
Abstract
Multiple sclerosis (MS) afflicts genetically predisposed individuals and is associated with T lymphocyte-mediated damage to the myelin sheath of neurons in the central nervous system, resulting in severely impaired signal transmission. The mechanisms of the induction and manifestation of MS are not entirely understood. The control of autoimmune disorders is accomplished by both central tolerance in which autoreactive T lymphocytes are eliminated in the thymus and by tolerance mechanisms that operate in the periphery. Among the many mechanisms described, T regulatory (Treg) cells derived from the thymus (tTregs) and induced (iTregs) in the periphery as well as T regulatory type 1 cells (Tr1) are involved in many disease models. However, the precise details of the generation and perpetuation of these various Treg subsets and their relevance to the regulation of autoimmune diseases remain elusive. In this review, we critically analyze the current knowledge of the tolerance mechanisms involved in the regulation of MS and its animal model, experimental autoimmune encephalomyelitis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Anergy
- Autoimmune diseases
- Blood-brain barrier
- Central nervous system
- Cerebrospinal fluid
- Foxp3
- GM-CSF
- Human leukocyte antigen
- Interferon-γ
- Interleukin 17
- Myelin basic protein
- Multiple sclerosis
- Myelin oligodendrocyte glycoprotein
- Neuromyelitis optica
- Proteolipid protein
- Th1
- Th17
- T regulatory cells
- Trichostatin A
- Tumor necrosis factor-α, Tolerance
Introduction
The tenet of the immune system is the protection of the host against both invading pathogens and autoimmune diseases that arise in genetically susceptible individuals. For the former, it is essential to mount robust immune responses, both T-cell-mediated immunity and antibody production, against a myriad of pathogens. After the elimination of the source of “foreign antigenic determinants,” the adaptive immune responses must contrive to restore the normal clonal T- and B-cell repertoire. Activation-induced cell death or apoptosis is credited with the restoration of the clonal size of primarily antigen-activated T lymphocytes at the end of productive immune responses [1]. The failure to do so can result in overt immune responses which can cause more harm than benefit to the host. An example of overt immune responses causing damage to endogenous tissues is the production of the multipotent, noxious cytokine, tumor necrosis factor-α (TNF-α) in inflammatory conditions including sepsis [2]. In addition to the need to control these adaptive immune responses, autoimmune responses need to be kept in check to minimize or eliminate adverse reactions against the host tissues. CD4+ T lymphocytes systematically control these seemingly opposite versions of immune responses. The various regulatory mechanisms involved in controlling autoimmune diseases have been the subject of intense investigation over many decades [3,4,5,6,7,8].
Among the 80 known autoimmune diseases, only a few are specifically directed against the central nervous system (CNS). Although evidence for the involvement of autoimmunity in many CNS disorders is weak or nonexistent, indications of autoimmunity exist in some cases. Neuromyelitis optica (NMO) is an autoimmune, demyelinating disorder of the CNS with typical clinical manifestations of optic neuritis and acute transverse myelitis attacks [9]. Although NMO was believed to be a variant of MS, it is now considered as an independent disorder. NMO is characterized by the presence of antibodies against the water channel proteins aquaporin-4 and aquaporin-1 and myelin oligodendrocyte glycoprotein (MOG). The characteristic phenotype is disruption of astrocyte function and demyelination of the spinal cord, optic nerves, and particular brain regions. Lack of self-tolerance to water channel proteins is likely to be the cause of NMO [10]. However, the underlying pathogenic mechanisms have not been fully elucidated. Alzheimer’s disease is characterized by dementia, immunoglobulin in the brain parenchyma, and deposition of complement components in neurons [11]. The autoimmune encephalitis is a group of disorders characterized by autoantibodies directed to synaptic surface antigens (NMDA- and AMPA-type glutamate receptors, GABA (B) receptor, and LGI1) resulting in severe neurological symptoms [12]. Antibodies against glutamic acid decarboxylase, the rate-limiting enzyme for GABA synthesis, are associated with the autoimmune neurological syndromes, namely, stiff person syndrome, cerebellar ataxia, epilepsy, limbic encephalitis, and abnormal eye movements [13]. Whereas these characteristics are indicative of the underlying autoimmunity, detailed studies are required to fully understand the autoimmune nature of these diseases and importantly regulatory mechanisms useful for controlling these diseases. Importantly, there is a paucity of experimental models to dissect the immunological mechanisms involved in these various neurological disorders.
Multiple sclerosis (MS) is a well-characterized neuronal disorder with an underlying autoimmune basis [14,15,16,17]. Whether MS is a non-immunological disorder and primarily a neurodegenerative disease with unknown etiology is heavily debated over the years [18, 19]. Accounts of various T-cell tolerance mechanisms involved in immune responses to nominal antigens and self-determinants including neuronal antigens have been described previously [reviewed in Ref. 3,4,5,6,7,8]. In this chapter, we focus on MS and its animal model, experimental autoimmune encephalomyelitis (EAE), to highlight the underlying common regulatory mechanisms and point out the disparity between these systems. Finally, we discuss the limitations of the data obtained in various EAE models to their translation into clinical practice for treatment of differing forms of MS.
Effector T Cells and Pathology of Multiple Sclerosis
MS is a chronic disease lasting over many decades and has highly variable presentations [14,15,16,17]. After the initial diagnosis, clinically isolated syndrome, a majority (85%) of patients display the common relapsing-remitting form of MS. After 10–15 years of diagnosis, 50% of untreated patients develop secondary progressive MS, whereas in 15% of patients, the disease progresses without remission, referred to as primary, progressive MS. Most current treatments are directed to relapsing-remitting MS, and none of them is effective on primary or secondary progressive MS [14, 16, 17, 20, 21]. These drugs are directed to cull the autoimmune component [14,15,16,17], whereas modalities to treat the neurodegenerative component of MS [18, 19] are scarce [20, 21]. The CNS is an immunologically privileged site and lacks a potent innate immune response in healthy individuals [22]. Immune cell infiltration across the blood-brain barrier (BBB) promotes inflammation, demyelination, gliosis, and neuroaxonal degeneration, resulting in disruption of neuronal signaling [14,15,16,17]. To better understand the impact of T-cell tolerance on MS, knowledge of the critical underlying pathological features is vital. MS is thought to be mediated by CD4+ T-cells, although CD8+ T cells and antibody-producing B cells are known to contribute to disease pathogenesis [14,15,16,17]. CNS-intrinsic events such as activation of microglia and astrocytes as well as chemokines have also been implicated in MS [23,24,25].
Autoreactive T lymphocytes are thought to mount aberrant immune responses against CNS autoantigens. Susceptibility to develop MS has been linked to the class II human leukocyte antigen HLA-DRB1∗15:01 allele, which is expressed on antigen-presenting cells, implying a role for distinct antigen presentation to T lymphocytes necessary for disease induction [26]. Although the etiology of MS is unknown, poorly understood stochastic events and environmental factors influence the disease penetrance. Infiltration of immune cells from the periphery is prominent in relapsing-remitting MS, and T cells appear early in lesion formation. Inflammation of the brain and spinal cord is evident in all MS patients, which declines with age and duration of the disease [14,15,16,17]. An open question is whether MS is triggered in the periphery such as in draining lymph nodes or originates in the CNS. Peripheral T lymphocytes specific to neuronal antigens are thought to be activated by “molecular mimicry” [26], and subsequent reactivation by the CNS resident antigen-presenting cells leads to a cascade of events resulting in neuronal damage [14,15,16,17, 27].
Although the specificity of autoreactive T cells in MS remains obscure, recognition of autoantigens such as MOG and myelin basic protein (MBP) by CD4+ T lymphocytes from both healthy individuals and MS patients has been demonstrated [28,29,30]. The relative frequency of these cells in healthy individuals and MS cases remains contentious. Thus, it is not clear whether the observed self-reactivity of T lymphocytes in MS patients reflects the underlying pathological events or a stochastic event resulting from molecular mimicry and breach of self-tolerance. Although earlier studies implicated interferon-γ (IFN-γ)-producing Th1 cells as the sole pathogenic T cells, recent investigations also support a role for Th17 cells expressing IL-17 as they are found in the peripheral blood, CNS, and cerebrospinal fluid (CSF) of MS patients. Furthermore, Th cells with a mixed phenotype (double-producers) expressing both IFN-γ and interleukin 17A (IL-17A) have a higher capacity to infiltrate the CNS as inferred from studies of postmortem MS brain tissues [14,15,16,17, 31,32,33]. These results indicate a pivotal role of double producers in MS pathogenesis.
