
Abstract
As all the enveloped viruses, the entry of influenza viruses includes a number of steps in host cell infection. This chapter summarizes the current knowledge of the entry pathway and the role of the fusion protein of influenza virus, hemagglutinin, in this process. Hemagglutinin (HA) is a trimeric glycoprotein that is present in multiple copies in the membrane envelope of influenza virus. HA contains a fusion peptide, a receptor binding site, a metastable structural motif, and the transmembrane domain. The first step of influenza virus entry is the recognition of the host cell receptor molecule, terminal α-sialic acid, by HA. This multivalent attachment by multiple copies of trimetric HA triggers endocytosis of influenza virus that is contained in the endosome. The endosome-trapped virus traffics via a unidirectional pathway to near the nucleus. At this location, the interior pH of the endosome becomes acidic that induces a dramatic conformational change in HA to insert the fusion peptide into the host membrane, induce juxtaposition of the two membranes, and form a fusion pore that allows the release of the genome segments of influenza virus. HA plays a key role in the entire entry pathway. Inhibitors of virus entry are potentially effective antiviral drugs of influenza viruses.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
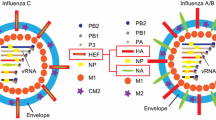
Influenza virus belongs to the family of Orthomyxoviridae. Infection of influenza virus causes a disease in humans with symptoms including high fever, cough, body ache, and runny nose. The word “influenza” was originated from the Italian language, meaning “influence.” Since the disease caused by influenza virus infection occurs more often in winter seasons, it was thought in the ancient world to be caused by the influence of astrological movements. The true reason that influenza returns each year is because of the unique structure of the influenza virus. The influenza virion contains an envelope of lipid membrane that is derived from the plasma membrane of the infected host cell, the site of influenza virus assembly (Fig. 9.1). The membrane envelope forms a barrier inside of which are the viral components protected from the environment when the virus particle is in circulation. Immediately underneath the membrane envelope there is a layer of matrix formed by the matrix protein. The matrix protein layer encloses the viral genome that is composed of eight segments of nucleoprotein-enwrapped single-stranded RNA in the negative sense. These eight genome segments encodes ten viral proteins, including three subunits, PA, PB1, and PB2, of the viral-specific RNA polymerase, two surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), the nucleoprotein (NP), the matrix protein (M), the proton channel protein M2 that is translated from the spliced mRNA of M, and two nonstructural proteins NS1 and NS2 that are the products of the two alternatively spliced eighth viral mRNA. NS2 is also known as nuclear export protein (O’Neill et al. 1998). The outcome of the influenza virus entry into the host cell is to release the eight viral genome segments into the nucleus to initiate virus transcription and replication. The entire virus entry process may be divided into a number of steps. First, the influenza virus particle needs to recognize a specific receptor molecule on the surface of the target host cell to allow the virus particle to gain access to the specific host cell in which influenza virus may replicate. The receptor molecule for influenza virus is a terminal α-sialic acid that is linked to saccharides anchored on the host cell surface by various mechanisms. In the second step, the tight association of the influenza virus particle with the host cell induces endocytosis that generates an endosome to encapsulate the entering virus particle. The endosome is then translocated in the next step to the site near the nucleus. The fourth step is to fuse the viral membrane with the host membrane that forms the endosome, which is mediated by the HA glycoprotein embedded in the virus surface. This membrane fusion step releases all eight segments of the RNA genome into the nucleus where the initiation of transcription of viral genes and the later viral replication takes place.

Cartoon showing the architecture of influenza virus
The key player in virus entry is the surface glycoprotein HA that contains the host receptor binding site to allow the virus particle to attach to specific host cells, the fusion peptide that is inserted into the target cellular membrane during membrane fusion, and other structural elements that may refold during the membrane fusion process. Since HA is a surface glycoprotein on the virus particle, it is readily recognized by host antibodies when influenza virus infects the host. However, influenza virus is quite capable of escaping the host defense system by unique mechanisms. During the replication cycle, the viral-specific RNA polymerase makes errors with a high frequency in its synthesis of viral RNAs. The errors during replication will generate a large number of variants of the HA glycoprotein. Some of them will allow the influenza virus to become resistant to neutralization by the existing host antibodies. By this selection mechanism, a new influenza virus that can escape the host immune system is generated and can replicate readily in the infected host. The novel influenza virus variant can spread to other hosts if they have not been exposed to this variant previously either through vaccination or through natural infection. This is the main reason that influenza virus comes back year after year because in each season new influenza virus strains become the circulating variants to which the human population has not acquired sufficient immunity. The same variations also occur by the same mechanism in the NA glycoprotein, the other surface glycoprotein in influenza virus. Such changes in the HA and NA glycoproteins are termed “antigenic drift” (Hay et al. 2001). During antigenic drifts, amino acid sequences are replaced in areas that are exposed to antibody recognition, and at the same time, the amino acid substitutions are restricted from locations that are required for receptor binding, maintaining a function fusion peptide, and proper structural changes for mediating membrane fusion. When the HA amino acid substitutions are accumulated in the repertoire of influenza virus strains, they are mostly on the exposed surface of the HA glycoprotein except for the receptor binding site, the fusion peptide, and the amino acids that accommodate the fusion peptide before HA structural changes for membrane fusion (Wilson et al. 1981). Besides antigenic drifts, there is also occasionally a dramatic change in HA antigenicity of influenza A virus. The HA glycoprotein of influenza A virus has been classified in 20 subtypes (H1–H20) based on a hemagglutination-inhibition test (Yen and Webster 2009). Subtypes H3 and H1 have been circulating globally in recent years. However, there could be a sudden change of HA subtype in a given year, termed “antigenic shift.” This type of change is the result of replacing one gene segment of a circulating influenza virus with another gene segment, a process called reassortment. Reassortment could occur in influenza virus because its genome prepresent in the virion is segmented. The HA glycoprotein must be compatible with other segments in the emerging influenza virus derived from the reassortment. If two influenza virus strains with totally different segmented genomes coinfect a common host, a novel influenza virus could emerge by regrouping a set of genome segments from the two sources. An antigenic shift usually leads to a global influenza pandemic, such as the pandemic of 1918 influenza that was the result of introducing a new H1N1 avian influenza virus in the human population (Compans et al. 1970). Throughout the antigenic changes over the many years, the HA-mediated virus entry remains robust in all influenza virus strains. The HA glycoprotein is fully functional despite many substitutions in its amino acid sequence.
2 Structure of Hemagglutinin
Influenza virus is divided into three types: influenza A virus, influenza B virus, and influenza C virus. For virus entry, hemagglutinin, the major glycoprotein on the surface of influenza virion, plays multiple roles in each step of the virus entry pathway. For influenza A virus, HA has been classified in 20 subtypes (H1–H20). The subtype of an influenza A virus is determined if the antisera corresponding to that subtype could inhibit the hemagglutination by the virus. Influenza B and C viruses have only one subtype and these two viruses have only been found circulating in humans.
The first glimpse of influenza virus HA was from electron micrographs. HA molecules appear as spikes covering the surface of the influenza virion (Compans et al. 1970). In the recent higher resolution three-dimensional structure reconstructed from tomographic images of influenza virion (Harris et al. 2006), the trimeric glycoprotein of HA is present as clearly distinct spikes scattered on the virion surface. The HA trimers are anchored through a transmembrane domain that penetrates the membrane envelope, which allows the C-terminal cytoplasmic tail of HA to interact directly with the matrix protein layer. The lollipop-shaped tetrameric NA is present among HA trimers in a small number (Fig. 9.1).
