
Abstract
Disruption of the Hedgehog (HH) signaling pathway underlies an increasingly large array of different human tumors. Consistent with the important role HH plays in cancer, the tumor burden of patients treated with a novel HH inhibitor was dramatically reduced in a recent clinical trial. This drug binds directly to and antagonizes activity of the seven-transmembrane protein Smoothened (SMO), attenuating downstream signaling events that remain largely unknown. While functional studies of SMO signaling have provided a basic roadmap of information flow following ligand stimulation, the exact routes of signaling, and how they communicate with each other and with other signaling pathways, are not well characterized. We recently demonstrated that one route of SMO-mediated signal transduction involves activation of the heterotrimeric guanine nucleotide binding protein (G-protein) Gαi, suggesting that SMO can signal, at least in part, as a canonical G-protein-coupled receptor (GPCR). In this chapter, we discuss structural, functional, and mechanistic aspects of SMO signaling that relate to its function as a GPCR, and provide insight into how G-protein-dependent signaling might impact HH pathway activity.
Similar content being viewed by others


Keywords
Introduction
The important role the Hedgehog (HH) signaling pathway plays in cancer was first revealed by patients diagnosed with the familial disorder known as Gorlin Syndrome, who harbor loss-of-function mutations in the HH receptor Patched (PTCH) [1–3]. Besides numerous developmental abnormalities, consistent with disruption of this important developmental signaling pathway, individuals afflicted with this disorder have an inherited predisposition to medulloblastoma, basal cell carcinoma, and rhabdomyosarcoma [2]. Similar mutations found in sporadic cases of these same tumor types implicated PTCH as an important tumor suppressor in human cancer [4]. Other components of the HH pathway, such as the gene encoding the seven-transmembrane (7TM) protein Smoothened (SMO), are also found mutated in sporadic forms of these same malignancies [5, 6]. More recently, constitutive activation of the HH pathway has been implicated in other human cancers including those of the breast, prostate, pancreas, and lung, where HH is thought to play a role as a tumor-survival factor [7]. Combined, it has been estimated that approximately 25% of all human tumors harbor a constitutively active HH signaling pathway [8]. As such, considerable effort has gone into identifying novel small-molecule inhibitors of HH signaling. Consistent with the rate-limiting role SMO plays in HH signaling, the vast majority of HH inhibitors isolated from numerous small-molecule screens appear to target SMO [9]. A number of these compounds are currently in clinical trials as anti-cancer agents, targeting tumors that are dependent on HH pathway activity [10]. Thus, a clear understanding of the mechanisms by which SMO communicates with downstream pathway components, and how such inhibitors affect these processes, will directly impact human health.
Hedgehog Signal Transduction
Much of what is understood about HH signaling originates from studies of this signaling pathway during the development of the fruit fly Drosophila melanogaster [11]. It is now well accepted that the major components, and how they communicate with each other, are highly conserved from Drosophila to man. Although there are significant differences in the importance of some of the signaling components across phyla, it is not yet clear if these variations are due to specific contextual differences or to evolutionary divergence. Thus, in this chapter we generalize about the HH signaling pathway from work derived from numerous animal models, mentioning specific biological contexts only where necessary to illustrate a particular point.
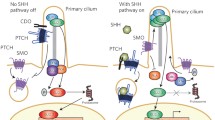
HH is produced and secreted by discrete compartments within a developing field of cells, where it elicits both short- and long-range effects on target cells [12]. The receiving cells interpret the level of HH activation through poorly defined, indirect interactions between the HH receptor PTCH and the signal transducer SMO. PTCH inhibits the activity of SMO in a manner that appears to be catalytic, whereas SMO is constitutively active in the absence of PTCH [13, 14]. One of the mechanisms by which PTCH inhibits SMO activity involves PTCH-dependent trafficking of SMO to lysosomes [15, 16]. In response to HH, PTCH is removed from the cell surface, thereby allowing SMO phosphorylation, stabilization, and accumulation at the plasma membrane [15, 17]. Ultimately all signaling downstream of SMO coalesces to regulate the stability and activity of the GLI/CI family of transcription factors [18]. In the absence of HH, these proteins exist as proteolyzed transcriptional repressors. HH blocks this proteolytic conversion, and stabilizes full-length transcriptional activators. The degree of HH a cell is exposed to ultimately determines the ratio of GLI/CI repressor and activator forms to regulate a spectrum of transcriptional targets that is thought to correlate with the concentration of the initial HH signal.
While the general flow of information through the HH signaling cascade is known, the direct effectors of SMO, and the mechanism(s) by which it communicates with them are still being characterized. The first clue as to how SMO transduces the signal from the plasma membrane to the intracellular effectors came with the observations that Drosophila SMO directly associates with the kinesin-related protein Costal2 (COS2) [19–22], and that mammalian SMO binds the COS2 ortholog KIF7 [23]. The functional consequence of KIF7-SMO binding in mammalian systems is not yet clear. However, in Drosophila, COS2 serves as a scaffold upon which a complex containing CI and the protein kinases Fused (FU), cyclic-AMP (cAMP)-dependent protein kinase A (PKA) and casein kinase 1 (CK1) assembles [24, 25]. As such, a direct association between COS2 and SMO connects the membrane signaling components with the cytoplasmic effectors. Subsequent to this finding, a direct association between SMO and FU that drives a feed-forward loop to facilitate high-level signaling was described [26]. We recently demonstrated that the intracellular molar concentration of SMO is significantly lower than that of CI, COS2, or FU, suggesting that direct association between SMO, COS2, and FU is unlikely to facilitate all aspects of HH signaling [27]. It is, therefore, likely that multiple pools of intracellular effectors exist; some that are in direct contact with SMO, and some that are regulated through the use of G-proteins and/or second messengers.
