
Abstract
Rapid development in genome-wide transcriptional analyses has led to the discovery of a large number of non-coding transcripts, also called long non-coding RNA (lncRNA). LncRNAs harbor biological activities including regulation of protein-coding gene expression at epigenetic, transcriptional and post-transcriptional levels. They also take a part in various physiological and pathological processes, participating in cell development, immunity, disease processes and oncogenesis.
Here I discuss and summarize, current knowledge about lncRNA origin, function and involvement in human disease.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others



Keywords
Introduction
Rapid developments in sequencing technologies during past decade revealed that protein coding genes represent only 2 % of the human genome [1]. This came across as a surprise considering that genomes of the other organisms (from yeast to Caenorhabditis elegans) are quite dense. Is the rest of the human genome merely “junk” DNA? This question was partially answered by experimental approaches like high-throughput sequencing or whole genome high density tailing arrays. Now it is known that this “junk” DNA is transcribed throughout mammalian genomes and because it lacks protein coding capacity it is referred to as long non-coding RNA (lncRNA) [2–7].
Non-coding transcripts have similar structure to messenger or coding RNA (mRNA): they are transcribed by RNA polymerase II (Pol II), they possess a cap and a polyA tail and they can be even spliced . However their function remains enigmatic [8]. Non-coding RNAs can be also divided into two groups based on their length: short and long non-coding RNA s (ncRNAs) or based on their primary function: structural and regulatory ncRNA (Fig. 4.1). Unlike mRNA and structural ncRNAs, most of lncRNAs are localized in nucleus [7].

Summary of various types of non-coding RNAs
Prior to the sequencing era, some lncRNA were discovered using old-fashioned gene cloning methods. Initially they were thought to be coding RNAs, however, deeper analyses revealed a lack of open reading frames (ORF). Furthermore they were thought to be random in nature more so than stable elements in genome . This opinion changed when FANTOM consortium analyzed over 60,000 full length cDNAs and identified over 11,000 lncRNAs in mouse [9]. Interestingly, a large portion of identified lncRNAs is transcribed in antisense orientation to protein-coding genes, thus are referred to as natural antisense transcripts (NATs). Another study extended these findings by identifying NATs in human genome [10]. Interestingly many cancer associated genes, particularly tumor suppressor genes have long antisense ncRNA.
More recently, it was shown that intergenic regions also express thousands of long non-coding RNA s, named large intervening non-coding RNAs (lincRNAs). These transcripts were discovered by analyses of active chromatin marks (H3K4 acetylation and H3K36 trimethylation) genome -wide and eliminating those regions corresponding to protein coding genes and microRNAs . This approach was followed by extended analyses using RNA -seq experiments [4–6, 11–13]. Up to date, there are more than 8000 lincRNAs identified. More than half one them are confirmed lincRNAs; they can be localized in nucleus, cytoplasm or both and they are multi-exonic, capped and polyadenylated.
A major interest now lies in functional analysis of lncRNA. The fact that they are not evolutionary conserved, even between related species could indicate that most of them are non functional and may represent just transcriptional noise. To date, only 200 lncRNAs have been studied functionally with variable outcome, although many of them show at least functional evidence in vitro. Only a few lncRNAs have been studied in animal models, suggesting that they are not essential for viability. For example, mouse homologue of HOTAIR is poorly conserved in sequence and its deletion, along with the deletion of the HoxC cluster, has only a little effect in vivo, neither on the expression pattern or transcription efficiency, nor on the amount of K27me3 coverage of different Hoxd target genes [14]. On the other hand it is possible that the lack of one particular lncRNA can substituted by another one.
Overall, it is certain that mammalian genomes, including humans, produce thousands of lncRNAs. Due to the complexity and variability of lncRNAs it may take several years to understand and clarify their functional identity. It is very likely that some lncRNA are involved in a range of biological processes. In this chapter, I will discuss a number of important topics regarding lncRNAs: their origin, function, and role in disease .
Origins and Genome Localization of lncRNA
Low degree of lncRNA conservation among species suggests their different evolutionary design to protein coding genes. The limited phylogenetic range of lncRNA could be also explained by specific existence, but rapid declination within particular lineages [15]. A first possible scenario for lncRNA emergence is metamorphosis of protein-coding gene into non-coding RNA seq uence. A protein-coding gene may under go mutations such as a frame shift that disrupts its open reading frame while maintaining the expression of the RNA transcript. For example, Xist gene encodes an lncRNA that is crucial for X chromosome inactivation. Recently, it’s been shown that several exons and the promoter of Xist are derived from the protein-coding gene Lnx3 that has acquired frame shift mutations during early mammalian evolution [16, 17]. It is possible that such a metamorphosis involved two steps: initial degeneration of the original sequence, followed by subsequent emergence of residual exons into newly formed Xist gene. Possibly these events occurred concurrently (Fig. 4.2a). A nother possibility includes chromosomal rearrangement, when two separate sequences are joined and together create an expressed non-coding sequence (Fig. 4.2b). One such example comes from the observation that a dog testis-derived non-coding RNA has arisen only recently following a lineage specific change. Also duplications in a non-coding RNA sequence could cause repeats, increasing the length of the transcript. Rare examples of duplicated lncRNA include Neat2 (mouse nuclear enriched abundant transcript) and mouse testis-derived lncRNA that are separate paralogous to non-exonic sequences elsewhere in the genome [18]. Local, tandem duplication might also lead to generation of repeats increasing the size of lncRNA (Fig. 4.2c). Insertions of transposable element s may also emerge as lncRNA. For example, BC1 and BC200 (brain cytoplasmic RNA1 and 200-nucleotide) ncRNAs derive from two separate transposons , in the rodent and anthropoid lineages. Despite their lack of common origin, BC1 and BC200 are involved in similar roles in translational regulation. Furthermore, a transposable element containing a transcriptional start site can be inserted into the genome to create a functional, but noncoding RNA sequence [19] (Fig. 4.2d).

Origins of lncRNAs. Block arrows represent genes. Black regions in block arrows are introns. (a) Mutations in protein coding gene. (b) Chromosomal rearrangements. (c) Duplications. (d) Insertions of transposable element s
In respect of protein-coding genes, lncRNA can overlap with a gene as well as being associated with the gene’s promoter region. They can be transcribed from intragenic sequence, exonic or intron, or from intergenic region (Fig. 4.3). In general, lncRNA are expressed at low levels and their expression varies with location, time, development and physiological stimuli.

Genomic localizations of lncRNAs. Schematic diagram illustrating organization of lncRNAs associated with protein-coding genes. Arrows pointing towards right represent sense transcription , arrows pointing towards left correspond to antisense transcription
Natural Antisense Transcripts (NATs)
Strand specific sequencing methods, like RNA -seq , reveal complex overlapping transcription with many lncRNA being transcribed from complementary DNA strands of protein coding genes. These are referred to as antisense transcripts (AS). AS can arise from novel transcription start sites as well as from bi-directional promoters , or through transcriptional read-through. In yeast, AS do not occur randomly, but have been linked to sexual differentiation or stress response genes [20–22] as well as genes with higher variability in expression. Furthermore, certain AS transcript pairs are conserved across several yeast species.
