to salt stress, yeast cells switch from a fermentative metabolism to fatty acid oxidation to cover the energetic needs during the environmental challenge. One important energy resource are intracellular lipid stores, which can be mobilized during sugar starvation by peroxisomal β-oxidation in yeast cells. Their results indicated that peroxisomal function was growth limiting upon salt stress especially during low sugar availability. They found that the HOG MAP kinase pathway was the master regulator of gene expression upon osmostress in yeast cells and confirmed that the Hog1 MAP kinase was indispensable for the increase of mRNA levels of genes involved in the mobilization of fatty acids. They confirmed that the Hog1 kinase function was indispensable for the NaCl induction of genes involved in fatty acid metabolism. Peroxisomal function was responsible for reinforced yeast mitochondrial respiration upon salt stress. For more details about fungal sensing of the host environment and stress, the reader is referred to Braunsdorf et al. (2016).
3.6 GPCRs and Fungal Pathogenicity Determinants
Only a handful of GPCRs have been identified in fungal genomes. However, in Saccharomyces cerevisiae and Schizosaccharomyces pombe only three and four receptors, respectively, are well characterized. In the Neurospora crassa genome, a total of ten receptors are predicted. A recent report for Aspergillus nidulans identified GPCRs similar to the yeast pheromone receptors, the glucose-sensing receptor GPR1, the nitrogen-starvation sensing STM1, and the Dictyostelium discoideum cAMP receptors (Kulkarni et al. 2005). Fungal pathogens sense nitrogen levels to control their rate of growth and changes in their morphology, processes that are important for host infection.
Magnaporthe grisea causes rice blast disease, the most destructive disease of rice worldwide. M. grisea is amenable to molecular genetic manipulation and the subject of large-scale genome-wide functional studies following the recent completion of its draft genome sequence. It worth noting that M. grisea and Magnaporthe oryzae are not interfertile and are considered different species (Couch and Kohn 2002). Cell-surface receptors, as previously mentioned, that perceive signals at critical times in the life cycle of M. grisea and other pathogenic fungi are strongly implicated as pathogenicity determinants. Signaling has a key role in appressorium formation and infection in M. grisea. The cAMP-dependent and pheromone response, as well as other mitogen-activated protein kinase (MAPK)-, phospholipase-, and calmodulin-dependent pathways, are essential for pathogenicity and are likely to involve perception of signals through GPCRs (Beckerman et al. 1997; Mitchell and Dean 1995). The three identified G-protein subunits required for different aspects of development and pathogenicity possibly transduce perceived signals to the aforementioned pathways (Liu and Dean 1997). The M. grisea G proteins probably receive signals from receptors such as PTH11, an integral membrane protein required for development of the appressorium and pathogenicity. It was proposed to act upstream of the cAMP pathway that restores both appressorium formation and pathogenicity (DeZwaan et al. 1999). A BLASTP search against the genome of the closely related filamentous fungus Neurospora crassa, using all the M. grisea GPCR-like proteins as query, revealed the presence of similar proteins in N. crassa, including PTH11 homologs (Galagan et al. 2003; Kulkarni et al. 2005). Analyses revealed putative homologs of the mPR-1 class in both yeasts in which they had not previously been identified (Kulkarni et al. 2005). They found three members of the mPR class and one (MG0532.4) with weak similarity to animal GPCRs. No members of these classes have been reported previously in fungi. Homologs of known fungal GPCRs were found in the M. grisea proteome, including the pheromone receptors STE2 and STE3 and the glucose-sensing receptor GPR1. In total, 76 GPCR-like proteins were identified in their study, of which 61 represent a large novel class related to PTH11, a receptor implicated in fungal development and pathogenicity that they proposed acts upstream of the cAMP-dependent pathway. Many of these novel receptors will have roles in known pathways or may define new pathways involved in fungal development. The cAMP, STM1, and mPR receptors are shared between fungi and other eukaryotic species. However, the fungal pheromone receptors (class D) and GPR1-like receptors appear to be fungus specific. For fungus-specific receptors, the conserved domain spanned almost the entire length of the seven transmembrane regions. Members of the large class of PTH11-related receptors were restricted to a fungal subphylum. In the rice blast fungus M. oryzae, a non-canonical G-protein-coupled receptor, Pth11, and membrane sensors MoMsb2 and MoSho1 are thought to function upstream of G-protein/cAMP signaling and the Pmk1 MAPK pathway to regulate appressorium formation and pathogenesis (Li et al. 2017). BLASTP of all the PTH11 class members, and PSI-BLAST using conserved regions, against the GenBank (nonredundant) and Swiss-Prot databases and publicly available fungal genomes, retrieved matches in members of the subphylum Pezizomycotina within the Ascomycota, including Podospora anserina, Blumeria graminis, Fusarium graminearum, and Aspergillus species. Other fungi belonging to the Ascomycota but not to this subphylum, such as Saccharomyces cerevisiae, Schizosaccharomyces pombe, Candida albicans, and Pneumocystis carinii lacked PTH11-related sequences. Also, no PTH11-related sequences were found in the genomes of the Basidiomycetes Cryptococcus neoformans, Ustilago maydis, and Phanerochaete chrysosporium. For fungus-specific receptors, the conserved domain spanned almost the entire length of the seven transmembrane regions (Kulkarni et al. 2005; Zhao et al. 2007), containing a conserved fungus-specific extracellular membrane-spanning domain (CFEM) at the amino terminus that is unique to filamentous fungi and contains eight conserved cysteine residues. It is similar to epidermal growth factor (EGF)-like domains that function as extracellular receptor or signal transducers or as adhesion molecules in host–pathogen interactions. PTH11 GPCRs containing CFEM domains have been identified in Trichoderma atroviride and Trichoderma reesei, and four in Trichoderma virens (Gruber et al. 2013). Sabnam and Barman (2017) selected and characterized a novel and unique conserved fungal-specific extracellular membrane-spanning (CFEM) domain containing a PTH11-like G-protein-coupled receptor (GPCR) in M. oryzae and called it WISH. The CFEM domain has been reported to function as a cell-surface transmembrane receptor or adhesion molecule during host–pathogen interactions. Their data suggest that WISH protein mediates appressorium differentiation and has a pleiotropic effect as well as acting as a negative regulator of conidium and conidiophore development in M. oryzae. They suggested that PTH11 and WISH might be functionally related to recognizing different chemical and physical signals present on the rice leaf surface; the WISH gene appears much more critical and vital than PTH11 in regulating commitment of the pathogen in the process of infection-related morphogenesis. In general, when microorganisms attempt to invade plants they are recognized by the plant immune system through pathogen-associated molecular patterns (PAMPs) or microorganism-associated molecular patterns (MAMPs), which are highly conserved molecules such as flagellin in bacteria, glucans in oomycetes, and chitin in fungi. Microbial invasions often trigger the production of host-derived endogenous signals referred to as danger- or damage-associated molecular patterns (DAMPs), which are also perceived by pattern recognition receptors (PRRs) to modulate PAMP-triggered immunity PTI responses (reviewed recently by Yu et al. 2017). The recognition of such molecules by PRRs that reside in the plant plasma membrane triggers an innate immune response known as PTI or MAMP-triggered immunity (MTI) (Maffei et al. 2012). Fungi change their global gene expression upon recognition of the host environment, leading to secretion of effectors, enzymes, and secondary metabolites; changes in metabolism; and defense against toxic compounds. During infection, pathogens secrete an arsenal of several hundred effectors, to target multiple host cell compartments and enable parasitic infection, which seem to be produced in consecutive expression waves during the course of an infection and/or are associated with pathogenic transitions (Toruno et al. 2016). Such effectors are the key elements of pathogenesis that modulate innate immunity of the plant and facilitate infection. Successful invaders are able to dampen or overcome PTI (or MTI) through the secretion of pathogen effectors that either remain at the plant–pathogen interface (apoplastic effectors) or are taken up by host cells (translocated or cytoplasmic effectors). Doehlemann et al. (2011) identified the PIT (proteins important for tumors) cluster, a novel gene cluster composed of four genes, two of which are predicted to encode secreted effectors. The apoplastic effectors Pep1 and Pit2 are functionally characterized (Doehlemann et al. 2009, 2011; Hemetsberger et al. 2012; Mueller et al. 2013) as well as the three cytoplasmic effectors Cmu1, Tin2, and See1 (Djamei et al. 2011; Tanaka et al. 2014; Redkar et al. 2015). Cmu1 is a protein rechannelling chorismate present in the phenylpropanoid pathway and thereby outcompeting the biosynthesis pathway of the plant defense salicylic acid. Pep1 is known to inhibit plant peroxidases and thereby interferes with the oxidative burst, whereas Pit2 inhibits cysteine proteases in the biotrophic interface. See1 is required for the reactivation of plant DNA synthesis, which is crucial for tumor progression in leaf cells and does not affect tumor formation in immature tassel floral tissues, where maize cell proliferation occurs independently of fungal infection. It interferes with the MAPK-triggered phosphorylation of maize SGT1 at a monocot-specific phosphorylation site, which identifies See1 as a fungal effector that directly and specifically contributes to the formation of leaf tumors in maize. Stirnberg and Djamei (2016) characterized ApB73, a protein transcriptionally upregulated during the biotrophic stage and essential for the successful colonization of Zea mays accession B73 by U. maydis. ApB73 is secreted by the fungus, but then localizes to the fungal surface during biotrophy. It seems to remain associated with fungal cell wall components or the fungal plasma membrane. Previous genomics studies revealed that the interaction with the respective host is largely determined by approximately 300 genes predicted to encode novel secreted protein effectors (Kämper et al. 2006). Further, genome analysis of several smut fungi including Ustilago maydis revealed a singular clustered organization of genes encoding secreted effectors. In U. maydis, many of these clusters have a role in virulence (Dutheil et al. 2016). They showed that the genome of Sporisorium scitamineum contains more and larger gene clusters encoding secreted effectors than any previously described species in this group [U. maydis, Sporisorium scitamineum, and Sphacelotheca (Sporisorium) reilianum]. Djamei et al. (2011) showed that U. maydis actively suppresses plant defense responses by secreted protein effectors. Its effector repertoire comprises at least 386 genes, mostly encoding proteins of unknown function and expressed exclusively during the biotrophic stage. The U. maydis secretome also contains about 150 proteins with probable functions in fungal nutrition, fungal cell wall modification, and host penetration as well as proteins unlikely to act in the fungal–host interface. Secreted proteins are critical virulence determinants in U. maydis. Schuster et al. (2017) compared the secretomes of 12 basidiomycete species with very different lifestyles to gain information about their composition and conservation. Of the secreted proteins with domains, they detected a small core secretome in all 12 species, whereas only one cluster with novel secreted proteins without domain was conserved. This finding does not necessarily mean that related novel secreted proteins with similar functions do not exist; rather, they may have diverged to an extent that their homology cannot be detected by the methods used. One way to approach the search for homologs with a very low sequence identity may lie in obtaining structural information. Their study revealed that a significant percentage of core effectors predicted to be secreted resided in gene families in one or several of the species they compared. The analyses of gene families have been lagging behind in most pathogen–host systems, mostly because of technical difficulties. Avirulence (avr) genes encoding effectors from a certain race of pathogens and their corresponding resistance (R) genes in specific host cultivars have been identified as major contributing factors for gene-for-gene resistance whereby the absence of products produced by any of these genes can cause disease. The recognition of a pathogen AVR protein triggers a set of immune responses grouped under the term effector-triggered immunity (ETI), frequently leading to a rapid localized cell death termed the hypersensitive response (HR). The interactions between five blast Avr effectors, namely AvrPita, Avr-Pik, AvrPiz-t, Avr-Pia, and AVr1-Co39, and their cognate rice R proteins have been reviewed by Liu et al. (2013a, b). AVR genes recognized by two distinct R genes both necessary for recognition were reported in the M. oryzae–rice pathosystem. M. oryzae, the causal agent of rice blast, is mostly controlled by using resistant rice cultivars harboring major R genes. Seven M. oryzae AVR genes have been cloned (Liu et al. 2013b; Petit-Houdenot and Fudal 2017). Some effectors can be recognized directly or indirectly by resistance (R) proteins from the plant and are then called avirulence (AVR) proteins. One class of R proteins corresponds to cell-surface (leucine-rich repeat LRR domain) LRR-containing R proteins that are anchored to the plasma membrane via a transmembrane (TM) domain and sometimes included an intracellular kinase domain: receptor-like proteins, RLP/receptor-like kinases (RLK) (Yang et al. 2012). R–AVRgene interactions are frequently exploited in the field to control diseases. Recently, the availability of fungal genomes has accelerated the identification of AVR genes in plant pathogenic fungi, including in those infecting agronomically important crops. Although single AVR genes recognized by their corresponding R gene were identified, more and more complex interactions between AVR and R genes are reported (e.g., AVR genes recognized by several R genes, R genes recognized several AVR genes in distinct organisms, one AVR gene suppressing recognition of another AVR gene by its corresponding R gene, and two cooperating R genes were both necessary for the recognition of AVR gene). These complex interactions were particularly reported in pathosystems showing a long co-evolution with their host plant but could also result from the way agronomic crops were obtained and improved (e.g., through interspecific hybridization or introgression of resistance genes from wild related species into cultivated crops). Petit-Houdenot and Fudal (2017) described some complex R—AVR interactions between plants and fungi that were recently reported and discussed their implications for AVR gene evolution and R gene management. Resistance genes recognizing several avirulence genes in distinct organisms were also covered. Under the selection pressure exerted by R genes, pathogens can become virulent through evolution of their AVR gene repertoire. The characterization of plant–fungal interactions that are emerging show some similarities (cooperating R genes, R genes recognizing distinct pathogens, AVR gene suppressing recognition of another AVR gene) but also specificities (bipartite AVR gene). Among the R genes displaying complex interaction with AVR genes, some of the most promising are those conferring broad-spectrum resistance because they guard key components of plant immunity and, as such, target essential effectors. Even if they exert a strong selection pressure on pathogen populations, they may remain effective through pyramiding with other specific or quantitative R genes. Another promising strategy to manage durable resistances would be to target antagonistic interactions between AVR genes and to combine the corresponding R genes in the same cultivars through pyramiding or to sequentially use the R genes in rotation. Established techniques for cataloguing effectors and identifying interactors have matured, and novel approaches to understand function have been applied or developed. Although much has been learned in recent years, we have only been chipping at the tip of the iceberg. Hundreds of putative effectors of unknown function have been identified, particularly in filamentous plant pathogens, and a challenge for the future is to develop high-throughput methods to validate and characterize these putative effectors to keep up with the pace of identification. It seems certain that novel activities of these proteins will be found, and there will be new insights into how pathogens remodel host cells for their own benefit and how plants recognize effectors and mount an immune response. Studies of effectors also continue to offer opportunities for the development of tools to probe host cell biology in the absence of disease. As our knowledge of fundamental effector biology develops, it will be important to consider how this information can be translated into policies and products to positively impact agriculture. Note for the future: everyone should look out for the sequels (Varden et al. 2017). As fungal pathogens invading mammalian hosts are exposed to systemic pH values above neutrality, it is unsurprising that the pal/RIM pathway has been identified as an important factor determining pathogenicity (Davis 2009; Cornet and Gaillardin 2014).
Despite their fungal-like morphology, oomycetes are related to photosynthetic red algae rather than to fungi. Multinucleated sporangia and uninucleate zoospores represent their most common dispersal forms. On the plant surface, sporangia germinate directly and enter host tissue via either stomata, wounds, hyphopodia, or appressoria; fungi can grow within the inner cell layers as extracellular hyphae, intracellular invasive hyphae, or extracellular hyphae that project feeding structures (haustoria) into host cells, in contrast to U. maydis, which does not develop haustoria. Both intracellular hyphae and haustoria are surrounded by the plant plasma membrane, termed EHM in haustoriated cells. Alternatively, sporangia may release zoospores that encyst, germinate, and develop appressoria which penetrate the epidermal cell layer (Tyler 2002). Despite significant advances in the field of effector biology, our understanding of the events leading to effector translocation is lagging behind. Oomycete effectors seem to share conserved motifs, whose exact role in effector uptake needs to be unequivocally demonstrated by in vivo and biochemical experiments; fungal effectors are highly divergent at the level of the primary amino acid sequence. Additionally, the lack of conserved motifs in candidate cytoplasmic effectors within a given fungal species could either suggest that the uptake mechanism is rather nonspecific or that a certain degree of conservation between effectors exists at the protein structure level that cannot be recognized at the primary amino acid sequence level. The data available for a subset of oomycete and fungal effectors suggest that effector entry into host cells may occur via endocytosis and depend on a membrane-binding motif, be it a conserved charged motif, a degenerate charged motif, or a solvent-exposed hydrophobic patch. Elucidating the composition of host–microbe interfaces, at both the molecular and ultrastructural level, promises to reveal important insights in the mechanism of effector uptake. The presence of exosomes in biotrophic interfaces, the identification of unconventionally secreted effectors in both oomycetes, and the possible involvement of exosomes in oomycete effector delivery all indicate that extracellular vesicles (EVs) might have a direct or indirect role in effector translocation and warrant further investigation. Lo Presti and Kahmann (2017) summarized current knowledge in the field of oomycete and fungal effector uptake and highlighted emerging themes that may unite rather than set apart these unrelated filamentous pathogens. How the endocytosed effectors escape host endosomes remains to be determined. Effector genes are not randomly distributed across the genome, but often reside in polymorphic regions of the genome, clustering with repetitive DNA. Despite the ubiquity and importance of fungal effectors, our mechanistic understanding of their transcriptional regulation and genome organization remains inadequate (Cook et al. 2017). As such, they are addressing two key questions: (1) How are in planta effectors transcriptionally regulated? (2) How does repetitive DNA contribute to the highly variable genomic regions that contribute to fungal virulence? They characterized how DNA modifications and chromatin structure (the organization of DNA in a cell) contribute to the evolution of virulence using the soil-borne fungal pathogen Verticillium dahliae. The genome of V. dahliae is predicted to express numerous homologs of known DNA and chromatin-modifying proteins, including three putative DNA methyltransferases. A single DNA methyltransferase was shown to control a significant portion of the observed DNA methylation at repetitive DNA. Interestingly, repetitive DNA arising from segmental genome duplications are devoid of DNA methylation and are more transcriptionally active relative to repetitive DNA at other loci. Additionally, the genome was assayed for open chromatin to develop a comprehensive view of how gene regulation and chromatin architecture impact the evolution of fungal virulence. Chitin-binding lysin motif (LysM) effectors contribute to the virulence of various plant-pathogenic fungi that are causal agents of foliar diseases (Kombrink et al. 2017). They reported that the LysM effectors of the soil-borne fungal vascular wilt pathogen V. dahliae have three core LysM effectors that are conserved in a collection of V. dahliae strains. Remarkably, and in contrast with the previously studied LysM effectors of other plant pathogens, no expression of core LysM effectors was monitored in planta in a taxonomically diverse panel of host plants. Moreover, targeted deletion of the individual LysM effector genes in V. dahliae strain JR2 did not compromise virulence in infections on Arabidopsis, tomato, or Nicotiana benthamiana. Interestingly, an additional LysM effector is encoded in the genome of V. dahliae strain VdLs17, but not in any other V. dahliae strain sequenced to date. Remarkably, this effector is expressed in planta and contributes to the virulence of V. dahliae strain VdLs17 on tomato, but on neither Arabidopsis nor N. benthamiana. Functional analysis revealed that this LysM effector binds chitin, is able to suppress chitin-induced immune responses, and protects fungal hyphae against hydrolysis by plant hydrolytic enzymes. Thus, in contrast with the core LysM effectors of V. dahliae, this LysM effector of strain VdLs17 contributes to virulence in planta. Pathogen effectors are considered to be secreted proteins that modify host cell structure and function to promote the success of a pathogen. In U. maydis, about 40% of all candidate secreted proteins are completely novel, as they lack known structural or functional domains (Schuster et al. 2017). Effector proteins can be grouped into functional groups, facilitating distinct steps of disease progression. The first group consists of general (core) effector proteins that facilitate breaking of the first defense barrier of the plants. After successful suppression of the epidermal defense barrier, the biotrophic interaction needs to be maintained while the fungus is progressing through the different tissues it encounters (Matei and Doehlemann 2016). The FLP-mediated selectable marker-removal technique was successfully applied to delete a family of 11 effector genes (eff1) using five sequential rounds of recombination. They showed that expression of all 11 genes is upregulated during the biotrophic phase. Strains carrying deletions of 9 or all 11 genes showed a significant reduction in virulence, and this phenotype could be partially complemented by the introduction of different members from the gene family, demonstrating redundancy (Khrunyk et al. 2010). Lanver et al. (2017) used the term effector for all U. maydis proteins that are predicted to be secreted through the classic endoplasmic reticulum (ER)–Golgi route and lack known domains (novel effectors), as well as for secreted proteins with a confirmed virulence function that contain a known domain, are transcriptionally induced following plant colonization, and are not required for growth or morphology when U. maydis is propagated in axenic culture. In their review, Lanver et al. summarized their current knowledge of the effector repertoire that has been gained from comparative genomics in smut fungi. They discussed the molecular mechanisms by which the effectors of U. maydis change host cell processes to dampen plant defenses, how the respective genes are regulated. Of the 6784 proteins expressed by U. maydis, 467 are potentially secreted (that is, they contain a signal peptide and lack transmembrane domains), and of these 203 are completely novel, which means that they lack functional or structural domains (Schuster et al. 2017). Many of these effectors were shown to contribute to virulence.
Aspergillus flavus, a soil-borne pathogen, represents a danger for humans and animals since it produces the carcinogenic mycotoxin Aflatoxin B1 (AFB1) in oil-rich crops such as maize (Zea mays L. ssp. mays) (Majumdar et al. 2017). The first full-genome assessment of fungal GPCRs through characterization of null mutants of all revealed 15 GPCRs encoded by the aflatoxin-producing fungus Aspergillus flavus. Most GPCR mutants were aberrant in one or more response processes, possibly indicative of crosstalk in downstream signaling pathways. Interestingly, the biological defects of the mutants did not correspond with assignment to established GPCR classes, likely because of the paucity of data for characterized fungal GPCRs. Many of the GPCR transcripts were differentially regulated under various conditions as well. Pathogenesis-related (PR) proteins serve as an important defense mechanism against invading pathogens by conferring systemic acquired resistance in plants (Affeldt et al. 2014). Among these, the production of the PR maize seed protein ZmPRms (AC205274.3_FG001) has been speculated to be involved in resistance to infection by A. flavus and other pathogens. Involvement of both elicitor-response element (ERE) and enhancing sequences in the PRms promoter were implicated in induction of the promoter by fungal elicitors. The presence of a specific motif in the ZmPRms gene promoter region showed strong association between promoter induction and biotic stressors. Involvement of both elicitor-response element (ERE) and enhancing sequences in the PRms promoter were implicated in induction of the promoter by fungal elicitors. In another study, transgenic expression of the ZmPRms gene in rice resulted in broad-spectrum resistance against fungal (Magnaporthe oryzae, Fusarium verticillioides, and Helminthosporium oryzae) pathogens suggesting involvement of this gene in the central defense mechanism in plants (Gómez-Ariza et al. 2007). In their study, Majumdar et al. (2017) have demonstrated a significant role for ZmPRms in resistance to A. flavus infection of maize kernels through global regulation of genes associated with biotic and abiotic stress responses in plants. This category includes genes associated with disease resistance, carbohydrate metabolism, and transcription factors that are known to be upregulated in plants under stress conditions. The observed increase in A. flavus growth and aflatoxin production in the ZmPRms-RNAi lines supports the RNA-seq interactome analysis that indicated ZmPRms might serve as a major network hub for regulation of downstream resistance-associated gene expression. ZmPRms–RNAi lines and their progenies were morphologically normal, suggesting that it will be a good candidate host-resistance gene for overexpression in maize for increased resistance to A. flavus and possibly against other pathogens. Their results are promising and it might be possible to fine-tune ZmPRms expression in a tissue-specific manner (using modern functional genomic tools) in the future, or use ZmPRms expression as a marker to screen for A. flavus-resistant maize genotypes to reduce aflatoxin contamination in maize and potentially in other economically important crop plants. Interestingly, El Khoury et al. (2017) demonstrated that an aqueous extract of the medicinal plant Micromeria graeca, known as hyssop, completely inhibits aflatoxin production by Aspergillus flavus.
