
Abstract
The nucleophosmin (NPM)/nucleoplasmin family of nuclear chaperones has three members: NPM1, NPM2, and NPM3. Nuclear chaperones serve to ensure proper assembly of nucleosomes and proper formation of higher order structures of chromatin. In fact, this family of proteins has such diverse functions in cellular processes such as chromatin remodeling, ribosome biogenesis, genome stability, centrosome replication, cell cycle, transcriptional regulation, apoptosis, and tumor suppression. Of the members of this family, NPM1 is the most studied and is the main focus of this review. NPM2 and NPM3 are less well characterized, and are also discussed wherever appropriate. The structure–function relationship of NPM proteins has largely been worked out. Other than the many processes in which NPM1 takes part, the major interest comes from its involvement in human cancers, particularly acute myeloid leukemia (AML). Its significance stems from the fact that AML with mutated NPM1 accounts for ∼30% of all AML cases and usually has good prognosis. Its clinical importance also comes from its involvement in virus replication, particularly in the era of outbreaks of infectious diseases.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Acute Myeloid Leukemia
- Japanese Encephalitis Virus
- Ribosome Biogenesis
- NPM1 Mutation
- Nucleolar Localization
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Nucleophosmin 1 (NPM1) is also known as nucleophosmin (NPM), nucleolar phosphoprotein B23, and numatrin in mammals, and NO38 in amphibians. NPM1 was first discovered as a nucleolar protein in rat liver cells and Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis (Table 10.1) (Orrick et al. 1973). It was first named as B23 because it was the 23rd protein in region B of the gel slab when the protein spots were numbered in order of decreasing mobility in both electrophoretic dimensions. It serves as a nuclear chaperone and has many other important functions (Grisendi et al. 2006). Nucleophosmin 2 (NPM2), originally called nucleoplasmin, was first identified and purified from the eggs of the African clawed frog Xenopus laevis (Laskey et al. 1978), in which it is the most abundant nuclear protein (Mills et al. 1980). It binds histones and transfers them to DNA, and facilitates the assembly of nucleosomes – the basic building block of chromatin. Nucleophosmin 3 (NPM3) was cloned and characterized as a novel gene (npm3) in mouse, and found to be very similar to human NPM1 and Xenopus npm2 in amino acid sequence, protein features, and exon organization (MacArthur and Shackleford 1997). In the same year, NPM3 was also discovered by sodium dodecyl sulfate polyacrylamide gel electrophoresis as a very acidic protein (NO29) immunolocalized in the nucleoli of Xenopus oocytes, and forming complex with NPM1 (Zirwes et al. 1997). It was proposed to be involved in the assembly of preribosomal particles.
NPM1, NPM2, NPM3, and invertebrate NPM-like proteins form the nucleophosmin/nucleoplasmin family of nuclear chaperones and are found throughout the animal kingdom (Eirín-López et al. 2006; Frehlick et al. 2007). Nuclear chaperones serve to ensure proper assembly of nucleosomes and proper formation of higher order structures of chromatin. In fact, this family of proteins has such diverse functions in cellular processes such as chromatin remodeling, ribosome biogenesis, genome stability, centrosome replication, and transcriptional regulation. Of the members of this family, NPM1 is the most studied because it is often altered in expression levels in tumors and mutated/translocated in hematological malignancies (Grisendi et al. 2006). It is the main focus of this review. NPM2 and NPM3 are less well characterized, and are also discussed wherever appropriate. Invertebrate NPM-like proteins are not discussed in this review.
2 Structure and Expression of NPM Genes
The human NPM1 gene spans a genomic region of about 23 kb at chromosome 5q35 and has 12 exons (Table 10.1). It can be transcribed as three variants. Transcript variant 1 is ubiquitously expressed and is the major and the longest transcript, giving rise to isoform 1 of 294 amino acids. Transcript variant 2 skips an in-frame exon (exon 8) and produces a shorter protein (isoform 2) of 265 amino acids, whose functions and expression pattern are not known. Transcript variant 3 utilizes an alternate 3′ exon (exon 10) and hence produces a 259-amino-acid protein with a C-terminus different from that of isoform 1. The corresponding isoforms 1 and 3 in rat are B23.1 and B23.2, and have different subcellular distribution patterns (Wang et al. 1993). Isoform 1 is localized in the nucleolus (Michalik et al. 1981; Spector et al. 1984) while isoform 3 is found in the nucleoplasm with low expression levels (Dalenc et al. 2002).
The human NPM2 gene is about 12 kb long at chromosome 8p21.3 and has nine exons (Table 10.1). Its single transcript produces a protein of 214 amino acids. NPM2 has a rather restricted tissue distribution and is found in the nucleus of oocytes and eggs only (Laskey et al. 1978; Mills et al. 1980; Burns et al. 2003). The human NPM3 gene is only about 2 kb long at chromosome 10q24.31 and has six exons (Table 10.1). Similar to NPM1, NPM3 is ubiquitously expressed, and is localized to the nucleolus (MacArthur and Shackleford 1997; Zirwes et al. 1997; Shackleford et al. 2001).
3 Structure–Function Relationship of NPM Proteins
The NPM proteins share strong sequence and structural homology. The sequence of NPM proteins can be divided into several domains with distinct biochemical, structural, and functional properties (Fig. 10.1) (Hingorani et al. 2000; Frehlick et al. 2007; Okuwaki 2008). In addition to these modular domains, the NPM proteins also contain several sequence motifs, including the nuclear localization signal (NLS), the nuclear export signal (NES), and the nucleolus localization signal (NoLS) that are critical for the localization of NPM proteins in the nucleolus as well as their nucleo-cytoplasmic shuttling (Grisendi et al. 2006; Frehlick et al. 2007; Okuwaki 2008). In this section, we intend to discuss the structural and biochemical properties of each individual domain and how these properties correlate with its respective function. The 294-amino-acid NPM1 protein is mostly used as the representative example.

Domain organization of NPM proteins. NES nuclear export signal; NLS nuclear localization signal; NoLS nucleolus localization signal; the NPM-ALK fusion protein that is involved in hematological malignancies
3.1 The N-Terminal Core Region (Residues 1–120)
The N-terminal region of NPM1 (residues 1–120) is commonly referred to as the “core” because this region is the most conserved among the NPM family of proteins. This region is largely hydrophobic and folds into a distinct structural domain that is protease resistant (Dutta et al. 2001). This core domain is responsible for oligomerization and the molecular chaperone activity of NPM1 by suppressing the misfolding and aggregation of target proteins in the crowded environment in the nucleolus (Szebeni and Olson 1999). This region can also interact with core histone proteins H2A, H2B, H3, and H4 to function as a histone chaperone and facilitate nucleosome assembly (Okuwaki et al. 2001b). A functional role of this domain in ribosomal biogenesis and p53-related tumor suppression has also been implicated (Colombo et al. 2002; Murano et al. 2008).
The X-ray crystal structure of the human NPM1 core region (residues 9–122) reveals a compact domain consisting of two four-stranded β-sheets packed in a β-sandwich topology (Fig. 10.2a) (Lee et al. 2007). The core region forms a tight pentameric assembly through hydrophobic interactions between the monomeric subunits (Fig. 10.2b). Moreover, in the crystallization environment, two pentameric complexes align along their fivefold symmetry axis and associate in head-to-head fashion to form a decameric assembly (Fig. 10.2c). The monomeric and pentameric structures of human NPM1 core domain are highly similar to that of the core region of several other NPM proteins including the Xenopus NO38 (i.e., npm1) (Namboodiri et al. 2004), the Xenopus nucleoplasmin (i.e., npm2) (Dutta et al. 2001), and the Drosophila nucleoplasmin-like protein (i.e., dNPL) (Namboodiri et al. 2003). All these structures share the same β-sandwich fold with root mean square deviation of ∼1.0 Å for the Cα atoms (Lee et al. 2007). Such an oligomeric assembly observed in crystal structures agrees with previous findings that NPM proteins have a propensity to oligomerize with the benefit of enhanced thermostability (Umekawa et al. 1993; Herrera et al. 1996). A structure-based model of the NPM-histone complex, mainly based on the structures of the Xenopus NO38 and nucleoplasmin core domain, was proposed to consist of the NPM decamer and the histone octamer, with either the H2A-H2B dimer or the H3-H4 tetramer contacting the NPM core on the lateral surface of the decameric ring (Fig. 10.2c) (Dutta et al. 2001; Namboodiri et al. 2004). However, many details of this modeled oligomeric assembly still await further clarification.

Structure of the N-terminal core region of NPM1. (a) Structure of the core region in monomeric form. The dotted line indicates the disordered surface loop. (b) Structure of the core region in pentameric form. The dot in the center indicates the fivefold axis. (c) Structure of the core region in decameric assembly as observed in crystal lattice. The histone-octamer is modeled to contact the lateral surface of NPM1 decameric ring to form the NPM-histone complex. The dotted lines indicate highly flexible and disordered loops. (d) Comparison of human NPM1 decameric complex (colored blue) with that of Xenopus npm1 (colored magenta). The two structures are aligned by superposing on the top pentamer of the decameric ring (left), and the rotational shift for the bottom pentamer as a result of such alignment is illustrated with an arrow (right). The structural files for the N-terminal pentameric domain of human NPM1 (PDB ID 2P1B; Lee et al. 2007) and Xenopus npm1 (PDB ID 1XE0; Namboodiri et al. 2004) are downloaded from the public database RCSB Protein Data Bank (www.rcsb.org), and the figures are prepared using the program CCP4mg (Potterton et al. 2004)
A notable difference between the structure of human NPM1 core domain and structures of the other NPM proteins is the plasticity of the pentamer–pentamer interface observed in crystal lattice. When the decameric structure of human NPM1 core region is superimposed onto that of Xenopus NO38 by aligning one of the pentamers (referred to as the top one), the other pentamer (referred to as the bottom one) in the decameric assembly shows a large rotational offset (∼20°) from that of Xenopus npm1 core (Fig. 10.2d) and a relative smaller rotational offset (∼10°) from that of Xenopus npm2 core (Lee et al. 2007). Such rotational offset does not change the pattern of which monomer of the top pentamer contacts the corresponding monomer of the bottom pentamer, but it does lead to different sets of polar interactions between these monomers to stabilize the pentamer–pentamer interface (Lee et al. 2007). In addition, the molecular composition of the lateral surface for the NPM decamer assembly would be affected by such rotational offset as well. This structural plasticity is likely due to the small interface area between the pentamers (∼560 Å2) and the limited number of residues directly involved in polar interactions at the interface (Lee et al. 2007). The differences in the decameric assembly of human and Xenopus NPM proteins could have a significant implication for their respective histone chaperone function because the histones were proposed in the above-mentioned structural model to contact the lateral surface of the decameric assembly. Indeed, some substrate preferences toward the various core histone proteins have been observed for NPM proteins, with Xenopus npm1 showing preference for the H3-H4 tetramers and the Xenopus npm2 preferring the H2A-H2B dimers (Dutta et al. 2001; Namboodiri et al. 2003).
The stable oligomeric assembly of the N-terminal core region is critical for mediating the interaction between NPM1 and the tumor suppressor ARF, which is the protein translated from the transcript produced by an alternate reading frame of the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene, and is also known as p14ARF in humans and p19Arf in mouse on the basis of their molecular weights (Gallagher et al. 2006). The NPM1–ARF interaction can lead to cell growth arrest and tumor suppression in either p53-dependent manner through the p53-Mdm2 pathway or p53-independent manner by inhibiting ribosome biogenesis and cell proliferation (Bertwistle et al. 2004; Brady et al. 2004; Itahana et al. 2003; Korgaonkar et al. 2005). Several conserved residues critical for NPM1–ARF interaction (Leu102, Gly105, and Gly107) are located on a short GS loop between β7 and β8 strand, in close proximity to the pentamer–pentamer interface of the decameric ring (Fig. 10.2a) (Enomoto et al. 2006). Mutations of these residues (Leu102Ala, Gly105Ala, and Gly107Ala) abolish the NPM1–ARF interaction and lead to loss of the NPM1 oligomeric state, probably by disturbing the pentamer–pentamer interactions and weakening the hydrophobic interactions between the monomer subunits within the pentameric assembly (Enomoto et al. 2006). The monomeric mutated NPM1 is delocalized from the nucleolus to the nucleoplasm and displays increased susceptibility to ubiquitination and proteasomal degradation (Enomoto et al. 2006).
The N-terminal core region domain contains two well-studied NES motifs, both of which are critical for the nucleo-cytoplasmic shuttling of NPM1 (Wang et al. 2005; Maggi et al. 2008). The first motif (residues 42–47) is located on the β3 strand forming part of inter-subunit interface within the pentameric ring (Fig. 10.2a). Mutation of two conserved leucine residues in this sequence motif (Leu42Ala and Leu44Ala) leads to a defective NPM1 that cannot be shuttled out of nucleus (Maggi et al. 2008). Such defect inhibits the nuclear export of both the 40S and 60S ribosomal units, which associate with NPM1 and depend on the shuttling of NPM1 for their export (Maggi et al. 2008). Thus, for this shuttling-defective NPM1 mutant, the available pool of cytoplasmic ribosome units would be reduced and the overall protein synthesis diminished, leading to inhibition of cell proliferation. The second NES motif (residues 94–102) is located on the β7 strand on the outer lateral surface of the pentameric ring. This motif leads to nuclear export of NPM1 by the Ran-Crm1 complex, and enables the association of NPM1 with the centrosome during mitosis to prevent centrosome reduplication (Wang et al. 2005). Removal of this NES motif by deleting residues 94–102 or mutating two conserved leucine residues (Leu100Ala and Leu102Ala) leads to nuclear retention of NPM1 and supernumerary centrosomes (Wang et al. 2005).
3.2 The Acidic Stretches A1 (Residues 121–132) and A2 (Residues 160–188)
Following the N-terminal hydrophobic core region, the NPM1 protein contains a long stretch of unstructured segment enriched in clusters of acidic residues, including the acidic stretch A1 (residues 121–132) and A2 (residues 160–188) (Grisendi et al. 2006; Frehlick et al. 2007; Okuwaki 2008). Dictated by the many acidic aspartate and glutamate residues, the distinct electrostatic property of this region suggests that it can possibly be engaged in interaction with basic proteins such as histones and sperm basic proteins to facilitate nucleosome assembly and chromatin remodeling. Indeed, the acidic stretch A1 was found to be critical for the histone chaperone activity of NPM1. While an NPM1 construct encompassing both the pentameric core domain and the acidic stretch A1 shows ∼97% chaperone activity, the pentameric domain alone shows only 30% chaperone activity and may be incapable of assembling nucleosomes (Hingorani et al. 2000). In the proposed model of NPM–histone complex mentioned above, the acidic stretch A1 was included as a critical element to bind to histone proteins and facilitate the assembly of the NPM–histone complex (Fig. 10.2c) (Dutta et al. 2001; Namboodiri et al. 2004).
