
Abstract
Cotton has a tremendous economic value worldwide due to its high-quality fiber, edible oil and protein contents. However, the intensifying scenario of human population expansion and global environmental changes demand a proportionate increase in cotton production. In the past, several successful attempts have been made by introgression of many quality- and yield-related traits into elite cotton cultivars through conventional breeding. However, those measures are time consuming due to the reliance on introgression of naturally-existing genetic variation through extensive backcrossing. Nonetheless, plant breeding can be accelerated through modern genome editing (GE) tools. Various GE techniques including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced palindromic repeats and CRISPR-associated proteins systems (CRISPR/Cas)-based approaches have been successfully employed for various crop plants. Among them, CRISPR/Cas-based approaches hold great GE potential due to their simplicity, competency and versatility. In cotton, this system can regulate the gene expression associated with quality traits, to circumscribe phytopathogens and/or to stack molecular traits at a desired locus. In gene stacking through site-specific endonucleases, the desired genes can be introduced in close proximity to a specific locus in the cotton genome with a low risk of segregation. However, such executions are tedious to achieve through classical breeding techniques. Moreover, through the CRISPR/Cas-based approaches, transgene-free cotton plants can easily be produced by selfing or backcrossing to meet the current genetically modified organisms (GMO) guidelines. In this chapter, we address the potential application of CRISPR/Cas-based approaches in available whole cotton genomes to sustain cotton productivity, and achieve genetic improvement, pathogen resistance and agronomic traits. Future prospects of GE applications in cotton breeding are also addressed.
Similar content being viewed by others

Keywords
1 Introduction
Cotton ( Gossypium hirsutum L.) is a major fiber crop and the sixth largest source of vegetable oil in the world. The genus Gossypium encompasses more than 52 species, but only 4 have been domesticated over a period of 1–2 million years of polyploidization (Wendel 1989). The widely cultivated New World (NW) allotetraploid (2n = 52), G. hirsutum and G. barbadense , consist of a pair of A and D sub-genomes; whereas, the Old World (OW) G. arboreum and G. herbaceum have diploid (2n = 26) A or D genomes (Chen et al. 2015). The primary source of more than 90% of fiber output is the most widely cultivated G. hirsutum.
The post-genomic era in agriculture faces a major challenge in the improvement of crop cultivars for higher productivity and protection against insects, pests and diseases. A continuous depletion of plant genetic resources coupled with global environmental calamities favors various biotic and abiotic stresses related to crop plants (Arzani and Ashraf 2016). The genetic manipulations of native genes to develop resistance in crop plants requires an efficient tool. Reverse genetics has been widely used to elucidate the functions and regulation of various genes and the metabolic pathways they control in crop plants (Canas and Beltran 2018). However, these approaches have limited application in plant breeding due to various shortcomings including instability and imperfect silencing, which have produced blurry interpretation of the phenotype. Unavailability of resistant germplasm against major biotic and abiotic stresses is the main reason for low productivity in different parts of the world. In this scenario, conventional plant breeding has been sharing its role to control insect, pests and diseases to sustain food security. However, the use of modern biotechnological tools has been integrated in different breeding programs to produce better crop varieties enabling plant breeders to cross unrelated species to introduce foreign genes into crop plants (Zhang 2015).
Genetic variation is an indispensable part of resistance breeding to enhance host plant resistance against various stresses. Random mutations with various physical and chemical mutagens and irradiations have long been employed to create genetic variation in crop plants (Laskar and Khan 2017). However, such mutations are random and generate a huge background mutation load on crop plants under study. In the modern era, different genome editing (GE) techniques are in use to create genetic variation in crop plants; however, clustered regular interspaced palindromic repeats and CRISPR-associated proteins system (CRISPR/Cas) is a versatile, robust and more suitable technique than its other counterparts. Various CRISPR-based GE systems are highly promising in crop genetic improvement because of their precision, specificity, multiplexing ability, minimized off-target activity, robustness and simplicity (Ding et al. 2016).
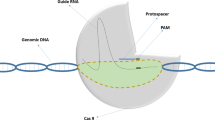
CRISPR is an RNA-based adaptive immune system of archaea and bacteria, which confront the invaded DNA phages and viruses. CRISPR/Cas system comprised of CRISPR-associated Cas genes, which encode RNA-based single-guide RNA (sgRNA) endonucleases, palindromic repeat sequences and non-coding short variable protospacers RNAs. The protospacers are created by inserting the short variable part of the invading nucleic acids that help the invaded cells in component recognition and to chop the foreign nucleic acids. Such CRISPR-based defense approaches have three fundamental steps: adaptation, expression and interference (Fig. 3.1). In adaptation, the spacer sequences are acquired and are chronologically arranged within the CRISPR-array. The expression phase involves the expression of Cas genes and precursor CRISPR-RNA to generate mature crRNA. The integration of crRNA and Cas proteins results in the cleavage of the foreign nucleic acids during the interference phase. The protospacer adjacent motif (PAM) is another important component of the CRISPR-system; PAM is the sequence which is recognized by the Cas protein to perform its functions and it is located either upstream or downstream of each protospacer sequence. It is not actually a part of the CRISPR locus, but represents a conserved sequence of the invading nucleic acids (Demirci et al. 2018). Depending upon their origin, various Cas orthologues recognize different sequences and vary for their requirement of PAM sequences. The Cas protein is guided to digest the target nucleic acid and results in a double-stranded break (DSB) ~3 bp upstream to the PAM sequence. This DSB is repaired by either non-homologous end joining (NHEJ) or homologous recombination (HR).

A generalized model for inducing CRISPR/Cas-based resistance in cotton. Invading nucleic acid is identified, recruited and cleaved by Cas protein. The whole model is based on three fundamental steps of acquisition, expression and interference. During the first step, the CRISPR-locus integrates and duplicates the invaded DNA, which triggers the expression of pre-CRISPR RNA machinery in the invaded cell during the expression step. The third and final step involves cleavage of the invaded nucleic acid in coordination with the CRISPR RNA machinery
2 Different Versions of CRISPR/Cas Systems and Their Applications
Ease of use and versatility of the CRISPR system has made it a rapidly- expanding programmable GE tool. The earlier, original and simple form of site-specific GE tool is CRISPR/Cas9 system. The Cas9 orthologue of CRISPR/Cas-system was obtained from Staphylococcus aureus (SaCas9) (Steinert et al. 2015). The CRISPR/Cas9-based system has been extensively used in plants to improve qualitative and quantitative traits and to confer resistance against various phytopathogens (Li et al. 2016; Nekrasov et al. 2017; Steinert et al. 2015). In order to exploit the unique binding ability of Cas9 to any complementary sequence, Cas9 was engineered to abolish its endonuclease activity and resulted into catalytically inactive protein, called dead Cas9 or dCas9. This dCas9 was successfully implicated to regulate the genes. Later, a transcriptional activator domain VP64 was fused to dCas9 and this system was applied to promote transcriptional activation in plants (Perez-Pinera et al. 2013).
