
Abstract
The role of microglial cells in neurodegenerative conditions is key to understanding the development and progression of the innate immune response during brain pathology. Although past and present research efforts have provided clues for understanding the contribution of microglia to neurodegeneration, the future offers new and exciting opportunities to study and modulate microglial biology in the degenerating brain. In this chapter we will summarize the main findings defining the role of microglia in neurodegenerative diseases, both in experimental animal models of disease and in studies with human brain tissue samples. We will also review the technical limitations to the study of microglia in neurodegenerative diseases and discuss the possible further lines of research to be pursued. In summary, we aim to provide a comprehensive picture of the role of microglial cells in the development, progression, and possible treatment of neurodegenerative diseases, to help build on the recent progress in this exciting field of neuroscience.
Similar content being viewed by others


Keywords
- Microglia
- Macrophages
- Inflammation
- Chronic neurodegeneration
- Alzheimer’s disease
- Parkinson’s disease
- Amyotrophic lateral sclerosis
- Prion disease
- Huntington’s disease
-
Evidence from genome-wide association and neuroimaging studies supports that inflammation is a driver of neurodegenerative diseases, rather than a consequence of ongoing pathology.
-
Microglial activation is influenced by systemic inflammation, accelerating the progression of chronic neurodegeneration.
-
The study of microglial biology in human samples of patients with chronic neurodegeneration is crucial, as current experimental models fail to reproduce all the pathological features observed in humans.
-
The composition and dynamics of the macrophages of the brain (microglia, perivascular macrophages, and meningeal macrophages) is not fully understood in neurodegenerative diseases, supporting further research in the field.
1 Introduction. Role of Microglia in Brain Pathology: Friend or Foe?
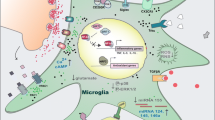
Microglial cells are active sensors of the disturbances in their microenvironment, capable of elaborating versatile responses to brain pathology (Kreutzberg 1996). These responses are neither linear, compartmentalized or binary, but plastic and multifaceted, i.e., finely tuned to the nature of the stimulus, the molecular repertoire that is engaged, and the prior state of the macrophage (Gordon 2003; Ransohoff and Perry 2009). When defining microglial activation during neurodegeneration, it is crucial to understand the temporal and spatial context of these diseases. Neurodegenerative diseases inexorably progress with the death of a slowly increasing number of neurons over years or even decades, to regional and cellular differential extents, determining the loss of disease-specific brain functions (memory, motor control, etc.). The slow degeneration of the neuronal components (synapses, axons, soma, and myelin sheath) may continue over many years, providing a stimulus that must lead to adaptive changes in the surrounding microglia. The adaptive changes may be then influenced by other comorbidities and systemic influences that communicate with the brain (Perry et al. 2007) (Fig. 18.1).

Impact of systemic inflammation on the progression of chronic neurodegeneration: microglial priming and proliferation. Schematic representation of the cross-talk of microglial cells with neurons and astrocytes in the healthy brain (left), during chronic neurodegeneration (middle) and when chronic neurodegeneration is combined with a systemic inflammatory event (right). The legend for the different cell types and phenotypes is provided in the top right corner. Figure adapted from Gomez-Nicola and Perry (2014)
A recurrent interest in the studies of microglial involvement in neuropathology is the dichotomy between their contributions to neurodegeneration versus neuroprotection. The current literature has extensively reviewed this issue, presenting microglial cells as “friend or foe” or a “double-edged sword”, trying to understand the determinants of the positive versus negative microglial contributions to brain pathology, with the goal of minimizing the harmful and favouring the beneficial (Crutcher et al. 2006; Popovich and Longbrake 2008). Achieving this ambitious objective becomes particularly difficult when taking into account the status of the immune-privilege of the brain that defines and tightly controls innate and acquired immune responses, and the influence of pathological processes from peripheral organs. For example, data from a genetically determined inflammatory demyelinating metabolic disorder, X-linked adrenoleukodystrophy, suggest a direct influence of the activation of microglia in the cerebral form of this disease (Eichler et al. 2008). Supporting the detrimental contribution of microglia to neurodegenerative diseases is the repeated observation that non-steroidal anti-inflammatory drugs (NSAIDs) delay the progression of Alzheimer’s disease (AD) (Vlad et al. 2008) and Parkinson’s disease (PD) (Chen et al. 2005), suggesting a major effect of prostaglandins and cyclooxygenase (COX) activation (Cunningham 2013). However, at the molecular level, the assumption that morphologically activated microglia show a classical pro-inflammatory phenotype, associated with the most detrimental effects during neurodegeneration, is no longer valid. On one hand, studies with transgenic models of AD report low gene expression of the pro-inflammatory cytokine interleukin-1β (IL-1β) or inducible nitric oxide synthase (iNOS), with low levels of protein production (Schwab et al. 2010). On the other, both mouse models and human cases of AD have been characterized to have limited pro-inflammatory polarization, but a consistent upregulation of anti-inflammatory markers, associated with potentially beneficial effects of microglia (Cunningham 2013).
