
Abstract
In this chapter we shall review recent developments in computational vaccinology, highlighting work in the area of epitope and antigen identification, and the discovery of delivery vectors and adjuvants. These diverse activities all have the potential to reduce significantly the laboratory resource needed for efficient vaccine discovery. We discuss how computational analysis of pathogenic genomes by epitope mapping and reverse vaccinology can provide viable vaccine targets.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Major Histocompatibility Complex
- Visceral Leishmaniasis
- Cell Epitope
- Major Histocompatibility Complex Molecule
- CCR4 Antagonist
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
For vaccines, it is the best of times and the worst of times. Mass vaccination and public sanitation are the two most effective prophylactic treatments forestalling the depredations of infectious disease. The greatest part of the vaccine story is that of smallpox. At its height, Smallpox killed 10% of Swedish children within their first year. In London, more than 3,000 died in a single smallpox epidemic in 1746, and during the period of 1760–1770, the city lost another 4% of its population to the disease. Even as recently as the late 1960s, there were 10–12 million cases in 31 countries, with two million deaths annually. Yet the disease is, apart from a few hopefully well-guarded stockpiles, a thing of the past. There have been no cases for most of last 30 years. It has been completely eradicated. Poliomyelitis or Polio has also been the target for a worldwide campaign designed to eradicate the disease by the year 2000. A program undertaken by the Pan American Health Organization eliminated polio in the Western Hemisphere in 1991. The Global Polio Eradication Program has dramatically reduced poliovirus transmission throughout the world. In 2003, only 784 confirmed cases of polio were reported globally. Today Polio remains endemic in only four countries: Nigeria, Afghanistan, Pakistan, and India.
Yet millions still die from disease preventable through vaccination. Infectious diseases are to blame for around 25% of global mortality, particularly in children under five. Annual figures are stark: pertussis accounts for 294,000 (aged under 5); tetanus for 198,000 (under 5) and 15,000 (aged over 5); Hib for 386,000 (under 5); Hepatitis B for 600,000 (over 5); Yellow Fever for 15,000 (under 5) and 15,000 (over 5); and diphtheria for 4,000 (under 5). Projections for as-yet-not-universally available vaccines estimate a consequently greater saving of human life: 449,000 for rotavirus vaccine and 1,612,000 for Pneumococcus vaccine. For example, influenza with an annual global estimate of half a million deaths. However, perhaps the most lamentable situation is Measles, which accounts for 540,000 (under 5) and 70,000 (over 5). Measles is an acute viral disease. The Arabic physician Rhazes referred to it, saying that it was ‘more dreaded than smallpox’. The eldest son of the famous late Victorian and Edwardian author, H Rider Haggard, who, among 68 novels, wrote Allan Quatermain and She, died from the disease aged 10. Oliva Dahl, daughter of Roald, died aged 7 from measles-induced encephalitis.
The leading annual causes of death are 2.9 millions for tuberculosis; 2.5 million for diarrhoeal illnesses, especially rotaviruses; a rapidly escalating 2.3 million for HIV/AIDS; and 1.08 millions of deaths for malaria. There are no effective vaccines for HIV (Girard et al. 2006) and Malaria (Vekemans and Ballou 2008), two of the WHO’s big three global killers; indeed, there is little hope that such vaccines will appear in the foreseeable future. And the only vaccine licensed for the third major world disease, tuberculosis, is of limited efficacy (de Lisle et al. 2005). Add to this the 35 new, previously unknown infectious diseases identified in the past 25 years: HIV, Marburg’s disease, SARS, Dengue, West Nile, and over 190 human infections with potentially pandemic H5N1 influenza.
Many infections caused by viruses have proved stubborn and recalcitrant threats to human health and wellbeing. 350 million people carry hepatitis B (HBV). 170 million carry Hepatitis C (HCV), and 40 million carry human immunodeficiency virus type 1 (HIV-1). Each year, 5–15% of the world’s population become infected by a new variant of the influenza virus, causing 250,000–500,000 deaths. Latent Bacterial infection can be even higher: there are, for example, over two billion people infected with TB. It is a commonly held conception that new infectious diseases will emerge continually throughout the twenty-first century. We are threatened by parasitic diseases such as malaria, visceral leishmaniasis, tuberculosis, and emerging zoonotic infections, such as H5N1; antibiotic-resistant bacteria; and bioterrorism; a threat compounded by a growing world population, overcrowded cities, increased travel, climate change, and intensive food production.