Smoothened as a G-Protein-Coupled Receptor
Much of what we know about G-protein-coupled receptor (GPCR) structure and function has resulted from studies of the prototypical GPCR rhodopsin, the first GPCR to be fully sequenced and to yield high-resolution structural data [28–30]. Sequence analysis of rhodopsin suggested the existence of several distinct functional domains, including seven predicted alpha-helical transmembrane segments, an extracellular amino-terminal domain, three extracellular loops, a carboxyl-terminal domain with multiple phosphorylation sites, and three intracellular loops [29]. Structural analysis of rhodopsin, and more recently of the β2-adrenergic receptor and A2A adenosine receptor, confirm the existence and conservation of these domains, underscoring their importance in GPCR function [28]. As such, proteins possessing these well-established functional domains in their primary amino acid sequence or predicted tertiary structure are classified as members of the GPCR superfamily, which is estimated to encompass more than 1% of all human genes [30].
SMO was originally identified as a gene necessary for proper organization of the early Drosophila embryo [31]. Subsequent genetic and molecular characterization of SMO revealed it to be a requisite component of the HH signal transduction cascade [32]. Primary sequence comparisons revealed that SMO and the Frizzled (FZ) family of GPCRs are quite similar across distinct functional domains: 37% similarity across the extracellular amino-terminal domains, and 52% similarity across the seven predicted transmembrane domains [32]. As such, SMO has been classified as a member of the FZ family of GPCRs. The specific contributions of conserved GPCR functional domains to SMO-mediated regulation of HH pathway activity are discussed below (Fig. 3.1).

Domains and effectors of smoothened (SMO). A schematic depicting the predicted topology and domains of SMO in the plasma membrane is shown. The seven predicted transmembrane domains of SMO are shown in black. The amino-terminal domain is shown in green and the cysteine-rich domain (CRD) in yellow. Three extracellular and one intracellular loops are shown in blue, but intracellular loops 2 and 3 – which are thought to couple to G-proteins – are shown in red. The carboxyl-terminal domain is shown in purple. The various known direct effectors of SMO are indicated below it
Cysteine-rich domain (CRD). A conserved CRD is situated in the extracellular amino terminus of all FZ family GPCRs [33–35]. Disulfide bonds between amino-terminal cysteine residues and/or cysteine residues in the extracellular loops of FZ drive receptor conformations that are necessary for its ligand binding and ligand-induced dimerization [34, 36]. Like FZ, SMO possesses multiple cysteines in its amino terminus and extracellular loops that are positionally conserved across species (http://www.gpcr.org). In vitro studies in mammalian cell culture suggested that the amino-terminus of SMO, which encompasses the CRD, is not required for GLI activation [37]. However, genetic analyses in both Drosophila and zebrafish support that conserved cysteine residues in the CRD are critical for SMO signaling and/or subcellular localization [38–40], suggesting that the CRD is a requisite functional domain. Further studies are needed to more clearly define contributions of the CRD to SMO signaling.
Transmembrane domains and intracellular loops. The topology of the 7TM regions dictates the activation state of a GPCR by directing the conformation of its intracellular loops and cytoplasmic tail [41]. In response to ligand, the TM domains of the receptor shift to allow changes in conformation of the intracellular portions of the protein that facilitate receptor phosphorylation and/or G-protein selectivity, docking, and activation [41]. The importance of SMO TM sequence/structure is underscored by known oncogenic SMO mutations, all of which are localized to predicted TM segments [6, 42]. It is likely that these mutations lock SMO TM and intracellular domains in an activated state, which is insensitive to PTCH-mediated inhibition.
The cytoplasmic tail along with intracellular loop 3 (ic3), and to a lesser extent ic2, constitute the G-protein docking site on the vast majority of GPCRs [43]. While extensive structure/function analysis of the SMO intracellular loops has not been reported, chimeric studies in cultured fibroblasts reveal a critical role for ic3 in activation of the signaling cascade [37]. These findings are supported by a loss-of-function SMO mutation in Drosophila of a highly conserved Arg residue localized to the carboxyl-terminal end of loop ic3 [39]. The importance of the intracellular loops is further supported by a study demonstrating that introduction of peptide analogs of either ic2 or ic3 into cultured cancer cell lines, which have an activated HH signaling pathway, attenuates their proliferation [44]. Further studies are needed to identify binding partners of SMO ic2 and ic3, and to determine whether these domains constitute a binding site for a partner G-protein.