Recently genome -wide transcriptome studies reveal that natural AS transcripts (NATs) frequently occur across mammalian genomes [23, 24]. The sizes and features of NATs can be variable. They can be classified based on their transcriptional start site, splicing , capping and polyadenylation. With respect to transcriptional start sites, NATs can be divided into three types: (1) overlapping, (2) intronic and (3) intergenic [25, 26]. Sequence of overlapping NATs partly overlaps with sense mRNA sequence with preference for the 3′-untranslated region (3′ UTR). Due to sequence complementarity, these regions can mutually interact by complete or partial hybridization. Thus NATs may function to regulate the expression of the overlapping sense mRNA [27–29].
Long Intergenic ncRNAs (lincRNAs)
Mammalian genomes encode >1000 long intergenic noncoding (linc)RNAs that are clearly conserved across mammals and are potentially functional. These lincRNAs have been implicated in diverse biological processes, including cell-cycle regulation, immune surveillance, and embryonic stem cell pluripotency. However, the mechanism by which these lincRNAs function remains elusive. To date, there are approximately 3300 of human lincRNAs identified by analyzing chromatin -state maps in various human cell types. It is known that one of the well-characterized lincRNA HOTAIR binds the polycomb repressive complex (PRC2). Remarkably, approximately 20 % of lincRNAs expressed in various cell types are bound by PRC2. Other lincRNA interact with different chromatin-modifying complexes. Furthermore depletion of certain lincRNAs associated with PRC2 leads to changes in gene express ion , causing the up-regulation of genes that are normally silenced by PRC2. Therefore it is suggested that some lincRNAs guide chromatin-modifying complexes to specific genomic loci to regulate gene expression [2, 5].
Promoter Associated ncRNAs (CUTs, PROMPTs)
Several genome -wide studies have revealed the unanticipated property of RNA polymerase II (Pol II) to initiate transcription in promoter regions in both directions. Such a bi-directional transcription results in so called cryptic unstable transcripts (CUTs) in budding yeast [30–32]. CUTs are Pol II-dependent transcripts produced from promoters in opposite direction to the coding gene, which are degraded by the nuclear exosome shortly after their synthesis. Another type of transcript derived from promoter regions are stable unannotated transcripts (SUTs), which are not processed by exosome [32]. Bidirectional transcription is not only limited to yeast species, but extends to higher eukaryotes too. Studies using exosome depletion in human fibroblasts have revealed lncRNAs, which correspond to upstream regions of protein coding genes. Such promoter-upstream transcripts are referred to as PROMPTs [33]. Furthermore, RNA-seq analysis from mouse embryonic stem cells has shown many promoter associated transcripts, which are transcribed in non-random, divergent orientation [34]. Another sequencing approach employed global run on sequencing (GRO-seq) to indentify nascent RNAs in human fibroblasts. This study revealed that almost 80 % of the active promoters display bidirectional transcriptional activity [35]. It is now known that bidirectional transcription is a widespread phenomenon, which is conserved across species. There are suggestions that this bidirectional transcription may be a type of gene express ion regulation that promotes an open chromatin structure at promoters by recruiting positive or negative transcriptional regulators.
Enhancer Associated ncRNAs (eRNAs)
Enhancers act to regulate expression of protein-coding genes from a distance in an orientation independent manner. A recent study, which analysed genome wide location of the enhancer binding protein CBP, showed over 12,000 positive loci in mouse neurons. Further transcriptional analysis also confirmed that Pol II is present at 25 % of those loci. Enhancer associated transcripts were identified as lncRNAs (termed eRNAs) produced in both directions and their expression does correlate with mRNA synthesis from nearby gene [36]. There is a possibility that eRNAs may be directly involved in enhancer function. eRNAs may facilitate recruitment of enhancer-associated proteins or enhance chromatin looping to provide contact between the enhancer and the promoter of a particular regulated gene [37].
Repeat Associated ncRNAs
Retrotransposons are genetic elements that can amplify themselves within a genome . They are ubiquitous components of the DNA of many eukaryotic organisms, and are particularly abundant in plants, where they are often a principal component of nuclear DNA. In mammals, almost half the genome (45–48 %) comprises retrotransposons, which possess extensive transcriptional activity. FANTOM 4 project has revealed that in human and mouse genomes, retrotransposons are expressed in a tissue specific manner. They are located close to promoter regions of protein-coding genes, suggesting that they may play a role in controlling alternative promoters or in the post-transcriptional regulation of gene express ion [38].
Pseudogenes are another type of repetitive elements that can be transcribed into lncRNA. These can regulate protein-coding genes through competition for regulatory miRNA binding [39, 40].
Biogenesis, Processing and Structure of lncRNAs
Biogenesis of lncRNA is very similar to mRNA. LncRNAs are produced from many regions across the genome by transcribing Pol II. Only some lncRNAs have been shown to be products of Pol III. The majority of lncRNAs are 5′ capped, polyadenylated and spliced . LncRNA can be divided into two categories based on their orientation: they can be encoded on positive or negative DNA strand (sense or antisense orientation).
Many lncRNA transcripts are not end products, but face to further processing into a final functional form. The presence of sense lncRNA, which contain exons from mRNA sequences and intronic lncRNA that are derived entirely from intronic sequence, has lead to the hypothesis that many lncRNA transcripts are unprocessed pre-mRNAs prior to splicing and that intronic lncRNAs are by-products of this processing step. However, this is not the case for all sense and intronic lncRNAs since the expression patterns of some of these transcripts are not the same as their associated protein-coding gene [41]. Another hypothesis suggests that some lncRNA sequences are precursors to short miRNAs. An example of this is the lncRNA H19 which encodes the miRNA miR-675 [4]. Based on this evidence, post-transcriptional processing may occur with many lncRNA transcripts, but until more of them are functionally defined this question remains open.
Secondary structure formation is an important consideration in lncRNAs because they are able to interact with proteins or genomic DNA via these structures. Recently, models used for prediction of secondary structure have redefined the question of lncRNA evolution by looking at sequence conservation or compensatory mutations that would maintain secondary structure motifs [42]. However, approaches that predict RNA secondary structures with high precision have yet to be developed. Therefore, the number of structured ncRNAs remains to be determined, but is expected in intergenic, intronic and UTR regions and lacking in exon sequence.
Mechanism of Action
Despite the fact that only a fraction of all identified lncRNAs has been examined experimentally, an emerging paradigm suggests that they are implicated in many biological contexts. To date, lncRNAs have been implicated in regulation of gene express ion , guidance of chromatin -modifying complexes, X chromosome inactivation, genomic imprinting, nuclear compartmentalization, nuclear-cytoplasmic trafficking, RNA splicing and translational control [43–46].
Regulation of Chromatin Structure
LncRNAs have been implicated in epigenetic gene regulation. Recent studies propose two basic models for lncRNA action at the chromatin level:
-
1.
epigenetic silencing in cis, where lncRNA transcripts coat gene clusters and silence their expression by making them inaccessible to transcription machinery. These lncRNAs can also recruit chromatin remodeling proteins to epigenetically mark the region for heritable gene silencing;
-
2.
epigenetic silencing in trans: lncRNAs can interact with chromatin modifying proteins to epigenetically silence genes at another locus (Fig. 4.4).