Therefore, a detailed and pathology-specific definition of the microglial response needs to be completed, to fully understand the roles of microglia during neurodegeneration.
2 Alzheimer’s Disease
AD is a chronic neurodegenerative disease and the most common form of dementia in Western countries. The clinical manifestations of AD include memory deficits, mood and behaviour changes, and disorientation. The pathological substrate of AD includes the generation of two abnormal structures: extracellular plaques or deposits of beta-amyloid (Aβ) protein and intracellular tangles of Tau protein. Despite a long-standing interest in the inflammatory response in AD, and the extensive research focused on understanding the role of microglia in this disease, the scientific community has failed to shed a clear and uniform light onto their contribution to the disease (Akiyama et al. 2000; Heneka and O’Banion 2007; Ransohoff and Perry 2009). The neuropathology of AD shows a robust innate immune response characterized by the presence of activated microglia, with increased or de novo expression of diverse macrophage antigens (Akiyama et al. 2000; Edison et al. 2008), and at least in some cases production of pro-inflammatory cytokines (Dickson et al. 1993; Fernandez-Botran et al. 2011). Evidence indicates that NSAIDs protect from the onset or progression of AD (Hoozemans et al. 2011), which is suggestive of the idea that inflammation is a causal component of the disease rather than simply a consequence of the neurodegeneration. There is, however, a growing body of evidence to suggest that systemic inflammation may interact with the innate immune response in the brain to act as a ‘driver’ of disease progression and exacerbate symptoms (Holmes et al. 2009, 2011) (Fig. 18.1). Studies in animal models show evidence of interactions between systemic inflammation and inflammation in the brain, and importantly provide biologically plausible mechanisms for its contribution to the progression of neurodegeneration. The impact of systemic inflammation means that any neuropathological studies on the inflammatory response in the AD brain must take into account systemic comorbidities that may influence the microglia phenotype.
The definition of the brain inflammatory profile of AD shows contrasting ideas in the literature, probably arising from the heterogeneity of the human postmortem samples and the difficult application of detection methods (for review see (Boche et al. 2013)). For example, AD has been associated with a pro-inflammatory phenotype, characterized by expression of interleukin IL-1β and complement proteins, with a direct association with Aβ plaques in human samples (Griffin et al. 1989, 1995; McGeer et al. 1989). By contrast, an upregulation of genes linked to an anti-inflammatory phenotype, arginase 1 or the transforming growth factor β (TGFβ), has been associated with AD in human samples and mouse models (Wang et al. 2003; Colton et al. 2006). However, the link with inflammation seems clear, as highlighted by a recent study on the gene signature of ageing and AD, using microarray technology (Cribbs et al. 2012). These results support the notion that an activation of the innate inflammatory response in microglia is a prelude to the subsequent development of AD (Cribbs et al. 2012). Furthermore, studies on incipient AD (iAD) post-mortem samples show a strong correlation between genes associated with the microglial response and the progression to AD (Blalock et al. 2004). The concept of the interconnection of AD and the innate immune response is further supported by evidence from genome-wide association studies (GWAS) implicating genes involved in innate immunity (Lambert et al. 2009). Recent studies link genetic variants of TREM2, a protein regulating the activation and phagocytic functions of myeloid cells, with the risk of developing AD (Guerreiro et al. 2013; Jonsson et al. 2013). TREM2 has been described to have a balancing role between phagocytic and pro-inflammatory microglial activities and is expressed in microglia around the plaques (Frank et al. 2008). Similarly, dysregulation of the complement system in humans has been associated with AD (McGeer and McGeer 2002; Lambert et al. 2009) and may influence the priming of microglia, defined as the conditioning of microglial response by a primary stimulus which results in an exaggerated response to a secondary challenge (Cunningham 2013). These promising studies are opening new avenues into the understanding of the impact of the innate immune response in AD, while supporting the need for future exploration.