Carboxyl-terminal intracellular tail. Multiple phosphorylation sites, which have been shown to be critical for pathway activation, have been identified in the SMO carboxyl-terminal tail [45–47]. Phosphorylation of such sites in response to ligand is a well-characterized event in canonical GPCR signaling, which generally serves to recruit various adaptor proteins and signaling effectors to the activated receptor [48]. Accordingly, HH-stimulated phosphorylation of SMO by PKA and G-protein regulated kinase 2 (GRK2) triggers both adaptor protein recruitment and SMO multimerization [49–51]. Mutations that prevent phosphorylation of any of these characterized phosphorylation sites compromise the ability of SMO to signal [45–47].
SMO Signaling Through Heterotrimeric G-Proteins
We recently demonstrated that Gαi overexpression in Drosophila triggers activation of HH target genes and wing patterning defects consistent with excessive HH signaling [52]. These phenotypes correlated with the activation state of the expressed Gαi transgene, as overexpression of a Gαi mutant that cannot bind GTP resulted in no observable phenotype. Conversely, overexpression of wild-type Gαi triggered modest gain of function phenotypes, and overexpression of a transgene encoding constitutively active Gαi resulted in strong HH gain of function phenotypes.
Activation of heterotrimeric G-proteins of the Gαi family frequently serves to decrease intracellular pools of cAMP through Gαi-mediated inhibition of adenylate cyclase (AC) [53]. Accordingly, in our study, we observed a SMO- and Gαi-dependent reduction in total intracellular cAMP within 5–10 min of HH stimulation [52]. Modulation of cAMP appears to be critical for in vivo HH signaling, as a mutant allele of the cAMP phosphodiesterase DUNCE [54] enhanced the HH loss-of-function phenotype induced by expression of a dominant negative SMO mutant in the Drosophila wing [52]. The ability of cAMP to modulate HH pathway-dependent patterning events is further supported by studies demonstrating that overexpression of an anthrax virulence factor, which functions as a potent bacterial AC, triggers wing phenotypes similar to HH loss-of-function mutations [55]. Further, modulation of cAMP by Sonic HH (SHH) has also been demonstrated in vertebrate systems: retinal ganglion cell axons exposed to recombinant SHH reduce their intracellular pools of cytoplasmic cAMP [56], while frog melanophores exposed to SHH aggregate their melanosomes, a process favored by low concentrations of intracellular cAMP [57]. Taken together, these studies support that one mechanism by which SMO initiates HH signal transduction is to regulate cAMP production through the activation of Gαi family heterotrimeric G-proteins.
Gαi as a context-specific modulator of HH signaling. In vitro studies on vertebrate SMO support the ability of SMO to activate a subset of heterotrimeric G-proteins, with strongest effects on those of the Gαi family, for which SHH-induced GTP binding has been demonstrated [57–60]. Activation of Gαi in these systems fulfills the requirements of canonical HH pathway induction, as it can be inhibited by PTCH and/or small-molecule SMO inhibitors, and can be activated by SHH stimulation [57, 60]. SHH target gene induction in cultured fibroblasts is sensitive to pertussis toxin (PTX), a potent Gαi inhibitor, further supporting that Gαi is engaged by vertebrate SMO in response to ligand stimulation [60].
The above studies provide support for involvement of Gαi in HH signal transduction. However, studies performed in differing developmental or cellular contexts failed to identify a role for Gαi in the HH pathway [37, 61–63]. RNAi screens in cultured Drosophila cells did not implicate Gαi as a component of the HH signaling pathway [62, 63], while studies performed in cultured 10T1/2 cells failed to detect changes in intracellular cAMP following SHH stimulation [37]. The latter might be explained by findings that the bulk of HH signaling in vertebrate cells appears to occur in the primary cilium, a small sensory organelle that is present on most vertebrate cell types [64]. Because the volume of the primary cilium is negligible when compared to the body of the cell, localized changes in ciliary cAMP may be undetectable in whole cell lysates.
Conflicting results have also been obtained from in vivo studies examining the role of Gαi in HH signaling. Uncoupling of SMO from Gαi by expression of the PTX catalytic subunit in chick neural tube did not demonstrate compromised SHH-dependent neural cell type specification, suggesting that Gαi is not required for SHH patterning events in this developmental context [61].
Taken together, these seemingly conflicting results raise the possibility that Gαi is required only in certain cellular or tissue contexts during development. This suggestion is supported by the observation that while chick retinal ganglion axon explants are sensitive to SHH-mediated cAMP modulation and growth suppression, chick neural tube explants are not [56]. Signaling redundancy in specific tissues and/or at distinct developmental time points may also account for the apparent lack of Gαi involvement in HH signaling in some in vivo systems. This possibility is supported by work in both Drosophila and cultured vertebrate cells, which show multiple activating signals and feed-forward loops originating from SMO following ligand stimulation [26, 65, 66]. Further studies are required to ascertain if these additional SMO signals are dominant, or can compensate when Gαi is compromised.