Fig. 4.4 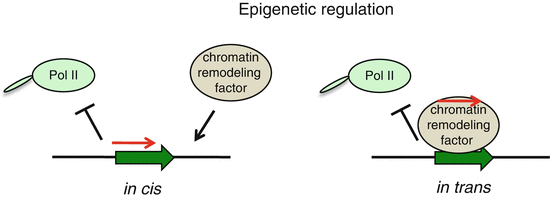
Epigenetic regulation of gene express ion by lncRNAs. Block arrow represents protein-coding gene, red arrow depicts lncRNA

Epigenetic Regulation in cis
One of the well studies lncRNAs to date is Xist, which is crucial for X chromosome inactivation in female somatic cells. It has been discovered in 1991 and despite of enormous effort, the exact mechanism of Xist-mediated X chromosome inactivation is still not fully understood. It is accepted that Xist is associated as an RNA compartment with the inactivated X chromosome [47]. The coating the chromatin that is silenced provides the first model for how lncRNA might function in stable epigenetic gene silencing in cis. Xist establishes a specialized, Pol II free, region, into which most of the X chromosome becomes localized during inactivation [48]. It should be noted that Xist coating of chromatin is stable even during metaphase, suggesting a form of epigenetic memory for the inactive X chromosome to remain silent over many cell divisions [49].
RepA, a small repeat region within Xist, is transcribed from both X chromosomes along with Tsix lncRNA (antisense partner of Xist) [50]. Tsix prevents RepA binding to either X chromosomes until post-cellular differentiation, when RepA in association with the chromatin -modifying complex PRC2 (polycomb repressive complex 2) binds to one of the two X chromosomes at the so called inactivation center [50]. Full-length Xist, produced from the X chromosome , destined to be inactivated, also binds to PRC2 and leads to the spreading of X inactivation from the center to the entire X chromosome in cis. Active chromatin status of the other X chromosome is protected by Tsix, which blocks transcription of Xist. It is not well understood, what prevents Xist from escaping the inactive X chromosome and acting on the active X chromosome in trans or how 20 % of X chromosome genes escape inactivation in human females [51, 52]. Once inactivation is established, X chromosome is condensed into facultative heterochromatin and forms a round body at the nuclear periphery [53]. The inactive chromosome possesses repressive chromatin marks and DNA methylation at CpG island s [52, 54, 55].
Genomic imprinting is another epigenetic phenomenon that utilizes lncRNA [56]. Imprinted genes play an important role in mammalian development and therefore their expression has to be tightly regulated [57]. Interestingly, many imprinted gene loci express lncRNAs that play a crucial role in regulating the expression of neighboring imprinted coding genes in cis [58]. One such lncRNA involved in genomic imprinting in cis is Air, which is mono-allelically expressed from the paternal allele. Air is known to bind to G9a histone methyltransferase and associate with chromatin to participate on silencing of three imprinted genes: Slc22a3, Slc22a2 and Igf2r. Loss of Air leads to bi-allelic expression of Slc22a3 and loss of G9a recruitment to imprinted genes. It has been suggested that Air acts to guide G9a to chromatin at the Slc22a3 promoter [59].
Epigenetic Regulation in trans
In contrast, to previous examples, a long intervening lncRNA, HOTAIR, regulates human genes expression in trans on a genome -wide scale by associating with chromatin modifying complexes such as polycomb repressive complex (PRC2), LSD1 and CoREST/REST [5, 60–62]. It has been shown that 5′ domain of HOTAIR binds PCR2, whereas a 3′ domain of HOTAIR binds LSD1/CoREST complexes. This way HOTAIR guides PCR2 and LSD1/CoREST to their endogenous targets. Consequently, PRC2 methylates histone H3 lysine 27, whilst LSD1/CoREST demethylates histone H3 at lysine 4. This collectively leads to the loss of active histone marks (H3K4 dimethylation) and the gain of a repressive histone marks (H3K27 trimethylation) at the target loci [62].
Gene Regulation Through lncRNA Transcription
Transcription of lncRNA itself can act as both, a positive (activation) or negative (repression) regulator of gene express ion (Fig. 4.5), affecting expression of neighboring genes.

Gene expression regulation through lncRNA transcription . Block arrows are protein-coding genes, red arrows are lncRNAs. Block crosses depict negative transcription factor
Activation: the act of lncRNA transcription can help to open the chromatin structure of a genetic locus to permit access of transcription machinery to neighboring protein-coding genes. In fission yeast, transcription of lncRNAs UAS1 and UAS2 have been shown to activate the expression of the fbp1 gene by this mechanism. Pol II transcribes several species of ncRNAs at the fbp1 locus during transcriptional activation. The chromatin is progressively converted to an open configuration, which is coupled to translocation of Pol II through the upstream region of the fbp1 transcriptional start site. It has been shown that transcription through the promoter region is required to make DNA sequence accessible to transcriptional activators and to Pol II [63]. Similar example of gene transcription regulation have been observed within β-globin locus [64].
Repression: transcription of lncRNAs near protein-coding loci can also act as a negative regulator. The presence of the transcription machinery on the lncRNA gene locus can physically prevent transcription machinery from binding to the protein-coding gene. In budding yeast, transcription of the lncRNA SRG1 inhibits transcription of the overlapping SER3 gene. This repression occurs by a transcription-interference mechanism in which SRG1 transcription across the SER3 promoter interferes with the binding of activators [65]. Such a transcriptional interference process may represent a widespread function for lncRNAs. There seems to be a strong conservation of their promoter regions in contrast to weaker conservation of their transcripts, which is consistent with the act of transcription itself having a greater biological impact than the transcript sequence [7, 66].
Transcriptional Regulation
Protein coding gene express ion is tightly regulated process, which involves direct interactions of proteins with other proteins or DNA. Another aspect of the regulation of gene expression comes from an additional layer of complexity consisting of dynamic interactions between RNA , DNA or proteins. Transcription of lncRNAs can regulate the expression of neighboring genes (in cis regulation) or can also target distant transcriptional activators or repressors (in trans regulation) (Fig. 4.6).

Types of transcriptional regulation by lncRNAs. Block arrows are protein-coding genes, red arrows are lncRNAs. Dashed line depicts nuclear membrane
Transcriptional Regulation in cis
If lncRNA sequence overlaps through complementarity with the binding site of a transcription factor, the lncRNA transcript can hybridize to this site and so prevent a transcription factor from binding. One such an example is a lncRNA that is transcribed from a minor promoter upstream of the human dihudrofolate reductase DHFR gene. Transcription of this full length lncRNA is thought to repress transcription from the major DHFR promoter [67] in an RNA dependent manner. The lncRNA binds to the major DHFR promoter and the general transcription factor IIB and leads to dissociation of the pre-initiation complex. It is proposed that the single-stranded lncRNA hybridizes to double-stranded DNA in the promoter region to form a triplex structure. Such a structure is predicted to be most concentrated around human promoters [68], but it is unclear whether this is a common mechanism for lncRNA transcriptional repression.
Another type of transcriptional gene regulation by lncRNA is the recruitment of transcription factors. When a lncRNA sequence is located near to transcription factor binding site, the lncRNA transcript may enhance the binding of the transcription factor to promoter region. An example of this type of regulation is the lncRNA called Evf2, which regulates two homeodomain genes, Dlx5 and Dlx6, involved in neuronal differentiation, migration and limb pattering [69]. Single-stranded Evf2 forms a complex with Dlx2, another homeodomain protein. This Evf2-Dlx2 complex activates Dlx5/6 enhancer, by a yet unknown mechanism.