HA is inserted into the lumen of the ER during translation. The trimeric HA precursor travels through the secretory pathway from ER, Golgi apparatus, and secretory vesicles to the plasma membrane. Along the secretory pathway, HA becomes glycosylated and is processed into HA1 and HA2 polypeptides by the host proteinase. The cleavage of HA into HA1 and HA2 generates the fusion peptide at the N terminus of HA2. Uncleaved HA molecules are incompetent to mediate membrane fusion. The ectodomains of the mature HA trimers can be released from the surface of influenza virion by a proteinase, bromelain. The crystal structure of the HA ectodomain from an influenza A virus revealed the features that are important for the functions of HA (Fig. 9.2) (Wilson et al. 1981). The HA1 molecule is primarily on the outside of the HA trimer. The first 18 amino acids participate in a β-sheet formed by amino acids in the N-terminal and C-terminal regions of the HA2 molecule. This motif appears to be the structure that tethers the HA1 molecule to the membrane-anchored HA2 molecule. The next 18 amino acids form part of the pocket that accommodates the hydrophobic fusion peptide in the metastable HA structure on the influenza virion prior to virus entry. The rest of the HA1 molecule continues to extend to the top of the trimetric HA spike where a ligand-binding domain of a β-fold is located at the extreme end. The conformation of the ligand-binding domain at the top of the trimeric HA spike is partially stabilized by the contacts between themselves around the threefold symmetry axis. The ligand-binding domain is similar to the common protein fold that binds saccharides. It is composed of two β-sheets folded as a barrel. Since the host cell receptor for influenza virus HA is sialic acid, a saccharide moiety, it is not surprising that HA structure contains such a protein fold at the strategic location where it can recognize the host receptor molecule. Following the ligand-binding domain, the HA1 molecule extends downward along the HA2 molecule to end near the fusion peptide at the N terminus of the HA2 molecule. This conformation of the HA1 C terminus is the result of the cleavage between HA1 and HA2. In the uncleaved precursor, this region forms a surface loop accessible to protease (Chen et al. 1998). The size of the loop and the number of positively charged amino acids in the loop determine how easy the HA molecule is processed. When there are more than three positively charged amino acids in this loop, the cleavage at this site is more efficient and the influenza virus becomes more virulent, often highly pathogenic (Zambon 2001). The HA2 molecule forms the core of the HA trimer. The fusion peptide leads to the amino acids that participate in the anchoring β-sheet involving the N terminus of the HA1 molecule. After the β-sheet, the first α-helix of the HA2 molecule stands up roughly parallel to the threefold symmetry axis of the HA trimer. A large loop underneath the HA1 structural motif that contains the ligand-binding domain links the first α-helix to the second long α-helix that forms a coiled coil with the other two HA2 molecules to form a three-helix bundle toward the membrane envelope. The three-helix bundle is firmly anchored by the transmembrane domain in the membrane envelope. The three-helix bundle is a common structural motif in all the type I fusion proteins of enveloped viruses, including human immunodeficiency virus (HIV) and SARS virus (Colman and Lawrence 2003). The length of the helices may vary from virus to virus, but the three-helix configuration is the same in all viruses with type I fusion proteins. The three-helix bundle coiled coil is the key structural element that keeps the trimeric association of HA molecules throughout its structural changes during the virus entry process. The overall structure of the trimeric HA molecule could be viewed as a crown of the three HA1 molecules that caps the HA2 trimer tied together by the three-helix bundle. The HA1 crown is essential in keeping the HA structure in the metastable conformation prior to membrane fusion. The interactions between the top structural motifs and those between HA1 and HA2 molecules all contribute to the metastability, which is required for storage of energy that will be needed for the structural changes of HA during membrane fusion.

Structure of hemagglutinin. (a) The crystal structure of the trimeric ectodomain of hemagglutinin (Wilson et al. 1981). Each subunit of HA is colored red, green, and blue, respectively, with all three fusion peptides colored yellow. The ribbon drawings in this and subsequent figures were prepared with the program PyMol (DeLano 2002). (b) A single subunit of the ectodomain is presented for clarity. The HA1 polypeptide is colored red, and the HA2, lighter red
Both HA1 and HA2 proteins are glycosylated at N-linked glycosylation sites, with the HA1 molecule having more sites glycosylated near the top of the HA spike. There are a lot of variations between influenza virus strains in terms of the number of the N-linked sequences, ranging from three to ten sites (Bragstad et al. 2008). The presence of a sequence for N-linked glycosylation is not sufficient for this site being glycosylated. For a given influenza virus strain, the degree of glycosylation may depend on what types of cells the virus infects. For instance, there are five N-linked sequences in HA of subtype H1. When the virus was propagated in human cells, four of five sites were glycosylated (Gamblin et al. 2004). When the virus of an avian origin was propagated in chicken embryos, only three of the five sites were glycosylated (Lin et al. 2009). The degree and location of glycosylation has an effect on the entry of influenza virus. In the first place, proper glycosylation may be required for proper folding of the HA molecule (Segal et al. 1992). For instance, glycosylation at Asn8 and Asn22 after the cleavage of signal peptide helps recruitment of chaperone proteins that facilitate HA folding. In other cases, the glycosylation near the receptor binding site on the top of the HA spike has a direct influence on the affinity of the HA molecule to the virus receptor sialic acid. As shown in the case of influenza virus A/Vietnam/1203/04, a highly pathogenic strain of avian influenza A virus, the HA of this virus recognizes an α2,3-linked sialyl receptor and can spread systematically in mice. Removal of the glycosylation sequence, Asn158, near the receptor binding site, contributed to the increase of HA affinity to an α2,6-linked sialyl receptor, which resulted in reduction in systematic virus spread (Wang et al. 2007). Similar increases of receptor affinities after removal of N-linked glycosylation sites, and subsequent reduction in virus growth and spread, were also observed in other studies involving a H7N1 influenza virus (Wagner et al. 2000).
In addition to glycosylation, formation of disulfide bonds in HA is also important for the structure of HA. There are six strictly conserved disulfide bonds in HA in all subtypes of influenza viruses, one of which is between HA1 and HA2 molecules (Segal et al. 1992). This high conservation indicates that the pattern of disulfide bonds plays a critical role in the structure and function of HA. Studies showed that without proper formation of the disulfide bonds, the folding of HA was greatly impaired. Similarly as glycosylation, disulfide bonds are also additional factors that facilitate proper folding of HA during translation.
Among different HA subtypes, the structural differences are mainly located in three regions. First, the loops on the surface of the spike crown formed by the HA1 molecule are among the most variable structural elements. These variations are probably mostly responsible for the antigenic differences among the HA subtypes, which might not have significant effects on virus entry. The other two regions, however, are probably directly related to the process of HA-mediated virus entry. One of the regions is the receptor binding site. The structure of each HA subtype in this region appears to favor one type or the other α-linked sialic acid, which may be a determinant in host specificity of influenza A viruses. Another region of structural differences is near the fusion peptide. As discussed above, the N-terminal and the C-terminal sequences of the HA1 molecule are part of the hydrophobic pocket that accommodates the fusion peptide at the N terminus of the HA2 molecule. The fusion peptide adopts the same structure among different HA subtypes, but the sequence and structure of the HA molecule surrounding the fusion peptide changes between the HA subtypes. More details about the two regions will be discussed in later sections with regard to receptor binding and membrane fusion.
For influenza B virus, there is no subtype of HA. The single serotype of influenza B virus HA has a similar overall structure as that of influenza A virus HA. The HA1 molecule in influenza B virus also forms a crown structure on top of the trimeric HA2 molecules. However, the exact three-dimensional structure of influenza B virus HA is quite different. For the HA1 molecule, the ligand-binding domain that contains the receptor binding site has a conformation that requires a significant rotation up to 60° in order to be superimposed with that in the HA1 molecule of influenza A virus HA (Wang et al. 2007). Such a large degree of rotation may reflect two aspects of the HA molecule. First, the crown-like structure formed by HA1 molecules must disassemble during HA-mediated fusion. One of the requirements is that the structure of the HA1 molecule must have a hinge region that allows the rotation of the structural motif at the top of the spike in order to disassemble from the metastable trimeric conformation. It is therefore an intrinsic property in HA that the structural motif at the top of the spike is allowed to rotate without any blockage during HA-mediated membrane fusion. It is not surprising that the structural motif at the top of the spike of the influenza B virus HA adopts a conformation that is, although different from that in influenza A virus HA, within the spatial range of the HA structure.
Influenza C virus is also a virus that is only found in humans, and it has a surface glycoprotein that is quite different from HA in other influenza viruses. This surface glycoprotein in influenza C virus is called the hemagglutinin-esterase-fusion (HEF) protein. In addition to the ability to bind the host receptor molecule and to mediate membrane fusion as influenza virus HA, HEF has an additional enzymatic activity that destroys the HEF receptor molecule, 9-O-acetylated sialic acid, by enzymatic removal of the acetyl group. Since the receptor destroying enzyme (RDE) and the receptor-binding activities are both in HEF, influenza C virus does not therefore have any NA activity or the gene. The overall three-dimensional structure of HEF is very similar to that of HA when the two structures are superimposed, except that there are additional structural motifs in HEF corresponding to the esterase (Rosenthal et al. 1998). The detailed structures are, however, very different between HEF and HA. HEF is also cleaved into HEF1 and HEF2 molecules in the mature influenza C virion. The N terminus of HEF1 participates in the similar β-sheet at the anchoring region, but the sequence following that does not have a structural element that forms part of the hydrophobic pocket accommodating the fusion peptide. Instead, the polypeptide directly extends to the structural motif at the top of the spike. Comparing to HA, there is one structural insertion prior to the ligand-binding domain, and one after, which are two of the three segments in the esterase. It appears that HA deleted two of the three segments in the esterase during evolution. The remaining structural element in HA helps to keep the ligand-binding domain at the same position as in HEF. The esterase in HEF is located below the receptor binding site. The C terminus of HEF1 is closely located near the N terminus of HEF1 that contains six amino acids in front of the fusion peptide. The fusion peptide in HEF is therefore not accommodated in a hydrophobic pocket as in HA. The location of the fusion peptide in HEF cannot be superimposed with that in HA. Another significant difference is in the loop that links the two long α-helices in the HEF2 molecule. This loop is much shorter in HEF, as well as the second α-helix that forms the coiled coil in the trimer. This difference makes HEF look hollow underneath the crown formed by the HEF1 trimer. The significance of this structural difference is not yet clear.