A SMO-Dependent G-Protein Signaling Network
Based on a series of elegant biochemical reconstitution experiments, heterotrimeric G-proteins are proposed to function as ligand-gated switches [53]. Ligand stimulation triggers the GPCR to serve as a guanine nucleotide exchange factor (GEF) for its partner heterotrimeric G-protein, allowing the GDP-bound Gα subunit to bind GTP and become activated. Upon activation, Gα was originally thought to dissociate from its Gβγ subunits and interact with its effector(s) through random collision along the plasma membrane. Initially the only known effector of G-proteins was adenylate cyclase (AC), the enzyme that converts ATP into cAMP. As such, the Gα subunit responsible for stimulating AC, and driving cAMP production was named Gαs, while the Gα subunit that inhibited AC activity to lower cAMP production was named Gαi [53]. Gα subunits were originally believed to be attenuated by their own intrinsic GTPase activity to return the Gα to its inactive GDP-bound form, thereby allowing it to reassociate with its partner Gβγ subunits.
The identification of additional G-proteins, the advent of molecular biology, and the subsequent investigation of G-protein function in vivo culminated to show that more regulators of the G-protein GTPase cycle were required than initially predicted by the in vitro model [67, 68]. These regulators consist of non-receptor GEFs that promote GDP release, novel inhibitors of GDP release (GDI), regulators of G-protein signaling (RGS) that significantly increase the GTPase activity of the Gα subunit, and GRKs that function to desensitize the activated GPCR and/or propagate receptor signaling [69]. Numerous Gα and Gγ subunits have now been described and demonstrated to be capable of signaling themselves, regulating their own spectrum of specific effectors [69].
The discovery that SMO can signal as a bona fide GPCR has the potential to quickly expand the number of signaling proteins regulated by SMO, to include those that act as part of a SMO regulated G-protein signaling network (SGN). Thus, we anticipate that like other GPCRs SMO might regulate a large network of signaling proteins, including other G-proteins, and modifiers and effectors of these G-proteins. We discuss below evidence for such a network of regulators, and what some of the novel components of this SGN might be.
G-protein modulators. Although overexpression of activated Gαi in vivo resulted in strong HH gain of function phenotypes, we found that attenuation of Gαi function triggered only mild HH loss of function phenotypes [52]. These weak phenotypes might indicate that another Gα gene product, of which there are five in Drosophila, functions in a redundant manner with Gαi during Drosophila development. A likely candidate gene for this redundant function is Gαo, a member of the Gαi family that can function redundantly with Gαi in other systems [53]. Further, the mammalian homologue of the Drosophila Concertina α subunit, Gα12/13, has been implicated in SHH-mediated regulation of the small GTP-binding protein Rho [58]. Although our survey of three Gα gene products did not implicate Gαs in HH pathway regulation, a genome-wide screen in cultured Drosophila cells showed that knocking down Gαs could enhance HH signaling activity [63]. Further work is needed to determine if Gαs might represent a feedback mechanism that resets the basal level of cAMP after HH induces a decrease in cellular cAMP concentration via Gαi.
Another group of G-proteins we anticipate will serve as novel SMO effectors are the Gβγ subunits of its partner heterotrimeric G-protein(s). At a minimum, these proteins could function as negative regulators of Gαi by acting as GDIs [69]. The Gβγ subunits might also have the capacity to regulate their own novel set of effectors in the HH signaling cascade and/or modulate effectors that they share with their Gα subunits. One such effector, AC, is stimulated by Gβγ proteins [69]. Therefore, like Gαs, Gβγ subunits activated in response to HH might be utilized to reverse Gαi-induced decreases in intracellular cAMP.
GPCR kinases and arrestins. As discussed above, phosphorylation of GPCRs on the cytoplasmic carboxyl-terminal tail is a common event following ligand stimulation. Phosphorylation regulates wide-ranging events including receptor subcellular localization, association with downstream pathway effectors, and commonly serves to recruit β-arrestin type adaptor proteins, which can propagate receptor signaling and/or drive receptor internalization and desensitization [70, 71]. Phosphorylation of activated receptors is driven primarily by the GRK family of kinases. GRK2 regulation of SMO follows a well-established GPCR paradigm: GRK2 phosphorylation triggers β-arrestin recruitment, which drives SMO to clathrin-coated pits where it undergoes activation-dependent internalization [72]. Interestingly, rather than desensitizing SMO to attenuate transduction of the HH signal, GRK-mediated phosphorylation and subsequent β-arrestin recruitment appear to regulate positive steps in HH signaling. Co-expression of GRK2 with SMO in cultured C3H10T1/2 cells enhances SMO-dependent activation of GLI, while GRK knockdown in cultured HEK293 cells attenuates SMO signaling in response to the SMO agonist SAG [50, 73]. An in vivo requirement for GRK2 in HH signaling was confirmed through studies analyzing zebrafish and mice lacking GRK2 function. In both cases, these animals demonstrated developmental phenotypes consistent with HH loss of function [73].