The morphological activation of microglia is evident in transgenic mouse models of AD, which reproduce the deposition of Aβ (LaFerla and Oddo 2005; Perry et al. 2007; Jucker 2010), but their associated cytokine profile is by no means clear, with the changes in expression level compounded by the various detection methods (for review about research methods to study microglial biology see (Ransohoff and Perry 2009)). An alternative approach to pinpoint the contribution of microglia to the progression of AD is to study their impact on the Aβ plaque load. The plaque burden in AD increases with age, in both mouse models and human patients, indicating the rather ineffective phagocytic activity of microglia. Aβ deposits have been shown to exert a potent chemoattractant activity on microglia, although their removal by phagocytosis has not been clearly evidenced in vivo (for review see (Sierra et al. 2013)) (see Chap. 4 for additional reading). However, the removal of Aβ can be improved by further activation of microglia with bacterial lipopolysaccharides (LPS) challenge (Herber et al. 2004) or the induction of IL-1β (Shaftel et al. 2007). Microglial activation in neurodegeneration is accompanied by an increase in their density, yet further amplifying the effects of systemic inflammation on the brain. In addition, other brain macrophages, perivascular macrophages (PVMs) and meningeal macrophages (MMs), play a critical role in signalling from the periphery to the brain.
A significant body of literature suggests that bone marrow-derived macrophages (BMCs) infiltrate the AD brain, playing a leading role in the removal of Aβ, therefore complementing the poor phagocytic activity of microglia (Simard and Rivest 2006; Simard et al. 2006) (Fig. 18.2). The relative contribution of BMCs to the PVMs, MMs, or microglial pool is a matter of intense debate, and recent studies support a minor or even absent contribution of BMCs to the microglial population in a mouse model of AD (Mildner et al. 2011). Although BMCs recruitment has been demonstrated in experimental models with a complete, partial, or no detectable blood-brain barrier (BBB) disruption (Davoust et al. 2008), several studies point to in situ microglial proliferation as the mechanism regulating microglial turnover, with little or no contribution from circulating progenitors (Lawson et al. 1992; Prinz and Mildner 2011). Recent studies support the notion that microglia are maintained and function independently of BMCs in health (Ginhoux et al. 2010) and disease, as evidenced by models of demyelination, neurodegeneration, or axotomy (Ajami et al. 2007; Mildner et al. 2007, 2011). But analyzing PVMs, MMs, and microglial proliferation under pathological conditions with widespread chronic neurodegeneration is critical for understanding how innate inflammation contributes to the onset and progression of disease (Gomez-Nicola et al. 2014). Recent studies have highlighted the ability of PVMs to clear Aβ in experimental models of AD (Mildner et al. 2011) and show the need for a better understanding of the differential contribution of BMCs, MMs, and PVMs to the expansion of the microglial population, thereby providing a key link with systemic inflammation (Fig. 18.2).

Microglial cells constantly scan the parenchyma of the central nervous system (CNS), seeking to detect changes in the functional or structural integrity and maintain homeostasis. In a normal brain, the microglial population is maintained by self-renewal, while the perivascular Fig. 18.2 (continued) macrophages (PVMs) can be renewed by bone-marrow-derived progenitors. In Alzheimer’s disease (AD) microglia proliferate and accumulate around Aβ plaques, participating in the removal of the misfolded protein, at which the PVMs are more efficient players. In AD, microglia are expanded without a contribution of circulating progenitors. Microglia are expanded and activated during the course of amyotrophic lateral sclerosis (ALS), without a contribution from circulating progenitors. In prion disease, the microglial population is expanded dramatically by local proliferation, being primed to give an exaggerated response to systemic inflammatory events. Little evidence is available regarding the expansion/renewal of the microglial population during Parkinson’s or Huntington’s diseases, or the dominant inflammatory phenotype. For all the considered neurodegenerative diseases, little evidence is available on the possible contribution of PVMs to the expansion/renewal of the microglial population. Figure adapted from Gomez-Nicola and Perry (2014)
Although proliferation was assumed to be responsible for the increased number of microglial cells observed in AD samples, direct evidence of proliferating microglial cells (Ki67, nuclear protein associated with the cell cycle, expression in Iba1+ microglia/macrophages) was reported only recently, together with the upregulation of the transcription factor PU.1 and the mitogen interleukin-34 (IL-34), key components of the pathway regulating microglial proliferation (Gomez-Nicola et al. 2013). Another determinant of microglial proliferation, colony-stimulating factor 1 receptor (CSF1R), has also been found to be upregulated in microglial cells from human postmortem samples of AD, indicating a prominent activity of this pathway (Akiyama et al. 1994). The expansion of the microglial population has been consistently documented in transgenic mouse models of AD, mainly accumulating around the plaques (Frautschy et al. 1998; Bolmont et al. 2008). However, direct evidence of microglial proliferation (incorporation of BrdU, a synthetic nucleoside analogue of thymidine, into Iba1+ cells; see Chap. 10 for more information) was only recently reported, suggesting direct effects of the plaque microenvironment on the regulation of microglial mitogenesis (Kamphuis et al. 2012). Therefore, these studies pinpoint the importance of controlling microglial proliferation during AD, offering new avenues for the regulation of the innate immune response in the brain.