Interestingly, CD8+ T cells found in the active lesions of MS patients produce IL-17, similar to mucosal-associated invariant T cells [34]. In addition to the consistent participation of CD4+ T cells, B cells have been proposed to be strong candidates for autoimmune effector cells in MS [35]. Although significant differences in B cells were found in the CSF, they were neither predictive of disease and disease progression (EDSS, expanded disability status scale) nor conversion to clinically definite MS following diagnosis of the clinically isolated syndrome [36].
MS pathology is characterized by confluent demyelinated areas known as plaques or lesions in the white and gray matter of the brain and spinal cord, indicating a loss of myelin sheaths and oligodendrocytes [14,15,16,17]. Damage of axons and neurons correlates with disease severity. Astrocytes form multiple sclerotic glial scars in white matter lesions. Demyelination of the gray matter of the cortex, nuclei, and spinal cord is also associated with MS. Inflammation is more pronounced in acute than in chronic phase. Invading immune cells and macrophages indicates BBB leakage. Macrophages, CD8+ cells, CD4+ cells, B cells, and plasma cells are represented in descending proportions. In the early stages, little damage outside of the plaques, called normal-appearing white matter, is present in both the brain and spinal cord despite general brain atrophy. During disease progression, diffuse T-cell and B-cell infiltrates, microglia and astrocyte activation, and diffuse myelin reduction and axonal damage are evident. Although the numbers of T cells do not change, the frequencies of B cells and plasma cells increase, whereas microglia and macrophages remain in a chronic state of activation throughout the disease. In secondary, progressive MS, tertiary lymphoid structures are evident in the meninges contributing to cortical demyelination and tissue damage at later stages [14,15,16,17].
Effector Mechanisms in EAE
The earliest description of EAE was the occurrence of acute disseminated encephalomyelitis in monkeys repeatedly given intramuscular injections of normal rabbit brain emulsions and extracts [37]. This observation explained that the induction of encephalomyelitis observed earlier in humans vaccinated with rabies virus grown on rabbit spinal cord was due to the immune response triggered by the spinal cord contaminant of the vaccine [38]. Since then, monophasic EAE has been induced in guinea pigs, rats, mice, and primates by immunization with spinal cord homogenates or peptides derived from MOG, MBP, and PLP [see Ref. 14 for citations; [39,40,41,42,43,44,45,46,47,48,49,50,51,52]. Complete Freund’s adjuvant along with pertussis toxin is required for EAE induction in guinea pigs, rats, and mice. However, incomplete adjuvant without Mycobacterium tuberculosis was sufficient to induce EAE in some strains of rats and marmosets.
Interestingly, EAE could be induced in the susceptible rat strain Dark Agouti without pertussis toxin. Several explanations have been put forward including breaching of BBB, breaking of self-tolerance, and enhancing immunogenicity of the inoculum to explain the dependence of pertussis toxin for EAE induction. Although questions were raised as to the validity of results obtained in EAE models for translation into the treatment of MS patients [53], arguments were also made in support of the fact that when used wisely EAE will provide beneficial information for clinical application [41].
Most rodent EAE models are characterized by ascending flaccid paralysis reflecting preferential targeting of inflammation to the spinal cord, referred to as classic EAE, which manifests in different forms. Immunization with the immunodominant MOG35–55 peptide induced a mild, monophasic form of EAE in the C57BL/6 strain (H-2b haplotype). In this model, the clinical symptoms peak around 9–12 days after immunization, followed by spontaneous resolution by 30 days [42, 43]. However, the pathology of monophasic EAE in C57BL/6 mice does not parallel that of an MS form [44, 45]. On the contrary, similar immunization induced a long-lasting (>75 days, Ref. 46,47,48,49,50,51), severe disease in NOD (H-2g7) mice characterized by paralysis of fore and hind limbs with [49,50,51] or without discernible remissions [46,47,48], respectively, representing secondary and primary progressive MS. Regardless, in NOD mice EAE occurs with a high frequency that shares unique features with MS including lifelong disease, prominent demyelination, axonal loss, and astrogliosis [46,47,48,49,50,51]. On the other hand, immunization with PLP139–151 peptide induced relapsing-remitting EAE in SJL/J (H-2s) mice characterized by the appearance of clinical signs 6–20 days after priming and relapses first appearing at 30–45 days [52].
In the common classic EAE induced by various peptide antigens in mice, inflammation is preferentially targeted to the spinal cord. In a small number of antigen-specific models, brain rather than the spinal cord is selectively targeted, referred to as atypical EAE [54,55,56]. It was initially observed in C3H/HeJ mice immunized with PLP190–209 peptide [54] and confirmed subsequently in IFN-γ knockout Balb/c mice immunized with MBP peptides and in C3HeB/FeJ mice immunized with MOG [54,55,56]. Atypical EAE is presented as a movement disorder, with proprioception defects, ataxia, spasticity, and axial rotation of the head and trunk, and characterized by predominant cerebellar or brainstem involvement. These various animal models are useful in gaining insights into the underlying immunological mechanisms of variant forms of MS. However, the display of complex and variable clinical features and spontaneous remission in certain models render the interpretation of results difficult. Since each variant of EAE recapitulates some but not all features of MS, it is useful to ascertain the efficacy of treatment procedures in a preclinical model that closely mimics the select form of MS in question.
Other models of demyelinating diseases include viral infections and administration of toxic substances [see Ref. 40 for citations]. Chronic demyelinating encephalomyelitis is induced by intracranial introduction of Theiler’s virus (BeAn strain or Daniel’s strain) or nasal infection with mouse hepatitis (Corona) virus. Inflammatory infiltrates consist of T cells and activated macrophages/microglia in the CNS. Although viral models may reflect critical features of MS-like inflammatory inflammation, it is complicated by the involvement of virus-induced immune-mediated mechanisms. Importantly, evidence for the role of viruses in MS pathogenesis has not yet been obtained. Demyelination induced by toxic models using cuprizone, a copper chelator, is useful for understanding mechanisms of demyelination and remyelination but does not fully reflect aspects of MS pathology and pathogenesis [40].
Although the role of CD4+ T cells in EAE is well established, controversy exists as to the identity of T helper subsets involved. Whereas IFN-γ-producing Th1 cells have been shown to be crucial early during EAE, IL-17A-expressing Th17 cells participate at a later stage [57–58]. In contrast, Th17 cells and double producers, those expressing both IFN-γ and IL-17A, migrate to the CNS before the arrival of Th1 cells [59] or ex-Th17 cells that lost the expression of IL-17A and gained IFN-γ during clinical disease manifestation [60]. Other studies indicated that Th1 [57] or Th17 cells [61] alone could mediate EAE upon adoptive transfer into naïve mice. However, contamination of various degrees of IFN-γ-producing cells in the Th17 cell preparations raised questions about the independent role of Th17 cells in EAE manifestation. In the primary, progressive EAE, Th1 cells were found in the spinal cord, whereas Th1 and Th17 cells but not Th1/Th17 cells infiltrated the spinal cord later during the disease [47]. However, Th1/Th17 cells were prominent in peripheral lymphoid tissues. The plasticity of Th17 cells further complicates the role of distinct T helper subsets in EAE manifestation [62]. Despite enormous effort to understand the role of various lymphokines, cytokines, and accessory cell-associated determinants, their roles in EAE pathogenesis remain obscure. Although Th17 cells have been dubbed as “encephalitogenic” T cells, neither IL-17A nor IL-17F contributes to EAE [63]. The only cytokine that has been attributed a role in EAE is GM-CSF [64]. In the primary progressive EAE model, amelioration of the disease by treatment with the most potent histone deacetylase inhibitor, trichostatin-A (TSA), was associated with repression of GM-CSF-producing CD4+ T cells in the secondary lymphoid organs and the CNS [47–48]. A closer analysis indicates that GM-CSF is dispensable for EAE induction but is essential for chronic tissue damage and neutrophil accumulation in the brain [48, 65, 66]. Blockade of the GM-CSF receptor α ameliorated relapses in mice [67]. Lesions from secondary progressive but not primary progressive MS patients contained GM-CSF receptor α+ myeloid cells, indicating a possible target for disease intervention [67]. Collectively, these data suggest an essential role for GM-CSF in the effector arm of the inflammatory disease. The identity of the cytokines crucial for the induction of the autoimmune CNS disease remains to be delineated.