The process of cell death is, itself, a way in which organisms control intracellular infections, removing the infected cell before the pathogen can replicate (Green 2017). Franco-Orozco et al. (2017) characterized a novel fungal PAMP, cell death inducing 1 (RcCDI1), identified in the Rhynchosporium commune transcriptome sampled at an early stage of barley (Hordeum vulgare) infection. R. commune is the causal agent of scald, one of the most destructive and economically important diseases of barley: it is a hemibiotroph with an extended asymptomatic phase. Following conidial germination and cuticle penetration, R. commune hyphae spread between the epidermal cells. As do several other important fungal pathogens of cereals, including M. oryzae, Zymoseptoria tritici, and Parastagonospora nodorum, R. commune belongs to the Ascomycota. This phylum also contains major pathogens of dicots, such as Botrytis cinerea and Sclerotinia sclerotiorum, as well as the model fungus Neurospora crassa. They identified a R. commune transcript, highly abundant at an early stage of barley colonization, and coding for a small secreted protein with four cysteine residues. Expression of the full-length coding sequence in Nicotania benthamiana triggered cell death, which led to a hypothesis that this protein, named cell death inducing 1 (RcCDI1), is a PAMP. They showed this protein to be conserved across different Ascomycetes, with RcCDI1 homologs from Neurospora crassa, Z. tritici, M. oryzae, Botrytis cinerea, and Sclerotinia sclerotiorum also capable of inducing cell death in Solanaceae. They showed that RcCDI1-triggered cell death is dependent on NbBAK1, NbSOBIR1, and NbSGT1. However, cell death was not suppressed by the effector PiAVR3a or PexRD2, suggesting that it does not require NbCMPG1 or NbMAPKKKe.
3.7 Fungal Mycotoxins
Among the 300 reported mycotoxins, AFB1 is the most dangerous one. In fact, AFB1 is a potent carcinogenic agent in humans, inducing liver cancer. For instance, piperine, a major active component of black and long peppers (Piper nigrum L. and Piper longum L.), has been previously demonstrated to be an effective inhibitor of aflatoxin production. The impact of piperine was analyzed by targeting a regulatory network of 20 genes including atfA, atfB, ap-1, msnA, srrA, sskA, and sakA coding for stress response transcription factors; genes involved in fungal antioxidant defense (catA, cat2, sod1, mnSOD); genes coding for oxylipins (ppoA, ppoB, ppoC); genes belonging to GPCRs (gprK, gprH, gprG, gprA, gprP); and the global regulator veA. Several of the GPCRs studied have been linked to aflatoxin production and fungal stress (Caceres et al. 2017). Overexpressed gprK levels were associated with AFB1 inhibition by piperine. As mentioned, GPCRs are responsible for oxylipin modulation. Within the analyzed oxylipins, ppoB have been shown to be the most impacted gene upon treatment. In A. nidulans, ppoB deletion resulted in a precocious production of sterigmatocystin, a precursor of the AFB1 metabolite. This result suggests that in A. flavus the overexpression of ppoB could also have a negative impact on aflatoxin production. Piperine significantly induced the overexpression of 11 genes (ppoA, ppoB, atfA, atfB, ap-1, catA, cat2, sod1, sskA, gprK, gprH) and 5 others (ppoC, veA, msnA, srrA and gprP) were downregulated. Results demonstrated that piperine inhibits almost all genes of the aflatoxin biosynthetic pathway, leading to inhibition of mycotoxin biosynthesis. Gene stress response was accompanied by an enhancement of catalase enzymatic activity. In conclusion, these findings strongly suggest that piperine inhibits AFB1 production by Aspergillus flavus via the perturbation of the oxidative stress balance (Caceres et al. 2017).
Both mycotoxins zearalenone (an estrogenic mycotoxin produced mainly by Fusarium fungi) and resveratrol (RES, 3,5,49-trihydroxystilbene, or RSV, a phytoalexin that participates in plant defense) are antagonistic on both estrogen receptors (ERα and ERβ) at high doses (Mueller et al. 2004). Resveratrol has been characterized as a phytoestrogen based on its ability to bind to and activate estrogen receptor (ER). ER is a nuclear steroid receptor that binds estrogens and regulates the transcription of estrogen-responsive genes by either binding directly to DNA, at particular sequences called estrogen response elements (EREs), or by interacting with other transcription factors, such as Sp1, bound to their cognate sites on DNA. When activated by an agonist ligand, ERα interacts with coactivators, such as SRC-1 and CBP, that either uses acetylate lysine residues in histones to alter chromatin conformation or interacts with components of the RNA polymerase II initiation complex to enhance target gene transcription. Ligands that bind ER can act as agonists, antagonists, or mixed agonist/antagonists. The archetype mixed agonist/antagonist is tamoxifen (TAM), used clinically to prevent breast cancer promotion and recurrence (Bowers et al. 2000). Although resveratrol was reported to interact with ERs, its agonist or antagonistic effects remain controversial (Le Corre et al. 2005). Gehm et al. (1997) showed that RES is a superagonist when combined with E2, and Lu and Serrero (1999) reported ER antagonism of RES (5 l M) in the presence of E2 and partial agonism in its absence. Bowers et al. (2000) observed partial to full agonism in CHO-K1 cells transfected with ERα or ERβ and reporter genes based on various estrogen receptor elements (EREs). They showed that RES acts as a mixed agonist/antagonist in cells transiently transfected with ER and mediates higher transcriptional activity when bound to ERb than to ERa. Moreover, RES showed antagonist activity with ERa, but not with ERb (Bowers et al. 2000).
Zearalenone (ZEA) is mainly produced by Fusarium graminearum Schwabe that causes ear rot of corn in many corn-producing areas of the world. The ascomycete F. graminearum (FG) is a filamentous fungus dwelling on and in a wide range of plant species, on crop debris and within soil, and is responsible for various corn and rice diseases. Zearalenone (ZEA), a nonsteroidal estrogenic mycotoxin, is widely present in cereals and agricultural products. This chemical and its metabolites, zearalanone, α-zearalanol (α-ZOL), and β-zearalanol (β-ZOL), particularly α-zearalenol (α-ZEL), which is used as a growth promoter in cattle, have been shown to competitively bind to estrogen receptors (ERα and Erβ), thereby interfering with the endogenous estrogenic response leading to activated transcription of estrogen-responsive genes and acting as strong ERα agonists. Their endocrine-disrupting behavior is tightly related to their capability to competitively bind the ligand-binding pocket (LBP) and to stabilize at least one of the functionally active conformational assets of the ligand-binding domain (LBD). ZEA, which could be used as an estrogenic effector, is implicated in reproductive disorders and hyperestrogenic syndromes in animals and humans exposed to contaminated food (Videmann et al. 2008). Zearalenone, the endocrine disruptor, has adverse effects such as genotoxicity, oxidative stress, alterations of immunological parameters, pituitary adenoma, and renal toxicity (Le Guevel and Pakdel 2001) and (EFSA Panel on Contaminants in the Food Chain Scientific 2011). ZEN as well as its metabolites exert harmful health effects via their strong estrogenic activities, resulting in decreased fertility, increased fetal resorption, and changes in the weight of endocrine glands and serum hormone levels (Lin et al. 2015; Ji et al. 2017). It has been reported that ZEA is associated with many mycotoxicosis diseases in farm animals; of course, the small intestine absorbs ZEA first, so it is exposed to high concentrations of the toxin, which will certainly influence intestinal tract health. Evidence indicates that a disruption of the epithelial cell integrity and functions induced by ZEA are well established. The review by Robert et al. (2017) aimed at providing a summary of DON, ZEN, OTA, FB1, AFB1, and PAT effects on intestinal barrier function, with special focus on mucus and microbiota. Their data showed that DON, ZEN, OTA, FB1, AFB1, and PAT were known to markedly affect epithelial cell integrity and functions. Previously, Fan et al. (2017) proved that ZEA could induce an accumulation of reactive oxygen species (ROS) in mitochondria and that elevated ROS levels can act as signaling molecules in pathological conditions. ZEA is also a genotoxic agent with DNA-adduct formation, DNA fragmentation, micronuclei production, and chromosome aberrations. Moreover, ZEA inhibits protein and DNA synthesis and triggers lipid peroxidation and apoptotic cell death (Sang et al. 2016). They reported also that the activity of the major antioxidant enzyme of MnSOD in mitochondria was decreased after ZEA treatment. The oxidative damage to cells as well as the lost mitochondrial membrane potential induced by ZEA are intimately associated with mitochondria-mediated apoptotic cell death. Fan et al. (2018) investigated inflammatory cytokine release and the activation of the NLRP3 inflammasome after ZEA treatment both in vitro and in vivo. First, intestinal porcine enterocyte cell line (IPEC-J2) cells and mouse peritoneal macrophages were treated with ZEA to detect NLRP3 inflammasome activation, and the role of ROS in ZEA-induced inflammation was investigated. Then, Balb/c mice were fed a gavage of ZEA, and the disease activity indices (DAIs) and histological analysis were used to assess intestinal inflammation. Their study showed that the mRNA expression of NLRP3 inflammasome, pro-interleukin-1b (pro-IL-1b), and pro-interleukin-18 (pro-IL-18) was upregulated 0.5- to 1 fold and that the release of IL-1b and IL-18 increased. However, the ROS inhibitor N-acetyl-l-cysteine (NAC) reduced IL-1b and IL-18 release. Moreover, the same phenomenon was observed in intestinal tissues of ZEA-treated mice. In addition, clinical parameters of treated mice showed the stools became loose and contained mucus. The presence of gross stool blood was observed in the last 2 days. Histological analysis showed obvious inflammatory cell infiltration and tissue damage in the colon. These findings uncovered a possible mechanism of intestinal mucosal innate immunity in response to mycotoxin ZEA, that it could activate the ROS-mediated NLRP3 inflammasome and, in turn, contribute to the caspase-1-dependent activation of the inflammatory cytokines IL-1b and IL-18.
The phenolic compound resveratrol (RSV) is one type of phytoalexin synthesized by plants in response to fungal attack or ultraviolet light exposure. Sang et al. (2016) treated the human embryo kidney (HEK) 293 cell line with RSV and found it caused a potential protective effect against ZEA cytotoxicity. It has been suggested that resveratrol displays several beneficial human health properties, including anti-high-fat diet-induced senescence and protective mycotoxin-induced cytotoxicity, reduced carbon monoxide-induced cardiotoxicity, and suppressed xenobiotic-induced toxicity through the regulation of xenobiotic-induced AhR activation. Exposure of plants to abiotic stresses or pathogenic attack induces RSV synthesis. Recent studies demonstrated that resveratrol acts a chemoprotective agent against a wide range of PAH-induced toxicities, which is mediated through reduced production of reactive metabolites and DNA adduct formation by specifically inhibiting CYP1A1 and -1B1 activation. Additionally, resveratrol opposed PAH-induced oxidative stress through activation of the nuclear factor E2-related factor 2 (Nrf2) signaling pathway to produce antioxidation molecules, such as superoxide dismutase, glutathione, glutathione reductase, and catalase (Wu et al. 2017). Pangeni et al. (2014) reported that it has been found to potentially exhibit anticancer, antiangiogenic, immunomodulatory, and cardioprotective activities as well as being an antioxidant, and that this is in addition to its usefulness in the treatment of neurodegenerative disease, diabetes, and cardiac ailments. RSV in combination with DON significantly stimulated the progesterone release by GCs at the highest doses (Kolesarova et al. 2012; Wu et al. 2017). The literature reports suggest that oxidative damage seems to be a key determinant of ZEA-induced toxicity, and the protective effect of resveratrol (RSV), an antioxidant phenolic compound, on ZEA-induced cytotoxicity to HEK293 cells was investigated. The experimental results showed that ZEA decreased cell viability in a dose-dependent manner and induced an increase in intracellular ROS in HEK293 cells. A remarkable elevation of MDA and decreased activity of manganese superoxide dismutase (MnSOD) were also observed. A decrease in mitochondrial membrane potential (MMP), cell-cycle arrest at the G0/G1 phase, and increased cell apoptosis indicate a mitochondria-mediated apoptosis. RSV pretreatment not only recovered the activity of MnSOD but also improved ZEA-induced cytotoxicity, evidenced by increased MMP and cell viability and decreased ROS. Furthermore, RSV pretreatment substantially upregulated the expression of the SIRT1 gene by 6.8 fold, reduced the acetylation level of the forkhead transcription factor FOXO3a, and decreased the expression ratio of Bax/Bcl-2. All these results demonstrated that RSV exhibited significant protective effects on ZEA-induced cell damage, and this effect may be attributed to the upregulation of SIRT1 and activation of FOXO3a-mediated pathways to enhance the resistance of cells to oxidative stress induced by ZEA exposure (Sang et al. 2016).
3.8 Fungal Effectors and Elicitors
Candidates for secreted effector proteins (CSEPs) in Sporisorium scitamineum are clustered into ten families (21 CSEPs). Most of the CSEPs in these ten families have more than 2.0% cysteine content and longer amino acids (Que et al. 2014). Many of the functions of CSEPs are unknown in biotrophic phytopathogenic fungi, such as Ustilago maydis (Kämper et al. 2006). Que et al. (2014) showed that 47% of CSEPs were expressed in the infection process. Among them, 7 expressed at all stages, whereas 12, 9, 5, 2, 2, and 1 CSEPs started to express at 0, 12, 24, 48, 96, and 120 hpi, respectively. Four of the aforementioned 7 CSEPs belong to conserved hypothetical proteins with unknown function and did not receive hits from any protein database. The remaining 3 CSEPs have a clan GH-D domain (IPR000111), a catalytic domain (IPR008258), and a Barwin-related endoglucanase domain (IPR009009), as well as sections encoding glycoside hydrolase and lytic transglycosylase-like protein. These CSEPs are mainly involved in carbohydrate degradation. It is unclear how these CSEPs facilitate infection or trigger defense responses, how they are secreted, and whether there is a specific invasion structure to help the secretion. These conundrums require additional endeavors and deeper research. During infection, U. maydis secretes hundreds of effector proteins to suppress plant defense responses and to reprogram plant signaling and metabolism (Djamei and Kahmann 2012). However, the function of most of these novel effectors is unknown (Djamei et al. 2011; Djamei and Kahmann 2012; Mueller et al. 2013). Endocytosis is a vesicular transport pathway in eukaryotic cells that internalizes extracellular fluid and particles, as well as plasma membrane molecules. A role of endocytosis in hyphal growth and development, and in fungal pathogenicity in particular, is still subject to debate (Read and Kalkman 2003). However, recent studies showed that a t-SNARE protein, Yup1, is required for endocytosis and pathogenicity in U. maydis. At later stages of host colonization, large plant tumors develop that provide the environment for massive fungal proliferation. During biotrophic growth, a high-affinity fungal sucrose H+ symporter, Srt1, is upregulated and is crucial for fungal nutrition (Wahl et al. 2010). Tanaka et al. (2014) revealed that an effector called Tin2, which is secreted by the corn smut fungus, causes the production of this anthocyanin pigment. Tin2 moves inside plant cells, where it blocks the breakdown of a protein-modifying enzyme that is necessary to ‘switch on’ the production of anthocyanin. As the building blocks for making lignin are also required for making anthocyanins, Tanaka et al. (2014) suggested a model whereby Tin2 compromises the ability of plants to protect themselves by diverting resources away from making lignin. The link between Tin2 and anthocyanin biosynthesis appears direct and occurs through the stabilization of the kinase ZmTTK1 via Tin2. They have shown that after U. maydis infection, when Tin2 is present, the stabilization of ZmTTK1 increases the biosynthesis of anthocyanin. Tin2 affected all steps of the anthocyanin pathway, but did not affect transcription of all genes coding for lignin pathway components. Conversely, when U. maydis is lacking tin2, more of the common precursor for anthocyanin and lignin is likely to be available for lignin biosynthesis, resulting in cell wall fortification in vascular tissue. The deposition of lignin in the plant cell wall is considered to provide an undegradable physical barrier to infection. At present, there is only a limited understanding of how the lignin biosynthetic pathway in maize is regulated. Several Myb and NAC transcriptional activators involved in secondary cell wall formation have been identified, but the mechanisms behind lignin tissue-specific and developmentally regulated gene expression, and the role of posttranscriptional regulation of gene expression, are yet to be fully elucidated. The increased susceptibility of maize bm mutants to U. maydis and the increased leaf areas displaying disease symptoms provide strong evidence for a role of lignification in disease resistance. A less lignified cell wall will be more easily penetrated by the fungus. The plant may also be less effective at producing defense-related lignin that could slow down the migration of the fungus. Last, because the fungus spreads via the vascular tissue and may rely on nutrients obtained from this tissue for massive proliferation, the altered physicochemical characteristics of the vascular tissue in the bm mutants, combined with altered dimensions of the xylem vessels, may influence entry and the rate of migration of the fungus. The bm2 mutant appears to be an exception in that it does not show increased susceptibility to U. maydis, and even appears to attenuate the effect of infection in bm1-bm2 and bm2-bm3 double mutants relative to the bm1 and bm3 single mutants. This effect may be the result of the accumulation of a fungitoxic (phenolic) compound in this mutant. Whether Tin2 is actively diverting the flux from lignin to anthocyanin remains speculative. Recently, a competition between anthocyanin and lignin pathways for their common precursor has also been detected in strawberry (Ring et al. 2013). In that system, it was demonstrated that plant class III peroxidase (FaPRX27), a gene connected with lignin biosynthesis, is linked to a region implied in decrease in fruit color. In addition, it was demonstrated that the downregulation of chalcone synthase led to an induction of FaPRX27 and that this diverted the flux from anthocyanins to lignin (Ring et al. 2013). Thus, there appears to be a metabolic connection between anthocyanin and lignin biosynthesis pathways in monocot as well as dicot systems, and U. maydis may be altering this with the help of Tin2. Future work is needed to address whether this connection is direct with lignin functioning as barrier or indirect with anthocyanin induction negatively affecting the accumulation of other defense compounds. Anthocyanin accumulation is frequently observed under abiotic and biotic stress conditions, provides UV-B stress protection, and allows scavenging of reactive oxygen species and enhanced resistance to microbial pathogens (Zhang et al. 2013). Tanaka et al. (2014) have uncovered a new positive connection between anthocyanin induction and the development of a biotrophic pathogen. They suggested that examples where anthocyanin induction is observed in response to biotic stress should be revised to see whether metabolic rewiring of the phenylpropanoid pathway is adopted as a common strategy by biotrophic microbes colonizing plants.
Elicitors are fast becoming the key instruments in plant disease control because of their competence in activating or inducing resistance in plants. In general, plants utilize systemic acquired resistance (SAR) or induced systemic resistance (ISR) to initiate systemic immunity during their interactions with elicitors (Chen et al. 2015; Hamid and Wong 2017). The term is now often used for any signal-inducing compounds recognized by the innate immune system involved in priming and/or induction of defense responses regardless of its origin. The fungal secretome consists of various functional groups of proteins, many of which participate in nutrient acquisition, self-protection, or manipulation of the environment and neighboring organisms. The least characterized component of the secretome is small secreted proteins (SSPs). Some SSPs have been reported to function as effectors, but most remain to be characterized. The composition of major secretome components, such as carbohydrate-active enzymes, proteases, lipases, and oxidoreductases, appears to reflect the lifestyle and ecological niche of individual species. Kim et al. (2016) hypothesized that many SSPs participate in manipulating plants as effectors. Obligate biotrophs likely encode more and diverse effector-like SSPs to suppress host defense compared to necrotrophs, which generally use cell wall-degrading enzymes and phytotoxins to kill hosts. Because different secretome prediction workflows have been used in different studies, available secretome data are difficult to integrate for comprehensive comparative studies to test this hypothesis. In their study, SSPs encoded by 136 fungal species were identified from data archived in the Fungal Secretome Database (FSD) via a refined secretome workflow. Subsequently, compositions of SSPs and other secretome components were compared in light of taxa and lifestyles. Those species that are intimately associated with host cells, such as biotrophs and symbionts, usually have higher proportion of species-specific SSPs (SSSPs) than hemibiotrophs and necrotrophs, but the latter groups displayed higher proportions of secreted enzymes. Their results established a foundation for functional studies on SSPs and will also help in understanding genomic changes potentially underpinning different fungal lifestyles (Kim et al. 2016). In most cases, adapted pathogens counteract the basal defense responses of the plants by secreting effector proteins into the plant cells (O’Leary et al. 2016). Successful suppression of PTI often results in effector-triggered susceptibility (ETS), as illustrated in the “zigzag” model by Jones and Dangl (2006) where the plants become susceptible to diseases. Nevertheless, effector-triggered immunity (ETI) is activated when a particular effector protein is recognized either directly or indirectly by its cognate resistance (R) protein in plants (Jones and Dangl 2006; Liu et al. 2013a, b). ETI is an accelerated and augmented response (Coll et al. 2011) that typically results in HR, a localized cell death at the site of infection that kills both the invading pathogen and the infected plant cells (Newman et al. 2013). Consequently, SAR in the host is activated (Jones and Dangl 2006) wherein the host plant becomes resistant to subsequent attacks from obligate biotrophic or hemibiotrophic pathogens. ETI is ineffective against necrotrophic pathogens (Glazebrook 2005). Several studies have shown that necrotrophic pathogens secrete a specialized group of necrotrophic effectors that are known as host-specific toxins (HSTs) which confer susceptibility to the pathogen upon recognition by host PRRs and lead to HST-induced programmed cell death (Friesen et al. 2008a, b; Oliver and Solomon 2010). It was reported that different responses (e.g., in rice: transient membrane depolarization, reactive oxygen generation, expression of typical pathogenesis-related PR genes as well as novel ‘early genes,’ and biosynthesis of jasmonic acid and phytoalexins) seemed to be a part of a complicated signal transduction cascade mediated by a single class of receptor molecule for chitin fragments elicitor (Tsukada et al. 2002). They conclude that the rice receptor for chitin fragments (N-acetyl chitooligosaccharide) elicitor did not couple to Gα, and thus not to the heterotrimeric G protein. For more details about elicitors and effectors in Basidiomycetes, refer to Hamid and Wong (2017).
Elicitins are structurally conserved extracellular proteins in Phytophthora and Pythium oomycete pathogen species (e.g., PiINF1) (reviewed by Derevnina et al. 2016). Abundant proteins in Phytophthora culture filtrates have the capacity to elicit hypersensitive (HR) cell death and disease resistance in tobacco. Later, they became well established as having features of MAMPs, a protein produced by oomycete pathogens known as elicitin that elicits defense in a variety of plant species. Research on elicitins culminated in the recent cloning of the elicitin response (ELR) cell-surface receptor-like protein, from the wild potato Solanum microdontum, which mediates response to a broad range of elicitins. Elicitins belong to complex multigene families and form diverse subclasses, defined by Jiang et al. (2006) as elicitin (ELI, class Ia, Ib, II) and elicitin-like (ELL, class III) genes. The number of ELI and ELL genes varies from one species to another, with each showing differential expression patterns and HR-inducing activities. With the exception of the canonical clade ELI-1, all other clades possess, in addition to a signal peptide and elicitin domain, C-terminal domains of variable length (17–291) that tend to be rich in threonine, serine, and proline residues (Jiang et al. 2006). Elicitins bind sterols and other lipids with varying affinities, and sterols are important for oomycete growth and sporulation. However, Phytophthora and Pythium species are sterol auxotrophs; this may be associated with the parasitic lifestyle of these organisms. Independent studies revealed that elicitins can act as sterol carriers by scavenging sterols from synthetic liposomes and plant plasma membranes. Further work demonstrated that elicitins form a diverse family of oomycete proteins, variously known as cryptogein (a.k.a. CRY-B elicitin), capsicein (CAP-A), parasiticein (PAR-A), and INF1 from Phytophthora cryptogea, Phytophthora capsici, Phytophthora parasitica, and Phytophthora infestans, respectively. These proteins induce potent defense responses in several plant species, particularly in the Solanaceae and Brassicaceae. Elicitin response (ELR), the first cell-surface receptor to mediate specific response to elicitins, was cloned from the wild potato species Solanum microdontum. ELR mediates HR cell death to classic elicitins, including INF1 and CRY-B, and also enhances resistance to the Irish potato famine pathogen Phytophthora infestans. Successful pathogens overcome MAMP-triggered immunity (MTI) by secreting effector molecules that suppress host immune responses. Elicitins carry many characteristic MAMP attributes: (1) they are structurally conserved; (2) they show no sequence similarity to plant proteins (Jiang et al. 2006), and therefore are viewed as nonself molecules by the host; (3) as sterol transporters, they fulfil an important biological function in oomycetes; (4) they are expressed during host interaction; and (5) they are recognized by cell-surface localized PRRs that trigger an immune response. In addition to tobacco, other reported elicitin-responsive species include tomato, potato, and pepper (Solanaceae), pigeon pea (Fabaceae), grapevine (Vitaceae), citrus (Rutaceae), oak (Fagaceae), and some radish and turnip cultivars (Brassicaceae). However, differential responses to elicitins can occur within a given plant taxon. For example, within the genus Solanum, some species respond to INF1 and variation in the response occurs between genotypes of the same species. This finding indicates that the genetic basis of the response to elicitins is likely to be highly variable across plant taxa. Derevnina et al. (2016) in review suggested that oomycete plant pathogens have evolved an effector toolbox to modulate host responses triggered by their elicitins. Many questions remain to be answered. What is the molecular basis of the response to INF1 in other species besides S. microdontum? Has elicitin recognition evolved independently in different plant taxa? How does the response of plants such as tomato differ from the typical cell death response observed in Nicotiana spp. and wild potato? How do oomycete effectors suppress elicitin-triggered responses? The degree to which these elicitin-like proteins can be detected by plant receptors, and the molecular basis of any potential plant response, remain to be determined. Ultimately, a better understanding of elicitin perception and plant response could help engineer crops with broad-spectrum resistance against oomycete pathogens.