This acidic region is also found to be critical for NPM1-mediated viral replication (see Sect. 10.6.1). One study showed that both acidic stretches A1 and A2 are critical for promoting in vitro replication of adenovirus DNA complexed with viral basic core proteins by mediating direct interaction between NPM1 and the viral basic proteins to enable them to serve as molecular chaperones and to facilitate the transfer of viral DNA onto these basic proteins (Okuwaki et al. 2001a; Samad et al. 2007). Similarly, another study demonstrated that the human hepatitis delta virus antigen molecules (HDVAgs) interact with NPM1 to up-regulate NPM1 expression and enhance viral replication (Huang et al. 2001). In vitro and in vivo studies mapped this NPM1–HDVAgs interaction to the N-terminal basic region of HDVAgs and the acidic stretch A1, probably because of their electrostatic complementarity (Huang et al. 2001). Lastly, the acidic region in general has been speculated to contribute to the relatively high affinity of NPM1 for peptides containing sequences of NLSs of the SV40 T-antigen type, such as the HIV Rev protein to stimulate its nuclear import and viral replication (Szebeni et al. 1997).
3.3 The Basic Domain (Residues 189–243)
This region refers to an unstructured segment (residues 189–243) that is enriched in the basic residues lysine and arginine. This basic domain is important for the nucleic acid binding activity of NPM1, likely because of its favorable electrostatic properties to associate with acidic DNA/RNA molecules (Hingorani et al. 2000). Such binding could be positive for NPM1’s function in nucleosome assembly by coordinating the packaging of DNA molecules onto core histone proteins. Moreover, this binding activity would be beneficial to the functional role of NPM1 in ribosomal biogenesis because NPM1 should recognize and associate with preribosomal particles to transport them from nucleus to cytoplasm. In fact, NPM1 has been found to interact with rRNA and several ribonuclear proteins in the 40S and 60S ribosome units (Yu et al. 2006; Maggi et al. 2008). This basic region also mediates the interaction of NPM1 with tumor suppressor p53. The NPM1–p53 interaction leads to enhanced stability of p53 and up-regulation of its tumor suppression function (Colombo et al. 2002).
3.4 The C-Terminal Aromatic Domain (Residues 244–294)
The C-terminal region of NPM1 (residue 244–294) is unique to the isoform 1 of NPM1 (corresponding to B23.1 variant in rat) but absent in isoform 3 (B23.2 in rat), which contains only residues 1–259 of NPM1 isoform 1 (corresponding to residues 1–257 of Npm1 protein in rat) (Chang and Olson 1989). This region contains the nucleolar localization signal (NoLS) that is critical for the nucleus–cytosol shuttling of NPM1 (Hingorani et al. 2000). Furthermore, this region contains several functionally important aromatic residues such as Trp288 and Trp290 that are important for the nucleolar localization of NPM1 (Nishimura et al. 2002) but are frequently mutated in acute myeloid leukemia (AML) (Falini et al. 2006). Such mutated NPM1 proteins are found to be aberrantly localized in the cytoplasm, instead of the nucleus, in leukemic cells (see Sect. 10.5.1.1).
Recently, the 3D structure for the C-terminal aromatic region of NPM1 was solved by nuclear magnetic resonance spectroscopy, which reveals a novel structural folding consisting of three helices packed tightly via a hydrophobic core formed by the conserved aromatic tryptophan and phenylalanine residues (Fig. 10.3) (Grummitt et al. 2008). Several well-conserved aromatic residues, including Trp288 and Trp290, are found to form a hydrophobic core and are critical to the structural folding of this domain. Mutation of these tryptophan residues to alanine results in collapse of the structure and loss of nucleolar localization (Grummitt et al. 2008). This result indicates that the structural integrity of this domain is critical to its nucleolar localization. Several surface lysine residues (Lys248, Lys263, and Lys267) have also been found to be critical to nucleolar localization of NPM1 although they are not critical to the structural integrity of this domain. Thus, these lysine residues could serve a functional, instead of a structural, role for nucleolar localization of NPM1.

Structure of the C-terminal aromatic domain of NPM1. Residues important for nucleolus localization are shown in stick model and colored in the cpk scheme. The structural file for human NPM1 C-terminal aromatic region (PDB ID 2VXD; Grummitt et al. 2008) is downloaded from the public database RCSB Protein Data Bank (www.rcsb.org), and the figure is prepared using the program CCP4mg (Potterton et al. 2004)
4 Physiological Functions of NPM Proteins
Elucidating the physiological functions of NPM1 has become an important and exciting research topic, particularly in the field of cancer research. Studies show that NPM1 is a multifunctional protein widely involved in many vital biological processes. The fact that NPM1 can influence both proliferative and growth-suppressive cellular events brings the scenario to a complex molecular situation that has to be completely resolved by future research efforts. Although the exact biological functions of NPM1 and its involved molecular mechanisms have yet to be fully unraveled, some clues on the physiological functions of NPM1 have been acquired since the discovery of this intriguing protein. This section mainly focuses on the physiological functions of NPM1: molecular chaperone function, ribosome biogenesis, transcriptional regulation, apoptosis inhibition, tumor suppressor modulation, genomic stability maintenance, and cell cycle regulation (Table 10.2). The physiological functions of NPM2 and NPM3 are also briefly described followed by the introduction of the identified posttranslational modifications of NPM proteins.
4.1 NPM1 as a Molecular Chaperone
NPM1 has been shown to have the ability to function as molecular chaperone for both proteins and nucleic acids (Okuwaki et al. 2001b; Szebeni and Olson 1999). The observed molecular chaperone activity of NPM1 is thought to prevent protein aggregation and protein misfolding (Szebeni and Olson 1999). Indeed, NPM1 has been shown to suppress the aggregation and misfolding of target proteins through the structural properties of its N-terminal core domain (Hingorani et al. 2000). During cell cycle, NPM1 works as a histone chaperone to control the assembly and disassembly of chromatin formation (Okuwaki et al. 2001b). To function as a molecular chaperone, NPM1 has to possess nucleo-cytoplasmic shuttling activities attributable to the presence of NES and NLS in NPM1 (Dingwall et al. 1987; Hingorani et al. 2000).
4.2 Involvement of NPM1 in Ribosome Biogenesis
NPM1 has all the necessary machinery features for the processing and assembly of ribosomes. These features include the abundant NPM1 localization in nucleolus and the ability to shuttle between nucleus and cytoplasm to bind nucleic acids and to transport preribosomal particles (Borer et al. 1989; Yun et al. 2003; Wang et al. 1994; Dumbar et al. 1989; Prestayko et al. 1974; Olson et al. 1986). In fact, NPM1 has been found to have intrinsic ribonuclease activity and be able to process preribosomal RNA in the internal transcribed spacer sequence (Savkur and Olson 1998; Herrera et al. 1995). This ribonuclease-mediated processing of ribosomal RNA is facilitated necessarily by the chaperone property of NPM1, which is important in preventing the aggregation of proteins in nucleolus during the process of ribosomal assembly. This proposition is supported by the finding that knockdown of NPM1 changes the ribosome profile and suppression of NPM1 inhibits the processing of preribosomal RNA (Grisendi et al. 2005; Itahana et al. 2003). One of the common morphological features of tumor cells is the enlarged nucleoli. This observation is conceivably linked to the proposed oncogenic role of NPM1 because the frequently observed aberrantly high NPM1 expression in rapidly proliferating tumor cells is generally consistent with the rapid ribosome biogenesis in maintaining the proliferative potential of tumor cells (Ruggero and Pandolfi 2003).
The physiological function of NPM1 in ribosomal biogenesis has been collectively proposed on the basis of the indirect evidence showing the intimate association of NPM1 with the synthetic machinery of ribosome. Nevertheless, this remains to be fully understood as it has been reported that NPM1-deficient embryos can survive to mid-gestation (Colombo et al. 2005; Grisendi et al. 2005), but embryonic lethality at very early stages has been demonstrated in embryos deficient in pescadillo, another nonribosomal protein participating in ribosome biogenesis (Lerch-Gaggl et al. 2002). These findings illustrate the difference of the survival time between NPM1-deficient and pescadillo-deficient embryos and raise the question whether NPM1 might not be an essential protein in the process of ribosomal biogenesis. Nonetheless, it is still possible that other factors such as ribosomal storage might have compensated for the loss of NPM1 in the NPM1-deficient embryos until the stage of mid-gestation (Grisendi et al. 2006).
4.3 Regulation of Gene Transcription by NPM1
NPM1 is also involved in transcriptional regulation and contributes to the cell growth control. The function of transcriptional regulation of NPM1 is related to the interaction of NPM1 with various transcription factors including NFκB, YY1, ARF, and IRF1 (Colombo et al. 2002; Dhar et al. 2004; Inouye and Seto 1994; Kondo et al. 1997; Korgaonkar et al. 2005). NPM1 has been shown to act as a coactivator for NFκB in regulating the expression of antioxidant enzyme MnSOD (Dhar et al. 2004). NPM1 can also alter the transcriptional activity of IRF1 and p53 (Colombo et al. 2002; Kondo et al. 1997). NPM1 is able to form a stable complex with YY1 and, interestingly, the transcriptional repressive function of YY1 can be relieved by the interaction with NPM1 (Inouye and Seto 1994). NPM1 can also modulate the binding of NFκB, E2F1, and pRB for the activation of E2F1 promoter (Lin et al. 2006).
A close interacting relationship is found between NPM1 and AP2α. In retinoic acid-induced differentiation in human leukemia HL-60 cells, a decline in c-myc, NPM1, and its promoter activity is observed (Yung 2004). The transcriptional mechanism underlying the down-regulation of NPM1 during retinoic acid-induced granulocytic differentiation involves dynamic changes in the promoter occupancy of different transcriptional regulators and these include NPM1 and AP2α (Liu et al. 2007a). It is intriguing that NPM1 is demonstrated to be recruited by AP2α to the promoters of some retinoic acid-responsive genes such as b-Myb, HSP60, and p120. These findings illustrate that NPM1 might be able to serve as a negative coregulator during retinoic acid signaling-induced gene expression. All these data indicate the potential important function of NPM1 as a molecular regulator of gene expression at the transcriptional level. By examining the U1 bladder cancer U4 premalignant cells, the intimate relationship of NPM1 with Ras and c-Myc has been further demonstrated in proliferation of cells associated with a high degree of malignancy (Yeh et al. 2006).
4.4 Inhibition of Apoptosis by NPM1
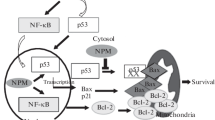
Overexpression of NPM1 can promote cell survival and this is accomplished partly through the inhibition of apoptosis (Ye 2005). Several pieces of evidenc support the anti-apoptotic role of NPM1. First of all, the oncogenic role of NPM1 hinges on its ability in inhibiting apoptosis in response to hypoxia (Li et al. 2004). As NPM1 is a transcriptional target of hypoxia inducible factor 1α (HIF1α), it has been reported that aberrantly increased expression of NPM1 might result in inhibition of the activation of tumor suppressor p53 and thus dampen p53-mediated activation of apoptosis during hypoxia-driven tumor progression (Li et al. 2004). Second, over-expression of NPM1 has been shown to induce resistance to ultraviolet irradiation-induced apoptosis, which is mediated by the tumor suppressor interferon regulatory factor-1 (IRF1, a transcription factor involved in DNA damage-induced apoptosis and cell cycle arrest) in NIH-3 T3 fibroblast cells (Wu et al. 2002b; Kondo et al. 1997). Third, as demonstrated in hematopoietic cells, overexpression of NPM1 has been shown to be related to the suppression of the activation of interferon-inducible, double-stranded RNA-dependent protein kinase (PKR), which normally induces apoptosis (Pang et al. 2003; Jagus et al. 1999). This observation has been associated with the aberrant proliferation of tumor cells. Fourth, NPM1 is shown to have a functional role as the receptor for second messenger phosphatidylinositol(3,4,5)-trisphosphate (PIP3) in nucleus and this NPM1–PIP3 complex is a downstream effector of phosphatidylinositol 3 kinase. The formation of nuclear NPM1–PIP3 complex has been shown to suppress apoptosis by inhibiting the DNA fragmentation activity of the pro-apoptotic factor caspase-activated DNase in PC12 cells treated with nerve growth factor (Ahn et al. 2005). Furthermore, the anti-apoptotic function of NPM1 has been related to its inhibitory effect on tumor suppressor p53. NPM1 interacts with p53 in hypoxic cells to hinder the hypoxia-induced activation of p53 phosphorylation (Li et al. 2004). There is evidence that NPM1 might inhibit the phosphorylation of p53 at serine 15 and thus abrogate the induction of p21 (Li et al. 2005). Last but not least, NPM1 interacts with the pro-apoptotic protein GADD45α, which is responsive to genotoxic stress. While GADD45α lacks a classical NLS, NPM1 has been shown to serve as the molecular chaperone for GADD45α in controlling its subcellular distribution. Most importantly, disruption of NPM1–GADD45α complex has been found to impair the cell cycle arrest and apoptotic functions of GADD45α (Gao et al. 2005).
NPM1 is one of the key elements in the down-regulation of nucleolar function for cellular apoptosis, as exemplified by the death of human promyelocytic leukemia HL-60 cells induced by sodium butyrate (BuONa; a cell growth inhibitor) and vanadate (a tyrosine phosphatase inhibitor) (Liu and Yung 1998). NPM1 is decreased via down-regulated transcriptional process during the BuONa/vanadate-induced apoptosis. As no decrease in NPM1 mRNA and the telomerase activities is observed during the growth arrest by serum-starvation, the decrease in NPM1 mRNA expression and telomerase activity in HL-60 cells subsequent to BuONa/vanadate treatment is suggested to be attributed to cellular apoptosis rather than the growth arrest induced by BuONa/vanadate. The anti-apoptotic role of NPM1 is further substantiated by the data that BuONa-induced apoptosis and inhibition of telomerase activity are significantly potentiated after NPM1 antisense oligomer treatment (Liu and Yung 1998).
It is intriguing that NPM1 might also serve as one of the key elements in the down-regulation of nucleolar function for cellular differentiation. The regulatory role of NPM1 in cellular differentiation has been demonstrated in the granulocytic differentiation of HL-60 cells induced by retinoic acid (Hsu and Yung 1998). NPM1 is reduced via transcriptionally mediated down-regulation during the retinoic acid-induced differentiation. Conversely, there is no decline of NPM1 mRNA during the growth arrest as induced by serum-starvation. These findings suggest that the decrease in NPM1 mRNA expression in HL-60 cells subsequent to retinoic acid treatment is attributed to cellular differentiation rather than the growth arrest induced by retinoic acid. The retinoic acid-induced cellular differentiation is shown to be potentiated by NPM1 antisense oligomer treatment (Hsu and Yung 1998).