The pathological mechanisms varied with the EAE models [14, 39,40,41, 44,45,46,47, 49, 51]. The acute monophasic EAE was characterized by multifocal, confluent areas of mononuclear inflammatory infiltration and demyelination in the peripheral white matter of the spinal cord. In the brain, in addition to meningitis, perivascular inflammatory cuffing in the cerebellum and hindbrain white matter was prominent. In the relapsing-remitting EAE induced by PLP139–151 immunization, lesions of the optic nerve, brainstem, spinal cord, cerebellum, and cerebral cortex along with perivascular and meningeal lymphocyte and neutrophil filtration were evident. Besides, white matter damage and gliosis and demethylated axons were observed. In the primary, progressive EAE induced by immunization of NOD mice, inflammatory cuffs around dilated blood vessels in the white matter with penetration into the gray matter were observed in the spinal cord during the acute phase of the disease [47]. The chronic phase was accompanied by severe damage of the periphery with numerous vacuoles in the dorsal and dorsolateral funiculus. Inflammatory cells were abundant in both the white and gray matter and in the subarachnoid space of ventral funiculus. Luxol fast blue staining indicated severe demyelination of neurons in the spinal cord. Bielschowsky’s silver impregnation method unraveled lack of neurofilaments in both the white and gray matter, indicative of severe axonal loss during this form of EAE [47]. Although controversy exists as to whether macrophages play a pathogenic or protective role in classic EAE, increased accumulation of neutrophils during the acute phase of EAE was evident in the spinal cord of mice with primary, progressive EAE [48], a feature attributed to atypical EAE [54,55,56]. Thus, it appears that the pathological mechanisms involved in monophasic, relapsing-remitting, primary, and secondary progressive EAE as well as atypical EAE are not remarkably distinct and seem to overlap.
Peripheral Immune Tolerance Mechanisms
Autoreactive T and B cells are deleted in the thymus, referred to as central tolerance, which accounts for the lack of overtly self-reactive lymphoid cells in apparently healthy individuals [68]. However, the deletional mechanism is not absolute, and a fraction of self-reactive T lymphocytes escape thymic selection and exit to the periphery. This may explain the presence of T cells specific to the MBP in the peripheral blood of asymptomatic relatives of MS patients [69, 70]. Similarly, MOG-specific CD4+ T cells were detected in the peripheral blood of healthy individuals albeit at a lower level compared to that of MS patients following in vitro expansion with a MOG peptide [71]. Although these self-reactive T cells can potentially trigger autoimmune diseases when appropriately activated by antigen presentation, they do not elicit autoimmunity in healthy people due to restraints imposed by peripheral tolerance mechanisms. Although reactivity to multiple neuronal determinants such as MBP, MOG, and PLP has been demonstrated in MS, the identity of the causative autoantigen involved in the induction of this disease remains obscure.
Similarly, the initiating autoantigen in most other autoimmune diseases including type 1 diabetes also remains unknown [72]. The lack of this critical information has negatively impacted on developing successful antigen-specific tolerance strategies for the manipulation of MS [73, 74]. However, the utility of other tolerance strategies to control the chronic neurodegenerative disease MS remains incompletely understood. Substantial data have been generated in EAE, a model of MS, supporting a role for immunoregulatory T cells in this autoimmune neurodegenerative disease. Although analogous CD4+ T-cell-mediated immunoregulation may operate in MS, the available data are limited and are often contentious. Herein, we discuss these data critically and evaluate their impact on MS.
One mechanism that was implicated in the prevention of autoimmune diseases is T-cell anergy, physical existence of T cells without displaying functional competence [75]. Exposure of cloned mouse Th1 cells expressing interleukin-2 (IL-2) and IFN-γ to chemically modified antigen-presenting cells pulsed with antigenic peptide induced a state of unresponsiveness, termed anergy. These anergic Th1 cells were unable to produce IL-2 when challenged subsequently with unmodified antigen-presenting cells pulsed with the specific peptide antigen in vitro. Interestingly, this form of tolerance is transient since activation with IL-2 reversed anergy and restored the ability of anergic Th1 cells to respond in an antigen-specific manner [76] subsequently. Demonstration of peripheral blood T lymphocytes reactive to self-antigens such as MBP and MOG in asymptomatic relatives of MS patients exemplifies the existence of potentially autoreactive T cells in the absence of overt autoimmunity, akin to anergic T cells [69,70,71]. Notably, antigen presentation by cloned murine thymic macrophages induced anergy in Th1 cells without requiring chemical modification, indicating the possibility that specific native antigen-presenting cells are capable of inducing anergy despite the optimal expression of co-stimulatory determinants necessary for T-cell activation [77]. However, the transient and reversible nature of T-cell anergy imposes severe restrictions in applying antigen-specific tolerance strategy to silence the autoreactive T cells in vivo. Another primary mechanism of peripheral tolerance intensely studied during the 1970s and 1980s involved the participation of subsets of antigen-specific T suppressor (Ts) cells governed by idiotype-anti-idiotype interactions and influenced by the unresolved genetic restriction element, I-J [3, 4]. The ensuing result was suppression of antigen-specific immune responses as well as linked suppression of unrelated immune responses [78]. However, the lack of robust biochemical and molecular evidence discouraged further studies of antigen-driven Ts cells.
Tolerance by T Regulatory Cells
During the decline of interest in antigen-specific Ts cells, the concept of regulation of autoimmune responses by Foxp3-expressing CD4+ T lymphocytes derived from the thymus, termed the thymic T regulatory (tTreg) cells, emerged [79,80,81,82,83]. Also, a subset of Treg cells called the induced Treg (iTreg) generated during the activation of conventional CD4+ T cells with antigen, IL-2 and transforming growth factor-β (TGF-β) [84], and type 1 Treg (Tr1) cells [85] have been described. The relationship between these Treg subsets and antigen-specific Ts cells described earlier as well as their relative contribution to the regulation of immune responses to nominal antigens and autoimmunity remains incompletely understood.
The notion that immunoregulation is accomplished by T lymphocytes distinct from conventional effector CD4+ T cells was fueled by the critical observation that neonatal thymectomy led to the emergence of many autoimmune diseases in mice due to the depletion of CD4+CD25+ tTreg cells [79, 80]. Notably, autoimmune neuronal diseases were not among those unleashed by the removal of tTreg cells. The tTreg cells are enriched for T-cell receptors and exhibit high affinity for self-peptides. The description of Foxp3 as the critical transcription factor for the development, function, and stability of Treg cells revitalized the study of tTreg cells in mice [81, 82] as well as humans [83]. Whereas tTreg cells control most autoimmune diseases [79, 80], iTreg cells generated from conventional CD4+ T cell with low affinity for self-antigens is thought to play a prominent role in general immune regulation [84]. Although these three Treg subsets are treated as distinct entities, confirmatory phenotypic features that can distinguish between them remain unknown [5,6,7,8, 79,80,81,82,83,84,85,86]. Although the Treg cells also exert non-specific immune suppression [87] similar to antigen-specific Ts cells [3, 78], the molecular nature of immunosuppression remains unclear in both cases.
Initially, the human counterparts of mouse Treg cells were identified in the thymus and peripheral blood of healthy individuals as CD4+CD25high cells which function similar to mouse Treg cells [88, 89]. The frequency, phenotype, and function of Treg cells have been extensively studied in MS patients. In some studies, the frequency of CD4+CD25hi Treg cells in the peripheral blood of MS patients was similar to that of healthy individuals irrespective of the disease activity [90,91,92,93]. Notably, removal of IL-7 receptor+ (CD127+) cells from the analysis unraveled that both the number and function of Treg cells in MS patients did not differ from that of healthy individuals [93]. Paradoxically, the number of Treg cells was higher in the cerebrospinal fluid than in the peripheral blood of MS patients [94]. Chronic MS patients had a higher frequency of memory CD4+CD25+CD127loCD45RO+ Treg cells in the peripheral blood [95]. Surprisingly, CD4+CD25+FOXP3+ Treg cells were also significantly increased in MS patients when compared to healthy individuals [96].
On the contrary, in relapsing-remitting MS, the numbers of CD4+CD25+FOXP3+ Treg cells were reduced in the peripheral blood [97]. Interestingly, both CD4+CD25+FOXP3+ cells and FOXP3 expression were lower during relapses than remission [98]. The numbers of CD31+ recent thymic emigrants of the CD4+CD25+CD45RA+CD45RO−FOXP3+ Treg phenotype within the peripheral blood decline with age and are significantly reduced in MS patients [99]. Interestingly, the Treg cells expressing CD39, an ectoenzyme that hydrolyzes ATP, were diminished in MS patients [100]. On the contrary, in another study the frequency of CD4+CD25+CD127loFOXP3+CD39+ Treg cells in MS patients was comparable to healthy controls [101]. To date, little consensus exists as to the phenotype and frequency of the Treg population in various clinical forms of MS , a disease that lasts for several decades. Longitudinal analysis of Treg cells using uniform phenotypic markers during different stages of the disease will help to delineate whether the Treg cell populations correlate with the clinical presentations.