Recently, a second CRISPR-based system, CRISPR/Cas12a (also referred as CRISPR/Cpf1), was introduced. This system belongs to the class II CRISPR systems, which differs from CRISPR/Cas9. In contrast to Cas9, Cpf1 requires a small crRNA to mediate its activity; it recognizes T-rich PAM and induces staggered double-standard breaks (DSBs) distal from the PAM (Lei et al. 2017). The successful application of Cas12a-mediated mutagenesis in rice and tobacco induced biallelic heritable mutation, proving its potential for mutagenesis in plants (Xu et al. 2017a, b). In rice, by using two different Cas12a versions i.e. FnCas12a and LbCas12a, targeted genomic insertions (via HR) were achieved with higher mutation rates as compared to SpCas9-based experiments (Begemann et al. 2017). Cas12a is an excellent choice for gene regulation as it can be fused with a variety of activators (such as VP16, p65, and Rta) to achieve excellent activation. Thus, CRISPR/Cpf1 system can provide an extremely important tool for cotton GE to induce alterations in the genome via HR.
Recently, a useful addition of another Cas version, Cas13 (previously known as C2C2 and C2C6), have been made into the CRISPR family. Cas13 is a class II effector protein that exclusively targets RNA molecules by its two higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domains (Abudayyeh et al. 2017). An important feature of this system is promiscuous RNA cleavage upon activation. Furthermore, similar to Cas12a, Cas13 proteins have the autonomous ability to process pre-crRNA without a tracrRNA. Such abilities of Cas13 have made it a best choice to target a wide range of RNA manipulations. Two of the most active orthologues are Cas13a from Leptotrichia wadei and Cas13b from Prevotella spp. (Abudayyeh et al. 2017; Cox et al. 2017). The superiority of RNA knockdown has been established in terms of specificity as compared to RNA interference (RNAi) technique. In rice plant protoplasts, more than 50% RNA knockdown at only 48 h after transformation has been achieved (Abudayyeh et al. 2017). Importantly, Cas13 has the matchless ability to target specific RNA splicing isoforms at the post-transcriptional level, while it indifferently affects all isoforms at the transcriptional level. This ability of Cas13 makes it an excellent choice to target RNA genome of various pathogens without affecting wild-type transcripts in crop plants (Mahas et al. 2018).
3 Functional Genomics and Limitations in Cotton Breeding
Up to now, a number of tools and resources have been applied to investigate large-scale functional genomic studies in cotton. During the past couple of decades, efforts to develop transgenic cotton cultivars have progressed impressively, to confer genetic resistance to insect, pests and various diseases (Yu et al. 2016). However, advancements to improve the overall physiology, plant architecture, early maturity, fiber traits and yield parameters are progressing at a slow pace. The D-genome of diploid cotton species G. raimondii was completely sequenced in 2012 (Wang et al. 2012), followed by 1694 Mb A-genome of G. arboreum (Li et al. 2014). Soon after the sequencing of A and D-progenitor species, the complete genomes of allotetraploid G. hirsutum and Sea Island cotton species (G. barbadense) were also sequenced (Li et al. 2015). After advancements in NGS technologies, different reference draft genomes are available for diploid and tetraploid Gossypium species. However, different erroneous interpretations have been observed in those genome assemblies regarding chromosomal lengths and the gene annotations (Ashraf et al. 2018). Such errors in genome assemblies need to be dealt using bioinformatics, transcriptomics and epigenomics tools to precisely address mutations, gene annotations and differential protein expression.
After whole-genome cotton sequencing, the major challenge could be the functional annotations and empirical investigations of the genome biology. Unlike other crop and model plants, a few genetic linkage maps are available for cotton (Li et al. 2016). Such genetic maps may provide a base for genetic marker development, gene mapping and effective characterization of different genes. During the last decade, marker-assisted selection has helped to identify ~2000 quantitative trait loci (QTLs) in the interspecific populations of G. hirsutum and G. barbadense (Said et al. 2015). However, those QTLs represented large genomic regions, and possibly, may miss several informative genes during functional genomic analysis based upon the QTL-studies. It is thus a prerequisite for cotton functional genomics to have a fine map of the crucial genomic regions along the cotton genome with an effectively large number of markers. In some recent studies, fine mapping of some crucial genes have been reported for the glandular gene (Cheng et al. 2016), leaf architecture (Andres et al. 2014) and some fiber-quality related traits (Xu et al. 2017a, b). The presence of copy-number variations (CNVs) in a plant genome may alter the genetic structure, gene dosage, gene regulations and ultimately may affect important traits. Fang et al. (2017) reported 989 CNV-affected genes in cotton, associated with the cell wall, plant morphology and translational regulation. Whole-transcriptome profiling is another important tool, which can be helpful for functional genomics in cotton. The transcriptome profiling for leaf senescence, fiber development and biotic and abiotic stresses (Zhang et al. 2016a, b) have been also studied. However, the comparative analysis and RNA-seq tools could not provide complete insight into the gene functions. Thus, functional genomic studies on a larger scale can efficiently assign gene functions through empirical corroborations.
Epigenetic modifications such as DNA-methylation may affect gene expression in plants and have a role in morphological diversity. In cotton, DNA-methylation has been reported to be affected by seasonal variations and fiber development (Osabe et al. 2014). Song et al. (2017) reported ~500 genes, which were epigenetically modified between wild and cultivated cotton cultivars.
The development of omics data in cotton functional genomics necessitates the development of a comprehensive database so that all genomic information may readily be available to cotton breeders. Many such databases have been developed (Table 3.1) and among them CottonGen offers a curated platform, which is easily accessible and has a user-friendly interface. Likewise, CottonFGD is another quick and readily available database to access whole genome sequences , functional annotations and transcriptomic data of most of the whole genomes of cotton. The ccNET database has been designed for coexpression networks and to display ~1155 G. arboreum and ~1884 G. hirsutum functional modules, respectively.
Innovations in omics platforms and the availability of diploid and tetraploid cotton whole genome sequences have significantly facilitated efforts to find candidate genes for many commercially-valuable traits. Nevertheless, understanding of the molecular basis of important traits (e.g. lint and fiber-quality traits) is very limited due to the complex nature of polyploidy (Chen et al. 2017). One major feature of polyploidy is an enormous variation in gene expression and functionality. For example, genome duplication in polyploid cotton may also duplicate the genes. Thus, either gene copies may be retained for a balance or one copy may be lost or silenced to create new functions (Wendel 2015). In allopolyploid cotton species, some of the homologous gene pairs (duplicated genes) are unequally expressed, possibly due to asymmetrical evolution (Zhang et al. 2015). These homologous genes may be expressed variably across species, tissues or even cells and represent a basic feature of allopolyploidy. Therefore, the genomic and transcriptomic information, together with modern biotechnological tools such as genome editing, can reshape future breeding programs to produce superior cotton cultivars.
Crop performance always depends on genotype, the environmental growing conditions and the interaction between genotype and environment. Genotypic expression of a cultivar in various environmental conditions is a main challenge faced by plant breeders as stability of the genotypic potential can be affected under diverse environments. The narrow genetic base of upland cotton germplasm being used in breeding programs is the main challenge to developing new cotton cultivars (Zhang et al. 2015).