4.5 Modulation of Tumor Suppressors by NPM1
NPM1 is also involved in the regulation of activity and stability of some key tumor suppressors such as ARF and p53. NPM1 is associated with the stabilization of ARF by retarding the turnover of ARF and this stabilization is essential to maintain the biological function of ARF (Kuo et al. 2004, 2008). The protective effect of NPM1 on ARF turnover involves both proteasome-dependent and -independent degradation. While ARF is known to suppress cell proliferation through both p53-dependent and -independent pathways (Bertwistle et al. 2004; Brady et al. 2004; Itahana et al. 2003; Korgaonkar et al. 2005), the deficiency of NPM1 has been demonstrated to result in acceleration of tumorigenesis and this is probably attributed to the destabilization of ARF (Sherr 2006). Taken together, NPM1 can possibly work with ARF in mediating the response to oncogenic stimulus. ARF is able to suppress cell proliferation by inhibiting the biogenesis of ribosome through the retardation of the production of rRNA (Sugimoto et al. 2003). Thus, the interaction of NPM1 and ARF in nucleolus is another way of controlling the cell proliferative activities.
Tumor suppressor p53 is another protein that is proposed to be modulated by NPM1, and is an important protein responsible for the prevention of cell growth and cell division when genomic stability is not achieved or DNA integrity is severely damaged (Levine 1997). A putative link has been established between the integrity of nucleolus and p53 stability, in which the nucleolus is believed to play a role in sensing the abundance of p53 in proliferating cells (Rubbi and Milner 2003). Indeed, NPM1 has been shown to promote p53 stability when undergoing nucleoplasmic relocalization (Horn and Vousden 2004). It has been demonstrated that NPM1 in the nucleolus can increase the stability of p53 by suppressing the physical binding interaction between MDM2 and p53 in response to ultraviolet irradiation (Kurki et al. 2004a, b). In response to cellular stress stimulus, disruption of nucleolar integrity induces the translocation of nucleolar NPM1 between subcellular compartments and the relocalized NPM1 can participate in the corresponding reaction mediated by p53 (Rubbi and Milner 2003; Horn and Vousden 2004).
4.6 Role of NPM1 in the Maintenance of Genomic Stability
NPM1 is implicated in the maintenance of genomic stability through participation in DNA repair process and control of cellular ploidy. In response to DNA double-strand breaks, NPM1 has been shown to act as a chromatin-binding factor (Lee et al. 2005). The early response of NPM1 to DNA damage involves rapid transcriptional up-regulation of NPM1 following ultraviolet irradiation (Wu and Yung 2002; Wu et al. 2002a). Increased DNA repair has been shown to be associated with elevated expression of NPM1 (Wu et al. 2002b). NPM1 also works to maintain the genomic stability during the cell cycle through the regulation of centrosome duplication, which is associated with the proposed physiological function of NPM1 in the regulation of cell cycle.
4.7 Regulation of the Cell Cycle by NPM1
The nucleolus undergoes reversible disassembly during mitosis and NPM1 is observed to translocate from nucleolar remnants to the cytoplasm (Hernandez-Verdun and Gautier 1994). NPM1 is found to translocate to the chromosome periphery and the cytoplasmic entities called nucleolus-derived foci (NDF) (Dundr et al. 2000). It then redistributes to the poles of the mitotic spindle to interact with a nuclear matrix protein called NuMA to control the formation of centrosomes in prometaphase and mitotic poles in metaphase (Compton and Cleveland 1994; Zatsepina et al. 1999). The consistent observation that NPM1 is present at the mitotic spindle poles suggests the protective role of NPM1 in preventing hyper-amplification of centrosome to ensure successful progression through G2-M phases (Tokuyama et al. 2001; Zatsepina et al. 1999; Zhang et al. 2004). Indeed, NPM1 is not classified as a centrosomal protein but it has been proposed to be involved in the duplication of centrosomes. This is supported by the data that NPM1 is associated specifically with the CDK2-cyclin E-mediated phosphorylation, which facilitates centrosome duplication (Okuda 2002; Tokuyama et al. 2001; Andersen et al. 2003).
Modification of the phosphorylation state of NPM1 by various protein kinases during cell cycle has been documented (Jiang et al. 2000). For instance, NPM1 has been identified as a substrate of cyclin-dependent kinase (CDK) 2-cyclin E complex in the regulatory process of centrosome duplication (Okuda et al. 2000). NPM1 has also been reported to be phosphorylated by cdc2 kinase during mitosis (Peter et al. 1990) and by nuclear casein kinase 2 during interphase (Chan et al. 1990). NPM1 has been suggested to be the candidate substrate for BRCA1-associated RING domain 1 (BRCA1-BARD1) ubiquitin ligase and the complex of BRCA1-BARD1-NPM1 has been shown to localize at centrosomes during mitosis (Sato et al. 2004). It is also proposed that the ubiquitylational interaction of NPM1 with BRCA1-BARD1 might be an important process in maintaining the integrity of spindle poles and genomic integrity (Grisendi et al. 2006).
4.8 Physiological Functions of NPM2 and NPM3
NPM2 can bind to histone proteins and is thus proposed to mediate the assembly of nucleosomes from histones and DNA (Earnshaw et al. 1980; Laskey et al. 1978). NPM2 has also been found to be involved in facilitating the postfertilization decondensation and remodeling of paternal chromosome by its binding activities with sperm nuclear basic proteins (Philpott and Leno 1992).
NPM3 is involved in the biogenesis of ribosomal RNA by interacting with NPM1 (Huang et al. 2005). It has been demonstrated that overexpression of NPM3 decreased the rates of pre-rRNA synthesis and processing, but overexpression of a NPM3 mutant that did not interact with NPM1 did not change the pre-rRNA synthesis and processing (Huang et al. 2005). Moreover, the expression level of NPM3 has been shown to be correlated with the process of decondensation of paternal chromosome after fertilization (McLay and Clarke 2003). Intriguingly, NPM3 has also been found to be associated with the histone tail peptides and serves as a histone-binding protein in mouse embryonic stem cells (Motoi et al. 2008). Thus, NPM3 is believed to be a chromatin-remodeling protein responsible for the unique chromatin structure and replicative capacity of embryonic stem cells. Recently, NPM3 was shown to interact with all the individual core histones and was able to enhance transcription via the modulation of the histone chaperone activities of its interacting partner NPM1 in vitro (Gadad et al. 2010).
4.9 Posttranslational Modification of NPM Proteins
Some physiological functions of NPM1 are regulated through posttranslational modification mechanisms including phosphorylation, dephosphorylation, acetylation, poly(ADP-ribosyl)ation, ubiquitination, and sumoylation (Table 10.2). However, the complete profile of posttranslational modifications of NPM1 remains to be fully elucidated.
For phosphorylation of NPM1, several kinases have been identified. Phosphorylation of NPM1 by cyclin E/cdk2 during G1 phase has been documented and this may be related to the initiation of centrosome duplication by dissociating NPM1 from centriole (Tarapore et al. 2006; Tokuyama et al. 2001). The RNA-binding activity of NPM1 is diminished after cdc2-mediated phosphorylation of Thr199 of NPM1 during mitosis, and this is suggested to link to the disassembly of nucleolus by disrupting the RNA-protein binding interaction of NPM1 (Hisaoka et al. 2010; Okuwaki et al. 2002). Phosphorylation of Thr199 has also been implicated in inhibiting GCN5-mediated histone acetylation (Zou et al. 2008). During mitosis, NPM1 is found to be phosphorylated by Polo-like kinase 1 (Plk1), which might be an important event in mediating mitosis (Zhang et al. 2004). During interphase, NPM1 has also been reported to be phosphorylated by casein kinase 2 (CK2) and this is thought to have a role in regulating the nucleolar structure by modulating the dynamic localization of NPM1 between nucleolus and nucleoplasm (Szebeni et al. 2003; Negi and Olson 2006). Dephosphorylation can be another mechanism in regulating the functions of NPM1. A serine/threonine protein phosphatase called PP1β has been shown to dephosphorylate NPM1 in response to DNA damage during ultraviolet irradiation and this process is suggested to facilitate the DNA repair process (Lin et al. 2010).
Acetylation of NPM1 increases its binding affinity to histone and this has been suggested to be involved in the NPM1-mediated regulation of chromatin transcription (Swaminathan et al. 2005). While histone acetyltransferase p300 activates transcription by acetylating the histones and “loosening” the tightly packed chromatin structure, the p300 enzyme also leads to acetylation of NPM1. Such acetylation potentiates the activating effect of p300 on transcription activation by ∼fourfold. In vitro experiments have mapped the acetylation sites of NPM1 mostly to the C-terminal region (Lys212, Lys229, Lys230, Lys248 or Lys250, Lys257 and Lys292), which need to be confirmed by further in vivo experiments.
Factors involved in poly(ADP-ribosyl)ation such as poly(ADP-ribose) polymerase 1 (PARP1) and PARP2 have been shown to have association with NPM1 (Meder et al. 2005). Poly(ADP-ribosyl)ation of NPM1 might contribute to the formation of chromatin because PARP1 can serve as a molecular linker that regulates the structure of chromatin (Kim et al. 2004). NPM1 has been shown to be the ubiquitination substrate of BRCA1-BARD1 ubiquitin ligase, but intriguingly the product is not targeted for proteasome-dependent protein degradation unless destabilized by tumor suppressor ARF (Sato et al. 2004; Itahana et al. 2003). Sumoylation is shown to be another mechanism that can modulate the activities of NPM1. In particular, NPM1 has been demonstrated to be sumoylated by ARF and this can increase the stability and modulate the subcellular localization of NPM1 (Liu et al. 2007b; Tago et al. 2005).
The biological activities of NPM2 are also regulated via phosphorylation. The activity of NPM2 that binds and removes sperm basic proteins, and replaces them with histones has been found to depend on the massive hyperphosphorylation of NPM2 that occurs when oocytes mature into eggs (Leno et al. 1996). The hyperphosphorylation of NPM2 is proposed to modulate the rapid changes in chromatin structure that accompany early development in Xenopus. The function of NPM2 to exchange the H2A-H2B heterodimers for sperm-specific proteins is shown to be mediated by adding 14–20 phosphates to each NPM2 monomer (Cotten et al. 1986).
5 Alteration of NPM1 in Human Cancers
Overexpression of NPM1 in general promotes cell growth and proliferation, particularly through enhancing ribosome biogenesis (see Sect. 10.4.2) (Grisendi et al. 2006). Another main effect of NPM1 overexpression is the inhibition of apoptosis (see Sect. 10.4.4) via several different pathways (Grisendi et al. 2006; Ye 2005). As such, NPM1 has been implicated in tumorigenesis. Indeed, NPM1 is overexpressed in many tumors of different origins (Table 10.2): gastric (Tanaka et al. 1992), colon (Nozawa et al. 1996), liver (Yun et al. 2007), breast (Skaar et al. 1998), ovarian (Shields et al. 1997), prostate (Léotoing et al. 2008; Subong et al. 1999), bladder (Tsui et al. 2004), thyroid (Pianta et al. 2010), brain (Gimenez et al. 2010), and multiple myeloma (Weinhold et al. 2010). In particular, NPM1 overexpression may be correlated with clinical features in some cases. Overexpression of NPM1 in hepatocellular carcinoma was found to be correlated with clinical prognostic parameters such as serum alpha fetal protein level, tumor pathological grading, and liver cirrhosis (Yun et al. 2007) – suggesting the potential of NPM1 overexpression as a marker of hepatocellular carcinoma. NPM1 overexpression was associated with recurrence and progression of bladder cancer (Tsui et al. 2004). Overall, the observation that NPM1 overexpression promotes tumor development tends to suggest its role as a proto-oncogene.
Genetic alteration of the NPM1 gene was not found in common solid cancers including lung, hepatocellular, breast, colorectal, and gastric carcinomas (Jeong et al. 2007). However, the NPM1 gene is a common target for genetic alteration in hematological malignancies (lymphomas and leukemias) (Naoe et al. 2006; Falini et al. 2007b; Rau and Brown 2009). The genetic alterations include frameshift mutations, translocations, and deletions (Table 10.2). The main focus of this section is on the NPM1 gene mutations in humans.
5.1 Acute Myeloid Leukemia Carrying Cytoplasmic NPM (NPMc+ AML)
The breakthrough in this field came with the first report by Falini et al. (2005) that aberrant cytoplasmic localization (instead of nucleolar) of the NPM1 protein (NPMc+) in leukemic blast cells was due to frameshift mutations in exon 12 of the NPM1 gene in patients with AML carrying a normal karyotype. NPMc+ AML accounts for ∼30% of all cases of adult AML, or ∼60% of all cases of adult AML with normal karyotype (Falini et al. 2007b; Rau and Brown 2009). The significance of the finding is that NPMc+ due to NPM1 frameshift mutations is the single most common somatic mutation in adult AML. Of note is the less frequent occurrence (∼7%) of NPMc+ in AML in children (Falini et al. 2007b; Rau and Brown 2009). This difference may reflect the difference in molecular pathogenesis of AML carrying normal karyotype in adults and children.
5.1.1 The NPM1 Mutations Producing NPMc+
A recent compilation documents over 50 reported somatic NPM1 mutations that include insertions, insertions and deletions (indels), base substitutions, and their combinations (Rau and Brown 2009). About 50% of the mutations are 4-base insertions between the second and the third base of the Trp288 codon (TG^G). About 20% of the mutations are insertions of 4–14 bases between Gln289 and Trp290 codons (CAG^TGG). Another 20% are insertions of 8–12 bases between the first and the second base of Trp290 codon (T^GG) followed by deletions of 2–5 bases. All these mutations produce a shift in the reading frame of the transcript from the point of insertion or deletion. Many more mutations are expected to be discovered. However, the most common mutation is a duplication (a type of insertion) of a 4-base sequence TCTG at positions 956–959 of the reference sequence (NM_002520, Table 10.1), and accounts for 70–80% of adult NPMc+ AML. This was designated as mutation A by Falini et al. (2005). About 15% of adult NPMc+ AML cases are due to mutations B (CATG insertion) or D (CCTG insertion) at the same position. The remaining mutations are all rare. Interestingly, a genome-wide computational analysis indicated that the generation of a new NES motif (see below) by a duplication of the TCTG sequence was unique to the NPM1 mutation – a genetic event specific to AML (Liso et al. 2008).
Despite such heterogeneity at the DNA sequence level, the mutations produce two alterations at the C-terminus of the mutated NPM1, both of which are crucial to the aberrant export of the mutated protein from the nucleolus to the cytoplasm (Falini et al. 2006). The first critical alteration is the loss of Trp288 and Trp290 residues or just Trp290 alone, which are essential to the nucleolar localization of the wildtype NPM1 (Nishimura et al. 2002). Loss of these tryptophan residues disrupts the triple helix structure of the NPM1 C-terminal domain (Fig. 10.3; see Sect. 10.3.4) and thus greatly reduces the NPM1 localization in the nucleolus (Grummitt et al. 2008). The second critical alteration is the generation of a new leucine-rich NES motif in the new C-terminus (Nakagawa et al. 2005), in addition to the original two NES motifs (residues 42–47 and 94–102) in the N-terminal core region (see Sect. 10.3.1).