The Complex Role of FOXP3 in Immunoregulation
The description of Foxp3 as a master regulator of tTreg cells led to its adoption as a surrogate marker of mouse [81, 82] and human Treg cells [7, 8, 83, 96,97,98, 100, 101]. The IPEX (immune regulation, polyendocrinopathy, enteropathy, and X-linked inheritance) syndrome is characterized by diarrhea, diabetes mellitus, hemolytic anemia, eczema, autodestruction of endocrine glands, and thyroiditis with absolutely no evidence of MS [102]. The IPEX syndrome is fatal and without aggressive immunosuppression or bone marrow transplantation, and male patients rarely survive beyond the second decade of life [103]. Although insulin-dependent diabetes was diagnosed in IPEX patients [102], association between variation of the FOXP3 gene and the common type 1 diabetes was not subsequently found [104]. IPEX is associated with mutations of the human FOXP3, the ortholog of the gene mutated in scurfy mice that develop autoimmune disorders [105, 106].
On the other hand, MS is diagnosed as clinically isolated syndrome typically during the second or third decade of life and requires several decades for full manifestation [14]. Therefore, it is highly unlikely that IPEX patients can develop the full spectrum of MS symptoms within two decades of life. Another unexpected complication is that FOXP3 mutation affects not only Treg cells but also the effector CD4+ T cells. This was indicated by the ability of CD4+CD25hiFOXP3+ cells from IPEX patients to suppress the proliferation of normal but not autologous responder T cells [107]. Thus, IPEX syndrome is associated with the development of resistance in responder T cells to suppressor signals and not impaired suppressor function of Treg cells [107]. Although there is no evidence linking IPEX syndrome and MS, sharing of similar defective functions of Treg cells is likely serendipitous (vide infra).
Studies in experimental models unraveled that the role of Treg cells in immunoregulation is complex and complicated. No spontaneous CNS inflammation was observed in Foxp3 mutant mice or after targeted depletion of Foxp3+ Treg cells in wild-type mice [108], indicating that the loss of Treg function alone is insufficient to cause EAE. Paradoxically, the same group observed that targeted and acute depletion of Foxp3+ Treg cells resulted in limited autoimmune inflammation by controlling the T effector cell proliferation and mobility within the CNS [109], indicating a role for Treg cells in EAE regulation. This finding is in contrast to another study which showed that the Treg cells accumulated in the CNS at the peak of EAE but were unable to suppress the proliferation of CNS-derived T effector cells in vitro [110]. In several studies, anti-CD25 antibody administration was used to demonstrate the participation of Treg cells in EAE [43, 111].
Interestingly, anti-CD25 antibody administration repressed the secondary but not primary remission [112]. However, CD25 is not a bona fide marker of Treg cells since these antibodies persist in the circulation for an extended period and also could affect the activated T effectors cells expressing CD25 [109]. Moreover, neither the administration of the anti-CD25 antibody nor adoptive transfer of Treg cells obtained during the resolution of monophasic EAE was determined to be antigen-specific [43]. Thus, further analysis is required for a full evaluation of the role of Treg cells in EAE.
Many other complications impede the understanding of the role of Treg cells in EAE. The expression of Foxp3 is not sufficient for the full expression of the suppressor phenotype and requires several “partner proteins” including the transcription factors Gata-3, NFAT, and Runx1, which influence Treg cell functions [113]. The critical importance of partner proteins in immunoregulation was demonstrated in Foxp3 reporter NOD mice in which the disruption of the interaction between Foxp3 and cofactors such as the histone acetyltransferase Tip60, histone deacetylase 7 (HDAC7), and Eos accelerated the development of autoimmune diabetes [114]. Another difficult aspect of Treg cells is their “plasticity.” In the peripheral blood of MS patients, increased frequency of CD4+CD25hiCD127low/− FOXP3+ IFN-γ+ secreting Th1-like Treg cells with lower suppressive ability was observed, indicating the instability of human Treg cells [115]. The conversion of Foxp3+ Treg cells into IL-17-producing Th17 cells has been implicated in the pathogenesis of collagen-induced arthritis in mice and rheumatoid arthritis [116]. Use of a dual lineage tracing model indicated that the conversion of effector Treg cells into central Treg cells was accompanied by increased Foxp3 stability in vivo [117]. Also, acquisition of Th2-like Treg cells expressing Gata-3, activation of STA6, and secretion of IL-4 have been reported [118]. Although Treg cells appear to populate specific tissues including muscles, the skin, lungs, and the gastrointestinal tract, their involvement in tissue homeostasis remains speculative [119]. An additional complication of Treg cells is the so-called ex-Foxp3 cells, a small proportion of tTreg cells that lost Foxp3 expression during EAE induction and express IFN-γ and the ability to mediate EAE [120]. Although the levels of FOXP3 mRNA and protein are decreased in MS patients [121], it is unclear whether this reflects the transition to “ex-FOXP3 cells” during the disease pathogenesis. Thus, despite extensive investigations on the phenotypic and functional characteristics of CD4+FOXP3+ Treg cells, several key issues await further elucidation for a better understanding of how these cells contribute to immune homeostasis in humans.
Defective Function of Treg Cells in MS Patients and its Clinical Relevance
Regardless of the complex nature of the phenotype of the Treg subsets, some investigations indicated that these cells are functionally impaired in MS patients [90, 92, 96,97,98, 100, 101]. Although both CD39+ and CD39− Treg subsets suppressed the proliferation of responder T cells and IFN-γ production, interestingly only the CD39+ Treg subset suppressed IL-17 production, which is also defective in MS patients [100, 101]. Since IL-17-producing T cells are enriched in active MS lesions and considered crucial for MS pathogenesis [14, 122], these data suggest that compromised Treg cell function may exaggerate IL-17-mediated disease symptoms. This observation could provide possible mechanistic insights into the control of MS. Interestingly, impaired suppressor function of CD4+CD25hi Treg cells was also noted without numerical reduction of these cells in the peripheral blood of some but not all MS patients [90, 92]. As mentioned above, impaired suppressor function correlated with diminished expression of FOXP3 protein and mRNA in MS patients [121]. Diminished suppression of MBP-induced proliferation of peripheral T cells observed in 45% of untreated MS patients was paradoxically associated with increased frequency of CD4+CD25intermediate Treg cells [96].
Interestingly, the ability of CD4+CD25+ Treg cells was depressed in relapsing-remitting but not secondary progressive MS patients, despite comparable number and phenotype of these cells [123]. However, the status of Treg cells in primary progressive MS in which neuronal deficits accrue without remission [14, 20] is currently not known. Thus, these studies suggest that despite the variability in number and phenotype, diminished suppressor function of Treg cells may have a significant impact on MS pathogenesis. However, the clinical value of depressed suppressor function of various Treg cells has been challenged [8, 124] and should be treated with caution. The uncertainty of the functional aspect of human Treg cells is attributed to technical issues associated with in vitro assay systems used for the functional assessment [8, 124]. These include the type of stimulation of responders (anti-CD3 antibody + antigen presenting cells along with anti-CD28 antibody vs. immobilized anti-CD3 antibody), blocking IL-2 production to maximize suppression in some cases, different cellular targets of suppression (responder T cells vs. antigen presenting cells such as dendritic cells), requirement for enormous numbers of Tregs (1:1 ratio of Treg cells and responders), and the lack of suitable animal models to determine the suppressive activity of human Treg cells in vivo. Importantly, using autologous responder and Treg cells, it will be hard to distinguish between the acquisition of resistance in responder cells to Treg cell-mediated suppression and compromised suppressor function of Treg cells. These technical issues render the results of in vitro suppression assays somewhat uninterpretable [8, 124].