Cerato-platanin (CP) is a noncatalytic, cysteine-rich protein; the first member of the cerato-platanin family is an elicitor of the primary defense response and it has been classified as MAMP/PAMP (Luti et al. 2017). It is a single-domain protein with a double Ψ/β barrel domain resembling the D1 domain of plant and bacterial expansins. Similar to expansins, CP shows a cell wall-loosening activity on cellulose and can be defined as an expanisin-like protein, in spite of the missing D2 domain normally present in plant expansins. The weakening activity shown on cellulose may facilitate the CP–host interaction, corroborating the role of CP in eliciting the plant defense response. Indeed, CP is an elicitor of primary defenses acting as a PAMP. So far, structure–function relationship studies have been mainly performed on the bacterial BsEXLX1 expansin, probably because of difficulties in expressing plant expansins in heterologous systems. Baccelli et al. (2014) reported a subcloning and purification method of CP in the engineered Escherichia coli Shuffle cells, which proved to be suitable to obtain the properly folded and biologically active protein. The method also enabled the production of the mutant D77A, rationally designed to be inactive. The wild-type and the mutated CP were characterized for cellulose-weakening activity and for PAMP activity (i.e., induction of ROS synthesis and phytoalexins production). Their analysis revealed that the carboxyl group of D77 is crucial for expansin-like and PAMP activities, thus permitting establishing a correlation between the ability to weaken cellulose and the capacity to induce defense responses in plants. Their results enable the structural and functional characterization of a mono-domain eukaryotic expansin and identified the essential role of a specific aspartic residue in cellulose weakening. Because CP lacks the D2 domain it can be, therefore, defined as a mono-domain expansin. The ease of producing the protein in high yield in Pichea pastoris, as well as the extraordinary heat stability, make this protein suitable for future potential applications in increasing the cellulose loosening rate as, for example, during biofuel production.
Secondary metabolite synthesis and accumulation in plant cell cultures can be stimulated by elicitors, signaling molecules that can boost the formation of secondary metabolites in cell cultures by initiating plant defense, hypersensitive responses, or pathogenesis-related proteins (Zhao et al. 2007). Among biotic elicitors, fungal elicitors have resulted in substantial augmentation in the production of a number of phytochemicals in plant tissue cultures of Withania somnifera, also known as Indian ginseng. W. somnifera is known to contain valuable bioactive compounds, called anolides, that structurally resemble the ginsenosides of Panax ginseng. The extracts as well as different isolated bioactive constituents of W. somnifera have been shown to possess adaptogenic, anticancer, anticonvulsant, antioxidative, immunomodulator, and neurological effects. Piriformospora indica, a root endophytic fungus that belongs to the family Sebacinaceae, was isolated from rhizosphere soil of xerophytic shrubs in the Thar desert in India; it is an axenically cultivable phytopromotional and biotrophic root endosymbiont. This fungus has multifaceted roles such as nutrient uptake, disease resistance, stress tolerance, and growth encouragement involving value addition. It has been reported to increase the growth of W. somnifera in vitro and in cell suspension and hairy root cultures (Ahlawat et al. 2014; Saxena et al. 2017). In the study by Ahlawat et al. (2014), a cell suspension culture of W. somnifera was elicited with cell homogenates of the following fungi (Alternaria alternata, Fusarium solani, Verticillium dahliae, and Piriformospora indica) in a shake flask; then, the major withanolides such as withanolideA, withaferin A, and withanone were analyzed. Among the biotic elicitor preparations from P. indica, the cell homogenate at 3% (v/v) affected the maximum enhancement on the production of withanolides in the cell cultures of W. somnifera in a 5.0-l bioreactor. In their study on the positive effect of P. indica on withanolides biosynthesis in W. somnifera, it appeared that the fungus has the potential of acting as a good source of elicitor in increasing the production of useful secondary metabolites in other plant cell cultures also, which might be worth investigating. Saxena et al. (2017) examined elicitation by cell homogenate of P. indica(CHP) and methyl jasmonate on withanolide accumulation in hairy root cultures as well as on the expression pattern of ten genes encoding enzymes in the withanolide biosynthetic pathway. The expression of all the genes encoding enzymes of withanolide biosynthesis and its related pathways supplying carbon, viz. HMGR, FPPS, DXS, DXR, SQS, SE, CAS, ODM, SDS, and SMT-1, was upregulated in CHP treatments compared with the control, although it was less than that observed with 15 μM MeJ during a 4-h treatment. Maximal expressions were found in 3% CHP for 48 h, which also had more biomass and withanolide content.
A wide range of external stress stimuli trigger plant cells to undergo complex network of reactions that ultimately lead to the synthesis and accumulation of secondary metabolites. Accumulation of such metabolites often occurs in plants subjected to stresses including various elicitors or signal molecules. Endophytic fungi, an important constituent in the environment of medicinal plants, have been known to form long-term stable and mutually beneficial symbiosis with these plants. The endophytic fungal elicitor can rapidly and specifically induce the expression of specific genes in medicinal plants, which can result in the activation of a series of specific secondary metabolic pathways resulting in the significant accumulation of active ingredients (Zhai et al. 2017). The plant–microbe interactions and plant defense responses, as well as the signal transduction pathways involved, have been studied extensively and continue to be topics of active research and discussion. The use of fungal elicitors has been reported to be one of the most effective strategies for improving the productivity of useful secondary metabolites in plant cell culture (Takeuchi et al. 2013). Recent studies of fungal elicitors focus on fungal elicitor recognition, G-protein, Ca2+, and hydrogen peroxide (H2O2) signal transduction, signal amplification of jasmonic acid (JA), nitric oxide (NO), salicylic acid (SA), abscisic acid (ABA), ethylene (ETH), signal crosstalk, gene expression, activation of the key enzymes, and the application of fungal elicitor. The results were summed up as follows: fungal elicitor, receptor to fungal elicitor, and the identification of fungal elicitor (Zhai et al. 2017). Although Fusarium, yeast, and Pythiumspecies can be used to elicit specific secondary metabolites in various medical plants, they may not be the best elicitor for the induction of a specific secondary metabolite. It is a remarkable fact that the combination of different elicitors may achieve a synergistic effect from the different action of different elicitors. There are some large-scale industrial productions of secondary metabolites in some medical plants.
3.9 Aspergillus GPCRs
The fungal genus Aspergillus is of critical importance to humankind because of the medical (A. fumigatus, A. terreus), food spoilage (A. flavus, A. parasiticus), and industrial (A. niger, A. aculeatus, A. oryzae) relevance of some of its species. Many Aspergillus species are included in important pathogens of humans, animals, and crops, a source of potent carcinogenic contaminants of food, and represent an important genetic model, especially that of Aspergillus nidulans, that have contributed broadly to our understanding of eukaryotic cell biology and molecular processes. The genome sequences of eight aspergilli have already been explored to investigate the aspects of fungal biology. Most filamentous fungi have three conserved Gα subunits (I, II, III), one Gβ protein, and one Gγ protein (de Vries et al. 2017). The number of predicted GPCRs varies widely, with a larger number identified in Ascomycetes than in Basidiomycetes (Han and Prade 2002; Li et al. 2007). Results of the domain-based identification of G proteins and GPCR orthologs in the Eurotiomycetes show that Gα proteins were found to be highly conserved. The Gα group I (fadA) regulates multiple pathways, and most filamentous fungi have a single copy of this gene (de Vries et al. 2017). In contrast, group II Gα proteins (ganA) are less conserved than the other Gα proteins, and as many as three copies were found in one species (Talaromyces stipitatus). The group II Gα proteins have a positive role in germination of conidia, possibly through cAMP signaling in carbon sensing (Chang et al. 2004; Lafon et al. 2005). The last Gα belongs to group III (ganB) and was found as a single copy in all Eurotiomycetes, where it is thought to act as a negative regulator (de Vries et al. 2017). Additionally, they found three different Gα groups of orthologs (Clusters FungiJGICTBE21135 only present in A. niger ATCC 1015, FungiJGICTBE20122 present in A. flavus NRRL 3557 and in A. oryzae RIB40, and FungiJGICTBE14195 present in A. flavus NRRL 3557) that appeared to encode genes which contain the Gα domain. There was also one copy of the Gβ-negative regulator of asexual reproduction (sfaD) in each genome (Rosen et al. 1999). Finally, all Eurotiomycetes genomes contained a single highly conserved Gγ gene (Downes and Gautam 1999). The pheromone/pheromone receptor genes, differentially regulated in ascomycetes and basidiomycetes, are discussed in detail in the review article by Xue et al. (2008). Aspergillus species are characterized by the unifying feature of the “aspergillum,” an asexual reproductive structure. A central regulatory pathway (brlA, abaA, wetA) controls conidiation-specific gene expression and asexual developmental processes. Proper activation of brlA requires upstream developmental activators (encoded by fluG, flbA-E) and removal of repression by several negative regulators including SfgA, VosA, NsdD, and two G-protein signaling pathways (de Vries et al. 2017). WetA is also required for normal vegetative growth, hyphal branching, production of aflatoxins, contribution to spore integrity and maturity by properly regulating the metabolic pathways of trehalose, chitin, α-(1,3)-glucan, β-(1,3)-glucan, melanin, hydrophobins, and secondary metabolism more generally. Light also regulates asexual developmental genes, including Eurotiomycetes, with several photoreceptors identified such as one red-light (FphA), which represses fruit-body formation and induces asexual spore formation, and three blue-light receptors represented by the white-collar complex (WCC) LreA (WC-1) and LreB (WC-2) and the photolyase/cryptochrome CryA. Full stimulation of conidia production was only achieved with a combination of red and blue light. In Neurospora crassa, WC-1 and WC-2 interact and form the white-collar complex (WCC). This complex, upon light exposure, binds transiently to the promoters of light-inducible genes, presumably to activate their transcription. LreA and LreB act as positive factors for the sexual cycle. LreB interacted not only with LreA, but also with the phytochrome FphA, which itself also interacted with the VeA regulatory protein. Light receptors transmit their signal to a number of other regulatory proteins including a bridging protein, VeA, as part of a trimeric complex. VeA has a central role in the balance of asexual and sexual development and in the coordination of morphogenesis and secondary metabolism. After light absorption, the phytochrome shuttles from the cytoplasm to the nucleus upon illumination, then interacts with transcription factors, for example, which in turn regulate the transcription of several genes in plants. In A. nidulans the phytochrome was found in the cytoplasm and in the nucleus, where it forms a light-sensing protein complex, but there is yet no evidence for a shuttling mechanism. All the presumed asexual species were found to contain either MAT1-1 alpha idiomorphs or MAT1-2 idiomorphs, ppgA homologs encoding a pheromone precursor, and preA and preB homologs encoding pheromone receptors; all of these are expressed in all the asexual species in the same way as known sexual species. Many regulatory genes for asexual sporulation in A. nidulans have been identified. A. nidulans forms asexual spores in light, but preferentially undergoes sexual reproduction and produces resistant and durable ascospores in the dark. However, it has to be considered that at least 75 other genes are required for completion of the sexual cycle in the aspergilli.
Aspergillus fumigatus is the most pathogenic species among the aspergilli, and is the major fungal agent of human pulmonary infection (Grice et al. 2013). In healthy individuals, mucociliary clearance and pulmonary immune defenses clear the hundreds of conidia inhaled daily. Beyond residual host immune responses, there are additional obstacles to successful colonization of the mammalian lung, including tolerance of host-facilitated stresses, such as iron starvation and alkaline pH. A screen of the predicted proteome using all GPCR sequences at the time available in the GPCR Database (GPCRDB) was applied to A. fumigatus and identified 15 putative GPCRs (Lafon et al. 2006). In aspergilli, putative GPCRs are classified by homology, and according to a convention established by Lafon et al. (2006) in A. nidulans, into nine groupings. In A. fumigatus, classes 1 and 2 are composed, respectively, of two putative pheromone receptors GprA (AFUA_3G14330) and GprB (AFUA_5G07880); class 3 is composed of two putative carbon sensors GprC (AFUA_7G04800), GprD (AFUA_2G12640); class 4 contains three putative nitrogen sensors: GprF (AFUA_5G04100), GprG (AFUA_1G11900), and GprJ (AFUA_1G06840); class 5 has three putative cAMP receptors: GprH (AFUA_5G04140), GprI (AFUA_3G00780), and GprL (AFUA_ 3G01750), the latter being unique to A. fumigatus; class 6 is composed of a single putative GPCR, GprK (AFUA_4G01350) having a regulator of G-protein signaling (RGS) domain, unique to filamentous fungi. Kim et al. (2019) characterized the functions of RgsD, one of the six RGS domain proteins present in the human pathogenic fungus A. fumigatus. Yeast two-hybrid assays reveal that RgsD can interact with the three Gα proteins GpaB, GanA, and GpaA, showing the highest interaction potential with GpaB. They concluded that RgsD attenuated the cAMP-PKA signaling pathway and negatively regulated asexual development, toxigenesis, melanin production, and virulence in A. fumigatus. In A. fumigatus, the rax1 gene encodes for a putative positively controlling growth and development, and modulates intracellular trehalose amount, cell wall melanin levels in conidia, and spore resistance to H2O2; class 7 includes two putative GPCRs with homology to rat growth hormone-releasing factor receptors, only one of which is found in A. fumigatus, GprM (AFUA_7G05300); class 8 contains three putative GPCRs with identity to yeast Izh zinc regulators, two of which are found in A. fumigatus GprO (AFUA_ 3G10570) and GprP (AFUA_6G07160); and Class 9 is composed of a single putative GPCR, NopA (AFUA_7g01430), having identity to bacterial opsins. The roles of some of these receptors have been identified in other species, although in A. fumigatus little is known. Among the 15 predicted GPCR-like proteins in A. fumigatus, only 2, GprC (AFUA_7G04800) and GprD (AFUA_2G12640), have been characterized. GprC and GprD have been noted as having homology to Gpr1p of Saccharomyces cerevisiae, which activates the cAMP pathway in response to glucose. As well, the A. nidulansGprD homolog mediates increase of intracellular cAMP in response to oxygenated polyunsaturated fatty acids (oxylipins), which act as autocrine and paracrine mediators in eukaryotic organisms (Affeldt et al. 2012). In contrast to most Aspergillus spp., wherein four predicted Gα-subunits occur, only three (GpaA, AFUA_1G13140, GpaB, AFUA_1G12930, and GpaC, AFUA_3G12400) have been identified for A. fumigatus, which presumably act via interaction with the Gβ- and Gγ-subunits (SfaD, AFUA_5G12210 and GpgA, AFUA_1G05210), which are undoubtedly significantly relevant to A. fumigatus viability and vegetative growth. Kulkarni et al. (2005) noted, based upon membrane topology, that the number of putative GPCR-like proteins encoded by the Magnaporthe grisea genome rose to 76 when the criteria were relaxed to include homologs of the Pth11 receptor (DeZwaan et al. 1999). The CFEM domain of seven-transmembrane protein Pth11 is necessary for proper development of appressoria, appressoria-like structures, and pathogenicity. Applying a more universal approach to A. fumigatus, Grice et al. (2013) identified 6496 proteins having putative transition-minimized differential signaling (TMDs). Among them, 161 proteins were found to encode seven predicted TMDs. Histidine kinases (HK) are phospho-relay protein sensors that transduce extracellular signals. HKs are common in the fungal kingdom and apparently absent in humans. Among Archaea, Bacteria, and Fungi, two classes of HK (two-component and hybrid) are found. HK activities have been associated with both the osmo- and peroxide-regulatory pathways in many fungi and have been most extensively characterized in Saccharomyces cerevisiae. However, RR proteins are not abundantly encoded by fungal genomes; Skn7 and Ssk1 are two examples of such proteins, which in S. cerevisiae and Candida albicans, account for the entire RR cohort of these species (Kaserer et al. 2009). The fungal phospho-transfer relay can involve three proteins, as exemplified by the S. cerevisiae HOG1 MAPK phospho-relay, where an HK (Sln1), a histidine phospho-intermediate (Ypd1) and an RR (Ssk1) collectively mediate a multistep phospho-transfer (Kaserer et al. 2009). In a study addressing the role of oxidative stress in A. fumigatus pathogenicity, Du et al. (2006) characterized the A. fumigatus TcsB protein, a putative homolog of Sln1 in S. cerevisiae.
3.10 Candida albicans GPCR
Although Candida albicans is commensal in humans, it often causes mucosal or systemic infections that contribute to substantial morbidity and life-threatening bloodstream infections in immunocompromised patients. Four classic C. albicanscell types are known, which switch among at least six distinct forms: yeast-like morphotypes, hyphae, pseudohyphae, and chlamydospores in response to environmental cues, and this adaptability is thought to contribute to its virulence. The chlamydospores are large, spherical, thick-walled cells that are observed in vitro under certain harsh conditions, such as starvation and hypoxia. Macrophages are important innate immune cells that limit the niches in the human body in which C. albicans can persist through phagocytic removal. However, following phagocytosis C. albicans readily escapes from the immune cell by differentiating into filamentous hyphae. In C. albicans, Tor1 has been implicated in the negative regulation of filamentous growth. Inhibition of TORC1 results in the activation of the GATA transcription factor Brg1, which is involved in the regulation of hypha-specific genes and blocking the recruitment of the Nrg1-Tip1 transcriptional repressor complex (Lu et al. 2012; Su et al. 2013). Hyphae express numerous cell type-specific virulence factors such as adhesins [for example, hyphal wall protein 1 (Hwp1), agglutinin-like protein 3 (Als3), Als10, factor activated 2 (Fav2), and Pga55], tissue-degrading enzymes [for example, secreted aspartyl protease 4 (Sap4), Sap5, Sap6], antioxidant defense proteins [for example, superoxide dismutase 5 (Sod5)], and even a recently described cytolytic peptide toxin (extent of cell elongation protein 1, Ece1. The CFEM domain-containing proteins in C. albicans are involved in binding and maintenance of iron, adherence, and virulence. Hyphal form has an important function in disease pathogenesis by invading epithelial cells and causing tissue damage by inducing their endocytic uptake by cultured human oral epithelial cells through a specific interaction between the hyphal adhesin, Als3, and host epithelial cadherin (E-cadherin); internalized hyphae then proceed to damage the host cells. They can also actively penetrate into oral epithelial cells, possibly through physical pressure and secreted enzymes. Thus, in a reconstituted model of human oral epithelial tissue, invading hyphae trigger several proinflammatory signaling pathways in the host, whereas yeasts, which merely colonize the surface of the tissue without causing damage, trigger a more muted inflammatory response. Yeasts, hyphae, and pseudohyphae were all present in infected tissues that were recovered from human patients and animals with disseminated candidiasis. Moreover, C. albicans mutants that are trapped as either yeasts or filaments are both defective in bloodstream infection models, which suggests that the ability to interconvert between different cell types is required for virulence. Although hyphae and pseudohyphae predominate in most virulence models, with white (a/α) yeasts also being essential in disseminated (bloodstream) infections, the newly described elongated yeasts may be more specialized for commensalism. For example, GUT a/α cells were identified based on their superior fitness in an intestinal commensalism model. Further, opaque (a/α) and opaque (a or α) cells have been reported to outperform other cell types during skin colonization (Noble et al. 2017). Therefore, inhibition of hyphal formation has significance in the prevention of candidiasis (Kurakado et al. 2017). During the survey of low molecular weight compounds, it was found that various steroids, including 17β-estradiol, inhibit hyphal formation without inhibiting growth. Previous reports have shown that several low molecular weight compounds inhibit the budded to hyphal form transition (Toenjes et al. 2005, 2009). The yeast to hyphae transition is central to C. albicans virulence through functions including tissue invasion, cell adhesion, evasion of macrophages, and development of clinically relevant biofilm communities. In response to stimuli such as temperature, nutrients, or serum, signal transduction pathways and transcription factors induce the hyphal gene expression program and filamentous growth (Sudbery 2011). The limited studies in which steroid response in yeasts has already been examined demonstrate that yeast cells are extremely responsive to steroids in affecting the morphology, growth and expression profile of several genes and proteins (Prasad et al. 2012). Interestingly, C. albicans has proteins capable of binding to steroids, including estrogen-binding protein (Ebp1). It also possesses corticosterone-binding protein (CBP) and progesterone-binding protein (PBP); however, the steroid signaling cascade does not appear to exist in yeast. Estrogen and progesterone are known to stimulate filamentation and biofilm formation, leading to vulvovaginal candidiasis. To determine whether EBP1 regulates a virulence factor, Kurakado et al. (2017) investigated the effect of 17β-estradiol on the morphological transition of C. albicans using an ebp1 deletion mutant. Treatment with 17β-estradiol inhibited hypha formation, whereas its effect on the ebp1 deletion mutant was decreased compared to that on the wild-type and revertant strains. These data suggest a new pathway for the yeast to hypha transition via EBP1 in C. albicans. To date it has been difficult to translate these basic science discoveries into new therapies to combat C. albicans infections (Vila et al. 2016, 2017).
The plasma membrane has key roles in virulence because it not only functions as a protective barrier but also mediates dynamic functions including secretion of virulence factors, cell wall synthesis, invasive hyphal morphogenesis, endocytosis, and nutrient uptake (Douglas and Konopka 2016). C. albicansbiofilms forming on implanted medical devices (abiotic surfaces) such as catheters serve as a source of infectious cells that can cause deep-seated and bloodstream infections and are associated with high levels of antifungal resistance (Nobile and Johnson 2015). Drug transporters are not only upregulated during planktonic growth but also remain upregulated during biofilm development, enabling C. albicans to persist in the presence of antifungals (Ramage et al. 2002). A typical biofilm architecture consists of layers of yeast and hyphal cells interlaced with each other and stabilized by adhesive interactions between these cell types (Nobile and Johnson 2015). The transcriptional network that controls biofilm formation contains six transcription factors (Efg1, Tec1, Bcr1, Ndt80, Brg1, Rob1), wherein all these excepting Bcr1 regulate hyphal morphogenesis (Schweizer et al. 2000). The downstream targets of these transcription factors constitute genes involved in adhesion, hyphal morphogenesis, matrix production, and drug resistance. Bcr1 regulates the expression of adhesins, including the agglutinin-like Als proteins Als1 and Als3, and the hyphal cell wall protein Hwp1 that promote adherence during biofilm formation (Nobile et al. 2008). Bcr1 thus serves as a positive regulator for cell–cell and cell–substrate adhesion (Finkel et al. 2012).