Transcriptional Regulation in trans
Another way that lncRNA may regulate transcription is through their affect on transcription factors trafficking in the cell. In particular, lncRNA can either enhance transcription factor access to DNA binding sites or prevent it, as in the case of the lncRNA NRON. This lncRNA prevents the transcription factor NFAT (nuclear factor of activated T cells) from entering the nucleus by directly interacting with importin-beta 1, one of the nuclear-cytoplasm transport factors [70]. The NRON gene contains three exons and can be alternatively spliced , producing variant transcripts ranging in size from 0.8 to 3.7 kb. Depletion of NRON leads to increased levels and activity of NFAT in nucleus. Interestingly NRON’s predicted secondary structure is rich in stem loops, which is conserved between diverse vertebrates and requires further study.
LncRNA can bind to accessory proteins to activate them allosterically, or induce their oligomerization and activation. One such lncRNA is HSR1 (heat shock RNA-1), which together with an eukaryotic translation-elongation factor 1A, stimulates trimerization of heat-shock factor 1 (HSF1) [71]. Trimeric HSF1 activates heat-shock proteins by binding to their promoters . Formation of HSR1-HSF1 is induced by heat shock and knockdown of HSR1 causes cells to become thermo-sensitive. This suggests that HSR1 may be a part of cellular thermo-sensing machinery, resembling a similar mechanism in bacteria.
Post-transcriptional Processing
In addition to all of the above transcriptional mechanisms, many lncRNAs are also involved in post-transcriptional processing of protein-coding mRNAs, including regulation of splicing , editing, transport, translation, and degradation of their corresponding mRNA transcripts.
Natural antisense transcripts (NATs) are typical example of lncRNAs that act to regulate mRNA dynamics. Unlike NATs associated with imprinting genes such as Tsix, Air or HOTAIR, which induce epigenetic changes in chromatin and lead to gene silencing, other NATs can form RNA duplexes to mask key cis regulatory elements. This can lead to an alternative splicing pattern of overlapping gene transcripts. For example, the Zeb2/Sip1 NAT is complementary to the 5′ splice site of an intron of the zinc finger Hox mRNA Zeb2, which is involved in epithelial-mesenchymal transition (EMT). Zeb2 NAT is expressed upon EMT and masks the splice site, so blocking splicesome function. This causes the translation machinery to recognize and bind to an internal ribosome entry site (IRES) in the retained intron resulting in more efficient Zeb2 translation (Fig. 4.7) [72].

Post-transcriptional regulation by lncRNA. Brown block arrow is Zeb2 gene. Intron is colored in orange. Hairpins correspond to miRNAs
More recently, a NAT specific for tyrosine kinase containing immunoglobulin and epidermal growth factor homology domain-1 (Tie-1) was identified in zebrafish, mouse and human. The tie-1 NAT specifically binds tie-1 mRNA in vivo, forming an RNA –RNA duplex. This leads to down-regulation of the Tie-1 protein with consequent specific defects in cellular endothelial contact junctions [73].
In contrast, the expression of beta-secretase-1 (BACE-1) NAT increases stability of BACE-1 mRNA and leads to high production of Abeta (amyloid-beta) 1-42 through a post-transcriptional feed-forward mechanism [74, 75]. In this way BACE-1 NAT acts a positive regulator of Abeta 1-42 through stabilization of BACE-1 mRNA.
Alternative splicing is a well-known mechanism of pre-mRNA processing in higher eukaryotes. The serine/arginine (SR) splicing factors regulate cell type specific alternative splicing in a concentration and phosphorylation-dependent manner. How levels of active SR proteins are regulated is not well understood. Recent studies on the long nuclear-retained regulatory RNA (nrRNA) called MALAT (metastasis-associated lung carcinoma transcript 1) implicated its role in alternative splicing [76, 77]. MALAT1 (also known as NEAT2) a 7 kb RNA is localized in nuclear speckles, where it interacts with SR splicing factor, SRSF1 and affects the distribution of other splicing factors. Depletion of MALAT1 changes the alternative splicing profile of multiple endogenous pre-mRNAs. More importantly, MALAT1 regulates the phosphorylation status of SR proteins, thereby regulating pre-mRNA processing via modulation of active SR proteins levels.
Furthermore, there is growing evidence showing that transcripts produced from pseudogenes play an important role in regulating mRNA stability of the gene paralogue. For example, transcripts from the tumor suppressor pseudogene of PTEN (PTENP1) and oncogenic KRAS (KRASP) regulate levels of their gene counterparts, PTEN and KRAS [39, 78]. It is biologically relevant to keep the right dosage of PTEN in the cell. A number of miRNA and pseudogene transcripts are also directly involved in PTEN dosage regulation at a post-transcriptional level. PTEN and PTENP1 3′ UTRs are highly conserved. PTENP1 RNA , which is also referred to as competing endogenous RNA (ceRNA), binds to common miRNA preventing their binding to miRNA response element in 3′ UTR of PTEN. This competitive binding of PTENP1 to miRNA results in increased levels of PTEN RNA and consequently PTEN protein (Fig. 4.8) [40, 79, 80].

Post-transcriptional regulation by lncRNA. Block arrows depict protein coding gene PTEN. Hairpins represent miRNAs
Additionally a similar mechanism has been shown for KRAS mRNA, which is increased by expression of ceRNAs of KRASP. Interestingly, some protein-coding genes, such as ZEB2, VAPA and CNOT6L can also act as ceRNAs.
The initial studies describing links between imprinting and X chromosome inactivation were based on discovery of the H19 and Xist RNAs. The H19 gene encodes a 2.3 kb lncRNA that is expressed exclusively from the maternal allele and is spliced , polyadenylated and exported into cytoplasm where it accumulates [81]. H19 cause imprinting of its counterpart protein-coding gene, the insulin-like growth factor 2, IGF2. Recent studies revealed that H19 is host to an exonic miRNA, miR-675 which is also imprinted and maternally expressed. H19 and miR675 are conserved across mammalian species, suggesting that both are selected [82]. It is proposed that H19 might act through the nonsense mediated RNA decay pathway. Indeed, a key component of this pathway has been shown to regulate the levels of H19 RNA during embryonic stem cell differentiation [83].
lncRNA and Disease
Recent and rapid progress in lncRNA research reveals a growing body of evidence that lncRNA play an important role in variety of normal physiological processes. Consequently their mis-regulated expression contributes to numerous diseases , including cancer .
lncRNA and Cancer
New technological approaches, such as genome -wide studies, RIP-RNA seq uencing, gene express ion screens, region-targeted assays and gene knock-down/knock-out experiments all contribute to the determination of lncRNA function in pathogenesis. Accumulating data show that lncRNAs are indeed involved in carcinogenesis, invasion and metastasis. Based on their function, lncRNAs can be divided into two major categories: oncogenic and tumor suppressor classes.
Oncogenic lncRNAs
Some lncRNAs, referred to as oncogenic transcripts, can regulate cellular pathways that lead to oncogenesis . Recent studies identify more and more of onco-lncRNAs such as KRASP, HULC, HOTAIR, MALAT1/NEAT1, p15AS, ANRIL, H19, SRA1, p21NAT or RICTOR. Some lncRNAs can act as oncogenic as well as tumor suppressor transcripts, depending on cellular context.