Disease-Modifying Therapies Failed to Impact Treg Cells
Since it is difficult to determine the impact of phenotype, defective number, and function of human Treg cells on disease progression during long duration such as in MS, a correlation was sought between these parameters and the health status of MS patients at specific time points. A majority of disease-modifying treatments available today are directed to relapsing-remitting MS, and there is a paucity of drugs to treat secondary and primary progressive MS [14, 20]. Treatment of MS patients with IFN-beta-1a reduces relapses without altering the circulating numbers of CD4+CD25hi Treg cells [95]. Another study demonstrated that treatment of relapsing-remitting MS patients with IFN-beta-1a increased the proportion of CD4+CD25+GITR+ Treg cells above the baseline [125]. Glatiramer acetate treatment of relapsing-remitting MS patients reconstituted naïve Treg cells and increased total Treg cell numbers [126]. Combined treatment with IFN-beta-1a and glatiramer acetate reduced the numbers of naïve (CD4+CD25+CD127lowCD45RA+) Treg cells without affecting the memory type Treg cells (CD4+CD25+CD127lowCD45RO+) in chronic MS patients [96]. Glatiramer acetate treatment of MS patients improved the Treg cell function by expanding CD4+CD25+FOXP3+ T cells [96].
Interestingly, IFN-1a-beta treatment redistributed tTreg subset to central memory-like Treg population expressing CCR27 and the increased Tr1-like subset that expressed IL-10 and CD46 mRNA [127]. Natalizumab, a monoclonal humanized antibody targeting the α-4 chain of the very late antigen 4 (VLA-4) integrin, reduces relapses independent of alterations in Treg cell frequency or function [128]. Although these disease-modifying therapies (IFN-1a-beta, glatiramer acetate, and natalizumab) reduce relapses in MS patients, they do not provide robust protection against MS or reverse axonal degeneration [20]. It will be exciting and appropriate to determine the alteration of the number and function of Treg cells during various stages of the disease and after treatment with more effective disease-modifying drugs when they become available.
Pharmacological Modulation of Treg Cells
Posttranslational modifications of histones by acetylation, phosphorylation, and ubiquitylation are powerful epigenetic modulations that have a substantial influence on gene expression [129]. Epigenetic markers including acetylation and methylation of histones and cytosine-guanosine (CpG) dinucleotide methylation have been reported at the Foxp3 locus [130]. Naive CD4+CD25− T cells, activated CD4+ T cells, and TGF-β-induced adaptive Treg cells, but not in natural Tregs, CpG dinucleotides are methylated at the Foxp3 locus. It has been proposed that Treg cells can be manipulated via epigenetic modification of the transcription factor Foxp3 in mice. In support of this notion, it was shown that treatment of mice with TSA enhanced Treg cell-mediated suppression of homeostatic proliferation and decreased inflammatory bowel disease [131]. In conjunction with low-dose rapamycin, TSA induced permanent Treg cell-dependent cardiac and islet allograft survival and donor-specific allograft tolerance [131].
In contrast to these data, attrition of the naturally occurring type 1 diabetes and primary, progressive EAE in autoimmune-prone NOD mice by TSA treatment was not associated with the modulation of CD4+CD25+Foxp3+ Treg cells or function [132, 47]. Global gene expression analysis indicated up- and downregulation of many genes in uninduced splenocytes from TSA-treated mice including novel proinflammatory genes specifically expressed in macrophages [133] but not Foxp3 transcription (unpublished data). On the contrary, TSA treatment induced histone hyperacetylation and reduced inflammation, demyelination, and axonal damage in the spinal cord [47]. Interestingly, drug treatment diminished the generation of CD4+ memory T cells and induced antigen-specific tolerance (Fig. 1) as indicated by abrogation of T-cell proliferation when draining lymph node cells and splenocytes were stimulated with MOG35–55 in vitro albeit normal proliferation to a T-cell mitogen. However, activation with IL-2 restored the ability of tolerized T cells to respond to antigen stimulation, indicating a reversal of anergy. Tolerance was characterized by the reduced antigen-induced production of IL-17A, IFN-γ, and GM-CSF but not IL-4.

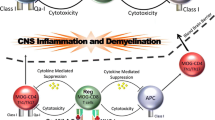
Overview of mechanisms involved in the regulation of autoimmune neurodegeneration. In the experimental model of MS, drug-mediated histone hyperacetylation induces anergy in effector IFN-γ-producing Th1 cells, IL-17A-expressing Th17 cells, and cells with mixed phenotype (Th1/Th17). In addition, the neutrophil expansion is also subject to regulation by the epigenetic modifier. Together, they contribute to the amelioration of neurodegeneration in the mouse model. It remains to be determined whether similar manipulation of IL-17A- and IFN-γ-producing Th1/Th17 cells found in MS patients by pharmacological intervention could benefit patients with MS. Defective numbers and function of Treg cells have been reported in MS patients, suggesting a role for these cells in disease pathogenesis. Restoration of functional Treg populations in MS patients may potentially provide therapeutic advantages. A role for Tr1 cells in MS disease pathogenesis is also possible
The consecutive appearance of double producers (IL-17A + IFN-γ) and Th1 cells occurred in peripheral lymphoid tissues and was susceptible to repression by TSA treatment. In the CNS, only Th1 cells appeared during the acute phase, while Th1 cells, Th17 cells, and GM-CSF-expressing cells were found in the chronic phase. Importantly, TSA treatment diminished the frequencies of these cells in the CNS [47]. In the primary, progressive EAE model, TSA treatment also reduced the abundance of mature CD11b+Ly-6Gdim-activated neutrophils in the secondary lymphoid tissues and their influx into the spinal cord [48]. Thus, in addition to myelin-specific T-cell tolerance induction, selective repression of mature neutrophils and PD-L1+ cells is critically involved in the epigenetic regulation of primary, progressive EAE. These data indicate that epigenetic regulation by histone acetylation ameliorates autoimmune diseases such as type 1 diabetes and EAE by modulating gene expression without involving the Treg population or Foxp3 transcription.
Interestingly, treatment with various small molecule inhibitors of histone deacetylases increased the suppressive function of both freshly isolated and in vitro-expanded human Treg cells [134]. This functional change was accompanied by increased expression of the negative regulator of immune response, CTLA-4, indicating that epigenetic drugs can be promising pharmacologic agents that can improve the immunosuppressive potential of T lymphocytes. Since TSA, the most potent inhibitor of histone deacetylases is effective in reversing naturally occurring type 1 diabetes and immunization-induced EAE without causing undesirable side effects [132–134, 48], this strategy may be of potential use to treat patients with autoimmunity.
Administration of the lipid-lowering drug atorvastatin alleviated EAE without increasing IL-4-producing Th2 cells or Treg population, implicated in protection against neurodegeneration [135]. This observation is consistent with the ability of statins, inhibitors of cholesterol biosynthesis, to affect IL-17-producing Th17 cells [136]. Since Th17 cells are pivotal for MS pathogenesis [14], inhibition of IL-17 production by statins could be useful for the treatment of MS.
Antigen-Specific Tolerance-Inducing Strategies Failed to Block MS Progression
Since MS is considered as an autoimmune disease, induction of antigen-specific tolerance is the best approach to annihilate this debilitating disease via inactivation of autoreactive T cells without causing undesirable side effects. As MS is genetically predisposed, self-reactive T lymphocytes recognizing neuronal antigens in the context of HLA-DRB1 hypothetically escape thymic deletion (central tolerance), which can be subsequently reactivated by the peripheral antigen-presenting cells causing dire consequences. As discussed above, the induction of anergy could restrain these autoreactive T cells from causing neuronal destruction. Toward this goal, several clinical trials were conducted in MS patients by administering peptides derived from MBP, MOG, and PLP via various routes [Ref. 73, 74 and citations therein]. Also, complexes of HLA class II molecule HLA-DR2 and MOG35–55, and myelin peptides (MBP85–99, MOG35–55, and PLP139–151), a plasmid containing MBP protein, referred to as DNA vaccine, attenuated autologous T cells specific to MBP, MOG, and PLP were candidates for tolerance induction in MS patients. Furthermore, peripheral blood mononuclear cells coupled with seven myelin peptides (MOG1–20, MOG35–55, MBP13–32, MBP83–99, MBP111–129, MBP146–170, and PLP139–154) were also tested for their efficacy to produce a favorable outcome in MS patients. Not surprisingly, oral administration of MBP failed to protect MS [73, 74], as observed previously in type 1 diabetes [137]. Altered ligand peptide analogs of immunogenic peptides that have been modified to interact with the T-cell receptor while retaining the ability to bind the HLA motifs proved fatal in clinical trials [138]. Although most of these approaches did not have safety issues, no breakthrough as to clinical benefits has been achieved. Recent efforts to establish an antigen-specific tolerance in autoimmune patients include administration of tolerogenic dendritic cells [139] and tolerogenic immune-modifying nanoparticles [140]. So far, successful “tolerance-inducing” strategies have not emerged as standard-of-care clinical use. The identification of the disease-instigating antigen(s) will pave the way for the successful design of antigen-specific tolerance for the treatment of the debilitating CNS disease.