Insertion in between the bases of the Trp288 codon results in the loss of both tryptophan residues while insertion (with or without concomitant deletion) at positions after the Trp288 codon removes the Trp290 residue only. There is a strong correlation between the loss of one or two tryptophan residues and the type of new NES motif generated at the C-terminus. Loss of both tryptophan residues is always found with the common NES motif Leu-xxx-Val-xx-Val-x-Leu, where x is any amino acid. On the contrary, loss of Trp290 alone is always found with the much less frequent variant NES motif Leu-Trp xx-X-xx-Val-x-Leu, where X replaces the Val residue and can be leucine, methionine, phenylalanine, or cysteine (Rau and Brown 2009). Experimentally, the variant NES motifs were found to provide a much stronger force than the common NES motif in driving the Trp288-containing mutated NPM1 from the nucleolus to the cytoplasm (Bolli et al. 2007). Tested with the same experimental system, the physiological N-terminal NES motifs were found to be weak in transporting the wildtype NPM1 protein to the cytoplasm, and this explains the dominant localization of the wildtype NPM1 protein in the nucleolus. The artificial combination of a common weak C-terminal NES motif with a Trp288-containing NPM1 mutant localized the mutated protein mainly in the nucleoplasm and nucleolus with much less export to the cytoplasm. Intriguingly, a weak C-terminal NES motif together the retention of the Trp288 residue has never been detected in any primary AML samples. Therefore, this strongly suggests that cytoplasmic mislocalization of the mutated NPM1 protein is critical to the development of AML (Bolli et al. 2007).
The critical role of NPMc+ in the development of AML carrying normal karyotype is further supported by the report of NPMc+ generated by rare mutations found outside exon 12, namely, a mutation affecting the splicing donor site of exon 9 (Mariano et al. 2006) and two different insertions in exon 11 (Albiero et al. 2007; Pitiot et al. 2007). In all three cases, the mutations produce truncated mutated NPM1 proteins and hence abolish both Trp288 and Trp290 residues, and simultaneously create new functional NES motifs at the new respective C-termini (Mariano et al. 2006; Albiero et al. 2007; Falini et al. 2007a). In other words, these rare mutants utilize the same mechanism of transporting the mutated NPM1 to the cytoplasm as those mutations occurring in exon 12.
In NPMc+ AML, the leukemic blast cells are heterozygous with one mutated NPM1 allele and one wildtype allele (Falini et al. 2007b). While the mutated NPM1 protein is strictly localized to the cytoplasm, the wildtype NPM1 protein can be detected in both nucleoplasm and cytoplasm (Falini et al. 2006; Bolli et al. 2009). All mutated NPM1 protein retains the N-terminal oligomerizaton domain (see Sect. 10.3.1), and hence can form heterodimers with wildtype NPM1 protein. As such, mutated NPM1 protein can recruit wildtype NPM1 protein from nucleolus to nucleoplasm and cytoplasm. Indeed, in vitro transfection studies demonstrated the coimmunoprecipitation of mutated and wildtype NPM1 proteins.
5.1.2 Putative Mechanisms Leading to NPMc+ AML
Cytoplasmic localization of mutated NPM1 protein is believed to play a critical role in leukemogenesis. However, how somatic NPM1 frameshift mutations lead to NPMc+ AML remains elusive. The putative underlying mechanisms can be explored from two perspectives (Falini et al. 2007c, 2011). First, the remaining single copy of wildtype NPM1 allele produces wildtype NPM1 protein, which is less than that in normal counterparts and is also dislocated to the nucleoplasm and cytoplasm as a result of forming heterodimers with mutated NPM1 protein. Second, the mutated NPM1 allele produces mutated NPM1 protein, which is dislocated to the cytoplasm by its very nature, may recruit and hence dislocate other interacting nuclear proteins to the cytoplasm, and may also interact with other new partners in the cytoplasm.
Mutant mice with only one functional Npm1 gene (Npm1 +/−) showed greater instability in their genome and developed a hematological syndrome analogous the myelodysplastic syndrome in humans (Grisendi et al. 2005). When compared to wildtype mice (Npm1 +/+), Npm1 +/− mice showed a much higher frequency of developing malignancies including hematological malignancies, particularly myeloid malignancies (Sportoletti et al. 2008). In addition, chromosomal abnormalities were also consistently found in these mice. This shows that Npm1 is a haploinsufficient tumor suppressor in hemopoiesis. Given that NPMc+ AML is mainly found in patients with normal karyotype, factors other than haploinsufficiency must also contribute to the development of AML.
Removal of the critical C-terminal tryptophan residues and generation of a new C-terminal NES motif dictate the localization of the mutated NPM1 protein in the cytoplasm, instead of in the nucleolus (Falini et al. 2006) (see Sect. 10.5.1.1). Intriguingly, mutated NPM1 protein may still be able to interact with other nuclear proteins that interact with the wildtype NPM1 protein in normal cells, and dislocate them to the cytoplasm. Indeed, at least four such nucleolar/nuclear interacting partners have been found to interact with the mutated NPM1 protein, be dislocated to the cytoplasm, and hence have their physiological functions abrogated: mouse p19Arf and human p14ARF (den Besten et al. 2005; Colombo et al. 2006; Bolli et al. 2009), hexamethylene bis-acetamide-inducible protein 1 (HEXIM1) (Gurumurthy et al. 2008), the F-box protein Fbw7γ (Bonetti et al. 2008), and Miz1 (Wanzel et al. 2008). Attenuation of the functions of these interacting proteins are suggested to contribute to the oncogenic effect of NPMc+, as briefly explained below one by one. First, ARF is a well-known tumor suppressor and is stabilized by wildtype NPM1 protein in the nucleolus (Gallagher et al. 2006; Sherr 2006). In vitro experiments have shown that mutated NPM1 protein can interact directly with ARF and shuttle it to the cytoplasm, but cannot protect it from degradation (den Besten et al. 2005; Colombo et al. 2006). Second, HEXIM1 is an inhibitor of the positive transcription elongation factor b (P-TEFb), which is itself an important transcriptional regulator of the enzyme RNA polymerase II (Dey et al. 2007). Wildtype NPM1 negatively regulates HEXIM1 via proteasome-mediated degradation while mutated NPM1 associates with and shuttles HEXIM1 to the cytoplasm and hence promotes P-TEFb-mediated transcription in the nucleus (Gurumurthy et al. 2008). Third, the F-box protein Fbw7γ is a component of the E3 ligase complex, which ubiquitinates and degrades the oncoprotein c-Myc via the proteasome pathway (Welcker and Clurman 2008). Wildtype NPM1 protein localizes and stabilizes Fbw7γ in the nucleolus, and hence regulates the turnover of c-Myc (Bonetti et al. 2008). Mutated NPM1 protein interacts with Fbw7γ and dislocates it to the cytoplasm, where it is degraded. As a result, c-Myc is stabilized – a situation that reflects the oncogenic potential of NPMc+ (Bonetti et al. 2008). Fourth, Miz1 is a Myc-associated zinc-finger protein and, when bound to Myc, enables Myc to suppress transcription of the genes encoding the cell cycle inhibitors p15Ink4b and p21Cip1 (Adhikary and Eilers 2005). Wildtype NPM1 localizes Miz1 to the nucleolus and is an essential coactivator of Miz1. However, mutated NPM1 protein re-directs Miz1 to the cytoplasm and exhibits dominant-negative effect on Miz1 (Wanzel et al. 2008). Thus, disruption of Miz1 function may contribute to the transforming potential of NPMc+.
NPMc+ may acquire new function in its new environment – the cytoplasm. This is indeed the case. Mutated NMP1 directly interacts with the active cell-death proteases caspase 6 and caspase 8, and inhibits their activities, and thereby protects the cells from apoptosis (Leong et al. 2010). In addition, mutated NPM1 also suppresses myeloid differentiation mediated by caspase 6 and caspase 8. This new data provide the first evidence for the myeloid-restricted leukemogenic property of NPMc+.
In transgenic zebrafish embryos with forced ubiquitous expression of human NPMc+, primitive early myeloid cells expand in numbers (Bolli et al. 2010). There are also increased numbers of definitive erythromyeloid progenitors in the posterior blood island and hematopoietic stem cells in the aorta ventral wall. In transgenic mice expressing human NPMc+ under the influence of a myeloid-specific promoter, expansion of myeloid cells is noted in bone marrow and spleen (Cheng et al. 2010). However, both transgenic models show no evidence of AML. This suggests that the current animal models are not adequate and may not mimic the condition in the human NPMc+ AML.
5.1.3 Cell of Origin in NPMc+ AML
Wildtype NPM1 is predominantly located in the nucleolus while the mutated NPM1 is aberrantly localized in the cytoplasm (NPMc+). Because of their uniqueness, the mutation and the cytoplasmic localization of the mutated protein can be used as clonal markers to study the cell lineage involved in NPMc+ AML. Clonal NPM1 mutations are found in myeloid, monocytic, erythroid, and megakaryocytic cells, but not in fibroblasts and endothelial cells (Pasqualucci et al. 2006). In addition, two or more myeloid hemopoietic cell lineages are affected in about 62% of NPMc+ AML cases while the remaining 38% involve only one myeloid cell lineage (Pasqualucci et al. 2006). On the other hand, B and T lymphoid cells are not part of the mutated clones in NPMc+ AML (Martelli et al. 2008). This indicates that NPMc+ AML arise from a common myeloid or an earlier progenitor that is incapable of differentiating into lymphoid lineages.
The leukemic blast cells are negative for CD34 (i.e., <10% CD34+ cells) in over 90% of NPMc+ AML cases (Falini et al. 2005, 2011). The surface marker CD34 is typically present on hematopoietic stem cells. This raises the question whether the NPM1 mutation arises in a CD34− multipotent hemotapoietic progenitor (Engelhardt et al. 2002) or whether there exists a small pool of CD34+/CD38− NPM1-mutated progenitor. CD34+/CD38− cells usually contain the leukemia stem cells that are capable of propagating and maintaining the leukemia phenotype in immuocompromised mice (Estrov 2010). Indeed, CD34+ cells from NPMc+ AML carry the NPM1 mutation and, when transplanted into immunocompromised mice, generate a leukemia phenotype that is the same as the original patient’s disease in all aspects (Martelli et al. 2010). On the other hand, the evidence for the engraftment capability of CD34− cells from NPMc+ AML is less consistent (Martelli et al. 2010; Taussig et al. 2010). These findings may reflect the heterogeneity of leukemia stem cells in NPMc+ AML.
Leukemic blast cells from NPMc+ AML show characteristic gene expression signature and microRNA signature. In general, gene expression profiling shows down-regulation of CD34 and up-regulation of several members of the homeodomain-containing family of transcription factors, which include HOX genes and TALE genes (Alcalay et al. 2005; Andreeff et al. 2008; Becker et al. 2010; Mullighan et al. 2007; Verhaak et al. 2005). Intriguingly, HOX and TALE genes are known to be important in the maintenance of stem cells – a finding supporting that the cell of origin for NPMc+ AML is a multipotent hematopoietic progenitor. On the other hand, unique microRNA signature includes up-regulation of miR-10a, miR-10b, miR-196a, miR-196b, several members of let-7, and miR-29 families (Debernardi et al. 2007; Garzon et al. 2008; Jongen-Lavrencic et al. 2008), and down-regulation of miR-204 and miR-128a (Garzon et al. 2008). It is of interest to note that miR-10a, -10b, 196a, and -196b are located within the genomic cluster of the HOX genes (Jongen-Lavrencic et al. 2008). Moreover, miR-204 has been shown to down-regulate HOXA10 and MEISI genes (Garzon et al. 2008), a finding linking the down-regulation of miR-204 to the up-regulation of HOXA10 and MEIS1 in NPMc+ AML.
5.1.4 Distinctive Features of NPMc+ AML
Since the first report of NPMc+ AML in 2005, many studies have been done on this group of acute leukemias. It was listed as a new provisional entity (AML with mutated NPM1) under the category of “AML with recurrent genetic abnormalities” in the 2008 World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissue (Swerdlow et al. 2008). As a group, NPMc+ AML has many distinctive features. NPM1 mutations are unique to AML, usually de novo AML, (Falini et al. 2005, 2007c; Liso et al. 2008) and mutually exclusive of other “AML with recurrent genetic abnormalities” listed in the 2008 WHO Classification (Falini et al. 2005, 2008b). They are usually detected in all cells of the leukemic population and are stable over the course of the disease (Chou et al. 2006; Falini et al. 2008a). As such, monitoring of minimal residual disease can easily be achieved by detection of the NPM1 mutations with real-time quantitative polymerase chain reaction assay (Wertheim and Bagg 2008). NPM1 mutations appear to precede other associated mutations like fms-like tyrosine kinase internal tandem duplication (FLT3-ITD), which is found in 40% of NPMc+ AML (Thiede et al. 2006; Gale et al. 2008). Moreover, NPMc+ AML cells have distinct gene and microRNA expression profiles (see Sect. 10.5.1.3). Taken together, these features suggest that the NPM1 mutation is a founder genetic alteration in NPMc+ AML (Falini et al. 2011).
NPMc+ AML is more frequent in adults (∼30% of cases) than in children (∼7% of cases) (Falini et al. 2005; Cazzaniga et al. 2005; Brown et al. 2007). Interestingly, no NPM1 mutation has been detected in AML patients younger than 3 years old (Brown et al. 2007). The type of NPM1 mutations is also different in adult and childhood AML (Thiede et al. 2007). NPMc+ AML is more common in AML patients with normal karyotype (∼85% of cases), and frequently involves multiple lineages (Falini et al. 2005, 2007c). It shows good response to induction therapy, and the prognosis is relatively good in the absence of FTL3-ITD mutations (Falini et al. 2005, 2007c, 2011).
5.2 Lymphomas and Leukemias Carrying NPM1 Gene Translocations
The NPM1 gene at chromosome 5q35 is translocated in anaplastic large-cell lymphoma (ALCL) and in rare variants of AML. Translocation produces an oncogenic fusion protein and a reduced level of the wildtype NPM1 protein encoded by the remaining copy of the functional allele (heterozygosity). The fusion protein is made up of the N-terminus of the NPM1 protein and the C-terminus of the partner protein encoded by the other gene involved in the translocation. The role of the NPM1 moiety in these fusion proteins has not been fully elucidated although it may just serve to promote heterodimer formation, and hence shuttle the fusion protein to the nucleus.
ALCL is a T-cell lymphoma characterized by CD30 expression (Falini 2001; Falini et al. 2007b). It accounts for ∼3% of adult non-Hodgkin’s lymphoma and 10–30% of childhood lymphoma. About 60% of ALCL cases express the tyrosine kinase gene ALK (anaplastic lymphoma receptor tyrosine kinase) and are known as ALK+ ALCL. In general, ALK+ ALCL show good response to induction therapy and has good prognosis. The majority (∼85%) of such cases carry the t(2;5)(p23;q35) chromosome translocation, which joins the NPM1 gene on 5q35 with the ALK gene on 2p23 to produce the chimeric gene NPM1-ALK and hence the fusion protein. The remaining 15% are heterogeneous at the molecular level because the translocations involve other chromosomal partners. The NPM1-ALK fusion protein consists of the N-terminal part of the NPM1 protein (the first 116 amino acids; carrying the oligomerization domain) and the entire cytoplasmic domain of the ALK protein (the last 563 amino acids; carrying the catalytic domain). Through the oligomerization domain, the NPM1-ALK fusion proteins form homodimers with each other, and heterodimers with the wildtype NPM1 protein. Because of the NPM1 promoter, the fusion protein is ectopically expressed in lymphoid cells and the constitutive activation of the tyrosine kinase domain is thought to contribute to the tumor formation. The fusion protein is expressed in the cytoplasm as expected but is also unexpectedly localized in the nucleus because of the shuttling of the heterodimers composed of the fusion protein and the wildtype NPM1, which still possesses the nucleus localization signal and hence imports the heterodimers into the nucleus.