Cotton stability and adaptability have been studied and diverse levels of variations in different morphological and yield related traits have been observed. In this regard, AMMI analysis (additive main effects and multiplicative interactions) is useful in supporting breeding program decisions such as specific adaptations to target (tolerance to disease, heat, drought and cold) and selection of environments or test-site locations (Maleia et al. 2017; Riaz et al. 2013). Heritability and the genetic potential of different cultivars for different morpho-yield traits are earnestly needed for the selection of parental breeding lines. Yield is a highly complex character and is directly influenced by different morphological and yield-contributing traits (Khan et al. 2009). Therefore, a thorough knowledge of the nature and genetic potential of different genotypes, inheritance pattern of different traits and the extent of relationship and correlation of yield with various agronomic characters is crucial for breeders to tackle the breeding challenges and to increase yield successfully (Thiyagu et al. 2010; Tonk et al. 2018)
4 Difference Between Genome Editing and Classical Breeding of Cotton
Conventional cotton breeding involves combining desired traits from elite cotton or wild types of Gossypium (Zhang 2015). There are certain drawbacks in using the conventional approach in cotton breeding such as the intraspecific crossing barrier between G. barbadense and G. hirsutum . Although these species have no barrier in terms of reproduction within the parental lines, and a hybrid F1 generation can be propagated, such a cross encounters hybrid breakdown in the F2 and subsequent generations (Zhang 2015).
Inadequate achievement in pioneering desired genes from diploid species to Gossypium hirsutum is because of obscurity in the F1 hybrid production, chromosome number resurgence due to recurring backcrossing and chromosome doubling. Various breeding approaches coupled with the classical genetics tools could help cotton breeders to improve cotton fiber, seed quality and enhance yield. In addition, coupling of these technologies can lead to early development of tolerant or resistant cultivars and better adaptability of cotton crops to local environments. CRISPR/Cas GE technology could be a promising tool in addressing these issues of classical breeding, which are still unresolved or need improvement to expedite breeding programs. There are many candidate genes, which need investigation such as genes involved in biosynthesis of cotton gossypol, development of cotton fibers with respect to its negative regulator and resistance against Verticillium wilt . CRISPR/Cas technology holds promise in investigating these candidate genes and can advance research based on functional genomics , and ameliorate the prospective for the molecular breeding of cotton (Chen et al. 2017; Li et al. 2017a, b), which conventional breeding approaches may not achieve in a short time span.
However, the CRISPR/Cas system has been successful in achieving genetically-edited mutants for gene function study in cotton as well as in allotetraploids, with high specificity and high efficiency, both in terms of protoplast level and in the generation of stable cotton transgenics (Chen et al. 2017).
Compared to conventional mutation breeding , CRISPR/Cas technology can create specific mutations at the desired loci and some number of off-targets loci can be envisaged. Additionally, in contrast to the conventional mutation breeding program, CRISPR/Cas technology has little unintended load of mutation in the gene-edited crops. With the help of the CRISPR/Cas method, just as a plant-breeding approach, backcrossing is always possible in the case of an off-target mutation. In conventional breeding as well as mutation breeding, nevertheless, unknown and unintended mutations arise similarly and can be eliminated with several crosses in successive cycles. Homology recombination facilitated CRISPR/Cas can lead to the introduction of traits for cotton improvement. The technology carries an advantage over the use of transgene insertion methods for the development of transgenic products. Thus, CRISPR/Cas is a promising plant-breeding tool with huge potential that can result in precision cotton breeding by working with native traits available within the cotton crop.
5 Layout Plan of CRISPR/Cas-Based Genome Editing
The application of CRISPR/Cas-based GE has already been applied in a few studies to genetically engineer the cotton genome (Table 3.2). However, successful execution of the CRISPR/Cas system in cotton GE necessarily requires the generalized mechanism discussed in the preceding section (Fig. 3.2).

Schematic execution of CRISPR/Cas genome editing system in cotton. Online web-based tools are used to select and design ~20 nucleotides (nt) sgRNAs complementary to the targeted region in the cotton genome. The specific sgRNAs and Cas protein are expressed either from a single binary vector as one cassette or expressed separately from different binary plasmids. The assembled cassettes are then transfected into the plant genome through a suitable transfection technique such as Agrobacterium-based inoculations, particle bombardment using gene guns and/or protoplast transformation. After the successful transfection, the mutants are evaluated for the targeted mutations and are analyzed further for the presence of any off-target activity through the expression of reporter genes, nuclease enzymes, gel electrophoresis and/or next generation sequencing approaches. The successfully screened mutants are then employed directly for downstream applications
5.1 Data Mining and Single Guide RNA (sgRNA) Designing for Target Sequence
For successful execution of cotton GE, selection of the target region of the genome is of primary importance. While selecting the target region in the cotton genome, major challenges could possibly be off-targets, genome polymorphism, polyploidy, transposons, single nucleotide polymorphisms (SNPs) and introns (Iqbal et al. 2016; Klein et al. 2018; Zhang et al. 2018). Although a very few studies are available to gauge off-targets in plants, these problems can be overcome in three steps by using different computer-based programs: (i) target genome site selection for the design of sgRNA, (ii) predicting off-target activities of the designed sgRNA and (iii) evaluation of on- and off-target cleavage rates (Chen et al. 2017; Lei et al. 2014). Several web-based tools and bioinformatics programs are now available to ascertain the cotton GE (e.g. ATUM, Alt-RTM CRISPR/Cas9 System, Cas-OFFinder, CCTop, CHOPCHOP, CRISPRpred, CRISPR-P, CROP-IT, GT-Scan, MIT CRISPR design and sgRNA Designer).
5.2 Choice of CRISPR/Cas System
Selection of an appropriate CRISPR/Cas system, sgRNA promoter, choice of binary vector for sgRNA delivery and mode of transformation are some of the prerequisites to successfully engineer the cotton genome. Expression of bacterial Cas into cotton requires the codon optimized Cas and a suitable plant promoter. Different constitutive and/or inducible promoters such as CaMV 35 s, CMV, EF1A, LTR and UBO have been successfully utilized to drive Cas protein in plants. Beside these, many plant RNAIII-based promoters, such as U3p and U6p, have also been used successfully (Belhaj et al. 2013).
Selection of an efficient Cas protein is another important requirement. Based on the experiment need, a codon optimized Cas protein has efficient GE ability with improved efficacy. Besides GE, gene silencing [CRISPR interference (CRISPRi)] and gene activation [CRISPR activation (CRISPRa)] has also been achieved by using the codon optimized dCas9 version lacking nuclease ability (Qi et al. 2013). Recently, a new endonuclease Cpf1 has been employed to cleave the target DNA via a staggered DNA double-stranded break, producing cohesive ends with 4–5 overhangs of nucleotides with much higher efficiency and specificity, as compared to its predecessor Cas9. Availability of different CRISPR-based GE tools will broaden the applications of cotton genome engineering.