The Future Perspectives
It is now well established that peripheral tolerance is of paramount importance in the homeostatic control of the T-cell repertoire and for curtailing autoimmunity. The concerted effort for over half a century has unraveled unexpectedly diverse types of Treg cells in the mouse. By analogy, Treg cells with multiple phenotypes have been reported in humans as well. Attempts to understand the mode of immunosuppression mediated by these cells yielded clues to several mechanisms that need to be carefully discerned. Limited studies conducted in EAE models do not support in vivo manipulation of Treg cells, mostly the thymic-derived, “naturally occurring” Treg cells, by epigenetic or pharmacological strategies (Fig. 1). Although disease-modifying therapies provide some benefits to MS patients, it is unclear whether disease protection accompanies changes in the Treg cell number, phenotype, or function. Adoption of uniform parameters for evaluation of phenotypic markers may help to improve our understanding of the status of the Treg subsets during the variable and prolonged duration of chronic neurodegeneration. It is important to emphasize the need for refining in vitro conditions required for assessing the “suppressive” function of human Treg cells. The fact that the Treg cells are scarce in the brain lesions but abundant in the cerebrospinal fluid of MS patients [141] suggests that anatomical constraints are responsible for this uneven distribution of Treg cells. It is essential to know whether pharmacological intervention or administration of biologicals to improve MS symptoms may influence the Treg cell trafficking to the brain. Finally, since the Treg cells producing IL-10, type 1 Treg (Tr1), were also reported to be impaired in MS patients [127, 142], it will be informative whether disease-modifying treatments can also impact this cellular compartment.
References
Alderson MR, Lynch DH. Receptors and ligands that mediate activation-induced death of T cells. Springer Semin Immunopathol. 1998;19(3):289–300. PMID:9540157
Tracey KJ, Cerami A. Tumor necrosis factor: an updated review of its biology. Crit Care Med. 1993;21(10 Suppl):S415–22. PMID:8403979
Dorf ME, Benacerraf B. Suppressor cells and immunoregulation. Annu Rev Immunol. 1984;2:127–57. PMID:6242348
Germain RN. Special regulatory T-cell review: A rose by any other name: from suppressor T cells to Tregs, approbation to unbridled enthusiasm. Immunology. 2008;123(1):20–7. PMID:18154615
Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev. 2014;259(1):231–44. PMID:24712469
Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation. 2017;14(1):117. PMID:28599652
Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19:665. https://doi.org/10.1038/s41590-018-0120-4. PMID:29925983
Shevach EM. Foxp3+ T regulatory cells: still many unanswered questions-A perspective after 20 years of study. Front Immunol. 2018;9:1048. PMID:29868011
Jasiak-Zatonska M, Kalinowska-Lyszczarz A, Michalak S, Kozubski W. The immunology of neuromyelitis optica-current knowledge, clinical implications, controversies and future perspectives. Int J Mol Sci. 2016;17(3):273. PMID:26950113
Bar-Or A, Steinman L, Behne JM, Benitez-Ribas D, Chin PS, Clare-Salzler M, et al. Restoring immune tolerance in neuromyelitis optica: Part II. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e277. PMID:27648464
D’Andrea MR. Add Alzheimer’s disease to the list of autoimmune diseases. Med Hypotheses. 2005;64(3):458–63. PMID:15617848
Fukata M, Yokoi N, Fukata Y. Neurobiology of autoimmune encephalitis. Curr Opin Neurobiol. 2018;48:1–8. PMID:28829986
Alexopoulos H, Dalakas MC. Immunology of stiff person syndrome and other GAD-associated neurological disorders. Expert Rev Clin Immunol. 2013;9(11):1043–53. PMID:24168411
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–58. PMID:26250739.
Legroux L, Arbour N. Multiple sclerosis and T Lymphocytes: an entangled story. J Neuroimmune Pharmacol. 2015;10(4):528–46. PMID:25946987
Lassmann H. Targets of therapy in progressive MS. Mult Scler. 2017;23(12):1593–9. PMID:29041864
Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 2017;140(3):527–46. PMID:27794524
Chaudhuri A, Behan PO. Multiple sclerosis is not an autoimmune disease. Arch Neurol. 2004;61(10):1610–2. PMID:15477520
Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–69. PMID:18558855
Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89(2):225–40. PMID:24485135
Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis - success from bench to bedside. Nat Rev Neurol. 2019;15:53–8. https://doi.org/10.1038/s41582-018-0082-z. PMID:30315270
Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol. 2017;18(2):123–31. PMID:28092374
Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016;4:71. PMID:27551677
Scheu S, Ali S, Ruland C, Arolt V, Alferink J. The C-C chemokines CCL17 and CCL22 and their receptor CCR4 in CNS autoimmunity. Int J Mol Sci. 2017;18(11):pii:E2306. PMID:29099057
Parnell GP, Booth DR. The Multiple Sclerosis (MS) genetic risk factors indicate both acquired and innate immune cell subsets contribute to MS pathogenesis and identify novel therapeutic opportunities. Front Immunol. 2017;8:425. PMID:28458668
Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, et al. T cell-mediated autoimmune disease due to low-affinity crossreactivity to common microbial peptides. Immunity. 2009;30(3):348–57. Erratum in: Immunity. 2009;30(4):610. PMID:19303388
Ramadan A, Lucca LE, Carrié N, Desbois S, Axisa PP, Hayder M, et al. In situ expansion of T cells that recognize distinct self-antigens sustains autoimmunity in the CNS. Brain. 2016;139.(Pt 5:1433–46.
Fredrikson S, Söderström M, Hillert J, Sun JB, Käll TB, Link H. Multiple sclerosis: occurrence of myelin basic protein peptide-reactive T cells in healthy family members. Acta Neurol Scand. 1994;89(3):184–9. PMID:7518178
Hellings N, Barée M, Verhoeven C, D’hooghe MB, Medaer R, Bernard CC, et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63(3):290–302. PMID:11170179
Raddassi K, Kent SC, Yang J, Bourcier K, Bradshaw EM, Seyfert-Margolis V, et al. Increased frequencies of myelin oligodendrocyte glycoprotein/MHC class II-binding CD4 cells in patients with multiple sclerosis. J Immunol. 2011;187(2):1039–46. PMID:21653833
Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, Arbour N, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66(3):390–402. PMID:19810097
Alvermann S, Hennig C, Stüve O, Wiendl H, Stangel M. Immunophenotyping of cerebrospinal fluid cells in multiple sclerosis: in search of biomarkers. JAMA Neurol. 2014;71(7):905–12. PMID:24818670
van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, et al. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain. 2018;141(5):1334–49. PMID:29659729
Willing A, Leach OA, Ufer F, Attfield KE, Steinbach K, Kursawe N, et al. CD8+ MAIT cells infiltrate into the CNS and alterations in their blood frequencies correlate with IL-18 serum levels in multiple sclerosis. Eur J Immunol. 2014;44(10):3119–28. PMID:25043505
Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol 2018. https://doi.org/10.1038/s41590-018-0135-x. [Epub ahead of print]. PMID:29925992.