3 Receptor Binding Site
As many animal viruses, influenza virus attaches to the host cell surface by recognizing a specific receptor. The commonly known receptor molecule for influenza virus is a sialic acid (also known as N-acetylneuraminic acid) that is covalently linked to the end of oligosaccharides on glycoproteins or glycolipids (Schauer 2009). Sialic acid is present on the surface of many cell types and also on cell-secreted products. Sialylation is an important mechanism of masking cells or microorganisms, as well as products they produce, to avoid being recognized by the host immune system. At the same time, the sialyl moiety is often used as ligands for attachment proteins that have important cellular functions, or as receptors (co-receptors) for microorganisms including viruses, bacteria, fungi, and parasites. Influenza virus is one of the best known example that uses sialic acid as the receptor for cell entry.
The sialic acid binding site of influenza virus is located in each subunit of the trimeric HA (Fig. 9.3). Since there are many copies of HA on each influenza virus particle, the attachment of influenza virus to the host cell surface may be considered as a multivalent binding event. As a result, the viral attachment is fairly tight even though the affinity of each individual sialyl moiety to its binding site on HA may be weak (Takemoto et al. 1996). The estimated dissociation constant (K d) for sialic acid binding by influenza virus HA is about 3 mM (Hanson et al. 1992; Sauter et al. 1989). The precise interactions between sialic acid and HA have been described by cocrystal structures (Lin et al. 2009; Takemoto et al. 1996; Ibricevic et al. 2006; Tumpey et al. 2007). The binding site is a depression on the surface of a structural domain at the top of the HA trimer. The structural domain is composed of a β-barrel motif and α-helices. This structural motif is similar to other lectin proteins in shape, but topologically different. At the bottom of the receptor binding site there are two aromatic residues, a tyrosine (Tyr98) and a tryptophan (Trp153), in influenza A virus (A/Aichi/2/1968 H3N2). The pyranose ring of sialic acid sits on top of the aromatic residues. The four different functional groups linked to the pyranose ring form specific interactions with residues surrounding the depression. The carboxyl group in its α-configuration at the C2 position of the pyranose forms three hydrogen bonds with the side chain of Gln226 and main-chain atoms. The hydroxyl group at the C4 position does not seem to be involved in binding to HA. The acetylamido group at the C5 position, on the other hand, forms both hydrophobic and hydrophilic interactions. The amide group in the acetylamido group forms a hydrogen bond with a main-chain carbonyl oxygen atom, whereas the methyl group in the acetylamido group is within van der Waals contact with the side chain of Trp153. Finally, the last two hydroxyl groups in the glycerol moiety at the C6 position form hydrogen bonds with the side chains of Gln226 and Glu190, and a main-chain carbonyl oxygen atom. The network of these interactions renders the specificity for sialic acid recognition by HA. However, there are variations in the sialic acid binding site among different subtypes of influenza virus. For instance (Ha et al. 2001), residue Leu226 is replaced by Gln226 in an avian influenza A virus H5 HA. The side chain of Gln226 in H5 appears to form a hydrogen bond with one of the carboxylate oxygen atoms and more importantly, a hydrogen bond with the oxygen atom at the C2 position that is in the glycosidic bond linking the sialic acid moiety to the oligosaccharides. The hydrogen bond formed by the hydroxyl group at the C9 position is through a water molecule to the main-chain amide group of residue Gly228, instead of directly with the side chain of Ser228 in the H3 HA. In an H9 HA, the hydrogen bond formed with the water molecule as observed in the H5 HA is conserved, but that with the side chain of Glu190 as observed in both H3 and H5 HA is lost because this residue is a valine (Val190) in the H9 HA. In addition, the left-side loop in which residues Leu/Gln226 and Ser/Gly228 are located has a more opened conformation in H3 and H9, compared to that in H5.

The receptor binding site of an avian influenza virus hemagglutinin (Liu et al. 2009). (a) LSTc (α2,6-linked galactose) mimicking the human receptor bound in the binding site. (b) LSTa (α2,3-linked galactose) mimicking the avian receptor bound in the binding site. Critical residues for binding the receptor are presented as stick models with labels
Changes in the sialic acid binding site are related to the receptor preference of different subtypes of influenza virus HA. Currently, human influenza A viruses contain H1, H2, and H3 subtypes of influenza virus HA, whereas the avian and other animal influenza A viruses contain H1–H16 subtypes (Yen and Webster 2009). One of the differences among HA subtypes is that the HA of human influenza A viruses prefers a sialic acid that has an α2,6 glycosidic linkage to glycoproteins and glycolipids as the receptor molecule, but HA of avian influenza A viruses prefers an α2,3 glycosidic linkage. The receptor preference may be one of the barriers to prevent the wide spread of highly pathogenic avian influenza A viruses in humans to cause a severe pandemic (Ibricevic et al. 2006; Stevens et al. 2006; Yamada et al. 2006). Sialic acids with an α2,6 glycosidic linkage are abundant on the human airway epithelial cells (Stray et al. 2000; Thompson et al. 2006). It has been postulated that an avian influenza A virus must adapt to the human sialic acid receptor in order to transmit widely from humans to humans. The binding of HA to different receptor molecules may explain the difference in receptor preference. The avian H5 HA has a glutamine residue at position 226. In the crystal structure of H5 in complex with an α2,3 glycosidic linkage to a galactose (Ha et al. 2001), the glycosidic bond has a trans conformation that allows the side chain of Gln226 to make hydrogen bonds with both the glycosidic oxygen and the hydroxyl group at the C4 position of the galactose. The receptor binding site in H5 HA is also narrower so that it can only accommodate a sialic acid with the α2,3 glycosidic linkage in trans conformation. In the case of the avian H9 HA, a sialic acid with the α2,3 glycosidic linkage or the α2,6 glycosidic linkage may fit the receptor binding site, and the interactions with sialic acid by H9 HA are similar in both cases. However, the H9 HA can form hydrogen bonds with sugar moieties as the fourth or fifth molecule in oligosaccharides. The oligosaccharide chain adopts different conformation when bound to each individual HA even though the terminal sialic acid is bound in the same position.
In influenza B virus, the sialic acid binding site in its HA appears to be very different. First of all, the left-side loop homologous to that in influenza A virus HA is substantially more closed so the sialic acid moiety is no longer above a depressed site. The carboxylate group forms hydrogen bonds similar to the main-chain amide nitrogen and the side chain of a serine as in influenza A virus HA. However, the acetylamido group now forms hydrogen bonds with the side chain of Thr139, and the hydroxyl group at the C9 position forms hydrogen bonds with the side chain of Asp193 and Ser240. The narrower sialic acid binding site seems to be responsible for discriminating against the avian α2,3 glycosidic linkage by reducing two hydrogen bonds. Influenza B virus is found only circulating in humans.
In influenza C virus, the receptor binding site is very different. In fact, the receptor molecule is no longer the sialic acid, but 9-O-acetylsialic acid. The surface glycoprotein in influenza C virus is actually a HEF protein (Rosenthal et al. 1998). By comparison with the structure of HA, there is an inserted esterase with structural motifs flanking the receptor binding domain. The esterase may perform the receptor destroying function of NA, which is missing in influenza C virus. The main difference in the receptor binding site of the HEF protein is that the left-side loop is further opened, and the 9-O-acetyl group as well as the hydroxyl groups at the C8 and C7 positions all participate in forming hydrogen bonds with side chains of three different residues. The acetylamido group at the C5 position also forms new interactions with side chains of two different residues.