Drosophila GRK2 (dGRK2) has been demonstrated to be both a positive regulator of SMO signaling as well as a HH target gene, suggesting that it functions in a ligand-induced feed-forward loop [74]. As is the case in vertebrate systems, phosphorylation of SMO by dGRK2 results in both β-arrestin recruitment and SMO internalization in HH receiving cells [49]. dGRK2 appears to function only on activated SMO that has transduced a signal in response to ligand, as dGRK2 overexpression in wing discs triggers the removal of SMO from the plasma membrane without attenuating HH target gene induction. dGRK2-mediated internalization of SMO is independent of PTCH-driven removal of SMO from the plasma membrane in cells not receiving the HH signal, further suggesting that dGRK2 functions solely to regulate activated SMO [49]. This supports that SMO plasma membrane localization is regulated in a manner similar to numerous other GPCRs: in the absence of ligand stimulation, SMO undergoes a tonic endocytosis that is regulated by PTCH [75], while ligand stimulated SMO is internalized by the combined activity of dGRK and β-arrestin [49, 72]. As is the case with vertebrate SMO, assembly of the dGRK/β-arrestin complex on dSMO appears to be a positive regulatory event, despite it resulting in the eventual removal of SMO from the plasma membrane [49, 72]. Taken together, these studies highlight the importance of an evolutionarily conserved regulatory complex that assembles in response to ligand-induced GRK phosphorylation of SMO, of which β-arrestin appears to be paramount.
Protein kinase A. PKA was originally identified as a cAMP stimulated protein kinase, consisting of two regulatory subunits and two catalytic subunits [76]. The regulatory subunit inhibits the activity of the catalytic subunit, and this repression is released when the regulatory subunit binds to cAMP. PKA phosphorylates a broad spectrum of substrates, resulting in many diverse biological outputs. The various functions of PKA are thought to be spatially distinct, with PKA binding to its substrates and regulators on scaffolding proteins called A kinase anchoring proteins (AKAPs) [77]. AKAPs cluster relevant GPCRs, G-proteins, kinases, and other downstream effectors to discrete localizations within a cell [69, 77]. COS2 has been demonstrated to associate with SMO, downstream HH effectors, as well as with PKA and CK1 [20, 21, 24, 25]. As such, we hypothesized that COS2 might act as a nexus for HH signaling in a manner akin to that of AKAP proteins. We tested this hypothesis and noted that Gαi and COS2 do associate, and that this association was enhanced by HH [52]. It is, therefore, likely that COS2 acts as a scaffolding protein to recruit SMO, Gαi, and PKA, and likely, analogous to how AKAPs function, might also act to locally modulate the levels of cAMP.
PKA was initially shown to function as a negative regulator of HH signaling, phosphorylating CI in order to convert it to its repressor form [78, 79]. It was later identified as a positive regulator of HH signaling, through its ability to phosphorylate and stabilize SMO to result in SMO enriching at the plasma membrane in a highly active form [25, 46, 80]. Thus, PKA plays two seemingly opposite roles in HH signaling – in the absence of HH it acts to keep the HH pathway in its off-state and in the presence of HH functions to convert SMO into its active form. Consistent with the important role PKA plays in HH signaling, Costal1 (COS1) mutations, which enhance the phenotype of COS2 mutations, were recently shown to encode mutations in both the regulatory and catalytic subunits of PKA [81].
It has been suggested that the role PKA plays in HH signaling is cAMP independent [78, 79]. This hypothesis was presented to explain the observation that a mutant mouse PKA catalytic subunit was able to rescue a PKA null mutation in Drosophila. These experiments assumed that the mutant PKA catalytic subunit would be unable to associate with the regulatory subunit of Drosophila PKA. However, recent demonstrations of SMO coupling to Gαi and regulating cAMP levels suggests that a cAMP independent role of PKA in HH signaling may not be correct [52, 60, 65]. Moreover, the identification of a COS1 mutation encoding a PKA regulatory subunit, which modulates HH signaling, is consistent with a cAMP-dependent activation of PKA [81].
Small-Molecule Modulators of SMO
Mice engineered to lack SHH die shortly after birth and exhibit a wide range of developmental defects, including cyclopia [82]. A similar phenotype was observed in offspring of livestock that ingested the corn lily Veratrum californica. Two groups recognized the similarities between these phenotypes and tested the hypothesis that a chemical derived from this plant, cyclopamine, functioned as an inhibitor of HH signaling [83, 84]. Cyclopamine turned out to be a potent inhibitor of HH signaling, in vitro and in vivo, and was subsequently shown to bind directly to the heptahelical bundle of SMO to functionally antagonize its signaling capability [85]. Consistent with SMO facilitating a rate-limiting step in HH pathway activation, numerous small-molecule screens for novel HH inhibitors have identified distinct SMO antagonists [9]. Many of these SMO inhibitors act in a competitive manner with cyclopamine for binding to SMO, supporting that they bind the 7TM segments. However, some of these SMO modulators bind to SMO in a non-competitive manner and/or activate SMO, suggesting that SMO may have a number of different small-molecule binding sites, as is the case with numerous GPCRs [43].
