
Abstract
This review summarizes key developments in the heparanase field obtained 20 years prior to cloning of the HPSE gene and nearly 20 years after its cloning. Of the numerous publications and review articles focusing on heparanase, we have selected those that best reflect the progression in the field as well as those we regard important accomplishments with preference to studies performed by scientists and groups that contributed to this book. Apart from a general ‘introduction’ and ‘concluding remarks’, the abstracts of these studies are presented essentially as published along the years. We apologize for not being objective and not being able to include some of the most relevant abstracts and references, due to space limitation. Heparanase research can be divided into two eras. The first, initiated around 1975, dealt with identifying the enzyme, establishing the relevant assay systems and investigating its biological activities and significance in cancer and other pathologies. Studies performed during the first area are briefly introduced in a layman style followed by the relevant abstracts presented chronologically, essentially as appears in PubMed. The second era started in 1999 when the heparanase gene was independently cloned by 4 research groups [1–4]. As expected, cloning of the heparanase gene boosted heparanase research by virtue of the readily available recombinant enzyme, molecular probes, and anti-heparanase antibodies. Studies performed during the second area are briefly introduced followed by selected abstracts of key findings, arranged according to specific topics.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Historical Introduction
Initially, several apparently different heparanase enzymes and activities have been described. However, it soon became surprisingly apparent and now well documented that there is a single unique gene encoding for heparanase and that the enzyme is the sole heparan sulfate (HS) degrading endoglycosidase expressed by normal and malignant cells and playing a role not only in cancer metastasis and angiogenesis but also in inflammatory and autoimmune conditions. Although only one confirmed endoglycosidase (heparanase) had been cloned and characterized, there are nine different exoglycosidases that are involved in the ordered disassembly of HS in the lysosomes of all cells [5]. Genetic deficiencies in these exoenzymes result in a range of lysosomal storage disorders [5]. It has been generally assumed that heparanase is the first enzyme involved in the intracellular turnover of HS [5].
Enzymatic activity capable of cleaving glucuronidic linkages and converting macromolecular heparin to physiologically active fragments was first identified by Ogren and Lindahl in mastocytoma cells [6]. Yet, developments in the field were slow due to conflicting reports regarding the physicochemical properties and substrate specificity of the enzyme. Heparanase activity has been attributed to molecules ranging in molecular mass from 8 to 134 kDa [7], [8], [9]. It was claimed, for example, that based on their substrate specificities there are at least three types of endo-beta-D-glucuronidases and that the melanoma heparanase (Mr approximately 96,000) differ from the platelet and mastocytoma enzymes [10]. There have also been claims that the enzyme is a heat shock protein [11] or related to the CXC chemokine, connective-tissue-activating peptide III (CTAP) [8]. Other studies distinguished between the secreted and intracellular enzymes and suggested that there may be a family of heparanase proteins with different substrate specificities and potential functions [12]. Studies performed by Bame et al. focused on intracellular heparanase(s) and their involvement in normal catabolism of HS. It was claimed that inside cells, these enzymes are important for the normal catabolism of HS proteoglycans (HSPG) , generating glycosaminoglycan fragments that are then transported to lysosomes and completely degraded. Characterization of the short glycosaminoglycans produced in Chinese hamster ovary (CHO) cells suggested that multiple heparanases are necessary for the formation of the short HS chains [13]. Based on their ability to bind ion-exchange resins and their elution from gel-filtration columns, four separate heparanase activities were partially purified. All four activities cleave free glycosaminoglycans over a broad pH range of 3.5–6.5, suggesting that they act in the endosomal/lysosomal pathway. It was further suggested that the formation of short HSPG inside CHO cells may be a result of the concerted action of multiple heparanases, and may depend on the proportions of the different enzymes and the environment in which the chains are degraded [13].
-
∗A large part of the lay summary presented below was taken from commentaries written by Drs. Eccles and Finkel [14, 15] in response to the heparanase cloning papers published in 1999 by Vlodavsky et al. [4] and Hulett et al. [1]. These commentaries were found suitable for the current ‘historic review’ by virtue of their accuracy and high relevance to heparanase research performed since then.
Tumor cell invasion and secondary spread through the blood and lymphatics is the hallmark of malignant disease and the greatest impediment to cancer cure. Two of the essential processes required for metastasis are neoangiogenesis and tumor cell invasion of the basement membrane (BM) and extracellular matrix (ECM) . Prior to cloning of the heparanase gene, attention focused on serine and cysteine proteases and matrix metalloproteinases (MMPs). Although metastasizing cancer cells may produce as many as 25 different matrix-digesting metalloproteinases, cloning of the same heparanase gene by different groups [1,2,3,4] indicated that there is only one heparanase so that if its activity can be inhibited, other heparanases shouldn’t be around to cover for it [15]. In addition to the structural proteins (i.e., collagens, laminin, fibronectin, vitronectin) cleaved by MMPs in the BM and ECM , the other chief components are glycosaminoglycans, mainly heparan sulfate proteoglycans (HSPG). HSPG are composed of a protein core covalently linked to HS glycosaminoglycan chains that interact closely with other ECM components. Endoglycosidase activity (heparanase) that degrades the HS side chains of HSPG is normally found mainly in platelets, placental trophoblasts, and leukocytes. Given their abundance in tumor tissues, it appears that the normal physiological functions of proteases and heparanases in embryonic morphogenesis, wound healing, tissue repair and inflammation have been effectively ‘hijacked’ by tumor cells.
Evidence indicates that heparanase not only assists in the breakdown of ECM and BM but also is involved in the regulation and bioavailability of growth factors and cytokines [16]. For example, basic fibroblast growth factor (bFGF) and other heparin-binding growth factors (i.e., HB-EGF, VEGF, HGF) are sequestered by HS, providing a localized, readily accessible depot, protected from proteolytic degradation, yet available to activate cells and promote angiogenesis after being released by heparanase. It was suggested that the release of tissue-specific growth factors may be involved in organ selectivity of metastasis and the formation of a metastatic niche [17, 18]. Although these phenomena were well-documented, it has taken about 20 years to purify the heparanase protein and clone the respective gene mainly because of instability of the enzyme(s), its low abundance in normal cells and tissues and the lack of a robust, accurate and rapid assay for enzyme activity [14].
Researchers first made the connection between metastasis and heparanase in the mid-1980s. Three groups [Garth Nicolson’s (M. D. Anderson Cancer Center in Houston), Christopher Parish (John Curtin School of Medical Research, Canberra) and Israel Vlodavsky (Hadassah-Hebrew University, Jerusalem)] were following up on the finding that the natural anticoagulant, heparin, inhibits the spread of cancer in animals [15]. The prevailing belief was that heparin worked because it prevented platelets from clotting around cancer cells, an event likely to help the cells lodge into, and ultimately penetrate the vessel wall. But “heparin” is a family of molecules, only some of which inhibit clot formation. The three groups independently showed that it still inhibits metastasis, even when depleted of its anticlotting activity [14]. Only about 15 years later it was demonstrated that heparin inhibits heparanase enzymatic activity through competition with HS on binding to the heparin/HS binding domains (HBD) of heparanase [19].
Both Parish [20] and Vlodavsky [21] had reported that heparanase helps immune cells traverse blood vessel walls on their way to infection sites. The evidence that it might be doing something similar for cancer cells immediately made researchers think about getting better heparanase inhibitors. But getting the pure enzyme proved to be difficult. Not only is heparanase unstable, but the only assay then available was slow and cumbersome [22, 23]. Nevertheless, the Israeli group finally managed to purify heparanase from a human liver cancer cell line and also from human placenta [4], while the Australian group purified it from human platelets [24]. In parallel, Toyoshima and Nakajima [3] reported the purification of a human heparanase from an SV40-transformed embryonic fibroblast cell line, and Kussie et al. [2] isolated and purified the enzyme from human SK-HEP-1 hepatoma cells. After determining peptide amino acid sequences derived from the purified proteins, the researchers then screened EST databases looking for gene sequences that could encode those amino acid sequences. Contrary to expectations that there might be more than one heparanase, the four groups found themselves with the same gene - the only one like it in the databases. Soon after, genomic organization and chromosome localization of the newly identified human heparanase gene was reported [25]. Cloning, expression, and purification of mouse heparanase that is 77% identical to the human enzyme was reported by Miao et al. [26]. The recombinant mouse heparanase protein was purified to homogeneity from cell lysates by a combination of Con-A affinity chromatography, heparin affinity chromatography, and size exclusion chromatography, purification steps that are commonly used nowadays. Experiments confirmed that the newly cloned gene aids the spread of cancer cells. When Vlodavsky and his colleagues introduced the heparanase gene into nonmetastatic mouse melanoma and lymphoma cancer cells, they turned into rampantly malignant cells that colonized the lung and liver when injected into mice [4]. Parish, looking at several different types of rat cancer cells, found that their invasiveness correlates with the activity of their heparanase gene [1]. Conversely, inhibiting the enzyme inhibits cancer metastasis. Parish reported that a previously identified inhibitor of heparanase called PI-88 [27] decreased by 90% the number of lung tumors formed by breast cancer cells injected into rats. It also cut the blood supply of the primary tumors and slowed tumor growth [28]. The encouraging animal results have led Progen (Brisbane, Australia) to test the safety of the inhibitor in healthy volunteers, followed by clinical trials in cancer patients [29].
Notably, studies aimed at determining the role of heparanase in tumor progression relied on the use of heparin-mimicking molecules to inhibit heparanase activity [30,31,32]. Because the reagents used in these studies lacked specificity, the conclusions were somewhat debatable and inconclusive. It took another 12 years to generate heparanase neutralizing monoclonal antibodies (mAb) and demonstrate their ability to inhibit lymphoma tumor growth and dissemination [33]. Notably, the inhibitory activity of the mAbs was lower than that of the heparin mimetics (i.e., PG545) [33], further raising the issue of drug specificity vs. efficiency. Nowadays, heparanase is well established as a cancer drug target, and several inhibitors have progressed to clinical trials [31, 34]. In recent years, heparanase has also been implicated in a range of other diseases, such as diabetes and its complications [35,36,37], kidney disease [38], atherosclerosis [39, 40] and viral infections [41], to name a few, which continues to fuel research into this protein.
Studies performed before cloning of the heparanase gene contributed immensely to our understanding of key features in the biology of the enzyme, its mode of action and involvement in cancer metastasis and inflammation. Abstracts of selected ‘Historical’ studies referred to in the introduction are presented below in chronological order. The purpose of selecting these abstracts is to provide a historic perspective of how heparanase research was developed and progressed with time, rather than to highlight the most prominent and influential papers.
1.1 Key Observations Made Prior to Cloning of the HPSE Gene (Chronological Order)
Cleavage of Macromolecular Heparin by an Enzyme From Mouse Mastocytoma
Heparinase was isolated from a transplantable mouse mastocytoma . The enzyme was shown to degrade macromolecular 35S-labeled, mastocytoma heparin to products similar in size to commercial heparin apparently by nonrandom cleavage of a limited number of glycosidic linkages per molecule. Prolonged incubation times did not result in further degradation of the product. No significant depolymerizing activity was observed with any other glycosaminoglycan tested, including chondroitin sulfate, dermatan sulfate, hyaluronic acid, heparan sulfate, and commercial heparin. The pH optimum for degradation of macromolecular heparin was around pH 5. Analysis of the degradation products showed a major radioactive component which behaved like L-gulonic acid. Since [3H]gulonic acid would be the expected reduction product of a polysaccharide molecule, containing a glucuronic acid residue in terminal position, these results tentatively suggest that the heparinase is an endoglucuronidase. It appears that cleavage occurs in regions more abundant in N-acetylated glucosamine residues than other portions of the molecule [6].
-
∗First paper stating that an endoglucuronidase is responsible for cleavage of heparin by mastocytoma cells. Notably, the presence of such heparin degrading activity was reported by the same authors already in 1971 [42].
A Heparan Sulfate-Degrading Endoglycosidase From Rat Liver Tissue
Incubation of a rat liver lysosomal fraction with [35S]heparan sulfate resulted in degradation of the polymer to oligosaccharides, demonstrating the presence of a heparan sulfate-degrading endoglycosidase . Judging from the size of the oligosaccharides, representing degradation end-products, only a limited number of the glycosidic linkages in the HS molecule would seem to be susceptible to the heparitinase. The pH-dependence of the enzyme (active at pH 5.6; inactive at pH 3.8) was found to differ from that of liver hyaluronidase (active at pH 3.8; inactive at pH 5.6), suggesting that the heparitinase is a previously unknown enzyme [43].
-
∗Early (1975) study on the presence of HS-degrading endoglycosidase in liver tissue.
Purification and Properties of Human Platelet Heparitinase
An endoglycosidase which cleaves heparin and HS was isolated from outdated human platelets. The overall extent of purification of the platelet heparitinase is about 240,000-fold and the overall yield of the enzyme is about 5.6% as compared to the initial freeze-thaw solubilization preparation. The final product is physically homogeneous and exhibits an apparent molecular weight of approximately 134,000. Furthermore, our results indicate that the above enzyme is present within platelet lysosomes. The biologic potency of the endoglycosidase was examined as a function of pH and is maximally active from pH 5.5 to pH 7.5. The substrate specificity of the platelet endoglycosidase was determined by identifying susceptible linkages within the heparin molecule that can be cleaved by the above component. Our studies indicate that this enzyme is only able to hydrolyze glucuronsylglucosamine linkages. Furthermore, investigation of the structure of the disaccharide which lies on the nonreducing end of the cleaved glucuronic acid residue suggests that N-sulfation of the glucosamine moiety or ester sulfation of the adjacent iduronic acid groups are not essential for bond scission [9].
-
∗Early (1982) study on the purification and properties of HS-degrading endoglycosidase in platelets.
Heparan Sulfate Degradation: Relation to Tumor Invasive and Metastatic Properties of Mouse B16 Melanoma Sublines
Mouse B16 melanoma sublines were used to determine the relation between metastatic properties and the ability of the sublines to degrade sulfated glycosaminoglycans present in the ECM of cultured vascular endothelial cells. Highly invasive and metastatic B16 sublines degraded matrix glycosaminoglycans faster than did sublines of lower metastatic potential. The main products of this matrix degradation were HS fragments. Intact B16 cells (or their cell-free homogenates) with a high potential for lung colonization degraded purified HS from bovine lung at higher rates than did B16 cells with poor potential for lung colonization. Analysis of the degradation fragments indicated that B16 cells have HS endoglycosidase. Thus the abilities of B16 melanoma cells to extravasate and successfully colonize the lung may be related to their capacities to degrade HS in the walls of pulmonary blood vessels [44].
-
∗First study (1983) showing that HS endoglycosidase is associated with the metastatic potential of melanoma cells.
Lymphoma Cell-Mediated Degradation of Sulfated Proteoglycans in the Subendothelial Extracellular Matrix: Relationship to Tumor Cell Metastasis
Cloned lines of the low-metastatic T-lymphoma Eb line and its highly metastatic variant ESb line were compared for the ability to degrade proteoglycans in the subendothelial ECM produced by cultured endothelial cells. The ECM was metabolically labeled with Na235SO4, and the tumor cell-mediated release of labeled degradation products was analyzed by gel filtration. More than 90% of the labeled material released upon incubation of ESb cells with the ECM, either when exposed or covered with vascular endothelial cells, was in the form of low-Mr, HS-containing fragments compared to high-Mr nearly intact sulfated proteoglycans released by incubation with the low-metastatic Eb cells. The same high- and low-Mr degradation products were obtained by incubation of the ECM with a serum-free medium conditioned by the low (Eb)- and high (ESb)-metastatic sublines, respectively. The high-Mr proteoglycans released by incubation of the ECM with Eb-conditioned medium was further degraded into low Mr. GAG fragments upon subsequent incubation with ESb-conditioned medium. These fragments were smaller than intact glycosaminoglycan side chains released by treatment of the ECM with papain or alkaline borohydride, suggesting an ESb-specific endoglycosidase activity. The higher ability of the ESb over the Eb cells to solubilize the GAG scaffolding of the sub-endothelial ECM may, among other properties, facilitate their hematogenous dissemination and extravasation [45].
-
∗First study (1983) showing that HS endoglycosidase is associated with the metastatic potential of lymphoma cells.
Activated T Lymphocytes Produce a Matrix-Degrading Heparan Sulfate Endoglycosidase
Circulating activated T lymphocytes specifically autosensitized to the basic protein of myelin (BP) penetrate blood vessels, accumulate in the nervous system and cause experimental autoimmune encephalomyelitis (EAE). To investigate how effector T cells reach targets outside the walls of blood vessels, we have studied the interaction of anti-BP effector T lymphocytes with the basement membrane-like ECM produced by vascular endothelial cells. It was found that activated but not resting T lymphocytes produce an endoglycosidase (heparanase) capable of degrading HS side chains of the proteoglycan scaffold of the ECM. Moreover, the anti-BP T lymphocytes respond to BP presented by ECM by markedly enhanced elaboration of the endoglycosidase. These results suggest that tissue-specific antigens on blood vessel walls could direct lymphocyte homing by activating enzymes that facilitate penetration of the subendothelial basal lamina. They also suggest that effector T lymphocytes can recognize antigen which is not associated with a major histocompatibility complex signal [21].
-
∗First study (1984) describing the expression and function of heparanase in activated T lymphocytes.
Sequential Degradation of Heparan Sulfate in the Subendothelial Extracellular Matrix by Highly Metastatic Tumor Cells
Both a protease and heparanase are synergistically involved in ESb-mediated degradation of ECM-bound HS and one enzyme produces a more accessible substrate for the next enzyme [46]. Briefly, it was found that degradation of HS in the ECM was markedly enhanced in the presence of plasminogen and inhibited by aprotinin, suggesting a role for plasminogen activator (PA) in sequential degradation of the ECM-HS [47]. Moreover, subsequent studies revealed that PA activity is residing in the ECM itself [48]. Thus, incubation of plasminogen on ECM resulted in plasmin generation and heating the ECM inactivated its ability to generate plasmin upon incubation with plasminogen. It was concluded that a proteolytic activity expressed by the tumor cells and/or residing in the tumor microenvironment (i.e., ECM) participate synergistically in sequential degradation of the EM’ HS. This sequential cleavage is characteristic of degradation of a multimolecular structure such as the subendothelial ECM and hence cannot be detected in studies with purified HS as a substrate . [46, 47].
-
∗First study showing that a proteolytic activity residing in the ECM is critical for subsequent cleavage of HS by heparanase.
Endothelial Cell-Derived Basic Fibroblast Growth Factor: Synthesis and Deposition into the Subendothelial Extracellular Matrix
The endothelium can store growth factors capable of autocrine growth promotion in two ways: by sequestering growth factor within the cell and by incorporating it into the underlying ECM. We hypothesized that release of ECM-bound basic FGF could stimulate the autocrine proliferation of adjacent endothelial cells. Moreover, tumor angiogenesis may in part be mediated by the action of tumor-derived HS-degrading enzymes, which would release basic FGF stored in capillary basement membrane. We further proposed that release of intracellular and extracellular stores of basic FGF may be a mechanism for the rapid mobilization of angiogenesis factors. This hypothesis was strengthened by the in situ experiments described below [49].
A Heparin-Binding Angiogenic Protein - Basic Fibroblast Growth Factor – Is Stored Within Basement Membrane
The basement membranes of bovine cornea were found to contain an angiogenic endothelial cell mitogen, basic fibroblast growth factor (FGF) that was bound to HS and released from the cornea by treatment with heparin, a hexasaccharide heparin fragment, HS, or heparanase. These findings indicate that basement membranes of the cornea may serve as physiologic storage depots for an angiogenic molecule. Abnormal release of this growth factor could be responsible for corneal neovascularization in a variety of ocular diseases. It was suggested that sequestration of heparin-binding proangiogenic mitogens in the basement membrane and their liberation by heparanase might be a general mechanism for regulating their accessibility to the vascular endothelium and that heparanase plays a key role in fulfilling this function [50].
-
∗By now, the concept of ECM as a reservoir for bioactive molecules is well recognized, providing a strong basis to the current appreciation of the tumor microenvironment and its significance in supporting tumor growth and metastasis. Additional studies on the pro-angiogenic activity of heparanase and the contribution of heparanase residing in the tumor microenvironment are described later under ‘key studies performed after cloning of the HPSE gene [50].
-
∗The above two manuscripts are the first to demonstrate that heparanase functions in the liberation and mobilization of HS-bound growth factors, an activity that underlies the high significance of this enzyme in promoting tumor angiogenesis and growth, among other biological effects .