3 Parkinson’s Disease
PD is a chronic neurodegenerative disease characterised by tremor, rigidity, and slowness of movement. The pathological basis of PD includes the death of neurons in the substantia nigra pars compacta (SNpc) and the subsequent loss of dopaminergic tone in addition to a more widespread loss of neurons. The activation of the innate immune response during the progression of PD has been evidenced by the presence of morphologically activated microglia in human postmortem samples (McGeer et al. 1988), and in vivo PET imaging, showing increased binding of the ligand [11C]PK-11195, considered as mainly labeling ‘activated’ microglia, without any correlation with the clinical symptoms, thus potentially dissociating microglial activation from the progression of the disease (Gerhard et al. 2006). Some studies suggest that microglia have a pro-inflammatory activation phenotype in PD, which is potentially driving neuronal injury (Mogi et al. 1994; Hunot et al. 1996), although no mechanistic study has yet addressed microglial contribution to the disease progression. The fact that PD has a late onset and that most studies analysed end-stage samples, representing a brain that has been suffering from the disease for many years, complicates the interpretation. Ageing alone has an impact on the phenotype of microglia, and systemic comorbidities, which can influence the microglial physiology, have not been taken into account in the previous studies focusing on PD (for review see (Perry 2012)). The clinical course of PD is often associated with other comorbidities, like chronic constipation or aspiration pneumonia, driving a peripheral inflammatory response that might impact the brain microglial responses and the progression of PD (for review see (Perry 2012); Fig. 18.1).
A significant body of knowledge regarding the role of microglial in PD comes from the study of experimental animal models. However, the current models fail to accurately reflect all aspects of the neuropathology of PD as described in humans. PD is characterised by a slowly evolving degeneration of the SNpc dopaminergic neurons, an aspect not replicated in the rodent models using either neurotoxic toxins or inflammatory challenges. The intracerebral use of neurotoxins, most commonly 6-hydroxydopamine (6OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), or rotenone, provides a rapid degeneration (within a few days) of the SNpc dopaminergic neurons. Microglial activation has been described in the 6OHDA and MPTP models of PD (McGeer et al. 2003; Walsh et al. 2011), although limited information is available regarding the inflammatory phenotype of these cells, contrarily to their morphological features which have been described in details. However, gene-expression studies defining the inflammatory profile in these animal models of PD are numerous, but limited evidence is available regarding the protein expression levels and the particular roles of these molecules in the disease (Hirsch and Hunot 2009). Studies modulating microglial activity with minocycline, an antibiotic having anti-inflammatory actions, provided contrasting results, model-dependent, about the contribution of innate inflammation to the acute neurodegeneration of dopaminergic neurons in the SNpc (Wu et al. 2002; Sriram et al. 2006). Systemic inflammation, induced by IL-1β administration, was shown to impact the survival of dopaminergic neurons in the 6OHDA model, providing a clear evidence of the influence of immune-to-brain communication on the progression of PD (Pott Godoy et al. 2008). Even though the studies with toxin models shed some light onto the understanding of microglial reaction during neurodegeneration in the SNpc, the translation of these findings into neuroprotective or regenerative strategies appears premature and requires further understanding of the complexity of the innate immune response in postmortem samples from well-characterised PD patients.
Additionally, the generation of transgenic mouse models of PD, based on the identification of genes linked to familial PD, also provides a promising approach to model chronic neurodegeneration (Dawson et al. 2010). Transgenic over-expression of α-synuclein, a presynaptic neuronal protein that is linked genetically and neuropathologically to PD, leads to microglial activation and production of tumour necrosis factor alpha (TNF-α) in the SNpc (Su et al. 2008), although little neuronal death is observed. The injection of inflammatory agents, such as LPS, into the SNpc induces the selective loss of dopaminergic neurons, mimicking the neuropathology of PD, albeit acutely (Herrera et al. 2000). The LPS-induced neuronal death involves TNF-α, indicating a direct contribution of microglial activation (McCoy et al. 2006). Both transgenic and inflammatory models of PD can capture aspects of the disease, but fail to provide a comprehensive picture in which to address the roles of the innate immune response, in the context of a slowly evolving neurodegenerative condition. To summarize, the contribution of microglial cells to the onset or progression of PD is not yet established. Further research into the effect of systemic comorbidities and in refining the experimental animal models will help to understand the roles of the innate immune response in PD.