Acute promyelocytic leukemia (APL) is characterized by a maturational block at the promyelocytic stage and classically carries the t(15;17) translocation, which generates the fusion protein PML-RARA (Zelent et al. 2001). Chromosomal translocation t(5;17)(q35;q12) fuses the NPM1 gene at 5q35 to the retinoic acid receptor alpha (RARA) gene at 17q12, and produces the fusion protein NPM1-RARA (Falini et al. 2007b). This translocation is extremely rare and has so far been reported in a few children with APL. Leukemic cells express NPM1-RARA fusion protein, and its reciprocal, the wildtype NPM1 and the wildtype RARA. The fusion protein affects the expression of retinoid-responsive genes, disrupts the retinoic acid signaling pathway, and arrests myeloid differentiation at the promyelocytic stage. Like other APL, APL with t(5;17) shows good response to differentiation therapy with all-trans retinoic acid.
The chromosomal translocation t(3;5)(q25;q35) is found in myelodysplastic syndrome and in <1% of AML (Raimondi et al. 1989; Falini et al. 2007b). It fuses the NPM1 gene at 5q35 to the myelodysplasia/myeloid leukemia factor 1 (MLF1) gene at 3q25. The fusion protein is composed of the N-terminal portion of the NPM1 protein and almost the entire MLF1 protein (only without the first 16 amino acids). Since wildtype MLF1 is not expressed in normal hematopoietic tissues, it is speculated that the NPM1-MLF1 fusion protein promotes malignant transformation via ectopic expression in hematopoietic cells (Hitzler et al. 1999). Like the NPM1-ALK fusion protein, the NPM1-MLF1 fusion protein is expectedly expressed in the cytoplasm, and unexpectedly in the nucleus.
6 Interactions Between Viruses and NPM1
Viruses are obligate intracellular parasites with small-sized genomes (in the form of either DNA or RNA) and very limited coding capacities, and hence their replication and metabolic activities rely on the host cellular machineries (Flint 2000). Most DNA viruses, negative-sense RNA viruses with segmented genomes, and retroviruses replicate in the nucleus and frequently interact with nuclear or nucleolar proteins. In contrast, most positive-sense RNA viruses replicate in the cytoplasm and are not much dependent on the nucleus. However, there are growing evidences that this group of viruses also relies on the nucleus because the proteins involved in viral replication and assembly are localized in the nucleolus (Mai et al. 2006; Perkins et al. 1989; Tamini et al. 2005; Tsuda et al. 2006). Interaction between viruses and nucleus requires trafficking of viral products in and out of the nucleus.
Apart from the diverse functions discussed in Sect. 10.4, NPM1 is also known to affect viral replication and assembly during infection (Hiscox 2007). More importantly, such interaction often results in dislocation and loss of normal functions of nucleolar proteins. This consequently leads to disruption of normal host cell functions. Most of the published studies concerning virus–NPM1 interaction focus on NPM1.1 (or B23.1), while the role of NPM1.3 (or B23.2) in viral activities is not well-understood. This section reviews some examples of virus–NPM1 interaction and discusses the influence of such interaction on the viruses and the host.
6.1 Influence of Virus–NPM1 Interaction on Virus Replication Cycle
The role of NPM1 in transporting human immunodeficiency virus type 1 (HIV-1) proteins to nucleolus is well-documented. HIV is the causative agent of acquired immunodeficiency syndrome (AIDS), and possesses two identical copies of positive-sense RNA genomes. Replication of HIV-1 is a complicated process and involves both nucleus and cytoplasm. During replication, the RNA genome is being reverse-transcribed into DNA in the cytoplasm and transported to the nucleus, where the DNA is transcribed into mRNA and the latter then returns to the cytoplasm for subsequent translation. The HIV-1 regulatory protein Rev is involved in the export of partially spliced or unspliced mRNA from the nucleus (Perkins et al. 1989). As Rev is localized in the nucleolus, it was speculated that certain host factors would be involved in the transportation of Rev in and out of the nucleolus. Using affinity chromatography, Fankhauser et al. (1991) confirmed the participation of NPM1 in the transit of Rev between cytoplasm and nucleus, which allowed for further rounds of export of HIV-1 mRNA. Apart from Rev, NPM1 also localizes the HIV Tat protein into nucleolus (Li 1997), where Tat will recruit cellular cofactor (positive transcription elongation factor b or P-TEFb) and transactivate proviral DNA transcription. This process is critical for HIV replication.
Japanese encephalitis virus (JEV) can cause acute encephalitis in humans. It belongs to the same Flaviviridae family as hepatitis C virus (HCV). It is transmitted among mosquitoes and pigs, and transmission to humans may occur when the number of infected mosquito vectors increases to a very high level. The RNA genome encodes the envelope, structural proteins, as well as core and the non-structural proteins. Similar to HCV and other flaviviridae, JEV core protein is localized in the cytoplasm and nucleus (Bulich and Aaskov 1992; Tsuda et al. 2006). According to mutation and animal inoculation studies, localization of core protein in the nucleus is crucial to the replication of JEV (Mori et al. 2005). During JEV infection, amino acids Gly42 and Pro43 of the JEV core protein interact with the N-terminal region of NPM1, and this interaction results in transportation of the viral core protein into the nucleus (Tsuda et al. 2006). Both chaperone and RNA binding activities of NPM1 are important for JEV replication. Besides, dislocation of NPM1 from nucleus to cytoplasm has also been noted. Although the precise mechanism is not well-defined, it is possible that NPM1 is retained in the cytoplasm by JEV-induced cytoplasmic factors or NPM1 is released into the cytoplasm on disruption of nuclear organization (Tsuda et al. 2006).
HDV is a negative-sense RNA virus that requires the presence of hepatitis B virus (HBV) as a helper virus for replication. Patients with chronic HBV infection and superinfected with HDV can suffer from much more severe complications such as fulminant hepatitis, cirrhosis, and hepatocellular carcinoma. The virus possesses an RNA genome and HDVAg in two isoforms; the small form is involved in HDV RNA replication and the large form in virus assembly (Casey 2006). Replication of HDV occurs in the nucleolus, and NPM1 interacts with HDV antigens and modulates viral RNA replication (Huang et al. 2001; Li et al. 2006). During HDV infection, NPM1 is up-regulated and interacts with the small and the large form (to a lesser extent) of HDVAg. The interaction domains of NPM1–HDVAg are within the NLS of HDVAg and NPM1 acts as a shuttle protein for transportation of HDVAg into the nucleus. Apart from this, NPM1 also plays a role in HDV RNA replication. Huang et al. (2001) demonstrated that exogenous NPM1 had stimulatory effect on HDV RNA replication, while deleting the HDV binding site in NPM1 impaired this effect. Besides, they also observed colocalization of the small HDVAg with NPM1 and nucleolin in the nucleolus. As nucleolin can serve as a transcriptional factor (Yang et al. 1994), this may in turn confer a regulatory role for HDVAg on HDV RNA replication.
Adenovirus is a double-stranded DNA virus causing a wide range of human infections including respiratory, ocular, and gastrointestinal tract infections (Lenaerts et al. 2008). Replication of the viral genome relies on three early proteins, namely, the viral polymerase (Adpol); preterminal protein (pTP), which primes DNA synthesis (Liu et al. 2003); and DNA-binding protein (DBP), which initiates DNA replication (de Jong et al. 2003). Two NPM1 isoforms are involved in adenovirus DNA replication (Hindley et al. 2007). During viral genome replication, NPM1.1 and NPM1.3 interact differently with the viral early proteins pTP and DBP, and NPM1.3 is initially localized in the DBP/viral DNA-rich regions. Once the viral pTP expression increases, NPM1.1 is localized in the pTP-rich regions and interacts with pTP. This is followed by recruiting NPM1.3 into the pTP-rich regions and interaction with the pTP/NPM1.1/viral DNA complex. Apart from genome replication, NPM1 also plays a role in viral assembly of adenovirus. During the late stage of viral infection, the viral protein V interacts with NPM1, which acts as a chaperone by transferring the newly synthesized core protein to the viral DNA genome (Matthews and Russell 1998; Samad et al. 2007).
HBV belongs to the Hepadnaviridae family and is associated with cirrhosis and hepatocellular carcinoma (Ganem and Schneider 2001). It possesses a partial double-stranded DNA genome which encodes four viral proteins, namely, the core protein, surface protein, polymerase, and the X protein (Ganem and Schneider 2001). The HBV core protein consists of an assembly domain at the N-terminal and a viral replication regulatory domain at the C-terminal (Kang et al. 2006; Zlotnick et al. 1996). Ning and Shih (2004) have reported the colocalization of HBV core antigen with nucleolin and NPM1 in the nucleolus, and cells with such colocalization exhibited binucleated and apoptotic morphology. Recently, Lee et al. (2009) demonstrated the involvement of NPM1 in the assembly of HBV, in which the N-terminal of the HBV core protein bound to NPM1 during viral encapsidation. Their study also showed that amino acid residues 259–294 of NPM1 were essential for the interaction with HBV core protein.
NPM1 also interacts with Kaposi sarcoma herpes virus (KSHV) to regulate viral latency. KSHV is a DNA virus belonging to family γ-herpesviruses. The virus is capable of cell transformation and is associated with Kaposi’s sarcoma and AIDS-related non-Hodgkin lymphoma (Renne et al. 1996; Zhong et al. 1996). KSHV remains in latency stage after infection but the virus can be reactivated by various intra- and extra-cellular factors, including cytokines, hypoxia, and chemical agents (Miller et al. 2007). In addition, interactions of the viral protein with host transcription factors and components of the host cellular signaling pathways may reactivate the virus. The KSHV latent protein v-cyclin and host cellular CDK6 kinase can phosphorylate NPM1 on threonine 199 (Sarek et al. 2010). Phosphorylation of NPM1 facilitates interaction of NPM1 with the latency-associated nuclear antigen, a repressor for viral lytic replication. Depletion of NPM1 causes KSHV reactivation; this demonstrated that NPM1 is a regulator of KSHV latency.
6.2 Influence of Virus–NPM1 Interaction on Host Cell Cycle
With an understanding of the interaction between NPM1 and the Rev protein of HIV-1, the clinical implication has also been studied. Miyazaki et al. (1996) reported that an overexpression of Rev altered the nucleolar architecture, and this correlated with the accumulation of NPM1. An elevated level of NPM1 may alter the cell cycle control as the nucleolar protein is involved in ribosome biogenesis (Okuda 2002). In fact, the T-lymphocytes from HIV-1-infected patients had changes in the nucleolar architecture and this correlated with a loss of cell cycle control (Galati et al. 2003).
HCV is associated with posttransfusion hepatitis, which may progress to cirrhosis and hepatocellular carcinoma (Allain 2000; Barazani et al. 2007). The positive-sense RNA genome of HCV encodes the viral envelope, core protein, and several other nonstructural proteins (Choo et al. 1991; Takamizawa et al. 1991). Among various HCV viral proteins, the core protein is the best studied. Instead of being a viral nucleocapsid, the core protein interacts with various host cellular factors and influences various host cell functions including apoptosis, signal transduction, and transcriptional regulation (Fischer et al. 2007; Koike 2007). One of the host cellular factors that interact with HCV is NPM1. The virus interacts with NPM1 through the NLSs located in the core protein amino acid 51–100 (Chen et al. 2003) and is transported into the nucleolus. During HCV infection, NPM1 together with YY1 and P300 forms complex with HCV (Mai et al. 2006). This relieves the suppression effect of YY1 on the NPM1 promoter, thereby up-regulating the expression of NPM1, which in turn activates RNA polymerase I transcription and results in higher rate of ribosome biogenesis. Overall, this promotes cell proliferation during viral infection, and consequently leads to cell transformation and hepatocellular carcinogenesis.
The SARS-coronavirus (SARS-CoV) is the pathogen responsible for severe acute respiratory syndrome (SARS), a human respiratory infection that first identified in Southern China in 2002 (Christian et al. 2004; Ksiazek et al. 2003). The 30-kb positive-sense RNA genome encodes the viral envelope, nucleocapsid, hemagglutinin, and membrane-associated proteins (Wang et al. 2003). The nucleocapsid protein is involved in viral assembly and also regulation of signal transduction. It is mainly localized in the cytoplasm of SARS-CoV-infected cells, but the protein is also present at low level in the nucleus (Tamini et al. 2005). The nucleocapsid protein of SARS-CoV is able to interact with various cellular proteins, including cyclophylin A (Luo et al. 2004), human ubiquitin-conjugating enzyme (Fan et al. 2006), and CDK-cyclin complex proteins (Surjit et al. 2006). NPM1 interacts with nucleocapsid protein of SARS-CoV and, during the interaction, the viral protein competitively inhibits the interaction of NPM1 with CDK2 kinase, and thereby inhibits the phosphorylation of NPM1 (Zeng et al. 2008). A decrease in phosphorylation inhibits the duplication of centromere, which leads to subsequent cell cycle arrest. In addition, interaction of the viral nucleocapsid protein with NPM1 may also cause defects in ribosome synthesis and results in suppression of gene expression, or leads to protein misfolding (Zeng et al. 2008).
Adenovirus mobilizes NPM1 from the nucleolus to nucleoplasm and cytoplasm for genome replication and viral assembly. This causes disruption of the nucleolus, which leads to inhibition of rRNA processing and transportation in the host cell (Castiglia and Flint 1983; Matthews 2001).
To date, much progress has been achieved in understanding the interaction between viruses and nucleolus. The study of viral interactions with the nucleolus enables a deeper understanding of the pathogenic mechanisms of the viral pathogens. The findings also provide new insights into various nucleolar activities and functions. More importantly, the knowledge on the biological pathways involved in virus and nucleolus interactions can be exploited for design of novel therapeutics against viral infections.
7 Concluding Remarks
NPM1 is truly a multifunctional protein whose functions have been unraveled in the past few decades through the work of many research groups while NPM2 and NPM3 have been less well studied. Other than the many processes in which NPM1 takes part, the major interest comes from its involvement in human cancers, particularly AML. Its significance stems from the fact that NPMc+ AML accounts for ∼30% of all AML cases and usually has good prognosis. Its clinical importance also comes from its involvement in virus replication, particularly in the era of outbreaks of infectious diseases. A lot more remain to be discovered and learned for all three NPM proteins in the years to come.