Inhibition of Heparanase-Mediated Degradation of Extracellular Matrix Heparan Sulfate by Non-Anticoagulant Heparin Species
The present study examined the heparanase inhibitory effect of nonanticoagulant species of heparin that might be of potential use in preventing heparanase mediated extravasation of blood-borne cells. For this purpose, we prepared various species of low-sulfated or LMW heparins, all of which exhibited less than 7% of the anticoagulant activity of native heparin. N-sulfate groups of heparin are necessary for its heparanase inhibitory activity but can be substituted by an acetyl group provided that the O-sulfate groups are retained. O-sulfate groups could be removed provided that the N positions were resulfated. Total desulfation of heparin abolished its heparanase inhibitory activity. Heparan sulfate was a 25-fold less potent heparanase inhibitor than native heparin. Efficiency of LMW heparins to inhibit degradation of HS in ECM decreased with their main molecular size, and a synthetic pentasaccharide, representing the binding site to antithrombin III, was devoid of inhibitory activity. Similar results were obtained with heparanase activities released from platelets, neutrophils, and lymphoma cells. We propose that heparanase inhibiting nonanticoagulant heparins may interfere with dissemination of blood borne tumor cells and development of experimental autoimmune diseases [51].
Evidence That Sulphated Polysaccharides Inhibit Tumour Metastasis by Blocking Tumour-Cell-Derived Heparanases
Rat mammary adenocarcinoma 13,762 MAT cells produce a HS-specific glycosidase (heparanase) that degrades the HS side-chains of the ECM. The action of this enzyme, rather than that of other ECM-solubilizing enzymes, was inhibited by 5 antimetastatic sulphated polysaccharides but not by 4 polysaccharides that failed to inhibit metastasis. Additional experiments indicated that the anti-coagulant activity of the polysaccharides probably plays a minor role in their anti-metastatic effects since heparin, almost completely depleted (98–99.5%) of heparin molecules with anti-coagulant activity by passage over an anti-thrombin III column, retained its ability to inhibit 13,762 MAT heparanases and was almost as effective as unfractionated heparin at inhibiting tumour-cell metastasis. Collectively, these data suggest that sulphated polysaccharides inhibit the metastasis of 13,762 MAT cells by inhibiting tumour-cell-derived heparanases involved in the penetration of the vascular endothelium and its underlying basement membrane by tumour cells. These results paved the way for the development and clinical testing of PI-88 (= phosphomannopentaose sulfate = Muparfostat ) [52].
-
∗The above two studies demonstrate that both heparanase enzymatic activity and experimental metastasis are inhibited by non-anticoagulant species of heparin and other sulfated polysaccharides.
Inhibition of Allergic Encephalomyelitis in Rats by Treatment With Sulfated Polysaccharides
A number of sulfated polysaccharides were tested for their ability to inhibit passively induced experimental allergic encephalomyelitis (EAE) in rats. Heparin and fucoidan both completely inhibited passive EAE even when treatment was begun 3 days after transfer of cells. Pentosan sulfate was partially inhibitory whereas chondroitin-4-sulfate had no effect. Inhibition was not merely due to killing of the cells since active sensitization 14 days after cell transfer resulted in an early onset of disease indicating the persistence of transferred cells as memory cells. Although all the inhibitory polysaccharides are anticoagulants, it would appear that this function alone is not the reason for inhibition since a heparin preparation devoid of anticoagulant activity also partially inhibited EAE. Actively induced EAE was also significantly delayed by treatment with heparin. The results are discussed in terms of the polysaccharides inhibiting the enzymatic dependent movement of lymphocytes across central nervous system vascular endothelium [53].
Suppression of Experimental Autoimmune Diseases and Prolongation of Allograft Survival by Treatment of Animals With Low Doses of Heparins
Heparanase-inhibiting , nonanticoagulant species of heparin markedly reduced the incidence of lung metastasis in experimental animals. Low doses of these species of heparin also significantly impaired the traffic of T lymphocytes and suppressed cellular immune reactivity and experimental autoimmune diseases (allograft rejection, adjuvant arthritis, experimental autoimmune encephalomyelitis). The ability of chemically modified heparins to inhibit these immune reactions was associated with their ability to inhibit expression of T lymphocyte heparanase. There was no relationship to anticoagulant activity. Thus heparins devoid of anticoagulant activity can be effective in regulating immune reactions when used at appropriate doses [54].
-
∗The above two studies demonstrate that both heparanase and experimental autoimmune diseases are inhibited by non-anticoagulant species of heparin and other sulfated polysaccharides.
Molecular Behavior Adapts to Context: Heparanase Functions as an Extracellular Matrix-Degrading Enzyme or as a T Cell Adhesion Molecule, Depending on the Local pH
Migration of lymphocytes into inflammatory sites requires their adhesion to the vascular endothelium and subendothelial ECM. Depending on the local pH, heparanase can function either as an enzyme or as an adhesion molecule. At relatively acidified pH conditions, heparanase performs as an enzyme, degrading HS. In contrast, at the hydrogen ion concentration of a quiescent tissue, heparanase binds specifically to HS molecules without degrading them, and thereby anchors CD4+ human T lymphocytes. Thus, the local state of a tissue can regulate the activities of heparanase and can determine whether the molecule will function as an enzyme or as a proadhesive molecule [55].
-
∗Early article demonstrating that heparanase may function as cell adhesive molecule.
CXC Chemokines Connective Tissue Activating Peptide-III and Neutrophil Activating Peptide-2 Are Heparin/Heparan Sulfate-Degrading Enzymes
In this study, purification of a HS-degrading enzyme from human platelets led to the discovery that the enzymatic activity resides in at least two members of the platelet basic protein (PBP) family known as connective tissue activating peptide-III (CTAP-III) and neutrophil activating peptide-2. PBP and its N-truncated derivatives, CTAP-III and neutrophil activating peptide-2 are CXC chemokines, a group of molecules involved in inflammation and wound healing. SDS-PAGE analysis of the purified heparanase resulted in a single broad band at 8–10 kDa, the known molecular weight of PBP and its truncated derivatives. Gel filtration chromatography of heparanase resulted in peaks of activity corresponding to monomers, dimers, and tetramers. N-terminal sequence analysis of the same preparation indicated that only PBP and truncated derivatives were present, and commercial CTAP-III from three suppliers had heparanase activity. Antisera produced in animals immunized with a C-terminal synthetic peptide of PBP inhibited heparanase activity by 95%, compared with activity of the purified enzyme in the presence of the preimmune sera. The enzyme was determined to be an endoglucosaminidase, and it degraded both heparin and HS with optimal activity at pH 5.8. Sequence analysis showed that the two peaks contained identical protein, suggesting that a post-translational modification activates the enzyme [8].
Partial Purification of Heparanase Activities in Chinese Hamster Ovary Cells: Evidence for Multiple Intracellular Heparanases
Our studies characterizing the short glycosaminoglycans produced in Chinese hamster ovary (CHO) cells suggested that multiple heparanases are necessary for the formation of the short HS chains. We examined whether this is the case by purifying heparanase activity from CHO cell homogenates. Based on their ability to bind ion-exchange resins and their elution from gel-filtration columns, four separate heparanase activities were partially purified. All four activities cleave free glycosaminoglycans over a broad pH range (3.5–6.5), suggesting that they act in the endosomal/lysosomal pathway. The sizes of the short HS chains generated by the partially purified heparanases ranged from 6 to 9 kDa. Interestingly, all four enzymes generate short glycosaminoglycans with a sulfate-rich, modified domain at the non-reducing end of the newly formed chain. Our findings suggest that the formation of short HS glycosaminoglycans inside CHO cells may be a result of the concerted action of multiple heparanases, and may depend on the proportions of the different enzymes and the environment in which the chains are degraded [13].
-
∗As demonstrated in the above two studies, there were several serious attempts to purify the heparanase enzyme, yet subsequent studies failed to confirm the results.
2 Heparanase Gene Cloning
∗Mammalian Heparanase: Gene Cloning, Expression and Function in Tumor Progression and Metastasis
We have purified a 50-kDa heparanase from human hepatoma and placenta, and now report cloning of the gene encoding this enzyme. Expression of the cloned cDNA in insect and mammalian cells yielded 65-kDa and 50-kDa recombinant heparanase proteins. The 50-kDa enzyme represents an N-terminally processed enzyme, at least 100-fold more active than the 65-kDa form. The heparanase mRNA and protein are preferentially expressed in metastatic cell lines and specimens of human breast, colon and liver carcinomas. Low metastatic murine T-lymphoma and melanoma cells transfected with the heparanase cDNA acquired a highly metastatic phenotype in vivo, reflected by a massive liver and lung colonization. This represents the first cloned mammalian heparanase and provides direct evidence for its role in tumor metastasis. Cloning of the heparanase gene enables the development of specific molecular probes for early detection and treatment of cancer metastasis and autoimmune disorders [4].
∗Cloning of Mammalian Heparanase, an Important Enzyme in Tumor Invasion and Metastasis
We report the cDNA sequence of the human platelet enzyme, which encodes a unique protein of 543 amino acids, and the identification of highly homologous sequences in activated mouse T cells and in a highly metastatic rat adenocarcinoma. Furthermore, the expression of heparanase mRNA in rat tumor cells correlates with their metastatic potential. Exhaustive studies have shown only one heparanase sequence, consistent with the idea that this enzyme is the dominant endoglucuronidase in mammalian tissues [1].
-
∗The above two papers were published side by side (Nat Med. 1999; 5:793–809) preceded by a commentary entitled: Heparanase: Breaking down barriers in tumors. Cloning and functional characterization of the long sought-after heparanase opens a new chapter in the understanding and potential manipulation of metastasis and inflammatory processes [14].
Cloning and Functional Expression of a Human Heparanase Gene
We have cloned a gene (HSE1) from a human placental cDNA library that encodes a novel protein exhibiting heparanase activity. The cDNA was identified through peptide sequences derived from purified heparanase isolated from human SK-HEP-1 hepatoma cells. HSE1 contains an open reading frame encoding a predicted polypeptide of 543 amino acids and possesses a putative signal sequence at its amino terminus. Northern blot analysis suggested strong expression of HSE1 in placenta and spleen. Transient transfection of HSE1 in COS7 cells resulted in the expression of a protein with an apparent molecular mass of 67–72 kDa. HSE1 protein was detectable in conditioned media but was also associated with the membrane fraction following cell lysis. The HSE1 gene product was shown to exhibit heparanase activity by specifically cleaving a labeled heparan sulfate substrate in a similar manner as purified native protein [2].
Human Heparanase: Purification, Characterization, Cloning, and Expression
We report the purification of a human heparanase from an SV40-transformed embryonic fibroblast cell line by four sequential column chromatographies. The enzyme was purified to homogeneity, yielding a peptide with an apparent molecular mass of 50 kDa when analyzed by SDS-polyacrylamide gel electrophoresis. Using the amino acid sequences of the N-terminal and internal heparanase peptides, a cDNA coding for human heparanase was cloned. NIH3T3 and COS-7 cells stably transfected with pBK-CMV expression vectors containing the heparanase cDNA showed high heparanase activitiy. The homology search revealed that no homologous protein had been reported [3].
Cloning, Expression, and Purification of Mouse Heparanase
A full-length heparanase gene was cloned from a mouse embryo cDNA library and determined to encode a protein of 535 amino acids that is 77% identical to human heparanase. The full-length mouse gene was stably expressed in NS0 myeloma cells. The recombinant mouse heparanase protein was purified to homogeneity from cell lysates by a combination of Con-A affinity chromatography, heparin affinity chromatography, and size exclusion chromatography. The purified protein consisted of a non-covalent heterodimer of 50- and 8-kDa polypeptides, similar to the human homolog. The protein was enzymatically active in assays using radiolabeled ECM and HS as substrates. The maximum heparanase activity was observed at acidic conditions; however, significant activity was also detected at neutral pH. The enzymatic activity of mouse heparanase was blocked by known heparanase inhibitors [26] (see Gaskin et al., Chap. 7 in this volume, for more information about heparanase gene cloning).
3 Studies Performed Following Cloning of the HPSE Gene
3.1 Introductory Notes
Cloning of the heparanase gene boosted heparanase research thanks to the readily available recombinant enzyme, molecular probes and anti-heparanase antibodies. Notes on early developments obtained soon after the cloning of the HPSE gene are presented below, followed by selected abstracts of key findings, arranged according to specific topics. Of the numerous publications focusing on heparanase, we have selected those we regard as important contributions to the heparanase field with preference to studies performed by scientists and groups that contributed to this book.
-
∗The ‘metastasis’ paragraph presented below was taken from a commentary written by Drs. Nakajima and Boyd [56] in response to the heparanase gene silencing paper published in JNCI by Edovitsky et al. [57]. This commentary was found suitable for this ‘historic review’ due to its high relevance to the current status of heparanase research and the related questions that were raised back in 2004.
Metastasis
Subsequent to the simultaneous cloning of the cDNA-encoding heparanase, Goldshmidt et al. [58] found that overexpression of the cDNA-encoding heparanase conferred a metastatic phenotype in lymphoma cells. Nevertheless, it was argued that the ability of increased heparanase levels to induce a metastatic phenotype does not necessarily imply that tumors make use of heparanase to drive tumor dissemination. In subsequent studies, Edovitsky et al. [57] have attempted to address this shortcoming by using ribozyme and small interfering RNA (siRNA) technology to knock down the levels of endogenous heparanase. The authors showed that, in models of experimental and spontaneous metastases, these strategies attenuated the ability of diverse tumor cells, including melanoma, mammary adenocarcinoma, lymphoma, and glioma cells, to invade in vitro and to colonize distant sites including the liver and lungs. This study [57] and an earlier study [59] performed by Uno et al. provided strong support for a role of heparanase in the metastatic process. Nevertheless, caution was taken in predicting clinical efficacy given that tumor cells have a remarkable system of redundant mechanisms that can efficiently overcome the targeting of single molecules [60]. This redundancy is one of the contributing mechanisms underlying the lack of clinical benefit seen with metalloproteinase inhibitors in cancer patients [61]. Taking into account that only one HS-degrading endoglycosidase was identified, redundancy is not considered a problem in targeting heparanase as compared to metalloproteinases.
An equally important issue raised by Boyed and Nakajima [56] relating to the utility of anti-heparanase and other anti-metastatic therapies in cancer treatment concerns the fact that, at the time of presentation, the majority of patients already have disseminated disease. Consequently, treatment of such patients with anti-heparanase regimens might be akin to closing the barn door after the horse has bolted [56]. Thus, how could a knock-out punch against heparanase be useful in the treatment of cancer patients? There are at least two options. First, with increasing public awareness, and as cancer screenings become more prevalent in the general population, the number of patients diagnosed with early-stage disease should increase. By definition, such tumors are still localized and are therefore more amenable to therapy with anti-metastasis agents. Second, considering the proangiogenic effects of heparanase first documented by Edovitsky et al. [62] and Elkin et al. [63], anti-heparanase drugs may have a static effect on both the primary tumor and distant tumor lesions by preventing the establishment of tumor vasculature necessary for tumor growth beyond 1 mm3. These considerations made a compelling case for the role of heparanase in tumor progression. The extent to which this information can be exploited in novel therapies depends on the development of specific inhibitors that target heparanase and the blockade of redundant mechanisms that compensate for the loss of heparanase in cancer. This statement was rightly written (2004) in an editorial to the gene silencing paper [56] and is relevant nowadays as well.
Gene Regulation
de Mestre et al. reported the identification of the serum-inducible zinc finger transcription factor human early growth response gene 1 (EGR1), as a key regulator of inducible HPSE transcription in T lymphocytes [64] and cancer cells [65]. EGR1 is a nuclear phospho-protein that is rapidly induced in response to a variety of extracellular and environmental signals (including growth factors, cytokines, vascular injury, and hypoxia) [66, 67]. Studies using knockdown strategies have confirmed that EGR1 binds the HPSE promoter in vivo and plays a central role in tumor angiogenesis, growth, and metastasis in breast, bladder, colon and prostate adenocarcinomas [65, 68, 69], supporting a central role for EGR1 in regulating HPSE transcription in tumor cells (Gaskin et al., Chap. 7 in this volume).
Elkin et al. identified putative estrogen response elements in the heparanase promoter and demonstrated their functionality applying a luciferase reporter gene driven by the heparanase promoter [70]. Physical association between estrogen receptor (ER) and the heparanase promoter was confirmed by ChIP analysis. ChIP analysis, also revealed that wild type p53 inhibits transcription of the heparanase gene by direct binding to its promoter, while mutated, tumor-derived variants of p53 lose this inhibitory ability and in some cases even up regulate heparanase gene expression [71]. Examining a series of tumor-derived cell lines, we have found that cells which exhibit heparanase activity also harbor at least one unmethylated allele [72], while cell lines which exhibit no heparanase expression or activity were found to harbor fully methylated alleles. Treating these cells with demethylating agents such as 5-azacytidine restored heparanase activity accompanied by augmented metastatic capacity in vivo [69, 72]. Cathepsin L plays a critical role in the processing and conversion of latent heparanase into its active form [73]. Interestingly, promoter methylation [74] and EGR family members are also involved in cathepsin L activation [75], suggesting that heparanase and cathepsin L share some regulatory aspects. Applying the RIP1-Tag2 tumor model, Joyce et al. demonstrated that while cathepsin L expression was restricted to tumor cells, the majority of heparanase appeared to originate from infiltrating immune cells [76]. Thus, pro-heparanase secreted from one cell type (inflammatory cells) can be activated by cathepsin L secreted by another compartment (cancer cells) in cooperation that drives tumor development [18]. Notably, regulation of both heparanase and cathepsin L does not rely solely on gene transcription but rather involves complex regulatory mechanisms. Important regulatory elements of cathepsin L were identified in 5′, and 3′ untranslated regions (UTR) of the gene [77]. Likewise, Arvatz et al. revealed post-transcriptional regulation of heparanase gene expression by a 3′ AU-rich element in the 3′UTR of the gene [78].
3.2 Key Observations Made After Cloning of the HPSE Gene
(Abstracts of papers arranged according to specific topics)
3.2.1 Structural Aspects
Processing of the Human Heparanase Precursor and Evidence That the Active Enzyme Is a Heterodimer
Human platelet heparanase has been purified to homogeneity and shown to consist of two, non-covalently associated polypeptide chains of molecular masses 50 and 8 kDa. Protein sequencing provided the basis for determination of the full-length cDNA for this novel protein. Based upon this information and results from protein analysis and mass spectrometry, we propose a scheme to define the structural organization of heparanase in relation to its precursor forms, proheparanase and pre-proheparanase. The 8- and 50-kDa chains which make up the active enzyme reside, respectively, at the NH (2)- and COOH-terminal regions of the inactive precursor, proheparanase. The heparanase heterodimer is produced by excision and loss of an internal linking segment. This paper is the first to suggest that human heparanase is a two-chain enzyme [79].
-
∗Among the earliest studies demonstrating that the active enzyme is a heterodimer.
Identification of Active-Site Residues of the Pro-Metastatic Endoglycosidase Heparanase
Using PSI-BLAST and PHI-BLAST searches of sequence databases, similarities were identified between heparanase and members of several of the glycosyl hydrolase families from glycosyl hydrolase clan A (GH-A), including strong local identities to regions containing the critical active-site catalytic proton donor and nucleophile residues that are conserved in this clan of enzymes. Furthermore, secondary structure predictions suggested that heparanase is likely to contain an (alpha/beta) (8) TIM-barrel fold, which is common to the GH-A families. Based on sequence alignments with a number of glycosyl hydrolases from GH-A, Glu(225) and Glu(343) of human heparanase were identified as the likely proton donor and nucleophile residues, respectively. The substitution of these residues with alanine and the subsequent expression of the mutant heparanases in COS-7 cells demonstrated that the HS-degrading capacity of both was abolished. In contrast, the alanine substitution of two other glutamic acid residues (Glu(378) and Glu(396)), both predicted to be outside the active site, did not affect heparanase activity. These data suggest that heparanase is a member of the clan A glycosyl hydrolases and has a common catalytic mechanism that involves two conserved acidic residues, a putative proton donor at Glu(225) and a nucleophile at Glu(343) [80].
-
∗First paper showing that heparanase is a member of the clan A glycosyl hydrolases and has a common catalytic mechanism that involves a putative proton donor at Glu(225) and a nucleophile at Glu(343).
Biochemical Characterization of the Active Heterodimer Form of Human Heparanase (Hpa1) Protein Expressed in Insect Cells
Hpa1 protein is initially synthesized as an inactive 65 kDa proenzyme that is then believed to be subsequently activated by proteolytic cleavage to generate an active heterodimer of 8 and 50 kDa polypeptides. By analysis of a series of Hpa1 deletion proteins we confirm that the 8 kDa subunit is essential for enzyme activity. We present here for the first time an insect cell expression system used for the generation of large amounts of recombinant protein of high specific activity. Individual subunits were cloned into baculoviral secretory vectors and co-expressed in insect cells. Active secreted heterodimer protein was recovered from the medium and isolated by a one-step heparin-Sepharose chromatography procedure to give protein of >90% purity. The recombinant enzyme behaved similarly to the native protein with respect to the size of HS fragments liberated on digestion, substrate cleavage specificity and its preference for acidic pH. A significant amount of activity, however, was also detectable at physiological pH values, as measured both by an in vitro assay and by in vivo degradation of cell-bound HS [81].