5.3 Delivering CRISPR/Cas Cassettes into Cotton Genome
After evaluating the designed sgRNAs and the selection of a CRISPR system, the next step is to assemble the whole cassette (including sgRNA, CRISPR, Cas/Cpf1) into a suitable plant-based binary vector. Delivery of the resultant recombinant vector into the cotton genome is a very crucial step as the success of entire system is dependent on the choice of a successful transformation system. Several techniques are being used to execute stable plant transformation such as polyethylene glycol (PEG)-mediated transformation, protoplast transformation, biolistic inoculations and transit peptides; besides these techniques, successful transformation can also be achieved by using various plant virus-based vectors (Butler et al. 2016). Nonetheless, the most tested way of stable genetic transformation in cotton is Agrobacterium-mediated transformation, and this method has been opted successfully in CRISPR-based cotton genome engineering (Chen et al. 2017; Li et al. 2017a, b; Wang et al. 2017a, b). In Agrobacterium-mediated stable transformation, cotton seeds are surface sterilized followed by germination on wet filter papers at 28 °C in darkness. After a couple of days, when the length of the seed root reaches 1–2 cm, the root apex is injured with a sterile syringe and are suspended in Agrobacterium culture. Afterward, the infected seedlings are cultivated for 48 h in darkness and 48 h in light and then transferred to the greenhouse. Transgenic plants are selected by growing them on suitable antibiotics.
5.4 Manipulation of Targeted Mutagenesis Through In Vitro Regeneration and Screening of Transgenic Plants
Stable transformation of cotton can be achieved by agro-inoculating the hypocotyl or cotyledonary petiole. After successful transfection, the inoculated material can be grown on the selection media and resulting transgenic plantlets subjected to confirmation either by Southern blotting or by sequencing the PCR amplicons (Fig. 3.2). However, the phenotypic and genotypic screening requires detection and confirmation of the targeted mutations, which may include, but are not limited to, resistance to insects, pests and diseases, tolerance to abiotic stresses, and improvement of agronomic features such as yield and quality traits (Hua et al. 2017).
5.5 Detection and Confirmation of Successful Genome Modification
The verification of a mutation induced by the CRISPR system and efficiency of GE is a crucial step for downstream processing of the transformants. An easy way to detect the CRISPR/Cas-induced putative mutation in the target plants is to amplify the target gene by using specific primers. The achieved PCR amplicons then can be purified and cloned into a vector. To map all the possible mutations, more than ten clonal amplicons of each transgenic plants should be sequenced by the Sanger method. (Chen et al. 2017; Li et al. 2017a, b). Sanger sequencing, developed by Fredrick Sanger et al. (1977), is based on the selective incorporation of dideoxynucleotides (ddNTPS) by DNA polymerase during in vitro DNA replication. This is quite simple, inexpensive and a convenient approach to determine simple or complicated chimeric mutations. Another approach to verify the GE event is the use of a reporter gene, like GUS, fluorescent proteins (GFP, YFP, RFP) and luciferase, where a reporter gene bearing a mutation could be corrected by the CRISPR/Cas system and vice versa (Feng et al. 2014). Use of T7 Endonuclease I is another promising way to evaluate and calculate the GE efficiency. In this method, PCR amplicons of the target region are annealed and digested with T7 Endonuclease I, only non-perfectly mismatched DNA fragments are digested, which are quantified through gel electrophoresis (Wang et al. 2017a, b). Alternatively, at the target loci, a restriction endonuclease site can be targeted (either corrected or introduced) during Cas9/sgRNA cleavage (Xie and Yang 2013). Polyacrylamide gel electrophoresis (PAGE) can also be used to confirm the GE events by single-stranded conformation polymorphism. In this technique, the single-stranded DNA (of the target gene) with mutation will show a different migration rate in the gel because of different DNA conformations (Zhang et al. 2016a, b). High throughput or next-generation sequencing of the whole genome is highly sensitive, robust and an efficacious method to detect the mutation in the target region and to evaluate any off-targets during a GE event (Feng et al. 2014).
6 Potential Applications of CRISPR/Cas in the Post-Genomic Era of Cotton Breeding
The availability of whole genomes of diploid and tetraploid cotton species has enabled scientists to assign gene functions to a few genes by comparative analysis with the known genes (Liu et al. 2018). These comparative analyses cannot provide an exact insight into the specific roles of the genes. Their precise roles can only be elucidated empirically and by reverse or forward genetics tools, which is the most difficult task in cotton breeding. To tackle this problem, GE can be a good solution to elucidate the functions of the genes, especially those which are involved in insect, pest and disease resistance, abiotic stresses, agronomic features, yield and quality traits. Among potential application of GE, the targeted interference (CRISPRi) or activation (CRISPRa) of a specific gene in upland cotton can provide precise role of the gene.
Cotton fibers are seed trichomes, which are made from long single integument cells and represent a good model to study plant growth. Although cotton fibers are biologically the most important trait in cotton development, understanding the cellular mechanisms regarding fiber initiation and elongation are still scanty. Genetic evidence has revealed that among 28 members of HD-ZIP IV subfamily the transcription factor GhHOX3 in the GL2 type domain is crucial for fiber elongation (Shan et al. 2014). According to Deng et al. (2016), phytoseterols and a balanced ratio between campestosterol:sistosterol plays a pivotal role in cotton fiber development. Several other studies also identify fiber-related genes such as E6, GhExp1, GhSusA1, PIP2s, and GA20ox (Bai et al. 2014). Moreover, many transcription factors in MYB, C2H2, bHLH, WRKY and HD-ZIP gene families are also differentially expressed during cotton fiber development.
The role of transposable elements (TEs) in cotton fiber development cannot be ruled out, apart from their role to diverge the genome sizes in cotton species. Cotton fiber development is thus a complex program and is a trait of primary importance. At this stage, GE can contribute to elucidate the mechanisms of fiber development in cotton. The gene knocking capability of CRISPR-based approaches can be utilized for the identification and functional characterization of certain genes in cotton. It has been successfully employed to knockdown OsERF922 gene against rice blast and TMS5 to study temperature sensitivity in rice (Wang et al. 2016a, b).
A robust CRISPR/Cas9 system has been developed for site-specific mutagenesis in G. hirsutum by generating multiplexed sgRNAs with 66–100% GE efficiency (Wang et al. 2018a, b). Li et al. (2017a, b) also successfully targeted two identical genomic regions GhMYB25-like A and GhMYB25-like D in upland cotton with >98% mutation efficiency and no off-target activity. Similarly, Chen et al. (2017) obtained 47.6–81% transformation efficiency by adopting GE in upland cotton.
Gossypol biosynthesis pathways enable cotton plants to resist against insect and pests. The GaWRKY1 transcription factor has recently been shown to be involved in the gossypol biosynthesis pathway (Tian et al. 2016). Moreover, Wu et al. (2017) identified at least nine genes directly involved in gossypol biosynthesis. The complete regulatory network for gossypol biosynthesis necessitates the CRISPR/Cas9 system to unravel the molecular basis of the whole pathway and to produce glandless cotton cultivars.
Cotton leaf curl disease (CLCuD) is a devastating threat caused by CLCuD-associated begomoviruses (genus Begomovirus, family Geminiviridae) to global cotton production (Sattar et al. 2013, 2017). CLCuD has been reported to be associated with at least five different begomovirus species and various DNA-satellites in cotton. Numerous conventional and nonconventional strategies have been utilized to circumvent this disease complex; however, none could achieve successful control. In this situation, a comprehensive CRISPR/Cas-based resistance strategy can be devised to control this important cotton disease (Iqbal et al. 2016).