Thus it is the best of times for vaccines because of their uncompromising success and the worst of times because so much remains to be done. There are now more than 30 licensed individual vaccines targeted against 26 infectious diseases, most of which are viral or bacterial in nature. About half of these 30 vaccines are in common use, and are, in the main, employed to prevent childhood infections. In the First World, annual mortality for diseases such as polio, diphtheria, or measles is less than 0.1%; and the lasting effects of vaccination work to greatly reduce the morbidity and mortality of disease, often conferring lifetime protection. Most vaccines target childhood infections or are used by travellers to tropical or sub-tropical regions; a significant minority combat disease in the developing world.
Within the pharmaceutical industry and academia, vaccines are also seeing the best and worst of times. Persistent infection, which includes HIV, Hepatitis B, hepatitis C, and TB, occurs when a pathogen evades or subverts T cell responses, is a key therapeutic target. At the other extreme are benign yet economically important infections, such as the common cold. Respiratory track infections remain the major cause of community morbidity and hospitalisation in the developed world: about 60% of GP referrals and cause the loss of a huge number of working days. Sporadic or epidemic respiratory infections are caused by over 200 distinct viruses, including coronaviruses, rhinoviruses, respiratory syncytial virus (better known as RSV), parainfluenza virus, influenza A and B, and cytomegalovirus. Anti-Allergy vaccination also offers great potential for successful commercial exploitation. This often relies on allergen-specific immunotherapy (STI) (Palma-Carlos et al. 2006), where a patient is administered increasing quantities of allergen to augment their natural tolerance. STI, though often effective, is very time consuming and is not universally applicable. Recombinant hypo-allergenic allergens are also of interest, as they can target specific immune cells. New agents for the prophylaxis and treatment of allergic disease are legion: recombinant proteins, peptides, immunomodulatory therapy, and DNA vaccines, which are particularly promising tools. Several anti-allergy DNA vaccines are being developed: including optimised expression of allergen genes, CpG-enrichment of delivery vectors, and the targeting of hypoallergenic DNA vaccines. Vaccines against the common cold or anti-allergy vaccines lie close to so-called life-style vaccines. None of these vaccines necessarily saves lives but does reduce hugely important economic effects of disease morbidity. Life-style vaccines target dental caries and drug addiction, as well as genetic and civilisation diseases, such as obesity.
Vaccination is also being used to tackle cancer. Gardasil, the new human papillomavirus vaccine (Hung et al. 2008), was licensed in 2006 with the goal of saving 4,000 deaths a year from cervical cancer. Cancer is the second greatest cause of death in the developed world after cardiovascular disease; yet most of the 250,000 deaths from cervical cancer occur in the Third World. Cancer treatment typically involves a combination of chemotherapy, radiotherapy, and surgery. While treating primary tumours this way is largely successful, preventing the metastatic spread of disease is not. Cancer vaccines are attractive, both clinically and commercially, since they exploit immunity’s ability to recognise and destroy tumours. Tumour cells express several mutated or differentially expressed antigens, enabling the immune system to discriminate between non-malignant and cancerous cells. Tumour antigens form the basis of both subunit and epitope-based vaccines. Host immune system responses to tumour-antigen cancer vaccines are often weak, necessitating the use of adjuvants.
Thus we can see that, biomedically speaking at least, vaccines are indeed having the best and worst of times: best because of the enormous opportunity for them to treat an ever-widening tranche of diseases and worst because of the inadequacies of existing techniques used to foster vaccine development. Vaccinology has, until relatively recently, been a primarily empirical science, relying on tried-and-tested – yet poorly understood – approaches to vaccine development. As a consequence of this, few effective vaccines were developed and deployed during the 150 years following Jenner: most targets remained inaccessible to science to the emerging science of vaccinology. The success of a vaccine can be measured by its strength, its specificity, the duration of the immune response, and its capacity to create immunological memory. A vaccine is a molecular or supramolecular agent which can elicit specific protective immunity and ultimately mitigate the effect of subsequent infection. Vaccination is the use of a vaccine, in whatever form, to produce active prophylactic immunity in a host organism.
Vaccines have taken many forms. Until recently, they have been attenuated or inactivated whole pathogen vaccines such as anti-tuberculosis BCG or Sabin’s vaccine against Polio. Safety difficulties have led to the subsequent development of other strategies for vaccine development. The most successful alternative has focused on the antigen – or subunit – vaccine, such as recombinant Hepatitis B vaccine (Ebo et al. 2008). Vaccines based around sets of epitopes have also gained ground in recent years. They can be delivered into the host in many ways: as naked DNA vaccines, using live viral or bacterial vectors, and via antigen-loaded antigen presenting cells. Adjuvants are substances, such as alum, which are used with weak vaccines to increase immune responses (O’Hagan et al. 2001).