-
∗Similar observations were reported at the same time by Levy-Adam et al. [82] in a paper entitled ‘Heterodimer formation is essential for heparanase enzymatic activity’. Few months afterward, Nardella et al [83] published a paper entitled: ‘Mechanism of activation of human heparanase investigated by protein engineering’ and concluded that (i) the heparanase heterodimer (alpha/beta) [8]-TIM barrel fold is contributed by both the 8 and 50 kDa subunits with the 6 kDa connecting fragment leading to inhibition of heparanase by possibly obstructing access to the active site, (ii) proteolytic excision of the 6 kDa fragment is necessary and sufficient for heparanase activation, and (iii) Substituting the 6 kDa fragment with a spacer of three glycine-serine pairs resulted in constitutively active, single-chain heparanase which was comparable to the processed, heterodimeric enzyme .
Involvement of Disulfide Bond Formation in the Activation of Heparanase
The link between disulfide bond formation and the activation of heparanase in human tumor cells was investigated. Mass spectrometry analysis of heparanase purified from a conditioned medium of human fibrosarcoma cells revealed two disulfide bonds, Cys127-Cys179 and Cys437-Cys542, and one S-cysteinylation at the Cys211 residue. It was shown that although the formation of the Cys127-Cys179 bond and S-cysteinylation at Cys211 have little effect on heparanase function, the disulfide bond between Cys437 and Cys542 is necessary for the secretion and activation of heparanase. Thus, the present findings will provide a basis for further refinement of heparanase structural studies and for the development of novel heparanase inhibitors [84].
Processing and Activation of Latent Heparanase Occur in Lysosomes
We generated an antibody (733) that preferentially recognizes the active 50 kDa heparanase form as compared to the non-active 65 kDa heparanase precursor. We have utilized this and other anti-heparanase antibodies to study the cellular localization of the latent 65 kDa and active 50 kDa heparanase forms during uptake and processing of exogenously added heparanase. Interestingly, not only the processed 50 kDa, but also the 65 kDa heparanase precursor was localized to perinuclear vesicles, suggesting that heparanase processing occurs in lysosomes. Indeed, heparanase processing was completely inhibited by chloroquine and bafilomycin A1, inhibitors of lysosome proteases. Similarly, processing of membrane-targeted heparanase was also chloroquine-sensitive, further ruling out the plasma membrane as the heparanase processing site. Finally, we provide evidence that antibody 733 partially neutralizes the enzymatic activity of heparanase, suggesting that the N-terminal region of the molecule is involved in assuming an active conformation [85].
-
∗Early paper on the localization of heparanase in late endosomes and lysosomes .
Cathepsin L Is Responsible for Processing and Activation of Proheparanase Through Multiple Cleavages of a Linker Segment
Applying cathepsin L knock-out tissues and cultured fibroblasts, as well as cathepsin L gene silencing and overexpression strategies, we have demonstrated that removal of the linker peptide and conversion of pro-heparanase into its active 8 + 50-kDa form is brought about predominantly by cathepsin L. Excision of a 10-amino acid peptide located at the C terminus of the linker segment between two functional cathepsin L cleavage sites (Y156Q and Y146Q) was critical for activation of proheparanase. Mass spectrometry revealed that the entire linker segment is susceptible to multiple endocleavages by cathepsin L and that an active 8-kDa subunit can be generated by several alternative adjacent endocleavages, yielding the precise 8-kDa subunit and/or slightly elongated forms. Altogether, the mode of action presented here demonstrates that processing and activation of proheparanase can be brought about solely by cathepsin L [73].
-
∗The above results corroborated an earlier publication by the same group entitled ‘Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase’ presenting, among other aspects, a predicted structural model of the heparanase protein including a 1 kDa peptide of the linker segment that hinder accessibility of the HS substrate to the active site of the enzyme and hence inhibits heparanase enzymatic activity [86].
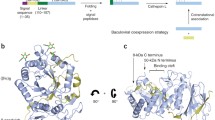
Structural Characterization of Human Heparanase Reveals Insights into Substrate Recognition
Wu and al presented crystal structures of human HPSE at 1.6-Å to 1.9-Å resolution that reveal how an endo-acting binding cleft is exposed by proteolytic activation of latent proHPSE. We used oligosaccharide complexes to map the substrate-binding and sulfate-recognition motifs. These data shed light on the structure and interactions of a key enzyme involved in ECM maintenance and provide a starting point for the design of HPSE inhibitors for use as biochemical tools and anticancer therapeutics [87].
-
∗Resolution of the heparanase crystal structure. In a related study entitled ‘Activity-based probes for functional interrogation of retaining β-glucuronidases’ the same group reported that both the active and supposedly inactive heparanase proenzyme can be labeled by the same activity-based (ABP) probes, leading to surprising insights regarding structural relationships between pro-heparanase, mature heparanase, and their bacterial homologs [88].
-
See Wu & Davies, Chap. 5 in this volume for more information on heparanase structural properties.
3.2.2 Gene Regulation
(See Gaskin et al., Chap. 7 in this volume, for more information on heparanase gene regulation)
Cloning and Characterization of the Human Heparanase-1 (HPR1) Gene Promoter: Role of GA-Binding Protein and Sp1 in Regulating HPR1 Basal Promoter Activity
To understand the mechanisms of heparanase-1 (HPR1) gene expression and regulation , we first mapped the transcription start site of the heparanase (HPR1) gene and found that HPR1 mRNA was transcribed from the nucleotide position 101 bp upstream of the ATG codon. A 3.5-kb promoter region of the HPR1 gene was cloned. Sequence analysis revealed that the TATA-less, GC-rich promoter of the HPR1 gene belongs to the family of housekeeping genes. This 3.5-kb promoter region exhibited strong promoter activity in two thyroid tumor cell lines. Truncation analysis of the HPR1 promoter identified a minimal 0.3-kb region that had strong basal promoter activity. Truncation and mutational analysis of the HPR1 promoter revealed three Sp1 sites and four Ets-relevant elements (ERE) significantly contributing to basal HPR1 promoter activity. Binding to the Sp1 sites by Sp1 and to the ERE sites by GA-binding protein (GABP) was confirmed by electrophoretic mobility shift assay and competition and supershift electrophoretic mobility shift assays. Co-transfection of Sp- and GABP-deficient Drosophila SL-2 cells with the HPR1 promoter-driven luciferase construct plus the expression vector encoding the Sp1, Sp3, or GABP gene induced luciferase gene expression. Mutation or truncation of the Sp1 or ERE sites reduced luciferase expression in both SL-2 cells and thyroid tumor cell lines. Co-expression of GABPalpha/beta and Sp1 or Sp3 further increased luciferase reporter gene expression. Our results collectively suggest that Sp1 cooperates with GABP to regulate HPR1 promoter activity [89].
-
∗Characterization of the human heparanase gene promoter and demonstration that Sp1 cooperates with GABP to regulate heparanase promoter activity.
Regulation of Heparanase Gene Expression by Estrogen in Breast Cancer
We identified four putative estrogen response elements in the heparanase promoter region and found that transcription of a luciferase reporter gene driven by the heparanase promoter was significantly increased in estrogen-receptor positive MCF-7 human breast carcinoma cells after estrogen treatment. Estrogen-induced heparanase mRNA transcription in estrogen receptor-positive, but not in estrogen receptor-negative, breast cancer cells, confirmed the promoter study data. The estrogen effects on heparanase mRNA expression levels were abolished in the presence of the pure antiestrogen ICI 182,780, indicating that the classic estrogen receptor pathway is involved in transcriptional activation of heparanase. In vivo, exposure to estrogen augmented levels of heparanase protein in MCF-7 cells embedded in Matrigel plugs and correlated with increased plug vascularization. Collectively, our data suggest a new molecular pathway through which estrogen, independent of its proliferative effect, may induce heparanase overexpression and, thus, promote tumor-stromal interactions, critical for breast carcinoma development and progression [70].
-
∗Estrogen induces heparanase overexpression and, thus, promotes tumor-stromal interactions.
Role of Promoter Methylation in Regulation of the Mammalian Heparanase Gene
To investigate the epigenetic regulation of the heparanase locus, methylation-specific and bisulfite PCR were performed on a panel of 22 human cancer cell lines. Cytosine methylation of the heparanase promoter was associated with inactivation of the affected allele. Despite lack of sequence homology, extensively methylated CpG islands were found both in human choriocarcinoma (JAR) and rat glioma (C-6) cells which lack heparanase activity. Treatment of these cells with demethylating agents (5-azacytidine, 5-aza-2′-deoxycytidine) resulted in stable dose- and time-dependant promoter hypomethylation accompanied by reappearance of heparanase mRNA, protein and enzymatic activity. An inhibitor of histone deacetylase, Trichostatin A, failed to induce either of these effects. Upregulation of heparanase expression and activity by demethylating drugs was associated with a marked increase in lung colonization by pretreated C-6 rat glioma cells. The increased metastatic potential in vivo was inhibited in mice treated with laminaran sulfate, a potent inhibitor of heparanase activity. We propose a model wherein expression of mammalian heparanase gene is modulated by the interplay between trans-activating genetic and cis-inhibitory epigenetic elements in its promoter [72].
-
∗Heparanase gene expression is modulated by the interplay between trans-activating genetic and cis-inhibitory epigenetic elements in its promoter.
Promoter CpG Hypomethylation and Transcription Factor EGR1 Hyperactivate Heparanase Expression in Bladder Cancer
We hypothesized that promoter CpG hypomethylation with increased EGR1 expression could determine heparanase expression during the pathogenesis of bladder cancer. Bladder cancer cell lines significantly restored heparanase expression after 5-Aza-dC treatment. Transfection of EGR1 siRNA into T24 bladder cancer cell line significantly downregulated heparanase expression compared to the control siRNA transfection. In 54 bladder cancer and paired normal bladder samples, heparanase expression was significantly higher in bladder cancer than in normal bladder (P < 0.01). We performed methylation-specific PCR targeting the CpG sites within the core-binding consensus motifs of EGR1 (GGCG) and Sp1 (GGGCGG). Methylation prevalence was significantly higher in normal bladder than in bladder cancer (P < 0.05) and inversely correlated with heparanase expression (P = 0.055). In the total series of bladder cancer and normal bladder samples, the combination of promoter CpG methylation and EGR1 expression regulated heparanase expression in a stepwise manner, where heparanase expression was the lowest in methylation-positive and EGR1-negative samples and the highest in methylation-negative and EGR1-positive samples. This is the first study demonstrating that increased heparanase expression during the pathogenesis of bladder cancer is due to promoter hypomethylation and transcription factor EGR1 [69].
Early Growth Response Gene 1 (EGR1) Regulates Heparanase Gene Transcription in Tumor Cells
We identified the transcription factor early growth response gene 1, EGR1 , as a key regulator of inducible heparanase transcription in T cells. Using chromatin immunoprecipitation, we demonstrate for the first time that EGR1 binds to the heparanase gene promoter in vivo. The important question of the role of EGR1 in regulating heparanase transcription in tumor cells was then assessed. Studies were carried out in four epithelial tumor lines of different tissue origin. Functional dissection of the heparanase promoter identified a 280-bp region that was critical for transcription of the heparanase gene. Transactivation studies using an EGR1 expression vector co-transfected with a reporter construct containing the 280-bp region showed EGR1-activated heparanase promoter activity in a dose-dependent manner in prostate or breast adenocarcinoma and colon carcinoma cell lines. In contrast, overexpression of EGR1 resulted in a dose-dependent repression of promoter activity in melanoma cells. Using site-directed mutagenesis the 280-bp region was found to contain two functional EGR1 sites and electrophoretic mobility shift assays showed binding of EGR1 to both of these sites upon activation of tumor cells. Furthermore, the heparanase promoter region containing the EGR1 sites was also inducible in tumor cells and induction corresponded to HPSE expression levels. These studies show that EGR1 regulates heparanase transcription in tumor cells and importantly, can have a repressive or activating role depending on the tumor type [65].
-
∗The above two studies indicate that heparanase gene expression is due to promoter hypomethylation and interaction with transcription factor EGR1 .
Tumor Suppressor p53 Regulates Heparanase Gene Expression
We demonstrate that wild-type (wt) p53 binds to heparanase promoter and inhibits its activity, whereas mutant p53 variants failed to exert an inhibitory effect. Moreover, p53-H175R mutant even activated heparanase promoter activity. Elimination or inhibition of p53 in several cell types resulted in a significant increase in heparanase gene expression and enzymatic activity. Trichostatin A abolished the inhibitory effect of wt p53, suggesting the involvement of histone deacetylation in negative regulation of the heparanase promoter. Altogether, our results indicate that the heparanase gene is regulated by p53 under normal conditions, while mutational inactivation of p53 during cancer development leads to induction of heparanase expression, providing a possible explanation for the frequent increase of heparanase levels observed in the course of tumorigenesis [71].
-
∗Wild-type p53 binds to heparanase promoter and inhibits its activity.
Post-Transcriptional Regulation of Heparanase Gene Expression by a 3’ AU-Rich Element
The purpose of the current study was to identify mechanisms responsible for heparanase induction. We provide evidence that heparanase expression is regulated at the post-transcriptional level by sequences at the 3′ untranslated region (3′ UTR) of the gene. Constructing the 3′ UTR immediately following the heparanase cDNA reduces heparanase enzymatic activity and protein levels, resulting in decreased cellular invasion capacity. We further identified a 185-bp sequence within the 3′ UTR that mediates heparanase down-regulation, and characterized an adenine (A)/uracil (U)-rich consensus element (ARE) within this region. Deletion of the entire 185-bp region or the ARE eliminated the inhibitory effect of the 3′ UTR, resulting in elevated heparanase levels and formation of larger tumor xenografts indistinguishable from those produced by heparanase-overexpressing cells in terms of size, vascularization, and Akt activation. These results suggest that loss of the ARE is an important regulatory mechanism contributing to heparanase induction in human cancer [78].
-
∗Heparanase expression is regulated at the post-transcriptional level by sequences at the 3′ untranslated region (3′ UTR) of the gene .
MicroRNA-1258 Suppresses Breast Cancer Brain Metastasis by Targeting Heparanase
Heparanase (HPSE) is a potent protumorigenic, proangiogenic, and prometastatic enzyme that is overexpressed in brain metastatic breast cancer (BMBC). We hypothesized that HPSE gene expression might be regulated by micro RNA that might be exploited therapeutically. Using miR and a RNAhybrid, we identified miR-1258 as a candidate micro RNA that may directly target HPSE and suppress BMBC. We found that miR-1258 levels inversely correlated with heparanase expression, enzymatic activity, and cancer cell metastatic propensities, being lowest in highly aggressive BMBC cell variants compared with either nontumorigenic or nonmetastatic human mammary epithelial cells. These findings were validated by analyses of miR-1258 and heparanase content in paired clinical specimens of normal mammary gland versus invasive ductal carcinoma, and primary breast cancer versus BMBC. In regulatory experiments, miR-1258 inhibited the expression and activity of heparanase in BMBC cells, whereas modulating heparanase blocked the phenotypic effects of miR-1258. In functional experiments, stable expression of miR-1258 in BMBC cells inhibited heparanase in vitro cell invasion and experimental brain metastasis. Together, our findings illustrate how micro RNA mechanisms are linked to brain metastatic breast cancer through heparanase control, offering a strong rationale to develop heparanase-based therapeutics for treatment of cancer patients with brain metastases, BMBC in particular [90].
Genetic Variations in the Heparanase Gene (HPSE) Associate With Increased Risk of GVHD Following Allogeneic Stem Cell Transplantation: Effect of Discrepancy Between Recipients and Donors
Graft-versus-host disease (GVHD) is the most common cause of nonrelapse mortality and morbidity after hematopoietic stem cell transplantation (HSCT). The well-documented involvement of heparanase in the process of inflammation and autoimmunity led us to investigate an association between HPSE gene single-nucleotide polymorphisms (SNPs) and the risk of GVHD. The present study indicates a highly significant correlation of HPSE gene SNPs rs4693608 and rs4364254 and their combination with the risk of developing acute GVHD. Moreover, the study revealed that discrepancy between recipient and donor in these SNPs may elevate significantly the risk of acute GVHD. This association was statistically significant when the recipients possessed genotype combinations dictating higher levels of heparanase compared with their human leukocyte antigen (HLA)-matched donors. In addition, HPSE gene SNPs disclosed a correlation with extensive chronic GVHD, nonrelapse mortality, and overall survival. Our study indicates involvement of heparanase in the development of acute and extensive chronic GVHD. Moreover, it suggests a possible mechanism for the aggressive behavior of T lymphocytes leading to GVHD when the recipients possess genotype combinations that dictate high levels of heparanase mRNA compared with their HLA-matched donors expressing low levels of heparanase [91].
-
∗See Ostrovsky et al., Chap. 8 in this volume for more information about heparanase gene SNPs.
3.2.3 Angiogenesis & Metastasis
Heparanase as Mediator of Angiogenesis: Mode of Action
We demonstrate that heparanase is intimately involved in angiogenesis and elucidate its mode of action. Apart from its direct involvement in ECM degradation and EC migration, heparanase releases active bFGF from the subendothelial ECM, as well as bFGF-stimulating HS degradation fragments from the EC surface. Interestingly, ECM-derived HS fragments induced little or no potentiation of the growth-promoting activity of bFGF. The angiogenic effect of heparanase was demonstrated in vivo (Matrigel plug assay) by showing a three- to four fold increase in neovascularization induced by murine T-lymphoma cells after stable transfection with the heparanase gene. Increased tissue vascularity was also observed in a mouse wound-healing model in response to topical administration of recombinant heparanase. Immunohistochemical staining of human colon carcinoma tissue revealed a high expression of the heparanase protein in the endothelium of sprouting capillaries and small vessels, but not of mature quiescent blood vessels. The ability of heparanase to promote tumor angiogenesis and its involvement in tumor metastasis make it a promising target for cancer therapy [63].
-
∗Early study demonstrating the ability of heparanase to promote tumor angiogenesis.
Cell Surface Expression and Secretion of Heparanase Markedly Promote Tumor Angiogenesis and Metastasis
The present study emphasizes the importance of cell surface expression and secretion of heparanase in tumor angiogenesis and metastasis. For this purpose, nonmetastatic Eb mouse lymphoma cells were transfected with the predominantly intracellular human heparanase or with a readily secreted chimeric construct composed of the human enzyme and the chicken heparanase signal peptide. Eb cells overexpressing the secreted heparanase invaded a reconstituted basement membrane to a much higher extent than cells overexpressing the intracellular enzyme. Cell invasion was inhibited in the presence of laminaran sulfate, a potent inhibitor of heparanase activity and experimental metastasis. The increased invasiveness in vitro was reflected in vivo by rapid and massive liver colonization and accelerated mortality. In fact, mice inoculated with cells expressing the secreted enzyme succumb because of liver metastasis and dysfunction, as early as 10 days after s.c. inoculation of the cells, when their tumor burden did not exceed 1% of body weight. Cell surface localization and secretion of heparanase markedly stimulated tumor angiogenesis, as demonstrated by a 4–six-fold increase in vessel density and functionality evaluated by MRI of tumors produced by cells expressing the secreted vs. the non-secreted heparanase, consistent with actual counting of blood vessels. Altogether, our results indicate that the potent proangiogenic and prometastatic properties of heparanase are tightly regulated by its cellular localization and secretion. The increased potency of the secreted enzyme makes it a promising target for anticancer drug development [58].
Heparanase Gene Silencing, Tumor Invasiveness, Angiogenesis, and Metastasis
Studies performed prior to HPSE gene cloning , have sought to determine the role of heparanase in tumor progression. However, such investigations relied on the use of heparin-mimicking molecules to inhibit heparanase activity. Because the reagents used in those previous studies lacked specificity, the conclusions drawn from the studies are somewhat debatable. Edovitsky et al. [57] applied ribozyme and small interfering RNA (siRNA) technology to knock down the levels of endogenous heparanase. The authors convincingly show that, in models of experimental and spontaneous metastases, these strategies attenuate the ability of diverse tumor cells, including melanoma, mammary adenocarcinoma, lymphoma, and glioma cells, to invade in vitro and to colonize distant sites including the liver and lungs. These and other results [59] provide strong support for a role for heparanase in the metastatic process. Moreover, these studies can be used to rationalize the development of anti-heparanase strategies for cancer patients .
∗
The above description was taken from a commentary written by Boyd & Nakajima [56].
Heparanase Induces Tissue Factor Pathway Inhibitor Expression and Extracellular Accumulation in Endothelial and Tumor Cells
We have reported that heparanase stimulates tissue factor (TF) expression in endothelial and cancer cells, resulting in elevation of coagulation activity. We hypothesized that heparanase regulates other coagulation modulators, and found that heparanase over-expression or exogenous addition stimulated tissue factor pathway inhibitor (TFPI) expression by 2–3 folds. TFPI accumulation in the cell culture medium exceeded in magnitude the observed induction of TFPI gene transcription reaching 5- to six-fold increase. Extracellular accumulation of TFPI correlated with increased coagulation activity. This effect was found to be independent of heparanase enzymatic activity and interaction with HS, and correlated with reduced TFPI levels on the cell surface. Interaction between heparanase and TFPI was evident by co-immunoprecipitation and resulted in TFPI displacement from the surface of the vascular endothelium. Thus, heparanase facilitates blood coagulation on the cell surface by two independent mechanisms: dissociation of TFPI from the vascular surface shortly after local elevation of heparanase levels, and subsequent induction of TF expression [92].