References
Adhikary S, Eilers M (2005) Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 6:635–645
Ahn JY, Liu X, Cheng D, Peng J, Chan PK, Wade PA, Ye K (2005) Nucleophosmin/B23, a nuclear PI(3,4,5)P(3) receptor, mediates the antiapoptotic actions of NGF by inhibiting CAD. Mol Cell 18:435–445
Albiero E, Madeo D, Bolli N, Giaretta I, Bona ED, Martelli MF, Nicoletti I, Rodeghiero F, Falini B (2007) Identification and functional characterization of a cytoplasmic nucleophosmin leukaemic mutant generated by a novel exon-11 NPM1 mutation. Leukemia 21:1099–1103
Alcalay M, Tiacci E, Bergomas R, Bigerna B, Venturini E, Minardi SP, Meani N, Diverio D, Bernard L, Tizzoni L, Volorio S, Luzi L, Colombo E, Lo Coco F, Mecucci C, Falini B, Pelicci PG (2005) Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood 106:899–902
Allain JP (2000) Emerging viral infections relevant to transfusion medicine. Blood Rev 14:173–181
Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426:570–574
Andreeff M, Ruvolo V, Gadgil S, Zeng C, Coombes K, Chen W, Kornblau S, Barón AE, Drabkin HA (2008) HOX expression patterns identify a common signature for favorable AML. Leukemia 22:2041–2047
Barazani Y, Hiatt JR, Tong MJ, Busuttil RW (2007) Chronic viral hepatitis and hepatocellular carcinoma. World J Surg 3:1243–1248
Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D, Whitman SP, Wu YZ, Schwind S, Paschka P, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD (2010) Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol 28:596–604
Bertwistle D, Sugimoto M, Sherr CJ (2004) Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol 24:985–996
Bolli N, Nicoletti I, De Marco MF, Bigerna B, Pucciarini A, Mannucci R, Martelli MP, Liso A, Mecucci C, Fabbiano F, Martelli MF, Henderson BR, Falini B (2007) Born to be exported: COOH-terminal nuclear export signals of different strength ensure cytoplasmic accumulation of nucleophosmin leukemic mutants. Cancer Res 67:6230–6237
Bolli N, De Marco MF, Martelli MP, Bigerna B, Pucciarini A, Rossi R, Mannucci R, Manes N, Pettirossi V, Pileri SA, Nicoletti I, Falini B (2009) A dose-dependent tug of war involving the NPM1 leukaemic mutant, nucleophosmin, and ARF. Leukemia 23:501–509
Bolli N, Payne EM, Grabher C, Lee JS, Johnston AB, Falini B, Kanki JP, Look AT (2010) Expression of the cytoplasmic NPM1 mutant (NPMc+) causes the expansion of hematopoietic cells in zebrafish. Blood 115:3329–3340
Bonetti P, Davoli T, Sironi C, Amati B, Pelicci PG, Colombo E (2008) Nucleophosmin and its AML-associated mutant regulate c-Myc turnover through Fbw7 gamma. J Cell Biol 182:19–26
Borer RA, Lehner CF, Eppenberger HM, Nigg EA (1989) Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56:379–390
Brady SN, Yu Y, Maggi LB Jr, Weber JD (2004) ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol Cell Biol 24:9327–9338
Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G, Alonzo TA, Chang M, Arceci RJ, Small D (2007) The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 110:979–985
Bulich R, Aaskov JG (1992) Nuclear localization of dengue 2 virus core protein detected with monoclonal antibodies. J Gen Virol 73:2999–3003
Burns KH, Viveiros MM, Ren Y, Wang P, DeMayo FJ, Frail DE, Eppig JJ, Matzuk MM (2003) Roles of NPM2 in chromatin and nucleolar organization in oocytes and embryos. Science 300:633–666
Casey JL (2006) Hepatitis delta virus. Springer, Berlin
Castiglia CL, Flint SJ (1983) Effects of adenovirus infection on rRNA synthesis and maturation in HeLa cells. Mol Cell Biol 3:662–671
Cazzaniga G, Dell’Oro MG, Mecucci C, Giarin E, Masetti R, Rossi V, Locatelli F, Martelli MF, Basso G, Pession A, Biondi A, Falini B (2005) Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 106:1419–1422
Chan PK, Liu QR, Durban E (1990) The major phosphorylation site of nucleophosmin (B23) is phosphorylated by a nuclear kinase II. Biochem J 270:549–552
Chang JH, Olson MO (1989) A single gene codes for two forms of rat nucleolar protein B23 mRNA. J Biol Chem 264:11732–11737
Chen SY, Kao CF, Chen CM, Shih CM, Hsu MJ, Chao CH, Wang SH, You LR, Lee YH (2003) Mechanisms for inhibition of hepatitis B virus gene expression and replication by hepatitis C virus core protein. J Biol Chem 278:591–607
Cheng K, Sportoletti P, Ito K, Clohessy JG, Teruya-Feldstein J, Kutok JL, Pandolfi PP (2010) The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 115:3341–3345
Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, Weiner AJ, Bradley DW, Kuo G, Houghton M (1991) Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci USA 88:2451–2455
Chou WC, Tang JL, Lin LI, Yao M, Tsay W, Chen CY, Wu SJ, Huang CF, Chiou RJ, Tseng MH, Lin DT, Lin KH, Chen YC, Tien HF (2006) Nucleophosmin mutations in de novo acute myeloid leukemia: the age-dependent incidences and the stability during disease evolution. Cancer Res 66:3310–3316
Christian MD, Poutanen SM, Loutfy MR, Muller MP, Low DE (2004) Severe acute respiratory syndrome. Clin Infect Dis 38:1420–1427
Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG (2002) Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol 4:529–533
Colombo E, Bonetti P, Lazzerini DE, Martinelli P, Zamponi R, Marine JC, Helin K, Falini B, Pelicci PG (2005) Nucleophosmin is required for DNA integrity and p19Arf protein stability. Mol Cell Biol 25:8874–8886
Colombo E, Martinelli P, Zamponi R, Shing DC, Bonetti P, Luzi L, Volorio S, Bernard L, Pruneri G, Alcalay M, Pelicci PG (2006) Delocalization and destabilization of the Arf tumor suppressor by the leukemia-associated NPM mutant. Cancer Res 66:3044–3050
Compton DA, Cleveland DW (1994) NuMA, a nuclear protein involved in mitosis and nuclear reformation. Curr Opin Cell Biol 6:343–346
Cotten M, Sealy L, Chalkley R (1986) Massive phosphorylation distinguishes Xenopus laevis nucleoplasmin isolated from oocytes or unfertilized eggs. Biochemistry 25:5063–5069
Dalenc F, Drouet J, Ader I, Delmas C, Rochaix P, Favre G, Cohen-Jonathan E, Toulas C (2002) Increased expression of a COOH-truncated nucleophosmin resulting from alternative splicing is associated with cellular resistance to ionizing radiation in HeLa cells. Int J Cancer 100:662–668
de Jong RN, van der Vliet PC, Brenkman AB (2003) Adenovirus DNA replication: protein priming, jumping back and the role of the DNA binding protein DBP. Curr Top Microbiol Immunol 272:187–211
Debernardi S, Skoulakis S, Molloy G, Chaplin T, Dixon-McIver A, Young BD (2007) MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 21:912–916
den Besten W, Kuo ML, iams RT, Sherr CJ (2005) Myeloid leukemia-associated nucleophosmin mutants perturb p53-dependent and independent activities of the Arf tumor suppressor protein. Cell Cycle 4:1593–1598
Dey A, Chao SH, Lane DP (2007) HEXIM1 and the control of transcription elongation: from cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle 6:1856–1863
Dhar SK, Lynn BC, Daosukho C, St CD (2004) Identification of nucleophosmin as an NF-kappaB co-activator for the induction of the human SOD2 gene. J Biol Chem 279:28209–28219
Dingwall C, Dilworth SM, Black SJ, Kearsey SE, Cox LS, Laskey RA (1987) Nucleoplasmin cDNA sequence reveals polyglutamic acid tracts and a cluster of sequences homologous to putative nuclear localization signals. EMBO J 6:69–74
Dumbar TS, Gentry GA, Olson MO (1989) Interaction of nucleolar phosphoprotein B23 with nucleic acids. Biochemistry 28:9495–9501
Dundr M, Misteli T, Olson MO (2000) The dynamics of postmitotic reassembly of the nucleolus. J Cell Biol 150:433–446
Dutta S, Akey IV, Dingwall C, Hartman KL, Laue T, Nolte RT, Head JF, Akey CW (2001) The crystal structure of nucleoplasmin-core: implications for histone binding and nucleosome assembly. Mol Cell 8:841–853
Earnshaw WC, Honda BM, Laskey RA, Thomas JO (1980) Assembly of nucleosomes: the reaction involving X. laevis nucleoplasmin. Cell 21:373–383
Eirín-López JM, Frehlick LJ, Ausió J (2006) Long-term evolution and functional diversification in the members of the nucleophosmin/nucleoplasmin family of nuclear chaperones. Genetics 173:1835–1850
Engelhardt M, Lubbert M, Guo Y (2002) CD34(+) or CD34(−): which is the more primitive? Leukemia 16:1603–1608
Enomoto T, Lindström MS, Jin A, Ke H, Zhang Y (2006) Essential role of the B23/NPM core domain in regulating ARF binding and B23 stability. J Biol Chem 281:18463–18472
Estrov Z (2010) The leukemia stem cell. Cancer Treat Res 145:1–17
Falini B (2001) Anaplastic large cell lymphoma: pathological, molecular and clinical features. Br J Haematol 114:741–760
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF, GIMEMA Acute Leukemia Working Party (2005) Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352:254–266
Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A, Bigerna B, Pasqualucci L, Mannucci R, Rosati R, Gorello P, Diverio D, Roti G, Tiacci E, Cazzaniga G, Biondi A, Schnittger S, Haferlach T, Hiddemann W, Martelli MF, Gu W, Mecucci C, Nicoletti I (2006) Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 107:4514–4523
Falini B, Albiero E, Bolli N, De Marco MF, Madeo D, Martelli M, Nicoletti I, Rodeghiero F (2007a) Aberrant cytoplasmic expression of C-terminal-truncated NPM leukaemic mutant is dictated by tryptophans loss and a new NES motif. Leukemia 21:2052–2054
Falini B, Nicoletti I, Bolli N, Martelli MP, Liso A, Gorello P, Mandelli F, Mecucci C, Martelli MF (2007b) Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica 92:519–532
Falini B, Nicoletti I, Martelli MF, Mecucci C (2007c) Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood 109:874–885
Falini B, Martelli MP, Mecucci C, Liso A, Bolli N, Bigerna B, Pucciarini A, Pileri S, Meloni G, Martelli MF, Haferlach T, Schnittger S (2008a) Cytoplasmic mutated nucleophosmin is stable in primary leukemic cells and in a xenotransplant model of NPMc+ acute myeloid leukemia in SCID mice. Haematologica 93:775–779
Falini B, Mecucci C, Saglio G, Lo Coco F, Diverio D, Brown P, Pane F, Mancini M, Martelli MP, Pileri S, Haferlach T, Haferlach C, Schnittger S (2008b) NPM1 mutations and cytoplasmic nucleophosmin are mutually exclusive of recurrent genetic abnormalities: a comparative analysis of 2562 patients with acute myeloid leukemia. Haematologica 93:439–442
Falini B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E, Haferlach T (2011) Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood 117:1109–1120
Fan Z, Zhuo Y, Tan X, Zhou Z, Yuan J, Qiang B, Yan J, Peng X, Gao GF (2006) SARS-CoV nucleocapsid protein binds to hUbc9, a ubiquitin conjugating enzyme of the sumoylation system. J Med Virol 78:1365–1373
Fankhauser C, Izaurralde E, Adachi Y, Wingfield P, Laemmli UK (1991) Specific complex of human immunodeficiency virus type 1 rev and nucleolar B23 proteins: dissociation by the Rev response element. Mol Cell Biol 11:2567–2575
Fischer R, Baumert T, Blum HE (2007) Hepatitis C virus infection and apoptosis. World J Gastroenterol 3:4865–4872
Flint SJ (2000) Principles of virology: molecular biology, pathogenesis, and control. ASM Press, Washington
Frehlick LJ, Eirín-López JM, Ausió J (2007) New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. Bioessays 29:49–59
Gadad SS, Shandilya J, Kishore AH, Kundu TK (2010) NPM3, a member of the nucleophosmin/nucleoplasmin family, enhances activator-dependent transcription. Biochemistry 49:1355–1357
Galati D, Paiardini M, Cervasi B, Albrecht H, Bocchino M, Costantini A, Montroni M, Magnani M, Piedimonte G, Silvestri G (2003) Specific changes in the posttranslational regulation of nucleolin in lymphocytes from patients infected with human immunodeficiency virus. J Infect Dis 188:1483–1491
Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, Linch DC, Medical Research Council Adult Leukaemia Working Party (2008) The impact of FLT3 internal tandem duplication mutant level, number, size and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111:2776–2784
Gallagher SJ, Kefford RF, Rizos H (2006) The ARF tumour suppressor. Int J Biochem Cell Biol 38:1637–1641
Ganem D, Schneider R (2001) Hepadnaviridae: the viruses and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (eds) Fields virology, 4th edn. Lippincott iams & Wilkins, Philadelphia
Gao H, Jin S, Song Y, Fu M, Wang M, Liu Z, Wu M, Zhan Q (2005) B23 regulates GADD45a nuclear translocation and contributes to GADD45a-induced cell cycle G2-M arrest. J Biol Chem 280:10988–10996
Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, Volinia S, Liu CG, Schnittger S, Haferlach T, Liso A, Diverio D, Mancini M, Meloni G, Foa R, Martelli MF, Mecucci C, Croce CM, Falini B (2008) Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA 105:3945–3950
Gimenez M, Souza VC, Izumi C, Barbieri MR, Chammas R, Oba-Shinjo SM, Uno M, Marie SK, Rosa JC (2010) Proteomic analysis of low- to high-grade astrocytomas reveals an alteration of the expression level of raf kinase inhibitor protein and nucleophosmin. Proteomics 10:2812–2821
Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP (2005) Role of nucleophosmin in embryonic development and tumorigenesis. Nature 437:147–153
Grisendi S, Mecucci C, Falini B, Pandolfi PP (2006) Nucleophosmin and cancer. Nat Rev Cancer 6:493–505
Grummitt CG, Townsley FM, Johnson CM, Warren AJ, Bycroft M (2008) Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J Biol Chem 283:23326–23332
Gurumurthy M, Tan CH, Ng R, Zeiger L, Lau J, Lee J, Dey A, Philp R, Li Q, Lim TM, Price DH, Lane DP, Chao SH (2008) Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. J Mol Biol 378:302–317
Hernandez-Verdun D, Gautier T (1994) The chromosome periphery during mitosis. Bioessays 16:179–185
Herrera JE, Savkur R, Olson MO (1995) The ribonuclease activity of nucleolar protein B23. Nucleic Acids Res 23:3974–3979
Herrera JE, Correia JJ, Jones AE, Olson MO (1996) Sedimentation analyses of the salt- and divalent metal ion-induced oligomerization of nucleolar protein B23. Biochemistry 35:2668–2673