With more and more pathogen genomes being fully or partially determined, it has become imperative to develop reliable in silico methods able to identify potential vaccine candidates within microbial genomes. While it is possible to assess in the laboratory those properties of vaccine which make it successfully, it is not practical to do on the scale of a large pathogen genome. Immunomics is a solution to this dilemma. It is a post-genomic systems biology approach to immunology that explores mechanistic aspects of the immune system (De Groot 2006). It subsumes immunoinformatics and computational vaccinology, combining several fields, including genomics, proteomics, immunology and clinical medicine. To date, a key focus of immunomics has been the development of algorithms for the design and discovery of new vaccines. Here we outline currently available techniques and software for vaccine discovery as well as examples of how such algorithms can be applied. We concentrate on four areas: antigen prediction, epitope prediction, vector design, and adjuvant identification.
Epitope Prediction
Complex microbial pathogens, such as Mycobacterium tuberculosis, can interact within the immune system in a multitude of ways (McMurry et al. 2005). For a vaccine to be effective it must invoke a strong response from both T Cells and B Cells; therefore, epitope mapping is a central issue in their design. In silico prediction methods can accelerate epitope discovery greatly. B Cell and T Cell epitope mapping has led to the predictive scanning of pathogen genomes for potential epitopes (Pizza et al. 2000a). There are over 4,000 proteins in the TB genome; this means that experimental analysis of host-pathogen interactions would be prohibitive in terms of time, labour, and expense.
T Cell Epitope Prediction
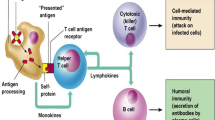
T cell epitopes are antigenic peptide fragments derived from a pathogen that, when bound to a Major Histocompatibility Complex (MHC) molecule, interact with T Cell receptors after transport to the surface of an Antigen-Presenting Cell. If sufficient quantities of the epitope are presented, the T Cell may trigger an adaptive immune response specific for the pathogen. MHC Class I and Class II molecules form complexes with different types of peptide. The Class I molecule binds a peptide of 8–15 amino acids in length within a single closed groove. The peptide is secured largely through interactions with anchoring residues at the N- and C-termini of the peptide, while the central region is more flexible (Rammensee et al. 1999). Class II peptides vary in length from 12 to 25 amino acids and are bound by the protrusion of peptide side chains into cavities within the groove and through a series of hydrogen bonds formed between the main chain peptide atoms and the side chains atoms of the MHC molecule (Jardetzky et al. 1996). Unlike the Class I molecule, where the binding site is closed at either end, the peptide can extend out of both open ends of the binding groove.
Experimentally determined IC50 and BL50 affinity data have been used to develop a variety of MHC-binding prediction algorithms, which can distinguish binders from non-binders based on the peptide sequence. These include motif-based systems, Support Vector Machines (SVMs) (Donnes and Elofsson 2002; Liu et al. 2006; Wan et al. 2006), Hidden Markov Models (HMMs) (Noguchi et al. 2002), QSAR analysis (Doytchinova et al. 2005), and structure-based approaches (Davies et al. 2006; Davies et al. 2003; Wan et al. 2004). MHC-binding motifs are a straightforward and easily comprehended method of epitope detection, yet produce many false-positive and many false-negative results. Support Vector Machines (SVMs) are machine learning algorithms based on statistical theory that seeks to separate data into two distinct classes (in this case binders and non-binders). HMMs are statistical models where the system being modelled is assumed to be a Markov process with unknown parameters. In an HMM, the internal state is not visible directly, but variables influenced by the state are. HMMs aim to determine the hidden parameters from observable ones. An HMM profile can be used to determine those sequences with ‘binder-like’ qualities. QSAR analysis techniques have been used to refine the peptide interactions with the MHC Class I groove by incrementally improving and optimising the individual residue-to-residue interactions within the binding groove. This has led to the design of so-called superbinders that minimise the entropic disruption in the groove and are therefore able to stabilise even disfavoured residues within so-called anchor positions. Finally, molecular dynamics has been used to quantify the energetic interactions between the MHC molecule and peptide for both Class I and Class II by analysis of the three-dimensional structure of the MHC-peptide complex.