The lncRNA, referred to as cancer metastasis-associated lung adenocarcinoma transcript, MALAT1, was identified in non-small-cell lung cancer [84]. MALAT1 is abundant and plays a key role in cell proliferation, migration and invasion. It localizes predominantly in nuclear speckles in a transcription dependent manner to regulate mRNAs post-transcriptional processing such as alternative splicing [76, 85]. MALAT1 is also up-regulated in other types of cancer, including breast, prostate, liver and colon [84, 86–90]. Furthermore, higher expression of MALAT1 is associated with metastatic tumors where it is correlated with poor prognosis. Recent studies demonstrate that MALAT1 is involved in cell mobility at it targets genes, required for cell migration, in order to regulate their gene express ion at both a transcriptional and post-transcriptional level. However, the underlying mechanism of MALAT1 in tumor metastatic process remains unclear.
A genome -wide study unveiled associations of multiple genetic variants in a large “gene-desert” region of chromosome 8q24 with susceptibility to prostate cancer (PC). Re-sequencing approaches helped to identify a 13 kb long intron-less lncRNA, termed PRNCR1 (prostate cancer non-coding RNA1) [91]. Depletion of PRNCR1 attenuated the viability of PC cells and the trans-activation activity of the androgen receptor. Therefore, it has been proposed that PRNCR1 is involved in prostate carcinogenesis through androgen receptor activity. These findings provide a novel insight into understanding the pathogenesis of genetic factors for prostate cancer.
The lincRNA HOTAIR is expressed in many posterior and distal sites during evolution and is highly conserved in vertebrates [60]. HOTAIR is over-expressed in metastatic breast cancer and correlates with poor prognosis [61]. De-regulation of HOTAIR represses the expression of a subset of cell-to-cell interaction promoting genes, including JAM2, PCDH10, PCDHB5 and EPHA1. Furthermore, the interaction of HOTAIR and PRC2, which leads to increased H3K27 trimethylation and silencing of metastasis suppressor genes, is responsible for HOTAIR mediated tumor cell invasion and subsequent metastasis. Additionally, increased levels of HOTAIR were detected in hepatocellular carcinoma (HCC), suggesting that HOTAIR could be a candidate biomarker for tumor recurrence prediction. Depletion of HOTAIR in liver cancer cells results in reduced cell viability and sensitized apoptosis induced by TNF-alpha [92]. These studies indicate that lincRNAs have active roles in modulating the cancer epigenome and could be an important predictor for cancer outcome as well as novel targets for cancer therapy.
Loss of imprinting (LOI) is involved in a number of human hereditary diseases and cancers [93]. Disruption in the expression of imprinted genes such as H19, p57, IGF2 and KvLQT1, results in almost 80 % of Beckwith–Wiedemann syndrome (BWS). About 5–10 % of BWS patients are predisposed to a number of childhood tumors. Kcnq1ot1 is an imprinted antisense lncRNA, which is about 60 kb long and possesses a silencing domain at its 5′ end [94]. Kcnq1ot1 transcript is associated with multiple chromosomal rearrangements in BWS. Its abnormal expression was observed in 50 % of BWS patients and 53 % of colorectal cancers [95–97]. Loss of Kcnq1ot1 imprinting is accompanied by loss of methylation of the control element, a CpG-island called KvDMR1. KvDMR1 contains the promoter for the paternally expressed Kcnq1ot1. Disruption of this promoter abolishes Kcnq1ot1 transcripts leading to activation of neighboring genes such as tumor repressor CDKN1C [98]. These data suggest that abnormal expression of Kcnq1ot1 contributes to carcinogenesis.
Recent studies report consistent differences in the expression of sense and antisense transcripts between normal and neoplastic cells. A group of genes that generate NATs in normal, but not cancer cells are involved in essential metabolic processes. Altered ratio of sense and antisense transcription contributes to tumorigenesis and cancer progression [99–103]. For example, leukemic cells express higher amounts of antisense p15 NATs and smaller amounts of its partner p15 sense mRNA than normal lymphocytes. Many NAT lncRNAs may have relevance to the cancer genes, including p21, p53, E-cadherin or myc [104]. Thus, it is proposed that tumorigenic NATs are a trigger for heterochromatin formation and DNA methylation in tumor suppressor silencing.
Tumor Suppressor lncRNAs
Some lncRNAs are found to function as tumor suppressors, resembling some protein-coding genes. This group of lncRNAs includes MEG3, GAS5, lincRNA-p21, PTENP1, TERRA , CCND1 and TUG1.
MEG3 is a lncRNA transcript of a maternally imprinted gene, which is expressed in normal human cells. Loss of MEG3 was found in meningiomas and adenomas of gonadotroph origins [105, 106]. MEG3 is a positive regulator of the tumor suppressor gene, p53. Ectopic expression of MEG3 up-regulates p53 protein levels and dramatically induces p53 transcription . Furthermore, MEG3 selectively enhances p53 binding to its target promoter, such as GDF15. Expression of MEG3 is able to inhibit cell proliferation in the absence of p53. All these data suggest that lncRNA MEG3 functions as a tumor suppressor in both a p53-dependent and p53-independent manner [107, 108].
LincRNA-p21 is another example of a tumor suppressor lncRNA, whose expression is directly induced by the p53-signaling pathway. LincRNA-p21 is required for global repression of genes that interfere with p53 function to regulate cellular apoptosis. This occurs through physical interaction with RNA -binding protein hnRNP-K leading to its localization on gene promoters , which are thought to be repressed in a p53-dependent manner [109].
GAS5 (growth arrest-specific 5) is tumor suppressor lncRNA, which regulates normal growth in lymphocytes. Depletion of GAS5 inhibits apoptosis and maintains rapid cell cycling, which indicates that its expression is necessary for normal growth arrest. GAS5 regulates the expression of a critical group of genes with tumor suppressive functions. Additionally, several snoRNAs are transcribed solely from GAS5 introns. Under starvation, GAS5 directly interacts with the DNA binding domain of glucocorticoid receptor (GR), leading to inhibition of GR binding to its target gene promoters . Such repression is not limited only to GR, but applies also to other members of the nuclear receptor family. Interestingly, GAS5 is significantly down-regulated in breast cancer cells [110–112].
Cis-acting lncRNA, CCND, originates from the promoter of the CCND1 gene encoding cyclin D1 protein. Upon induction, CCND lncRNA transcript is tethered to the CCND1 promoter and so inhibits CCND1 expression. Cyclin D1 is frequently over-expressed in human tumors. Therefore, it is proposed that CCND1 transcript functions as a tumor suppressor to repress tumorigenesis [113].
Expression of the telomere-related lncRNA, TERRA , is highly dependent on development, nuclear reprogramming, telomere length, cellular stresses and chromatin structure. Many abnormal telomere phenotypes in aging and cancer cells are linked to mis-regulated expression of TERRA. Low levels of TERRA have been observed in the tumor-derived and in vitro immortalized cell lines. It has been proposed that TERRA-regulated telomere length plays an important role in tumor development [114–117].
lncRNA and Other Diseases
LncRNAs in the context of their cellular function can also be involved in diseases other than cancer .
Patients with SCA8 have a trinucleotide expansion in an lncRNA called ataxin 8, which is antisense to the KLHL1 gene. Involvement of this type of mutation in disease progression was confirmed in mouse model transgenic mice with this repeat expansion displaying a progressive neurological phenotype similar to human SCA8 [118].