Although the diploid fungusCandida albicans, a human pathogen, has been thought to have no sexual cycle, it normally possesses mating-type-like orthologs (MTL) of both the Saccharomyces cerevisiae mating-type genes (MAT) a and alpha on chromosome 5 (Magee and Magee 2000). Evidence for mating included formation of stable prototrophs from strains with complementing auxotrophic markers; these contained both MTL alleles and molecular markers from both parents and were tetraploid in DNA content and mononucleate. However, mating-competent forms of the organism were recently described that produced tetraploid mating products. Bennett and Johnson (2003) described the conditions in which growth of a tetraploid strain of C. albicans on S. cerevisiae ‘pre-sporulation’ medium induced efficient, random chromosome loss in the tetraploid. They proved evidence for a chromosome loss pathway that, combined with mating, completes the parasexual cycle for C. albicans that can be readily carried out in the laboratory. Their study leaves open the possibility that C. albicans is able to undergo meiosis. They suggested that if meiosis does occur in C. albicans, it may occur by a different signaling pathway, perhaps requiring a stimulus from its mammalian host. The C. albicans genome encodes CaSte2p, a homolog of the S. cerevisiae alpha-mating pheromone receptor Ste2p, and two potential pheromones, alpha-F13 (GFRLTNFGYFEPG) and alpha-F14 (GFRLTNFGYFEPGK). Janiak et al. (2005) indicated that CaSte2p is effectively coupled to the S. cerevisiae signal transduction pathway. Functional expression of CaSte2p in S. cerevisiae provides a well-defined system for studying the biochemistry and molecular biology of the C. albicans pheromone and its receptor. Analyzing more than 500 genes important for sexual differentiation in S. cerevisiae, Janiak et al. found many homologs of genes that are implicated in the initiation of meiosis (SPO11, DMC1, NDT80), chromosome recombination, and the formation of synaptonemal complexes. However, others (ME1, ZIP2, and SPO13, which are strictly required for meiosis in S. cerevisiae) are striking by their absence. C. albicans seems to have homologs of all the elements of a functional pheromone response pathway involved in mating in S. cerevisiae (STE2, STE3, Gα(Gpa1), Gβ(Ste4), Gγ(Ste18), (MAP)STE20, STE11, STE7, FUS3, Ste12p) but lacks many homologs of S. cerevisiae genes for meiosis (Tzung et al. 2001). However, the identification of a mating-type-like (MTL) locus and genes such as CPH1, CAG1, DLH1, NDT80, and HST6 in C. albicans (C. albicans genome project information http://alces.med.umn.edu/Candida.html and http://www-sequence.stanford.edu/group/candida), which participate in meiotic differentiation in S. cerevisiae, suggests that the classification of this diploid fungus belies the existence of a sexual cycle. The investigation of pathogenesis-related underlying processes, which are mediated by protein–protein interactions (PPI), is restricted to a limited number of genetic tools available in C. albicans (Subotić et al. 2017). Unique features of C. albicans, such as its alternative codon usage and incomplete meiosis, have enforced the optimization of standard genetic methods as well as development of novel approaches. Hence, Subotić et al. (2017) have successfully established an additional tool for PPI studies in C. albicans by creating an optimized set of vectors allowing the use of the BiFC assay to detect PPI, and confirmed and visualized the interaction of the membrane-bound receptors, Gpr1 and Gpa2. Full-length Gpr1 interacted with Gpa2 in the membrane, whereas the C-tail, devoid of the membrane-anchoring part, formed a complex with Gpa2 in the cytosol. The BiFC assay proved to be useful in visualizing in vivo, for the first time, the interaction of PKA subunits Bcy1-Tpk1 and Bcy1-Tpk2 in C. albicans. Rta3, a member of the Rta1-like family of lipid-translocating exporters, has a 7-transmembrane domain topology, similar to the G-protein-coupled receptors, and is unique to the fungal kingdom. Transcriptome analysis revealed that Rta3 regulates the expression of Bcr1 target genes involved in cell-surface properties, adhesion, and hyphal growth. The identification of this novel Rta3-dependent regulatory network that governs biofilm formation and PC asymmetry across the plasma membrane will provide important insights into C. albicans pathogenesis (Srivastava et al. 2017). Their findings point toward a role for the plasma membrane localized Rta3 in providing tolerance to miltefosine, an analog of alkylphosphocholine, by maintaining mitochondrial energetics. There is a complex relationship among the Cek1 and Cek2 MAP kinases (redundant for mating in C. albicans) and the MAP kinase phosphatase Cpp1 functioning in the C. albicans mating response (Rastghalam et al. 2019). Evidence provided in their study suggests that the Cek1 kinase is the major MAP kinase required for the pheromone response. Loss of this kinase greatly compromises all aspects of the mating response, reducing processes such as induction of pheromone-responsive genes, projection formation, and mating, far below the wild-type level. However, the loss of Cek1 function does not create complete sterility, as a residual level of morphological response and mating can be detected in cek1Δ/Δ mutant strains that is dependent on the related MAP kinase Cek2. The deletion of both kinases completely blocked pheromone response and mating. This pattern of two kinases, with distinct roles but having overlapping functions that require both to be deleted to generate sterility, is similar to that seen for the MAP kinases Fus3 and Kss1 of the post-whole-genome duplication (WGD) yeast S. cerevisiae (Marcet-Houben and Gabaldón 2015). The fact that Cek1/Cek2 paralogs are present in the pre-WGD yeast C. albicans establishes that the MAPK duplication preceded the WGD, occurring coincidentally with the appearance of an Ste5 equivalent (Cote et al. 2011). Therefore, during the WGD, duplication of the Cek1/Cek2 paralogs should have created four paralogous genes, a pair of CEK1 orthologs, and a pair of Cek2 orthologs. To get to the two-paralog pattern that exists in S. cerevisiae, two of these duplicated genes must be lost. Synteny, sequence similarity, and functional relatedness all are consistent with FUS3 and CEK1 being orthologs. The function of the Cek1 MAP kinase in the pheromone response appears regulated by the Cpp1 phosphatase. In response to pheromone treatment, the activation loop of Cek1 shows enhanced phosphorylation, and the level of phosphorylation in both pheromone-treated and -untreated cells was enhanced by the loss of the Cpp1 phosphatase. Phenotypic analysis of the cpp1Δ/Δ strain showed that all aspects of pheromone response were modified in such strains: the cells showed enhanced projection formation and pheromone-induced gene expression in response to pheromone application, they showed greatly increased pheromone-mediated cell-cycle arrest measured by halo assays, and they showed significantly reduced mating compared to nonmutant cells. As well, pheromone-treated cells or cells lacking Cpp1 showed a greater concentration of Cek1 in the nucleus, consistent with activation loop phosphorylation being involved, directly or indirectly, in nuclear localization. Rastghalam et al. (2019) provided a speculative model for the roles of the MAP kinases and MAP kinase phosphatase in the C. albicans pheromone response. It appears that the major signaling pathway involved the receptor–G-protein module activating the upstream elements leading to Hst7 phosphorylating Cek1 on the activation loop, and this in turn enhanced nuclear localization of the kinase and activation of downstream events. This activation is opposed by the action of the Cpp1 phosphatase; in the absence of Cpp1, Cek1 activation is enhanced and sustained, leading to increases in pheromone-mediated gene induction, projection production, and halo formation. In the absence of Cek1, a low level of mating still occurs, and this mating is dependent on Cek2. However, overall Cek2 appears to have a minor role in the mating process: its most significant role in this process in wild-type cells appears to be regulating reentry into the cell cycle after pheromone-mediated arrest. Overall, loss of Cek2 causes only a minor decrease in mating and in morphological changes in response to pheromone and has essentially no effect on pheromone-mediated gene induction. However, loss of Cek2 leads to a significant increase in pheromone-mediated cell-cycle arrest as measured by halo assays. Because the frequency of projections in the mutant strain is not dramatically enhanced relative to WT cells, it is likely the larger halos result from an inability to adapt to pheromone treatment and to reenter the cell cycle. They concluded that further work will be necessary to determine the details of MAP kinase phosphorylation/dephosphorylation and cellular function, with the ultimate goal of understanding how a single MAP kinase with roles in different pathways is properly directed and regulated and how closely related kinases are connected to unique regulatory events.
Under laboratory conditions, the human fungal pathogenCandida albicans can undergo both heterothallic and homothallic (opposite- and same-sex) mating. However, both mating modes require the presence of cells with two opposite mating types (MTLa/a and α/α) in close proximity. Given the predominant clonal feature of this yeast in the human host, both opposite- and same-sex mating would be rare in nature. Glucose starvation and oxidative stress, common environmental stresses encountered by the pathogen, induce the development of mating projections and efficiently permit same-sex mating in C. albicans with an “a” mating type (MTLa/a). This induction bypasses the requirement for the presence of cells with an opposite mating type and allows efficient sexual mating between cells derived from a single progenitor. Glucose starvation causes an increase in intracellular oxidative species, overwhelming the heat-shock transcription factor-1 (Hsf1)- and heat-shock protein (Hsp) 90-mediated stress-response pathway (Guan et al. 2019). Candida transactivating protein 4 (Cta4) and cell wall transcription factor 1 (Cwt1), downstream effectors of the Hsf1–Hsp90 pathway, regulate same-sex mating in C. albicans through the transcriptional control of the master regulator of a-type mating, MTLa2, and the pheromone precursor-encoding gene mating α-factor precursor (MFα). Their results suggest that mating could occur much more frequently in nature than was originally appreciated and that same-sex mating could be an important mode of sexual reproduction in C. albicans.
C. albicans exhibits several survival mechanisms to evade attack by antifungals and colonize host tissues; therefore, their importance derives not only from the severity of their infections but also from their ability to develop resistance against antifungals, such as azoles, in patients undergoing long-term or prophylactic treatment (Saini et al. 2005). In C. albicans, upregulation of genes encoding the Tac1-regulated ABC drug transporters (CDR1 and CDR2) is a predominant cause for the development of antifungal resistance. With the increasing number of Candida infections in the recent years, multidrug resistance (MDR) has become a growing health concern. Three efflux pumps are responsible for decreasing the intracellular concentration of azoles in C. albicans: these pumps are encoded by genes for Candida drug resistance (CDR1 and CDR2) and multidrug resistance (MDR1). Notably, human steroid hormones are also shown to be substrates of yeast and mammal ABC multidrug transporters. The fact that C. albicans often come in direct contact with these hormones suggests that these could indeed be the physiological substrates of this transporter protein. Notably, their competition data revealed that progesterone was unable to compete with the steroid transport and extrapolated that progesterone may not be the substrate of Cdr1p. However, it is very likely that progesterone might interact with a different set of residues of Cdr1p and has different binding sites. Mdr1p, a MFS H+/drug antiporter that is also a multidrug transporter and relevant in clinical drug resistance of Candida, cannot export steroid hormones. It should be noted that although both are promiscuous multidrug transporters having many common substrates, yet they remain selective toward steroid transport.
Invading hyphae trigger several proinflammatory signaling pathways; during inflammation host- and microbial-derived proteases trigger the activation of protease-activated receptors (PARs), a family of G-protein-coupled receptors, that are encoded in the mammalian genome. The activation of PARs occurs through proteolytic cleavage of the extracellular N-terminal domain, which generates a new N-terminus that functions as a tethered ligand and binds to the receptor through an intramolecular interaction to trigger transmembrane signaling. Once cleaved, activated PARs undergo conformational changes within transmembrane helices that facilitate interaction with heterotrimeric G proteins. PARs display biased agonism or functional selectivity, which refers to the capacity of different ligands to stabilize unique active conformations of a GPCR that facilitates activation of distinct signaling responses. PAR1 is promiscuous and interacts with multiple distinct types of these G proteins, including Gαi, Gαq, and Gα12/13. PARs are expressed differentially in distinct cell types in a species-specific manner. Moretti et al. (2008) reported that activation of Toll-like receptors (TLRs) by fungi unmasks an essential and divergent role for PAR1 and PAR2 in downstream signaling and inflammation. TLRs activated PARs and triggered distinct signal transduction pathways involved in inflammation and immunity to C. albicans and Aspergillus fumigatus. Inflammation is promoted by PAR1 and PAR2 activation in response to Candida and by PAR2 inhibition in response to Aspergillus, which occurs by TLR regulation of PAR signaling, with TLR2 promoting PAR1 activity and TLR4 suppressing PAR2 activity. Thus, tissue injury and pathogens induce signals that are integrated at the level of distinct TLR/PAR-dependent pathways, the exploitation or subversion of which contributes to divergence in microbial promotion of the inflammatory response. The only current clinical agent that targets PARs is the PAR1 antagonist vorapaxar for the prevention of thrombotic cardiovascular events (Hamilton and Trejo 2017).
3.11 GPCRs Used in the Pharmaceutical Industry
Endogenous natural ligands Fig. 3.4 (a) are those that are produced in the body, not those introduced into the body, such as certain drugs.
Antagonist ligands include these:
-
Competitive antagonists Fig. 3.4 (b), which are drugs that bind to the same site as the natural ligand, agonists, or partial agonist, and inhibit their effects: these would be analogous to competitive inhibitors of an enzyme. One could also imagine a scenario in which an “allosteric” antagonist binds to an allosteric site on the receptor, inducing a conformational change in the receptor so the ligand, agonist, or partial agonist could not bind.
-
Noncompetitive antagonists (or perhaps more generally mixed antagonists) Fig. 3.4 (c) are drugs that bind to a different site on the receptor than the natural ligand, agonist, or partial agonist, and inhibit the biological function of the receptor. In analogy to noncompetitive and mixed enzyme inhibitors, the noncompetitive antagonist may change the apparent dissociation constant (Kd) for the ligand, agonist, or partial agonist (the ligand concentration required to achieve half-maximal biological effects), but will change the maximal response to the ligand (as mixed inhibitors change the apparent Vmax). The following figure shows the action of a competitive and noncompetitive antagonist.
-
Irreversible agonists, or ‘negative antagonists,’ arise from covalent modification of the receptor.
|
Fig. 3.4 (a) Natural ligand at the upper left hand side. (b) competitive antagonist at the upper right hand side. (c) noncompetitive antagonist at the bottom of the figure. (Source: By courtesy and permission of Jakubowski, Henry) |

As pockets or binding sites of the receptors are of great importance in the pharmaceutical point of view for designing the proper ligand, it is valuable to look at a relevant review. Gao and Skolnick (2012) reported a comprehensive study of the distribution of protein–ligand interaction sites, namely, ligand-binding pockets, around protein–protein interfaces where protein–protein interactions occur. They inspected a representative set of 1611 representative protein–protein complexes and identified pockets with a potential for binding small molecule ligands. The majority of these pockets are within a 6 Å distance from protein interfaces. Accordingly, in about half of ligand-bound protein–protein complexes, amino acids from both sides of a protein interface are involved in direct contact with at least one ligand. Statistically, ligands are closer to a protein–protein interface than a random surface patch of the same solvent-accessible surface area. Similar results were obtained in an analysis of the ligand distribution around domain–domain interfaces of 1416 nonredundant, two-domain protein structures. Furthermore, comparably sized pockets as observed in experimental structures are present in artificially generated protein complexes, suggesting that the prominent appearance of pockets around protein interfaces is mainly a structural consequence of protein packing and, thus, is an intrinsic geometric feature of protein structure. Humankind may take advantage of such a structural feature by selecting and further optimizing for biological function. The authors propose that packing nearby protein–protein or domain–domain interfaces is a major route to the formation of ligand-binding pockets. The author refers the reader to Landry and Gies (2007) for more details.
An important role in the study of ligand-binding sites (pockets) is the role of the N-terminus of the GPCR. This topic is elegantly covered by Coleman et al. (2017), who concluded that recent advances in the field of GPCRs show the humble N-terminus to be more than a ligand-binding site, a substrate for glycosylation, or an anchor for a signal peptide in the diverse examples summarized in their work. The protease-activated receptors are exemplars of this, revealing that GPCRs can harbor their own agonists within their N-termini, uncovered following a regulated proteolytic event. Similarly, the MC4 receptor N-terminus contains an agonist for its own receptor, allowing evolution of agouti-related peptide as an endogenous inverse agonist, through its ability to antagonize the N-terminus. In contrast, they reported the orphan receptor GPR37L1 to have constitutive activity maintained by its N-terminus and abolished by metalloprotease-mediated removal of the region. This differs again from the N-terminus of yeast pheromone α-factor receptor Ste2p, which reportedly serves to constrain the receptor response to its agonist. The receptors that have been discussed in their review represent emerging examples of novel modalities of GPCR signaling whereby the N-terminus is shown to be a crucial and dynamic contributor to signal transduction.
G-protein-coupled receptors (GPCRs) are the starting point for the control of several signaling pathways and are therefore considered a potentially rich source of innovation as drug targets and for drug design to alleviate many human diseases of genetic or biotic origin. The most appropriate GPCR candidate target for developing new fungicides needs searching for new compounds blocking this particular target and requires the knowledge of its 3D structure. Second, to identify potential new fungicide target(s), an important step is to verify that the identified target(s) are not present in host organisms and humans. Bresso et al. (2016) provided a detailed example of an integrated process with detailed steps merging genomics, structural bioinformatics, and drug design for Fusarium graminearum, leading to propose valuable and innovative solutions to a worldwide threat to wheat grain producers and consumers.
The trehalose biosynthetic pathway is found in a wide variety of organisms, including human-pathogenic fungi, but not in humans. Genes encoding proteins involved in trehalose biosynthesis are mechanistically linked to the metabolism, cell wall homeostasis, stress responses, and virulence of Candida albicans, Cryptococcus neoformans, and Aspergillus fumigatus. By further defining the biology and functions of trehalose and its biosynthetic pathway components in pathogenic fungi, an opportunity exists to leverage this pathway as a potent antifungal drug target (Thammahong et al. 2017). It will be necessary to identify antagonists or inverse agonists for these receptors, which will require good screening systems and large compound libraries (Van Dijck 2009). Considering the abundant expression of several nuclear receptors, it has also been suggested that one and the same nuclear receptor may have distinct endogenous ligands in distinct tissues or cell types.
Analysis of the human genome in 2002 led to the estimation of 6000–8000 targets of pharmacological interest. Only a small part of these targets relates to approved drugs. Lagerström and Schiöth (2008) overviewed a comprehensive overview of the five main human GPCR families–Rhodopsin, Secretin, Adhesion, Glutamate and Frizzled/Taste2–with a focus on gene repertoire, general ligand preference, common and unique structural features, and the potential for future drug discovery. A consensus number of 324 drug targets for all classes of approved therapeutic drugs was proposed by Overington et al. (2006). Of these, 266 are human genome-derived proteins, and 58 are bacterial, viral, fungal or other pathogenic organism targets. A large number of druggable targets have been recently proposed from preclinical and first clinical data, but a huge reservoir of putative drug targets, possibly several thousands, remains to be explored. The review of Landry and Gies (2008) considered the different types of ligands and their selectivity in the main superfamilies of drug targets, enzymes, membrane transporters, and ion channels, and the various classes of membrane and nuclear receptors with their signaling pathway. Recently approved drugs such as monoclonal antibodies, tyrosine kinase, and proteasome inhibitors, and major drugs under clinical studies, are reviewed with their molecular target and therapeutic interest. The druggability of emerging targets such as multidrug resistance transporters and cystic fibrosis transmembrane conductance regulator (CFTR), hyperpolarization-activated cyclic nucleotides-gated (HCN), cyclic nucleotide-gated (CNG) and transient receptor potential (TRP) ion channels, tumor necrosis factor (TNF) and receptor activator of NFjB (RANK) receptors, integrins, and orphan or recently deorphanized G-protein-coupled and nuclear receptors were discussed. Large advances have been made in the therapeutic use of recombinant cytokines and growth factors [i.e., tasonermin, TNFa-1a; becaplermin, platelet-derived growth factor (PDGF); dibotermin-alpha, bone morphogenetic proteins (BMP)2; anakinra, interleukin-1 receptor antagonist protein (IRAP); and in enzyme replacement therapy, i.e., algasidase (alpha-galactosidase) and laronidase (alpha-l-iduronidase)]. New receptor classes are emerging, such as membrane aminopeptidases, and novel concepts are stimulating drug research, such as epigenetic therapy, but the molecular target of some approved drugs, such as paracetamol and imidazolines, must be identified (Landry and Gies 2008). G-protein-coupled receptors are the largest class of receptors mediating the effects of small neurotransmitters, all known neuropeptides, many peptide hormones and inflammatory mediators, some lipids, and even calcium for the control of its blood concentration. Almost 30% of all marketed drugs act on GPCRs. The most familiar GPCRs as historical drug targets are the muscarinic acetylcholine receptors, the alpha- and beta-adrenergic, dopaminergic, histaminergic, and opioid receptors. Some other GPCR ligands have been developed as drugs during the past three decades: serotonin 5-HT, prostaglandins, leucotrienes, ADP, or calcium receptor ligands. The actual top-selling GPCRs ligands are clopidogrel (ADP-P2Y12 antagonist, platelet antiaggregant), olanzapin (mixed serotonin-5HT2/dopamine-D2 antagonist, neuroleptic), valsartan (angiotensin-AT1 antagonist, antihypertensive), fexofenadine (histamine-H1 antagonist, antiallergic), sumatriptan (serotonin-5HT1D antagonist, anti-migrainous), and leuprorelin (GnRH/LH-RH peptidic agonist, antihormone-dependent cancer). Cellular differentiation, mating, and filamentous growth are regulated in many fungi by environmental and nutritional signaling cascades. The mitogen-activated protein kinase (MAPK) pathway is a network of signaling pathways that allow adaptation to environmental changes and is evolutionarily conserved from yeasts to mammals. The divergent point between fungal and mammalian MAPK pathways is their upstream signaling module. Based on this, several MAPK signaling pathways in fungi have been identified that may have therapeutic implications: the Mkc1 pathway, the Cek1 pathway, the Cek2 pathway, and the high-osmolarity glycerol (HOG) pathway. In C. albicans, these pathways are involved in invasive hyphal growth, morphogenesis, biogenesis of the cell wall, dimorphism, and the stress response, all of which likely have an important role in virulence and serve as potential targets for new drugs. Among these potential targets, the HOG pathway appears to be the most promising as it is the main signal transduction system that is responsible for cellular stress responses (Rutherford et al. 2019). The Ssk2-Pbs2-Hog1 MAPK module is mainly regulated by the multicomponent phospho-relay system, which consists of sensor hybrid histidine kinases (HHKs), histidine-containing phospho-transfer (HPt) protein, and response regulator RRs in C. albicans. Its transduction network is composed of a two-component-system-like phospho-relay system and the Hog1-type MAPK cascade. Overactivation of the HOG pathway can be devastating to an organism, as witnessed by the action of fludioxonil, which overactivates the HOG pathway and confers lethal effects. This observation suggests that drugs targeting the negative regulators of the HOG pathway could be more efficient at killing a pathogen than those targeting its positive regulators. Other compounds targeting the HOG system have been identified including the ambruticins, a family of cyclopropyl-pyran acids with broad antifungal activity. Ambruticin S, an antifungal cyclopropyl-pyran acid, has long been known to have curative effects against murine pulmonary coccidioidal infection. Recently, two analogs of ambruticin S with greater in vitro potency, KOSN-2079 and KOSN-2089, were tested against lethal murine Coccidioides infection; both improved the survival of mice over that of controls, suggesting that larger studies in animals may be warranted (Landry and Gies 2008). In their review article, McCarthy et al. (2017) reported that recent advances in our understanding of the fungal life cycle, functional genomics, proteomics, and gene mapping have enabled the identification of multiple potential new drug targets that could bolster the arsenal of available options to treat resistant invasive fungal infections (IFI). In their review, they examined those targets mechanistically and described how promising new therapies might be developed, with special attention paid to molecules that promote growth inhibition. They explored fungal architecture, the anchoring of proteins to the plasma membrane through covalent attachment to glycosylphosphatidylinositol (GPI), which provides an appealing potential target for the development of antifungal agents. E1210 is a novel isoxazolyl bis-pyridine wall-active antifungal compound that inhibits an early step in the GPI-dependent anchoring of mannosylated cell wall proteins attached to the polysaccharide wall component. Its target is Gwp1p, the fourth enzyme in the GPI-anchoring pathway, responsible for inositol acylation (the mammalian homolog is not inhibited). A second GPI inhibitor has recently been discovered. Gepinacin, a monocarboxylic amide, selectively inhibits Gwt1, a critical acyltransferase required for the biosynthesis of fungal GPI anchors. In contrast to all three major classes of antifungal agents in current use, its direct antifungal activity results from its ability to induce overwhelming stress to the endoplasmic reticulum (ER) and does not affect the viability of mammalian cells, nor does it inhibit their orthologous acyltransferase. It was noted to increase the immunogenicity of Candida albicans, further enhancing its antifungal activity by disrupting the mannoprotein outer layer of the cell wall and unmasking the more immunogenic inner β-glucan layer. Glycosphingolipids (GSLs) are a type of glycolipid found in cell membranes that have emerged as key regulators of pathogenicity in a variety of fungi. When yeast and mold lack the GSL called glucosylceramide (GlcCer), they are unable to replicate. Two newly identified compounds have been found to decrease levels of fungal, but not mammalian, GlcCer. Metabolic pathways are essential for fungal viability and virulence. The glyoxylate cycle, a modified tricarboxylic acid cycle, bypasses the carbon dioxide-generating steps to conserve carbons as substrates for gluconeogenesis, which enables fungi to survive in nutrient-limited host niches. The cycle consists of five enzymes, including isocitrate lyase and malate synthase, which are unique to this cycle, and three others that are essential for the virulence of both yeasts (including C. albicans) and molds (including Aspergillus fumigatus), making it a promising target for the development of novel antifungal agents. The pyrimidine salvage pathway offers another potential target for antifungal therapies. Discovered as the result of an Aspergillus nidulans library screen, it targets the enzyme dihydroorotate dehydrogenase, which catalyzes the fourth enzymatic step of pyrimidine biosynthesis. Cytochrome P450 enzymes (CYPs) are a superfamily of proteins that catalyze a wide range of metabolic reactions and present an attractive site for drug development. The commonly used triazole-class drugs work by selectively inhibiting fungal cytochrome P450 14α-demethylase. The MAPK pathway is a network of signaling pathways is their upstream signaling module. Several MAPK signaling pathways in fungi that have been identified may have therapeutic implications: the Mkc1 pathway, the Cek1 pathway, the Cek2 pathway, and the high-osmolarity glycerol (HOG) pathway. In C. albicans these pathways are involved in invasive hyphal growth, morphogenesis, biogenesis of the cell wall, dimorphism, and the stress response, all of which likely have an important role in virulence and serve as potential targets for new drugs. Others have sought to develop novel antifungal agents by interrupting the modification of nucleic acids. Epigenetic therapy is being widely developed for the treatment of human neoplastic disease and has recently found a role in the treatment of invasive candidiasis, using histone deacetylases (HDACs) as a target, which are involved in a wide range of cellular functions, ranging from cell proliferation to cell death, by regulating chromatin structure and transcription through lysine deacetylation of histones. The report ended by anticipating future directions: IFIs continue to be an important cause of morbidity and mortality, particularly in immunocompromised patients. The emergence of antifungal resistance is a growing global health problem, underscoring the need for new therapeutic options. Significant progress has been made in the identification of potential targets for novel agents that exploit inherent differences between fungi and humans. Although many of these targets remain theoretical, several molecules have recently entered early clinical development. Complementing these therapies are advances in innate host defense: a pioneering clinical study using adoptive transfer of Aspergillus-stimulated T cells showed significant efficacy in treating stem cell transplant recipients with invasive aspergillosis. Other investigators have generated T-cytotoxic cells expressing Dectin-1 chimeric antigen receptors (CARs) that recognize surface fungal glucans. New treatment options for IFIs are urgently needed, but it remains a challenge to bring novel agents to the market. The discovery of a new therapy is difficult, chemical genetic-based screens are labor intensive, traditional regulatory requirements are stringent, and the financial reward may be limited if the IFI is uncommon. In the face of these hurdles, patients continue to suffer. We must address this unmet need by bolstering support for research, considering alternative approval pathways, repurposing existing non-antifungal drugs, strengthening ties between academia and industry, and providing financial incentives for the development of new therapeutic options to treat these uncommon yet devastating infections. Beyond small molecules, new approaches for augmenting host responses are critical and include the development of vaccines, immunomodulators, and novel approaches for reducing immunosuppression and exploiting the immunopharmacology of existing antifungal agents.