4 Amyotrophic Lateral Sclerosis
The pathogenesis of ALS involves the gain-of-function of a mutant protein, with approximately 5–10 % of the cases having a familial inheritable form. ALS is characterised by the progressive loss of motor neurons in the spinal cord, the brain stem, and the motor cortex, caused by increased levels of reactive oxygen species (ROS), leading to an increased muscle weakness and atrophy, with the malfunction of the muscles controlling respiratory functions being the most common cause of death. A significant proportion of the familial ALS subjects have mutations in the enzyme superoxide dismutase-1 (SOD-1), facilitating the modelling of the disease in experimental animals. Microglial cells produce pro-inflammatory molecules and ROS in the regions showing motor neurons degeneration, suggesting an impact on the progression of the disease (Troost et al. 1990; Clement et al. 2003). The modulation of microglial activity in ALS, either by removing the specific downregulatory influence of CX3CL1, mainly produced by neurons and signalling through microglial CX3CR1 (Cardona et al. 2006) or by lowering the levels of mutant SOD-1 in microglia (Boillee et al. 2006), indicates a detrimental role of these cells in the pathogenesis. These results were independently confirmed in microglia-devoid PU.1−/− mice, using a repopulation method with SOD-1-expressing bone-marrow cells, thus leading to a shortening of mouse survival (Beers et al. 2006). A recent report supports the detrimental role of microglia in the pathology of ALS, evidencing that microglia induce motor neuron death via activation of the NFκB pathway (Frakes et al. 2014). Microglial cells carrying the G93A SOD-1 mutation show exaggerated responses to stimulation with LPS and interferon-γ (IFNγ), associated with the activity of C/EBPβ, a transcription factor which regulates genes such as TNF-α, IL-1β, or iNOS (Valente et al. 2012). These results are suggestive of a priming effect on microglia, also taking place in ALS, which is supported by experiments using repeated systemic dosing with LPS on mice carrying the G37R SOD-1 mutation (Nguyen et al. 2004). Other approaches, aiming at removing the contribution of the microglial population from the equation, by either the transgenic expression of thymidine kinase (TK), leading to the “suicide” of proliferating CD11b+ cells (Gowing et al. 2008) (see also Chap. 6 for further discussion of this method) or by the administration of the non-specific blocker of mitosis Ara-C (Audet et al. 2012), indicated a neutral or benign role of microglia in ALS. However, the methods used in these studies lead to a massive and uncontrolled death of microglia, or to a switch of microglial phenotype following treatment with Ara-C (Gomez-Nicola et al. 2013), which does not provide a ‘physiologically silent’ way to address the contribution of microglial cells in the context of on-going neurodegeneration. Additionally, the activation of the TK transgene in CD11b-TK mice is achieved by administration of ganciclovir: this agent was recently identified to have a potent anti-proliferative impact on microglia during brain pathology (Ding et al. 2014). Other alternative approaches boosting the intrinsic proliferative activity of microglia with recombinant CSF1 have supported a detrimental role of microglia in the pathophysiology of ALS (Gowing et al. 2009), although these experiments also affected the contribution from CSF1-responsive peripheral cells.
In conclusion, the picture of the microglial contribution seems much clearer in ALS than in other neurodegenerative diseases, but further research is necessary to address the specific involvement of the different macrophage populations (peripheral, perivascular, microglia) (Fig. 18.2), in order to understand the mechanisms underlying neurodegeneration. Once again it is particularly important to precisely define the microglial phenotype, and other aspects of the innate inflammatory response, at different stages of disease evolution in mouse models as well as in clinically characterised postmortem human brain samples.