One basic tenet of pharmacology is that drugs themselves do not possess intrinsic biological properties, but rather can only act to modify existing biological processes [86]. Thus, the identification of small-molecule modulators of SMO implied the existence of endogenous SMO modulators. Furthermore, it has been known for a number of years that the HH receptor PTCH has significant homology with a family of physiological pumps in bacteria, leading to the speculation that PTCH functions to regulate the concentration of such an endogenous SMO modulator [13, 87]. Consistent with this homology, a recent study using a mixed-cell culturing system provided evidence for a lipophilic molecule being pumped into the culture medium in a PTCH-dependent manner [88]. This molecule was identified as the oxysterol, pro-vitamin D3, which was demonstrated to bind SMO in manner similar to that of cyclopamine. Purified pro-vitamin D3 inhibited HH activity, both in vitro and in vivo, with a potency similar to that of cyclopamine [88]. This was one of the first identifications of an endogenous SMO modulator, in this case an antagonist. Two other groups subsequently identified oxysterol molecules that functioned as HH activators [89, 90], suggesting that, like numerous GPCRs, SMO activity is controlled by endogenous small-molecule ligands.
Future Directions
As the critical role(s) that HH signaling plays in tumor growth and progression continues to emerge, and the clinical use of SMO antagonists increases, the impact of on-target adverse effects is likely to become evident. For example, one SMO antagonist was recently demonstrated to have significant efficacy against medulloblastoma [91], but to induce growth defects when administered to young mice [92]. This study was performed to reveal potential problems that may be encountered by inhibiting a developmentally relevant signaling pathway in a pediatric population, the most common class of patients presenting with medulloblastoma [93]. Long bones of the limbs of animals exposed to this compound during early development were found to be significantly shortened, an effect likely due to specific inhibition of Indian HH-regulated bone growth [92]. This observation underscores the importance of delineating all the signaling events immediately downstream of SMO, as one could anticipate that various classes of small-molecule SMO inhibitors might affect distinct signaling arms. Moreover, classes of SMO antagonists that only inhibit a distinct subset of SMO effectors, such as the SGN effectors, might be used clinically for specific classes of cancer patients. Ideally, such compounds would inhibit the effectors relevant to tumor growth while having minimal impact on effectors more relevant to the role HH plays in tissue homeostasis. Such drugs would be particularly useful to medulloblastoma patients, whose ability to take at least a subset of the SMO antagonist currently in clinical trials would be severely compromised by on-target developmental defects.
References
Johnson RL et al (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272(5268):1668–1671
Hahn H et al (1996) Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85(6):841–851
Unden AB et al (1996) Mutations in the human homologue of Drosophila patched (PTCH) in basal cell carcinomas and the Gorlin syndrome: different in vivo mechanisms of PTCH inactivation. Cancer Res 56(20):4562–4565
Wolter M, Reifenberger J, Sommer C, Ruzicka T, Reifenberger G (1997) Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 57(13):2581–2585
Lam CW et al (1999) A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene 18(3):833–836
Xie J et al (1998) Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391(6662):90–92
Teglund S, Toftgard R (1805) Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta 2:181–208
Lum L, Beachy PA (2004) The Hedgehog response network: sensors, switches, and routers. Science 304(5678):1755–1759
Robbins DJ, Goetz JA, Yuan Z, Stegman MA (2005) Inhibitors of the hedgehog signal transduction pathway. Curr Cancer Ther Rev 1:227–288
Xie J (2008) Hedgehog signaling pathway: development of antagonists for cancer therapy. Curr Oncol Rep 10(2):107–113
Jia J, Jiang J (2006) Decoding the Hedgehog signal in animal development. Cell Mol Life Sci 63(11):1249–1265
Ingham PW, McMahon AP (2001) Hedgehog signaling in animal development: paradigms and principles. Genes Dev 15(23):3059–3087
Taipale J, Cooper MK, Maiti T, Beachy PA (2002) Patched acts catalytically to suppress the activity of Smoothened. Nature 418(6900):892–897
Alcedo J, Noll M (1997) Hedgehog and its patched-smoothened receptor complex: a novel signalling mechanism at the cell surface. Biol Chem 378(7):583–590
Incardona JP, Gruenberg J, Roelink H (2002) Sonic hedgehog induces the segregation of patched and smoothened in endosomes. Curr Biol 12(12):983–995
Martin V, Carrillo G, Torroja C, Guerrero I (2001) The sterol-sensing domain of Patched protein seems to control Smoothened activity through Patched vesicular trafficking. Curr Biol 11(8):601–607
Denef N, Neubuser D, Perez L, Cohen SM (2000) Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102(4):521–531
Robbins DJ, Hebrok M (2007) Hedgehogs: la dolce vita. Workshop on Hedgehog-Gli signaling in cancer and stem cells. EMBO Rep 8(5):451–455
Jia J, Tong C, Jiang J (2003) Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev 17(21):2709–2720