-
∗See Nadir, Chap. 33 in this volume for more information on heparanase and the coagulation system.
3.2.4 Animal Models
Transgenic Expression of Mammalian Heparanase Uncovers Physiological Functions of Heparan Sulfate in Tissue Morphogenesis, Vascularization, and Feeding Behavior
We have generated homozygous transgenic mice (hpa-tg) overexpressing human heparanase in all tissues and characterized the involvement of the enzyme in tissue morphogenesis, vascularization, and energy metabolism. Biochemical analysis of HS isolated from newborn mice and adult tissues revealed a profound decrease in the size of HS chains derived from hpa-tg vs. control mice. Despite this, the mice appeared normal, were fertile, and exhibited a normal life span. A significant increase in the number of implanted embryos was noted in the hpa-tg vs. control mice. Overexpression of heparanase resulted in increased levels of urinary protein and creatinine, suggesting an effect on kidney function, reflected also by electron microscopy examination of the kidney tissue. The hpa-tg mice exhibited a reduced food consumption and body weight compared with control mice. The effect of heparanase on tissue remodeling and morphogenesis was best demonstrated by the phenotype of the hpa-tg mammary glands, showing excess branching and widening of ducts associated with enhanced neovascularization and disruption of the epithelial basement membrane. The hpa-tg mice exhibited an accelerated rate of hair growth, correlated with high expression of heparanase in hair follicle keratinocytes and increased vascularization [93].
Transgenic or Tumor-Induced Expression of Heparanase Upregulates Sulfation of Heparan Sulfate
In Hpa-tg liver showing excessive heparanase overexpression, HSPG turnover is accelerated along with upregulation of HS N- and O-sulfation, thus yielding heparin-like chains without the domain structure typical of HS. Heparanase overexpression in other mouse organs and in human tumors correlated with increased 6-O-sulfation of HS, whereas the domain structure was conserved. The heavily sulfated HS fragments strongly promoted formation of ternary complexes with fibroblast growth factor 1 (FGF1) or FGF2 and FGF receptor 1. Heparanase thus contributes to regulation of HS biosynthesis in a way that may promote growth factor action in tumor angiogenesis and metastasis [94].
Newly Generated Heparanase Knock-out Mice Unravel Co-Regulation of Heparanase and Matrix Metalloproteinases
We report that targeted disruption of the murine heparanase gene eliminated heparanase enzymatic activity, resulting in accumulation of long HS chains. Unexpectedly, the heparanase knockout (Hpse-KO) mice were fertile, exhibited a normal life span and did not show prominent pathological alterations. The lack of major abnormalities is attributed to a marked elevation in the expression of matrix metalloproteinases (primarily MMP2 and MMP14) compensating for the lack of heparanase. Co-regulation of heparanase and MMPs was also noted by a marked decrease in MMP (primarily MMP-2,-9 and 14) expression following transfection and over-expression of the heparanase gene in cultured human mammary carcinoma (MDA-MB-231) cells. Generation of viable Hpse-KO mice lacking significant abnormalities may provide a promising indication for the use of heparanase as a target for drug development [95].
Mice Deficient in Heparanase Exhibit Impaired Dendritic Cell Migration and Reduced Airway Inflammation
In this study, constitutive heparanase-deficient (Hpse(−/−)) mice were generated on a C57BL/6 background using the Cre/loxP recombination system, with a complete lack of heparanase mRNA, protein and activity. Although heparanase has been implicated in embryogenesis and development, Hpse(−/−) mice are anatomically normal and fertile. Interestingly, the trafficking of dendritic cells from the skin to the draining lymph nodes was markedly reduced in Hpse(−/−) mice. Furthermore, the ability of Hpse(−/−) mice to generate an allergic inflammatory response in the airways, a process that requires dendritic cell migration, was also impaired. These findings establish an important role for heparanase in immunity and identify the enzyme as a potential target for regulation of an immune response [96].
-
∗The above four studies describe the generation and properties of heparanase over-expressing and knockout mice.
3.2.5 Heparanase Uptake and Cellular Traffic
Heparanase Uptake Is Mediated by Cell Membrane Heparan Sulfate Proteoglycans
We provide evidence that HS is not only a substrate for, but also a regulator of, heparanase. Addition of heparin or xylosides to cell cultures resulted in a pronounced accumulation of heparanase in the culture medium, whereas sodium chlorate had no such effect. Moreover, cellular uptake of heparanase was markedly reduced in HS-deficient CHO-745 mutant cells, heparan sulfate proteoglycan-deficient HT-29 colon cancer cells, and heparinase-treated cells. Notably, heparanase resides in the endosomal/lysosomal compartment for a relatively long period of time and is likely to play a role in the normal turnover of HS. Co-localization studies and cell fractionation following heparanase addition have identified syndecan family members as candidate molecules responsible for heparanase uptake, providing an efficient mechanism that limits extracellular accumulation and function of heparanase [97].
-
∗First paper describing syndecan-mediated cellular uptake of heparanase .
Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy
The protumorigenic properties of heparanase were found to be mediated , in part, by its proautophagic function, as demonstrated in tumor xenograft models of human cancer and through use of inhibitors of the lysosome (chloroquine) and heparanase (PG545), both alone and in combination. Notably, heparanase-overexpressing cells were more resistant to stress and chemotherapy in a manner associated with increased autophagy, effects that were reversed by chloroquine treatment. Collectively, our results establish a role for heparanase in modulating autophagy in normal and malignant cells, thereby conferring growth advantages under stress as well as resistance to chemotherapy [98].
-
∗First paper describing a role for heparanase in autophagy.
Heparanase Regulates Secretion, Composition, and Function of Tumor Cell-Derived Exosomes
Emerging evidence indicates that exosomes play a key role in tumor-host cross-talk and that exosome secretion, composition, and functional capacity are altered as tumors progress to an aggressive phenotype. We have discovered that in human cancer cells (myeloma, lymphoblastoid, and breast cancer), when expression of heparanase is enhanced or when tumor cells are exposed to exogenous heparanase, exosome secretion is dramatically increased. Heparanase enzyme activity is required for robust enhancement of exosome secretion because enzymatically inactive forms of heparanase do not dramatically increase exosome secretion. Heparanase also impacts exosome protein cargo as reflected by higher levels of syndecan-1, VEGF, and hepatocyte growth factor in exosomes secreted by heparanase-high expressing cells as compared with heparanase-low expressing cells. In functional assays, exosomes from heparanase-high cells stimulated spreading of tumor cells on fibronectin and invasion of endothelial cells through ECM better than did exosomes secreted by heparanase-low cells. These studies reveal that heparanase helps drive exosome secretion, alters exosome composition, and facilitates production of exosomes that impact both tumor and host cell behavior, thereby promoting tumor progression [99].
-
∗First paper describing a role for heparanase in exosome formation and secretion.
Heparanase Activates the Syndecan-Syntenin-ALIX Exosome Pathway
We showed that syndecans control the biogenesis of exosomes through their interaction with syntenin-1 and the endosomal-sorting complex required for transport accessory component ALIX. We investigated the role of heparanase in the syndecan-syntenin-ALIX exosome biogenesis pathway. Our findings identify heparanase as a modulator of the syndecan-syntenin-ALIX pathway, fostering endosomal membrane budding and the biogenesis of exosomes by trimming the heparan sulfate chains on syndecans. In addition, our data suggest that this mechanism controls the selection of specific cargo to exosomes [100]. It was further reported that heparanase tailors syndecan for exosome production and suggested that upregulation of syntenin and heparanase in cancers may support the suspected roles of exosomes in tumor biology [101].
-
∗See: David & Zimmermann and Purushothaman and Sanderson Chap. 10 and 12 in this volume for more information on heparanase and exosomes.
-
∗∗A recent review written by Sanderson et al. [102] focuses on the emerging role of exosomal surface enzymes in disease progression. Briefly, the review demonstrates that enzymatically active proteases and glycosidases are present on the surface of some exosomes. These enzymes can degrade the ECM, liberate growth factors and alter cell invasion, resulting in remodeling the extracellular space and regulating cancer progression, inflammation, and Alzheimer’s disease .
Chemotherapy Induces Secretion of Exosomes Loaded with Heparanase that Degrades Extracellular Matrix and Impacts Tumor and Host Cell Behavior
We investigated the impact of anti-myeloma drugs on exosome biogenesis. When myeloma cells were exposed to the commonly utilized anti-myeloma drugs bortezomib, carfilzomib or melphalan, exosome secretion by the cells was dramatically enhanced. These chemotherapy-induced exosomes (chemoexosomes) have a proteome profile distinct from cells not exposed to drug including a dramatic elevation in the level of heparanase present as exosome cargo. Heparanase was present on the exosome surface where it was capable of degrading heparan sulfate embedded within an ECM. When exposed to myeloma cells, chemoexosomes transferred their heparanase cargo to those cells, enhancing their HS degrading activity and leading to activation of ERK signaling and an increase in shedding of the syndecan-1 proteoglycan. Exposure of chemoexosomes to macrophages enhanced their secretion of TNF-α, an important myeloma growth factor. Moreover, chemoexosomes stimulated macrophage migration and this effect was blocked by H1023, a monoclonal antibody that inhibits heparanase enzymatic activity. These data suggest that anti-myeloma therapy ignites a burst of exosomes having a high level of heparanase that remodels ECM and alters tumor and host cell behaviors that likely contribute to chemoresistance and eventual patient relapse [103].
-
∗Anti-myeloma chemotherapy dramatically stimulates secretion of exosomes and alters exosome composition. Exosomes secreted during therapy (chemoexosomes) contain high levels of heparanase on their surface that can degrade ECM and also can be transferred to both tumor and host cells, altering their behavior in ways that may enhance tumor survival and progression [103].
3.2.6 Nuclear Heparanase and Its Transcriptional Activity
Heparanase-Mediated Loss of Nuclear Syndecan-1 Enhances Histone Acetyltransferase (HAT) activity to Promote Expression of Genes that Drive an Aggressive Tumor Phenotype
Heparanase acts as a master regulator of the aggressive tumor phenotype in part by enhancing expression of proteins known to drive tumor progression (e.g. VEGF, MMP-9, hepatocyte growth factor (HGF), and RANKL). However, the mechanism whereby this enzyme regulates gene expression remains unknown. We previously reported that elevation of heparanase levels in myeloma cells causes a dramatic reduction in the amount of syndecan-1 in the nucleus. Because HS has been shown to inhibit the activity of histone acetyltransferase (HAT) enzymes in vitro, we hypothesized that the reduction in nuclear syndecan-1 in cells expressing high levels of heparanase would result in increased HAT activity leading to stimulation of protein transcription. We found that myeloma cells or tumors expressing high levels of heparanase and low levels of nuclear syndecan-1 had significantly higher levels of HAT activity when compared with cells or tumors expressing low levels of heparanase. High levels of HAT activity in heparanase-high cells were blocked by SST0001, an inhibitor of heparanase. Restoration of high syndecan-1 levels in heparanase-high cells diminished nuclear HAT activity, establishing syndecan-1 as a potent inhibitor of HAT. Exposure of heparanase-high cells to anacardic acid, an inhibitor of HAT activity, significantly suppressed their expression of VEGF and MMP-9, two genes known to be up-regulated following elevation of heparanase. These results reveal a novel mechanistic pathway driven by heparanase expression, which leads to decreased nuclear syndecan-1, increased HAT activity, and up-regulation of transcription of multiple genes that drive an aggressive tumor phenotype [104].
The Endoglycosidase Heparanase Enters the Nucleus of T Lymphocytes and Modulates H3 Methylation at Actively Transcribed Genes Via the Interplay with Key Chromatin Modifying Enzymes
The methylation of histones is a fundamental epigenetic process regulating gene expression programs in mammalian cells. Here, we report the unexpected finding that heparanase enters the nucleus of activated human T lymphocytes and regulates the transcription of a cohort of inducible immune response genes by controlling histone H3 methylation patterns. It was found that nuclear heparanase preferentially associates with euchromatin. Genome-wide ChIP-on-chip analyses showed that heparanase is recruited to both the promoter and transcribed regions of a distinct cohort of transcriptionally active genes. Knockdown and overexpression of the heparanase gene also showed that chromatin-bound heparanase is a prerequisite for the transcription of a subset of inducible immune response genes in activated T cells. Furthermore, the actions of heparanase seem to influence gene transcription by associating with the demethylase LSD1, preventing recruitment of the methylase MLL and thereby modifying histone H3 methylation patterns. These data indicate that heparanase belongs to an emerging class of proteins that play an important role in regulating transcription in addition to their well-recognized extra-nuclear functions [105].
-
∗Two papers describing the involvement of nuclear heparanase in gene transcription.
3.2.7 Heparanase Non-Enzymatic and Signaling Function
Heparanase Induces Vascular Endothelial Growth Factor Expression: Correlation With p38 Phosphorylation Levels and Src Activation
We examined the possibility that heparanase directly participates in VEGF gene regulation. We provide evidence that heparanase overexpression in human embryonic kidney 293, MDA-MB-435 human breast carcinoma, and rat C6 glioma cells resulted in a 3- to six-fold increase in VEGF protein and mRNA levels, which correlated with elevation of p38 phosphorylation. Moreover, heparanase down-regulation in B16 mouse melanoma cells by a specific siRNA vector was accompanied by a decrease in VEGF and p38 phosphorylation levels, suggesting that VEGF gene expression is regulated by endogenous heparanase. Interestingly, a specific p38 inhibitor did not attenuate VEGF up-regulation by heparanase whereas Src inhibitors completely abrogated this effect. These results indicate that heparanase is actively involved in the regulation of VEGF gene expression, mediated by activation of Src family members [106].
-
∗First paper on heparanase and VEGF gene expression.
Structure-Function Approach Identifies a COOH-Terminal domain that Mediates Heparanase Signaling
Heparanase exerts biological functions apparently independent of its enzymatic activity, enhancing the phosphorylation of selected protein kinases and inducing gene transcription. A predicted three-dimensional structure of constitutively active heparanase clearly delineates a TIM-barrel fold previously anticipated for the enzyme. Interestingly, the model also revealed the existence of a COOH-terminal domain (C-domain) that apparently is not an integral part of the TIM-barrel fold. We provide evidence that the C-domain is critical for heparanase enzymatic activity and secretion. Moreover, the C-domain was found to mediate nonenzymatic functions of heparanase, facilitating Akt phosphorylation, cell proliferation, and tumor xenograft progression. These findings support the notion that heparanase exerts enzymatic activity-independent functions, and identify, for the first time, a protein domain responsible for heparanase-mediated signaling. Inhibitors directed against the C-domain, combined with inhibitors of heparanase enzymatic activity, are expected to neutralize heparanase functions and to profoundly affect tumor growth, angiogenesis, and metastasis [107].
-
∗First paper on the heparanase C-terminal domain and signal transduction.
Heparanase Augments Epidermal Growth Factor Receptor Phosphorylation: Correlation With Head and Neck Tumor Progression
We provide evidence that enzymatically active and inactive heparanase enhance epidermal growth factor receptor (EGFR) phosphorylation. Enhanced EGFR phosphorylation was associated with increased cell migration, cell proliferation, and colony formation, which were attenuated by Src inhibitors. Similarly, heparanase gene silencing by means of siRNA was associated with reduced Src and EGFR phosphorylation levels and decreased cell proliferation. Moreover, heparanase expression correlated with increased phospho-EGFR levels and progression of head and neck carcinoma, providing a strong clinical support for EGFR modulation by heparanase. Thus, heparanase seems to modulate two critical systems involved in tumor progression, namely VEGF expression and EGFR activation. Neutralizing heparanase enzymatic and nonenzymatic functions is therefore expected to profoundly affect tumor growth, angiogenesis, and metastasis [108].
-
∗First paper describing the involvement of heparanase in epidermal growth factor receptor (EGFR) phosphorylation. The enzyme was also shown to induce Stat phosphorylation [109].
3.2.8 Heparanase Inhibitors
Development of a Colorimetric Assay for Heparanase Activity Suitable for Kinetic Analysis and Inhibitor Screening
We have developed a convenient assay based on the cleavage of the synthetic heparin oligosaccharide fondaparinux. The assay measures the appearance of the disaccharide product of heparanase-catalyzed fondaparinux cleavage colorimetrically using the tetrazolium salt WST-1. Because this assay has a homogeneous substrate with a single point of cleavage, the kinetics of the enzyme can be reliably characterized, giving a K(m) of 46 microM and a k(cat) of 3.5 s(−1) with fondaparinux as substrate. The inhibition of heparanase by the published inhibitor, PI-88, was also studied, and a K(i) of 7.9 nM was determined. The simplicity and robustness of this method, should, not only greatly assist routine assay of heparanase activity but also could be adapted for high-throughput screening of compound libraries, with the data generated being directly comparable across studies [110].
-
∗The colorimetric assay is being used for screening of heparanase-inhibiting molecules.
A Functional Heparan Sulfate mimetic Implicates both Heparanase and Heparan Sulfate in Tumor Angiogenesis and Invasion in a Mouse Model of Multistage Cancer
Heparanase mRNA and protein expression are increased in the neoplastic stages progressively unfolding in a mouse model of multistage pancreatic islet carcinogenesis. Notably, heparanase is delivered to the neoplastic lesions in large part by infiltrating Gr1+/Mac1+ innate immune cells. A sulfated oligosaccharide mimetic of HS, PI-88 (= Mupafostat), was used to inhibit simultaneously both heparanase activity and HS effector functions. PI-88 had significant effects at distinct stages of tumorigenesis, producing a reduction in the number of early progenitor lesions and an impairment of tumor growth at later stages. These responses were associated with decreased cell proliferation, increased apoptosis, impaired angiogenesis, and a substantive reduction in the number of invasive carcinomas. In addition, we show that the reduction in tumor angiogenesis is correlated with a reduced association of VEGF-A with its receptor VEGF-R2 on the tumor endothelium, implicating heparanase in the mobilization of matrix-associated VEGF. These data encourage clinical applications of inhibitors such as PI-88 for the many human cancers where heparanase expression is elevated or mobilization of HS-binding regulatory factors is implicated [76].
SST0001, a Chemically Modified Heparin, Inhibits Myeloma Growth and Angiogenesis Via Disruption of the Heparanase/Syndecan-1 Axis
The ability of SST0001 (glycol-split heparin = Roneparstat) to inhibit growth of myeloma tumors was assessed using multiple animal models and a diverse panel of human and murine myeloma cell lines. SST0001 effectively inhibited myeloma growth in vivo, even when confronted with an aggressively growing tumor within human bone. In addition, SST0001 treatment causes changes within tumors consistent with the compound’s ability to inhibit heparanase, including downregulation of HGF, VEGF, and MMP-9 expression and suppressed angiogenesis. SST0001 also diminishes heparanase-induced shedding of syndecan-1, and inhibited the heparanase-mediated degradation of syndecan-1 HS side chains, thus confirming the anti-heparanase activity of this compound. In combination with dexamethasone, SST0001 blocked tumor growth in vivo presumably through dual targeting of the tumor and its microenvironment [30].
-
∗See Noseda & Barberi Chap. 21 in this volume for more information on Roneparstat.
PG545, a Dual Heparanase and Angiogenesis Inhibitor, Induces Potent Anti-Tumour and Anti-Metastatic efficacy in Preclinical Models
PG545 (HS mimetic = Pixatimod) was shown to inhibit angiogenesis in vivo and induce anti-tumour or anti-metastatic effects in murine models of breast, prostate, liver, lung, colon, head and neck cancers and melanoma. Enhanced anti-tumour activity was also noted when used in combination with sorafenib in a liver cancer model. PK data revealed that the half-life of PG545 was relatively long, with pharmacologically relevant concentrations of radiolabeled PG545 observed in liver tumours. The anti-metastatic property of PG545, likely due to the inhibition of heparanase, may prove to be a critical attribute as the compound enters phase I clinical trials [111].
-
∗See Hammond & Dredge, Chap. 22 in this volume for more information on Pixatimod.
M402, a Novel Heparan Sulfate Mimetic, Targets Multiple Pathways Implicated in Tumor Progression and Metastasis
M402 (= Necuparanib) is a rationally engineered, non-cytotoxic HS mimetic, designed to inhibit multiple factors implicated in tumor-host cell interactions, including VEGF, FGF2, SDF-1α, P-selectin, and heparanase. A single s.c. dose of M402 effectively inhibited seeding of B16F10 murine melanoma cells to the lung in an experimental metastasis model. Fluorescent-labeled M402 demonstrated selective accumulation in the primary tumor. Immunohistological analyses of the primary tumor revealed a decrease in microvessel density in M402 treated animals. M402 treatment also normalized circulating levels of myeloid derived suppressor cells in tumor bearing mice. Chronic administration of M402, alone or in combination with cisplatin or docetaxel, inhibited spontaneous metastasis and prolonged survival in an orthotopic 4 T1 murine mammary carcinoma model [112].