Wurth S, Kuenz B, Bsteh G, Ehling R, Di Pauli F, Hegen H, et al. Cerebrospinal fluid B cells and disease progression in multiple sclerosis – A longitudinal prospective study. PLoS One. 2017;12(8):e0182462. PMID:28777826
Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med. 1933;58(1):39–53. PMID:19870180
Sabin AB, Wright AM. Acute ascending myelitis following a monkey bite, with the isolation of a virus capable of reproducing the disease. J Exp Med. 1934;59(2):115–36. PMID:19870235
Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079–106. PMID:21371012
Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. 2017;133(2):223–44. PMID:27766432
Baker D, Amor S. Experimental autoimmune encephalomyelitis is a good model of multiple sclerosis if used wisely. Mult Scler Relat Disord. 2014;3(5):555–64. PMID:26265267
McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175(5):3025–32. PMID:16116190
Shetty A, Gupta SG, Varrin-Doyer M, Weber MS, Prod’homme T, Molnarfi N, et al. Immunodominant T-cell epitopes of MOG reside in its transmembrane and cytoplasmic domains in EAE. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):e22. PMID:25340074
Pham H, Doerrbecker J, Ramp AA, D’Souza CS, Gorasia DG, Purcell AW, Ayers MM, Orian JM. Experimental autoimmune encephalomyelitis (EAE) in C57Bl/6 mice is not associated with astrogliosis. J Neuroimmunol. 2011;232(1–2):51–62. PMID:21056916
Kipp M, Nyamoya S, Hochstrasser T, Amor S. Multiple sclerosis animal models: a clinical and histopathological perspective. Brain Pathol. 2017;27(2):123–37. PMID:27792289
Basso AS, Frenkel D, Quintana FJ, Costa-Pinto FA, Petrovic-Stojkovic S, Puckett L, et al. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;118(4):1532–43. PMID:18340379
Jayaraman A, Soni A, Prabhakar BS, Holterman M, Jayaraman S. The epigenetic drug Trichostatin A ameliorates experimental autoimmune encephalomyelitis via T cell tolerance induction and impaired influx of T cells into the spinal cord. Neurobiol Dis. 2017;108:1–12. PMID:28736194
Jayaraman A, Sharma M, Prabhakar B, Holterman M, Jayaraman S. Amelioration of progressive autoimmune encephalomyelitis by epigenetic regulation involves selective repression of mature neutrophils during the preclinical phase. Exp Neurol. 2018;304:14–20. PMID:29453977
Slavin A, Ewing C, Liu J, Ichikawa M, Slavin J, Bernard CC. Induction of a multiple sclerosis-like disease in mice with an immunodominant epitope of myelin oligodendrocyte glycoprotein. Autoimmunity. 1998;28(2):109–20. PMID:9771980
Hidaka Y, Inaba Y, Matsuda K, Itoh M, Kaneyama T, Nakazawa Y, et al. Cytokine production profiles in chronic relapsing-remitting experimental autoimmune encephalomyelitis: IFN-γ and TNF-α are essential participants in the first attack but not in the relapse. J Neurol Sci. 2014;340(1–2):117–22. PMID:24655735
Dang PT, Bui Q, D’Souza CS, Orian JM. Modelling MS: chronic-relapsing EAE in the NOD/Lt mouse strain. Curr Top Behav Neurosci. 2015;26:143–77. PMID:26126592
McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD. Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol. 1992;38(3):229–40. PMID:1376328
Behan PO, Chaudhuri A. EAE is not a useful model for demyelinating disease. Mult Scler Relat Disord. 2014;3(5):565–74.PMID:26265268.
Muller DM, Pender MP, Greer JM. A neuropathological analysis of experimental autoimmune encephalomyelitis with predominant brain stem and cerebellar involvement and differences between active and passive induction. Acta Neuropathol. 2000;100(2):174–82. PMID:10963365
Abromson-Leeman S, Bronson R, Luo Y, Berman M, Leeman R, Leeman J, et al. T-cell properties determine disease site, clinical presentation, and cellular pathology of experimental autoimmune encephalomyelitis. Am J Pathol. 2004;165(5):1519–33. PMID:15509523
Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14(3):337–42. PMID:18278054
Lowther DE, Chong DL, Ascough S, Ettorre A, Ingram RJ, Boyton RJ, et al. Th1 not Th17 cells drive spontaneous MS-like disease despite a functional regulatory T cell response. Acta Neuropathol. 2013;126(4):501–15. PMID:23934116
O’Connor RA, Prendergast CT, Sabatos CA, Lau CW, Leech MD, Wraith DC, et al. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181(6):3750–4. PMID:18768826
Murphy AC, Lalor SJ, Lynch SJ, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24(4):641–51. PMID:20138983
Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12(3):255–63. PMID:21278737
Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–7. PMID:21516112
Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. 2018;87:1–15. PMID:29275836
Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. 2009;119(1):61–9. PMID:19075395
McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194(7):873–82. PMID:11581310
Pierson ER, Goverman JM. GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight. 2017;2(7):e92362. PMID:28405624
Duncker PC, Stoolman JS, Huber AK, Segal BM. GM-CSF promotes chronic disability in experimental autoimmune encephalomyelitis by altering the composition of central nervous system-infiltrating cells, but is dispensable for disease induction. J Immunol. 2018;200(3):966–73. PMID:29288202
Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J, et al. Targeting the GM-CSF rece2017ptor for the treatment of CNS autoimmunity. J Autoimmun. 84:1–11. PMID:28641926
Marrack P, Lo D, Brinster R, Palmiter R, Burkly L, Flavell RH, et al. The effect of thymus environment on T cell development and tolerance. Cell. 1988;53(4):627–34. PMID:3259471
Fredrikson S, Söderström M, Hillert J, Sun JB, Käll TB, Link H. Multiple sclerosis: occurrence of myelin basic protein peptide-reactive T cells in healthy family members. Acta Neurol Scand. 1994;89(3):184–9. PMID:7518178
Hellings N, Barée M, Verhoeven C, D’hooghe MB, Medaer R, Bernard CC, et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63(3):290–302. PMID:11170179
Raddassi K, Kent SC, Yang J, Bourcier K, Bradshaw EM, Seyfert-Margolis V, et al. Increased frequencies of myelin oligodendrocyte glycoprotein/MHC class II-binding CD4 cells in patients with multiple sclerosis. J Immunol. 2011;187(2):1039–46. PMID:21653833
Jayaraman S. Novel methods of type 1 diabetes treatment. Discov Med. 2014;17(96):347–55. PMID:24979255
Lutterotti A, Martin R. Antigen-specific tolerization approaches in multiple sclerosis. Expert Opin Investig Drugs. 2014;23(1):9–20. PMID:24151958
Steinman L. The re-emergence of antigen-specific tolerance as a potential therapy for MS. Mult Scler. 2015;21(10):1223–38. PMID:25921045
Schwartz RH, Mueller DL, Jenkins MK, Quill H. T-cell clonal anergy. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 2:605–10. PMID:2534840
Beverly B, Kang SM, Lenardo MJ, Schwartz RH. Reversal of in vitro T cell clonal anergy by IL-2 stimulation. Int Immunol. 1992;4(6):661–71. PMID:1616898
Jayaraman S, Luo Y, Dorf ME. Tolerance induction in T helper (Th1) cells by thymic macrophages. J Immunol. 1992;148(9):2672–81. PMID:1533409
Jayaraman S, Bellone CJ. Interaction of idiotype-specific T suppressor factor with the hapten-specific third-order T suppressor subset results in antigen-nonspecific suppression. Cell Immunol. 1986;101(1):72–81. PMID:3489537
Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161(1):72–87. PMID:3871469
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3):1151–64. PMID:21422251
Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–61. PMID:12522256
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. PMID: 12612578
Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899–911. PMID:19464196
Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. 2016;37(11):803–11. PMID:27623114
Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. PMID:11722624
Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626–35. PMID:19464985
Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164(1):183–90. PMID:10605010
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167(3):1245–53. PMID:11466340
Stephens LA, Mottet C, Mason D, Powrie F. Human CD4 + CD25 + thymocytes and peripheral T cells have immune suppressive activity in vitro. Eur J Immunol. 2001;31(4):1247–54. PMID:11298351
Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4 + CD25 + regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199(7):971–9. PMID: 15067033
Putheti P, Pettersson A, Soderstorm M, Link H, Huang YM. Circulating CD4 + CD25 + T regulatory cells are not altered in multiple sclerosis and unaffected by disease-modulating drugs. J Clin Immunol. 2004;24(2):155–61. PMID:15024182
Haas J, Hug A, Viehöver A, Fritzsching B, Falk CS, Filser A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35(11):3343–52. PMID:16206232
Michel L, Berthelot L, Pettré S, Wiertlewski S, Lefrère F, Braudeau C, et al. Patients with relapsing-remitting multiple sclerosis have normal Treg function when cells expressing IL-7 receptor α-chain are excluded from the analysis. J Clin Invest. 2008;118(10):3411–9. PMID:18769633
Feger U, Luther C, Poeschel S, Melms A, Tolosa E, Wiendl H. Increased frequency of CD4 + CD25 + regulatory T cells in the cerebrospinal fluid but not in the blood of multiple sclerosis patients. Clin Exp Immunol. 2007;147(3):412–8. PMID:17302889
Venken K, Hellings N, Broekmans T, Hensen K, Rummens JL, Stinissen P. Natural naive CD4 + CD25 + CD127 low regulatory T cell (Treg) development and function are disturbed in multiple sclerosis patients: recovery of memory T reg homeostasis during disease progression. J Immunol. 2008;180(9):6411–20. PMID:18424765
Kumar M, Putzki N, Limmroth V, Remus R, Lindemann M, Knop D, et al. CD4 + CD25 + FoxP3 + T lymphocytes fail to suppress myelin basic protein-induced proliferation in patients with multiple sclerosis. J Neuroimmunol. 2006;180(1–2):178–84. PMID:17011048
Venken K, Hellings N, Thewissen M, Somers V, Hensen K, Rummens JL, et al. Compromised CD4 + CD25 high regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with a reduced frequency of FOXP3-positive cells and reduced FOXP3 expression at the single-cell level. Immunology. 2008;123(1):79–89. PMID:17897326
Frisullo G, Nociti V, Iorio R, Patanella AK, Caggiula M, Marti A, et al. Regulatory T cells fail to suppress CD4T+-bet+ T cells in relapsing multiple sclerosis patients. Immunology. 2009;127(3):418–28. PMID:19016907
Haas J, Fritzsching B, Trübswetter P, Korporal M, Milkova L, Fritz B, et al. Prevalence of newly generated naive regulatory T cells (T reg) is critical for T suppressive function and determines T reg dysfunction in multiple sclerosis. J Immunol. 2007;179(2):1322–30. PMID:17617625
Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3 + Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4):1225–32. PMID: 17449799
Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O’Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. 2009;183(11):7602–10. PMID: 19917691
Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediar. 1982;100(5):731–7. PMID:7040622
Tan QKG, Louie RJ, Sleasman JW. IPEX Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington; 2004. Seattle; 1993–2018. [updated 2018 Jul 19]. PMID:20301297.