There are two main families of membrane proteins involved in drug transport, the major facilitator superfamily (MFS) and the ATP-binding cassette (ABC) proteins. Both types of protein possess multiple membrane spanning α-helices in transmembrane domains (TMDs), and ABC proteins, in addition, contain cytosolic nucleotide-binding domains (NBDs) involved in ATP hydrolysis. ABC proteins, and to a lesser extent MFS proteins, have broad substrate specificities that are determined by the structure and arrangement of the transmembrane α-helices (Lamping et al. 2017b). In an effort to understand the molecular basis of drug recognition, binding, and release, several studies are being pursued, giving an insight into its drug-binding pocket (Baghel et al. 2017). One of the most clinically significant mechanisms of azole resistance in the pathogenic yeast Candida albicans is overexpression of the multidrug transporter protein Cdr1p (Candida drug resistance), belonging to the ABC superfamily of transporters (MDR), which is a phenotype where cells in parallel develop resistance to multiple chemically unrelated chemotherapeutic agents. Drug efflux pumps belonging to either the ATP-binding cassette (ABC) family or major facilitator superfamily (MFS) transporters have a major role in the development and sustenance of drug tolerance. The induced overexpression of genes encoding these transporter proteins does not allow cells to retain toxic concentrations of the drug, because of their rapid extrusion, thus making it ineffective. Lamping et al. (2010) reported that the overexpression of pleiotropic drug resistance (PDR) efflux pumps of the ATP-binding cassette transporter frequently correlates with multidrug resistance. Fungal, plant, and human ABCG-family Pdrps possess a nucleotide-binding domain (NBD) and a transmembrane domain (TMD) in a family-defining ‘reverse’ ABC transporter topology (NBD–TMD) that is duplicated (NBD–TMD2) in full-size fungal and plant Pdrps. The Human Genome Organization (HUGO) classified ABC superfamily members into eight subfamilies in which Cdr1p, as the pleiotropic drug resistance (PDR) family in Saccharomyces cerevisiae, belongs to the ABCG subfamily. The subfamily contains a N- and a C-terminal nucleotide-binding domain (NBD), each followed by a trans-membrane domain (TMD) (N- and C-TMDs). Each TMD is composed of six TMHs; although NBDs are highly conserved and participate in ATP catalysis to power drug extrusion, TMDs are more variable in structure and are involved in the formation of overlapping multiple substrate-binding sites. These TMHs are linked to each other via six extracellular loops (ECLs) and four intracellular loops (ICLs). The recent publication of the heterodimer ABCG5/ABCG8 X-ray structure allowed establishing that proteins belonging to the G-subfamily constitute a new type of transporters, called type II, compared to B- and C-subfamilies transporters that belong to the type I. Type II deeply differ from type I by the absence of domain swapping in the TMD between the N- and C-moieties, together with the absence of a large extension of the membrane helices toward the cytoplasm to connect each NBD. These features let us suppose a distinct transport mechanism. Notably, Rawal et al. (2013) in a recent study probed the nature of the drug-binding pocket of Cdr1p by performing a full alanine scanning mutagenesis of its membrane region, wherein each of the residues of 12 TMHs was replaced with an alanine (or a glycine when being an alanine). Their study revealed multiple overlapping mini drug-binding sites within a large centrally located polyspecific drug-binding cavity. Baghel et al. (2017) examined the kinetics of steroids efflux mediated by the Candida drug resistance protein 1 (Cdr1p) and evaluated their interaction with the protein. They exploited their in-house mutant library for targeting the 252 residues forming the 12 transmembrane helices (TMHs) of Cdr1p. The selective structural arrangement of the steroid-binding pockets in the core region and at the lipid–TMD interface, which was never reported previously, together with the possible rotation of some TMHs may be the structural basis of the drug-transport mechanism achieved by these type II ABC transporters. Baghel et al. (2017) showed that the central binding pocket in which β-estradiol and R6G competitively bind is made of 14 residues; among them, 10 are identified as critical for the former and 8 for the latter. It mainly engages the N-half of Cdr1p with 7 residues brought by TMHs 2 and 5 while only 3 are recruited on the C-side, brought by TMHs 7 and 8. In this binding pocket F1233 from TMH 8, although highly critical, remains rather distant whereas, in contrast, residues I1237 and N1240, although never found critical, are part of the binding pocket. All this information suggests that the position of TMH 8 in the current model/conformation may probably slightly differ, with F1233 closer to the substrate and I1237 and N1240 further. By contrast, corticosterone engages only 3 critical residues in that pocket, which strongly suggests that it does not interact with Cdr1p as β-estradiol or R6G in that region, and consequently may be transported by the protein with a distinct mechanism. Cdr1p has not only acquired significant clinical importance but is considered an important player in any design of strategies to combat antifungal resistance. The CDR1 gene encodes an integral plasma membrane (PM) protein. On the basis of its amino acid sequence, Cdr1p is predicted to consist of two homologous halves, each containing one N-terminal hydrophilic domain followed by a C-terminal hydrophobic domain (Shukla et al. 2003). Their data showed for the first time that the drug substrate-binding sites of Cdr1p exhibit striking similarities with those of mammalian drug-transporting P-glycoproteins and, despite differences in topological organization, the transmembrane segment 6 in Cdr1p is also a major contributor to drug substrate-binding site(s). It is clear that much remains to be learned of the signals, fungal signaling pathways, and transcriptional regulatory networks that control morphogenesis in C. albicans (Noble et al. 2017). ATP-binding cassette drug transporters are not only considered determinants of multidrug resistance but also have crucial functions in controlling lipid levels. Adenosine triphosphate-binding cassette (ABC) transporters constitute a group of highly conserved cellular transmembrane transporter proteins. ABCA1, ABCA7, and ABCA4 are members of the ABCA subfamily and share extensive sequence and structural similarity. Several studies have shown that ABCA proteins are involved in lipid transport. ABCA7 is thought to have an important role in lipid homeostasis in cells of the immune system. It is a full-size single-subunit ABC transporter consisting of 12 trans-membrane-spanning domains (Nowyhed et al. 2017). Within cells, it is expressed predominantly on the plasma membrane, but it is also detected in intracellular membranes. More recently, ABCA7 was found to be significantly associated with phagocytosis in macrophages both in vivo and in vitro (Jehle et al. 2006; Linsel-Nitschke et al. 2005). Nowyhed et al. (2017) data show that ABCA7 is required for efficient phagocytosis of apoptotic cells. In C. albicans, which causes infections associated with venous catheters, urinary catheters, and several other implanted devices, upregulation by genes encoding the Tac1-regulated ABC drug transporters (CDR1 and CDR2) is a predominant cause for the development of antifungal resistance (Coste et al. 2004; Prasad et al. 2015). As a consequence, drug-resistant isolates display enhanced efflux of azole antifungals, leading to reduced inhibition of their target enzyme, lanosterol 14α-demethylase of the ergosterol biosynthesis pathway. The presence of gain-of-function mutations in Tac1 renders it hyperactive, resulting in the simultaneous induction of CDR1 and CDR2 along with RTA3, IFU5, and HSP12 in azole-resistant isolates (Coste et al. 2004; Liu et al. 2007). The significance of coordinate regulation has been inferred from studies with S. cerevisiae, wherein Pdr5, the major ABC drug efflux pump, and genes involved in sphingolipid biosynthesis are induced coordinately via the transcription factors Pdr1/Pdr3 (Devaux et al. 2002). These studies argue for the role of the pleiotropic drug resistance (Pdr) pathway in coordinately controlling lipid levels on the plasma membrane and multidrug resistance in the budding yeast (Johnson et al. 2010; Khakhina et al. 2015). Studies on the noteworthiness of Tac1-coregulated genes have not been established. Thus, among the Tac1-coregulated genes, RTA3 (named for resistance to 7-aminocholesterol) caught the attention of Srivastava et al. (2017) in particular as it was annotated as a putative lipid translocase in the Candida Genome Database (CGD). Lipid translocators can direct inward or outward transbilayer movement of phospholipids and are referred to as flippases and floppases, respectively. The balanced action of these proteins is crucial for generating plasma membrane asymmetry, such that the aminophospholipids are sequestered in the cytoplasmic leaflet of the membrane, whereas choline lipids are enriched in the outer leaflet (Devaux et al. 2008). Any perturbation in membrane asymmetry serves as a signal for activating multiple cellular events. Other than the identification of Cdr1 and Cdr2 as floppases, molecular entities that can function as flippases or signals which regulate the activity of these lipid translocators to generate membrane asymmetry in C. albicans remain unidentified. The problem of drug resistance is not only limited to free-living planktonic forms of Candida but also extends to surface attached communities such as biofilms (Nobile and Johnson 2015). Microorganisms naturally exist primarily in association with surfaces in communities called biofilms. Central to the biofilm formation is the ability of microbial cells to adhere to substrates. Adherence of biofilms is clinically significant as the basis for infections associated with implanted medical devices. Adherence mechanisms are diverse, and involve specific cell-surface proteins (adhesins), more complex surface structures such as pili, and secreted extracellular matrix material. Adherence is often found to be highly regulated, reflecting the need for biofilms to release cells to colonize new sites. Biofilms are clinically significant as the basis for infections associated with implanted medical devices. For device-associated biofilms the definition of the mechanisms that regulate cell–substrate adherence provides insight into how these biofilms form (Finkel et al. 2012). Besides the Bcr1 that functions in yeast form cells and is among the best characterized biofilm regulators, its adhesin targets Als1/3 and Hwp1 mediate cell–cell interaction in biofilms as well as genes induced upon hyphal development. The other best characterized biofilm regulator Ace2 is known to affect adherence, biofilm formation, and hyphal morphogenesis. Much of the phenotypic impact of Snf5, a subunit of the eukaryotic SWI/SNF chromatin remodeling complex, stems from its role in ACE2 expression. Their findings also have significant implications on a more global scale: they define a regulatory network through which 12 transcription factors govern expression of more than one quarter of the C. albicans cell-surface protein genes. Zinc response regulator Zap1 is among the 12 transcription factors that also govern biofilm matrix accumulation and quorum sensing molecule production. Zap1 is thus positioned to coordinate multiple steps in biofilm formation. Finally, many adherence regulators do not have clear functional targets, based on their analysis. However, their unifying target gene classes will help to direct future studies. In addition to the identification of transcription factors, the role of Ras- and Tor1-mediated signaling pathways in regulating cell–cell adhesion during biofilm formation via Bcr1, Efg1, Nrg1, Tec1, and Tup1 has also been established (Inglis and Sherlock 2013). In aggregate, these studies point toward the complexity of the gene regulatory programs that control biofilm formation in the fungal pathogen C. albicans. The predicted topology of Rta3 revealed the presence of 7-transmembrane domains (7TMDs) similar to the family of 7-TM receptor proteins often associated with the G-protein-coupled receptors (GPCRs). The homologs of RTA3 are referred to as the Rta1-family of proteins or the lipid translocating exporters in S. cerevisiae (Manente and Ghislain 2009). In addition to RTA3, C. albicans has three additional genes, orf19.6224, RTA2, and RTA4, coding for the Rta1-family of proteins. Noteworthy is that these proteins lack an overall sequence conservation with the classical GPCRs. The role of Rta2 in modulating azole tolerance and in endoplasmic reticulum (ER) stress resistance has been established, and the others remain uncharacterized (Thomas et al. 2015). These proteins are unique to the fungal kingdom and can be considered as potential therapeutic targets. In agreement with its 7TMD topology, Srivastava et al. (2017) showed that C. albicans Rta3 is localized at the plasma membrane similar to Rsb1 of S. cerevisiae and thus is most likely to function as a regulatory protein rather than a transporter or translocator. Moreover, Rta3 and proteins of this family represent targets for new antifungals owing to their exclusivity in the fungal kingdom. RTA3 is a downstream target of the transcription factor Tac1 and is coregulated with CDR1 and CDR2, two well-documented drug efflux pumps in C. albicans. Except for a single study where Rta3 was implicated to affect susceptibility to fluconazole in an azole-resistant clinical isolate (Whaley et al. 2016), its functional relevance remains unexplored in C. albicans. Srivastava et al. (2017) findings indicate that absence of Rta3 affects a spectrum of biological processes highlighting the role of this 7-TM receptor protein in multiple regulatory pathways. The loss of Rta3 not only affects the tolerance to miltefosine, an alkylphosphocholine analog, but also alters mitochondrial membrane energetics. Miltefosine-induced apoptosis-like cell death in the budding yeast is caused by its direct interaction with COX9, a subunit of the complex IV of the electron transport chain in S. cerevisiae. This action results in the disassembly of complex IV, causing disruption of the electron transport chain, which in turn affects the MMP. Consistent with this, they proposed that altered mitochondrial parameters in rta3Δ/Δ cells might sensitize it to a lower dose of miltefosine than is required for the wild-type cells. Furthermore, a reduced flop or export of miltefosine in rta3Δ/Δ as a cause for the increased susceptibility to miltefosine also cannot be ruled out. Additionally, Tac1-regulated RTA3 is essential for curbing miltefosine-induced stress as transcript levels of RTA3 are elevated in wild-type cells treated with miltefosine. Recent studies demonstrate the ability of miltefosine to kill azole-resistant clinical isolates and inhibit biofilm formation by C. albicans (Vila et al. 2016, 2017). In this context, identification of Rta3 as one of the determinants of miltefosine tolerance may lead to the identification of its target in C. albicans. Srivastava et al. (2017) demonstrated that the loss of Rta3 perturbs the dynamic equilibrium of PC by increasing its inwardly directed (flip) movement across the plasma membrane; this results in the enrichment of PC on the inner leaflet of the plasma membrane and consequently its transfer to intracellular membranes by passive diffusion. They thus proposed that the increased flip of PC may be attributed to the role of Rta3 as a negative regulator of PC-specific flippase activity. In conclusion, given the 7TMD topology of Rta3, their study shows that functional Rta3, instead of functioning as a lipid translocator itself, may have a role in maintaining the asymmetrical distribution of PC across the plasma membrane, possibly by modulating regulatory pathway(s) that signal unidentified PC-specific flippase(s) or floppase(s) in C. albicans. The proposal that Rta3 is a positive regulator of biofilm formation and functions upstream to Bcr1 stems from the following observations. First, the catheter inoculated with the rta3Δ/Δ cells was essentially devoid of any material, similar to bcr1Δ/Δ, suggesting that there was a defect in early events of biofilm formation in vivo. Second, the overexpression of BCR1 in rta3Δ/Δ cells partially rescues the biofilm defect of the mutant. Third, the biofilm defect phenotype of the bcr1Δ/Δ cells was not rescued by the increased expression of RTA3. Their results therefore argue that Rta3 is pivotal for biofilm formation and that Bcr1 is one of the key downstream effector molecules of the Rta3-dependent regulatory pathway that contributes to biofilm formation. The biofilm formation in vivo involves dynamic interactions with host factors. Their transcript analyses are indicative of a positive transcriptional feedback loop between Rta3 and downstream Bcr1 transcription factor. These signaling pathways are modulated by feedback loops where the downstream components of the pathway regulate upstream elements of the same pathway. Positive feedback regulation amplifies the signal, whereas negative feedback regulation helps in attenuation of the response. It is possible that in unique niches of the mammalian host, expression of Rta3 and BCR1 may be interdependent. The mutual regulation of Bcr1 and Rta3 may regulate the magnitude and duration of the signaling event entailing response to host specific cues and thus biofilm formation. Their study also showed a mitochondrial membrane energetics-dependent expression of BCR1. Based on these data, it is evident that Rta3 governs biofilm formation largely by regulating Bcr1. Tac1-regulated drug transporters (Cdr1 and Cdr2) are responsible for the asymmetrical distribution of phosphoglycerides across the plasma membrane, in addition to contributing to the development of azole resistance in C. albicans. Considering the role of Rta3 in maintaining the asymmetrical distribution of PC across the plasma membrane, the physiological role of Tac1-coregulated genes under unstressed conditions may be to maintain phospholipid asymmetry in the plasma membrane. Furthermore, the function of RTA3 in conferring tolerance to another broad-spectrum antifungal, miltefosine, highlights the physiological relevance of Tac1-regulated genes in defending Candida cells against xenobiotics present in the environment. The fact that Rta3 is unique to the fungal kingdom opens the possibility of interfering with the ability of C. albicans to thrive as a biofilm on indwelling devices in the human host. They also propose that the principal purpose of coregulation of Rta3 with drug transporters in C. albicans may be to enable this pathogen to efficiently switch to the highly drug-resistant biofilm mode of growth in the human host, upon perceiving a host-specific stimulus. As a consequence, this pathogenic fungus is empowered to resist the onslaught of antifungals in both planktonic as well as the biofilm modes of growth in the host environment. In view of the fact that biofilm-specific drugs do not exist for C. albicans, identification of Rta3 as a novel component that primarily regulates Bcr1-dependent adhesion may lead to new strategies to prevent biofilm formation in this human fungal pathogen. C. albicans cells with CDR1 deletion showed a significant increase in sensitivity to styrylquinolines in contrast to 8-hydroxyquinoline and 8-hydroxyquinaldine, suggesting that styrylquinolines are Cdr1p substrates. Styrylquinolines consist of two aromatic parts connected by an unsaturated ethylene linker that results in a flat rigid structure with relatively high lipophilicity. Such molecules are generally expected to penetrate cell membranes effectively; moreover, they may interact with lipid structures such as ergosterol, indicating a possible mode of action as competitive inhibitors of this transporter (Szczepaniak et al. 2017). As Cdr1p has a higher impact on fluconazole resistance than Cdr2p, the strain without Cdr1p transporters is characterized by a lower fluconazole minimum inhibitory concentration (MIC) than strains that express Cdr1p transporters. However, the simultaneous presence of fluconazole and styrylquinolines abolishes the Cdr1p-induced resistance. It was confirmed that the cell membrane of C. albicans with treated with styrylquinolines was not disrupted. The treatment of C. albicans cells with ABC transporter substrates increases their expression. The Cdr1p-GFP protein level in crude extract was also higher in treated cells. Very similar results to wild-type strain (MICs) were obtained in cytotoxicity testing using normal human fibroblast cells, which suggests excluding the use of higher concentrations of styrylquinolines as antifungal drugs. The structures of styrylquinolines and their lipophilic properties predispose them to accumulate in cellular membranes. One of the mechanisms of action could be reducing the activity of Cdr1p and its relocation by disrupting the construction of the membrane. These findings are promising for designing novel antifungal agents. The identification of the dopamine G-protein-coupled receptor (GPCR) antagonists clozapine and fluspirilene as budded to hyphal transition (BHT) inhibitors was very interesting, especially given that clozapine and a number of its bioactive derivatives are FDA approved for the treatment of atypical schizophrenia (Toenjes et al. 2009). There are only three annotated GPCRs in C. albicans, with two being the STE2 and STE3 pheromone receptors. The remaining GPCR is Gpr1p, which has been implicated in a nutrient-regulated BHT signaling pathway upstream of PKA (Maidan et al. 2005a; Miwa et al. 2004). It remains to be determined whether clozapine and fluspirilene function through the Gpr1p GPCR. It is worth noting that the side effects of clozapine increase the disability of patients with schizophrenia and should be monitored regularly (Gürcan et al. 2017). Clozapine and olanzapine were consistently more strongly associated with metabolic adverse events than were other second-generation antipsychotic (SGAs) currently available (Hirsch et al. 2017). Vasudev et al. (2017) showed that about two thirds of the patients on clozapine were overweight or obese and had abnormal triglycerides and HDL cholesterol, with about half the patients having metabolic syndrome. The evidence-based report of the Canadian Agency for Drugs and Technologies in Health (CADTH) clearly indicated that high-dose or combination antipsychotic strategies are not known to be more effective and may be more harmful. Their study confirms that concurrent use of medications known to increase body mass index (BMI) significantly contributed to the increased BMI in patients on clozapine (Hirsch et al. 2017). It is now clear that Gpr1 is the founding member of a novel seventh class of GPCRs. Functional homologues seem to be present in most fungi with the C. neoformans Gpr4 receptor as a clear example (Van Dijck 2009). This receptor functions similarly to the C. albicans Gpr1 receptor, despite the absence of sequence homology. As this class of GPCRs is fungal specific and GPCRs form the largest groups of therapeutic targets, two important criteria for a good drug target are fulfilled (specificity and possibility to screen for drugs). In humans, histatins are produced by the submandibular and parotid glands and secreted in saliva. They form part of the innate immune system and have an important role in maintaining oral health. Histatins are small, cationic, histidine-rich peptides of 3–4 kDa with potent antimicrobial activity. The most important members of the histatin family are histatin3 and histatin5 (32 and 24 amino acids, respectively). Histatin5 has potent activity against the pathogenic fungi Candida albicans, Cryptococcus neoformans, and Aspergillus fumigatus. Recent data seem to indicate that it would initiate an osmotic stress response by activation of the Hog1 pathway. Van Dijck (2009) showed that cell defense to histatin 5 is dependent on Gpr1: a homozygous gpr1 mutant is very sensitive to histatin 5. At higher concentrations of histatin 5, this strain no longer grows, whereas a heterozygous or wild-type strain still grows, which would indicate that a combination of histatin 5 and an antagonist of Gpr1 would be an interesting antifungal cocktail. Once the ligand is known, it may be important to identify the exact binding sites. Another Candida showing drug efflux pumps and multidrug efflux pump, C. krusei is an innately azole-resistant diploid fungal pathogen with a tendency to create aneuploid strains or triploid hybrids when exposed to environmental stress (see Knöppel et al. 2017). The experimental evidence indicates that, although all three genes are under very strong purifying selection, ABC1, ABC11, and ABC12 are not dosage-sensitive genes, and one gene copy appears to be enough for the survival of C. krusei (Lamping et al. 2017a). In C. krusei, ABC1 and ABC11 are encoding for drug efflux pumps, and a third close homolog, multidrug efflux pump ABC12 was discovered (Lamping et al. 2017a). ABC11 and ABC1 orthologs are found in the closest sequenced relative, Pichia membranifaciens in the three C. krusei family members. Although C. krusei, also known as Issatchenkia orientalis or Pichia kudriavzevii, is a relatively poorly studied ascomycetous yeast, it is important for natural food fermentation. Overexpression of Abc1p in S. cerevisiae AD∆ caused multidrug resistance, is most frequently caused by active transporters, such as P-glycoprotein (ABCB-1), that pump a broad spectrum of chemically distinct molecules out of cells, which could be reversed with known efflux pump inhibitors. Strains overexpressing Abc1p or Abc11p were found to be resistant to a number of xenobiotics: resistant to azole antifungals (FLC, CLT, ITC, MCZ, and VRZ), fluorescent substrates (R6G and R123), large ionophores (NIG), translation inhibitors (CHX and ANI), and anticancer drugs (DOX and DAU). The discovery of a third C. krusei multidrug efflux transporter, ABC12, which had the same substrate specificities as Abc1p and Abc11p [the seven Abc1/11p: milbemycins α11; α26; β9; and β11; FK506 (FK); ENI; beauvericin (B) or OLI] were also Abc12p substrates. Although all three multidrug efflux pumps transported the same range of substrates, their transport activities for individual compounds differed significantly. Abc1p appeared to pump the selected test compounds best overall. However, Abc12p was the best transporter of the smaller test compounds [i.e., low molecular weight (MW); ANI, CHX, FLC, and CLT], and Abc11p was the best transporter of the larger (i.e., high MW) test compounds (R6G and NIG). Perhaps surprisingly, Abc11p of 89,102 (pump strain A), a chimera with two Abc1p-specific amino acids in TMS6 (patch III) and one in TMS11 (patch V), was an even better multidrug efflux pump (i.e., it was more resistant to most test compounds) than Abc1p. Additional variations in substrate specificities were observed for the remaining ABC11-1 and ABC1-11 chimeras (pump strains B–E and F–G, respectively). Although all chimeras were clearly functional, some effluxed certain compounds even more efficiently than wild-type Abc11p or Abc1p (e.g., FLC efflux by Abc1-11p chimeras; pump strains F and G), whereas some Abc11-1p chimeras showed severely reduced drug efflux, especially of ANI, KTC, or NIG (pump strains B, C, and E). Differences were also observed in relative sensitivities conferred by Abc1p and Abc11p to known efflux pump inhibitors. Milbemycins are acaricides, insecticides, and anthelmintics widely used in agriculture and veterinary medicine. FK506 is a commonly used immunosuppressor, ENI and beauvericin are ionophoric depsipeptides with antibiotic and insecticidal activities, and OLI is an ATPase inhibitor. Abc1p was more sensitive than Abc11p to all eight efflux pump inhibitors tested, especially milbemycin β11, and, as expected, Abc11p of 89102 conferred inhibitor sensitivities that were between those for Abc1p and Abc11p of B2399 because it contains elements of Abc1p. The investigators have previously shown that the constitutive expression of Abc1p may tip the balance in favor of innate azole resistance for C. krusei (Lamping et al. 2009). In their report, Lamping et al. (2017a) showed that there may yet be other factors contributing to the innate azole resistance of C. krusei, namely, the expression of additional multidrug efflux transporters Abc11p and Abc12p. Lamping et al. (2009) described the isolation and characterization of Abc1p, a constitutively expressed multidrug efflux pump, and investigated ERG11 and ABC1 expression in C. krusei. The examination of the ERG11 promoter revealed a conserved azole responsive element that has been shown to be necessary for the transcription factor Upc2p mediated upregulation by azoles in related yeast functional overexpression of ERG11 and ABC1 in S. cerevisiae and conferred high levels of resistance to azoles and a range of unrelated Abc1p pump substrates, whereas small molecule inhibitors of Abc1p chemosensitized C. krusei to azole antifungals. It is worth noting to have a look at Wingard et al. (1991) in the context of drug resistance and prophylaxis precautions. They have previously noted an increasing prevalence of C. krusei colonization in their renowned center, although it remained only a sporadic systemic pathogen. The data in their report indicated the emergence of C. krusei during 1990 as the chief systemic pathogen in patients with marrow transplants, but not in patients with leukemia. Although they cannot exclude the possibility that there was a common source of introduction of these organisms, they believed that such an explanation is unlikely. The same kitchen did provide food to both the bone marrow transplantation and leukemia services; however, these wards had different rates of colonization and infection. The transplant recipients were cared for by three different teams of nurses and physicians. In contrast, the increased prevalence of C. krusei was associated with the use of prophylactic fluconazole. Recent case reports describe a similar inability of fluconazole to suppress C. krusei infection. Although susceptibility testing is in the early stages of development, the lack of in vitro activity of fluconazole against C. krusei supports these clinical findings. This lack of susceptibility was probably not the only reason for the emergence of C. krusei. This species has been noted to be at a disadvantage with respect to adherence, an important determinant of colonization. Unfortunately, the factors that lead to colonization, tissue invasion, and systemic infection were poorly understood. It was concluded that although fluconazole prophylaxis was associated with a significant decline in the number of disseminated infections caused by C. albicans and C. tropicalis, and the emergence of a less virulent organism C. krusei as a systemic pathogen, they proposed that the suppression of more virulent Candida species can contribute to the emergence of other, less virulent Candida species that are not susceptible to fluconazole, such as C. krusei, as systemic pathogens. Although drug resistance is one of the human major concerns, unexpectedly the findings of Knöppel et al. (2017) showed that antibiotic resistance can evolve in bacteria in response to a novel selection pressure without any antibiotic exposure. The molecular mechanisms that cause drug resistance are naturally occurring in less susceptible species and are acquired in strains of susceptible organisms (Perlin et al. 2017). Another unexplored possibility is that resistance evolves coincidentally in response to other selective pressures. They showed that selection in the absence of antibiotics can coselect for decreased susceptibility to several antibiotics. Thus, genetic adaptation of bacteria to natural environments may drive resistance by generating a pool of resistance mutations on which selection could act to enrich resistant mutants when antibiotic exposure occurs. Prigitano et al. (2015) arrived at a similar conclusion.