-
∗The above four papers represent heparin/HS mimetics that were or are being examined in clinical trials. (See Chapters 19, 23, 22, 21 by Chhabra & Ferro; Hammond & Dredge; Gianini et al., and Noseda & Barbieri, for more information on heparanase-inhibiting compounds).
3.2.9 Various Tumors
Inhibition of Heparanase in Pediatric Brain Tumor Cells Attenuates their Proliferation, Invasive Capacity, and in Vivo Tumor Growth
Levels of heparanase (HPSE) in pediatric brain tumors are higher than in healthy brain tissue and treatment of pediatric brain tumor cells with HPSE stimulated their growth. Notably, the latent form of HPSE enhanced cell viability and rapidly activated the ERK and AKT signaling pathways, before enzymatically active HPSE was detected. The HPSE inhibitor PG545 efficiently killed pediatric brain tumor cells, but not normal human astrocytes, and this compound also reduced tumor cell invasion in vitro and potently reduced the size of flank tumors in vivo. These results indicate that HPSE in malignant brain tumors affects both the tumor cells themselves and their ECM [113].
-
∗See Chapter 14 by Karin-Forsberg-Nillson for more information on heparanase and gliomas.
Involvement of Heparanase in the Pathogenesis of Mesothelioma: Basic Aspects and Clinical Applications
Mesothelioma tumor growth was markedly attenuated by heparanase gene silencing and by heparanase inhibitors (PG545 and defibrotide). A marked increase in survival of the mesothelioma-bearing mice was recorded. Heparanase inhibitors were more potent in vivo than conventional chemotherapy. Clinically, heparanase levels in patients’ pleural effusions could distinguish between malignant and benign effusions, and heparanase H-score above 90 was associated with reduced patient survival. Given these preclinical and clinical data, heparanase appears to be an important mediator of mesothelioma, and heparanase inhibitors are worthy of investigation as a new therapeutic modality in mesothelioma clinical trials [114].
3.2.9.1 Multiple Myeloma
Heparanase Promotes the Spontaneous Metastasis of Myeloma Cells to Bone
Using a SCID mouse model , we demonstrate that enhanced expression of heparanase by myeloma cells dramatically up-regulates their spontaneous metastasis to bone. This occurs from primary tumors growing subcutaneously and also from primary tumors established in bone. Interestingly, tumors formed by subcutaneous injection of cells metastasize not only to bone, but also to other sites including spleen, liver, and lung. In contrast, tumors formed by injection of cells directly into bone exhibit a restricted pattern of metastasis that includes dissemination of tumor to other bones but not to extramedullary sites. In addition, expression of heparanase by myeloma cells (1) accelerates the initial growth of the primary tumor, (2) increases whole-body tumor burden as compared with controls, and (3) enhances both the number and size of microvessels within the primary tumor. These studies indicate that heparanase is a critical determinant of myeloma dissemination and growth in vivo [115].
Heparanase Influences Expression and Shedding of Syndecan-1, and its Expression by the Bone Marrow Environment Is a Bad Prognostic Factor in Multiple Myeloma
Using Affymetrix microarrays we show that the gene encoding heparanase (HPSE) is expressed by 11 of 19 myeloma cell lines (HMCLs). In HSPE positive HMCLs, syndecan-1 gene expression and production of soluble syndecan-1 were significantly increased. Knockdown of HPSE by siRNA resulted in a decrease of syndecan-1 gene expression and soluble syndecan-1 production without affecting membrane syndecan-1 expression. Thus, HPSE influences expression and shedding of syndecan-1. Contrary to HMCLs, HPSE is expressed in only 4 of 39 primary MMC samples, whereas it is expressed in 36 of 39 bone marrow (BM) microenvironment samples. In the latter, HPSE is expressed at a median level in polymorphonuclear cells and T cells; it is highly expressed in monocytes and osteoclasts. Affymetrix data were validated at the protein level, both on HMCLs and patient samples. We report that a gene’s expression mainly in the BM environment (i.e, HSPE) is associated with a shorter event-free survival of patients with newly diagnosed myeloma treated with high-dose chemotherapy and stem cell transplantation. Our study suggests that clinical inhibitors of HPSE could be beneficial for patients with MM [116].
Heparanase Enhances Myeloma Progression Via CXCL10 Downregulation
In order to explore the mechanism(s) underlying the pro-tumorigenic capacity of heparanase , we established an inducible Tet-on system. Heparanase expression was markedly increased following addition of doxycycline (Dox) to the culture medium of CAG human myeloma cells infected with the inducible heparanase gene construct, resulting in increased colony number and size in soft agar. Moreover, tumor xenografts produced by CAG-heparanase cells were markedly increased in mice supplemented with Dox in their drinking water compared with control mice maintained without Dox. Consistently, we found that heparanase induction is associated with decreased levels of CXCL10, suggesting that this chemokine exerts tumor-suppressor properties in myeloma. Indeed, recombinant CXCL10 attenuated the proliferation of CAG, U266 and RPMI-8266 myeloma cells. Similarly, CXCL10 attenuated the proliferation of human umbilical vein endothelial cells, implying that CXCL10 exhibits anti-angiogenic capacity. Strikingly, development of tumor xenografts produced by CAG-heparanase cells overexpressing CXCL10 was markedly reduced compared with control cells. Moreover, tumor growth was significantly attenuated in mice inoculated with human or mouse myeloma cells and treated with CXCL10-Ig fusion protein, indicating that CXCL10 functions as a potent anti-myeloma cytokine [117].
Chemotherapy Induces Expression and Release of Heparanase Leading to Changes Associated with an Aggressive Tumor Phenotype
We discovered that drugs used in the treatment of myeloma upregulate heparanase expression. Frontline anti-myeloma drugs, bortezomib and carfilzomib activate the NF-κB pathway to trigger heparanase expression in tumor cells. Blocking the NF-κB pathway diminished this chemotherapy-induced upregulation of heparanase expression. Activated NF-κB signaling was also found to drive high heparanase expression in drug resistant myeloma cell lines. In addition to enhancing heparanase expression, chemotherapy also caused release of heparanase by tumor cells into the conditioned medium. This soluble heparanase was taken up by macrophages and triggered an increase in TNF-α production. Heparanase is also taken up by tumor cells where it induced expression of HGF, VEGF and MMP-9 and activated ERK and Akt signaling pathways. These changes induced by heparanase are known to be associated with the promotion of an aggressive tumor phenotype. Importantly, the heparanase inhibitor Roneparstat diminished the uptake and the downstream effects of soluble heparanase. Together, these discoveries reveal a novel mechanism whereby chemotherapy upregulates heparanase, a known promoter of myeloma growth, and suggest that therapeutic targeting of heparanase during anti-cancer therapy may improve patient outcome [118].
-
∗See Chapter 12 by Sanderson et al., for more information on heparanase and myeloma.
Antitumor Efficacy of the Heparanase Inhibitor SST0001 Alone and in Combination with Antiangiogenic Agents in the Treatment of Human Pediatric Sarcoma Models
The present study focuses on the effect of SST0001 in a panel of pediatric sarcoma models, representative of various tumor histotypes (soft tissue and bone sarcomas). SST0001 treatment downregulated several angiogenic factors in the conditioned media of sarcoma cells, inhibited the pro-invasive effect of heparin-binding factors (VEGF, bFGF, HGF, PDGF), and abrogated PDGF receptor tyrosine phosphorylation. Subcutaneous administration of SST0001 was very effective, resulting in a significant growth inhibition (range, 64–95%) of all tested tumor xenografts. The efficacy of SST0001 was enhanced in combination with antiangiogenic agents (bevacizumab, sunitinib) as documented by the high rate of complete response. The synergistic effect of SST0001 in combination with antiangiogenic agents is consistent with the heparanase mode of action and with the relevant role of heparin-binding proangiogenic/growth factors in the malignant behavior of sarcoma cells [119].
-
∗See Chapter 15 by Cassinelly & Lanzi for more information on heparanase in sarcomas.
3.2.10 Tumor Microenvironment
Heparanase Cooperates with Ras to Drive Breast and Skin Tumorigenesis
Hpa-Tg mice overexpressing heparanase were far more sensitive than control mice to DMBA/TPA skin carcinogenesis, exhibiting a ten-fold increase in the number and size of tumor lesions. Conversely, DMBA/TPA-induced tumor formation was greatly attenuated in Hpa-KO mice lacking heparanase, pointing to a critical role of heparanase in skin tumorigenesis. In support of these observations, the heparanase inhibitor PG545 potently suppressed tumor progression in this model system. Our findings establish that heparanase exerts protumorigenic properties at early stages of tumor initiation, cooperating with Ras to dramatically promote malignant development [120].
-
∗This study emphasizes the co-operation of heparanase with master oncogenes such as Ras.
Heparanase-Neutralizing Antibodies Attenuate Lymphoma Tumor Growth and Metastasis
We provide evidence that heparanase is expressed by human follicular and diffused non-Hodgkin’s B-lymphomas, and that heparanase inhibitors restrain the growth of tumor xenografts produced by lymphoma cell lines. Furthermore, we describe the development and characterization of heparanase-neutralizing monoclonal antibodies that inhibit cell invasion and tumor metastasis, the hallmark of heparanase activity. Using luciferase-labeled Raji lymphoma cells, we show that the heparanase-neutralizing monoclonal antibodies profoundly inhibit tumor load in the mouse bones, associating with reduced cell proliferation and angiogenesis. Notably, we found that Raji cells lack intrinsic heparanase activity, but tumor xenografts produced by this cell line exhibit typical heparanase activity, likely contributed by host cells composing the tumor microenvironment [33].
-
∗Anti-heparanase neutralizing monoclonal antibodies attenuate lymphoma growth by targeting heparanase in the tumor microenvironment.
3.2.11 Inflammation and Cells of the Immune System
Heparanase Powers a Chronic Inflammatory Circuit that Promotes Colitis-Associated Tumorigenesis in Mice
The research focuses on the importance of heparanase in sustaining the immune-epithelial crosstalk underlying colitis-associated tumorigenesis . Using histological specimens from ulcerative colitis (UC) patients and a mouse model of dextran sodium sulfate-induced colitis, we found that heparanase was constantly overexpressed and activated throughout the disease. We demonstrate, using heparanase-overexpressing transgenic mice, that heparanase overexpression markedly increased the incidence and severity of colitis-associated colonic tumors. We found that highly coordinated interactions between the epithelial compartment (contributing heparanase) and mucosal macrophages preserved chronic inflammatory conditions and created a tumor-promoting microenvironment characterized by enhanced NF-κB signaling and induction of STAT3. Our results indicate that heparanase generates a vicious cycle that powers colitis and the associated tumorigenesis: heparanase, acting synergistically with the intestinal flora, stimulates macrophage activation, while macrophages induce production (via TNF-α-dependent mechanisms) and activation (via secretion of cathepsin L) of heparanase contributed by the colon epithelium. Thus, disruption of the heparanase-driven chronic inflammatory circuit is highly relevant to the design of therapeutic interventions in colitis and the associated cancer [121].
-
∗Highly coordinated interactions between the epithelial compartment and mucosal macrophages generate a vicious cycle that powers colitis and the associated tumorigenesis.
Soluble Heparan Sulfate Fragments Generated by Heparanase Trigger the Release of pro-Inflammatory Cytokines through TLR-4
The study focuses on the role of heparanase in regulating the expression and release of cytokines from human and murine immune cells. Ex vivo treatment of human peripheral blood mononuclear cells with heparanase resulted in the release of a range of pro-inflammatory cytokines including IL-1β, IL-6, IL-8, IL-10 and TNF. A similar pattern of cytokine release was also observed when cells were treated with soluble HS. Furthermore, heparanase-induced cytokine release was abolished by enzymatic-inhibitors of heparanase, suggesting that this process is mediated via the enzymatic release of cell surface HS fragments. As soluble HS can signal through the Toll-like receptor (TLR) pathway, heparanase may promote the upregulation of cytokines through the generation of heparanase-cleaved fragments of HS. In support of this hypothesis, mouse spleen cells lacking the key TLR adaptor molecule MyD88 demonstrated an abolition of cytokine release after heparanase stimulation. Furthermore, TLR4-deficient spleen cells showed reduced cytokine release in response to heparanase treatment, suggesting that TLR4 is involved in this response. Consistent with these observations, the pathway involved in cytokine upregulation was identified as being NF-κB-dependent. These data identify a new mechanism for heparanase in promoting the release of pro-inflammatory cytokines that is likely to be important in regulating cell migration and inflammation [122].
-
∗Heparanase triggers upregulation of pro-inflammatory cytokines through the generation of heparanase-cleaved fragments of HS.
Heparanase Is Required for Activation and Function of Macrophages
We applied a genetic approach and examined the behavior and function of macrophages isolated from wild-type (WT) and heparanase-knockout (Hpa-KO) mice. Hpa-KO macrophages express lower levels of cytokines (e.g., TNFα, IL1-β) and exhibit lower motility and phagocytic capacities. Intriguingly, inoculation of control monocytes together with Lewis lung carcinoma (LLC) cells into Hpa-KO mice resulted in nearly complete inhibition of tumor growth. In striking contrast, inoculating LLC cells together with monocytes isolated from Hpa-KO mice did not affect tumor growth, indicating that heparanase is critically required for activation and function of macrophages. Mechanistically, we describe a linear cascade by which heparanase activates Erk, p38, and JNK signaling in macrophages, leading to increased c-Fos levels and induction of cytokine expression in a manner that apparently does not require heparanase enzymatic activity. [123].
-
∗Heparanase is a key mediator of macrophage activation and function in tumorigenesis and cross-talk with the tumor microenvironment.
Macrophage Polarization in Pancreatic Carcinoma: Role of Heparanase Enzyme
Overexpression of heparanase is associated with increased TAM infiltration in both experimental and human PDAC. Moreover, macrophages derived from heparanase-rich tumors (which grew faster in mouse hosts), display pronounced procancerous phenotype, evidenced by overexpression of MSR-2, IL-10, CCL2, VEGF, and increased production of IL-6, an important player in PDAC pathogenesis. Furthermore, in vitro heparanase enzyme-rendered macrophages (stimulated by necrotic cells which are often present in PDAC tissue) pro-cancerous, as exemplified by their enhanced production of key cytokines implicated in PDAC (including IL-6), as well as by their ability to induce STAT3 signaling and to augment pancreatic carcinoma cell proliferation. In agreement, we observed activation of STAT3 in experimental and clinical specimens of heparanase-overexpressing PDAC. These findings underscore a novel function of heparanase in molecular decision-making that guides cancer-promoting action of TAM and imply that heparanase expression status may become highly relevant in defining a target patient subgroup that is likely to benefit the most from treatment modalities targeting TAM/IL-6/STAT3 [124].
-
∗Heparanase plays a key role in molecular decision-making that guides the cancer-promoting action of tumor associated macrophages .
Heparan Sulfate Mimetic PG545-Mediated Anti-Lymphoma Effects Require TLR9-Dependent NK Cell Activation
HS mimetics, such as PG545 , have been developed as antitumor agents and are designed to suppress angiogenesis and metastasis by inhibiting heparanase and competing for the HS-binding domain of angiogenic growth factors. However, how PG545 exerts its antitumor effect remains incompletely defined. Here, using murine models of lymphoma, we determined that the antitumor effects of PG545 are critically dependent on NK cell activation and that NK cell activation by PG545 requires TLR9. We demonstrate that PG545 does not activate TLR9 directly but instead enhances TLR9 activation through the elevation of the TLR9 ligand CpG in DCs. Specifically, PG545 treatment resulted in CpG accumulation in the lysosomal compartment of DCs, leading to enhanced production of IL-12, which is essential for PG545-mediated NK cell activation. Overall, these results reveal that PG545 activates NK cells and that this activation is critical for the antitumor effect of PG545. Moreover, our findings may have important implications for improving NK cell-based antitumor therapies [125].
-
∗PG545 activates NK cells and thereby exerts an antitumor effects.
Heparanase Augments Inflammatory Chemokine Production from Colorectal Carcinoma Cell Lines
To explore possible roles of heparanase in tumor-host crosstalk, we examined whether heparanase influences expression of inflammatory chemokines in colorectal cancer cells . Murine colorectal carcinoma cells incubated with heparanase upregulated MCP-1, KC, and RANTES genes and released MCP-1 and KC proteins. Heparanase-dependent production of IL-8 was detected in two human colorectal carcinoma cell lines. Addition of a heparanase inhibitor Heparastatin (SF4) did not influence MCP-1 production, while both latent and mature forms of heparanase augmented MCP-1 release, suggesting that heparanase catalytic activity was dispensable for MCP-1 production. In contrast, addition of heparin to the medium suppressed MCP-1 release in a dose-dependent manner. Similarly, targeted suppression of Ext1 by RNAi significantly suppressed cell surface expression of HS and MCP-1 production in colon 26 cells. Taken together, it is concluded that colon 26 cells transduce the heparanase-mediated signal through HS binding. We propose a novel function for heparanase independent of its endoglycosidase activity, namely as a stimulant for chemokine production [126].
NK Cell Heparanase Controls Tumor Invasion and Immune Surveillance
NK cells are highly efficient at preventing cancer metastasis but are infrequently found in the core of primary tumors. Here, have we demonstrated that freshly isolated mouse and human NK cells express low levels of heparanase that increase upon NK cell activation. Heparanase deficiency did not affect development, differentiation, or tissue localization of NK cells under steady-state conditions. However, mice lacking heparanase specifically in NK cells (Hpsefl/fl NKp46-iCre mice) were highly tumor prone when challenged with the carcinogen methylcholanthrene (MCA). Hpsefl/fl NKp46-iCre mice were also more susceptible to tumor growth than were their littermate controls when challenged with the established mouse lymphoma cell line RMA-S-RAE-1β, which overexpresses the NK cell group 2D (NKG2D) ligand RAE-1β, or when inoculated with metastatic melanoma, prostate carcinoma, or mammary carcinoma cell lines. NK cell invasion of primary tumors and recruitment to the site of metastasis were strictly dependent on the presence of heparanase. Cytokine and immune checkpoint blockade immunotherapy for metastases was compromised when NK cells lacked heparanase. Our data suggest that heparanase plays a critical role in NK cell invasion into tumors and thereby tumor progression and metastases. This should be considered when systemically treating cancer patients with heparanase inhibitors, since the potential adverse effect on NK cell infiltration might limit the antitumor activity of the inhibitors [127].
Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes
Adoptive transfer of chimeric antigen receptor (CAR)-redirected T lymphocytes (CAR-T cells) has had less striking therapeutic effects in solid tumors than in lymphoid malignancies. Although active tumor-mediated immunosuppression may have a role in limiting the efficacy of CAR-T cells, functional changes in T lymphocytes after their ex vivo manipulation may also account for the reduced ability of cultured CAR-T cells to penetrate stroma-rich solid tumors compared with lymphoid tissues. We therefore studied the capacity of human in vitro-cultured CAR-T cells to degrade components of the ECM. In contrast to freshly isolated T lymphocytes, we found that in vitro-cultured T lymphocytes lack expression of the enzyme heparanase (HPSE), which degrades heparan sulfate proteoglycans, the main components of ECM. We found that HPSE mRNA is downregulated in in vitro-expanded T cells, which may be a consequence of p53 (officially known as TP53, encoding tumor protein 53) binding to the HPSE gene promoter. We therefore engineered CAR-T cells to express HPSE and showed their improved capacity to degrade the ECM, which promoted tumor T cell infiltration and antitumor activity. The use of this strategy may enhance the activity of CAR-T cells in individuals with stroma-rich solid tumors [128].
-
∗The above two papers show that heparanase plays a critical role in NK- and T- cell invasion into tumors. This might adversely limit the antitumor effectiveness of heparanase-inhibiting compounds.
-
See Chapter 17 in this volume for more information on heparanase in inflammation and cells of the immune system.
3.2.12 Vaccination
Heparanase: A New Metastasis-Associated Antigen Recognized in Breast Cancer Patients by Spontaneously Induced Memory T Lymphocytes
Increased expression and secretion of heparanase (Hpa) by tumor cells promotes tumor invasion, tissue destruction, angiogenesis, and metastasis. Here, we show the existence in breast cancer patients of Hpa-specific T lymphocytes by fluorescence-activated cell sorting flow cytometry using Hpa peptide-MHC class I tetramers. We furthermore show memory T-cell responses in a high proportion of breast cancer patients to Hpa-derived HLA-A2-restricted peptides, leading to production of IFN-gamma and to generation of antitumor CTLs lysing breast cancer cells. Such CTLs recognized endogenously processed respective Hpa peptides on Hpa-transfected and Hpa-expressing untransfected breast carcinoma cells. According to these results and to the fact that such cells were not found in healthy people, Hpa seems to be an attractive new tumor-associated antigen and its HLA-A2-restricted peptides ought to be good candidates for peptide vaccination to reactivate memory immune responses to invasive and metastatic cancer cells [129].