Lum L et al (2003) Hedgehog signal transduction via Smoothened association with a cytoplasmic complex scaffolded by the atypical kinesin, Costal-2. Mol Cell 12(5):1261–1274
Ogden SK et al (2003) Identification of a functional interaction between the transmembrane protein Smoothened and the kinesin-related protein Costal2. Curr Biol 13(22):1998–2003
Ruel L, Rodriguez R, Gallet A, Lavenant-Staccini L, Therond PP (2003) Stability and association of Smoothened, Costal2 and Fused with Cubitus interruptus are regulated by Hedgehog. Nat Cell Biol 5(10):907–913
Endoh-Yamagami S et al (2009) The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr Biol 19(15):1320–1326
Robbins DJ et al (1997) Hedgehog elicits signal transduction by means of a large complex containing the kinesin-related protein costal2. Cell 90(2):225–234
Zhang W et al (2005) Hedgehog-regulated Costal2-kinase complexes control phosphorylation and proteolytic processing of Cubitus interruptus. Dev Cell 8(2):267–278
Claret S, Sanial M, Plessis A (2007) Evidence for a novel feedback loop in the Hedgehog pathway involving Smoothened and Fused. Curr Biol 17(15):1326–1333
Farzan SF et al (2009) A quantification of pathway components supports a novel model of Hedgehog signal transduction. J Biol Chem 284(42):28874–28884
Costanzi S, Siegel J, Tikhonova IG, Jacobson KA (2009) Rhodopsin and the others: a historical perspective on structural studies of G protein-coupled receptors. Curr Pharm Des 15(35):3994–4002
Hofmann KP et al (2009) A G protein-coupled receptor at work: the rhodopsin model. Trends Biochem Sci 34(11):540–552
Morris MB, Dastmalchi S, Church WB (2009) Rhodopsin: structure, signal transduction and oligomerisation. Int J Biochem Cell Biol 41(4):721–724
Nusslein-Volhard C, Wieschaus E, Kluding H (1984) Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster: zygotic loci on the second chromosome. Roux’s Arch Dev Biol 192:267–282
Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE (1996) The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 86(2):221–232
Bhanot P et al (1996) A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 382(6588):225–230
Dann CE et al (2001) Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 412(6842):86–90
Wang Y et al (1996) A large family of putative transmembrane receptors homologous to the product of the Drosophila tissue polarity gene frizzled. J Biol Chem 271(8):4468–4476
Moro S, Hoffmann C, Jacobson KA (1999) Role of the extracellular loops of G protein-coupled receptors in ligand recognition: a molecular modeling study of the human P2Y1 receptor. Biochemistry 38(12):3498–3507
Murone M, Rosenthal A, de Sauvage FJ (1999) Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr Biol 9(2):76–84
Aanstad P et al (2009) The extracellular domain of Smoothened regulates ciliary localization and is required for high-level Hh signaling. Curr Biol 19(12):1034–1039
Nakano Y et al (2004) Functional domains and sub-cellular distribution of the Hedgehog transducing protein Smoothened in Drosophila. Mech Dev 121(6):507–518
van den Heuvel M, Ingham PW (1996) Smoothened encodes a receptor-like serpentine protein required for hedgehog signalling. Nature 382(6591):547–551
Yeagle PL, Albert AD (2003) A conformational trigger for activation of a G protein by a G protein-coupled receptor. Biochemistry 42(6):1365–1368
Reifenberger J et al (1998) Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 58(9):1798–1803
Bockaert J, Pin JP (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J 18(7):1723–1729
Remsberg JR, Lou H, Tarasov SG, Dean M, Tarasova NI (2007) Structural analogues of smoothened intracellular loops as potent inhibitors of Hedgehog pathway and cancer cell growth. J Med Chem 50(18):4534–4538
Apionishev S, Katanayeva NM, Marks SA, Kalderon D, Tomlinson A (2005) Drosophila Smoothened phosphorylation sites essential for Hedgehog signal transduction. Nat Cell Biol 7(1):86–92
Jia J, Tong C, Wang B, Luo L, Jiang J (2004) Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432(7020):1045–1050
Zhang C, Williams EH, Guo Y, Lum L, Beachy PA (2004) Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc Natl Acad Sci USA 101(52):17900–17907
Pierce KL, Lefkowitz RJ (2001) Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci 2(10):727–733
Cheng S, Maier D, Neubueser D, Hipfner DR (2010) Regulation of smoothened by Drosophila G-protein-coupled receptor kinases. Dev Biol 337(1):99–109
Meloni AR et al (2006) Smoothened signal transduction is promoted by G protein-coupled receptor kinase 2. Mol Cell Biol 26(20):7550–7560
Zhao Y, Tong C, Jiang J (2007) Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450(7167):252–258
Ogden SK et al (2008) G protein Galphai functions immediately downstream of Smoothened in Hedgehog signalling. Nature 456(7224):967–970
Gilman AG (1987) G proteins: transducers of receptor-generated signals. Annu Rev Biochem 56:615–649
Davis RL, Kiger JA Jr (1981) Dunce mutants of Drosophila melanogaster: mutants defective in the cyclic AMP phosphodiesterase enzyme system. J Cell Biol 90(1):101–107
Guichard A, Park JM, Cruz-Moreno B, Karin M, Bier E (2006) Anthrax lethal factor and edema factor act on conserved targets in Drosophila. Proc Natl Acad Sci USA 103(9):3244–3249
Trousse F, Marti E, Gruss P, Torres M, Bovolenta P (2001) Control of retinal ganglion cell axon growth: a new role for Sonic hedgehog. Development 128(20):3927–3936