Esquivel and White (2017) compared the filamentous fungus Magnaporthe oryzae to yeast species such as Saccharomyces cerevisiae, Candida albicans, Cryptococcus neoformans, Candida glabrata, and Candida krusei, and found it to contain an unusually high number of genes encoding predicted membrane transporters, including at least 50 ATP-binding cassettes (ABC) and at least 250 major facilitator superfamily (MFS) transporters (Kim et al. 2013). The majority of these transporter genes are still uncharacterized to date. Most filamentous fungal species uncharacterized to date and molds are intrinsically resistant to fluconazole (FLC) because they possess multiple targets of azoles: one is CYP51, an enzyme localized on the endoplasmic reticulum (ER), and required for fungal ergosterol biosynthesis. Most filamentous fungal species and molds are intrinsically resistant to FLC because they possess multiple target CYP51 paralogs: two in M. oryzae, A. fumigatus, and A. nidulans, and three in A. flavus and species of Fusarium. This CYP51 redundancy allows for slight changes to occur in the active site of one or all Cyp51 copies that affect the binding affinity to azoles. The structure of FLC in particular allows multiple Cyp51 active site-binding conformations that are weak or transitory so that there is incomplete inhibition of the Cyp51 target enzyme compared to other azoles that have stronger Cyp51 binding affinity. Regardless, the E-tests confirmed that even azole drugs used to treat human fungal pathogens are taken up by the plant pathogen M. oryzae as evidenced by growth inhibition seen with the medical azoles ITC, KTC, POS, and VRC. Overall, the susceptibility and resistance patterns illustrated by the E-tests suggest a common mechanism of action of azoles on all fungal species, including the requirement for entry into the cell, passing through the cell wall and plasma membrane. This report may be the first of the minimum inhibitory concentration (MICs) of medical antifungals on a distinctly plant-based pathogen. A common mechanism of action of azoles on all fungal species is that it must enter the fungal cell through the cell wall and plasma membrane to inhibit the intracellular Cyp51 target enzyme. Therefore, reduced or modified drug imports may help to explain why some pathogenic fungi are more resistant to azoles than others. Their assay can be used to compare drug import in agricultural, medical, and other pathogenic fungi. Experiments thus far have demonstrated that azole entry into the fungal cell is not solely by a passive diffusion mechanism. There may be some baseline level of azole passive diffusion into the cell, but their evidence suggests azole import into M. oryzae is more substantially through a plasma membrane-localized protein channel or carrier that recognizes a specific moiety found in azole drugs. This finding is in agreement with studies on the human pathogenic fungi C. albicans, C. neoformans, and A. fumigatus as well as the model yeast S. cerevisiae (Esquivel et al. 2015). Similarly, there may be a certain amount of azole import from uncharacterized ATP-dependent importers that was masked by the high activity of ATP-dependent efflux transporters. Import of azoles did not require a proton gradient, as no change was observed in uptake over a range of buffered pHs. There was a trend toward alkaline sensitivity for drug uptake as seen by a decrease in labeled FLC (3H-FLC) uptake in samples in pH 7 media. However, a deficiency in cell growth was observed in M. oryzae cells at pH 7, so import at this pH may be affected by other cellular factors directly or indirectly related to pH and proton gradients. They did find significant differences in drug accumulation in M. oryzae depending on the growth media used. Environmental adaptations that prevent or enhance azole uptake are an important aspect for drug resistance and treatment analysis, and it is difficult to identify a single factor that would cause such a dramatic difference in uptake between the different medias used. Cell adaptation to the different media that affect FLC uptake is probably a complex mixture of differences in protein synthesis, lipid storage, transcriptional activity, and other metabolic activities that may alter the cell membrane composition. The altered azole uptake between the samples argues against passive diffusion entry into the cell, in which case one would expect only minimal reduction or increase in drug accumulation between the samples. The evidence indicates that the efflux of azoles is stimulated by energy, potentially via ABC efflux transporters, which suggests there are distinct transporters for influx and efflux of azoles, as opposed to a single transporter that functions in both directions. Resistance to commonly used antifungals has frequently been shown to develop as a result of overexpression or increased activity of ABC transporters in human fungal pathogens (Sanglard 2016). More recently, ABC transporters are being recognized for their role in pathogenicity, virulence, stress tolerance, and drug resistance in M. oryzae and other plant pathogens.
Nutrient receptors could represent a potentially rich source of targets for antifungal drug development. About 40% of all available drugs target GPCRs, making these receptors the most important drug target group. Transceptors are often important for fungal development and virulence, and hence could be an excellent drug target group as well. Indeed, receptors that sense different stress responses have been proposed as drug targets. Current antifungal therapies are limited to drugs such as amphotericins, azoles, and echinocandins that inhibit the growth of C. albicans cells rather than specific virulence processes. Unfortunately, these drugs can have inhibitory effects on human cells, leading to serious side effects for the host. The problem of drug resistance should be manipulated with innovative and unconventional approaches and strategies at the population levels. McCarthy et al. (2017) reviewed the challenges associated with identifying broad-spectrum antifungal drugs and highlighted novel targets that could enhance the armamentarium of agents available to treat drug-resistant invasive fungal infections. Unfortunately, none of the novel targets that they proposed was using GPCR and ligand antagonists in fugal therapy. The emergence of antifungal resistance is a growing global health problem, underscoring the need for new therapeutic options. Significant progress has been made in the identification of potential targets for novel agents that exploit inherent differences between fungi and humans. Many of these targets remain theoretical, but several molecules have recently entered early clinical development. Complementing these therapies are advances in innate host defense: a pioneering clinical study using adoptive transfer of Aspergillus-stimulated T cells showed significant efficacy in treating stem cell transplant recipients with invasive aspergillosis. Other investigators have generated T-cytotoxic cells expressing Dectin-1 chimeric antigen receptors (CARs) that recognize surface fungal glucans. Infusion of D-CAR+ T cells reduced both fungal burden and mortality. Beyond small molecules, new approaches for augmenting host responses are critical and include the development of vaccines, immunomodulators, and novel approaches for reducing immunosuppression and exploiting the immunopharmacology of existing antifungal agents. Lundstrom (2017) stated, in his article “G-protein-coupled receptor-based drugs rediscovered,” that it seems the drug development process has become more complex, longer in duration, and more expensive. Today, generating a commercially available drug requires synthesis of 10,000 to 30,000 compounds, extensive fine tuning, and thorough testing before being approved. The efficacy requirements and the safety standards have reached new levels.
The author pinpointed that GPCR–ligand antagonists have not yet been adopted as a plan for producing antifungal drugs, at the industrial level, after surveying many scientific publication to date; to his best knowledge, perhaps such work is still under scrutinized scientific research, whether academic or in pharmaceutical factories. The author would like also to take the advantage of writing this chapter about GPCRs to launch a novel and original idea. As population and quantitative geneticists we can statistically partition the phenotypic variance to its components: additive, dominance, epistatic, and environmental. The problem that we face is the genotype × environment interaction. I believe that using marker-assisted selection and the marker in such cases will be the highly responsive GPCR that represent(s) the interface between the cell or organism and its surrounding environment and will leads us to stability in performance, which will be a new approach in breeding. We all know that traits for which breeders have never selected have a great amount of potential encrypted variance, which means that it provides a rich row material for high selection response. Such an idea was handled in another way by Botella (2012), who showed that a recent report identified two important yield quantitative trait loci (QTLs), GS3 and DEP1, cloned in rice (Oryza sativa), that were found to be heterotrimeric G-protein γ-subunits. This identification has profound consequences on our current understanding of both QTLs and the plant G-protein signaling network. Botella discussed how the manipulation of G-protein signaling may lead to yield improvements in rice and other crop species.
References
Affeldt KJ, Brodhagen M, Keller NP (2012) Aspergillus oxylipin signaling and quorum sensing pathways depend on G protein-coupled receptors. Toxins (Basel) 4:695–717
Affeldt KJ, Carrig J, Amare M, Keller NP (2014) Global survey of canonical Aspergillus flavus G protein-coupled receptors. mBio 5(5):e01501–e01504. https://doi.org/10.1128/mBio.01501-14
Ahlawat S, Saxena P, Alam P, Wajid S, Abdin MZ (2014) Modulation of artemisinin biosynthesis by elicitors, inhibitor, and precursor in hairy root cultures of Artemisia annua L. J Plant Interact 9(1):811–824. https://doi.org/10.1080/17429145.2014.949885
Aksam EB, Pinkse MWH, Verhaert PDEM (2013) Molecular characterization of Saccharomyces cerevisiae α-pheromone by mass spectrometry-based peptidomics. FEMS Yeast Res 13:350–353
Andersson AK, Cohn M (2017) Naumovozyma castellii: an alternative model for budding yeast molecular biology. Yeast 34:95–109
Atay O, Skotheim JM (2017) Spatial and temporal signal processing and decision making by MAPK pathways. J Cell Biol 2:317. https://doi.org/10.1083/jcb.201609124
Attfield PV (1997) Stress tolerance: the key to effective strains of industrial baker’s yeast. Nat Biotechnol 15:1351–1357
Baccelli I, Luti S, Bernardi R, Scala A, Pazzagli L (2014) Cerato-platanin shows expansin-like activity on cellulosic materials. Appl Microbiol Biotechnol 98:175–184. https://doi.org/10.1007/s00253-013-4822-0. PMID: 23512479
Baghel P, Rawal MK, Firoz Khan M, Sobhan Sen S, Siddiqui MH, Chaptal V, Falson P, Rajendra Prasad R (2017) Multidrug ABC transporter Cdr1 of Candida albicans harbors specific and overlapping binding sites for human steroid hormones transport. Biochim Biophys Acta 1859:1778–1789
Bahn YS, Xue C, Idnurm A, Rutherford JC, Heitman J, Cardenas ME (2007) Sensing the environment: lessons from fungi. Nat Rev Microbiol 5:57–69
Bakkeren G, Kämper J, Schirawski J (2008) Sex in smut fungi: structure, function and evolution of mating-type complexes. Fungal Genet Biol 45(suppl 1):15–21
Banuett F, Herskowitz I (1989) Different a alleles of Ustilago maydis are necessary for maintenance of filamentous growth but not for meiosis. Proc Natl Acad Sci USA 88:5878–5882
Bardwell L (2004) A walk-through of the yeast mating pheromone response pathway. Peptides 25:1465–1476
Bardwell L, Cook JG, Inouye CJ, Thorner J (1994) Signal propagation and regulation in the mating pheromone response pathway of the yeast Saccharomyces cerevisiae. Dev Biol 166:363–379
Barnett JA (2007) A history of research on yeasts. 10: Foundations of yeast genetics. Yeast 24:799–845
Beckerman JL, Naider F, Ebbole DJ (1997) Inhibition of pathogenicity of the rice blast fungus by Saccharomyces cerevisiae α-factor. Science 276:1116–1119
Bennett RJ, Johnson AD (2003) Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. EMBO J 22:2505–2515
Bermejo C, Haerizadeh F, Sadoine MSC, Chermak D, Frommer WB (2013) Differential regulation of glucose transport activity in yeast by specific cAMP signatures. Biochem J 452:489–497. https://doi.org/10.1042/BJ20121736
Bölker M, Urban M, Kahmann R (1992) The a mating-type locus of Ustilago maydis specifies cell signalling components. Cell 68:441–450
Booth LN, Tuch BB, Johnson AD (2010) Intercalation of a new tier of transcription regulation into an ancient circuit. Nature (Lond) 468:959–963
Borkovich KA, Alex LA, Yarden O (2004) Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol Mol Biol Rev 68:1–108
Botella JR (2012) Can heterotrimeric G proteins help to feed the world? Trends Plant Sci 17(10):563–568. https://doi.org/10.1016/j.tplants.2012.06.002
Botstein D (2004) Ira Herskowitz: 1946–2003. Genetics 166(2):653–660. https://doi.org/10.1534/genetics.166.2.653
Böttcher B, Pöllath C, Staib P, Hube B, Brunke S (2016) Candida species rewired hyphae developmental programs for chlamydospore formation. Front Microbiol 7:1697. https://doi.org/10.3389/fmicb.2016.01697
Bowers JL, Tyulmenkov VV, Jernigan SC, Klinge CM (2000) Resveratrol acts as a mixed agonist/antagonist for estrogen receptors α and β. Endocrinology 141:3657–13667
Braunsdorf C, Mailänder-Sánchez D, Schaller M (2016) Fungal sensing of host environment. Cell Microbiol 18:1188–1200. https://doi.org/10.1111/cmi.12610
Brefort T, Doehlemann G, Mendoza-Mendoza A, Reissmann S, Djamei A, Kahmann R (2009) Ustilago maydis as a pathogen. Annu Rev Phytopathol 47:423–445
Bresso E, Togawa R, Hammond-Kosack K, Urban M, Maigret B, Martins NF (2016) GPCRs from Fusarium graminearum detection, modeling and virtual screening – the search for new routes to control head blight disease. BMC Bioinf 17(18):39
Broach JR (2012) Nutritional control of growth and development in yeast. Genetics, 192:73–105. https://doi.org/10.1534/genetics.111.135731
Burkholder AC, Hartwell LH (1985) The yeast α-factor receptor: structural properties deduced from the sequence of the STE2 gene. Nucleic Acids Res 13:8463–8475
Butler G, Kenny C, Fagan A, Kurischko C, Gaillardin C, Wolfe KH (2004) Evolution of the MAT locus and its Ho endonuclease in yeast species. Proc Natl Acad Sci USA 101:1632–1637
Caceres I, El Khoury R, Bailly S, Oswald IP, Puel O, Bailly J (2017) Piperine inhibits aflatoxin B1 production in Aspergillus flavus by modulating fungal oxidative stress response. Fungal Genet Biol 107:77–85
Chachisvilis M, Zhang YL, Frangos JA (2006) G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci USA 103:15463–15468
Chang MH, Chae KS, Han DM, Jahng KY (2004) The GanB Galpha-protein negatively regulates asexual sporulation and plays a positive role in conidial germination in Aspergillus nidulans. Genetics 167:1305–1315
Chen X, Mou Y, Ling J, Wang N, Wang X, Hu J (2015) Cyclic dipeptides produced by fungus Eupenicillium brefeldianum HMP-F96 induced extracellular alkalinization and H2O2 production in tobacco cell suspensions. World J Microbiol Biotechnol 31:247–253
Choudhury SR, Pandey S (2016) Interaction of heterotrimeric G-protein components with receptor-like kinases in plants: an alternative to the established signaling paradigm? Mol Plant 9(8):1093–1095
Chung KS, Won M, Lee SB, Jang YJ, Hoe KL, Kim DU, Lee JW, Kim KW, Yoo HS (2001) Isolation of a novel gene from Schizosaccharomyces pombe: stm1 encoding a seven-transmembrane loop protein that may couple with the heterotrimeric G-alpha 2 protein, Gpa2. J Biol Chem 276:40190–40201
Coelho MA, Bakkeren G, Sun S, Hood ME, Giraud T (2017) Fungal sex: the Basidiomycota. Microbiol Spectrum 5(3):147–175. https://doi.org/10.1128/microbiolspec.FUNK-0046-2016
Coleman JLJ, Ngo T, Smith NJ (2017) The G protein-coupled receptor N-terminus and receptor signaling. Cell Signal 33:1–9
Coll NS, Epple P, Dangl JL (2011) Programmed cell death in the plant immune system. Cell Death Differ 18:1247–1256
Conrad M, Schothorst J, Kankipati HN, Zeebroeck GV, Rubio-Texeira M, Thevelein JM (2014) Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 38(2):254–299
Cook DE, Seidl MF, Kramer HM, Thomma BPHJ (2017) DNA methylation and chromatin architecture contribute to pathogenic fungal genome organization and adaptation. In: Abstract book, 29th fungal genetics conference, Asilomar 17, Pacific Grove, CA, 14–19 March 2017. Genetics Society of America, pp 166–167
Cornet M, Gaillardin C (2014) pH signaling in human fungal pathogens: a new target for antifungal strategies. Eukaryot Cell 13:342–352. Correction. Eukaryot Cell. 13:691
Coste AT, Karababa M, Ischer F, Bille J, Sanglard D (2004) TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652
Cote P, Sulea T, Dignard D, Wu C, Whiteway M (2011) Evolutionary reshaping of fungal mating pathway scaffold proteins. mBio 2:e00230–e00210. https://doi.org/10.1128/mBio.00230-10
Couch BC, Kohn LM (2002) A multilocus gene genealogy concordant with host preference indicates segregation of a new species, Magnaporthe oryzae, from M. grisea. Mycologia 94(4):683–693
Coughlan AY, Hanson SJ, Byrne KP, Wolfe KH, (2016) Centromeres of the Yeast Komagataella phaffii (Pichia pastoris) Have a simple inverted-repeat structure. Genome Biol Evol 8(8):2482–2492. https://doi.org/10.1093/gbe/evw178
Crawford RA, Pavitt DP (2018) Translational regulation in response to stress in Saccharomyces cerevisiae. Yeast 36:5–21. https://doi.org/10.1002/yea.3349
Cyert MS (2003) Calcineurin signaling in Saccharomyces cerevisiae: how yeast go crazy in response to stress. Biochem Biophys Res Commun 311(4):1143–1150
Davis DA (2009) How human pathogenic fungi sense and adapt to pH: the link to virulence. Curr Opin Microbiol 12:365–370
de Vries RP, Riley R, Wiebbenga Ad, Aguilar-Osorio G, Amilis S, et al (2017) Genome Biology 18:28. https://doi.org/10.1186/s13059-017-1151-0
Derevnina L, Dagdas YF, de la Concepcion JC, Bialas A, Kellner R, Petre B, Domazakis E, Du J, Wu C-H, Lin X, Aguilera-Galvez C, Cruz-Mireles N, Vleeshouwers VGAA, Kamoun S (2016) Nine things to know about elicitins. New Phytol 212(4):888–895. https://doi.org/10.1111/nph.14137
Devaux F, Carvajal E, Moye-Rowley S, Jacq C (2002) Genome-wide studies on the nuclear PDR3-controlled response to mitochondrial dysfunction in yeast. FEBS Lett 515:25–28
Devaux PF, Herrmann A, Ohlwein N, Kozlov MM (2008) How lipid flippases can modulate membrane structure. Biochim Biophys Acta 1778:1591–1600
DeZwaan TM, Carroll AM, Valent B, Sweigard JA (1999) Magnaporthe grisea Pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell 11:2013–2030
Di Segni G, Gastaldi S, Tocchini-Valentini GP (2008) cis- and trans-splicing of mRNAs mediated by tRNA sequences in eukaryotic cells. Proc Natl Acad Sci USA 105:6864–6869
Di Segni G, Gastaldi S, Zamboni M, Tocchini-Valentini GP (2011) Yeast pheromone receptor genes STE2 and STE3 are differently regulated at the transcription and polyadenylation level. PNAS 108(41):17082–17086
Diallinas G (2017) Transceptors as a functional link of transporters and receptors. Microb Cell 4(3):69–73
Díez E, Álvaro J, Espeso EA, Rainbow L, Suárez T, Tilburn J, Arst HN, Peñalva MÁ (2002) Activation of the Aspergillus PacC zinc finger transcription factor requires two proteolytic steps. EMBO J 21:1350–1359
Djamei A, Kahmann R (2012) Ustilago maydis: dissecting the molecular interface between pathogen and plant. PLoS Pathog 8:e1002955. https://doi.org/10.1371/journal.ppat.1002955
Djamei A, Schipper K, Rabe F, Ghosh A, Vincon V, Kahnt J, Osorio S, Tohge T, Fernie AR, Feussner I, Feussner K, Meinicke P, Stierhof Y, Schwarz H, Macek B, Mann M, Kahmann R (2011) Metabolic priming by a secreted fungal effector. Nature (Lond) 478:395–398. https://doi.org/10.1038/nature10454
Doehlemann G, van der Linde K, Aßmann D, Schwammbach D, Hof A, Mohanty A, Jackson D, Kahmann R (2009) Pep1, a secreted effector protein of Ustilago maydis, is required for successful invasion of plant cells. PLoS Pathog 5:e1000290. https://doi.org/10.1074/jbc.M113.488353
Doehlemann G, Reissmann S, Assmann D, Fleckenstein M, Kahmann R (2011) Two linked genes encoding a secreted effector and a membrane protein are essential for Ustilago maydis-induced tumour formation. Mol Microbiol 81:751–766
Douglas LM, Konopka JB (2016) Plasma membrane organization promotes virulence of the human fungal pathogen Candida albicans. J Microbiol 54:178. https://doi.org/10.1007/s12275-016-5621-y
Downes GB, Gautam N (1999) The G protein subunit gene families. Genomics 62:544–552
Du C, Sarfati J, Latge JP, Calderone R (2006) The role of the sakA (Hog1) and tcsB (sln1) genes in the oxidant adaptation of Aspergillus fumigatus. Med Mycol 44:211–218
Dutheil JY, Mannhaupt G, Schweizer G, Sieber CMK, Münsterkötter M, Güldener U, Schirawski J, Kahmann R (2016) A tale of genome compartmentalization: the evolution of virulence clusters in smut fungi. Genome Biol Evol 8(3):681–704
El Khoury R, Caceres I, Puel O, Bailly S, Atoui A, Oswald IP, El Khoury A, Bailly J (2017) Identification of the anti-aflatoxinogenic activity of Micromeria graeca and elucidation of its molecular mechanism in Aspergillus flavus. Toxins 9(3):87
EFSA Panel on Contaminants in the Food Chain (CONTAM). https://doi.org/10.2903/j.efsa.2011.2197
Eskes E, Deprez M, Wilms T, Winderickx J (2017) pH homeostasis in yeast, the phosphate perspective. Curr Genet 64(1):155–161. https://doi.org/10.1007/s00294-017-0743-2
Espeso E, Roncal T, Díez E, Rainbow L, Bignell E, Álvaro J, Suárez T, Denison SH, Tilburn J, Arst HN, Peñalva MA (2000) On how a transcription factor can avoid its proteolytic activation in the absence of signal transduction. EMBO J 19:719–728. https://doi.org/10.1093/emboj/19.4.719
Esquivel BD, White TC (2017) Accumulation of azole drugs in the fungal plant pathogen Magnaporthe oryzae is the result of facilitated diffusion influx. Front Microbiol 8(1320):1–12
Esquivel BD, Smith AR, Zavrel M, White TC (2015) Azole drug import into the pathogenic fungus Aspergillus fumigatus. Antimicrob Agents Chemother 59:3390–3398. https://doi.org/10.1128/AAC.0500314
Fan W, Shen T, Ding Q, Lv Y, Li L, Huang K, Yan L, Song S (2017) Zearalenone induces ROS-mediated mitochondrial damage in porcine IPEC-J2 cells. J Biochem Mol Toxicol 31(10):e21944. https://doi.org/10.1002/jbt.21944
Fan W, Lv Y, Ren S, Shao M, Shen T, Huang K, Zhou J, Yan L (2018) Zearalenone (ZEA)-induced intestinal inflammation is mediated by the NLRP3 inflammasome. Chemosphere 190:272–279
Feldbrügge M, Kamper J, Steinberg G, Kahmann R (2004) Reglation of mating and pathogenic development in Ustilago maydis. Curr Opin Microbiol 7:666–672
Finkel JS, Xu W, Huang D, Hill EM, Desai JV, Woolford CA (2012) Portrait of Candida albicans adherence regulators. PLoS Pathog 8(2):1002525
Franco-Orozco B, Berepili A, Ruiz O, Gamble L, Griffe LL, Wang S, Birch PR, Janyuka K, Avrova A (2017) New Phytologist 214:1657–1672.