Many programs that are able to facilitate the design of optimised vaccines are now available. In this section, some of the most effective algorithms for each form of vaccine design are discussed. For T Cell epitope prediction, many programs are available. A sensible approach for a new user would be to use MHCBench (Salomon and Flower 2006), an interface developed specifically for evaluating the various MHC-binding peptide prediction algorithms. MHCBench allows users to compare the performance of various programs with both threshold-dependent and -independent parameters. The server can also be extended to include new methods for different MHC alleles.
B Cell Epitope Prediction
B cells generate antibodies when stimulated by helper T cells as part of the adaptive immune response. The antibodies act to bind and neutralise pathogenic material from a virus or bacterium. Individual antibodies are composed of two sets of heavy and light chains. Each B cell produces a unique antibody due to the effects of somatic hypermutation and gene segment rearrangement. Those cells, within the primary repertoire whose antibodies convey antigen recognition, are selected for clonal expansion, an iterative process of directed hypermutation and antigen-mediated selection. This facilitates the rapid maturation of antigen-specific antibodies with a high affinity for a specific epitope. A B cell appropriate to deal with a specific infection is selected and cloned to deal with the primary infection, and a population of the B cell is then maintained in the body to combat secondary infection. It is the capacity to produce a huge variety of different antibodies that allows the immune system to deal with a broad range of infections.
B Cell prediction is more problematic due to the difficulties in correctly defining both linear and discontinuous epitopes from the rest of the protein. The epitope of a B Cell is defined by the discrete surface region of an antigenic protein bound by the variable domain of an antibody. The production of specific antibodies for an infection can boost host immunity in the case of both intracellular and extracellular pathogens. The antibody’s binding region is composed of three hypervariable loops that can vary in both length and sequence so that the antibodies generated by an individual cell present a unique interface (Blythe and Flower 2004). All antibodies contain two antigen-binding sites, composed of complementary determining (CDR) loops. The three CDR loops of the heavy and light chains form the ‘paratope’, the protein surface which binds to the antigen. The molecular surface that makes specific contact with the residues of the paratope is termed an ‘epitope’. A B cell epitope can be an entire molecule or a region of a larger structure. The study of the paratope–epitope interaction is a crucial part of immunochemistry, a branch of chemistry that involves the study of the reactions and components on the immune system.
Despite the extreme variability of the region, the antibody-binding site is more hydrophobic than most protein surfaces with a significant predilection for tyrosine residues. B Cell epitopes can be divided into continuous (linear) and discontinuous (conformational), the latter being regions of the antigen separated within the sequence but brought together in the folded protein to form a three-dimensional interface. Another problem with B cell epitopes relates to the fact that they are commonly divided into two groups: continuous epitopes and discontinuous epitopes. Continuous epitopes correspond to short peptide fragments of a few amino acid residues that can be shown to cross-react with antibodies raised against the intact protein. Since the residues involved in antibody binding represent a continuous segment of the primary sequence of the protein they are also referred to as ‘linear’ or ‘sequential’ epitopes. Studies have shown that this class of epitope often contains residues that are not implicated in antibody interaction, while some residues play a more important role than others in antibody binding. Discontinuous epitopes are composed of amino acid residues that are not sequential in the primary sequence of a protein antigen but brought into spatial proximity by the three-dimensional folding of the peptide chain (Greenbaum et al. 2007).
There is considerable interest in developing reliable methods for predicting B cell epitopes. However, to date, the amino acid distribution of the complementary antigen surface has been difficult to characterise, presenting no unique sequential or structural features upon which to base a predictive system. It is partly for this reason that B Cell epitope has lagged far behind T Cell prediction in terms of accuracy but also because most of the data upon which predictions are based remain open to question due to the poorly understood recognition properties of cross-reactive antibodies. One of the central problems with B cell epitope prediction is that the epitopes themselves are entirely context dependent. The surface of a protein is, by definition, a continuous landscape of potential epitopes that is without borders. Therefore both epitope and paratope are fuzzy recognition sites, forming not a single arrangement of specific amino acids but a series of alternative conformations. In this instance, a binary classification of binder and non-binder may simply not reflect the nature of the interaction. A factor also to be considered is that the average paratope consists of only a third of the residues within the CDR loops, suggesting the remaining two-thirds could potentially bind to an antigen with an entirely different protein surface.