5 Prion Disease
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are a group of rare progressive neurodegenerative disorders which show similar temporal and neuropathological profiles in both humans and animals models, characterised by depression and cognitive impairments followed by problems with motor control. The pathological substrate of prion diseases is the misfolding of the prion protein (PrP), leading to spongiform changes and neuronal loss. Murine models of prion disease are tractable laboratory models showing a slowly progressing chronic and fatal neurodegeneration, mimicking human prion diseases and presenting features in common with other neurodegenerative conditions. Prion-diseased brains show large numbers of microglia with a morphologically activated phenotype (Perry et al. 2010) and a cytokine profile similar to that of AD (Perry et al. 2002; Cunningham et al. 2003), with low levels of pro-inflammatory cytokines but high levels of TGF-β and prostaglandin E2 (PGE2), associated with a phagocytosing phenotype (Perry et al. 2002), although limited abilities for microglial removal of the misfolded prion protein have been described (Hughes et al. 2010). Following systemic inflammatory challenge the microglia are switched to adopt a pro-inflammatory phenotype (Cunningham et al. 2007, 2009), exacerbating the acute symptoms and accelerating the disease progression. Microglia in chronic neurodegeneration were therefore proposed to be ‘primed’ by the on-going pathology and then switched by systemic inflammation to produce tissue damaging inflammatory mediators (Perry et al. 2007) (Fig. 18.1). Previous studies in prion disease suggested that microglia arose from bone marrow precursors (BMPs), as evidenced by the use of bone marrow chimeras (Priller et al. 2006). However, recent studies have shown that the expansion of the microglial population during prion disease is maintained by local proliferation in a mouse model of prion disease (Gomez-Nicola et al. 2013) (Fig. 18.2). The expansion of the pool of parenchymal microglial cells is independent from the recruitment of circulating monocytes, while the population of PVMs is expanded by infiltrated cells, in a CCR2-dependent manner (Gomez-Nicola et al. 2014). Furthermore, microglial proliferation in prion disease is maintained by the activity of the CSF1R signaling pathway, and specific antagonism of the receptor, using either blocking antibodies or the selective CSF1R inhibitor GW2580, highlights the contribution of microglial cells as detrimental to the disease (Gomez-Nicola et al. 2013). A reduction in the numbers of proliferating microglia delayed the onset of behavioural deficits and modestly extended the time to terminal disease. The components of this mitogenic pathway are common to the human prion disease (variant Creutzfeldt-Jakob disease; vCJD) and to AD, suggesting that common pathways are controlling microglial proliferation and activation in chronic neurodegeneration (Gomez-Nicola et al. 2013). The analysis of the experimental models of prion disease offers an attractive perspective for the future, as they exhibit the main pathological features observed in many human neurodegenerative conditions (prion disease, AD, PD): protein misfolding, synaptic dysfunction, neurodegeneration, and an innate inflammatory reaction (for review see (Ransohoff and Perry 2009)).
6 Huntington’s Disease
Huntington’s disease (HD) is an inherited disorder characterized by the progressive degeneration of medium spiny striatal GABAergic interneurons, leading to a wide spectrum of clinical symptoms including impairment of movement control, cognitive deficits, and psychiatric symptoms. Progressive morphological activation of microglia and increase in their number has been evidenced in human brain from early pre-symptomatic stages of HD, suggestive of a causative role for these cells in the disease (Sapp et al. 2001; Tai et al. 2007). Binding studies of PK11195 using PET imaging in HD patients suggests that microglial activation correlates with the severity of the disease (Pavese et al. 2006), leading to the suggestion that they might provide a useful diagnostic tool to predict the disease onset (Politis et al. 2011). Recent studies further suggest that microglial activation during HD is a cell-autonomous mechanism, as mutant Huntingtin directly promotes the activation of microglial cells (Crotti et al. 2014).
Microglial activation can be exacerbated by systemic LPS administration in a mouse model of HD, however, without any impact on the neurological symptoms (Franciosi et al. 2012) (Fig. 18.1). The impact of systemic inflammatory events is clear during the progression of HD, as peripheral myeloid cells have been shown to produce altered levels of inflammatory cytokines (Trager and Tabrizi 2013; Trager et al. 2014). A detrimental contribution of microglia in HD has been suggested, through complement-mediated neuronal damage, although supporting mechanistic evidence is limited (Singhrao et al. 1999). Other in vitro studies have evidenced microglial proliferation and pro-inflammatory activation in HD, suggesting a reparative role in the removal of dysfunctional neurites at early and middle stages of the pathology (Kraft et al. 2012). Microglia have been shown to present defective chemotactic responses, in parallel with a reduced migration of immune cells, in a mouse model of HD (Kwan et al. 2012). The current evidence supports the idea that microglial cells are activated in HD, but the question of whether the innate immune response is a bystander consequence or whether they have a direct effect on the disease progression is still a matter of debate which needs further research (for review see (Moller 2010)).
7 Concluding Remarks
The role of microglia in neurodegenerative diseases and their contribution to the neuropathology is still a matter of intense debate. Although a significant effort has been directed towards unveiling the phenotype and activity of microglia in different models of experimental neurodegeneration, correlation with the human diseases is still to be determined. There is a need for improving the experimental models to better reflect the molecular neuropathology and inflammatory processes seen in human neurodegenerative diseases, in order to achieve a full understanding of the multi-faceted nature of chronic neurodegeneration. A more precise definition of the correlation between the clinical and molecular pathology and the systemic comorbidities in well-defined cohorts of patients will help us to understand the role of the innate immune response. With an increasing awareness of the potential importance of the innate immune response in neurodegenerative diseases, further research is needed in order to bridge the gap from bench to bedside, and finally understand the beautiful complexity of their cellular effectors, including particularly, the microglia.