An inherited form of alpha-thalassaemia is caused by the translocation of an antisense lncRNA to a neighboring region of the alpha-globin gene (HBA2). Induction of this lncRNA results in epigenetic silencing of HBA2 leading to anemia [119].
The expression of the antisense transcript to BACE1 gene, as a response to cell stress, leads to progression of the well-studied Alzheimer’s disease [74, 120].
Also, psoriasis-associated RNA induced by stress, called PRINS, is up-regulated in skin cells of patients with psoriasis. It acts through down-regulation of G1P3, gene encoding a protein with anti-apoptotic effect in keratinocytes leading to psoriasis progression [121].
A study using a single-nucleotide polymorphism marker identified a lncRNA called MIAT (myocardial infarction-associated transcript) on chromosome 22 in patients with myocardial infarction [122]. Furthermore, genome -wide analysis identified a region encompassing a lncRNA, ANRIL, which is linked to coronary artery disease [123, 124].
Overall, identified lncRNAs play a clear role in pathology of various diseases . It remains to be determined what is their specific function and how they are associated with human pathology.
lncRNA as Biomarkers
To date, although our understanding on how lncRNAs cause disease is far from complete, certain features of lncRNAs make them ideal candidates for therapeutic intervention. For example, only a minority of lncRNAs are unstable. LncRNA half-lives vary over a wide range, comparable to that of mRNAs. Combining half-lives with comprehensive lncRNA annotations hundreds of unstable (half-life < 2 h) intergenic, cis-antisense, and intronic lncRNAs, as well as lncRNAs showing extreme stability (half-life > 16 h) were identified. Intergenic and cis-antisense RNAs are more stable than those derived from introns [125].
LncRNA expression is elevated in several types of cancers , including human prostate cancer , renal cell carcinomas, breast and ovarian cancer, as well as human lung cancer, suggesting that lncRNAs may become a promising biomarker in disease diagnostics. For example, the prostate specific lncRNA, DD3, shows higher specificity than serum prostate-specific antigen (PSA), suggesting that is could be developed into highly specific biomarker [126]. HCC-associated lncRNA, HULC, is also upregulated in the blood of hepatocarcinomas, implying its potential use in diagnosis of this type of cancer [127]. Expression of HOX specific antisense RNA , HOTAIR, is increased in breast tumor cells, suggesting that it may become a powerful predictor of patient outcome such as metastasis and death. SNORD-host RNA, Zfas1 is an antisense transcript of the protein-coding gene Znfx1. Zfas1 is highly expressed in mammary glands and it’s obviously down-regulated in breast cancer cells, suggesting its potential for diagnosis of breast cancer [128].
LncRNAs-based biomarkers could also be developed for diseases other than cancer . For example, noncoding transcript for beta-secretase-1 (BACE1), which regulates BACE1 mRNA and protein production, is upregulated in Alzheimer’s disease and thus could be exploited as a biomarker [129]. ANRIL lncRNA, is expressed in tissues and cells affected by atherosclerosis, which makes it a potential biomarker for coronary artery disease [130].
Overall it is clear that lncRNAs possess a significant potential for development of new approaches in diagnostics and therapy.
lncRNA and Stem Cell Development
Cellular reprogramming demonstrates the plasticity of cell fates. LncRNAs, whose expressions are linked to pluripotency, are direct targets of key transcription factors [131]. One such a lncRNA (RoR) modulates cellular reprogramming, which has been identified by loss-of function and gain-of function approaches. This provided first evidence for the critical function of a lncRNA in the derivation of pluripotent stem cells. LncRNAs also help to regulate development by physically interacting with proteins to coordinate gene express ion in embryonic stem cells (ESCs) [132]. This is contrary to the dogma that proteins alone are the key regulators of this process. LncRNA determine the fate of ESCs by keeping them in their unspecialized state or by directing them along a pathway to cellular differentiation.
lncRNA and Immunity
Whole-transcriptome analysis has shown that lncRNAs are associated with diverse biological processes in different tissues and are also involved in the host response to viral infection and innate immunity [6]. Also a recent study revealed altered expression of lncRNA during CD8+ T cell differentiation upon antigen recognition [133]. Likewise, eight mRNA-like lncRNAs were differentially expressed in virus-infected birds [134]. Whole-transcriptome analysis of severe acute respiratory syndrome in coronavirus-infected lung samples shows that there is a widespread differential regulation of lncRNAs in response to viral infection [135]. All of this suggests that lncRNAs are involved in regulating the host response in virus-infected cells, including innate immunity.
Concluding Remarks
The discovery of lncRNAs has changed our view of the complexity of the mammalian transcriptome. LncRNAs are becoming widely recognized as key regulators of protein-coding gene express ion and so provide an additional layer of transcriptional control. To date, lncRNAs have been shown to be involved in many different stages of gene expression regulation . This diversity in function suggests that lncRNAs will ultimately be found to participate at all levels of transcriptional control, from nuclear localization of transcription factors to transcriptional termination. Several lncRNAs have been implicated in the mediation of chromatin structure. Enabling the accessibility of the genome to Pol II and its associated factors is the most efficient way to activate or repress transcription. LncRNAs also function in X chromosome inactivation and genomic imprinting through chromatin remodeling.
Future discoveries may struggle to identify additional transcriptional regulatory lncRNA that share a function with known lncRNAs, because different RNAs can have similar functions even though they lack detectable sequence similarity. Real challenges lie in determining the biological significance of lncRNAs-protein interaction. Scientists have to clearly demonstrate biological roles of particular lncRNAs and relate them to their associated transcriptional units. It is peculiar that lncRNAs are not evolutionary conserved, they are expressed in very low levels and their knock-out don’t show a clear phenotype. Therefore, their biological significance remains an open topic for a further analysis.
The de-regulation of lncRNA expression in the context of cell pathology represents a new layer of complexity in the molecular architecture of human disease . Several lines of evidence have suggested that even small-scale mutations can affect lncRNA structure and function. Future studies need to elucidate the mechanism by which disease-causing mutations in lncRNA functional motifs can affect its regulatory domains and thereby contribute to disease pathology.
Future research of lncRNA may lead to discoveries of their biological functions and ultimately propose new RNA -based targets for the prevention and treatment of human disease .