The receptor preference of a sialic acid with the α2,6 glycosidic linkage may be one of the requirements for influenza virus to infect the human airway efficiently, but may not be the only requirement. The 1918 pandemic influenza A virus has been shown to be originated from an avian influenza A virus (Compans et al. 1970; Lin et al. 2009; Nicholls et al. 2008; Shen et al. 2009; Hayden 2009). Its receptor preference is the a 2,6-linked sialic acid. As shown in (Liu et al. 1995), the change of one amino acid from aspartic acid to glutamate at position 190 would switch the receptor preference from the α2,6 glycosidic linkage to α2,3 glycosidic linkage. An additional change from aspartic acid to glycine at position 225 would diminish the transmission of the pandemic virus between model animals such as ferrets (Tumpey et al. 2007). However, the influenza virus carrying HA that has the α2,3 glycosidic linkage preference could cause the same lethal infection in mice as the 1918 pandemic influenza virus (Qi et al. 2009), suggesting that viral factors in addition to HA also contribute to the pandemic virulence of the 1918 influenza virus. For instance, the polymerase subunit gene, PB2, from the 1918 influenza virus was also required for transmission between ferrets (Lakadamyali et al. 2003). The NS1 gene was also shown to be an important virulence factor (Konig et al. 2010). The adaptation of the avian influenza viruses may begin with the avian influenza virus strain that could bind both types of receptors (Matlin et al. 1981). Additional adaptation may be required in other influenza virus genes in order for an influenza virus strain to be transmitted efficiently among humans or to increase virulence. This adaptation may take place in animal hosts, such as pigs or birds, or humans. The most threatening pandemic influenza virus strain is likely to have the receptor preference for the α2,6 glycosidic linkage and contain highly virulent genes in addition to HA.
The exact role of sialic acid binding by HA, however, may not be as clear cut as most literature described (Nicholls et al. 2008). First of all, avian H5N1 influenza viruses that prefer the α2,3 glycosidic linkage infect humans frequently since 1997, even though the transmission from humans to humans has not been confirmed for this type of influenza virus. Furthermore, it has been shown by a number of laboratories that cells could still be infected by influenza virus after the sialic acid was removed by sialidase treatment (Kogure et al. 2006; Stray et al. 2000; Thompson et al. 2006). One of the possible explanations is that the entry of influenza virus into the host cell employs a sialic acid independent mechanism, such as an alternative primary receptor molecule. The binding to sialic acid by HA may be a pre-entry step to attach the virus particle to the host cell surface. The properties of binding to sialic acid with a specific glycosidic linkage appear to be associated more closely with production of infectious progeny viruses and the ability of transmission from one host to another by influenza virus (Qi et al. 2009; Giannecchini et al. 2007; Nunes-Correia et al. 2004). This suggests the potential that a novel influenza virus variant may not need to bind sialic acid at all to become transmissible among humans.
Amino acid sequence changes in HA happen frequently because the changes allow influenza virus to escape host immune responses. However, these changes must not compromise the entry function carried by HA. It has been observed that when influenza virus was passaged in mice that had been immunized with inactivated wild-type virus, the selected escape mutants also exhibited increased cellular receptor binding avidity by mutated HA (Rust et al. 2004). Mutation sites could be near the sialic acid binding site or not close at all. Some escape mutations did not diminish polyclonal antibody binding, but simply increased virus binding avidity for cellular glycan receptors. These mutant viruses could still effectively agglutinate erythrocytes treated with Vibrio cholerae NA RDE that removes terminal sialic acids. The increase in receptor avidity by HA was toward sialyl glycan molecules with both the α2,3 and α2,6 glycosidic linkages. When the escape mutants were passaged in naïve mice, the receptor avidity was restored to the normal level, but the resistance to polyclonal antibody neutralization was retained. However, additional mutations were also identified in some cases that render the new mutants further resistance to polyclonal antibody neutralization. These studies suggest that under neutralizing immune pressure, influenza virus increases its receptor avidity by changing amino acid sequences throughout the HA globular domain. Additional amino acid changes may accumulate in naïve hosts in which high receptor avidity is not required for influenza virus infection. These further mutations compound on antibody resistance. Indeed, the sequence data of isolates from human and animal hosts since 1918 revealed a close relationship between receptor binding and escaping host immune response (Shen et al. 2009).
Currently, the popular antiviral drugs for therapeutic treatment of influenza virus infection are NA inhibitors (NAIs) (Hayden 2009). It is inevitable that drug resistance variants would arise after a period of time, some of which may have mutations in HA. The key function of NA is to remove sialic acid moieties from oligosaccharides attached to HA on newly assembled influenza virus particles when they bud out of the infected cells. If the NA activity is not present, the progeny virus particles form aggregates on the cell surface because the sialylated HA binds to each other between two influenza virus particles, and to cell surface (Liu et al. 1995). The aggregation prevents virus release, which is the mechanism used by NAIs to stop influenza virus infection. When influenza virus was passaged in cell culture in the presence of NAIs mutations in HA were identified in some drug-resistant mutants, in addition to drug-resistant mutants that had mutations in NA (Giannecchini et al. 2007; Hurt et al. 2009). It is obvious that influenza virus can replicate in the presence of NAIs if the sialic acid binding properties of HA have been changed. NAI-resistant mutants with mutations in HA have not been reported from clinical isolates, but the potential still exists.
4 Entry Pathway
After the virus is attached to the host cell surface via binding to sialic acid by HA, the process of importing the incoming virus particle to a location inside the cell begins. There is a large number of HA molecules on influenza virus particles, which may be required for adequate binding to the cell surface to trigger the internalization process. The entry process follows a specific pathway with a number of cellular factors involved (Fig. 9.4). At the cell plasma membrane, the influenza virus is taken up by endocytosis (Bottcher et al. 1999; Daniels et al. 1985). The virus particle quickly enters coated vesicles (endosomes) inside the cell. The internalized virus particles are transported inside the endosomes to locations near the nucleus (Lakadamyali et al. 2003). The fusion of viral membranes with endosomes is triggered by acidification of late endosomes. The result of fusion is the release of viral RNA-dependent RNA polymerase-associated nucleocapsid (RNP) into the nucleus where viral transcription is initiated.

A model for the entry pathway of influenza virus
Studies by Lakadamyali et al. show that the trafficking pathway of influenza virus particles to the nucleus has three distinct stages (Lakadamyali et al. 2003). In Stage I, the endocytic vesicles are transported in an actin-dependent manner within the cytoplasm leading to the early endosomes. In Stage II, the early endosomes containing virus particles are rapidly transported by dynein-directed movement on microtubules toward the perinuclear region. At this location, the first acidification (pH = ≈6.0) takes place and the endosomes mature. The maturing endosomes continues to move on microtubules in Stage III. Finally, the second acidification (pH ≈ 5.0) of matured endosomes occurs and HA-mediated membrane fusion results in release of RNP into the nucleus.
The description of the entry pathway clearly indicates that the entry of influenza virus is a process directed by cellular endocytosis and transportation mechanisms. These functions are essential for the cell life cycle. Influenza virus utilizes some of the endocytosis and transportation mechanisms to support its own entry to infect the host cell. By systematic RNAi knockdown of host factors, up to 23 proteins have been shown to be related to the entry of influenza virus (Konig et al. 2010). One of the most obvious protein groups is the host proteins that form the endosomal coat protein complex (Chen and Zhuang 2008; Doxsey et al. 1987; Matlin et al. 1981; Roy et al. 2000). The clathrin-coated pits are formed around the attached virus particle once it is attached and the particle is then internalized. A number of other host factors are also involved in the initial internalization. Dynamin, a GTPase that play an important role in clathrin-mediated endocytosis, was shown to facilitate formation of virus-loaded coated vesicles (Roy et al. 2000). In the absence of dynamin’s functions, the pinch off of the coated pits was incomplete. Epsin 1 is also a host factor that has been identified to be involved in influenza virus endocytosis via coated pits (Chen and Zhuang 2008). Epsin 1 is an adaptor protein that interacts with proteins such as clathrin, AP-2, and Eps15, all of which play important roles in assembly of the coated pits. Association of epsin 1 induces membrane curvature. On the other hand, influenza virus could also be endocytosed by other clathrin-independent pathways, even though clathrin-dependent endocytosis appeared to be the main route used by influenza virus entry (Nunes-Correia et al. 2004; Rust et al. 2004; Sieczkarski and Whittaker 2002). Analogous to using multiple receptor molecules, endocytosis of influenza virus may also utilize multiple routes.