DeCamp DL, Thompson TM, de Sauvage FJ, Lerner MR (2000) Smoothened activates Galphai-mediated signaling in frog melanophores. J Biol Chem 275(34):26322–26327
Kasai K et al (2004) The G12 family of heterotrimeric G proteins and Rho GTPase mediate Sonic hedgehog signalling. Genes Cells 9(1):49–58
Masdeu C et al (2006) Identification and characterization of Hedgehog modulator properties after functional coupling of Smoothened to G15. Biochem Biophys Res Commun 349(2):471–479
Riobo NA, Saucy B, Dilizio C, Manning DR (2006) Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci USA 103(33):12607–12612
Low WC et al (2008) The decoupling of Smoothened from Galphai proteins has little effect on Gli3 protein processing and Hedgehog-regulated chick neural tube patterning. Dev Biol 321(1):188–196
Lum L et al (2003) Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science 299(5615):2039–2045
Nybakken K, Vokes SA, Lin TY, McMahon AP, Perrimon N (2005) A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat Genet 37(12):1323–1332
Huangfu D, Anderson KV (2005) Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA 102(32):11325–11330
Chinchilla P, Xiao L, Kazanietz MG, Riobo NA (2010) Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 9(3):570–579
Ogden SK et al (2006) Smoothened regulates activator and repressor functions of Hedgehog signaling via two distinct mechanisms. J Biol Chem 281(11):7237–7243
Birnbaumer L (2007) Expansion of signal transduction by G proteins. The second 15 years or so: from 3 to 16 alpha subunits plus betagamma dimers. Biochim Biophys Acta 1768(4):772–793
Gurevich VV, Gurevich EV (2008) Rich tapestry of G protein-coupled receptor signaling and regulatory mechanisms. Mol Pharmacol 74(2):312–316
Malbon CC (2005) G proteins in development. Nat Rev Mol Cell Biol 6(9):689–701
Lefkowitz RJ, Whalen EJ (2004) Beta-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol 16(2):162–168
Tobin AB (2008) G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol 153(Suppl 1):S167–S176
Chen W et al (2004) Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science 306(5705):2257–2260
Philipp M et al (2008) Smoothened signaling in vertebrates is facilitated by a G protein-coupled receptor kinase. Mol Biol Cell 19(12):5478–5489
Molnar C, Holguin H, Mayor F Jr, Ruiz-Gomez A, de Celis JF (2007) The G protein-coupled receptor regulatory kinase GPRK2 participates in Hedgehog signaling in Drosophila. Proc Natl Acad Sci USA 104(19):7963–7968
Zhu AJ, Zheng L, Suyama K, Scott MP (2003) Altered localization of Drosophila Smoothened protein activates Hedgehog signal transduction. Genes Dev 17(10):1240–1252
Krebs EG, Beavo JA (1979) Phosphorylation-dephosphorylation of enzymes. Annu Rev Biochem 48:923–959
Dessauer CW (2009) Adenylyl cyclase – A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol 76(5):935–941
Jiang J, Struhl G (1995) Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80(4):563–572
Li W, Ohlmeyer JT, Lane ME, Kalderon D (1995) Function of protein kinase A in hedgehog signal transduction and Drosophila imaginal disc development. Cell 80(4):553–562
Zhou Q, Apionishev S, Kalderon D (2006) The contributions of protein kinase A and smoothened phosphorylation to hedgehog signal transduction in Drosophila melanogaster. Genetics 173(4):2049–2062
Collier LS, Suyama K, Anderson JH, Scott MP (2004) Drosophila Costal1 mutations are alleles of protein kinase A that modulate hedgehog signaling. Genetics 167(2):783–796
Chiang C et al (1996) Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383(6599):407–413
Cooper MK, Porter JA, Young KE, Beachy PA (1998) Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280(5369):1603–1607
Incardona JP, Gaffield W, Kapur RP, Roelink H (1998) The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 125(18):3553–3562
Chen JK, Taipale J, Cooper MK, Beachy PA (2002) Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 16(21):2743–2748
Hardman J, Limbird L, Gilman A (2001) Goodman & Gilman’s the pharmacological basis of therapeutics, 10th edn. McGraw-Hill, New York
Ma Y et al (2002) Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell 111(1):63–75
Bijlsma MF et al (2006) Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol 4(8):e232
Corcoran RB, Scott MP (2006) Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci USA 103(22):8408–8413
Dwyer JR et al (2007) Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J Biol Chem 282(12):8959–8968
Romer JT et al (2004) Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell 6(3):229–240
Kimura H, Ng JM, Curran T (2008) Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 13(3):249–260
Gottardo NG, Gajjar A (2008) Chemotherapy for malignant brain tumors of childhood. J Child Neurol 23(10):1149–1159
Acknowledgments
We thank C. Carroll, D. Fei, S. Marada, and S. Singh for their comments on the manuscript. This work was supported by NIH grants 1RO1CA82628 and 1RO1GM064011 (DJR), March of Dimes MOD5-FY10-6 (SKO), and by the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Robbins, D.J., Ogden, S.K. (2011). Smoothened Signaling Through a G-Protein Effector Network. In: Xie, J. (eds) Hedgehog signaling activation in human cancer and its clinical implications. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-8435-7_3
Download citation
DOI: https://doi.org/10.1007/978-1-4419-8435-7_3
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-8434-0
Online ISBN: 978-1-4419-8435-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)