Often a short length of amino acids can be classified as a continuous epitope, though in fact it may be a component of a larger discontinuous epitope; this can be a result of the peptide representing a sufficient proportion of the discontinuous epitope to enable cross-reaction with the antibody. Since the majority of antibodies raised against complete proteins do not cross-react with peptide fragments derived from the same protein it is thought that the majority of epitopes are discontinuous. It is estimated that approximately 10% of epitopes on a globular protein antigen are truly continuous in nature. In spite of the this, the majority of research into B cell epitope prediction has focussed largely on linear peptides on the grounds that they are discrete sequences and easier to analyse. This can only be resolved by examination of the three-dimensional structure of the protein where the distinction between the continuous and discontinuous forms is not relevant.
Initial research into B cell epitope prediction looked for common patterns of binding or ‘motifs’ that characterise epitope from non-epitopes. Unfortunately, the wide variety of different epitope surfaces that can be bound made it impossible to determine any such motifs. More sophisticated machine learning approaches such as Artificial Neural Networks have also been applied but never with an accuracy exceeding 60%. More recently, structural analysis of known antigens has been used to determine the surface accessibility of residues as a measure of the probability that they are part of an epitope site. Despite these fundamental limitations, several B Cell epitope prediction programs are available including Discotope (Andersen et al. 2006), 3DEX (Schreiber et al. 2005) and CEP (Kulkarni-Kale et al. 2005). Both CEP and Discotope measure the surface accessibility of residues although neither has been developed to the point where they can identify coherent epitope regions rather than individual residues. A recent review of B cell epitope software (Ponomarenko and Bourne 2007) calculated the AROC curves for the evaluated methods were about 0.6 (indicating 60% accuracy) for DiscoTope, ConSurf (which identifies functional regions in proteins), and PPI-PRED (protein–protein interface analysis) methods, while protein–protein docking methods were in the region of 65% accuracy, never exceeding 70%. The remaining prediction methods assessed were all close to random. In spite of this, the increasing number of available antigen–antibody structures combined with sophisticated techniques for structural analysis suggests a more methodical approach to the study interface will yield a better understanding of what surfaces can and cannot form stable epitopes. The proposed research will take several different approaches to this problem that will lead to a more comprehensive understanding of antibody–antigen interactions.
Computational Identification of Virulence Factors
The word antigen has a wide meaning in immunology. We use it here to mean a protein, specifically one from a pathogenic micro-organism, which evokes a measurable immune response. Pathogenic proteins in bacterial are often acquired, through a process summarised by the epithet horizontal transfer, in groups. Such groups are known as pathogenicity islands. The unusual G+C content of genes and particularly large gene clusters is tantamount to a signature characteristic of genes acquired by horizontal transfer. Genome analyses at the nucleic acid level can thus allow the discovery of pathogenicity islands and the virulence genes they encode.
Perhaps the most obvious antigens are virulence factors (VF): proteins which enable a pathogen to colonise a host or induce disease. Analysis of pathogens – such as Vibrio cholerae or Streptococcus pyogenes – has identified coordinated ‘systems’ of toxins and virulence factors which may comprise over 40 distinct proteins. Traditionally, VFs have been classified as adherence/colonisation factors, invasions, exotoxins, Transporters, iron-binding Siderophores, and miscellaneous cell surface factors. A broader definition, groups VFs into three: ‘true’ VF genes, VFs associated with the expression of ‘true’ VF genes, and VF ‘life-style’ genes required for colonisation of the host (Guzmán et al. 2008).
Several databases exist which archive VFs. The Virulence Factors Database (VFDB) contains 16 characterised bacterial genomes with an emphasis on functional and structural biology and can be searched using text, BLAST, or functional queries (Yang et al. 2008). The ClinMalDB-US database is being establishing following the discovery of multi-gene families encoding VFs within the subtelomeric regions of P. falciparum (Mok et al. 2007) and P. vivax (Merino et al. 2006). TVFac (Los Alamos National Laboratory Toxin & Virulence Factor database) contains genetic information on over 250 organisms and separate records for thousands of virulence genes and associated factors. The Fish Pathogen Database, set up by the Bacteriology & Fish Diseases Laboratory, has identified over 500 virulence genes using fish as a model system. Pathogens studied include Aeromonas hydrophila, Edwardsiella tarda, and many Vibrio species. Candida albicans Virulence Factor (CandiVF) is a small species-specific database that contains VFs which may be searched using BLAST or a HLA-DR Hotspot Prediction server (Tongchusak et al. 2008). PHI-BASE is a noteworthy development, since it seeks to integrate a wide range of VFs from a variety of pathogens of plants and animals (Winnenburg et al. 2008). Obviously, antigens need not be virulence factors and another nascent database is intending to capture a wider tranche of data. We are helping to develop the AntigenDB database [http://www.imtech.res.in/raghava/antigendb/] which will aid considerably this endeavour.