Trafficking inside the host cell begins after the internalization of the influenza virus particle. Actin was shown to be required for the initial movement of the virus particle in polarized epithelial cells (Daniels et al. 1985; Bullough et al. 1994; Floyd et al. 2008; Kemble et al. 1994). When cytochalasin D, an inhibitor that disrupts actin polymerization, was added, influenza virus entry into the apical phase of polarized epithelial cells was inhibited. This inhibitor, however, has no effects on influenza virus entry into non-polarized cells or through the basolateral phase of polarized epithelial cells. Inhibition of myosin VI function, an actin motor protein, also decreased influenza virus infection. The coated vesicles containing influenza virus were arrested at the plasma membrane. These observations suggest that an active actin skeleton is required for directing influenza virus entry, which transports the coated vesicles to microtubules where the early endosomes are found (Lakadamyali et al. 2003). The movement of the virus-containing early endosome is rapid in Stage II before the first acidification (pH ≈ 6.0) occurs. The host cell protein sorting system appears to be selectively required for directional movement of early endosomes to late endosomes along microtubules. The virus-containing endosomes were shown to be colocalized with Rab9 and VPS4, suggesting that the ubiquitin/vacuolar protein sorting pathway was utilized by influenza virus (Khor et al. 2003). The lysosomal pathway for recycling could not be used for influenza virus infection. This specificity may be related to the functional requirement that virus-containing endosomes need to traffic through the early endosomes to the late endosomes along microtubules before the final membrane fusion takes place near the nucleus (Lakadamyali et al. 2003; Sieczkarski and Whittaker 2003). Before the final membrane fusion, the integrity of HA is not grossly affected during trafficking from endocytosis to the late endosomes through a directed, specific pathway to near the nucleus (Sieczkarski and Whittaker 2003). Since HA is the major surface glycoprotein that makes contact with the endosome membrane, it may very well play a role in selecting the specific trafficking pathway. This specificity may be related to recruitment of specific host proteins by HA or the unique envelope structure organized by HA.
5 Low pH-Induced Conformational Change in HA
In the late endosome, the HA mediates the fusion of the viral membrane with the endosome when the pH inside the endosome drops before 5.0. HA undergoes an extended conformational change at the acidic pH to insert the fusion peptide into the target membrane (Fig. 9.5). This is a common process used by many viral fusion proteins to mediate membrane fusion for viral entry. What has been learnt for influenza virus HA may have a broad application.

A ribbon drawing to compare the metastable conformation of a HA subunit (left) with the refolded HA subunit at low pH. HA1 is colored red, and HA2, lighter red. The fusion peptide is colored yellow. The helix-loop-helix region in HA2 is colored green, blue, and magenta, for the conformation at neural and low pH. The magenta helix region at low pH is leveled with that in the metastable HA
As discussed above, the native structure of HA is stabilized by trimeric association of HA1 and its interactions with HA2. Conformational changes of HA as well as membrane fusion could be prevented by introducing additional disulfide bonds at the top to stabilize the trimeric HA1 (Wharton et al. 1986), suggesting that the conformational change of the trimeric HA1 induced by low pH is required for HA-mediated membrane fusion. Upon binding to sialic acid, a step before the virus is exposed to low pH in the entry pathway, the HA1 trimer is stabilized and the conformational change of HA at low pH is actually restricted by sialic acid binding (Leikina et al. 2000). Since the virus particle is inside the endosome after endocytosis, there are multivalent interactions between trimeric HA and the receptor molecules on the endosome membranes. In such a structure, the fusion peptide at the N terminus of HA2 may be released when pH is lowered prior to HA refolding. In order to allow full refolding of HA2, it may be necessary for a step to dissociate HA from binding to sialic acid on the endosome membrane. Lowering pH may actually be a mechanism for such dissociation. Further downstream of the refolding pathway, there may be stepwise conformational changes that proceed sequentially at low pH. Before protonation of HA surface residues when the pH is lowered, the crown formed by the HA1 trimer has a positively charged surface both inside and outside (Huang et al. 2002). On the contrary, the HA2 trimer in its metastable conformation has a negatively charged surface. This negatively charged surface of the HA2 trimer electrostatically matches the positively charged interior surface of the trimeric HA1 crown. These attractive interactions allow HA to maintain its metastable conformation. When the pH is lowered, some surface residues in HA1 become more positively charged, such as residues His75, Lys259, and Lys299 (Huang et al. 2003). These extra charges increase the force of repulsion among HA1 subunits so the association within the HA1 trimer is weakened. Indeed, an obvious expansion of the HA1 trimer was observed at pH 4.9 by cryo-EM imaging (Bottcher et al. 1999). Furthermore, the structure corresponding to HA2 also appeared to move away from the native position, leaving an appearance of a channel through the center of the HA trimer. This may suggest that there is also significant destabilization of the HA2 trimer at low pH before the extended refolding. Mutagenesis studies support this notion (Daniels et al. 1985). Mutations that rearrange the interface of the HA2 molecule resulted in a higher pH needed for triggering HA2 refolding. These mutations are located in the helix that forms a coiled coil to hold the tree HA2 subunits together. This is consistent with the suggestion that there is a threshold of structural destabilization of the HA trimer before refolding. Protonation of surface residues, especially those on HA1, induces structural changes that begin to destabilize the HA trimer. The loosen-up of the HA trimer may allow more residues to be protonated until the complete HA2 refolding.
The refolding of HA2 under low pH is a remarkable structural phenomenon observed for any protein structure (Bullough et al. 1994). In the metastable native structure of HA, the central region of HA2 forms a helix-loop-helix structure, with residues 40–55 corresponding to the first helix; residues 56–74, the loop; and residues 75–125, the second helix. The second helix forms a coiled coil in HA2 trimer. The fusion peptide, the first 20 residues of HA2, is in front of the first helix, but oriented near the viral membrane envelope. At acidic pH, residues 40–105, which cover the first helix, the loop, and the first half of second helix in the native helix-loop-helix motif, become a continuous helix of about 100 Å in length. This extended refolding will bring the fusion peptide from its location near the viral envelope to the top of the refolded HA2 trimer, allowing it to reach the target endosome membrane. This may be the most important event during HA-mediated membrane fusion. Many viral fusion proteins have similar structural refolding to connect the viral membrane with the target membrane in the host cell. The second half of the second helix in the native helix-loop-helix motif undergoes a different conformational change. Residues 106–112 switch from a helical structure to an extended loop, and residues 113–125, although still exiting as a helix, has a 180° reorientation from its original orientation. This new orientation was suggested to represent the final collapsed “hairpin” structure of HA2 to bring the two merging membranes close to each other.
In addition to triggering the extended refolding of HA2, low pH also induces other critical structural changes that affect the fusion process. The release of the fusion peptide from the native pocket near the viral envelope is pH related and could influence the HA2 refolding. For instance, the carboxylate side chains of Asp109 and Asp112 form hydrogen bonds with residues 2–6 of the fusion peptide. Mutating either Asp109 or Asp112 to Ala increased the pH required for HA-mediated membrane fusion, suggesting that deionization of Asp109 and Asp112 at low pH could contribute to destabilization of HA (Heider et al. 1985). Biochemical analyses conformed the extended refolding of mutant HA at the elevated pH. Other residues in the vicinity of the fusion peptide binding pocket have similar effects when mutated (Heider et al. 1985; Ghendon et al. 1986). The fusion peptide appears to play a major role in stabilizing the metastable conformation of HA trimer and participates in low-pH triggering of HA2 refolding.
In addition to structural changes of HA, low pH also induces structural changes in the matrix protein (M1) that may be required for completion of membrane fusion and RNP release. It was proposed that the matrix protein, which constitutes a protein layer beneath the viral membrane envelope to anchor HA and NA (Harris et al. 2006), is the last barrier that was broken at low pH in the fusion process (Lee 2010). Furthermore, low pH dissociates the matrix protein from RNP to unveil the nuclear import mechanism (Bui et al. 1996). The synchronized structural changes induced by low pH in the endosomes make up the process to complete the ultimate task to release RNP into the nucleus for initiation of virus replication.
6 HA-Mediated Membrane Fusion
The low pH-triggered conformational refolding of HA projects the fusion peptide to the target endosomal membrane. This is the first step in HA-mediated membrane fusion (Fig. 9.6). The fusion peptide is generated after the HA precursor, HA0, is cleaved into HA1 and HA2 during influenza virus assembly. Before the cleavage, an extended loop consisting of residues 323–329 in HA1 and 1–12 in HA2 is exposed on the outer surface of the HA trimer (Chen et al. 1998). Residue 319 is usually a positively charged residue that allows the site to be cleaved by a trypsin-like host protease. Residues preceding 329, usually including a number of positively charged residues, would enlarge the loop to facilitate protease cleavage as found in case of highly pathogenic avian influenza viruses (Nicholls et al. 2008). The polybasic residues that precede the cleavage site are postulated to allow a broad spectrum of host proteases to carry out the cleavage so that the virus gains increased virulence. However, the loop did not have an exposed conformation in HA of the 1918 H1N1 virus, a highly pathogenic human influenza virus (Luo et al. 1996). Other structural features in the pocket where the fusion peptide is tucked in after cleavage also contribute to the pathogenicity of influenza virus. Normally, the side chains of ionizable residues in the pocket provide a number of hydrogen bond interactions with main chain amides of the fusion peptide to stabilize it in the pocket (Wilson et al. 1981). In highly pathogenic strains, the structural changes near the pocket may allow the fusion peptide to be released from the pocket more easily.