References
Ponting CP, Belgard TG (2010) Transcribed dark matter: meaning or myth? Hum Mol Genet 19:R162–R168
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B et al (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A et al (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136:629–641
Khalil AM, Guttman M, Huarte M, Garber M, Raj A et al (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106:11667–11672
Guttman M, Amit I, Garber M, French C, Lin MF et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC et al (2005) The transcriptional landscape of the mammalian genome. Science 309:1559–1563
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159
Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H et al (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420:563–573
He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW (2008) The antisense transcriptomes of human cells. Science 322:1855–1857
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E et al (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129:823–837
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J et al (2010) Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28:503–510
Schorderet P, Duboule D (2011) Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet 7:e1002071
Wang J, Zhang J, Zheng H, Li J, Liu D et al (2004) Mouse transcriptome: neutral evolution of ‘non-coding’ complementary DNAs. Nature 431:1 p (following 757; discussion following 757)
Duret L, Chureau C, Samain S, Weissenbach J, Avner P (2006) The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 312:1653–1655
Elisaphenko EA, Kolesnikov NN, Shevchenko AI, Rogozin IB, Nesterova TB et al (2008) A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One 3:e2521
Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB et al (2007) A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8:39
Conley AB, Miller WJ, Jordan IK (2008) Human cis natural antisense transcripts initiated by transposable elements. Trends Genet 24:53–56
Ni T, Tu K, Wang Z, Song S, Wu H et al (2010) The prevalence and regulation of antisense transcripts in Schizosaccharomyces pombe. PLoS One 5:e15271
Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ et al (2011) Comparative functional genomics of the fission yeasts. Science 332:930–936
Yassour M, Pfiffner J, Levin JZ, Adiconis X, Gnirke A et al (2010) Strand-specific RNA sequencing reveals extensive regulated long antisense transcripts that are conserved across yeast species. Genome Biol 11:R87
Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M et al (2005) Antisense transcription in the mammalian transcriptome. Science 309:1564–1566
Kiyosawa H, Mise N, Iwase S, Hayashizaki Y, Abe K (2005) Disclosing hidden transcripts: mouse natural sense-antisense transcripts tend to be poly(A) negative and nuclear localized. Genome Res 15:463–474
Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S et al (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308:1149–1154
Kiyosawa H, Yamanaka I, Osato N, Kondo S, Hayashizaki Y (2003) Antisense transcripts with FANTOM2 clone set and their implications for gene regulation. Genome Res 13:1324–1334
Gingeras TR (2007) Origin of phenotypes: genes and transcripts. Genome Res 17:682–690
Wahlestedt C (2006) Natural antisense and noncoding RNA transcripts as potential drug targets. Drug Discov Today 11:503–508
Sun M, Hurst LD, Carmichael GG, Chen J (2005) Evidence for a preferential targeting of 3′-UTRs by cis-encoded natural antisense transcripts. Nucleic Acids Res 33:5533–5543
Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME et al (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121:725–737
Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM et al (2009) Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457:1038–1042
Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S et al (2009) Bidirectional promoters generate pervasive transcription in yeast. Nature 457:1033–1037
Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS et al (2008) RNA exosome depletion reveals transcription upstream of active human promoters. Science 322:1851–1854
Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB et al (2008) Divergent transcription from active promoters. Science 322:1849–1851
Core LJ, Waterfall JJ, Lis JT (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322:1845–1848
Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM et al (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature 465:182–187
Orom UA, Shiekhattar R (2011) Long non-coding RNAs and enhancers. Curr Opin Genet Dev 21:194–198
Faulkner GJ, Carninci P (2009) Altruistic functions for selfish DNA. Cell Cycle 8:2895–2900
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ et al (2010) A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465:1033–1038
Tay Y, Kats L, Salmena L, Weiss D, Tan SM et al (2011) Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 147:344–357
Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS (2008) Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A 105:716–721
Babak T, Blencowe BJ, Hughes TR (2007) Considerations in the identification of functional RNA structural elements in genomic alignments. BMC Bioinformatics 8:33
Wang KC, Chang HY (2011) Molecular mechanisms of long noncoding RNAs. Mol Cell 43:904–914
Nagano T, Fraser P (2011) No-nonsense functions for long noncoding RNAs. Cell 145:178–181
Clark MB, Mattick JS (2011) Long noncoding RNAs in cell biology. Semin Cell Dev Biol 22:366–376
Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF (2009) RNA regulation of epigenetic processes. Bioessays 31:51–59
Clemson CM, McNeil JA, Willard HF, Lawrence JB (1996) XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol 132:259–275
Chaumeil J, Le Baccon P, Wutz A, Heard E (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20:2223–2237
Jonkers I, Monkhorst K, Rentmeester E, Grootegoed JA, Grosveld F et al (2008) Xist RNA is confined to the nuclear territory of the silenced X chromosome throughout the cell cycle. Mol Cell Biol 28:5583–5594
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322:750–756
Carrel L, Willard HF (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434:400–404
Khalil AM, Driscoll DJ (2007) Trimethylation of histone H3 lysine 4 is an epigenetic mark at regions escaping mammalian X inactivation. Epigenetics 2:114–118
Barr ML, Bertram EG (1949) A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 163:676
Panning B (2004) X inactivation in mouse ES cells: histone modifications and FISH. Methods Enzymol 376:419–428
Heard E, Rougeulle C, Arnaud D, Avner P, Allis CD et al (2001) Methylation of histone H3 at Lys-9 is an early mark on the X chromosome during X inactivation. Cell 107:727–738
Reik W, Murrell A (2000) Genomic imprinting. Silence across the border. Nature 405:408–409
Li Y, Sasaki H (2011) Genomic imprinting in mammals: its life cycle, molecular mechanisms and reprogramming. Cell Res 21:466–473
Mohammad F, Mondal T, Kanduri C (2009) Epigenetics of imprinted long noncoding RNAs. Epigenetics 4:277–286
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC et al (2008) The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322:1717–1720
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM et al (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK et al (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693
Hirota K, Miyoshi T, Kugou K, Hoffman CS, Shibata T et al (2008) Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature 456:130–134
Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P (2000) Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol Cell 5:377–386
Martens JA, Laprade L, Winston F (2004) Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429:571–574
Ponjavic J, Ponting CP (2007) The long and the short of RNA maps. Bioessays 29:1077–1080
Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A (2007) Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 445:666–670
Goni JR, de la Cruz X, Orozco M (2004) Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res 32:354–360
Feng J, Bi C, Clark BS, Mady R, Shah P et al (2006) The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev 20:1470–1484
Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG et al (2005) A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 309:1570–1573
Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E (2006) RNA-mediated response to heat shock in mammalian cells. Nature 440:556–560
Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB et al (2008) A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 22:756–769
Li K, Blum Y, Verma A, Liu Z, Pramanik K et al (2010) A noncoding antisense RNA in tie-1 locus regulates tie-1 function in vivo. Blood 115:133–139
Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG et al (2008) Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med 14:723–730
Jiang Y, Mullaney KA, Peterhoff CM, Che S, Schmidt SD et al (2010) Alzheimer’s-related endosome dysfunction in down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 107:1630–1635
Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T et al (2010) A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J 29:3082–3093
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q et al (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39:925–938
He L (2010) Posttranscriptional regulation of PTEN dosage by noncoding RNAs. Sci Signal 3:pe39