Hindley CE, Davidson AD, Matthews DA (2007) Relationship between adenovirus DNA replication proteins and nucleolar proteins B23.1 and B23.2. J Gen Virol 88:3244–3248
Hingorani K, Szebeni A, Olson MO (2000) Mapping the functional domains of nucleolar protein B23. J Biol Chem 275:24451–24457
Hisaoka M, Ueshima S, Murano K, Nagata K, Okuwaki M (2010) Regulation of nucleolar chromatin by B23/nucleophosmin jointly depends upon its RNA binding activity and transcription factor UBF. Mol Cell Biol 30:4952–4964
Hiscox JA (2007) RNA viruses: hijacking the dynamic nucleolus. Nat Rev Microbiol 5:119–127
Hitzler JK, Witte DP, Jenkins NA, Copeland NG, Gilbert DJ, Naeve CW, Look AT, Morris SW (1999) cDNA cloning, expression pattern, and chromosomal localization of Mlf1, murine homologue of a gene involved in myelodysplasia and acute myeloid leukemia. Am J Pathol 155:53–59
Horn HF, Vousden KH (2004) Cancer: guarding the guardian? Nature 427:110–111
Hsu CY, Yung BY (1998) Down-regulation of nucleophosmin/B23 during retinoic acid-induced differentiation of human promyelocytic leukemia HL-60 cells. Oncogene 16:915–923
Huang WH, Yung BY, Syu WJ, Lee YH (2001) The nucleolar phosphoprotein B23 interacts with hepatitis delta antigens and modulates the hepatitis delta virus RNA replication. J Biol Chem 276:25166–25175
Huang N, Negi S, Szebeni A, Olson MO (2005) Protein NPM3 interacts with the multifunctional nucleolar protein B23/nucleophosmin and inhibits ribosome biogenesis. J Biol Chem 280:5496–5502
Inouye CJ, Seto E (1994) Relief of YY1-induced transcriptional repression by protein-protein interaction with the nucleolar phosphoprotein B23. J Biol Chem 269:6506–6510
Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y (2003) Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell 12:1151–1164
Jagus R, Joshi B, Barber GN (1999) PKR, apoptosis and cancer. Int J Biochem Cell Biol 31:123–138
Jeong EG, Lee SH, Yoo NJ, Lee SH (2007) Absence of nucleophosmin 1 (NPM1) gene mutations in common solid cancers. APMIS 115:341–234
Jiang PS, Chang JH, Yung BY (2000) Different kinases phosphorylate nucleophosmin/B23 at different sites during G(2) and M phases of the cell cycle. Cancer Lett 153:151–160
Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Löwenberg B (2008) MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 111:5078–5085
Kang HY, Lee S, Park SG, Yu J, Yim K, Jung G (2006) Phosphorylation of hepatitis B virus Cp at Ser87 facilitates core assembly. Biochem J 398:311–317
Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL (2004) NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119:803–814
Koike K (2007) Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J Gastroenterol Hepatol 22(suppl 1):S108–S111
Kondo T, Minamino N, Nagamura-Inoue T, Matsumoto M, Taniguchi T, Tanaka N (1997) Identification and characterization of nucleophosmin/B23/numatrin which binds the anti-oncogenic transcription factor IRF-1 and manifests oncogenic activity. Oncogene 15:1275–1281
Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, Quelle DE (2005) Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol 25:1258–1271
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ; SARS Working Group (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348:1953–1966
Kuo ML, den Besten W, Bertwistle D, Roussel MF, Sherr CJ (2004) N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev 18:1862–1874
Kuo ML, den Besten W, Thomas MC, Sherr CJ (2008) Arf-induced turnover of the nucleolar nucleophosmin-associated SUMO-2/3 protease Senp3. Cell Cycle 7:3378–3387
Kurki S, Peltonen K, Laiho M (2004a) Nucleophosmin, HDM2 and p53: players in UV damage incited nucleolar stress response. Cell Cycle 3:976–979
Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M (2004b) Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 5:465–475
Laskey RA, Honda BM, Mills AD, Finch JT (1978) Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 275:416–420
Lee SY, Park JH, Kim S, Park EJ, Yun Y, Kwon J (2005) A proteomics approach for the identification of nucleophosmin and heterogeneous nuclear ribonucleoprotein C1/C2 as chromatin-binding proteins in response to DNA double-strand breaks. Biochem J 388:7–15
Lee HH, Kim HS, Kang JY, Lee BI, Ha JY, Yoon HJ, Lim SO, Jung G, Suh SW (2007) Crystal structure of human nucleophosmin-core reveals plasticity of the pentamer-pentamer interface. Proteins 69:672–678
Lee SJ, Shim HY, Hsieh A, Min JY, Jung G (2009) Hepatitis B virus core interacts with the host cell nucleolar protein, nucleophosmin 1. J Microbiol 47:746–752
Lenaerts L, De Clercq E, Naesens L (2008) Clinical features and treatment of adenovirus infections. Rev Med Virol 18:357–374
Leno GH, Mills AD, Philpott A, Laskey RA (1996) Hyperphosphorylation of nucleoplasmin facilitates Xenopus sperm decondensation at fertilization. J Biol Chem 271:7253–7256
Leong SM, Tan BX, Bte Ahmad B, Yan T, Chee LY, Ang ST, Tay KG, Koh LP, Yeoh AE, Koay ES, Mok YK, Lim TM (2010) Mutant nucleophosmin deregulates cell death and myeloid differentiation through excessive caspase-6 and -8 inhibition. Blood 116:3286–3296
Léotoing L, Meunier L, Manin M, Mauduit C, Decaussin M, Verrijdt G, Claessens F, Benahmed M, Veyssière G, Morel L, Beaudoin C (2008) Influence of nucleophosmin/B23 on DNA binding and transcriptional activity of the androgen receptor in prostate cancer cell. Oncogene 27:2858–2867
Lerch-Gaggl A, Haque J, Li J, Ning G, Traktman P, Duncan SA (2002) Pescadillo is essential for nucleolar assembly, ribosome biogenesis, and mammalian cell proliferation. J Biol Chem 277:45347–45355
Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88:323–331
Li YP (1997) Protein B23 is an important human factor of the nucleolar localization of the human immunodeficiency virus protein Tat. J Virol 71:4098–4102
Li J, Zhang X, Sejas DP, Bagby GC, Pang Q (2004) Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J Biol Chem 279:41275–41279
Li J, Zhang X, Sejas DP, Pang Q (2005) Negative regulation of p53 by nucleophosmin antagonizes stress-induced apoptosis in human normal and malignant hematopoietic cells. Leuk Res 29:1415–1423
Li YJ, Macnaughton T, Gao L, Lai MM (2006) RNA-templated replication of hepatitis delta virus: genomic and antigenomic RNAs associate with different nuclear bodies. J Virol 80:6478–6486
Lin CY, Liang YC, Yung BY (2006) Nucleophosmin/B23 regulates transcriptional activation of E2F1 via modulating the promoter binding of NF-kappaB, E2F1 and pRB. Cell Signal 18:2041–2048
Lin CY, Tan BC, Liu H, Shih CJ, Chien KY, Lin CL, Yung BY (2010) Dephosphorylation of nucleophosmin by PP1β facilitates pRB binding and consequent E2F1-dependent DNA repair. Mol Biol Cell 21:4409–4417
Liso A, Bogliolo A, Freschi V, Martelli MP, Pileri SA, Santodirocco M, Bolli N, Martelli MF, Falini B (2008) In human genome, generation of a nuclear export signal through duplication appears unique to nucleophosmin (NPM1) mutations and is restricted to AML. Leukemia 22:1285–1289
Liu WH, Yung BY (1998) Mortalization of human promyelocytic leukemia HL-60 cells to be more susceptible to sodium butyrate-induced apoptosis and inhibition of telomerase activity by down-regulation of nucleophosmin/B23. Oncogene 17:3055–3064
Liu H, Naismith JH, Hay RT (2003) Adenovirus DNA replication. Curr Top Microbiol Immunol 272:131–164
Liu H, Tan BC, Tseng KH, Chuang CP, Yeh CW, Chen KD, Lee SC, Yung BY (2007a) Nucleophosmin acts as a novel AP2alpha-binding transcriptional corepressor during cell differentiation. EMBO Rep 8:394–400
Liu X, Liu Z, Jang SW, Ma Z, Shinmura K, Kang S, Dong S, Chen J, Fukasawa K, Ye K (2007b) Sumoylation of nucleophosmin/B23 regulates its subcellular localization, mediating cell proliferation and survival. Proc Natl Acad Sci USA 104:9679–9684
Luo C, Luo H, Zheng S, Gui C, Yue L, Yu C, Sun T, He P, Chen J, Shen J, Luo X, Li Y, Liu H, Bai D, Shen J, Yang Y, Li F, Zuo J, Hilgenfeld R, Pei G, Chen K, Shen X, Jiang H (2004) Nucleocapsid protein of SARS coronavirus tightly binds to human cyclophilin A. Biochem Biophys Res Commun 321:557–565
MacArthur CA, Shackleford GM (1997) Npm3: a novel, widely expressed gene encoding a protein related to the molecular chaperones nucleoplasmin and nucleophosmin. Genomics 42:137–140
Maggi LB Jr, Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, Pandolfi PP, Weber JD (2008) Nucleophosmin serves as a rate-limiting nuclear export chaperone for the mammalian ribosome. Mol Cell Biol 28:7050–7065
Mai RT, Yeh TS, Kao CF, Sun SK, Huang HH, Wu Lee YH (2006) Hepatitis C virus core protein recruits nucleolar phosphoprotein B23 and coactivator p300 to relieve the repression effect of transcriptional factor YY1 on B23 gene expression. Oncogene 25:448–462
Mariano AR, Colombo E, Luzi L, Martinelli P, Volorio S, Bernard L, Meani N, Bergomas R, Alcalay M, Pelicci PG (2006) Cytoplasmic localization of NPM in myeloid leukemias is dictated by gain-of-function mutations that create a functional nuclear export signal. Oncogene 25:4376–4380
Martelli MP, Manes N, Pettirossi V, Liso A, Pacini R, Mannucci R, Zei T, Bolli N, di Raimondo F, Specchia G, Nicoletti I, Martelli MF, Falini B (2008) Absence of nucleophosmin leukaemic mutants in B and T cells from AML with NPM1 mutations: implications for the cell of origin of NPMc+ AML. Leukemia 22:195–198
Martelli MP, Pettirossi V, Thiede C, Bonifacio E, Mezzasoma F, Cecchini D, Pacini R, Tabarrini A, Ciurnelli R, Gionfriddo I, Manes N, Rossi R, Giunchi L, Oelschlägel U, Brunetti L, Gemei M, Delia M, Specchia G, Liso A, Di Ianni M, Di Raimondo F, Falzetti F, Del Vecchio L, Martelli MF, Falini B (2010) CD34+ cells from AML with mutated NPM1 harbor cytoplasmic mutated nucleophosmin and generate leukemia in immunocompromised mice. Blood 116:3907–3922
Matthews DA (2001) Adenovirus protein V induces redistribution of nucleolin and B23 from nucleolus to cytoplasm. J Virol 75:1031–1038
Matthews DA, Russell WC (1998) Adenovirus core protein V is delivered by the invading virus to the nucleus of the infected cell and later in infection is associated with nucleoli. J Gen Virol 79:1671–1675
McLay DW, Clarke HJ (2003) Remodelling the paternal chromatin at fertilization in mammals. Reproduction 125:625–633
Meder VS, Boeglin M, de Murcia G, Schreiber V (2005) PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci 118:211–222
Michalik J, Yeoman LC, Busch H (1981) Nucleolar localization of protein B23 (37/5.1) by immunocytochemical techniques. Life Sci 28:1371–1379
Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L (2007) Lytic cycle switches of oncogenic human gammaherpesviruses. Adv Cancer Res 97:81–109
Mills AD, Laskey RA, Black P, De Robertis EM (1980) An acidic protein which assembles nucleosomes in vitro is the most abundant protein in Xenopus oocyte nuclei. J Mol Biol 139:561–568
Miyazaki Y, Nosaka T, Hatanaka M (1996) The posttranscriptional regulator Rev of HIV: implications for its interaction with the nucleolar protein B23. Biochimie 78:1081–1086
Mori Y, Okabayashi T, Yamashita T, Zhao Z, Wakita T, Yasui K, Hasebe F, Tadano M, Konishi E, Moriishi K, Matsuura Y (2005) Nuclear localization of Japanese encephalitis virus core protein enhances viral replication. J Virol 79:3448–3458
Motoi N, Suzuki K, Hirota R, Johnson P, Oofusa K, Kikuchi Y, Yoshizato K (2008) Identification and characterization of nucleoplasmin 3 as a histone-binding protein in embryonic stem cells. Dev Growth Differ 50:307–320
Mullighan CG, Kennedy A, Zhou X, Radtke I, Phillips LA, Shurtleff SA, Downing JR (2007) Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 21:2000–2009
Murano K, Okuwaki M, Hisaoka M, Nagata K (2008) Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol Cell Biol 28:3114–3126
Nakagawa M, Kameoka Y, Suzuki R (2005) Nucleophosmin in acute myelogenous leukemia. N Engl J Med 352:1819–1820
Namboodiri VM, Dutta S, Akey IV, Head JF, Akey CW (2003) The crystal structure of Drosophila NLP-core provides insight into pentamer formation and histone binding. Structure 11:175–186
Namboodiri VM, Akey IV, Schmidt-Zachmann MS, Head JF, Akey CW (2004) The structure and function of Xenopus NO38-core, a histone chaperone in the nucleolus. Structure 12:2149–2160
Naoe T, Suzuki T, Kiyoi H, Urano T (2006) Nucleophosmin: a versatile molecule associated with hematological malignancies. Cancer Sci 97:963–969
Negi SS, Olson MO (2006) Effects of interphase and mitotic phosphorylation on the mobility and location of nucleolar protein B23. J Cell Sci 119:3676–3685
Ning B, Shih C (2004) Nucleolar localization of human hepatitis B virus capsid protein. J Virol 78:13653–13668
Nishimura Y, Ohkubo T, Furuichi Y, Umekawa H (2002) Tryptophans 286 and 288 in the C-terminal region of protein B23.1 are important for its nucleolar localization. Biosci Biotechnol Biochem 66:2239–2242
Nozawa Y, Van Belzen N, Van der Made AC, Dinjens WN, Bosman FT (1996) Expression of nucleophosmin/B23 in normal and neoplastic colorectal mucosa. J Pathol 178:48–52
Okuda M (2002) The role of nucleophosmin in centrosome duplication. Oncogene 21:6170–6174
Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K (2000) Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 103:127–140
Okuwaki M (2008) The structure and functions of NPM1/nucleophsmin/B23, a multifunctional nucleolar acidic protein. J Biochem 143:441–448
Okuwaki M, Iwamatsu A, Tsujimoto M, Nagata K (2001a) Identification of nucleophosmin/B23, an acidic nucleolar protein, as a stimulatory factor for in vitro replication of adenovirus DNA complexed with viral basic core proteins. J Mol Biol 311:41–55
Okuwaki M, Matsumoto K, Tsujimoto M, Nagata K (2001b) Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett 506:272–276
Okuwaki M, Tsujimoto M, Nagata K (2002) The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol Biol Cell 13:2016–2030