7 Multiplexed Gene Stacking Using CRISPR/Cas9
Simultaneous modification of multiple genes and controlling a particular trait can be a useful method in functional genomics study and resistance against various pathogens. CRISPR system is a powerful tool, which can target member of multiple gene families in one go. Several successful examples are known where multiplex GE has been employed in different plant species such as rice, maize, Arabidopsis, tomato, tobacco and wheat (Li et al. 2017a, b; Wang et al. 2017a, b). The majority of these studies followed the polycistronic tRNA-sgRNA (PTG)-based approach (Ma et al. 2015; Xie et al. 2015). In this approach, sgRNAs scaffold constructs are arrayed with a specific spacer, which can be separated by conserved tRNA for multiplexing (Fig. 3.3). Varying efficiencies of GE have been obtained through the PTG-based system; PTG showed a 3–31 fold higher GE efficiency with 15–19% higher mutation as compared to other CRISPR/Cas9-based multiplexing approaches (Xie et al. 2015). However, a new multiplexing method referred as simplified single transcriptional unit (SSTU) CRISPR system has been developed (Wang et al. 2018a, b). In the SSTU-based method, different endonucleases such as FnCpf1, LbCpf1 orCas9 and their sgRNA array were coexpressed in rice from a single Pol II promoter, without any additional processing machinery. A higher magnitude of GE efficiency was achieved by this simple and efficient method. The SSTU-based multiplex CRISPR system can be an option for cotton GE as it is advantageous because of the simplified construction of the cassettes and higher efficiency compared to other multiplexing techniques. By opting for the SSTU-based approach, successful manipulation of different cotton genomic loci can be engineered with improved efficiency against different abiotic and biotic stresses (Fig. 3.3).

A schematic representation of multiplex CRISPR/Cas-based genome editing in cotton. The multiplex cassette can be assembled for multiple sgRNAs following the tRNA, with spacer sequences insertions followed by the NOS terminator at the end. Finally, the Cas protein can be expressed through a separate Pol-III promotor and NOS terminator sequences sharing the same cassette in the binary vector. These multi-components will be transcribed and expressed independently from each other in the cotton genome to commence GE
8 Countering Off-targets During Genome Editing
Off-target activity is a major concern during GE events, which causes chopping of non-targeted sequences in the plant genome leading to the disruption of normal functioning of non-target genes. Many web-based tools (such as CROP-IT, CCTop and/or CRISPOR) are available now to predict and evaluate the off-target activity of the designed sgRNAs. Genome-wide off-target mutations have been implicated to the seed sequence in the single-guided RNA, which is specific for the intended target DNA, due to the observation that the first eight nucleotides at the distal end to PAM motif of seed sequence are relatively tolerant to the target site compared to the 12 nucleotides at the proximal end to PAM motif. Off-target incidence in plants has been rare according to the data available in the literature; however, it has been suggested that elimination of off-target mutation can be achieved with the use of backcrossing. Whole genome analysis of sgRNA can be performed in order to minimize the chance of off-target mutation and to achieve highly specific GE (Tang et al. 2018).
In cotton, whole genome analysis of sgRNA during its design has been done to avoid potential off-targets. Potential off-targets of various sgRNAs with few base pairs of mismatches in the cotton genome in GE transgenic cotton plants have been checked using online analysis, such as CRISPR-P website with the database containing the G. raimondii genome and the G. hirsutum genome database website; this was validated using the restriction enzyme digestion-suppressed PCR (RE-PCR) assay and sequence analysis. The verified mutations in the plant genes were only used for the analysis of off-target mutations. No off-target mutation was observed for these sgRNAs using CRISPR/Cas9 technology in cotton (Chen et al. 2017; Wang et al. 2018a, b). Li et al. (2017a, b) also reported that there was no off-target mutation after sequencing the putative off-target sites for the sgRNA1 and sgRNA2 of GhMYB25-like gene with 3 and 1 mismatched nucleotides, respectively.
Other GE strategies have been developed to offset off-target repercussions, such as a D10A mutation in the RuvC nuclease domain of Cas9, which can only create a single strand break using two gRNAs, simultaneously. FokI nucleases can be fused to catalytically inactive Cas9 protein to form dimers in order to cleave the DNA. These strategies will effectively address the common and widespread practical issues to adopt CRISPR/Cas systems in crop plants (Bortesi and Fischer 2015).
In rice, it has been shown that the Cas9-paired nickases effectively suppress off-target mutations, as two guide RNAs are used simultaneously. However, it has been reported that decreased frequency of on-target mutation has occurred (Mikami et al. 2016). The use of ribonucleoproteins (RNPs) has been known to mitigate the effect of off-target repercussions during GE application. In plants, it has been shown that the use of SpCas9 in amalgamation with a single sgRNA has been shown to lessen the effect of off-target repercussions. The RNP strategy has also been shown to eliminate the cytotoxicity effect, which is often associated when performing the transfection of DNA. In addition, RNPs also eliminate the possibility of small DNA fragments arising from the plasmids to be integrated (Kim et al. 2017).
9 Production of Transgene-Free Cotton
Unlike genetically modified (GM) crops, the genetic makeup of GE crops is not disturbed by a native genetic change in the form of deletion or insertion and thus resembles the naturally-mutated crop plant (Nekrasov et al. 2017). However, the GE crops will have to satisfactorily overcome many regulations to be labelled as non-GMO. In general, the complete CRISPR/Cas-based system is expressed in the plant cell through intermediate DNA machinery, including T-DNA of the vector, Cas9 protein and adjoining components, which act as transgenes. The social acceptance of GE crops demands the production of stable lines of heritable CRISPR-mutated genes free of the expression cassettes. Genetic segregation is used during T0 to T2 generations for stable inheritance of the mutants of interest, free of transgenes. Yang et al. (2017) reported that the T1 Brassica napus GE plants were free of T-DNA (76.2%) and Cas9 (11.3%), respectively. The more advanced approach is the use of DNA-free GE using RNP complex delivery system (Ricroch et al. 2017) to get transgene-free plants in a single generation. The RNP approach is equally efficient as plasmid-based expression cassettes to knockout or edit a gene in those plants where protoplast transfection is successful, such as Arabidopsis, lettuce, petunia, rice, tobacco and wheat (Zhang et al. 2017). Alternatively, fluorescent proteins can be utilized as marker genes to assist in the selection of transgene-free crop plants in which protoplast transfection is difficult.
GE crops can be obtained by either selfing or backcrossing to the original parental line where the segregation of gametes takes place and homozygous GE mutants with desired alteration in the intended loci of gene can be obtained without any RNA-guided GE nuclease transgene construct. The GE0 plants can be selected for the selfing process wherein the GE1 generation, the transgenes become segregated during the segregation of gametes. RE-PCR genotyping as well as the DNA sequencing method can be used to select the GE plants, whereas the transgene-free plants can be obtained by negative selection with intended modification only in the first generation. The conventional molecular approaches are known for the elimination of transgenes with the use of Cre/loxP piggyback transposon systems and FLP/FRT systems (Khatodia et al. 2016; Zaidi et al. 2018).