A model for the membrane fusion process. Possible steps are attachment of the fusion peptide at initial refolding of HA2, hemifusion mediated by the fusion peptide and HA2, completion of membrane fusion with final refolding of HA2
The released fusion peptide is capable of penetrating into the target membrane, especially when it is lifted by the refolded HA. To fuse the two membranes, the fusion peptide first catalyses the hemifusion, mixing the outer leaflets of the two lipid bilayers, followed by formation of a fusion pore where both leaflets are fused. The fusion peptide forms an α-helix and inserts into the lipid bilayer at an angle (Lear and DeGrado 1987; Luneberg et al. 1995). The fusion peptide helix in the lipid bilayer has a kink that divides the structure into two halves (Yoshimoto et al. 1999; Bodian et al. 1993). At the fusion pH of 5.0, residues 2–10 form an α-helix in the first half and residues 13–18 form a 310-helix in the second half. The two helices make almost a right angle. Such a tight structure allows the fusion peptide to insert deeper in the lipid bilayer with charged side chains in the fusion peptide left out of the membrane (Bodian et al. 1993). Insertion of the tightly folded fusion peptide likely induces membrane perturbation to facilitate lipid exchanges between juxtaposed membranes. Studies showed that the insertion of the fusion peptide into the lipid bilayer is reversible at neutral pH at which the fusion peptide could not form tightly folded two helices (Chang and Cheng 2006). Only at low pH, insertion of the fusion peptide would lead to the following steps toward irreversible membrane fusion to form a functional fusion pore. Prior to hemifusion of the lipid bilayer, the fusion peptide undergoes a process involving diffusion in the lipid bilayer in order to become self-associated, which may be a rate-limiting step and require the participation of multiple copies of HA (Chernomordik et al. 1998; Floyd et al. 2008).
In the in vitro experiments, the fusion peptide alone could facilitate the complete fusion of two membranes. However, the fusion peptide is presumably covalently linked to the refolded HA2 at the fusogenic pH during influenza virus infection. At this stage, it is conceivable that the viral and endosomal membranes are still apart, but hemifusion could already occur (Chernomordik et al. 1998; Kemble et al. 1994). Studies suggest that both the fusion peptide and the exposed part of HA2 after low pH refolding contribute to hemifusion under low pH (Leikina et al. 2001). First of all, the polypeptide following the fusion peptide is responsible for the formation of HA2 trimer. Fusion activities are dependent upon effective concentrations of HA. The contribution of HA2 trimer to fusion can at least in part attribute to concentrating the fusion peptide. Second, the low pH conformation of HA2 prior to the final refolded hairpin structure is required for catalysis of hemifusion. In fact, the whole HA1−HA2 ectodomain, released from the influenza virus particle by bromelain treatment, could not induce lipid mixing or membrane fusion (Leikina et al. 2001; Wharton et al. 1986; White et al. 1982). In addition, deletion of residues 91–127 or mutations in this region also diminished its activities to induce hemifusion (Leikina et al. 2001). These observations suggest that the entire fusogenic HA2 molecule plays a role to induce hemifusion in addition to the fusion peptide. Furthermore, it has been found that small pores are induced by insertion of the fusion peptide. They flicker at this stage, i.e., they are open or closed reversibly. The HA2 molecules could aggregate membrane-inserted fusion peptides to enlarge the pores, but the system did not proceed to complete fusion in the absence of the viral membrane. This leads to a hypothetical model that HA induces hemifusion and large pores in the target endosomal membrane before final refolding (Bonnafous and Stegmann 2000).
Expansion of the initial fusion pore leads to complete fusion of the two membranes and release of the contents in the viral particle. This last step requires the participation of the transmembrane domain in HA. The main function of the transmembrane domain is to package and anchor HA in the viral membrane envelope. In addition, studies showed that the transmembrane domain also participates in the final step of membrane fusion (Kemble et al. 1994). If HA is anchored to a cell membrane through GI-linked lipids, it may induce hemifusion with other cell membranes, but not complete fusion. This shows that only a full-length HA with the transmembrane domain could function to mediate membrane fusion. The transmembrane domain of HA could be replaced by a different transmembrane domain derived from another virus, and the complete fusion could still be induced by the chimeric HA. This suggests that the role of transmembrane domain is more biophysical in membrane fusion. The specific amino acid sequence of the transmembrane domain is not required for membrane fusion as long as its biophysical properties are suitable for this process. It was shown that the transmembrane domain forms helices that can aggregate in lipids and increases the order of acyl chains in lipid bilayers, which may be required for both targeting HA to the lipid “raft” during assembly and complete fusion (Tatulian and Tamm 2000). Mutations in the transmembrane domain not only reduced the copy number of HA in virus particles, but also the efficiency of HA-mediated membrane fusion (Takeda et al. 2003). Changes of the amino acid sequence altered the interactions of the transmembrane domain with lipid bilayer in terms of biophysical interactions, which is the reason that the functions of HA were affected in both the anchoring and membrane fusion. The transmembrane domain forms tight oligomers in the lipid bilayer, and also insert deeper into the lipid bilayer at low pH (Chang et al. 2008). However, the transmembrane domain itself could not disturb membrane structure or induce hemifusion. Its contribution to membrane fusion must be in association with the fusion peptide. As the result of refolding, the fusion peptide and the transmembrane domain are brought close to each other. The residues at the N terminus of HA2 in the refolded structure form a cap connecting the ends of the three helices to close off the helices in the refolded HA2 trimer (Chen et al. 1999). The residues preceding the transmembrane domain are also structured next to the cap. The region between the cap and the fusion peptide, as well as the region between the end of the structured C-terminal part of HA2 and the transmembrane domain, appear to be very flexible, which allows the fusion peptide and the transmembrane domain to assume their conformation independent of the rigid HA2 trimer. Evidence suggested that the fusion peptide could associate with the oligomerized transmembrane domain in lipid bilayer (Chang et al. 2008). The association of the fusion peptide with the transmembrane domain oligomer may be one mechanism that the transmembrane domain facilitates fusion pore expansion and complete fusion since the transmembrane domain can insert deeper in the lipid bilayer at low pH, which brings the fusion peptide further into the lipid bilayer because of the association between the two sequences.
The complete process of HA-mediated membrane fusion was captured as snapshots using EM cryo-tomography (Lee 2010). Liposomes were incubated with influenza virus particles at different pH for different time periods. Images showed that the fusion peptide was first released and inserted into the target liposomal membrane as the pH was lowered. The insertion of the fusion peptide from multiple copies of HA glycoproteins on the virus surface deformed the target membrane, but the membrane structure of the virus particle was still intact at this stage. Following the initial insertion of the fusion peptide, formation of pores was observed in the target membrane and the refolding of HA brought the target membrane in touch with the viral membrane. Opening in the membranes was observed while the fusion process was in progress. However, a protection layer was still present to protect the content of the virus particle. This layer was identified as the matrix protein layer, which gave way as the fusion process went further at low pH. The layer of the matrix protein provided not only the last protection of viral contents, but also a platform for HA to anchor on during the initial deformation of the target membrane and subsequent joining of the two membrane parties for fusion. This may be another reason that the transmembrane domain of HA is required for complete fusion because the anchoring of HA on the layer of the matrix protein underneath the viral membrane envelope is by interactions of the transmembrane domain with the matrix protein. The final stage would require expansion of pores at low pH and release of the viral contents. The EM cryo-tomography snapshots could only resolve certain steps in the membrane fusion process. The limitation of the image resolution does not allow the fine details of membrane structures to be fully revealed. The exact details of lipid exchange, hemifusion, and fusion pores may only be found out with images of higher resolution.
7 Entry Inhibitors as Antiviral Drugs
Since entry of influenza virus is an essential step in its replication cycle, inhibitors that block this step could be effective antiviral drugs. An entry inhibitor of HIV, Enfuvirtide, is a drug currently used for treatment of HIV infection (Kilby et al. 1998). There is no reason why entry inhibitors of influenza virus would not be effective antiviral drugs. In light of emergence of mutant viruses resistant to current drugs, such as NAIs, the need for novel antiviral drugs is obvious for treatment of influenza virus infection, especially as combination therapy.