References
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543
Akiyama H, Nishimura T, Kondo H, Ikeda K, Hayashi Y, McGeer PL (1994) Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res 639:171–174
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM et al (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Audet JN, Gowing G, Paradis R, Soucy G, Julien JP (2012) Ablation of proliferating cells in the CNS exacerbates motor neuron disease caused by mutant superoxide dismutase. PLoS One 7:e34932
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA et al (2006) Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 103:16021–16026
Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW (2004) Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A 101:2173–2178
Boche D, Perry VH, Nicoll JA (2013) Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39:3–18
Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G et al (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE et al (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28:4283–4292
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924
Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ et al (2005) Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol 58:963–967
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M et al (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP (2006) Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation 3:27
Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ et al (2012) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation 9:179
Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C et al (2014) Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci 17:513–521
Crutcher KA, Gendelman HE, Kipnis J, Perez-Polo JR, Perry VH, Popovich PG et al (2006) Debate: “is increasing neuroinflammation beneficial for neural repair?”. J Neuroimmune Pharmacol 1:195–211
Cunningham C (2013) Microglia and neurodegeneration: the role of systemic inflammation. Glia 61:71–90
Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP et al (2003) Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci 17:2147–2155
Cunningham C, Campion S, Teeling J, Felton L, Perry VH (2007) The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C). Brain Behav Immun 21:490–502
Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM et al (2009) Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 65:304–312
Davoust N, Vuaillat C, Androdias G, Nataf S (2008) From bone marrow to microglia: barriers and avenues. Trends Immunol 29:227–234
Dawson TM, Ko HS, Dawson VL (2010) Genetic animal models of Parkinson’s disease. Neuron 66:646–661
Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7:75–83
Ding Z, Mathur V, Ho PP, James ML, Lucin KM, Hoehne A et al (2014) Antiviral drug ganciclovir is a potent inhibitor of microglial proliferation and neuroinflammation. J Exp Med 211:189–198
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE et al (2008) Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis 32:412–419
Eichler FS, Ren JQ, Cossoy M, Rietsch AM, Nagpal S, Moser AB et al (2008) Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann Neurol 63:729–742
Fernandez-Botran R, Ahmed Z, Crespo FA, Gatenbee C, Gonzalez J, Dickson DW et al (2011) Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism Relat Disord 17(9):683–688
Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ et al (2014) Microglia induce motor neuron death via the classical NF-kB pathway in amyotrophic lateral sclerosis. Neuron 81:1009–1023
Franciosi S, Ryu JK, Shim Y, Hill A, Connolly C, Hayden MR et al (2012) Age-dependent neurovascular abnormalities and altered microglial morphology in the YAC128 mouse model of Huntington disease. Neurobiol Dis 45:438–449
Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I et al (2008) TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56:1438–1447
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K et al (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152:307–317
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A et al (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21:404–412
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Gomez-Nicola D, Perry VH (2014) Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist [2014 Apr 10, Epub ahead of print]
Gomez-Nicola D, Fransen NL, Suzzi S, Perry VH (2013) Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33:2481–2493
Gomez-Nicola D, Schetters ST, Perry VH (2014) Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 62(7):1041–1052
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35
Gowing G, Philips T, Van Wijmeersch B, Audet JN, Dewil M, Van Den Bosch L et al (2008) Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci 28:10234–10244
Gowing G, Lalancette-Hebert M, Audet JN, Dequen F, Julien JP (2009) Macrophage colony stimulating factor (M-CSF) exacerbates ALS disease in a mouse model through altered responses of microglia expressing mutant superoxide dismutase. Exp Neurol 220:267–275
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ et al (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A 86:7611–7615
Griffin WS, Sheng JG, Roberts GW, Mrak RE (1995) Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J Neuropathol Exp Neurol 54:276–281
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E et al (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127
Heneka MT, O'Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184:69–91
Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D et al (2004) Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol 190:245–253
Herrera AJ, Castano A, Venero JL, Cano J, Machado A (2000) The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis 7:429–447
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S et al (2009) Systemic inflammation and disease progression in Alzheimer disease. Neurology 73:768–774
Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH (2011) Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology 77:212–218
Hoozemans JJ, Veerhuis R, Rozemuller JM, Eikelenboom P (2011) Soothing the inflamed brain: effect of non-steroidal anti-inflammatory drugs on Alzheimer’s disease pathology. CNS Neurol Disord Drug Targets 10:57–67