Karreth FA, Tay Y, Perna D, Ala U, Tan SM et al (2011) In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 147:382–395
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O et al (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147:358–369
Bartolomei MS, Zemel S, Tilghman SM (1991) Parental imprinting of the mouse H19 gene. Nature 351:153–155
Smits G, Mungall AJ, Griffiths-Jones S, Smith P, Beury D et al (2008) Conservation of the H19 noncoding RNA and H19-IGF2 imprinting mechanism in therians. Nat Genet 40:971–976
Ciaudo C, Bourdet A, Cohen-Tannoudji M, Dietz HC, Rougeulle C et al (2006) Nuclear mRNA degradation pathway(s) are implicated in Xist regulation and X chromosome inactivation. PLoS Genet 2:e94
Ji P, Diederichs S, Wang W, Boing S, Metzger R et al (2003) MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 22:8031–8041
Bond CS, Fox AH (2009) Paraspeckles: nuclear bodies built on long noncoding RNA. J Cell Biol 186:637–644
Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G et al (2009) A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics 10:163
Lin R, Maeda S, Liu C, Karin M, Edgington TS (2007) A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 26:851–858
Luo JH, Ren B, Keryanov S, Tseng GC, Rao UN et al (2006) Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 44:1012–1024
Schmidt LH, Spieker T, Koschmieder S, Humberg J, Jungen D et al (2011) The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol 6:1984–1992
Tano K, Mizuno R, Okada T, Rakwal R, Shibato J et al (2010) MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett 584:4575–4580
Chung S, Nakagawa H, Uemura M, Piao L, Ashikawa K et al (2011) Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci 102:245–252
Yang Z, Zhou L, Wu LM, Lai MC, Xie HY et al (2011) Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol 18:1243–1250
Feinberg AP, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4:143–153
Mohammad F, Pandey RR, Nagano T, Chakalova L, Mondal T et al (2008) Kcnq1ot1/Lit1 noncoding RNA mediates transcriptional silencing by targeting to the perinucleolar region. Mol Cell Biol 28:3713–3728
Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S et al (1999) Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci U S A 96:5203–5208
Mitsuya K, Meguro M, Lee MP, Katoh M, Schulz TC et al (1999) LIT1, an imprinted antisense RNA in the human KvLQT1 locus identified by screening for differentially expressed transcripts using monochromosomal hybrids. Hum Mol Genet 8:1209–1217
Nakano S, Murakami K, Meguro M, Soejima H, Higashimoto K et al (2006) Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the KvDMR1 in colorectal cancers. Cancer Sci 97:1147–1154
Horike S, Mitsuya K, Meguro M, Kotobuki N, Kashiwagi A et al (2000) Targeted disruption of the human LIT1 locus defines a putative imprinting control element playing an essential role in Beckwith-Wiedemann syndrome. Hum Mol Genet 9:2075–2083
Maruyama R, Shipitsin M, Choudhury S, Wu Z, Protopopov A et al (2012) Altered antisense-to-sense transcript ratios in breast cancer. Proc Natl Acad Sci U S A 109:2820–2824
Grigoriadis A, Oliver GR, Tanney A, Kendrick H, Smalley MJ et al (2009) Identification of differentially expressed sense and antisense transcript pairs in breast epithelial tissues. BMC Genomics 10:324
Numata K, Osada Y, Okada Y, Saito R, Hiraiwa N et al (2009) Identification of novel endogenous antisense transcripts by DNA microarray analysis targeting complementary strand of annotated genes. BMC Genomics 10:392
Kohno K, Chiba M, Murata S, Pak S, Nagai K et al (2010) Identification of natural antisense transcripts involved in human colorectal cancer development. Int J Oncol 37:1425–1432
Monti L, Cinquetti R, Guffanti A, Nicassio F, Cremona M et al (2009) In silico prediction and experimental validation of natural antisense transcripts in two cancer-associated regions of human chromosome 6. Int J Oncol 34:1099–1108
Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J et al (2008) Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 451:202–206
Gejman R, Batista DL, Zhong Y, Zhou Y, Zhang X et al (2008) Selective loss of MEG3 expression and intergenic differentially methylated region hypermethylation in the MEG3/DLK1 locus in human clinically nonfunctioning pituitary adenomas. J Clin Endocrinol Metab 93:4119–4125
Zhang X, Gejman R, Mahta A, Zhong Y, Rice KA et al (2010) Maternally expressed gene 3, an imprinted noncoding RNA gene, is associated with meningioma pathogenesis and progression. Cancer Res 70:2350–2358
Benetatos L, Vartholomatos G, Hatzimichael E (2011) MEG3 imprinted gene contribution in tumorigenesis. Int J Cancer 129:773–779
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL et al (2007) Activation of p53 by MEG3 non-coding RNA. J Biol Chem 282:24731–24742
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ et al (2010) A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142:409–419
Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT (2009) GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 28:195–208
Mourtada-Maarabouni M, Hedge VL, Kirkham L, Farzaneh F, Williams GT (2008) Growth arrest in human T-cells is controlled by the non-coding RNA growth-arrest-specific transcript 5 (GAS5). J Cell Sci 121:939–946
Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP (2010) Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal 3:ra8
Kim JK, Diehl JA (2009) Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol 220:292–296
Feuerhahn S, Iglesias N, Panza A, Porro A, Lingner J (2010) TERRA biogenesis, turnover and implications for function. FEBS Lett 584:3812–3818
Caslini C (2010) Transcriptional regulation of telomeric non-coding RNA: implications on telomere biology, replicative senescence and cancer. RNA Biol 7:18–22
Luke B, Lingner J (2009) TERRA: telomeric repeat-containing RNA. EMBO J 28:2503–2510
Ng LJ, Cropley JE, Pickett HA, Reddel RR, Suter CM (2009) Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res 37:1152–1159
Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK et al (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38:758–769
Tufarelli C, Stanley JA, Garrick D, Sharpe JA, Ayyub H et al (2003) Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 34:157–165
Khalil AM, Faghihi MA, Modarresi F, Brothers SP, Wahlestedt C (2008) A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One 3:e1486
Sonkoly E, Bata-Csorgo Z, Pivarcsi A, Polyanka H, Kenderessy-Szabo A et al (2005) Identification and characterization of a novel, psoriasis susceptibility-related noncoding RNA gene, PRINS. J Biol Chem 280:24159–24167
Ishii N, Ozaki K, Sato H, Mizuno H, Saito S et al (2006) Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet 51:1087–1099
McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R et al (2007) A common allele on chromosome 9 associated with coronary heart disease. Science 316:1488–1491
Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D et al (2007) Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 67:3963–3969
Clark MB, Johnston RL, Inostroza-Ponta M, Fox AH, Fortini E et al (2012) Genome-wide analysis of long noncoding RNA stability. Genome Res 22:885–898
Hessels D, Klein Gunnewiek JM, van Oort I, Karthaus HF, van Leenders GJ et al (2003) DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur Urol 44:8–15, discussion 15–16
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M et al (2007) Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132:330–342
Askarian-Amiri ME, Crawford J, French JD, Smart CE, Smith MA et al (2011) SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA 17:878–891
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L et al (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32:232–246
Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H et al (2008) Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet 17:806–814
Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K et al (2010) Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 42:1113–1117
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK et al (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477:295–300
Pang M, Woodward AW, Agarwal V, Guan X, Ha M et al (2009) Genome-wide analysis reveals rapid and dynamic changes in miRNA and siRNA sequence and expression during ovule and fiber development in allotetraploid cotton (Gossypium hirsutum L.). Genome Biol 10:R122
Ahanda ML, Ruby T, Wittzell H, Bed’Hom B, Chausse AM et al (2009) Non-coding RNAs revealed during identification of genes involved in chicken immune responses. Immunogenetics 61:55–70
Peng X, Gralinski L, Armour CD, Ferris MT, Thomas MJ et al (2010) Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. MBio 1:e00206-10
Acknowledgement
I am grateful to Prof Nick J Proudfoot for constructive comments on this manuscript.
This work was supported by MRC Career Development Award and by L’Oreal/UNESCO Woman in Science UK and Ireland award to Monika Gullerova.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gullerova, M. (2015). Long Non-coding RNA. In: Felekkis, K., Voskarides, K. (eds) Genomic Elements in Health, Disease and Evolution. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-3070-8_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3070-8_4
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-3069-2
Online ISBN: 978-1-4939-3070-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)