Friesen TL, Faris JD, Solomon PS, Oliver RP (2008a) Host-specific toxins: effectors of necrotrophic pathogenicity. Cell Microbiol 10:1421–1428
Friesen TL, Zhang ZC, Solomon PS, Oliver RP, Faris JD (2008b) Characterization of the interaction of a novel Stagonospora nodorum host-selective toxin with a wheat susceptibility gene. Plant Physiol 146:682–693
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, Fitzhugh W, Ma L, Smirnov S, Purcell S (2003) The genome sequence of the filamentous fungus Neurospora crassa. Nature (Lond) 422:859–868
Galgoczy DJ, Cassidy-Stone A, Llinás M, O’Rourke SM, Herskowitz I, DeRisi JL, Johnson AD (2004) Genomic dissection of the cell-type-specification circuit in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 101(52):18069–18074. https://doi.org/10.1073/pnas.0407611102
Van Dijck P (2009) Nutrient sensing G protein-coupled receptors: interesting targets for antifungals? Med Mycol 47(7):671–680
Gao M, Skolnick J (2012) The distribution of ligand-binding pockets around protein-protein interfaces suggests a general mechanism for pocket formation. PNAS 109(10):3784–3789. https://doi.org/10.1073/pnas.1117768109
García-Muse T, Steinberg G, Pérez-Martín J (2003) Pheromone-induced G2 arrest in the phytopathogenic fungus Ustilago maydis. Eukaryot Cell 2:494–500
Gehm BD, McAndrews JM, Chien PY, Jameson JL (1997) Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc Natl Acad Sci USA 94:14138–14143
Gillissen B, Bergemann J, Sandmann C, Schroeer B, Bölker M, Kahmann R (1992) A two-component regulatory system for self/non-self recognition in Ustilago maydis. Cell 68:647–657
Glazebrook J (2005) Contrasting mechanisms of defence against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol 43:205–227
Gómez-Ariza J, Campo S, Rufat M, Estopà M, Messeguer J, San Segundo B, Coca M (2007) Sucrose-mediated priming of plant defense responses and broad-spectrum disease resistance by overexpression of the maize pathogenesis-related PRms protein in rice plants. Mol Plant-Microbe Interact 20(7):832–842
Green R (2017) Cell death and the immune system: getting to how and why. Immunol Rev 277:4–8
Grice CM, Bertuzzi M, Bignell EM (2013) Receptor-mediated signaling in Aspergillus fumigatus. Front Microbiol 4:1–12. https://doi.org/10.3389/fmicb.2013.00026
Gruber S, Omann M, Zeilinger S (2013) Comparative analysis of the repertoire of G protein-coupled receptors of three species of the fungal genus Trichoderma. BMC Microbiol 13:1
Guan G, Tao L, Yue H, Liang W, Gong J, Bing J, Zheng Q, Veri AO, Fan S, Robbins N, Cowen LE, Huang G (2019) Environment-induced same-sex mating in the yeast Candida albicans through the Hsf1–Hsp90 pathway. PLoS Biol 17(3):1–25. https://doi.org/10.1371/journal
Gürcan G, Şenol SH, Yağcıoğlu AEA, Ertuğrul A (2017) Effect of clozapine on psychiatric comorbidities in patients with schizophrenia. Eur Psychiatry 41(suppl):S197. https://doi.org/10.1016/j.eurpsy.2017.01.2140
Haber JE (2012) Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 191:33–64
Hahn J-S, Santoro N, Thiele DJ (2001) Snf1 protein kinase: a key player in the response to cellular stress in yeast. Biochem Soc Trans 31(pt 1):178–181
Hamid S, Wong MY (2017) Elicitors and their roles in plant defence against pathogens particularly basidiomycetes. In: Abdullah S, Chai-Ling H, Wagstaff C (eds) Crop improvement. Springer, Cham
Hamilton JR, Trejo J (2017) Challenges and opportunities in protease-activated receptor drug development. Annu Rev Pharmacol Toxicol 57:349–373
Han KH, Prade RA (2002) Osmotic stress-coupled maintenance of polar growth in Aspergillus nidulans. Mol Microbiol 43:1065–1078
Han KH, Seo JA, Yu JH (2004) A putative G protein-coupled receptor negatively controls sexual development in Aspergillus nidulans. Mol Microbiol 51:1333–1345
Hanson SJ, Byrne KP, Wolfe KH (2014) Mating-type switching by chromosomal inversion in methylotrophic yeasts suggests an origin for the three-locus Saccharomyces cerevisiae system. Proc Natl Acad Sci USA 111:E4851–E4858
Hanson SJ, Wolfe KH (2017) An evolutionary perspective on yeast mating-type switching. Genetics 206(1):9–32. https://doi.org/10.1534/genetics.117.202036
Hartwell LH (1980) Mutants of Saccharomyces cerevisiae unresponsive to cell division control by polypeptide mating hormone. J Cell Biol 85(3):811–822
Harmar AJ (2001) Family-B G-protein-coupled receptors. Genome Biol 2(12):3013.1–3013.10
Hartig A, Holly J, Saari G, MacKay VL (1986) Multiple regulation of STE2, a mating-type-specific gene of Saccharomyces cerevisiae. Mol Cell Biol 6:2106–2114
Hemetsberger C, Herrberger C, Zechmann B, Hillmer M, Doehlemann G (2012) The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog 8:e1002684
Herskowitz I (1995) MAP kinase pathways in yeast: for mating and more. Cell 80:187–197
Hicks WM, Yamaguchi M, Haber JE (2011) Real-time analysis of double-strand DNA break repair by homologous recombination. Proc Natl Acad Sci USA 108:3108–3115
Hirsch L, Yang J, Bresee L, Jette N, Patten S, Pringsheim T (2017) Second-generation antipsychotics and metabolic side effects: a systematic review of population-based studies. Drug Saf 40(9):771–781. https://doi.org/10.1007/s40264-017-0543-0
Holliday R (1961) The genetics of Ustilago maydis. Genet Res 2:204–230
Hsieh HM, Chung MC, Chen PY, Hsu FM, Liao WW, Sung AN, Lin CR, Wang CJR, Kao YH, Fang MJ, Lai CY, Huang CC, Chou JC, Chou WN, Chang BCH, Ju YM (2017) A termite symbiotic mushroom maximizing sexual activity at growing tips of vegetative hyphae. Bot Stud 58:39. https://doi.org/10.1186/s40529-017-0191-9
Inglis DO, Sherlock G (2013) Ras signaling gets fine-tuned: regulation of multiple pathogenic traits of Candida albicans. Eukaryot Cell 12:1316–1325
Ira G, Satory D, Haber JE (2006) Conservative inheritance of newly synthesized DNA in double-strand break-induced gene conversion. Mol Cell Biol 26:9424–9429
Janiak AM, Sargsyan H, Russo J, Naider F, Hauser M, Becker JM (2005) α-Factor receptor in Saccharomyces cerevisiae. Fungal Genet Biol 42(4):328–338
Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Vandivier RW, Wang N, Greenberg S, Dale BM, Qin C, Henson PM, Tall AR (2006) ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 174(4):547–556
Jenness DD, Burkholder AC, Hartwell LH (1983) Binding of α-factor pheromone to yeast a-cells: chemical and genetic evidence for an α-factor receptor. Cell 35:521–529
Ji J, Zhu P, Cui F, Pi F, Zhang Y, Li Y, Wang J, Sun X (2017) The antagonistic effect of mycotoxins deoxynivalenol and zearalenone on metabolic profiling in serum and liver of mice. Toxins 9(1):28. https://doi.org/10.3390/toxins9010028
Jiang RHY, Tyler BM, Whisson SC, Hardham AR, Govers F (2006) Ancient origin of elicitin gene clusters in Phytophthora genomes. Mol Biol Evol 23:338–351
Johnson SS, Hanson PK, Manoharlal R, Brice SE, Cowart LA, Moye-Rowley WS (2010) Regulation of yeast nutrient permease endocytosis by ATP-binding cassette transporters and a seven-transmembrane protein, RSB1. J Biol Chem 285:35792–35802
Johnson JM, Jin M, Lew DJ (2011) Symmetry breaking and the establishment of cell polarity in budding yeast. Curr Opin Genet Dev 21(6):740–746. https://doi.org/10.1016/j.gde.2011.09.007
Jones JD, Dangl JL (2006) The plant immune system. Nature (Lond) 444:323–329
Josefsson LG (1999) Evidence for kinship between diverse G-protein coupled receptors. Gene (Amst) 239:333–340
Kaserer AO, Andi B, Cook PF, West AH (2009) Effects of osmolytes on the SLN1-YPD1-SSK1 phosphorelay system from Saccharomyces cerevisiae. Biochemistry 48:8044–8050
Kämper J, Kahmann R, Bölker M, Ma L, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Müller O, Perlin MH, Wösten HAB, de Vries R, Ruiz-Herrera J, Reynaga-Peña CG, Snetselaar K, McCann M, Pérez-Martín J, Feldbrügge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, González-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Münch K, Rössel N, Scherer M, Vraneš M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho ECH, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li W, Sanchez-Alonso P, Schreier PH, Häuser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schlüter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Güldener U, Münsterkötter M, Haase D, Oesterheld M, Mewes H, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature (Lond) 444:97–101. https://doi.org/10.1038/nature05248
Khakhina S, Johnson SS, Manoharlal R, Russo SB, Blugeon C, Lemoine S, Sunshine AB, Dunham MJ, Cowart LA, Devaux F, Moye-Rowley WS (2015) Control of plasma membrane permeability by ABC transporters. Eukaryot Cell 14:442–453
Khrunyk Y, Münch K, Schipper K, Lupas AN, Kahmann R (2010) The use of FLP-mediated recombination for the functional analysis of an effector gene family in the biotrophic smut fungus Ustilago maydis. New Phytol 187:957–968
Kim K, Jeon J, Choi J, Cheong K, Song H, Choi G, Kang S, Lee Y (2016) Kingdom-wide analysis of fungal small secreted proteins (SSPs) reveals their potential role in host association. Front Plant Sci 7:186
Kim Y, Lee M, Jun S, Choi Y, Yu J, Shin K (2019) RgsD negatively controls development, toxigenesis, stress response, and virulence in Aspergillus fumigatus. Sci Rep 9:811. https://doi.org/10.1038/s41598-018-37124-2
Kim Y, Park SY, Kim D, Choi J, Lee YH, Lee JH, Choi W (2013) Genome-scale analysis of ABC transporter genes and characterization of the ABCC type transporter genes in Magnaporthe oryzae. Genomics 101:354–361. https://doi.org/10.1016/j.ygeno.2013.04.003
Klar AJ (2007) Lessons learned from studies of fission yeast mating-type switching and silencing. Annu. Rev. Genet. 41:213–236
Klar AJ (2010) The yeast mating-type switching mechanism: a memoir. Genetics 186:443–449
Knöppel A, Näsvall J, Andersson DI (2017) Evolution of antibiotic resistance without antibiotic exposure. Antimicrob Agents Chemother 61(1):01495–01417
Kolesarova A, Capcarova M, Maruniakova N, Lukac N, Ciereszko RE, Sirotkin A (2012) Resveratrol inhibits reproductive toxicity induced by deoxynivalenol. J Environ Sci Health A Tox Hazard Subst Environ Eng 47:1329–1334
Kombrink A, Rovenich H, Shi-Kinne X, Rojas-Padilla E, Van den Berg GCM, Domazakis E, De Jonge R, Valkenburg D, Sánchez-Vallet A, Seidl MF, Thomma BPHJ (2017) Verticillium dahliae LysM effectors differentially contribute to virulence on plant hosts. Mol Plant Pathol 18(4):596–608
Kraakman L, Lemaire K, Ma P, Teunissen AW, Donaton MC, Dijck PV (1999) A Saccharomyces cerevisiae G-protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol Microbiol 32:1002–1012
Krishnan A, Almén MS, Fredriksson R, Schiöth HB (2012) The origin of GPCRs: identification of mammalian-like rhodopsin, adhesion, glutamate and frizzled GPCRs in fungi. PLoS One 7(1):e29817. https://doi.org/10.1371/journal.pone.0029817
Kronstad JW, Leong SA (1990) The b mating-type locus of Ustilago maydis contains variable and constant regions. Genes Dev 4:1384–1395
Kües U, James TY, Heitman J (2011) Mating type in basidiomycetes: unipolar, bipolar, and tetrapolar patterns of sexuality. In: Pöggeler S, Wöstemeyer J (eds) Evolution of fungi and fungal-like organisms. The Mycota (a comprehensive treatise on fungi as experimental systems for basic and applied research), vol 14. Springer, Berlin, Heidelberg
Kulkarni RD, Thon MR, Pan H, Dean RA (2005) Novel G-protein-coupled receptor-like proteins in the plant pathogenic fungus Magnaporthe grisea. Genome Biol 6(3):R24. https://doi.org/10.1186/gb-2005-6-3-r24
Kumamoto CA (2008) Molecular mechanisms of mechanosensing and their roles in fungal contact sensing. Nat Rev Microbiol 6:667–673
Kurakado S, Kurogane R, Sugita T (2017) 17β-Estradiol inhibits estrogen binding protein-mediated hypha formation in Candida albicans. Microb Pathog 109:151–155
Kurjan J (1985) Alpha-factor structural gene mutations in Saccharomyces cerevisiae: effects on alpha-factor production and mating. Mol Cell Biol 5:787–796
Lanver D, Tollot M, Schweizer G, Presti LL, Reissmann S, Ma L, Schuster M, Tanaka S, Liang L, Ludwig N, Kahmann R (2017) Ustilago maydis effectors and their impact on virulence. Nat Rev Microbiol 15:409–421
Lafon A, Han KH, Seo JA, Yu JH, d’Enfert C (2006) G-protein and cAMP-mediated signaling in aspergilli: a genomic perspective. Fungal Genet Biol 43:490–502
Lafon A, Seo JA, Han KH, Yu JH, d’Enfert C (2005) The heterotrimeric G-protein GanB(a)-SfaD(b)-GpgA(g) is a carbon source sensor involved in early cAMP-dependent germination in Aspergillus nidulans. Genetics 171:71–80
Lagerstrom MC, Schiöth HB (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7:339–357
Lamping E, Ranchod A, Nakamura K, Tyndall JD, Niimi K, Holmes AR, Niimi M, Cannon RD (2009) Abc1p is a multidrug efflux transporter that tips the balance in favor of innate azole resistance in Candida krusei. Antimicrob Agents Chemother 53:354–369
Lamping E, Baret PV, Holmes AR, Monk BC, Goffeau A, Cannon RD (2010) Fungal PDR transporters: phylogeny, topology, motifs and function. Fungal Genet Biol 47(2):127–142
Lamping E, Zhu J, Niimi M, Cannon RD (2017a) Role of ectopic gene conversion in the evolution of a Candida krusei pleiotropic drug resistance transporter family. Genetics 205:1619–1639
Lamping E, Madani G, Lee HJ, Niimi M, Cannon RD (2017b) Structure–function analyses of multidrug transporters. In: Candida albicans: Cellular and molecular biology. Springer, Cham, pp 379–406
Landry Y, Gies J (2008) Drugs and their molecular targets: an updated overview. Fundam Clin Pharmacol 22:1–18. https://doi.org/10.1111/j.1472-8206.2007.00548.x
Le Corre L, Chalabi N, Delort L, Bignon Y-J, Bernard-Gallon DJ (2005) Resveratrol and breast cancer chemoprevention: molecular mechanisms. Mol Nutr Food Res 49:462–471
Le Guevel R, Pakdel F (2001) Assessment of oestrogenic potency of chemicals used as growth promoter by in vitro methods. Hum Reprod 16:1030–1036
Lee CS, Haber JE (2015) Mating-type gene switching in Saccharomyces cerevisiae. Microbiol Spectr 3(2):MDNA3-0013-2014. https://doi.org/10.1128/microbiolspec.MDNA3-0013-2014
Leeuw T, Wu C, Schrag JD, Whiteway M, Thomas DY, Leberer E (1998) Interaction of a G-protein beta-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature (Lond) 391(6663):191–195
Le Marquer M, San Clemente H, Roux C, Savelli B, Nicolas Frey F (2019) Identification of new signalling peptides through a genome-wide survey of 250 fungal secretomes. BMC Genomics 20:64. https://doi.org/10.1186/s12864-018-5414-2
Lemaire K, Van de Velde S, Van Dijck P, Thevelein JM (2004) Glucose and sucrose act as agonist and mannose as antagonist ligands of the G protein-coupled receptor Gpr1 in the yeast Saccharomyces cerevisiae. Mol Cell 16:293–299
Lesage G, Bussey H (2006) Cell wall assembly in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 70:317–343
Li L, Wright SJ, Krystofova S, Park G, Borkovich KA (2007) Heterotrimeric G protein signaling in filamentous fungi. Annu Rev Microbiol 61:423–452
Li X, Gao C, Li L, Liu M, Yin Z, Zhang H, Zheng X, Wang P, Zhang Z (2017) MoEnd3 regulates appressorium formation and virulence through mediating endocytosis in rice blast fungus Magnaporthe oryzae. PLoS Pathog 13(8):e1006554
Lin P, Chen F, Sun J, Zhou J, Wang X, Wang N, Li X, Zhang Z, Wang A, Jin Y (2015) Mycotoxin zearalenone induces apoptosis in mouse Leydig cells via an endoplasmic reticulum stress-dependent signalling pathway. Reprod Toxicol 52:71–77
Linsel-Nitschke P, Jehle AW, Shan J, Cao G, Bacic D, Lan D, Wang N, Tall AR (2005) Potential role of ABCA7 in cellular lipid efflux to apoA-I. J Lipid Res 46(1):86–92
Lismaa TP, Shine J (1992) G protein-coupled receptors. Curr Opin Cell Biol 4:195–202. https://doi.org/10.1016/0955-0674(92)90033-9
Liu S, Dean RA (1997) G protein alpha subunit genes control growth, development, and pathogenicity of Magnaporthe grisea. Mol Plant Microbe Interact 10:1075–1086
Liu B, Li JF, Ao Y, Li Z, Liu J, Feng D, Qi K, He Y, Zeng L, Wang J, Wang HB (2013a) OsLYP4 and OsLYP6 play critical roles in rice defence signal transduction. Plant Signal Behav 8:e22980
Liu J, Men J, Chang M, Feng C, Yuan L (2017) iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress. J Proteome 156:75–84
Liu TT, Znaidi S, Barker KS, Xu L, Homayouni R, Saidane S, Morschhäuser J, Nantel A, Raymond M, Rogers PD (2007) Genome-wide expression and location analyses of the Candida albicans Tac1p regulon. Eukaryot Cell 6:2122–2138
Liu W, Liu J, Ning Y, Ding B, Wang X, Wang Z, Wang GL (2013b) Recent progress in understanding PAMP-and effector-triggered immunity against the rice blast fungus Magnaporthe oryzae. Mol Plant 6:605–620
Lo Presti L, Kahmann R (2017) How filamentous plant pathogen effectors are translocated to host cells. Curr Opin Plant Biol 38:19–24
Lorenz MC, Pan X, Harashima T, Cardenas ME, Xue Y, Hirsch JP, Heitman J (2000) The G protein-coupled receptor Gpr1 is a nutrient sensor that regulates pseudo-hyphal differentiation in Saccharomyces cerevisiae. Genetics 154:609–622
Lu R, Serrero G (1999) Resveratrol, a natural product derived from grape, exhibits antiestrogenic activity and inhibits the growth of human breast cancer cells. J Cell Physiol 179:297–304
Lu Y, Su C, Liu H (2012) A GATA transcription factor recruits Hda1 in response to reduced Tor1 signaling to establish a hyphal chromatin state in Candida albicans. PLoS Pathog 8:e1002663. https://doi.org/10.1371/journal.ppat.1002663
Lundstrom K (2017) G-protein-coupled receptor-based drugs rediscovered. Future Med Chem 9(7):633–636. https://doi.org/10.4155/fmc-2017-0045
Luti S, Martellini F, Bemporad F, Mazzoli L, Paoli P, Pazzagli L (2017) A single amino acid mutation affects elicitor and expansins-like activities of cerato-platanin, a non-catalytic fungal protein. PLoS One 12(5):e0178337. https://doi.org/10.1371/journal.pone.0178337
MacKay V, Manney R (1974) Mutations affecting sexual conjugation and related processes in Saccharomyces cerevisiae. II. Genetic analysis of nonmating mutants. Genetics 76:273–288
Maffei ME, Arimura GI, Mithöfer A (2012) Natural elicitors, effectors and modulators of plant responses. Nat Prod Rep 29:1288–1303
Magee BB, Magee PT (2000) Induction of mating in Candida albicans by construction of MTLa and MTLalpha strains. Science 289(5477):310–303
Maidan MM, De Rop L, Serneels J, Exler S, Rupp S, Tournu H, Thevelein JM, Van Dijck P (2005b) The G protein-coupled receptor Gpr1 and the G{alpha} protein Gpa2 act through the cAMP-protein kinase A pathway to induce morphogenesis in Candida albicans. Mol Biol Cell 16:1971–1986
Maidan MM, Thevelein JM, Van Dijck P (2005a) Carbon source induced yeast-to-hypha transition in Candida albicans is dependent on the presence of amino acids and on the G-protein-coupled receptor Gpr1. Biochem Soc Trans 33:291–293
Majumdar R, Rajasekaran K, Sickler C, Lebar M, Musungu BM, Fakhoury AM, Payne GA, Geisler M, Carter-Wientjes C, Wei Q, Bhatnagar D, Cary JW (2017) The pathogenesis-related maize seed (PRms) gene plays a role in resistance to Aspergillus flavus infection and aflatoxin contamination. Front Plant Sci 8:1758. https://doi.org/10.3389/fpls.2017.01758
Martin T, Lu SW, van Tilbeurgh H, Ripoll DR, Dixelius C, Turgeon BG, Debuchy R (2010) Tracing the origin of the fungal alpha1 domain places its ancestor in the HMG-box superfamily: implication for fungal mating-type evolution. PLoS One 5(12):e15199
Maller JL (2003) Signal transduction. Fishing at the cell surface. Science 300:594–595
Manente M, Ghislain M (2009) The lipid-translocating exporter family and membrane phospholipid homeostasis in yeast. FEMS Yeast Res 9:673–687
Manzanares-Estreder S, Espí-Bardisa J, Alarcón B, Amparo Pascual-Ahuir A, Proft M (2017) Multilayered control of peroxisomal activity upon salt stress in Saccharomyces cerevisiae. Mol Microbiol 104(5):851–868
Marcet-Houben M, Gabaldón T (2015) Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker’s yeast lineage. PLoS Biol 13:e1002220. https://doi.org/10.1371/journal.pbio.1002220
Matei A, Doehlemann G (2016) Cell biology of corn smut disease Ustilago maydis as a model for biotrophic interactions. Curr Opin Microbiol 34:60–66
McCarthy MW, Kontoyiannis DP, Cornely OA, Perfect JR, Walsh TJ (2017) Novel agents and drug targets to meet the challenges of resistant fungi. J Infect Dis 216(suppl 3):S474–S483. https://doi.org/10.1093/infdis/jix130
Mitchell TK, Dean RA (1995) The cAMP-dependent protein kinase catalytic subunit is required for appressorium formation and pathogenesis by the rice blast pathogen Magnaporthe grisea. Plant Cell 7:1869–1878
Miwa T, Takagi Y, Shinozaki M, Yun CW, Schell WA, Perfect JR, Kumagai H, Tamaki H (2004) Gpr1, a putative G-protein-coupled receptor, regulates morphogenesis and hypha formation in the pathogenic fungus Candida albicans. Eukaryot Cell 3:919–931