Identifying Antigens In Silico Using Subcellular Location Prediction
Historically, antigens have been supposed to be secreted or exposed membrane proteins accessible to surveillance of the immune system. Subcellular location prediction is thus a key approach to predicting antigens. There are two basic kinds of prediction method: manual construction of rules of what determines subcellular location and the application of data-driven machine learning methods, which determine factors that discriminate between proteins from different known locations. Accuracy differs markedly between different methods and different compartments, mostly due to a paucity of data. Data used to discriminate between compartments include: the amino acid composition of the whole protein; sequence derived features of the protein, such as hydrophobic regions; the presence of certain specific motifs; or a combination thereof.
Different organisms evince different locations. PSORT is a knowledge-based, multi-category prediction method, composed of several programs, for subcellular location (Rey et al. 2005); it is often regarded as a gold standard. PSORT I predicts 17 different subcellular compartments and was trained on 295 different proteins, while PSORT II predicts ten locations and was trained on 1,080 yeast proteins. Using a test set of 940 plant proteins and 2,738 non-plant proteins, the accuracy of PSORT I and II was 69.8% and 83.2%, respectively. There are several specialised versions of PSORT. iPSORT deals specifically with secreted, mitochondrial and chloroplast locations; its accuracy is 83.4% for plants and 88.5% for non-plant. PSORT-B only predicts bacterial subcellular locations. It reports precision values of 96.5% and recall values of 74.8%. PSORT-B is a multi-category method which combines six algorithms using a Bayesian Network.
Among binary approaches, arguably the best method is SignalP, which employs neural networks and predicts N-terminal Spase-I-cleaved secretion signal sequences and their cleavage site (Emanuelsson et al. 2007). The signal predicted is the type-II signal peptide common to both eukaryotic and prokaryotic organisms, for which there is wealth of data, in terms of both quality and quantity. A recent enhancement of SignalP is a Hidden Markov Model version able to discriminate uncleaved signal anchors from cleaved signal peptides.
One of the limitations of SignalP is over-prediction, as it is unable to discriminate between several very similar signal sequences, regularly predicting membrane proteins and lipoproteins as type-II signals. Many other kinds of signal sequence exist. A number of methods have been developed to predict lipoproteins, for example. The prediction of proteins that are translocated via the TAT-dependent pathway is also important but is not addressed yet in any depth.
The Many Successes of Reverse Vaccinology
Reverse vaccinology is a principal means of identifying subunit vaccines and involves a considerable computational contribution. Conventional experimental approaches cultivate pathogens under laboratory conditions, dissecting them into their components, with proteins displaying protective immunity identified as antigens. However, it is not always possible to cultivate a particular pathogen in the lab nor are all proteins expressed during infection are easily expressed in vitro, meaning that candidate vaccines can be missed. Reverse vaccinology, by contrast, analyses a pathogen genomes to identify potential antigens and is typically more effective for prokaryotic than eukaryotic organisms.
Initially, an algorithm capable of identifying Open Reading Frames (ORFs) scans the pathogenic genome. Programs that can do this include ORF-FINDER (Rombel et al. 2003), Glimmer (Delcher et al. 1999), and GS-finder (Ou et al. 2004). Once all ORFs have been identified, proteins with the characteristics of secreted or surface molecules must be identified. Unlike the relatively straightforward task of identifying ORFs, selecting proteins liable to immune system surveillance is challenging. Programs such as ProDom (Servant et al. 2002), Pfam (Bateman et al. 2000), and PROSITE (Falquet et al. 2002) can identify sequence motifs characteristic of certain protein families and can thus help predict if a protein belongs to an extracellular family of proteins.
We have developed VaxiJen [http://www.jenner.ac.uk/VaxiJen/] that implements a statistical model able to discriminate between candidate vaccines and non-antigens, using an alignment-free representation of the protein sequence (Doytchinova and Flower 2007). Rather than concentrate on epitope and non-epitope regions, the method used bacterial, viral, and tumour protein datasets to derive statistical models for predicting whole protein antigenicity. The models showed prediction accuracy up to 89%, indicating a far higher degree of accuracy than has, for example, been obtained previously for B Cell epitope prediction. Such a method is an imperfect beginning; future research will yield significantly more insight as the number of known protective antigens increases.