H-2Kb-Restricted CTL Epitopes from Mouse Heparanase Elicit an Antitumor Immune Response In Vivo
Heparanase is broadly expressed in various advanced tumors and can serve as a universal tumor-associated antigen. Although several epitopes of heparanase antigen are known in humans, the corresponding knowledge in mice is still rather limited. The present study was designed to predict and identify the CTL epitopes in the mouse heparanase protein. The results showed that, of the tested peptides, effectors induced by peptides of mouse heparanase at residue positions 398 to 405 (LSLLFKKL; mHpa398) and 519 to 526 (FSYGFFVI; mHpa519) lysed three kinds of carcinoma cells expressing both heparanase and H-2 K(b) (B16 melanoma cells, EL-4 lymphoma cells, and Lewis lung cancer cells). In vivo experiments indicated that mHpa398 and mHpa519 peptides offered the possibility of not only immunizing against tumors but also treating tumor-bearing hosts successfully. Our results suggest that the mHpa398 and mHpa519 peptides are novel H-2 K(b)-restricted CTL epitopes capable of inducing heparanase-specific CTLs in vitro and in vivo. These epitopes may serve as valuable tools for the preclinical evaluation of vaccination strategies [130].
-
∗Two representative papers indicating that heparanase is an attractive new tumor-associated antigen and its HLA-restricted peptides are good candidates for peptide vaccination to reactivate memory immune responses to invasive and metastatic cancer cells .
3.2.13 Diabetes, Diabetic Complications and Other Disorders
Heparan Sulfate and Heparanase Play Key Roles in Mouse β Cell Survival and Autoimmune Diabetes
The autoimmune type 1 diabetes (T1D) that arises spontaneously in NOD mice is considered to be a model of T1D in humans. It is characterized by the invasion of pancreatic islets by mononuclear cells (MNCs), which ultimately leads to destruction of insulin-producing β cells. Although T cell dependent, the molecular mechanisms triggering β cell death have not been fully elucidated. Here, we report that HS is expressed at extraordinarily high levels within mouse islets and is essential for β cell survival. In vitro, β cells rapidly lost their HS and died. β Cell death was prevented by HS replacement, a treatment that also rendered the β cells resistant to damage from ROS. In vivo, autoimmune destruction of islets in NOD mice was associated with production of catalytically active heparanase by islet-infiltrating MNCs and loss of islet HS. Furthermore, in vivo treatment with the heparanase inhibitor PI-88 preserved intra-islet HS and protected NOD mice from T1D. These results identified HS as a critical molecular requirement for islet β cell survival and HS degradation as a mechanism for β cell destruction. Hence, preservation of islet HS could be a therapeutic strategy for preventing T1D [131].
-
∗See Chapter 24 by Simeonovic et al. for more information on heparanase and immune diabetes.
Heparanase Is Essential for the Development of Diabetic Nephropathy in Mice
Diabetic nephropathy (DN) is the major life-threatening complication of diabetes. Abnormal permselectivity of glomerular basement membrane (GBM) plays an important role in DN pathogenesis. Loss of GBM HS in diabetic kidney was associated with increased glomerular expression of heparanase pointing to the essential involvement of heparanase in DN. With the use of Hpse-KO mice, we found that deletion of the heparanase gene protects diabetic mice from DN. Furthermore, by investigating the molecular mechanism underlying induction of the enzyme in DN, we found that transcription factor early growth response 1 (Egr1) is responsible for activation of heparanase promoter under diabetic conditions. The specific heparanase inhibitor SST0001 markedly decreased the extent of albuminuria and renal damage in mouse models of DN. Collectively these results underscore the crucial role of heparanase in the pathogenesis of DN and its potential as a highly relevant target for therapeutic interventions in patients with DN [132].
Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx
Diabetic nephropathy (DN) is the leading cause of CKD in the Western world. Endothelin receptor antagonists have emerged as a novel treatment for DN, but the mechanisms underlying the protective effect remain unknown. We previously showed that both heparanase and endothelin-1 are essential for the development of DN. Here, we further investigated the role of these proteins in DN, and demonstrated that endothelin-1 activates podocytes to release heparanase. Furthermore, conditioned podocyte culture medium increased glomerular transendothelial albumin passage in a heparanase-dependent manner. In mice, podocyte-specific knockout of the endothelin receptor prevented the diabetes-induced increase in glomerular heparanase expression, consequent reduction in heparan sulfate expression and endothelial glycocalyx thickness, and development of proteinuria observed in wild-type counterparts. Our data suggest that in diabetes, endothelin-1 signaling, as occurs in endothelial activation, induces heparanase expression in the podocyte, damage to the glycocalyx, proteinuria, and renal failure. Thus, prevention of these effects may constitute the mechanism of action of endothelin receptor blockers in DN [133].
-
∗These and other studies underscore the crucial role of heparanase in the pathogenesis of diabetic nephropathy.
-
∗See Chapters 27 and 28 by Massola et al., and by Abassi & Goligorsky, for more information on heparanase in fibrosis and kidney dysfunction [134, 135].
Endothelial Cell Heparanase Taken up by Cardiomyocytes Regulates Lipoprotein Lipase Transfer to the Coronary Lumen after Diabetes
After diabetes, the heart has a singular reliance on fatty acid (FA) for energy production, which is achieved by increased coronary lipoprotein lipase (LPL) that breaks down circulating triglycerides. Coronary LPL originates from cardiomyocytes, and to translocate to the vascular lumen, the enzyme requires liberation from myocyte surface HS, an activity that needs to be sustained after chronic hyperglycemia. We investigated the mechanism by which endothelial cells (EC) and cardiomyocytes operate together to enable continuous translocation of LPL after diabetes. EC were co-cultured with myocytes, exposed to high glucose, and uptake of endothelial heparanase into myocytes was determined. Upon uptake, the effect of nuclear entry of heparanase was also investigated. A streptozotocin model of diabetes was used to expand our in vitro observations. In high glucose, EC-derived latent heparanase was taken up by cardiomyocytes by a caveolae-dependent pathway using HSPGs. This latent heparanase was converted into an active form in myocyte lysosomes, entered the nucleus, and upregulated gene expression of matrix metalloproteinase-9. The net effect was increased shedding of HSPGs from the myocyte surface, releasing LPL for its onwards translocation to the coronary lumen. EC-derived heparanase regulates the ability of the cardiomyocyte to send LPL to the coronary lumen. This adaptation, although acutely beneficial, could be catastrophic chronically because excess FA causes lipotoxicity. Inhibiting heparanase function could offer a new strategy for managing cardiomyopathy observed after diabetes [136].
-
∗See also comment entitled ‘Heparanase shakes hands with lipoprotein lipase: a tale of two cells’ [137]. See Chapter 30 by Chang et al., for more information on heparanase and cardiomyocytes.
In Vivo Fragmentation of Heparan Sulfate by Heparanase Overexpression Renders Mice Resistant to Amyloid Protein a Amyloidosis
Amyloid diseases encompass >20 medical disorders that include amyloid protein A (AA) amyloidosis, Alzheimer’s disease, and type 2 diabetes. A common feature of these conditions is the selective organ deposition of disease-specific fibrillar proteins, along with the sulfated glycosaminoglycan, HS. We have tested the susceptibility of Hpa-tg mice to amyloid induction. Drastic shortening of HS chains was observed in heparanase-overproducing organs, such as liver and kidney. These sites selectively escaped amyloid deposition on experimental induction of inflammation-associated AA amyloidosis, whereas the same tissues from control animals were heavily infiltrated with amyloid. By contrast, the spleens of transgenic mice that failed to significantly overexpress heparanase remained susceptible to amyloid deposition. Our findings provide direct in vivo evidence that heparan sulfate is essential for the development of amyloid disease [138].
∗See Chapter 25 by Li & Zhang, for more information on heparanase and amyloidosis.
3.2.14 Aterosclerosis & Thrombosis
Heparanase Regulates Thrombosis in Vascular Injury and Stent-Induced Flow Disturbance
The purpose of this study was to examine the role of heparanase in controlling thrombosis following vascular injury or endovascular stenting. In the absence of vascular injury, wild type and heparanase overexpressing (HPA-Tg) mice had similar times to thrombosis in a laser-induced arterial thrombosis model. However, in the presence of vascular injury, the time to thrombosis was dramatically reduced in HPA-Tg mice. An ex vivo system was used to flow blood from wild type and HPA-Tg mice over stents and stented arterial segments from both animal types. These studies demonstrate markedly increased thromboses on stents with blood isolated from HPA-Tg mice in comparison to blood from wild type animals. We found that blood from HPA-Tg animals had markedly increased thrombosis when applied to stented arterial segments from either wild type or HPA-Tg mice [139].
-
∗These results indicate that heparanase is a powerful mediator of thrombosis in the context of vascular injury and stent-induced flow disturbance.
The Pulmonary Endothelial Glycocalyx Regulates Neutrophil Adhesion and Lung injury during Experimental Sepsis
Sepsis , a systemic inflammatory response to infection, commonly progresses to acute lung injury (ALI), an inflammatory lung disease with high morbidity. We postulated that sepsis-associated ALI is initiated by degradation of the pulmonary endothelial glycocalyx, leading to neutrophil adherence and inflammation. Using intravital microscopy, we found that endotoxemia in mice rapidly induced pulmonary microvascular glycocalyx degradation via tumor necrosis factor-α (TNF-α)-dependent mechanisms. Glycocalyx degradation involved the specific loss of heparan sulfate and coincided with activation of endothelial heparanase, a TNF-α-responsive, heparan sulfate-specific glucuronidase. Glycocalyx degradation increased the availability of endothelial surface adhesion molecules to circulating microspheres and contributed to neutrophil adhesion. Heparanase inhibition prevented endotoxemia-associated glycocalyx loss and neutrophil adhesion and, accordingly, attenuated sepsis-induced ALI and mortality in mice. These findings are potentially relevant to human disease, as sepsis-associated respiratory failure in humans was associated with higher plasma heparan sulfate degradation activity; moreover, heparanase content was higher in human lung biopsies showing diffuse alveolar damage than in normal human lung tissue [140].
3.2.15 Viral Infection
Heparanase Is a Host Enzyme Required for Herpes Simplex Virus-1 Release from Cells
Herpesviruses exemplified by herpes simplex virus-1 (HSV-1) attach to cell surface HS for entry into host cells. However, during a productive infection, the HS moieties on parent cells can trap newly exiting viral progenies and inhibit their release. Here we demonstrate that a HS-degrading enzyme of the host, heparanase (HPSE), is upregulated through NF-kB and translocated to the cell surface upon HSV-1 infection for the removal of HS to facilitate viral release. We also find a significant increase in HPSE release in vivo during infection of murine corneas and that knockdown of HPSE in vivo inhibits virus shedding. Overall, we propose that HPSE acts as a molecular switch for turning a virus-permissive ‘attachment mode’ of host cells to a virus-deterring ‘detachment mode’. Since many human viruses use HS as an attachment receptor, the HPSE-HS interplay may delineate a common mechanism for virus release [142].
-
∗See Chapter 32 by Agelidis & Shukla, for more information on heparanase and viral infection.
Note added in Proofs: Given that many human viruses use HS as an attachment receptor, non-anticoagulant heparin/HS mimicking compounds (i.e., Roneparstat) may compete with HS and thereby inhibit viral infection. This may be relevant to the recent coronavirus (COVID-19) pandemic as HS has been found to function as adhesion molecule that increases the virus density on the cell surface, possibly facilitating the interaction between the virus (HCoV-NL63) and its receptor [143].
4 Concluding Remarks and Perspectives
Heparanase exerts strong pro-tumorigenic properties, promoting all aspects of tumor development (tumor initiation, growth, and metastasis) and chemo-resistance. The enzyme is expressed by tumor cells and cells of the tumor microenvironment and functions extracellularly as well as inside the cell. Collectively, the emerging premise is that heparanase expressed by tumor cells and cells of the tumor microenvironment is a dominant regulator of the aggressive phenotype of cancer, an important contributor to the poor outcome of cancer patients and a prime target for therapy.
As the investigation of heparanase proceeds, new roles for the enzyme in diverse processes such as signal transduction, gene regulation, exosome formation, autophagy, activation of innate immune cells, chemo-resistance, are emerging and thus widening the impact of this enzyme [144]. It appears that heparanase functions extracellularly to remodel the ECM and regulate the bioavailability of ECM-bound factors, as well as inside the cell, augmenting, among other effects, gene transcription, and HS turnover. Unraveling these and other aspects of heparanase biology is ongoing and is critical to our understanding of its multiple functions in health and disease. Central to some of the downstream effects of heparanase is the enzyme’s ability to regulate gene transcription. At the molecular level, nuclear heparanase appears, among other effects, to regulate histone 3 lysine 4 (H3K4) methylation by influencing the recruitment of demethylases to transcriptionally active genes [105]. Yet, our understanding of heparanase nuclear accessibility and mode of action is far from being complete. An important challenge in the field rests on structure-based rational development of clinically effective inhibitors (heparin mimics, neutralizing antibodies, small molecules) of heparanase that will be applied to treat cancer, inflammation and other diseases. A limiting factor in anti-heparanase drug development is the lack of a high throughput screening and validation assays. Most assays continue to use heterogeneous substrates prepared by derivatization of HS or heparin/LMWH in various ways. These assays are limited by their heterogeneity, semi-quantitative nature, multiple enzyme cleavage sites and inappropriateness for use in biological samples. Advances in the synthesis of simple synthetic oligosaccharide substrates with a single point of cleavage will ultimately lead to a “gold standard” assay for detailed kinetic analyses [23]. Also, undesirable effects of anti-heparanase therapy should be considered. For example, it was recently reported that heparanase plays a critical role in NK cell invasion into tumors and thereby tumor progression and metastases. It was shown that cytokine and immune checkpoint blockade immunotherapy for metastases were compromised when NK cells lacked heparanase [127]. Likewise, it was found that in contrast to freshly isolated T lymphocytes, HPSE mRNA is downregulated in in vitro-expanded T cells. This may explain the reduced ability of cultured CAR-T cells to penetrate stroma-rich solid tumors compared with lymphoid tissues. In fact, engineering the CAR-T cells to express HPSE resulted in their improved capacity to degrade the ECM, which promoted tumor T cell infiltration and antitumor activity [128]. It was suggested that the use of this strategy might enhance the activity of CAR-T cells in individuals with stroma-rich solid tumors. These results should be considered when systemically treating cancer patients with heparanase inhibitors, since the potential adverse effect on NK and CAR-T cells on cell infiltration might limit the antitumor activity of the inhibitors [128]. Yet, these effects appear negligible given that heparanase knock-out mice exhibit no obvious immunological and other deficits [95], implying that inhibition of heparanase will cause minimal side effects in cancer patients. Remarkably, heparanase inhibitors were effective even when the xenografted tumor cells were devoid of heparanase, emphasizing the significance of heparanase contributed by cells residing in the tumor microenvironment. It appears that targeting the tumor microenvironment by heparanase inhibitors enhances the antitumor activity of approved therapies, further providing a strong rationale for applying anti-heparanase therapy in combination with conventional anti-cancer drugs.
Another topic of interest is the crosstalk between heparanase and heparanase 2 (Hpa2), a close homolog of heparanase devoid of heparanase enzymatic activity [145]. Unlike heparanase, the role played by Hpa2 in normal physiology and pathological disorders is still largely obscure. Hpa2 appears to attenuate tumor growth via inhibition of heparanase, decreasing the expression of pro-angiogenic mediators and inducing the manifestation of genes involved in tumor suppression and cell differentiation (see chapters by Mckenzie and by Roberts & Woolf in this book). It appears that Hpa2 functions in heparanase activity- and HS- dependent and independent manners, and regulates the expression of selected genes that affect tumor vascularity, tumor fibrosis, cell differentiation, and apoptosis of cancer cells, resulting in tumor suppression [144, 145,145,146,147,149]. Clearly, further research is needed in order to appreciate the scope of Hpa2 function and crosstalk with heparanase.
Personal Notes
I would like to close with a more personal note. I became interested in cell interaction with the ECM about 40 years ago just before finishing my postdoctoral research under the supervision of Prof. Denis Gospodarowicz (UCSF). We were among the first to realize that the ECM plays an active role in orchestrating cellular responses to both normal and pathological situations [148,149,150,152]. The emerging concept was one of an active interplay between cells and the ECM where cells synthesize matrix components which in turn dictate and regulate cell shape and function. The impact of these studies is clearly indicated by the current awareness of the ECM and the tumor microenvironment as key elements in the regulation of cell survival and cancer progression. It was only a few years after my return to Israel that I became interested in degradation of the ECM’ HS as a mean to understand better how tumor cells enter and exit blood vessels in the process of cancer metastasis. I decided to focus on HS knowing that this polyanionic polysaccharide binds to other constituents of the ECM and plays an important role in assembling and stabilizing the entire supramolecular structure of the ECM. It was then that I became interested in the heparanase enzyme, a topic that kept me busy since then. Our first papers on heparanase and cancer metastasis were published in 1983 [152], in parallel to studies reported by Dr. Nicolson et al. [44]. Next, we reported on the sequential mode of heparanase action in degrading the ECM [46], its inhibition by species of heparin [51, 153] and expression by platelets [154], neutrophils [155] and activated cells of the immune system. An important concept was brought about in studies performed during a sabbatical with Michael Klagsbrun and the late Judah Folkman (Harvard Medical Center) revealing that HS in the ECM provides a storage depot for FGF2, and hence heparanase regulates its bioavailability [16, 49, 50]. The concept of ECM as a reservoir for bioactive molecules became well recognized and is of prime importance to the current appreciation of the tumor microenvironment and its significance in cancer progression and treatment.
These and other studies further emphasized the need to purify the heparanase protein and clone the respective gene, an objective that took a long time and was finally achieved at 1999 [4] and published back to back with a study performed by the group of Cristopher Parish in Canberra [1]. Gene cloning enabled studies on the modes of heparanase gene regulation, cellular uptake, lysosomal storage and activation [31, 34, 144, 156, 157]. We and others further revealed the modes of heparanase action in eliciting angiogenesis, tumorigenesis and signal transduction. Thus, 20 years following its cloning, the functional repertoire of heparanase and the complexity of the system are still being revealed. From activity mainly implicated in cell invasion associated with tumor metastasis, heparanase has turned into a multifaceted protein that appears to participate in essentially all major aspects of tumor progression and in the pathogenesis of other diseases. Importantly, the crystal structure of the heparanase protein was resolved [87], paving the way for rational design and optimization of site-directed heparanase-inhibiting small molecules and monoclonal antibodies. As a direct result of these and subsequent studies performed by other groups and us, heparanase was advanced from being an obscure enzyme with a poorly understood function to a highly promising, novel drug target, offering new treatment strategies for various cancers and other diseases (i.e., chronic inflammation, diabetic nephropathy). The significance of heparanase as a valid target for anti-cancer drug development was reinforced by studies indicating a marked inhibition of human myeloma, lymphoma, glioma, sarcoma, mesothelioma and pancreatic tumor growth in mice treated with heparin-like heparanase-inhibiting compounds (i.e., SST0001 = Roneparstat, PG545 = Pixatimod) that are being examined in clinical trials alone and in combination with other drugs [159, 160]. Unfortunately, we are not yet there raising the question/dilemma if it was wise to focus and invest so much time, energy and effort in the study of a single molecule. Do we really understand the mode of heparanase action in tumorigenesis?; Should we adopt another way of thinking?; Should we focus on other diseases?; Where we objective and unbiased in our interpretations of the results?; What mistakes, if any, were done along the way?; Should the final goal of a basic scientist is to translate his findings into a drug?; Was it worth? etc. (see also Ilan et al., Chap. 9 in this volume). Obviously, there are no simple and clear cut answers. Focusing on one molecule is risky and, nowadays in the era of ‘big science’, is regarded as an ‘old fashion’ approach. Yet, the heparanase journey is actively ongoing and should be evaluated in the broad context of cell interaction with the ECM and tumor microenvironment. In fact, heparanase research markedly reinforced the significance of the ECM in the control of cell proliferation and differentiation [148,149,150,152]. It led to important and often unexpected observations in diverse normal and pathological processes including, wound healing, angiogenesis [63], autophagy [98], signal transduction [107, 156, 161], protein trafficking [162], lysosomal secretion [144, 163], blood coagulation [92, 164], epithelial-mesenchymal transition [165], activation of immune cells [21, 121,122,123], exosome formation [99, 100, 102], drug resistance [34, 166], gene transcription [35, 104] and others. While most studies emphasize the involvement of heparanase in cancer progression, other pathologies were investigated. Among these are diabetes [35, 131], diabetic complications (i.e., diabetic nephropathy, diabetic cardiomyopathy) [132, 136, 167], kidney dysfunction [38], fibrosis [134, 168], inflammatory disorders (i.e., neuroinflammation, pancreatitis, ulcerative colitis, arthritis, sepsis) [121, 140, 169, 170], amyloidosis [138], atherosclerosis [39, 40, 171] and others. Interestingly, heparanase accomplishes all these by exerting both enzymatic and non-enzymatic functions that are mostly HS-dependent yet in some cases are HS-independent [172]. In comparison with other ECM degrading enzymes (i.e., MMPs, cathepsins) [173, 174] the heparanase field is small, leaving enough room for other research groups to join and make significant contributions to basic and translational aspects. For example, little is known about a putative non-HS heparanase receptor [107, 174,175,176,178] that binds the enzyme and mediates its downstream signaling function. Likewise, in-depth research is needed to elucidate better the mode of heparanase nuclear translocation and transcriptional activity [104, 105], as well as its ability to activate macrophages and mediate their polarization [121,122,123,124]. An open area, not referred to in this review, has to do with heparanase-2 (Hpa2), a heparanase homolog devoid of heparanase enzymatic activity [146, 147, 178]. Of particular interest is to resolve the crystal structure of Hpa2, elucidate its mode of action as a tumor suppressor and reveal the significance of its crosstalk with heparanase [144]. Of prime importance from a translational point of view, is to apply the crystal structure of heparanase for rational design of heparanase-inhibiting small molecules, neutralizing monoclonal antibodies and HS-mimicking compounds endowed with good pharmacokinetics and examine their efficiency in animal models and cancer patients.