Argonaute 18 (ZmAgo18a and ZmAgo18b) gene has been studied in maize using CRISPR/Cas9 technology via the selfing method to eliminate the Cas9 transgene. To remove the Cas9/gRNA trasngene, selfing in the T0 population was conducted. The segregation of Cas9 transgene is expected to occur at 3:1 (Cas9 positive: Cas9 negative) ratio. This ratio was veracious in Ago18a #2, Ago18b #19, Ago18b #20, but not in Ago18a #15 where all 20 seedlings from Ago 18a#15 were found to contain Cas9/gRNA transgene (Char et al. 2017). The selfing method was conducted by Tang et al. (2018) in rice to eliminate the Cas9 transgene where they elucidated the role of OsNramp5 mutation on accretion of cadmium and other related metals.
Moreover, He et al. (2018) used a set of suicide transgenes, which eliminates all the pollen as well as the embryos produced by T0 plants that contain the CRISPR/Cas9 transgene. The authors point out that this offsets the time required in the labor-intensive selection of GE crops free of transgenes through selfing and backcrossing. The GE crops and the GMOs have been compared over time and many studies concluded that the GE crops must be declared as non-GMOs. In many other countries, the fate of GE crops regarding GMO regulation is still under review (Davison and Ammann 2017). The highest court of the European Union (EU) has recently adjudicated stringent regulations for GE crops, as has been practiced for GMOs, such as routine regulations, health assessment, impact on environment and proper labeling. Responding to this decision, many scientists presumed plummeted funding on GE crop research. This may unfortunately provoke similar regulations in other countries however, US officials are not prepared to impose such a regulation for GE crops so far (Callaway 2018; Stokstad 2018).
The production of transgene-free cotton is particularly important to convince the public about it being a non-GMO product and to obtain cotton crops using the CRISPR/Cas9 system to commercialize the GE crop without encountering any GMO laws in the future. The use of backcrossing, selfing, as well depending upon breeders’ choice, and other methods mentioned above could be crucial in obtaining GE cotton plants without the presence of the Cas9 T-DNA cassette transgene.
10 Genome Editing Bottlenecks in Polyploid Cotton
In contrast to rice and Arabidopsis, cotton functional genomics is intricate due to its allopolyploid genetic structure. Additionally, cotton’s relatively inefficient genetic transformation compared to other crops, and low number of mutants, position cotton as a challenging crop in generating stable cotton transgenics and mutants.
Due to polyploidy, homologous sequences and the presence of larger repeat sequences in the allotetraploid cotton genome, many economically-significant traits governing fiber quality, resistance to biotic stresses, tolerance to abiotic stresses and/or yield related traits are regulated by multiple genes or alleles. The polyploidization of A and D-diploid genomes generate highly repetitive DNA contents in the tetraploid Gossypium hirsutum genome (Li et al. 2015). Therefore, it is a prerequisite to select multiple homoeoalleles for site-directed mutagenesis with the CRISPR/Cas9-based system. It is a critical step to choose the best candidate sgRNAs for targeted mutagenesis in cotton. Moreover, the experimental validation of the sgRNAs is also another crucial step towards the generation of a GE-cotton crop. Gao et al. (2017) has recently introduced a reliable transient transformation assay to validate sgRNAs in cotton within a stipulated short time period.
Most genes involved in various pathways have not been assigned any proven functions and their functional annotation has largely been inferred by comparison with known genes from other plants. Only a few studies are available in which most of the functional genomics relied upon RNA-interference. Somehow, such studies were limited due to gene redundancy and the presence of highly homologous genes. The effective application of the CRISPR/Cas9 system necessitates stable targeted mutation to accomplish the generation of stable homozygous mutants. The selection of an appropriate sgRNA is a key factor, which may affect the mutagenic efficacy of CRISPR/Cas9 because use of different sgRNAs targeting a single gene may produce variable results (Ma et al. 2016). One of the major bottlenecks in cotton transformation is the generation of stably-inherited cotton mutants, which itself is a laborious and lengthy process. Moreover, stable transformation is particularly needed for practical application of the CRISPR/Cas9-based system to study important agronomical traits. The genetically-stable execution of GE events can only be validated by producing successive generations of transformed cotton plants.
11 Conclusions and Prospects
Since the advent of the first CRISPR/Cas-based GE system, many new versions of CRISPR/Cas have been developed, which have revolutionized biotechnology. Contemporary CRISPR-based technologies have opened new horizons in disease management, insect resistance and functional genomics , especially in assigning gene(s) functions through a gain- and/or loss-of function approach. Although conventional breeding has developed many new elite, disease-resisting and high-yielding cotton varieties, the pace of these approaches is insufficient to meet the present-day requirements. Henceforth, the post-genomic era challenged scientists to develop efficient tools for quick, high-yielding and abiotic/biotic-stress resistant crops. Given the whole-genome sequence of G. hirsutum, much information is now available to engineer the cotton genome, study gene functions and improve different traits pertinent to biotic and abiotic stresses. The majority of the genes have multiple copies, homologous sequences and a large proportion of repeats. Therefore, cotton genome engineering poses a tedious task, which can produce undesirable results due to high similarity and gene redundancy. It demands careful and precise investigations to engineer the cotton genome. However, the availability of cotton genome information will promote cotton genome engineering and functional genomic research. CRISPR/Cas-based GE approaches can help in promoting yield, lint amelioration, mitigating major biotic (such as fungal, bacterial and viral diseases) and abiotic stresses (Fig. 3.4). Additionally, CRISPR/Cas-based systems can be explored to activate resistance genes, and to curtail susceptible genes involved in fungal, bacterial and viral diseases of cotton. Additional applications may include the regulation of secondary metabolites to downregulate the gossypol production and for the hyperproduction of genes involved in lint amelioration. Likewise, other characters such as upregulation of antioxidants, fiber length, pests and disease resistance can be promoted through GE in cotton. Thus, the CRISPR/Cas-based GE approaches hold substantial potential to accelerate the genetic improvements in cotton. Together with conventional breeding , it will be a valuable addition in the cotton breeder’s toolbox.