The NERVE program has been developed to further automate and refine the process of reverse vaccinology, in particular the process of identifying surface proteins. In NERVE, the processing of potential ORFs is a six-step process. It begins with the prediction of subcellular localisation, followed by the calculation of probability of the protein being adhesion, the identification of TM domains, a comparison with the human proteome and then with that of the selected pathogen, after which the protein is assigned a putative function. The vaccine candidates are then filtered and ranked based upon these calculations. While it is generally accepted that determining ORFs is a relatively straightforward process, the algorithm used to define extracellular proteins from other proteins needs to be carefully selected. One of the most effective programs that can be used for this purpose is HensBC, a recursive algorithm for predicting the subcellular location of proteins. The program constructs a hierarchical ensemble of classifiers by applying a series of if-then rules. HensBC is able to assign proteins to one of four different types (cytoplasmic, mitochondrial, nuclear, or extracellular) with approximately 80% accuracy for Gram-negative bacterial proteins. The algorithm is non-specialised and can be applied to any genome. Any protein identified as being extracellular could be a potential vaccine candidate.
The technique of reverse vaccinology was pioneered by a group investigating Neisseria meningitides, the pathogen responsible for sepsis and Meningococcal meningitis. Vaccines based upon the capsular proteins have been developed for all the serotypes with the exception of subgroup B. The Neisseria meningitides genome was scanned for potential ORFs (Tettelin et al. 2000; Pizza et al. 2000b). Out of the 570 proteins that were identified, 350 could be successfully expressed in vitro and 85 of these were determined to be surface exposed. Seven identified proteins conferred immunity over a broad range of strains within the natural N. meningitidis population, demonstrating the viability of in silico analysis as an aid to finding candidates for the clinical development of a MenB vaccine. Another example of the successful application of reverse vaccinology is Streptococcus pneumoniae, a major cause of sepsis, pneumonia, meningitis, and otitis media in young children (Wizemann et al. 2001; Maione et al. 2005). Mining of the genome identified 130 potential ORFs with significant homology to other bacterial surface proteins and virulence factors. 108 of 130 ORFs were successfully expressed and purified; six proteins were found to induce protective antibodies against pneumococcal challenge in a mouse sepsis model. All six of these candidates showed a high degree of cross-reactivity against the majority of capsular antigens expressed in vivo and which are believed to be immunogenic in humans.
Another example is Porphyromonas gingivalis is a gram-negative anaerobic bacterium present in subgingival plaques present in chronic adult periodontitis, an inflammatory disease of the gums. Shotgun sequences of the genome identified approximately 370 ORFS (Ross et al. 2001). Seventy-four of these had significant global homology to known surface proteins or an association with virulence. Forty-six had significant similarity with other bacterial outer membrane proteins. Forty-nine proteins were identified as surface proteins using PSORT and 22 through motif analysis. This generated 120 unique proteins sequences, 40 of which were shown to be positive for at least one of the sera. These were used to vaccinate mice, with only two of the antigens demonstrating significant protection. Chlamydia pneumoniae is an obligate intracellular bacterium associated with respiratory infections, cardiovascular and atherosclerotic disease. 141 ORFS were selected through in silico analysis (Montigiani et al. 2002) and 53 putative surface-exposed proteins identified. If reverse vaccinology is applied appropriately in vaccine design, it can save enormous amounts of money, time, and wasted labour.
Developing Vectors for Vaccines Delivery
Safe and effective methods of gene delivery have been sought for 30 years. Viral delivery of genes has effectively targeted inter alia haemophilia, coronary heart disease, muscular dystrophy, arthritis, and cancer. Despite their immanent capacity to transfer genes into cells, concerns over safety, manufacturing, restricted targeting ability and plasmid size have limited deployment of effective and generic gene therapy approaches. This remains a key objective for vaccinology. Vectors for gene therapy and vaccines differ in their requirements, yet both must overcome issues of targeting, plasmid cargo, and adverse immunogenicity. For example, up to 10% of the vaccinia genome can be replaced by DNA coding for antigens from other pathogens. The resulting vector generates strong antibody and T cell responses, and is protective. Viruses commonly used as vectors include Poxviruses, Adeno, varicella, polio, and influenza. Bacterial vectors include both Mycobacterium bovis and Salmonella. Adding extra DNA coding for large molecule adjuvants greatly can exacerbate antibody or T cell responses.