Moretti S, Bellocchio S, Bonifazi P, Bozza S, Zelante T, Bistoni F, Romani L (2008) The contribution of PARs to inflammation and immunity to fungi. Mucosal Immunol 1:156–168. https://doi.org/10.1038/mi.2007.13
Mueller AN, Ziemann S, Treitschke S, Aßmann D, Doehlemann G (2013) Compatibility in the Ustilago maydis–maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog 9:e1003177. https://doi.org/10.1371/journal.ppat.1003177
Mueller SO, Simon S, Chae K, Metzler M, Korach KS (2004) Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor α (ERα) and ERβ in human cells. Toxicol Sci 80(1):14–25. https://doi.org/10.1093/toxsci/kfh147
Navarathna DHMLP, Pathirana RU, Lionakis MS, Nickerson KW, Roberts DD (2016) Candida albicans ISW2 regulates chlamydospore suspensor cell formation and virulence in vivo in a mouse model of disseminated candidiasis. PLoS One 11(10):e0164449. https://doi.org/10.1371/journal.pone.0164449
Newman MA, Sundelin T, Nielsen JT, Erbs G (2013) MAMP (microbe-associated molecular pattern) triggered immunity in plants. Front Plant Sci 4:139
Nieuwenhuis BP, Immler S (2016) The evolution of mating-type switching for reproductive assurance. BioEssays 38:1141–1149
Nobile CJ, Johnson AD (2015) Candida albicans biofilms and human disease. Annu Rev Microbiol 69:71–92
Nobile CJ, Schneider HA, Nett JE, Sheppard DC, Filler SG, Andes DR, Mitchell AP (2008) Complementary adhesin function in C. albicans biofilm formation. Curr Biol 18:1017–1024
Noble SM, Gianetti BA, Witchley JN (2017) Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol 15:96–108
Nowyhed HN, Chandra S, Kiosses W, Marcovecchio P, Andary F, Zhao M, Fitzgerald ML, Kronenberg M, Hedrick CC (2017) ATP binding cassette transporter ABCA7 regulates NKT cell development and function by controlling CD1d expression and lipid raft content. Sci Rep 7:40273–40284. https://doi.org/10.1038/srep40273
O’Leary BM, Neale HC, Geilfus CM, Jackson RW, Arnold DL, Preston GM (2016) Early changes in apoplast composition associated with defence and disease in interactions between Phaseolus vulgaris and the halo blight pathogen Pseudomonas syringae pv. phaseolicola. Plant Cell Environ 39:2172–2184
Oliver RP, Solomon PS (2010) New developments in pathogenicity and virulence of necrotrophs. Curr Opin Plant Biol 13:415–419
Oshima Y (1993) Homothallism, mating-type switching, and the controlling element model in Saccharomyces cerevisiae. In: Hall MN, Linder P (eds) The early days of yeast genetics. Cold Spring Harbor Laboratory Press, New York, pp 291–304
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996
Panadero J, Pallotti C, Rodrı́guez-Vargas S, Randez-Gil F, Prieto JA (2006) A downshift in temperature activates the high osmolarity glycerol (HOG) pathway, which determines freeze tolerance in Saccharomyces cerevisiae. J Biol Chem 281(8):4638–4645
Pangeni R, Pharm B, Sahni JK, Ali J, Sharma S, Baboota S (2014) Resveratrol: review on therapeutic potential and recent advances in drug delivery. Expert Opin Drug Deliv 11(8):1285–1298
Perlin DS, Rautemaa-Richardson R, Alastruey-Izquierdo A (2017) The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis 17(12):e383–e392. https://doi.org/10.1016/S1473-3099(17)30316-X
Petit-Houdenot Y, Fudal I (2017) Complex interactions between fungal avirulence genes and their corresponding plant resistance genes and consequences for disease resistance management. Front Plant Sci 8:1072. https://doi.org/10.3389/fpls.2017.01072
Prasad R, Banerjee A, Khandelwal NK, Dhamgaye S (2015) The ABCs of Candida albicans multidrug transporter Cdr1. Eukaryot Cell 14:1154–1164
Prasad R, Devaux F, Dhamgay S, Banerjee D (2012) Response of pathogenic and non-pathogenic yeasts to steroids. J Steroid Biochem Mol Biol 129:61–69
Prigitano A, Esposto MC, Biffi A, De Lorenzis G, Favuzzi V, Koncan R, Lo Cascio G, Ocampo MB, Colombo C, Pizzamiglio G, Romanò L, Tortorano AM (2015) Triazole resistance in Aspergillus fumigatus isolates from patients with cystic fibrosis in Italy. J Cyst Fibros 16:64–69
Pryciak PM, Huntress FA (1998) Membrane recruitment of the kinase cascade scaffold protein Ste5 by the Gbetagamma complex underlies activation of the yeast pheromone response pathway. Genes Dev 12:2684–2697
Puhalla JE (1968) Compatibility reactions on solid medium and interstrain inhibition in Ustilago maydis. Genetics 66:461–474
Que Y, Xu L, Wu Q, Liu Y, Ling H, Liu Y, Zhang Y, Guo J, Su Y, Chen J, Wang S, Zhang C (2014) Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genet 15:996
Rajagopal S, Shenoy SK (2018) GPCR desensitization: acute and prolonged phases. Cell Signal 41(201):9–16. https://doi.org/10.1016/j.cellsig.2017.01.024
Ramage G, Bachmann S, Patterson TF, Wickes BL, Lopez-Ribot JL (2002) Investigation of multidrug efflux pumps in relation to fluconazole resistance in Candida albicans biofilms. J Antimicrob Chemother 49:973–980
Rastghalam G, Omran RP, Alizadeh M, Fulton D, Mallick J, Whiteway M (2019) MAP kinase regulation of the Candida albicans pheromone pathway. Mol Biol Physiol 4(1):1–14
Rawal MK, Khan MF, Kapoor K, Goyal N, Sen S, Saxena AK, Lynn AM, Tyndall JD, Monk BC, Cannon RD, Komath SS, Prasad R (2013) Insight into pleiotropic drug resistance ATP-binding cassette pump drug transport through mutagenesis of Cdr1p transmembrane domains. J Biol Chem 288(34):24480–24493
Read ND, Kalkman ER (2003) Does endocytosis occur in fungal hyphae? Fungal Genet Biol 39:199–203
Redkar A, Villajuana-Bonequi M, Doehlemann G (2015) Conservation of the Ustilago maydis effector See1 in related smuts. Plant Signal Behav 10(12):e1086855. https://doi.org/10.1080/15592324.2015.1086855
Ring L, Yeh SY, Hücherig S, Hoffmann T, Blanco-Portales R, Fouche M, Villatoro C, Denoyes B, Monfort A, Caballero JL, Muñoz-Blanco J, Gershenson J, Schwab W (2013) Metabolic interaction between anthocyanin and lignin biosynthesis associated with peroxidase FaPRX27 in strawberry fruit. Plant Physiol 163:43–60. https://doi.org/10.1104/pp.113.222778
Robert H, Payros D, Pinton P, Théodorou V, Mercierbonin M, Oswald IP (2017) Impact of mycotoxins on the intestine: are mucus and microbiota new targets? J Toxicol Environ Health Part B 20(5):249–275
Roelants FM, Leskoske KL, Marshall MNM, Locke MN, Thorner J (2017) The TORC2-dependent signaling network in the yeast Saccharomyces cerevisiae. Biomol Ther 7(3):66. https://doi.org/10.3390/biom7030066
Rosen S, Yu JH, Adams TH (1999) The Aspergillus nidulans sfaD gene encodes a G protein beta subunit that is required for normal growth and repression of sporulation. EMBO J 18:5592–5600
Rowell JB, DeVay JE (1954) Genetics of Ustilago zeae in relation to basic problems of its pathogenicity. Phytopathology 44:356–362
Rubio-Texeira M, Zeebroeck GV, Voordeckers K, Thevelein JM (2010) Saccharomyces cerevisiae plasma membrane nutrient sensors and their role in PKA signaling. FEMS Yeast Res 10(2):134–149
Rutherford JC, Bahn Y, van den Berg B, Heitman J, Xue C (2019) Nutrient and stress sensing in pathogenic yeasts. Front Microbiol 10(442):1–17. https://doi.org/10.3389/fmicb.2019.00442
Sabnam N, Barman NSR (2017) WISH, a novel CFEM GPCR, is indispensable for surface sensing, asexual and pathogenic differentiation in rice blast fungus. Fungal Genet Biol 105:37–51
Saini P, Prasad T, Gaur NA, Shukla S, Jha S, Komath SS, Khan LA, Rizwanul Haq Q, Prasad R (2005) Alanine scanning of transmembrane helix 11 of Cdr1p ABC antifungal efflux pump of Candida albicans: identification of amino acid residues critical for drug efflux. J Antimicrob Chemother 56:7–86. https://doi.org/10.1093/jac/dki183
Sang Y, Li W, Zhang G (2016) The protective effect of resveratrol against cytotoxicity induced by mycotoxin, zearalenone. Food Funct 7:3703–3715. https://doi.org/10.1039/C6FO00191B
Sanglard D (2016) Emerging threats in antifungal-resistant fungal pathogens. Front Med (Lausanne) 3:11. https://doi.org/10.3389/fmed.2016.00011
Sanz P (2003) Snf1 protein kinase: a key player in the response to cellular stress in yeast. Biochem Soc Trans 31(1):178–181. (In AMPK 2002: 2nd International Meeting on AMP-activated Protein Kinase, a Biochemical Society-sponsored meeting held at University of Dundee, Scotland, 12–14 September 2002)
Saxena P, Ahlawat S, Ali A, Khan S, Zainul Abdin M (2017) Gene expression analysis of the withanolide biosynthetic pathway in hairy root cultures of Withania somnifera elicited with methyl jasmonate and the fungus Piriformospora indica. Symbiosis 71:143–154. https://doi.org/10.1007/s13199-016-0416-9
Schulz B, Banuett F, Dahl M, Schlesinger R, Schäfer W, Martin T, Herskowitz I, Kahmann R (1990) The b alleles of U. maydis, whose combinations program pathogenic development, code for polypeptides containing a homeodomain-related motif. Cell 60:295–306
Schuster M, Schweizer G, Kahmann R (2017) Comparative analyses of secreted proteins in plant pathogenic smut fungi and related basidiomycetes. Fungal Genet Biol 112:21–30. https://doi.org/10.1016/j.fgb.2016.12.003
Schweizer A, Rupp S, Taylor BN, Rollinghoff M, Schroppel K (2000) The TEA/ATTS transcription factor CaTec1p regulates hyphal development and virulence in Candida albicans. Mol Microbiol 38:435–445
Seo JA, Han KH, Yu JH (2004) The gprA and gprB genes encode putative G protein-coupled receptors required for self-fertilization in Aspergillus nidulans. Mol Microbiol 53:1611–1623
Seraj Uddin M, Hauser M, Naider F, Becker JM (2016) The N-terminus of the yeast G protein-coupled receptor Ste2p plays critical roles in surface expression, signaling, and negative regulation. Biochim Biophys Acta 1858:715–724
Shellhammer JP, Morin-Kensicki E, Matson JP, Yin G, Isom DG, Campbell SL (2017) Amino acid metabolites that regulate G protein signaling during osmotic stress. PLoS Genet 13(5):e1006829. https://doi.org/10.1371/journal.pgen.1006829
Shi L, Ren A, Zhu J, Yu H, Jiang A, Zheng H, Zhao M (2019) 14-3-3 Proteins: a window for a deeper understanding of fungal metabolism and development. World J Microbiol Biotechnol 35:24. https://doi.org/10.1007/s11274-019-2597-x
Shukla S, Saini P, Jha S, Ambudkar SV, Prasad R (2003) Functional characterization of Candida albicans ABC transporter Cdr1p. Eukaryot Cell 2:1361–1375
Singh A, Chen EY, Lugovoy JM, Chang CN, Hitzeman RA, Seeburg PH (1983) Saccharomyces cerevisiae contains two discrete genes coding for the α-factor pheromone. Nucleic Acids Res 11(12):4049–4063
Snetselaar KM, Mims CW (1992) Sporadically fusion and infection of maize seedlings by the smut fungus Ustilago maydis. Mycologia 84:193–203
Snetselaar K, McCann M (2017) Ustilago maydis, the corn smut fungus, has an unusual diploid mitotic stage. Mycologia 109(1):140–152
Sorrells TR, Booth LN, Tuch BB, Johnson AD (2015) Intersecting transcription networks constrain gene regulatory evolution. Nature (Lond) 523:361–365
Srivastava A, Sircaik S, Husain F, Thomas E, Ror S, Rastogi S, Alim D, Bapat P, Andes DR (2017) Distinct roles of the 7-transmembrane receptor protein Rta3 in regulating the asymmetric distribution of phosphatidylcholine across the plasma membrane and biofilm formation in Candida albicans. Cell Microbiol 19(12):e12767. https://doi.org/10.1111/cmi.12767
Steinberg G, Perez-Martin J (2008) Ustilago maydis, a new fungal model system for cell biology. Trends Cell Biol 18(2):61–67
Stirnberg A, Djamei A (2016) Characterization of ApB73, a virulence factor important for colonization of Zea mays by the smut Ustilago maydis. Mol Plant Pathol 17(9):1467–1479. https://doi.org/10.1111/mpp.12442
Subotić A, Swinnen E, Demuyser L, De Keersmaecker H, Mizuno H, Tournu H, Van Dijck P (2017) A bimolecular fluorescence complementation tool for identification of protein-protein interactions in Candida albicans. G3: Genes Genomes Genet 7(10):3509–3520. https://doi.org/10.1534/g3.117.300149
Sudbery PE (2011) Growth of Candida albicans hyphae. Nat Rev Microbiol 9:737–748
Su C, Lu Y, Liu H (2013) Reduced TOR signaling sustains hyphal development in Candida albicans by lowering Hog1 basal activity. Mol Biol Cell 24:385–397. https://doi.org/10.1091/mbc.E12-06-0477
Sun S, Heitman J (2016) Running hot and cold: recombination around and within mating-type loci of fungi and other eukaryotes. In: Druzhinina I, Kubicek C (eds) Environmental and microbial relationships. The Mycota: a comprehensive treatise on fungi as experimental systems for basic and applied research, vol IV. Springer, Cham
Szczepaniak J, Cieślik W, Romanowicz A, Musioł R, Krasowska A (2017) Blocking and dislocation of Candida albicans Cdr1p transporter by styrylquinolines. Int J Antimicrob Agents 50:171–176
Takeuchi C, Nagatani K, Sato Y (2013) Chitosan and a fungal elicitor inhibit tracheary element differentiation and promote accumulation of stress lignin-like substance in Zinnia elegans xylogenic culture. J Plant Res 126:811–821
Tanaka S, Brefort T, Neidig N, Djamei A, Kahnt J, Vermerris W, Koenig S, Feussner K, Feussner I, Kahmann R (2014) A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. eLife 3:e01355
Thammahong A, Puttikamonkul S, Perfect JR, Brennan RG, Cramer RA (2017) Central role of the trehalose biosynthesis pathway in the pathogenesis of human fungal infections: opportunities and challenges for therapeutic development. Microbiol Mol Biol Rev 81(2):00053–00016. https://doi.org/10.1128/MMBR.00053-16
Thomas E, Sircaik S, Roman E, Brunel JM, Johri AK, Pla J, Panwar SL (2015) The activity of RTA2, a downstream effector of the calcine-urin pathway, is required during tunicamycin-induced ER stress response in Candida albicans. FEMS Yeast Res 15:29
Thompson-Jaeger S, François J, Gaughran JP, Tatchell K (1991) Deletion of SNF1 affects the nutrient response of yeast and resembles mutations which activate the adenylate cyclase pathway. Genetics 129(3):697–706
Toenjes KA, Stark BC, Brooks KM, Johnson DI (2009) Inhibitors of cellular signalling are cytotoxic or block the budded-to-hyphal transition in the pathogenic yeast Candida albicans. J Med Microbiol 58:779–790
Toenjes KA, Munsee SM, Ibrahim AS, Jeffrey R, Edwards JE, Johnson DI (2005) Small-molecule inhibitors of the budded-to-hyphal-form transition in the pathogenic yeast Candida albicans. Antimicrob Agents Chemother 49:963–972
Toruno TY, Stergiopoulos I, Coaker G (2016) Plant-pathogen effectors: cellular probes interfering with plant defenses in spatial and temporal manners. Annu Rev Phytopathol 54:419–441
Tsukada K, Ishizaka M, Fujisawa Y, Iwasaki Y, Yamaguchi T, Minami E, Shibuya N (2002) Rice receptor for chitin oligosaccharide elicitor does not couple to heterotrimeric G-protein: elicitor responses of suspension cultured rice cells from Daikoku dwarf (d1) mutants lacking a functional G-protein α-subunit. Physiol Plant 116(3):373–382
Tyler BM (2002) Molecular basis of recognotion between Phytophthora pathogens and their hosts. Annu Rev. Phytopathol 40:137–167.
Tzung KW, Williams RM, Scherer S, Federspiel N, Jones T, Hansen N, Bivolarevic V, Huizar L, Komp C, Surzycki R, Tamse R, Davis RW, Agabian N (2001) Genomic evidence for a complete sexual cycle in Candida albicans. PNAS 98(6):3249–3253
Varden FA, De la Concepcion JC, Maidment JHR, Banfield MJ (2017) Taking the stage: effectors in the spotlight. Curr Opin Plant Biol 38:25–33
Vasudev K, Choi YH, Norman R, Kim RB, Schwarz UI (2017) Genetic determinants of clozapine-induced metabolic side effects. Can J Psychiatry 62(2):138–149. https://doi.org/10.1177/0706743716670128
Vauquelin G, Von Mentzer B (2007) G protein-coupled receptors: molecular pharmacology from academic concept to pharmaceutical research. Wiley, Chichester
Videmann B, Mazallon M, Tep J, Lecoeur S (2008) Metabolism and transfer of the mycotoxin zearalenone in human intestinal Caco-2 cells. Food Chem Toxicol 46(10):3279–3286
Vila T, Ishida K, Seabra SH, Rozental S (2016) Miltefosine inhibits Candida albicans and non-albicans Candida spp. biofilms and impairs the dispersion of infectious cells. Int J Antimicrob Agents 48:512–520
Vila T, Romo JA, Pierce CG, McHardy SF, Saville SP, Lopez-Ribot JL (2017) Targeting Candida albicans filamentation for antifungal drug development. Virulence 8(2):150–158
Wahl R, Wippel K, Goos S, Kämper J, Sauer N (2010) A novel high-affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis. PLoS Biol 8:e1000303. https://doi.org/10.1371/journal.pbio.1000303
Wang X, Stone DE (2017) Mating yeast cells concentrate the pheromone receptor and its G protein as polarized crescents at the default polarity site that then track to the eventual chemotropic site. FASEB J 31(1 suppl):930–932
Whaley SG, Tsao S, Weber S, Zhang Q, Barker KS, Raymond M, Rogers PD (2016) The RTA3 gene, encoding a putative lipid translocase, influences the susceptibility of Candida albicans to fluconazole. Antimicrob Agents Chemother 60:6060–6066
Whiteway M, Hougan L, Dignard D, Thomas DY, Bell L, Saari GC, Grant FJ, O’Hara P, MacKay VL (1989) The STE4 and STE18 genes of yeast encode potential beta and gamma subunits of the mating factor receptor-coupled G protein. Cell 56:467–477
Wingard JR, Merz WG, Rinaldi MG, Johnson TR, Karp JE, Saral R (1991) Increase in Candida krusei infection among patients with bone marrow transplantation and neutropenia treated prophylactically with fluconazole. N Engl J Med 325:1274–1277. https://doi.org/10.1056/NEJM199110313251803
Wu JC, Lai CS, Tsai ML, Ho CT, Wang YJ, Pan MH (2017) Chemopreventive effect of natural dietary compounds on xenobiotic-induced toxicity. J Food Drug Anal 25:176–186
Xue Y, Batlle M, Hirsch JP (1998) GPR1 encodes a putative G protein-coupled receptor that associates with the Gpa2p Galpha subunit and functions in a Ras-independent pathway. EMBO J 17:1996–2007
Xue C, Bahn YS, Cox GM, Heitman J (2006) G protein-coupled receptor Gpr4 senses amino acids and activates the cAMP-PKA pathway in Cryptococcus neoformans. Mol Biol Cell 17:667–679
Xue C, Hsueh YP, Heitman J (2008) Magnificent seven: roles of G protein-coupled receptors in extracellular sensing in fungi. FEMS Microbiol Rev 32:1010–1032
Yang X, Deng F, Ramonell KM (2012) Receptor-like kinases and receptor-like proteins: keys to pathogen recognition and defense signaling in plant innate immunity. Front Biol 7:155–166. https://doi.org/10.1007/s11515-011-1185-8
Yu JH (2006) Heterotrimeric G protein signaling and RGSs in Aspergillus nidulans. J Microbiol 44(2):145–154
Yu X, Feng B, He P, Shan L (2017) From chaos to harmony: responses and signaling upon microbial pattern recognition. Annu Rev Phytopathol 55:109–137
Yu RC, Pesce CG, Colman-Lerner A, Lok L, Pincus D, Serra E, Holl M, Benjamin K, Gordon A, Brent R (2008) Fus3 generates negative feedback that improves information transmission in yeast pheromone response. Nature (Lond) 456(7223):755–761. https://doi.org/10.1038/nature07513
Yun C, Tamaki H, Nakayama R, Yamamoto K, Kumagai H (1997) G-protein coupled receptor from yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun 240(2):287–292
Zhang Y, Butelli E, de Stefano R, Schoonbeek HJ, Magusin A, Pagliarani C, Wellner N, Hill L, Orzaez D, Granell A, Jones JD, Martin C (2013) Anthocyanins double the shelf life of tomatoes by delaying overripening and reducing susceptibility to gray mold. Curr Biol 23:1094–1100. https://doi.org/10.1016/j.cub.2013.04.072
Zhai X, Jia M, Chen LM, Zheng C, Rahman K, Han T, Quin L (2017) The regulatory mechanism of fungal elicitor-induced secondary metabolite biosynthesis in medical plants. Journal Critical Reviews in Microbiology 43:238–261
Zhao X, Mehrabi R, Xu J (2007) Mitogen-activated protein kinase pathways and fungal pathogenesis. Eukaryot Cell 6(10):1701–1714
Acknowledgments
The author expresses his sincere gratitude to Prof. Kenneth H. Wolfe for his permission to use his figures concerning the cassette model of mating-type switching (the Genetics Society of America is the source of such figures), and to Prof. Henry Jakubowski for his permission to use his figure about antagonist ligands and competitive and noncompetent drugs and ligand. Also, to my colleague Muhammad A. Youssef, for providing other figures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
El-Defrawy, M.M.H., Hesham, A.EL. (2020). G-protein-coupled Receptors in Fungi. In: Hesham, AL., Upadhyay, R., Sharma, G., Manoharachary, C., Gupta, V. (eds) Fungal Biotechnology and Bioengineering. Fungal Biology. Springer, Cham. https://doi.org/10.1007/978-3-030-41870-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-41870-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-41869-4
Online ISBN: 978-3-030-41870-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)