The potential application of current and future CRISPR/Cas based genome editing in cotton
References
Abudayyeh OO, Gootenberg JS, Essletzbichler P et al (2017) RNA targeting with CRISPR-Cas13. Nature 550:280–284
Andres RJ, Bowman DT, Kaur B, Kuraparthy V (2014) Mapping and genomic targeting of the major leaf shape gene (L) in Upland cotton (Gossypium hirsutum L). Theor Appl Genet 127:167–177
Arzani A, Ashraf M (2016) Smart engineering of genetic resources for enhanced salinity tolerance in crop plants. Crit Rev Plant Sci 35:146–189
Ashraf J, Zou D, Qang Q et al (2018) Recent insights into cotton functional genomics: progress and future perspectives. Plant Biotechnol J 16:699–713
Bai WQ, Xiao YH, Zhao J et al (2014) Gibberellin overproduction promotes sucrose synthase expression and secondary cell wall deposition in cotton fibers. PLoS One 9:e96537
Begemann MB, Gray BN, January E et al (2017) Precise insertion and guided editing of higher plant genomes using Cpf1 CRISPR nucleases. Sci Rep 7:11606
Belhaj K, Chaparro-Garcia A, Kamoun S, Nekrasov V (2013) Plant genome editing made easy: targeted mutagenesis in model and crop plants using the CRISPR/Cas system. Plant Methods 9:39–47
Bortesi L, Fischer R (2015) The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol Adv 33:41–52
Butler NM, Baltes NJ, Voytas DF, Douches DS (2016) Geminivirus-mediated genome editing in potato (Solanum tuberosum L) using sequence-specific nucleases. Front Plant Sci 7:1045
Callaway E (2018) CRISPR plants now subject to tough GM laws in European Union. Nature 560:16
Canas LA, Beltran JP (2018) Model legumes: functional genomics tools in Medicago truncatula. Methods Mol Biol 1822:11–37
Char SN, Neelakandan A, Nahampun H et al (2017) An Agrobacterium-delivered CRISPR/cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol J 15:257–268
Chen ZW, Caoa JF, Zhanga XF et al (2015) Cotton genome: challenge into the polyploidy. Sci Bull 62:1622–1623
Chen X, Lu X, Shu N et al (2017) Targeted mutagenesis in cotton (Gossypium hirsutum L) using the CRISPR/Cas9 system. Sci Rep 7:44304
Cheng H, Lu C, Yu JZ et al (2016) Fine mapping and candidate gene analysis of the dominant glandless gene Gl2 e in cotton (Gossypium spp). Theor Appl Genet 129:1347–1355
Cox DBT, Gootenberg JS, Abudayyeh OO et al (2017) RNA editing with CRISPR-Cas13. Science 358:1019–1027
Davison J, Ammann K (2017) New GMO regulations for old: determining a new future for EU crop biotechnology. GM Crops Food 8:13–34
Demicri Y, Zhang B, Unver T (2018) CRISPR/Cas9: an RNA-guided highly precise synthetic tool for plant genome editing. J Cell Physiol 233:1844–1859
Deng SS, Wei T, Tan KL et al (2016) Phytosterol content and the campesterol: sitosterol ratio influence cotton fiber development: role of phytosterols in cell elongation. Sci China Life Sci 59:183–193
Ding Y, Li H, Chen LL, Xie K (2016) Recent advances in genome editing using CRISPR/Cas9. Front Plant Sci 7:703
Fang L, Wang Q, Hu Y et al (2017) Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits. Nat Genet 49:1089–1098
Feng Z, Mao Y, Xu N et al (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci USA 111:4632–4637
Gao W, Long L, Tian X et al (2017) Genome editing in cotton with the CRISPR/Cas9 system. Front Plant Sci 8:1364
He Y, Zhu M, Wang L et al (2018) Programmed self-elimination of the CRISPR/Cas9 construct greatly accelerates the isolation of edited and transgene-free rice plants. Mol Plant. https://doi.org/10.1016/jmolp201805005
Hua Y, Wang C, Huang J, Wang K (2017) A simple and efficient method for CRISPR/Cas9-induced mutant screening. J Genet Genomics 44:207–213
Iqbal Z, Sattar MN, Shafiq M (2016) CRISPR/Cas9: a tool to circumscribe cotton leaf curl disease. Front Plant Sci 7:475
Khan NU, Hassan G, Marwat KB et al (2009) Genetic variability and heritability in upland cotton. Pak J Bot 41:1695–1705
Khatodia S, Bhatotia K, Passricha N et al (2016) The CRISPR/Cas genome-editing tool: application in improvement of crops. Front Plant Sci 7:506
Kim H, Kim ST, Ryu J et al (2017) CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun 8:14406
Klein M, Eslami-Mossallam B, Arroyo DG, Depken M (2018) Hybridization kinetics explains CRISPR-Cas off-targeting rules. Cell Rep 22:1413–1423
Laskar RA, Khan S (2017) Assessment on induced genetic variability and divergence in the mutagenized lentil populations of microsperma and macrosperma cultivars developed using physical and chemical mutagenesis. PLoS One 12:e0184598
Lei Y, Lu L, Liu H-Y et al (2014) CRISPR-P: a web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol Plant 7:1494–1496
Lei C, Li S-Y, Liu J-K et al (2017) The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res 45:e74
Li F, Fan G, Wang K et al (2014) Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46:567–572
Li F, Fan G, Lu C et al (2015) Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33:524–530
Li X, Jin X, Wang H et al (2016) Structure, evolution, and comparative genomics of tetraploid cotton based on a high-density genetic linkage map. DNA Res 23:283–293
Li R, Li R, Li X et al (2017a) Multiplexed CRISPR/Cas9-mediated metabolic engineering of γ-aminobutyric acid levels in Solanum lycopersicum. Plant Biotechnol J 16:415–427
Li C, Unver T, Zhang B (2017b) A high-efficiency CRISPR/Cas9 system for targeted mutagenesis in cotton (Gossypium hirsutum L.). Sci Rep 7:43902
Liu Z, Liu Y, Liu F et al (2018) Genome-wide survey and comparative analysis of long terminal repeat (ltr) retrotransposon families in four Gossypium species. Sci Rep 8:9399
Ma X, Zhang Q, Zhu Q et al (2015) A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol Plant 8:1274–1284
Ma X, Zhu Q, Chen Y, Liu YG (2016) CRISPR/Cas9 platforms for genome editing in plants: developments and applications. Mol Plant 9:961–974
Mahas A, Neal SC, Mahfouz MM (2018) Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnol Adv 36:295–310
Maleia MP, Raimundo A, Moiana LD et al (2017) Stability and adaptability of cotton (Gossypium hirsutum L.) genotypes based on AMMI analysis. Aust J Crop Sci 11:367–372
Mikami M, Toki S, Endo M (2016) Precision targeted mutagenesis via Cas9 paired nickases in rice. Plant Cell Physiol 57:1058–1068
Nekrasov V, Wang C, Win J et al (2017) Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci Rep 7:482
Osabe K, Clement JD, Bedon F et al (2014) Genetic and DNA methylation changes in cotton (Gossypium) genotypes and tissues. PLoS One 9:e86049
Perez-Pinera P, Kocak DD, Vockley CM et al (2013) RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10:973–976
Qi LS, Larson MH, Gilbert LA et al (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152:1173–1183
Riaz M, Naveed M, Farooq A et al (2013) AMMI analysis for stability, adaptability and GE interaction studies in cotton (Gossypium hirsutum L.). J Anim Plant Sci 23:865–871
Ricroch A, Clairand P, Harwood W (2017) Use of CRISPR systems in plant genome editing: toward new opportunities in agriculture. Emerg Top Life Sci 1:169–182
Said JI, Song M, Wang H et al (2015) A comparative meta-analysis of QTL between intraspecific Gossypium hirsutum and interspecific G hirsutum x G barbadense populations. Mol Genet Genomics 290:1003–1025