The efforts to develop effective entry inhibitors of influenza virus have been attempted previously by a number of groups. One of the early reports is on triperiden (Fig. 9.7) that was shown to inhibit influenza virus replication at a concentration of 20 μg/ml (Heider et al. 1985). This compound was later shown to inhibit hemolysis of red blood cells and the sensitivity of HA1 to trypsin after low pH treatment of HA (Ghendon et al. 1986). Reassortment of the drug-sensitive strain with a strain that was not sensitive confirmed that the gene sensitive to triperiden was HA. Triperiden-resistant mutations were mapped to HA as well (Prosch et al. 1988). In a more recent report, it was shown that inhibition of influenza virus replication by triperiden may be due to its ability to lower the internal pH in the prelysosomal compartment (Ott and Wunderli-Allenspach 1994). In a later effort, a group in BMS Pharmaceutical Research Institute discovered a novel HA inhibitor (BMY-27709) that has an EC50 of 6–8 μM against influenza viruses that have HA subtypes H1 and H2, but not H3 (Luo et al. 1996). The compound was shown to inhibit virus replication at an early stage and the inhibition was reversible. These data suggest that the inhibitor target is HA. This was further confirmed by the inhibitor-resistant mutations found in HA and inhibition of hemolysis by the compound (Luo et al. 1997). Similarly, a group in Lilly Research Laboratories found a novel inhibitor, a podocarpic acid derivative (180299), by screening a chemical library (Staschke et al. 1998). The EC50 of 180299 is 0.01 μg/ml against A/Kawasaki/86 but ≥10 μg/ml against other trains. The target of action was also confirmed to be HA by inhibition of cell fusion and positions of inhibitor-resistant mutations. By screen, another inhibitor was also discovered, Stachyflin, that has an EC50 in the μM range against H1 and H2 viruses, but not H3 (Yoshimoto et al. 1999). HA as the target for Stachyflin was also confirmed by time of addition, inhibition of hemolysis, and reassortment between subtype H1 and H3. Another group initiated a structure-aided approach to identify inhibitors of HA-mediated membrane fusion (Bodian et al. 1993; Hoffman et al. 1997). The most effective compound identified (S19) has an EC50 of 0.8 μM against influenza virus X-31, and activities on other strains were not reported. What was remarkable about their work is that they found another moderate inhibitor (C22). Unlike other inhibitors that prevented the conformational change of HA, C22 facilitated the conformational change at fusion pH and its effect was irreversible. C22 destabilizes HA and also inhibits hemolysis, fusion, and viral infectivity. The authors concluded that since C22 does not induce the conformational change at neutral pH, it was conceivable that it might facilitate fusion by destabilizing HA as an effector.

The chemical structure of some published fusion inhibitors of influenza virus
The mutations identified in the process of verifying the mechanism of action by the fusion inhibitors of HA may provide insights on how HA fusion may be inhibited. The mutations may occur throughout HA1 and HA2 molecules, but most of them are in HA2. They can generally be classified in two groups. The first group includes the mutations that destabilize HA so the conformational change of HA2 can still occur even in the presence of the inhibitor. These mutations mostly were found in HA2 and occurred more often as found in resistant mutants to BMS-27709, 180299, and S19 (Hoffman et al. 1997; Luo et al. 1997; Staschke et al. 1998). Mutations reduce either the stabilizing interactions with the fusion peptide or the interfaces that stabilize the trimer association of HA. The mutant HAs are therefore more fusogenic at a pH higher than that required by the fusion of wild-type HA. Such mutations in HA were also identified in mutant viruses that are resistant to high concentrations of amantadine, an antiviral compound that elevates intracellular pH (Hoffman et al. 1997; Staschke et al. 1998). These data indicate that the fusion inhibitors discovered previously primarily stabilize HA when binding to HA. The stabilization by these inhibitors makes the conformational change of HA more energetically unfavorable so the chance of fusion is reduced, which leads to reduction of virus replication. The second group of mutations was identified by selection of resistant mutants to C22 that is an effector of the conformational change in HA, instead of an inhibitor (Hoffman et al. 1997). As expected, the mutant HAs have a downward shift in fusion pH, and the inhibitor resistant viruses in the first group are actually more sensitive to C22.
The results support the notion that entry inhibitors can inhibit influenza virus replication effectively. The challenge for developing an antiviral drug that can be used clinically is to discover entry inhibitors that have a high potency against all strains of human influenza virus. Since the conservation in amino acid sequence is low among influenza virus HA, a clear picture on how to discover a commonly efficacious entry inhibitor has not yet emerged. Since HA-mediated membrane fusion is the common mechanism for influenza virus entry, there is the potential to discover an inhibitor that blocks influenza virus entry through the common mechanism, if not a common inhibitor binding site in HA per se.
References
Bodian DL, Yamasaki RB, Buswell RL, Stearns JF, White JM, Kuntz ID (1993) Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 32:2967–2978
Bonnafous P, Stegmann T (2000) Membrane perturbation and fusion pore formation in influenza hemagglutinin-mediated membrane fusion. A new model for fusion. J Biol Chem 275:6160–6166
Bottcher C, Ludwig K, Herrmann A, van Heel M, Stark H (1999) Structure of influenza haemagglutinin at neutral and at fusogenic pH by electron cryo-microscopy. FEBS Lett 463:255–259
Bragstad K, Nielsen LP, Fomsgaard A (2008) The evolution of human influenza A viruses from 1999 to 2006: a complete genome study. Virol J 5:40
Bui M, Whittaker G, Helenius A (1996) Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J Virol 70:8391–8401
Bullough PA, Hughson FM, Skehel JJ, Wiley DC (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43
Chang DK, Cheng SF (2006) pH-dependence of intermediate steps of membrane fusion induced by the influenza fusion peptide. Biochem J 396:557–563
Chang DK, Cheng SF, Kantchev EA, Lin CH, Liu YT (2008) Membrane interaction and structure of the transmembrane domain of influenza hemagglutinin and its fusion peptide complex. BMC Biol 6:2
Chen C, Zhuang X (2008) Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc Natl Acad Sci USA 105:11790–11795
Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC (1998) Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409–417
Chen J, Skehel JJ, Wiley DC (1999) N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc Natl Acad Sci USA 96:8967–8972
Chernomordik LV, Frolov VA, Leikina E, Bronk P, Zimmerberg J (1998) The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol 140:1369–1382
Colman PM, Lawrence MC (2003) The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309–319
Compans RW, Klenk HD, Caliguiri LA, Choppin PW (1970) Influenza virus proteins. I. Analysis of polypeptides of the virion and identification of spike glycoproteins. Virology 42:880–889
Daniels RS, Downie JC, Hay AJ, Knossow M, Skehel JJ, Wang ML, Wiley DC (1985) Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431–439
DeLano WL (2002) The PyMOL Molecular Graphics System. DeLano Scientific, Palo Alto, CA
Doxsey SJ, Brodsky FM, Blank GS, Helenius A (1987) Inhibition of endocytosis by anti-clathrin antibodies. Cell 50:453–463
Floyd DL, Ragains JR, Skehel JJ, Harrison SC, van Oijen AM (2008) Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci USA 105:15382–15387
Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, Wiley DC, Skehel JJ (2004) The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303:1838–1842
Ghendon Y, Markushin S, Heider H, Melnikov S, Lotte V (1986) Haemagglutinin of influenza A virus is a target for the antiviral effect of Norakin. J Gen Virol 67(Pt 6):1115–1122
Giannecchini S, Campitelli L, Bandini G, Donatelli I, Azzi A (2007) Characterization of human H1N1 influenza virus variants selected in vitro with zanamivir in the presence of sialic acid-containing molecules. Virus Res 129:241–245
Ha Y, Stevens DJ, Skehel JJ, Wiley DC (2001) X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc Natl Acad Sci USA 98:11181–11186
Hanson JE, Sauter NK, Skehel JJ, Wiley DC (1992) Proton nuclear magnetic resonance studies of the binding of sialosides to intact influenza virus. Virology 189:525–533
Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, Steven AC (2006) Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci USA 103:19123–19127
Hay AJ, Gregory V, Douglas AR, Lin YP (2001) The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci 356:1861–1870