References
Hulett, M. D., Freeman, C., Hamdorf, B. J., Baker, R. T., Harris, M. J., & Parish, C. R. (1999). Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nature medicine., 5, 803–809.
Kussie, P. H., Hulmes, J. D., Ludwig, D. L., Patel, S., Navarro, E. C., Seddon, A. P., et al. (1999). Cloning and functional expression of a human heparanase gene. Biochemical and Biophysical Research Communications, 261, 183–187.
Toyoshima, M., & Nakajima, M. (1999). Human heparanase. Purification, characterization, cloning, and expression. The Journal of Biological Chemistry, 274, 24153–24160.
Vlodavsky, I., Friedmann, Y., Elkin, M., Aingorn, H., Atzmon, R., Ishai-Michaeli, R., et al. (1999). Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nature medicine., 5, 793–802.
Hopwood, J. J. (1989). In D. W. Lane & U. Lindahl (Eds.), Heparin-Chemical and biological properties and clinical applications (pp. 191–229). London: Edward Arnold.
Ogren, S., & Lindahl, U. (1975). Cleavage of macromolecular heparin by an enzyme from mouse mastocytoma. The Journal of Biological Chemistry, 250, 2690–2697.
Oldberg, A., Heldin, C. H., Wasteson, A., Busch, C., & Hook, M. (1980). Characterization of a platelet endoglycosidase degrading heparin-like polysaccharides. Biochemistry, 19, 5755–5762.
Hoogewerf, A. J., Leone, J. W., Reardon, I. M., Howe, W. J., Asa, D., Heinrikson, R. L., et al. (1995). CXC chemokines connective tissue activating peptide-III and neutrophil activating peptide-2 are heparin/heparan sulfate-degrading enzymes. The Journal of biological chemistry., 270, 3268–3277.
Oosta, G. M., Favreau, L. V., Beeler, D. L., & Rosenberg, R. D. (1982). Purification and properties of human platelet heparitinase. The Journal of biological chemistry., 257, 11249–11255.
Nakajima, M., Irimura, T., & Nicolson, G. L. (1988). Heparanases and tumor metastasis. Journal of Cellular Biochemistry, 36, 157–167.
Graham, L. D. (1994). Tumour rejection antigens of the hsp90 family (gp96) closely resemble tumour-associated heparanase enzymes. The Biochemical Journal, 301(Pt 3), 917–918.
Bame, K. J. (2001). Heparanases: Endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology, 11, 91R–98R.
Bame, K. J., Hassall, A., Sanderson, C., Venkatesan, I., & Sun, C. (1998). Partial purification of heparanase activities in Chinese hamster ovary cells: Evidence for multiple intracellular heparanases. The Biochemical Journal, 336(Pt 1), 191–200.
Eccles, S. A. (1999). Heparanase: Breaking down barriers in tumors. Nature medicine., 5, 735–736.
Finkel, E. (1999). Potential target found for antimetastasis drugs. Science, 285, 33–34.
Vlodavsky, I., Korner, G., Ishai-Michaeli, R., Bashkin, P., Bar-Shavit, R., & Fuks, Z. (1990). Extracellular matrix-resident growth factors and enzymes: Possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Reviews, 9, 203–226.
Chambers, A. F., & Matrisian, L. M. (1997). Changing views of the role of matrix metalloproteinases in metastasis. Journal of the National Cancer Institute, 89, 1260–1270.
Arvatz, G., Shafat, I., Levy-Adam, F., Ilan, N., & Vlodavsky, I. (2011). The heparanase system and tumor metastasis: Is heparanase the seed and soil? Cancer Metastasis Reviews, 30, 253–268.
Levy-Adam, F., Abboud-Jarrous, G., Guerrini, M., Beccati, D., Vlodavsky, I., & Ilan, N. (2005). Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. The Journal of Biological Chemistry, 280, 20457–20466.
Bartlett, M. R., Underwood, P. A., & Parish, C. R. (1995). Comparative analysis of the ability of leucocytes, endothelial cells and platelets to degrade the subendothelial basement membrane: Evidence for cytokine dependence and detection of a novel sulfatase. Immunology and Cell Biology, 73, 113–124.
Naparstek, Y., Cohen, I. R., Fuks, Z., & Vlodavsky, I. (1984). Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature, 310, 241–244.
Vlodavsky I. Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. Current protocols in cell biology. 2001;Chapter 10:Unit 10 4.
Chhabra, M., & Ferro, V. (2018). The development of assays for Heparanase enzymatic activity: Towards a gold standard. Molecules, 23.
Freeman, C., & Parish, C. R. (1998). Human platelet heparanase: Purification, characterization and catalytic activity. The Biochemical Journal, 330, 1341–1350.
Dong, J., Kukula, A. K., Toyoshima, M., & Nakajima, M. (2000). Genomic organization and chromosome localization of the newly identified human heparanase gene. Gene, 253, 171–178.
Miao, H. Q., Navarro, E., Patel, S., Sargent, D., Koo, H., Wan, H., et al. (2002). Cloning, expression, and purification of mouse heparanase. Protein Expression and Purification, 26, 425–431.
Parish, C. R., Freeman, C., Brown, K. J., Francis, D. J., & Cowden, W. B. (1999). Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Research., 59, 3433–3441.
Parish, C. R., Freeman, C., & Hulett, M. D. (2001). Heparanase: A key enzyme involved in cell invasion. Biochimica et Biophysica Acta, 1471, 99–108.
Basche, M., Gustafson, D. L., Holden, S. N., O’Bryant, C. L., Gore, L., Witta, S., et al. (2006). A phase I biological and pharmacologic study of the heparanase inhibitor PI-88 in patients with advanced solid tumors. Clinical Cancer Research, 12, 5471–5480.
Ritchie, J. P., Ramani, V. C., Ren, Y., Naggi, A., Torri, G., Casu, B., et al. (2011). SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clinical Cancer Research, 17, 1382–1393.
Rivara, S., Milazzo, F. M., & Giannini, G. (2016). Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Medicinal Chemistry, 8, 647–680.
Vlodavsky, I., Ilan, N., Naggi, A., & Casu, B. (2007). Heparanase: Structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Current Pharmaceutical Design, 13, 2057–2073.
Weissmann M, Arvatz G, Horowitz N, Feld S, Naroditsky I, Zhang Y, et al. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:704–709.
Vlodavsky, I., Singh, P., Boyango, I., Gutter-Kapon, L., Elkin, M., Sanderson, R. D., et al. (2016). Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resistance Updates, 29, 54–75.
Parish, C. R., Freeman, C., Ziolkowski, A. F., He, Y. Q., Sutcliffe, E. L., Zafar, A., et al. (2013). Unexpected new roles for heparanase in type 1 diabetes and immune gene regulation. Matrix Biology, 32, 228–233.
Simeonovic, C. J., Ziolkowski, A. F., Wu, Z., Choong, F. J., Freeman, C., & Parish, C. R. (2013). Heparanase and autoimmune diabetes. Frontiers in Immunology, 4, 471.
Wang, F., Jia, J., Lal, N., Zhang, D., Chiu, A. P., Wan, A., et al. (2016). High glucose facilitated endothelial heparanase transfer to the cardiomyocyte modifies its cell death signature. Cardiovascular Research, 112, 656–668.
Rabelink, T. J., van den Berg, B. M., Garsen, M., Wang, G., Elkin, M., & van der Vlag, J. (2017). Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nature Reviews. Nephrology, 13, 201–212.
Blich M, Golan A, Arvatz G, Sebbag A, Shafat I, Sabo E, et al. Macrophage activation by heparanase is mediated by TLR-2 and TLR-4 and associates with plaque progression. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33:e56–65.
Vlodavsky, I., Blich, M., Li, J. P., Sanderson, R. D., & Ilan, N. (2013). Involvement of heparanase in atherosclerosis and other vessel wall pathologies. Matrix Biology: Journal of the International Society for Matrix Biology., 32, 241–251.
Thakkar, N., Yadavalli, T., Jaishankar, D., & Shukla, D. (2017). Emerging roles of heparanase in viral pathogenesis. Pathogens., 6.
Ogren, S., & Lindahl, U. (1971). Degradation of heparin in mouse mastocytoma tissue. The Biochemical Journal, 125, 1119–1129.
Hook, M., Wasteson, A., & Oldberg, A. (1975). A heparan sulfate-degrading endoglycosidase from rat liver tissue. Biochemical and Biophysical Research Communications, 67, 1422–1428.
Nakajima, M., Irimura, T., DiFerrante, D., DiFerrante, N., & Nicolson, G. L. (1983). Heparan sulfate degradation: relation to tumor invasion and metastatic properties of Mouse B 16 Melanoma sublines. Science (Wash DC)., 6220, 611–613.
Vlodavsky, I., Fuks, Z., Bar-Ner, M., Ariav, Y., & Schirrmacher, V. (1983). Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: Relationship to tumor cell metastasis. Cancer Research., 43, 2704–2711.
Bar-Ner, M., Kramer, M. D., Schirrmacher, V., Ishai-Michaeli, R., Fuks, Z., & Vlodavsky, I. (1985). Sequential degradation of heparan sulfate in the subendothelial extracellular matrix by highly metastatic lymphoma cells. International Journal of Cancer, 35, 483–491.
Bar-Ner, M., Mayer, M., Schirrmacher, V., & Vlodavsky, I. (1986). Involvement of both heparanase and plasminogen activator in lymphoma cell-mediated degradation of heparan sulfate in the subendothelial extracellular matrix. Journal of Cellular Physiology, 128, 299–306.
Korner, G., Bjornsson, T. D., & Vlodavsky, I. (1993). Extracellular matrix produced by cultured corneal and aortic endothelial cells contains active tissue-type and urokinase-type plasminogen activators. Journal of Cellular Physiology, 154, 456–465.
Vlodavsky I, Folkman J, Sullivan R, Fridman R, Ishai-Michaeli R, Sasse J, et al. Endothelial cell-derived basic fibroblast growth factor: Synthesis and deposition into subendothelial extracellular matrix. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2292–2296.
Folkman, J., Klagsbrun, M., Sasse, J., Wadzinski, M., Ingber, D., & Vlodavsky, I. (1988). A heparin-binding angiogenic protein–basic fibroblast growth factor–is stored within basement membrane. The American Journal of Pathology, 130, 393–400.
Bar-Ner, M., Eldor, A., Wasserman, L., Matzner, Y., Cohen, I. R., Fuks, Z., et al. (1987). Inhibition of heparanase-mediated degradation of extracellular matrix heparan sulfate by non-anticoagulant heparin species. Blood, 70, 551–557.
Parish, C. R., Coombe, D. R., Jakobsen, K. B., Bennett, F. A., & Underwood, P. A. (1987). Evidence that sulphated polysaccharides inhibit tumour metastasis by blocking tumour-cell-derived heparanases. International Journal of Cancer, 40, 511–518.
Willenborg, D. O., & Parish, C. R. (1988). Inhibition of allergic encephalomyelitis in rats by treatment with sulfated polysaccharides. Journal of Immunology., 140, 3401–3405.
Lider, O., Baharav, E., Mekori, Y. A., Miller, T., Naparstek, Y., Vlodavsky, I., et al. (1989). Suppression of experimental autoimmune diseases and prolongation of allograft survival by treatment of animals with low doses of heparins. The Journal of Clinical Investigation., 83, 752–756.
Gilat, D., Hershkoviz, R., Goldkorn, I., Cahalon, L., Korner, G., Vlodavsky, I., et al. (1995). Molecular behavior adapts to context: Heparanase functions as an extracellular matrix-degrading enzyme or as a T cell adhesion molecule, depending on the local pH. The Journal of Experimental Medicine., 181, 1929–1934.
Boyd, D. D., & Nakajima, M. (2004). Involvement of heparanase in tumor metastases: A new target in cancer therapy? Journal of the National Cancer Institute, 96, 1194–1195.
Edovitsky, E., Elkin, M., Zcharia, E., Peretz, T., & Vlodavsky, I. (2004). Heparanase gene silencing, tumor invasiveness, angiogenesis, and metastasis. Journal of the National Cancer Institute, 96, 1219–1230.
Goldshmidt, O., Zcharia, E., Abramovitch, R., Metzger, S., Aingorn, H., Friedmann, Y., et al. (2002). Cell surface expression and secretion of heparanase markedly promote tumor angiogenesis and metastasis. PNAS, 99, 10031–10036.
Uno, F., Fujiwara, T., Takata, Y., Ohtani, S., Katsuda, K., Takaoka, M., et al. (2001). Antisense-mediated suppression of human heparanase gene expression inhibits pleural dissemination of human cancer cells. Cancer Research., 61, 7855–7860.
Kruger, A., Soeltl, R., Sopov, I., Kopitz, C., Arlt, M., Magdolen, V., et al. (2001). Hydroxamate-type matrix metalloproteinase inhibitor batimastat promotes liver metastasis. Cancer Research., 61, 1272–1275.
Holst-Hansen, C., Low, J. A., Stephens, R. W., Johnson, M. D., Carmeliet, P., Frandsen, T. L., et al. (2001). Increased stromal expression of murine urokinase plasminogen activator in a human breast cancer xenograft model following treatment with the matrix metalloprotease inhibitor, batimastat. Breast Cancer Research and Treatment, 68, 225–237.
Edovitsky, E., Lerner, I., Zcharia, E., Peretz, T., Vlodavsky, I., & Elkin, M. (2006). Role of endothelial heparanase in delayed-type hypersensitivity. Blood, 107, 3609–3616.
Elkin, M., Ilan, N., Ishai-Michaeli, R., Friedmann, Y., Papo, O., Pecker, I., et al. (2001). Heparanase as mediator of angiogenesis: Mode of action. The FASEB Journal, 15, 1661–1663.
de Mestre, A. M., Khachigian, L. M., Santiago, F. S., Staykova, M. A., & Hulett, M. D. (2003). Regulation of inducible heparanase gene transcription in activated T cells by early growth response 1. The Journal of Biological Chemistry, 278, 50377–50385.
de Mestre, A. M., Rao, S., Hornby, J. R., Soe-Htwe, T., Khachigian, L. M., & Hulett, M. D. (2005). Early growth response gene 1 (EGR1) regulates Heparanase gene transcription in tumor cells. The Journal of Biological Chemistry, 280, 35136–35147.
Yan, S. F., Lu, J., Zou, Y. S., Soh-Won, J., Cohen, D. M., Buttrick, P. M., et al. (1999). Hypoxia-associated induction of early growth response-1 gene expression. The Journal of biological chemistry., 274, 15030–15040.
Fahmy, R. G., Dass, C. R., Sun, L. Q., Chesterman, C. N., & Khachigian, L. M. (2003). Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nature Medicine., 9, 1026–1032.
Ogishima, T., Shiina, H., Breault, J. E., Tabatabai, L., Bassett, W. W., Enokida, H., et al. (2005). Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clinical Cancer Research, 11, 1028–1036.
Ogishima, T., Shiina, H., Breault, J. E., Terashima, M., Honda, S., Enokida, H., et al. (2005). Promoter CpG hypomethylation and transcription factor EGR1 hyperactivate heparanase expression in bladder cancer. Oncogene, 24, 6765–6772.
Elkin, M., Cohen, I., Zcharia, E., Orgel, A., Guatta-Rangini, Z., Peretz, T., et al. (2003). Regulation of heparanase gene expression by estrogen in breast cancer. Cancer Research., 63, 8821–8826.
Baraz, L., Haupt, Y., Elkin, M., Peretz, T., & Vlodavsky, I. (2006). Tumor suppressor p53 regulates heparanase gene expression. Oncogene, 25, 3939–3947.
Shteper, P. J., Zcharia, E., Ashhab, Y., Peretz, T., Vlodavsky, I., & Ben-Yehuda, D. (2003). Role of promoter methylation in regulation of the mammalian heparanase gene. Oncogene, 22, 7737–7749.
Abboud-Jarrous, G., Atzmon, R., Peretz, T., Palermo, C., Gadea, B. B., Joyce, J. A., et al. (2008). Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. The Journal of Biological Chemistry, 283, 18167–18176.
Jean, D., Rousselet, N., & Frade, R. (2006). Expression of cathepsin L in human tumor cells is under the control of distinct regulatory mechanisms. Oncogene, 25, 1474–1484.
Ishidoh, K., Taniguchi, S., & Kominami, E. (1997). Egr family member proteins are involved in the activation of the cathepsin L gene in v-src-transformed cells. Biochemical and Biophysical Research Communications, 238, 665–669.
Joyce, J. A., Freeman, C., Meyer-Morse, N., Parish, C. R., & Hanahan, D. (2005). A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene, 24, 4037–4051.
Jean, D., Rousselet, N., & Frade, R. (2008). Cathepsin L expression is up-regulated by hypoxia in human melanoma cells: Role of its 5′-untranslated region. The Biochemical Journal, 413, 125–134.
Arvatz, G., Barash, U., Nativ, O., Ilan, N., & Vlodavsky, I. (2011). Post-transcriptional regulation of heparanase gene expression by a 3' AU-rich element. The FASEB Journal, 24, 4969–4976.
Fairbanks, M. B., Mildner, A. M., Leone, J. W., Cavey, G. S., Mathews, W. R., Drong, R. F., et al. (1999). Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. The Journal of Biological Chemistry, 274, 29587–29590.
Hulett, M. D., Hornby, J. R., Ohms, S. J., Zuegg, J., Freeman, C., Gready, J. E., et al. (2000). Identification of active-site residues of the pro-metastatic endoglycosidase heparanase. Biochemistry, 39, 15659–15667.
McKenzie, E., Young, K., Hircock, M., Bennett, J., Bhaman, M., Felix, R., et al. (2003). Biochemical characterization of the active heterodimer form of human heparanase (Hpa1) protein expressed in insect cells. The Biochemical Journal, 373, 423–435.
Levy-Adam, F., Miao, H. Q., Heinrikson, R. L., Vlodavsky, I., & Ilan, N. (2003). Heterodimer formation is essential for heparanase enzymatic activity. Biochemical and Biophysical Research Communications, 308, 885–891.
Nardella, C., Lahm, A., Pallaoro, M., Brunetti, M., Vannini, A., & Steinkuhler, C. (2004). Mechanism of activation of human heparanase investigated by protein engineering. Biochemistry, 43, 1862–1873.
Simizu, S., Suzuki, T., Muroi, M., Lai, N. S., Takagi, S., Dohmae, N., et al. (2007). Involvement of disulfide bond formation in the activation of heparanase. Cancer Research., 67, 7841–7849.
Zetser, A., Levy-Adam, F., Kaplan, V., Gingis-Velitski, S., Bashenko, Y., Schubert, S., et al. (2004). Processing and activation of latent heparanase occurs in lysosomes. Journal of Cell Science., 117, 2249–2258.
Abboud-Jarrous, G., Rangini-Guetta, Z., Aingorn, H., Atzmon, R., Elgavish, S., Peretz, T., et al. (2005). Site-directed mutagenesis, proteolytic cleavage, and activation of human proheparanase. The Journal of Biological Chemistry, 280, 13568–13575.
Wu, L., Viola, C. M., Brzozowski, A. M., & Davies, G. J. (2015). Structural characterization of human heparanase reveals insights into substrate recognition. Nature Structural & Molecular Biology, 22, 1016–1022.
Wu, L., Jiang, J., Jin, Y., Kallemeijn, W. W., Kuo, C. L., Artola, M., et al. (2017). Activity-based probes for functional interrogation of retaining beta-glucuronidases. Nat Chem Biol.
Jiang, P., Kumar, A., Parrillo, J. E., Dempsey, L. A., Platt, J. L., Prinz, R. A., et al. (2002). Cloning and characterization of the human heparanase-1 (HPR1) gene promoter: Role of GA-binding protein and Sp1 in regulating HPR1 basal promoter activity. The Journal of Biological Chemistry., 277, 8989–8998.