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Sattar MN, Kvarnheden A, Saeed M, Briddon RW (2013) Cotton leaf curl disease - an emerging threat to cotton production worldwide. J Gen Virol 94:695–710
Sattar MN, Iqbal Z, Tahir MN, Ullah S (2017) The prediction of a new CLCuD epidemic in the old world. Front Microbiol 8:631
Shan CM, Shangguan XX, Zhao B et al (2014) Control of cotton fibre elongation by a homeodomain transcription factor GhHOX3. Nat Commun 5:5519
Song Q, Zhang T, Stelly DM, Chen ZJ (2017) Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol 18:99
Steinert J, Schiml S, Fauser F (2015) Highly efficient heritable plant genome engineering using Cas9 orthologues from Streptococcus thermophilus and Staphylococcus aureus. Plant J 84:1295–1305
Stokstad E (2018) European court ruling raises hurdles for CRISPR crops. Science. http://www.sciencemag.org/news. Accessed 25 July 2018
Tang X, Liu G, Zhou J et al (2018) A large-scale whole-genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 nucleases in rice. Genome Biol 19:84
Thiyagu K, Nadarajan N, Rajarathinam S et al (2010) Association and path analysis for seed cotton yield improvement in interspecific crosses of cotton (Gossypium spp). Electron J Plant Breed 1:1001–1005
Tian X, Ruan J, Huang J et al (2016) Gossypol: phytoalexin of cotton. Sci China Life Sci 59:122–129
Tonk FA, Istipliler D, Tosun M et al (2018) Correlation and path Analysis in yield and quality traits in F3 and F4 generation of Carmen x Devetüyü-176. J Anim Plant Sci 28:107–112
Wang K, Wang Z, Li F et al (2012) The draft genome of a diploid cotton Gossypium raimondii. Nat Genet 44:1098–1103
Wang K, Huang G, Zhu YX (2016a) Transposable elements play an important role during cotton genome evolution and fiber cell development. Sci China Life Sci 59:112–121
Wang F, Wang C, Liu P et al (2016b) Enhanced rice blast resistance by CRISPR/Cas9-targeted mutagenesis of the ERF transcription factor gene OsERF922. PLoS One 11:e0154027
Wang M, Mao Y, Lu Y et al (2017a) Multiplex gene editing in rice using the CRISPR-Cpf1 system. Mol Plant 10:1011–1013
Wang Y, Meng Z, Liang C et al (2017b) Increased lateral root formation by CRISPR/Cas9-mediated editing of arginase genes in cotton. Sci China Life Sci 60:524–527
Wang M, Mao Y, Lu Y et al (2018a) Multiplex gene editing in rice with simplified CRISPR-Cpf1 and CRISPR-Cas9 systems. J Integr Plant Biol. https://doi.org/10.1111/jipb.12667
Wang P, Zhang J, Sun L et al (2018b) High efficient multi sites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol J 16:137–150
Wendel JF (1989) New world tetraploid cottons contain old world cytoplasm. Proc Natl Acad Sci U S A 86:4132–4136
Wendel JF (2015) The wondrous cycles of polyploidy in plants. Am J Bot 102:1753–1756
Wu Z, Yang Y, Huang G (2017) Cotton functional genomics reveals global insight into genome evolution and fiber development. J Genet Genomics 44:511–518
Xie K, Yang Y (2013) RNA-guided genome editing in plants using a CRISPR-Cas system. Mol Plant 6:1975–1983
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA 112:3570–3575
Xu P, Gao J, Cao Z et al (2017a) Fine mapping and candidate gene analysis of qFL-chr1, a fiber length QTL in cotton. Theor Appl Genet 130:1309–1319
Xu R, Qin R, Li H et al (2017b) Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol J 15:713–717
Yang H, Wu J, Tang T et al (2017) CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA in Brassica napus. Sci Rep 7:7489
Yu LH, Wu SJ, Peng YS et al (2016) Arabidopsis EDT1/HDG11 improves drought and salt tolerance in cotton and poplar and increases cotton yield in the field. Plant Biotechnol J 14:72–84
Zaidi SS, Mukhtar MS, Mansoor S (2018) Genome editing: targeting susceptibility genes for plant disease resistance. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2018.04.005
Zhang J (2015) Transgenic cotton breeding. In: David DF, Richard GP (eds) Cotton, 2nd edn. American Society of Agronomy, Madison, pp 229–254
Zhang TZ, Hu Y, Jiang WK et al (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L acc TM-1) provides a resource for fiber improvement. Nat Biotechnol 33:531–537
Zhang Y, Liang Z, Zong Y et al (2016a) Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat Commun 7:12617
Zhang F, Zhu G, Du L et al (2016b) Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci Rep 6:20582
Zhang K, Raboanatahiry N, Zhu B, Li M (2017) Progress in genome editing technology and its application in plants. Front Plant Sci 8:177
Zhang Q, Xing H-L, Wang Z-P et al (2018) Potential high-frequency off-target mutagenesis induced by CRISPR/Cas9 in Arabidopsis and its prevention. Plant Mol Biol 96:445–456
Acknowledgments
We thank TUBITAK 2215 for providing a fully-funded PhD scholarship to Sarbesh Das Dangol.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix I: Some Research Institutes Relevant to Cotton Molecular Breeding
Appendix I: Some Research Institutes Relevant to Cotton Molecular Breeding
Institution | Specialization and research activities | Contact information and website |
|---|---|---|
Australian Cotton Research Institute | Cotton pathology and Biotechnology, Insecticide resistance management, Integrated pest management | Locked Bag 1000Narrabri NSW 2390 Narrabri, NSW, Australia Email: narrabri.office@dpi.nsw.gov.au https://www.dpi.nsw.gov.au/about-us/research-development/centres/narrabri |
The Institute of Cotton Research (ICR), CAAS | Cotton molecular biology, genetic and breeding, cotton pathology | Chinese Academy of Agricultural Sciences, No.12 Zhongguancun South St. Haidian District Beijing, China Email: diccaas@caas.cn |
Cotton Research Institute | Cotton breeding and Genetics; biotechnology, cytogenetics | Nanjing Agricultural University NO.1 Weigang, Nanjing 210,095, China Email: moelab@njau.edu.cn |
Central Institute for Cotton Research (CICR) | Cotton breeding and genetics, pathology, molecular breeding | PB.No.2, Shankarnagar PO, Nagpur 440,010, India. Email:cicrnagpur@gmail.com |
Central Cotton Research Institute (CCRI) | Cotton breeding and genetics, cytogenetics, pathology, molecular breeding | Central Cotton Research Institute (CCRI) Old Shuja Abad Road, Multan 60500, Pakistan. Email: dtmalik@gmail.com |
Cotton Research Institute (CRI) | Cotton breeding, molecular genetics | 9 El-Gamaa St., Giza, Cairo, Egypt. Email: naserradwan1936@yahoo.com |
Nazilli Cotton Research Institute, Ministry of Agriculture | Cotton breeding and Genetics and molecular breeding | Directorate, Bozdoğan Yolu Üzeri 5. Km Nazilli, Aydin, Turkey Email: pamuk@tarim.gov.tr |
USDA-ARS, Southern Regional Research Center | Cotton breeding and molecular genetics | New Orleans, LA 70179, USA Email: jpjordan@nola.srrc.usda.gov |
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sattar, M.N., Iqbal, Z., Dangol, S.D., Bakhsh, A. (2019). CRISPR/Cas9: A New Genome Editing Tool to Accelerate Cotton (Gossypium spp.) Breeding. In: Al-Khayri, J., Jain, S., Johnson, D. (eds) Advances in Plant Breeding Strategies: Industrial and Food Crops. Springer, Cham. https://doi.org/10.1007/978-3-030-23265-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-23265-8_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-23264-1
Online ISBN: 978-3-030-23265-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)