Zavattari P, Deidda E, Pitzalis M, Zoa B, Moi L, Lampis R, et al. No association between variation of the FOXP3 gene and common type 1 diabetes in the Sardinian population. Diabetes. 2004;53(7):1911–4. PMID:15220219
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27(1):18–20. PMID:11137992
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–1. PMID:11137993
Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, et al. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006;116(6):1713–22. PMID:16741580
Krishnamoorthy G, Holz A, Wekerle H. Experimental models of spontaneous autoimmune disease in the central nervous system. J Mol Med (Berl). 2007;85(11):1161–73. PMID:17569024
Koutrolos M, Berer K, Kawakami N, Wekerle H, Krishnamoorthy G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol Commun. 2014;2:163.. PMID:25476447.
Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13(4):423–31.
Montero E, Nussbaum G, Kaye JF, Perez R, Lage A, Ben-Nun A, Cohen IR. Regulation of experimental autoimmune encephalomyelitis by CD4+, CD25+ and CD8+ T cells: analysis using depleting antibodies. J Autoimmun. 2004;23(1):1–7. PMID:15236747
Gärtner D, Hoff H, Gimsa U, Burmester GR, Brunner-Weinzierl MC. CD25 regulatory T cells determine secondary but not primary remission in EAE: impact on long-term disease progression. J Neuroimmunol. 2006;172(1–2):73–84. PMID:16360886
Rudra D, de Roos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol. 2012;13(10):1010–9. PMID:22922362
Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity. 2012;36(5):717–30. PMID:22579476
Dominguez-Villar M, Baecher-Allan CM, Hafler DA. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat Med. 2011;17(6):673–5. PMID:21540856
Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3 + T cells into T H 17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–8. PMID: 24362934
Zhang Z, Zhang W, Guo J, Gu Q, Zhu X, Zhou X. Activation and functional specialization of regulatory T cells Lead to the generation of Foxp3 instability. J Immunol. 2017;198(7):2612–25. PMID:28228556
Jin HS, Park Y, Elly C, Liu YC. Itch expression by Treg cells controls Th2 inflammatory responses. J Clin Invest. 2013;123(11):4923–34. PMID:24135136
Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. 2016;34:609–33. PMID:27168246
Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity. 2013;39(5):949–62. PMID:24238343
Huan J, Culbertson N, Spencer L, Bartholomew R, Burrows GG, Chou YK, et al. Decreased FoxP3 levels in multiple sclerosis patients. J Neurosci Res. 2005;81(1):45–52. PMID:15952173
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172(1):146–55.
Venken K, Hellings N, Hensen K, Rummens JL, Medaer R, D’hooghe MB, et al. Secondary progressive in contrast to relapsing-remitting multiple sclerosis patients show a normal CD4+CD25+ regulatory T-cell function and FoxP3 expression. J Neurosci Res. 2006;83(8):1432–46. PMID:16583400
Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–59. PMID:21107346
de Andrés C, Aristimuño C, de Las Heras V, Martínez-Ginés ML, Bartolomé M, Arroyo R, et al. Interferon β-1a therapy enhances CD4 + regulatory T-cell function: an ex vivo and in vitro longitudinal study in relapsing-remitting multiple sclerosis. J Neuroimmunol. 2007;182(1–2):204–11. PMID:17157927
Haas J, Korporal M, Balint B, Fritzsching B, Schwarz A, Wildemann B. Glatiramer acetate improves regulatory T-cell function by expansion of naive CD4(+)CD25(+)FoxP3(+)CD31(+) T-cells in patients with multiple sclerosis. J Neuroimmunol. 2009;216(1–2):113–7. PMID:19646767
Chiarini M, Serana F, Zanotti C, Capra R, Rasia S, Rottoli M, et al. Modulation of the central memory and Tr1-like regulatory T cells in multiple sclerosis patients responsive to interferon-beta therapy. Multi Scler. 2012;18(6):788–98. PMID:22086901
Stenner MP, Waschbisch A, Buck D, Doerck S, Einsele H, Toyka KV, et al. Effects of natalizumab treatment on Foxp3+ T regulatory cells. PLoS One. 2008;3(10):e3319.. PMID:18836525
Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. PMID:27346641
Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. 2009;114(18):3727–35. PMID:19641188
Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13(11):1299–307. PMID:17922010
Patel T, Patel V, Singh R, Jayaraman S. Chromatin remodeling resets the immune system to protect against autoimmune diabetes in mice. Immunol Cell Biol. 2011;89(5):640–9. PMID:21321581
Jayaraman S, Patel A, Jayaraman A, Patel V, Holterman M, Prabhakar B. Transcriptome analysis of epigenetically modulated genome indicates signature genes in manifestation of type 1 diabetes and its prevention in NOD mice. PLoS One. 2013;8(1):e55074. PMID:23383062
Akimova T, Ge G, Golovina T, Mikheeva T, Wang L, Riley JL, et al. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin Immunol. 2010;136(3):348–63. PMID:20478744
Weber MS, Prod’homme T, Youssef S, Dunn SE, Steinman L, Zamvil SS. Neither T-helper type 2 nor Foxp3+ regulatory T cells are necessary for therapeutic benefit of atorvastatin in treatment of central nervous system autoimmunity. J Neuroinflammation. 2014;11:29. PMID:24498870
Ulivieri C, Baldari CT. Statins: from cholesterol-lowering drugs to novel immunomodulators for the treatment of Th17-mediated autoimmune diseases. Pharmacol Res. 2014;88:41–52. PMID:24657239
Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group, Krischer JP, Schatz DA, Bundy B, Skyler JS, Greenbaum CJ. Effect of Oral insulin on prevention of diabetes in relatives of patients with type 1 diabetes: a randomized clinical trial. JAMA. 2017;318(19):1891–902. PMID:29164254
Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6(10):1167–75. PMID:11017150
Phillips BE, Garciafigueroa Y, Trucco M, Giannoukakis N. Clinical Tolerogenic dendritic cells: exploring therapeutic impact on human autoimmune disease. Front Immunol. 2017;8:1279. PMID:29075262
Pearson RM, Podojil JR, Shea LD, King NJ, Miller SD, Getts DR. Overcoming challenges in treating autoimmuntity: development of tolerogenic immune-modifying nanoparticles. Nanomedicine. 2018; pii: S1549–9634(18)30538–0. PMID:30352312.
Fritzsching B, Haas J, König F, Kunz P, Fritzsching E, Pöschl J, et al. Intracerebral human regulatory T cells: analysis of CD4+ CD25+ FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. PLoS One. 2011;6(3):e17988. PMID:21437244
Martinez-Forero I, Garcia-Munoz R, Martinez-Pasamar S, Inoges S, Lopez-Diaz de Cerio A, Palacios R, et al. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur J Immunol. 2008;38(2):576–86. PMID:18200504
Acknowledgments
Arathi Jayaraman is acknowledged for comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jayaraman, S., Prabhakar, B.S. (2019). Immune Tolerance in Autoimmune Central Nervous System Disorders. In: Mitoma, H., Manto, M. (eds) Neuroimmune Diseases. Contemporary Clinical Neuroscience. Springer, Cham. https://doi.org/10.1007/978-3-030-19515-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-19515-1_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-19514-4
Online ISBN: 978-3-030-19515-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)