Olson MO, Wallace MO, Herrera AH, Marshall-Carlson L, Hunt RC (1986) Preribosomal ribonucleoprotein particles are a major component of a nucleolar matrix fraction. Biochemistry 25:484–491
Orrick LR, Olson MO, Busch H (1973) Comparison of nucleolar proteins of normal rat liver and Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 70:1316–1320
Pang Q, Christianson TA, Koretsky T, Carlson H, David L, Keeble W, Faulkner GR, Speckhart A, Bagby GC (2003) Nucleophosmin interacts with and inhibits the catalytic function of eukaryotic initiation factor 2 kinase PKR. J Biol Chem 278:41709–41717
Pasqualucci L, Liso A, Martelli MP, Bolli N, Pacini R, Tabarrini A, Carini M, Bigerna B, Pucciarini A, Mannucci R, Nicoletti I, Tiacci E, Meloni G, Specchia G, Cantore N, Di Raimondo F, Pileri S, Mecucci C, Mandelli F, Martelli MF, Falini B (2006) Mutated nucleophosmin detects clonal multilineage involvement in acute myeloid leukemia: impact on WHO classification. Blood 108:4146–4155
Perkins A, Cochrane AW, Ruben SM, Rosen CA (1989) Structural and functional characterization of the human immunodeficiency virus rev protein. J Acquir Immune Defic Syndr 2:256–263
Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA (1990) Identification of major nucleolar proteins as candidate mitotic substrates of cdc2 kinase. Cell 60:791–801
Philpott A, Leno GH (1992) Nucleoplasmin remodels sperm chromatin in Xenopus egg extracts. Cell 69:759–767
Pianta A, Puppin C, Franzoni A, Fabbro D, Di Loreto C, Bulotta S, Deganuto M, Paron I, Tell G, Puxeddu E, Filetti S, Russo D, Damante G (2010) Nucleophosmin is overexpressed in thyroid tumors. Biochem Biophys Res Commun 397:499–504
Pitiot AS, Santamaría I, García-Suárez O, Centeno I, Astudillo A, Rayón C, Balbín M (2007) A new type of NPM1 gene mutation in AML leading to a C-terminal truncated protein. Leukemia 21:1564–1566
Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, Murshudov GN, Cohen S, Perrakis A, Noble M (2004) Developments in the CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr 60:2288–2294
Prestayko AW, Klomp GR, Schmoll DJ, Busch H (1974) Comparison of proteins of ribosomal subunits and nucleolar preribosomal particles from Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Biochemistry 13:1945–1951
Raimondi SC, Dubé ID, Valentine MB, Mirro J Jr, Watt HJ, Larson RA, Bitter MA, Le Beau MM, Rowley JD (1989) Clinicopathologic manifestations and breakpoints of the t(3;5) in patients with acute nonlymphocytic leukemia. Leukemia 3:42–47
Rau R, Brown P (2009) Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol Oncol 27:171–181
Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D (1996) Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346
Rubbi CP, Milner J (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 22:6068–6077
Ruggero D, Pandolfi PP (2003) Does the ribosome translate cancer? Nat Rev Cancer 3:179–192
Samad MA, Okuwaki M, Haruki H, Nagata K (2007) Physical and functional interaction between a nucleolar protein nucleophosmin/B23 and adenovirus basic core proteins. FEBS Lett 581:3283–3288
Sarek G, Järviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM (2010) Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog 6:e1000818
Sato K, Hayami R, Wu W, Nishikawa T, Nishikawa H, Okuda Y, Ogata H, Fukuda M, Ohta T (2004) Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J Biol Chem 279:30919–30922
Savkur RS, Olson MO (1998) Preferential cleavage in pre-ribosomal RNA by protein B23 endoribonuclease. Nucleic Acids Res 26:4508–4515
Shackleford GM, Ganguly A, MacArthur CA (2001) Cloning, expression and nuclear localization of human NPM3, a member of the nucleophosmin/nucleoplasmin family of nuclear chaperones. BMC Genomics 2:8
Sherr CJ (2006) Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 6:663–673
Shields LB, Gerçel-Taylor C, Yashar CM, Wan TC, Katsanis WA, Spinnato JA, Taylor DD (1997) Induction of immune responses to ovarian tumor antigens by multiparity. J Soc Gynecol Investig 4:298–304
Skaar TC, Prasad SC, Sharareh S, Lippman ME, Brünner N, Clarke R (1998) Two-dimensional gel electrophoresis analyses identify nucleophosmin as an estrogen regulated protein associated with acquired estrogen-independence in human breast cancer cells. J Steroid Biochem Mol Biol 67:391–402
Spector DL, Ochs RL, Busch H (1984) Silver staining, immunofluorescence, and immunoelectron microscopic localization of nucleolar phosphoproteins B23 and C23. Chromosoma 90:139–148
Sportoletti P, Grisendi S, Majid SM, Cheng K, Clohessy JG, Viale A, Teruya-Feldstein J, Pandolfi PP (2008) Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood 111:3859–3862
Subong EN, Shue MJ, Epstein JI, Briggman JV, Chan PK, Partin AW (1999) Monoclonal antibody to prostate cancer nuclear matrix protein (PRO:4-216) recognizes nucleophosmin/B23. Prostate 39:298–304
Sugimoto M, Kuo ML, Roussel MF, Sherr CJ (2003) Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell 11:415–424
Surjit M, Liu B, Chow VT, Lal SK (2006) The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin–cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J Biol Chem 281:10669–10681
Swaminathan V, Kishore AH, Febitha KK, Kundu TK (2005) Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol Cell Biol 25:7534–7545
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds) (2008) WHO classification of tumours of haematopoietic and lymphoid tissues. IARC, Lyon
Szebeni A, Olson MO (1999) Nucleolar protein B23 has molecular chaperone activities. Protein Sci 8:905–912
Szebeni A, Mehrotra B, Baumann A, Adam SA, Wingfield PT, Olson MO (1997) Nucleolar protein B23 stimulates nuclear import of the HIV-1 Rev protein and NLS-conjugated albumin. Biochemistry 36:3941–3949
Szebeni A, Hingorani K, Negi S, Olson MO (2003) Role of protein kinase CK2 phosphorylation in the molecular chaperone activity of nucleolar protein b23. J Biol Chem 278:9107–9115
Tago K, Chiocca S, Sherr CJ (2005) Sumoylation induced by the Arf tumor suppressor: a p53-independent function. Proc Natl Acad Sci USA 102:7689–7694
Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H (1991) Structure and organization of the hepatitis C virus genome isolated from human carriers. J Virol 65:1105–1113
Tanaka M, Sasaki H, Kino I, Sugimura T, Terada M (1992) Genes preferentially expressed in embryo stomach are predominantly expressed in gastric cancer. Cancer Res 52:3372–3377
Tarapore P, Shinmura K, Suzuki H, Tokuyama Y, Kim SH, Mayeda A, Fukasawa K (2006) Thr199 phosphorylation targets nucleophosmin to nuclear speckles and represses pre-mRNA processing. FEBS Lett 580:399–409
Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, Lister TA, Gribben JG, Bonnet D (2010) Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(−) fraction. Blood 115:1976–1984
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, Ehninger G (2006) Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 107:4011–4020
Thiede C, Creutzig E, Reinhardt D, Ehninger G, Creutzig U (2007) Different types of NPM1 mutations in children and adults: evidence for an effect of patient age on the prevalence of the TCTG-tandem duplication in NPM1-exon 12. Leukemia 21:366–367
Tamini KA, Liao Q, Ye L, Zeng Y, Liu J, Zheng Y, Ye L, Yang X, Lingbao K, Gao J, Zhu Y (2005) Nuclear/nucleolar localization properties of C-terminal nucleocapsid protein of SARS coronavirus. Virus Res 114:23–34
Tokuyama Y, Horn HF, Kawamura K, Tarapore P, Fukasawa K (2001) Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J Biol Chem 276:21529–21537
Tsuda Y, Mori Y, Abe T, Yamashita T, Okamoto T, Ichimura T, Moriishi K, Matsuura Y (2006) Nucleolar protein B23 interacts with Japanese encephalitis virus core protein and participates in viral replication. Microbiol Immunol 50:225–234
Tsui KH, Cheng AJ, Chang PL, Pan TL, Yung BY (2004) Association of nucleophosmin/B23 mRNA expression with clinical outcome in patients with bladder carcinoma. Urology 64:839–844
Umekawa H, Chang JH, Correia JJ, Wang D, Wingfield PT, Olson MO (1993) Nucleolar protein B23: bacterial expression, purification, oligomerization and secondary structures of two isoforms. Cell Mol Biol Res 39:635–645
Verhaak RG, Goudswaard CS, van Putten W, Bijl MA, Sanders MA, Hugens W, Uitterlinden AG, Erpelinck CA, Delwel R, Löwenberg B, Valk PJ (2005) Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 106:3747–3754
Wang D, Umekawa H, Olson MO (1993) Expression and subcellular locations of two forms of nucleolar protein B23 in rat tissues and cells. Cell Mol Biol Res 39:33–42
Wang D, Baumann A, Szebeni A, Olson MO (1994) The nucleic acid binding activity of nucleolar protein B23.1 resides in its carboxyl-terminal end. J Biol Chem 269:30994–30998
Wang J, Ji J, Ye J, Zhao X, Wen J, Li W, Hu J, Li D, Sun M, Zeng H, Hu Y, Tian X, Tan X, Xu N, Zeng C, Wang J, Bi S, Yang H (2003) The structure analysis and antigenicity study of the N protein of SARS-CoV. Genomics Proteomics Bioinformatics 1:145–154
Wang W, Budhu A, Forgues M, Wang XW (2005) Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat Cell Biol 7:823–830
Wanzel M, Russ AC, Kleine-Kohlbrecher D, Colombo E, Pelicci PG, Eilers M (2008) A ribosomal protein L23-nucleophosmin circuit coordinates Mizl function with cell growth. Nat Cell Biol 10:1051–1061
Weinhold N, Moreaux J, Raab MS, Hose D, Hielscher T, Benner A, Meissner T, Ehrbrecht E, Brough M, Jauch A, Goldschmidt H, Klein B, Moos M (2010) NPM1 is overexpressed in hyperdiploid multiple myeloma due to a gain of chromosome 5 but is not delocalized to the cytoplasm. Genes Chromosomes Cancer 49:333–341
Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8:83–93
Wertheim G, Bagg A (2008) Nucleophosmin (NPM1) mutations in acute myeloid leukemia: an ongoing (cytoplasmic) tale of dueling mutations and duality of molecular genetic testing methodologies. J Mol Diagn 10:198–202
Wu MH, Yung BY (2002) UV stimulation of nucleophosmin/B23 expression is an immediate-early gene response induced by damaged DNA. J Biol Chem 277:48234–48240
Wu MH, Chang JH, Chou CC, Yung BY (2002a) Involvement of nucleophosmin/B23 in the response of HeLa cells to UV irradiation. Int J Cancer 97:297–305
Wu MH, Chang JH, Yung BY (2002b) Resistance to UV-induced cell-killing in nucleophosmin/B23 over-expressed NIH 3 T3 fibroblasts: enhancement of DNA repair and up-regulation of PCNA in association with nucleophosmin/B23 over-expression. Carcinogenesis 23:93–100
Yang TH, Tsai WH, Lee YM, Lei HY, Lai MY, Chen DS, Yeh NH, Lee SC (1994) Purification and characterization of nucleolin and its identification as a transcription repressor. Mol Cell Biol 14:6068–6074
Ye K (2005) Nucleophosmin/B23, a multifunctional protein that can regulate apoptosis. Cancer Biol Ther 4:918–923
Yeh CW, Huang SS, Lee RP, Yung BY (2006) Ras-dependent recruitment of c-Myc for transcriptional activation of nucleophosmin/B23 in highly malignant U1 bladder cancer cells. Mol Pharmacol 70:1443–1453
Yu Y, Maggi LB Jr, Brady SN, Apicelli AJ, Dai MS, Lu H, Weber JD (2006) Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol 26:3798–3809
Yun JP, Chew EC, Liew CT, Chan JY, Jin ML, Ding MX, Fai YH, Li HK, Liang XM, Wu QL (2003) Nucleophosmin/B23 is a proliferate shuttle protein associated with nuclear matrix. J Cell Biochem 90:1140–1148
Yun JP, Miao J, Chen GG, Tian QH, Zhang CQ, Xiang J, Fu J, Lai PB (2007) Increased expression of nucleophosmin/B23 in hepatocellular carcinoma and correlation with clinicopathological parameters. Br J Cancer 96:477–484
Yung BY (2004) c-Myc-mediated expression of nucleophosmin/B23 decreases during retinoic acid-induced differentiation of human leukemia HL-60 cells. FEBS Lett 578:211–216
Zatsepina OV, Rousselet A, Chan PK, Olson MO, Jordan EG, Bornens M (1999) The nucleolar phosphoprotein B23 redistributes in part to the spindle poles during mitosis. J Cell Sci 112:455–466
Zelent A, Guidez F, Melnick A, Waxman S, Licht JD (2001) Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene 20:7186–7203
Zeng Y, Ye L, Zhu S, Zheng H, Zhao P, Cai W, Su L, She Y, Wu Z (2008) The nucleocapsid protein of SARS-associated coronavirus inhibits B23 phosphorylation. Biochem Biophys Res Commun 369:287–291
Zhang H, Shi X, Paddon H, Hampong M, Dai W, Pelech S (2004) B23/nucleophosmin serine 4 phosphorylation mediates mitotic functions of polo-like kinase 1. J Biol Chem 279:35726–35734
Zhong W, Wang H, Herndier B, Ganem D (1996) Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc Natl Acad Sci USA 93:6641–6646
Zirwes RF, Schmidt-Zachmann MS, Franke WW (1997) Identification of a small, very acidic constitutive nucleolar protein (NO29) as a member of the nucleoplasmin family. Proc Natl Acad Sci USA 94:11387–11392
Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT (1996) Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry 35:7412–7421
Zou Y, Wu J, Giannone RJ, Boucher L, Du H, Huang Y, Johnson DK, Liu Y, Wang Y (2008) Nucleophosmin/B23 negatively regulates GCN5-dependent histone acetylation and transactivation. J Biol Chem 283:5728–5737
Acknowledgments
Our work on nucleophosmin is currently supported by grants from The Hong Kong Polytechnic University (1-BD03, G-U702 and G-U915), and was also supported by ROC National Science Council Grants and Chang Gung Research Grants.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Yip, S.P., Siu, P.M., Leung, P.H.M., Zhao, Y., Yung, B.Y.M. (2011). The Multifunctional Nucleolar Protein Nucleophosmin/NPM/B23 and the Nucleoplasmin Family of Proteins. In: Olson, M. (eds) The Nucleolus. Protein Reviews, vol 15. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-0514-6_10
Download citation
DOI: https://doi.org/10.1007/978-1-4614-0514-6_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-0513-9
Online ISBN: 978-1-4614-0514-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)