Zhang, L., Sullivan, P. S., Goodman, J. C., Gunaratne, P. H., & Marchetti, D. (2011). MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Research., 71, 645–654.
Ostrovsky, O., Shimoni, A., Rand, A., Vlodavsky, I., & Nagler, A. (2010). Genetic variations in the heparanase gene (HPSE) associate with increased risk of GVHD following allogeneic stem cell transplantation: Effect of discrepancy between recipients and donors. Blood, 115, 2319–2328.
Nadir, Y., Brenner, B., Gingis-Velitski, S., Levy-Adam, F., Ilan, N., Zcharia, E., et al. (2008). Heparanase induces tissue factor pathway inhibitor expression and extracellular accumulation in endothelial and tumor cells. Thrombosis and Haemostasis, 99, 133–141.
Zcharia, E., Metzger, S., Chajek-ShaulL, T., Aingorn, H., Elikn, M., Friedmann, Y., et al. (2004). Transgenic expression of mammalian heparanase uncovers physiological functions of heparan sulfate in tissue morphogenesis, vascularization, and feeding behavior. The FASEB Journal, 18, 252–263.
Escobar Galvis, M. L., Jia, J., Zhang, X., Jastrebova, N., Spillmann, D., Gottfridsson, E., et al. (2007). Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nature Chemical Biology, 3, 773–778.
Zcharia, E., Jia, J., Zhang, X., Baraz, L., Lindahl, U., Peretz, T., et al. (2009). Newly generated heparanase knock-out mice unravel co-regulation of heparanase and matrix metalloproteinases. PLoS One, 4, e5181.
Poon, I. K., Goodall, K. J., Phipps, S., Chow, J. D., Pagler, E. B., Andrews, D. M., et al. (2014). Mice deficient in heparanase exhibit impaired dendritic cell migration and reduced airway inflammation. European Journal of Immunology, 44, 1016–1030.
Gingis-Velitski, S., Zetser, A., Kaplan, V., Ben-Zaken, O., Cohen, E., Levy-Adam, F., et al. (2004). Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. The Journal of Biological Chemistry, 279, 44084–44092.
Shteingauz, A., Boyango, I., Naroditsky, I., Hammond, E., Gruber, M., Doweck, I., et al. (2015). Heparanase enhances tumor growth and chemoresistance by promoting autophagy. Cancer research., 75, 3946–3957.
Thompson, C. A., Purushothaman, A., Ramani, V. C., Vlodavsky, I., & Sanderson, R. D. (2013). Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. The Journal of Biological Chemistry, 288, 10093–10099.
Roucourt, B., Meeussen, S., Bao, J., Zimmermann, P., & David, G. (2015). Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Research, 25, 412–428.
David, G., & Zimmermann, P. (2016). Heparanase tailors syndecan for exosome production. Molecular & Cellular Oncology, 3, e1047556.
Sanderson RD, Bandari SK, Vlodavsky I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix biology 2019;75–76:160–9.
Bandari, S. K., Purushothaman, A., & Ramani, V. C. (2017). Brinkley GJ. Varambally S, et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix biology: Chandrashekar DS.
Purushothaman, A., Hurst, D. R., Pisano, C., Mizumoto, S., Sugahara, K., & Sanderson, R. D. (2011). Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. The Journal of Biological Chemistry, 286, 30377–30383.
He, Y. Q., Sutcliffe, E. L., Bunting, K. L., Li, J., Goodall, K. J., Poon, I. K., et al. (2012). The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates H3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription, 3, 130–145.
Zetser, A., Bashenko, Y., Edovitsky, E., Levy-Adam, F., Vlodavsky, I., & Ilan, N. (2006). Heparanase induces vascular endothelial growth factor expression: Correlation with p38 phosphorylation levels and Src activation. Cancer Research., 66, 1455–1463.
Fux, L., Feibish, N., Cohen-Kaplan, V., Gingis-Velitski, S., Feld, S., Geffen, C., et al. (2009). Structure-function approach identifies a COOH-terminal domain that mediates heparanase signaling. Cancer Research., 69, 1758–1767.
Cohen-Kaplan, V., Doweck, I., Naroditsky, I., Vlodavsky, I., & Ilan, N. (2008). Heparanase augments epidermal growth factor receptor phosphorylation: Correlation with head and neck tumor progression. Cancer Research., 68, 10077–10085.
Cohen-Kaplan, V., Jrbashyan, J., Yanir, Y., Naroditsky, I., Ben-Izhak, O., Ilan, N., et al. (2012). Heparanase induces signal transducer and activator of transcription (STAT) protein phosphorylation: Preclinical and clinical significance in head and neck cancer. The Journal of Biological Chemistry, 287, 6668–6678.
Hammond, E., Li, C. P., & Ferro, V. (2010). Development of a colorimetric assay for heparanase activity suitable for kinetic analysis and inhibitor screening. Analytical Biochemistry, 396, 112–116.
Dredge, K., Hammond, E., Handley, P., Gonda, T. J., Smith, M. T., Vincent, C., et al. (2011). PG545, a dual heparanase and angiogenesis inhibitor, induces potent anti-tumour and anti-metastatic efficacy in preclinical models. British Journal of Cancer, 104, 635–642.
Zhou, H., Roy, S., Cochran, E., Zouaoui, R., Chu, C. L., Duffner, J., et al. (2011). M402, a novel heparan sulfate mimetic, targets multiple pathways implicated in tumor progression and metastasis. PLoS One, 6, e21106.
Spyrou, A., Kundu, S., Haseeb, L., Yu, D., Olofsson, T., Dredge, K., et al. (2017). Inhibition of heparanase in pediatric brain tumor cells attenuates their proliferation, invasive capacity, and in vivo tumor growth. Molecular Cancer Therapeutics, 16, 1705–1716.
Barash, U., Lapidot, M., Zohar, Y., Loomis, C., Moreira, A., Feld, S., et al. (2018). Involvement of Heparanase in the pathogenesis of mesothelioma: Basic aspects and clinical applications. Journal of the National Cancer Institute.
Yang, Y., Macleod, V., Bendre, M., Huang, Y., Theus, A. M., Miao, H. Q., et al. (2005). Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood, 105, 1303–1309.
Mahtouk, K., Hose, D., Raynaud, P., Hundemer, M., Jourdan, M., Jourdan, E., et al. (2007). Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood, 109, 4914–4923.
Barash, U., Zohar, Y., Wildbaum, G., Beider, K., Nagler, A., Karin, N., et al. (2014). Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia, 28, 2178–2187.
Ramani, V. C., Vlodavsky, I., Ng, M., Zhang, Y., Barbieri, P., Noseda, A., et al. (2016). Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biology, 55, 22–34.
Cassinelli, G., Lanzi, C., Tortoreto, M., Cominetti, D., Petrangolini, G., Favini, E., et al. (2013). Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochemical Pharmacology, 85, 1424–1432.
Boyango, I., Barash, U., Naroditsky, I., Li, J. P., Hammond, E., Ilan, N., et al. (2014). Heparanase cooperates with Ras to drive breast and skin tumorigenesis. Cancer Research., 74, 4504–4514.
Lerner, I., Hermano, E., Zcharia, E., Rodkin, D., Bulvik, R., Doviner, V., et al. (2011). Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. The Journal of Clinical Investigation., 121, 1709–1721.
Goodall, K. J., Poon, I. K., Phipps, S., & Hulett, M. D. (2014). Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS One, 9, e109596.
Gutter-Kapon L, Alishekevitz D, Shaked Y, Li JP, Aronheim A, Ilan N, et al. Heparanase is required for activation and function of macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E7808-E7E17.
Hermano, E., Meirovitz, A., Meir, K., Nussbaum, G., Appelbaum, L., Peretz, T., et al. (2014). Macrophage polarization in pancreatic carcinoma: Role of heparanase enzyme. Journal of the National Cancer Institute, 106.
Brennan, T. V., Lin, L., Brandstadter, J. D., Rendell, V. R., Dredge, K., Huang, X., et al. (2016). Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. The Journal of Clinical Investigation, 126, 207–219.
Tsunekawa, N., Higashi, N., Kogane, Y., Waki, M., Shida, H., Nishimura, Y., et al. (2016). Heparanase augments inflammatory chemokine production from colorectal carcinoma cell lines. Biochemical and Biophysical Research Communications, 469, 878–883.
Putz, E. M., Mayfosh, A. J., Kos, K., Barkauskas, D. S., Nakamura, K., Town, L., et al. (2017). NK cell heparanase controls tumor invasion and immune surveillance. The Journal of Clinical Investigation., 127, 2777–2788.
Caruana, I., Savoldo, B., Hoyos, V., Weber, G., Liu, H., Kim, E. S., et al. (2015). Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature Medicine, 21, 524–529.
Sommerfeldt, N., Beckhove, P., Ge, Y., Schutz, F., Choi, C., Bucur, M., et al. (2006). Heparanase: A new metastasis-associated antigen recognized in breast cancer patients by spontaneously induced memory T lymphocytes. Cancer Research, 66, 7716–7723.
Tang, X. D., Wan, Y., Chen, L., Chen, T., Yu, S. T., Xiong, Z., et al. (2008). H-2Kb-restricted CTL epitopes from mouse heparanase elicit an antitumor immune response in vivo. Cancer Research, 68, 1529–1537.
Ziolkowski, A. F., Popp, S. K., Freeman, C., Parish, C. R., & Simeonovic, C. J. (2012). Heparan sulfate and heparanase play key roles in mouse beta cell survival and autoimmune diabetes. The Journal of Clinical Investigation., 122, 132–141.
Gil, N., Goldberg, R., Neuman, T., Garsen, M., Zcharia, E., Rubinstein, A. M., et al. (2012). Heparanase is essential for the development of diabetic nephropathy in mice. Diabetes, 61, 208–216.
Garsen, M., Lenoir, O., Rops, A. L., Dijkman, H. B., Willemsen, B., van Kuppevelt, T. H., et al. (2016). Endothelin-1 induces proteinuria by Heparanase-mediated disruption of the glomerular Glycocalyx. Journal of the American Society of Nephrology: JASN., 27, 3545–3551.
Masola, V., Zaza, G., Onisto, M., Lupo, A., & Gambaro, G. (2015). Impact of heparanase on renal fibrosis. Journal of Translational Medicine, 13, 181.
van den Hoven, M. J., Rops, A. L., Vlodavsky, I., Levidiotis, V., Berden, J. H., & van der Vlag, J. (2007). Heparanase in glomerular diseases. Kidney International, 72, 543–548.
Wang, Y., Chiu, A. P., Neumaier, K., Wang, F., Zhang, D., Hussein, B., et al. (2014). Endothelial cell heparanase taken up by cardiomyocytes regulates lipoprotein lipase transfer to the coronary lumen after diabetes. Diabetes, 63, 2643–2655.
Chakrabarti, S. (2014). Heparanase shakes hands with lipoprotein lipase: A tale of two cells. Diabetes, 63, 2600–2602.
Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, et al. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein a amyloidosis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:6473–6477.
Baker, A. B., Gibson, W. J., Kolachalama, V. B., Golomb, M., Indolfi, L., Spruell, C., et al. (2012). Heparanase regulates thrombosis in vascular injury and stent-induced flow disturbance. Journal of the American College of Cardiology., 59, 1551–1560.
Schmidt, E. P., Yang, Y., Janssen, W. J., Gandjeva, A., Perez, M. J., Barthel, L., et al. (2012). The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nature medicine. 18(8):1217–23
Garsen, M., Rops, A. L., Rabelink, T. J., Berden, J. H., & van der Vlag, J. (2014). The role of heparanase and the endothelial glycocalyx in the development of proteinuria. Nephrology, Dialysis, Transplantation, 29, 49–55.
Hadigal, S. R., Agelidis, A. M., Karasneh, G. A., Antoine, T. E., Yakoub, A. M., Ramani, V. C., et al. (2015). Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nature Communications, 6, 6985.
Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K. (2014). Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J Virol. 88:13221–30.
Vlodavsky, I., Gross-Cohen, M., Weissmann, M., Ilan, N., & Sanderson, R. D. (2018). Opposing functions of Heparanase-1 and Heparanase-2 in Cancer progression. Trends in Biochemical Sciences, 43, 18–31.
McKenzie, E., Tyson, K., Stamps, A., Smith, P., Turner, P., Barry, R., et al. (2000). Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochemical and Biophysical Research Communications, 276, 1170–1177.
Gross-Cohen, M., Feld, S., Doweck, I., Neufeld, G., Hasson, P., Arvatz, G., et al. (2016). Heparanase 2 attenuates head and neck tumor vascularity and growth. Cancer Research., 76, 2791–2801.
Gross-Cohen, M., Feld, S., Naroditsky, I., Nativ, O., Ilan, N., & Vlodavsky, I. (2016). Heparanase 2 expression inversely correlates with bladder carcinoma grade and stage. Oncotarget, 7, 22556–22565.
Levy-Adam, F., Feld, S., Cohen-Kaplan, V., Shteingauz, A., Gross, M., Arvatz, G., et al. (2010). Heparanase 2 interacts with heparan sulfate with high affinity and inhibits heparanase activity. The Journal of Biological Chemistry, 285, 28010–28019.
Gospodarowicz D, Delgado D, Vlodavsky I. Permissive effect of the extracellular matrix on cell proliferation in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:4094–4098.
Vlodavsky, I., & Gospodarowicz, D. (1981). Respective roles of laminin and fibronectin in adhesion of human carcinoma and sarcoma cells. Nature, 289, 304–306.
Vlodavsky, I., Lui, G. M., & Gospodarowicz, D. (1980). Morphological appearance, growth behavior and migratory activity of human tumor cells maintained on extracellular matrix versus plastic. Cell, 19, 607–616.
Vlodavsky, I., Fuks, Z., Bar-Ner, M., Ariav, Y., & Schirrmacher, V. (1983). Lymphoma cells mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: Relation to tumor cell metastasis. Cancer Research., 43, 2704–2711.
Naggi, A., Casu, B., Perez, M., Torri, G., Cassinelli, G., Penco, S., et al. (2005). Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. The Journal of Biological Chemistry, 280, 12103–12113.
Yahalom, J., Eldor, A., Fuks, Z., & Vlodavsky, I. (1984). Degradation of sulfated proteoglycans in the subendothelial extracellular matrix by human platelet heparitinase. The Journal of Clinical Investigation., 74, 1842–1849.
Matzner, Y., Bar-Ner, M., Yahalom, J., Ishai-Michaeli, R., Fuks, Z., & Vlodavsky, I. (1985). Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. The Journal of Clinical Investigation., 76, 1306–1313.
Ilan, N., Elkin, M., & Vlodavsky, I. (2006). Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. The International Journal of Biochemistry & Cell Biology., 38, 2018–2039.
Vreys, V., & David, G. (2007). Mammalian heparanase: What is the message? Journal of Cellular and Molecular Medicine., 11, 427–452.
Dredge, K., Brennan, T. V., Hammond, E., Lickliter, J. D., Lin, L., Bampton, D., et al. (2018). A phase I study of the novel immunomodulatory agent PG545 (pixatimod) in subjects with advanced solid tumours. British Journal of Cancer, 118, 1035–1041.
Galli, M., Chatterjee, M., Grasso, M., Specchia, G., Magen, H., Einsele, H., et al. (2018). Phase I study of the heparanase inhibitor roneparstat: An innovative approach for multiple myeloma therapy. Haematologica, 103, e469–ee72.
Hammond E, Haynes NM, Cullinane C, Brennan TV, Bampton D, Handley P, Karoli T, Lanksheer F, Lin L, Yang Y, Dredge K. (2018). Immunomodulatory activities of pixatimod: emerging nonclinical and clinical data, and its potential utility in combination with PD-1 inhibitors. J Immunother Cancer, 6(1), 54
Riaz, A., Ilan, N., Vlodavsky, I., Li, J. P., & Johansson, S. (2013). Characterization of heparanase-induced phosphatidylinositol 3-kinase-AKT activation and its integrin dependence. The Journal of Biological Chemistry, 288, 12366–12375.
Nadav, L., Eldor, A., Yacoby-Zeevi, O., Zamir, E., Pecker, I., Ilan, N., et al. (2002). Activation, processing and trafficking of extracellular heparanase by primary human fibroblasts. Journal of Cell Science., 115, 2179–2187.
Shafat, I., Vlodavsky, I., & Ilan, N. (2006). Characterization of mechanisms involved in secretion of active heparanase. The Journal of Biological Chemistry, 281, 23804–23811.
Nadir, Y., Brenner, B., Fux, L., Shafat, I., Attias, J., & Vlodavsky, I. (2010). Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica, 95, 1927–1934.
Masola, V., Zaza, G., Gambaro, G., Onisto, M., Bellin, G., Vischini, G., et al. (2016). Heparanase: A potential new factor involved in the renal epithelial mesenchymal transition (EMT) induced by ischemia/reperfusion (I/R) injury. PLoS One, 11, e0160074.
Sanderson, R. D., Elkin, M., Rapraeger, A. C., Ilan, N., & Vlodavsky, I. (2017). Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. The FEBS Journal., 284, 42–55.
Wan, A., & Rodrigues, B. (2016). Endothelial cell-cardiomyocyte crosstalk in diabetic cardiomyopathy. Cardiovascular Research, 111, 172–183.
Masola, V., Zaza, G., Secchi, M. F., Gambaro, G., Lupo, A., & Onisto, M. (1843). Heparanase is a key player in renal fibrosis by regulating TGF-beta expression and activity. Biochimica et Biophysica Acta, 2014, 2122–2128.
Li, J. P., & Vlodavsky, I. (2009). Heparin, heparan sulfate and heparanase in inflammatory reactions. Thrombosis and Haemostasis, 102, 823–828.
Zhang, X., Wang, B., & Li, J. P. (2014). Implications of heparan sulfate and heparanase in neuroinflammation. Matrix Biology: Journal of the International Society for Matrix Biology., 35, 174–181.
Osterholm, C., Folkersen, L., Lengquist, M., Ponten, F., Renne, T., Li, J., et al. (2012). Increased expression of heparanase in symptomatic carotid atherosclerosis. Atherosclerosis.
Gingis-Velitski, S., Zetser, A., Flugelman, M. Y., Vlodavsky, I., & Ilan, N. (2004). Heparanase induces endothelial cell migration via protein kinase B/Akt activation. The Journal of Biological Chemistry, 279, 23536–23541.
Kessenbrock, K., Plaks, V., & Werb, Z. (2010). Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell, 141, 52–67.
Olson, O. C., & Joyce, J. A. (2015). Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nature Reviews. Cancer, 15, 712–729.
Fux, L., Ilan, N., Sanderson, R. D., & Vlodavsky, I. (2009). Heparanase: Busy at the cell surface. Trends in Biochemical Sciences, 34, 511–519.
Wood, R. J., & Hulett, M. D. (2008). Cell surface-expressed cation-independent mannose 6-phosphate receptor (CD222) binds enzymatically active heparanase independently of mannose 6-phosphate to promote extracellular matrix degradation. The Journal of Biological Chemistry, 283, 4165–4176.
Vreys, V., Delande, N., Zhang, Z., Coomans, C., Roebroek, A., Durr, J., et al. (2005). Cellular uptake of mammalian heparanase precursor involves low density lipoprotein receptor-related proteins, mannose 6-phosphate receptors, and heparan sulfate proteoglycans. The Journal of Biological Chemistry., 280, 33141–33148.
Woolf, A. S., Stuart, H. M., Roberts, N. A., McKenzie, E. A., Hilton, E. N., & Newman, W. G. (2014). Urofacial syndrome: A genetic and congenital disease of aberrant urinary bladder innervation. Pediatric Nephrology, 29, 513–518.
Acknowledgments
This work was supported by grants from the National Institutes of Health CA138340 and CA211752 (RDS), the United States - Israel Binational Science Foundation (jointly to RDS and IV), the Israel Science Foundation (601/14 and 2277/15) and the Israel Cancer Research Fund (ICRF). Israel Vlodavsky is a Research Professor of the ICRF. We gratefully acknowledge the contribution, motivation, and assistance of the Tumor Biology research team at the Technion Integrated Cancer Center (TICC). The authors thank the many members of the Vlodavsky laboratory and collaborators that have contributed over the years to much of the knowledge reviewed in this chapter. We sincerely apologize for not being able to include all the significant studies focusing on heparanase and would like to profoundly acknowledge the vital contributions of groups whose studies are not presented in the current review. We also apologize for not citing many relevant articles, due to space limitation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vlodavsky, I., Ilan, N., Sanderson, R.D. (2020). Forty Years of Basic and Translational Heparanase Research. In: Vlodavsky, I., Sanderson, R., Ilan, N. (eds) Heparanase. Advances in Experimental Medicine and Biology, vol 1221. Springer, Cham. https://doi.org/10.1007/978-3-030-34521-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-34521-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-34520-4
Online ISBN: 978-3-030-34521-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)