Hughes MM, Field RH, Perry VH, Murray CL, Cunningham C (2010) Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia 58:2017–2030
Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y et al (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 72:355–363
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J et al (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368:107–116
Jucker M (2010) The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med 16:1210–1214
Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM (2012) Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 60:615–629
Kraft AD, Kaltenbach LS, Lo DC, Harry GJ (2012) Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol Aging 33(621):e17–e33
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R et al (2012) Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest 122:4737–4747
LaFerla FM, Oddo S (2005) Alzheimer’s disease: Abeta, tau and synaptic dysfunction. Trends Mol Med 11:170–176
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41:1094–1099
Lawson LJ, Perry VH, Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48:405–415
McCoy MK, Martinez TN, Ruhn KA, Szymkowski DE, Smith CG, Botterman BR et al (2006) Blocking soluble tumor necrosis factor signaling with dominant-negative tumor necrosis factor inhibitor attenuates loss of dopaminergic neurons in models of Parkinson’s disease. J Neurosci 26:9365–9375
McGeer PL, McGeer EG (2002) The possible role of complement activation in Alzheimer disease. Trends Mol Med 8:519–523
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346
McGeer PL, Schwab C, Parent A, Doudet D (2003) Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann Neurol 54:599–604
Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M et al (2007) Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 10:1544–1553
Mildner A, Schlevogt B, Kierdorf K, Bottcher C, Erny D, Kummer MP et al (2011) Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci 31:11159–11171
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M et al (1994) Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from Parkinsonian patients. Neurosci Lett 180:147–150
Moller T (2010) Neuroinflammation in Huntington’s disease. J Neural Transm 117:1001–1008
Nguyen MD, D’Aigle T, Gowing G, Julien JP, Rivest S (2004) Exacerbation of motor neuron disease by chronic stimulation of innate immunity in a mouse model of amyotrophic lateral sclerosis. J Neurosci 24:1340–1349
Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA et al (2006) Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology 66:1638–1643
Perry VH (2012) Innate inflammation in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009373
Perry VH, Cunningham C, Boche D (2002) Atypical inflammation in the central nervous system in prion disease. Curr Opin Neurol 15:349–354
Perry VH, Cunningham C, Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7:161–167
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6:193–201
Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ et al (2011) Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp 32:258–270
Popovich PG, Longbrake EE (2008) Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci 9:481–493
Pott Godoy MC, Tarelli R, Ferrari CC, Sarchi MI, Pitossi FJ (2008) Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 131:1880–1894
Priller J, Prinz M, Heikenwalder M, Zeller N, Schwarz P, Heppner FL et al (2006) Early and rapid engraftment of bone marrow-derived microglia in scrapie. J Neurosci 26:11753–11762
Prinz M, Mildner A (2011) Microglia in the CNS: immigrants from another world. Glia 59:177–187
Ransohoff RM, Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 27:119–145
Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K et al (2001) Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 60:161–172
Schwab C, Klegeris A, McGeer PL (2010) Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim Biophys Acta 1802:889–902
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK (2007) Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117:1595–1604
Sierra A, Abiega O, Shahraz A, Neumann H (2013) Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 7:6
Simard AR, Rivest S (2006) Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer’s disease. Mol Psychiatry 11:327–335
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49:489–502
Singhrao SK, Neal JW, Morgan BP, Gasque P (1999) Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol 159:362–376
Sriram K, Miller DB, O’Callaghan JP (2006) Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem 96:706–718
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29:1690–1701
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ et al (2007) Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 130:1759–1766
Trager U, Tabrizi SJ (2013) Peripheral inflammation in neurodegeneration. J Mol Med (Berl) 91:673–681
Trager U, Andre R, Lahiri N, Magnusson-Lind A, Weiss A, Grueninger S et al (2014) HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFkappaB pathway dysregulation. Brain 137:819–833
Troost D, Van den Oord JJ, Vianney de Jong JM (1990) Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol 16:401–410
Valente T, Mancera P, Tusell JM, Serratosa J, Saura J (2012) C/EBPbeta expression in activated microglia in amyotrophic lateral sclerosis. Neurobiol Aging 33:2186–2199
Vlad SC, Miller DR, Kowall NW, Felson DT (2008) Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70:1672–1677
Walsh S, Finn DP, Dowd E (2011) Time-course of nigrostriatal neurodegeneration and neuroinflammation in the 6-hydroxydopamine-induced axonal and terminal lesion models of Parkinson’s disease in the rat. Neuroscience 175:251–261
Wang G, Zhang Y, Chen B, Cheng J (2003) Preliminary studies on Alzheimer’s disease using cDNA microarrays. Mech Ageing Dev 124:115–124
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C et al (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gomez-Nicola, D., Perry, V.H. (2014). Neurodegenerative Diseases. In: Tremblay, MÈ., Sierra, A. (eds) Microglia in Health and Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1429-6_18
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1429-6_18
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1428-9
Online ISBN: 978-1-4939-1429-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)