Hayden F (2009) Developing new antiviral agents for influenza treatment: what does the future hold? Clin Infect Dis 48(Suppl 1):S3–S13
Heider H, Markushin S, Schroeder C, Ghendon Y (1985) The influence of Norakin on the reproduction of influenza A and B viruses. Arch Virol 86:283–290
Hoffman LR, Kuntz ID, White JM (1997) Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: irreversible inhibition of infectivity. J Virol 71:8808–8820
Huang Q, Opitz R, Knapp EW, Herrmann A (2002) Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys J 82:1050–1058
Huang Q, Sivaramakrishna RP, Ludwig K, Korte T, Bottcher C, Herrmann A (2003) Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: stability and energetics of the hemagglutinin. Biochim Biophys Acta 1614:3–13
Hurt AC, Holien JK, Barr IG (2009) In vitro generation of neuraminidase inhibitor resistance in A(H5N1) influenza viruses. Antimicrob Agents Chemother 53:4433–4440
Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL (2006) Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80:7469–7480
Kemble GW, Danieli T, White JM (1994) Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 76:383–391
Khor R, McElroy LJ, Whittaker GR (2003) The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4:857–868
Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS (1998) Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307
Kogure T, Suzuki T, Takahashi T, Miyamoto D, Hidari KI, Guo CT, Ito T, Kawaoka Y, Suzuki Y (2006) Human trachea primary epithelial cells express both sialyl(alpha2-3)Gal receptor for human parainfluenza virus type 1 and avian influenza viruses, and sialyl(alpha2-6)Gal receptor for human influenza viruses. Glycoconj J 23:101–106
Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, Gao Q, Andrews SE, Bandyopadhyay S, De Jesus P, Tu BP, Pache L, Shih C, Orth A, Bonamy G, Miraglia L, Ideker T, Garcia-Sastre A, Young JA, Palese P, Shaw ML, Chanda SK (2010) Human host factors required for influenza virus replication. Nature 463:813–817
Lakadamyali M, Rust MJ, Babcock HP, Zhuang X (2003) Visualizing infection of individual influenza viruses. Proc Natl Acad Sci USA 100:9280–9285
Lear JD, DeGrado WF (1987) Membrane binding and conformational properties of peptides representing the NH2 terminus of influenza HA-2. J Biol Chem 262:6500–6505
Lee KK (2010) Architecture of a nascent viral fusion pore. EMBO J 29:1299–1311
Leikina E, LeDuc DL, Macosko JC, Epand R, Shin YK, Chernomordik LV (2001) The 1-127 HA2 construct of influenza virus hemagglutinin induces cell-cell hemifusion. Biochemistry 40:8378–8386
Leikina E, Markovic I, Chernomordik LV, Kozlov MM (2000) Delay of influenza hemagglutinin refolding into a fusion-competent conformation by receptor binding: a hypothesis. Biophys J 79:1415–1427
Lin T, Wang G, Li A, Zhang Q, Wu C, Zhang R, Cai Q, Song W, Yuen KY (2009) The hemagglutinin structure of an avian H1N1 influenza A virus. Virology 392:73–81
Liu C, Eichelberger MC, Compans RW, Air GM (1995) Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J Virol 69:1099–1106
Liu J, Stevens DJ, Haire LF, Walker PA, Coombs PJ, Russell RJ, Gamblin SJ, Skehel JJ (2009) Structures of receptor complexes formed by hemagglutinins from the Asian Influenza pandemic of 1957. Proc Natl Acad Sci USA 106:17175–17180
Luneberg J, Martin I, Nussler F, Ruysschaert JM, Herrmann A (1995) Structure and topology of the influenza virus fusion peptide in lipid bilayers. J Biol Chem 270:27606–27614
Luo G, Colonno R, Krystal M (1996) Characterization of a hemagglutinin-specific inhibitor of influenza A virus. Virology 226:66–76
Luo G, Torri A, Harte WE, Danetz S, Cianci C, Tiley L, Day S, Mullaney D, Yu KL, Ouellet C, Dextraze P, Meanwell N, Colonno R, Krystal M (1997) Molecular mechanism underlying the action of a novel fusion inhibitor of influenza A virus. J Virol 71:4062–4070
Matlin KS, Reggio H, Helenius A, Simons K (1981) Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91:601–613
Nicholls JM, Chan RW, Russell RJ, Air GM, Peiris JS (2008) Evolving complexities of influenza virus and its receptors. Trends Microbiol 16:149–157
Nunes-Correia I, Eulalio A, Nir S, Pedroso de Lima MC (2004) Caveolae as an additional route for influenza virus endocytosis in MDCK cells. Cell Mol Biol Lett 9:47–60
O’Neill RE, Talon J, Palese P (1998) The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J 17:288–296
Ott S, Wunderli-Allenspach H (1994) Effect of the virostatic Norakin (triperiden) on influenza virus activities. Antiviral Res 24:37–42
Prosch S, Heider H, Schroeder C, Kruger DH (1988) Mutations in the hemagglutinin gene associated with influenza virus resistance to norakin. Arch Virol 102:125–129
Qi L, Kash JC, Dugan VG, Wang R, Jin G, Cunningham RE, Taubenberger JK (2009) Role of sialic acid binding specificity of the 1918 influenza virus hemagglutinin protein in virulence and pathogenesis for mice. J Virol 83:3754–3761
Rosenthal PB, Zhang X, Formanowski F, Fitz W, Wong CH, Meier-Ewert H, Skehel JJ, Wiley DC (1998) Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 396:92–96
Roy AM, Parker JS, Parrish CR, Whittaker GR (2000) Early stages of influenza virus entry into Mv-1 lung cells: involvement of dynamin. Virology 267:17–28
Rust MJ, Lakadamyali M, Zhang F, Zhuang X (2004) Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11:567–573
Sauter NK, Bednarski MD, Wurzburg BA, Hanson JE, Whitesides GM, Skehel JJ, Wiley DC (1989) Hemagglutinins from two influenza virus variants bind to sialic acid derivatives with millimolar dissociation constants: a 500-MHz proton nuclear magnetic resonance study. Biochemistry 28:8388–8396
Schauer R (2009) Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol 19:507–514
Segal MS, Bye JM, Sambrook JF, Gething MJ (1992) Disulfide bond formation during the folding of influenza virus hemagglutinin. J Cell Biol 118:227–244
Shen J, Ma J, Wang Q (2009) Evolutionary trends of A(H1N1) influenza virus hemagglutinin since 1918. PLoS One 4:e7789
Sieczkarski SB, Whittaker GR (2002) Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76:10455–10464
Sieczkarski SB, Whittaker GR (2003) Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 4:333–343
Staschke KA, Hatch SD, Tang JC, Hornback WJ, Munroe JE, Colacino JM, Muesing MA (1998) Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 248:264–274
Stevens J, Blixt O, Tumpey TM, Taubenberger JK, Paulson JC, Wilson IA (2006) Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 312:404–410
Stray SJ, Cummings RD, Air GM (2000) Influenza virus infection of desialylated cells. Glycobiology 10:649–658
Takeda M, Leser GP, Russell CJ, Lamb RA (2003) Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc Natl Acad Sci USA 100:14610–14617
Takemoto DK, Skehel JJ, Wiley DC (1996) A surface plasmon resonance assay for the binding of influenza virus hemagglutinin to its sialic acid receptor. Virology 217:452–458
Tatulian SA, Tamm LK (2000) Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin. Biochemistry 39:496–507
Thompson CI, Barclay WS, Zambon MC, Pickles RJ (2006) Infection of human airway epithelium by human and avian strains of influenza a virus. J Virol 80:8060–8068
Tumpey TM, Maines TR, Van Hoeven N, Glaser L, Solorzano A, Pappas C, Cox NJ, Swayne DE, Palese P, Katz JM, Garcia-Sastre A (2007) A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 315:655–659
Wagner R, Wolff T, Herwig A, Pleschka S, Klenk HD (2000) Interdependence of hemagglutinin glycosylation and neuraminidase as regulators of influenza virus growth: a study by reverse genetics. J Virol 74:6316–6323
Wang Q, Tian X, Chen X, Ma J (2007) Structural basis for receptor specificity of influenza B virus hemagglutinin. Proc Natl Acad Sci USA 104:16874–16879
Wharton SA, Skehel JJ, Wiley DC (1986) Studies of influenza haemagglutinin-mediated membrane fusion. Virology 149:27–35
White J, Helenius A, Gething MJ (1982) Haemagglutinin of influenza virus expressed from a cloned gene promotes membrane fusion. Nature 300:658–659
Wilson IA, Skehel JJ, Wiley DC (1981) Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289:366–373
Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y (2006) Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444:378–382
Yen HL, Webster RG (2009) Pandemic influenza as a current threat. Curr Top Microbiol Immunol 333:3–24
Yoshimoto J, Kakui M, Iwasaki H, Fujiwara T, Sugimoto H, Hattori N (1999) Identification of a novel HA conformational change inhibitor of human influenza virus. Arch Virol 144:865–878
Zambon MC (2001) The pathogenesis of influenza in humans. Rev Med Virol 11:227–241
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Luo, M. (2012). Influenza Virus Entry. In: Rossmann, M., Rao, V. (eds) Viral Molecular Machines. Advances in Experimental Medicine and Biology, vol 726. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-0980-9_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-0980-9_9
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-0979-3
Online ISBN: 978-1-4614-0980-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)