Successful transfection is hampered by DNA degradation within and outside the cell, inadequate cell penetration, poor intracellular trafficking, and inefficient nuclear localisation. Gene delivery requires both vector escape from digestion in late endosomes and nuclear translocation. Caveolin-dependent endocytosis, phagocytosis, and macropinocytosis do not transfer of material to the endo-lysosomal pathway. Some internalised material is released into the cytosol, through unknown mechanisms. However creating vectors with such desirable properties is difficult and their effectiveness may be compromised by their capacity to down-regulate other immune responses. The efficient and rational design of effective vaccine vectors is an area where informatic techniques could play a large role.
Similar to, yet simpler than, viral vectors are so-called DNA vaccines; they are plasmids capable of expressing antigenic peptide within the host (Babiuk et al. 2000). They are an attractive alternative to conventional vaccines, generating both a cellular and a humoral immune response, which are effective versus intracellular pathogens. The efficiency of a DNA vaccine has been successfully enhanced using codon optimisation (Babiuk et al. 2003), CpG motif engineering (Uchijima et al. 1998), (Klinman et al. 1997), and the introduction of promoter sequences (Booth et al. 2007), (Lee et al. 1997). Codon optimisation has been most effective in enhancing protein expression efficiency. Codons optimal for Translation are those recognised by abundant tRNAs (Xu et al. 2001). Within a phylogenetic group, codon frequency is highly correlated with gene expression levels. Immunogenicity depends upon effective translation and transcription of the antigen; it is possible to enhance this by selecting optimal codons for the vaccine.
The most comprehensive approach to vaccine optimisation is taken by DyNAVacs, an integrative bioinformatics tool that optimises codons for heterologous expression of genes in bacteria, yeasts, and plants (Henry and Sharp 2007). The program is also capable of mapping restriction enzyme sites, primer design, and designing therapeutic genes. The program calculates the optimal code for each amino acid encoded by a stretch of DNA by using codon usage table, which contains codon frequencies for a variety of different genomes.
A similar technique, CpG optimisation, may be used to optimise the codons in respect to CG dinucleotides. Pattern recognition receptors that form part of the innate immune system can often distinguish prokaryotic DNA from eukaryotic DNAs by detecting unmethylated CpG dinucleotides in particular base contexts, which are termed ‘CpG motifs’. The presence of such motifs in the sequence can be highly advantageous so long as it does not interfere with the process of codon optimisation.
Discovery of Adjuvants and Immunomodulators
Another technique for optimising the efficacy of vaccines is to develop an efficient adjuvant. Adjuvants are defined as any chemical which is able to enhance an immune response when applied simultaneously with a vaccine and thus improve the efficacy of vaccination (Harish et al. 2006); Singh and O’Hagan 2002. It is possible that some adjuvants act as immune potentiators, triggering an early innate immune response that enhances the vaccine effectiveness by increasing the vaccine uptake. Adjuvants may also enhance vaccination by improving the depot effect, the co-localisation of the antigen, and immune potentiators by delaying the spread of the antigen from the site of infection so that absorption occurs over a prolonged period (Stills 2005). Aluminium hydroxide or Alum is the only adjuvant currently licensed in humans. Aluminium-based adjuvants prolong antigen persistence due to the depot effect, as well as stimulating the production of IgG1 and IgE antibodies (Gupta 1998) and triggering the secretion of interleukin-4. There are also several small-molecule, drug-like adjuvants, such as imiquimod, resiquimod, and other imidazoquinolines (Singh and Srivastava 2003; Schijns 2003; Iellem et al. 2001). Other small molecules that have been investigated for adjuvant properties include Monophosphoryl-Lipid A, muramyl dipeptide, QS21, PLG, and Seppic ISA-51 (Schijns 2003). In many cases, the adjuvant molecules have displayed toxic properties or showed poor adsorption making them unsuitable for use. Thus there is a great demand for new compounds that can be used as adjuvants.
Chemokine receptors are a family of G-protein-coupled receptors (GPCRs) that transduce chemokines, leukocyte chemoattractant peptides that are secreted by several cell types in response to inflammatory stimuli (Charoenvit et al. 2004a; Hedrick and Zlotnik 1996; Luster 1998). GPCRs are a superfamily of transmembrane proteins responsible for the transduction of a variety of endogenous extracellular signals into an intracellular response (Locati and Murphy 1999; Christopoulos and Kenakin 2002; Gether et al. 2002). Activation of the chemokine receptors triggers an inflammatory response by inducing migration of the leukocytes from circulation to the site of injury or infection. The receptors play a pivotal role in angiogenesis, haematopoiesis, brain and heart development, and there is also evidence that CCR5 precipitates the entry of HIV-1 into CD4+ T cells by the binding of the viral envelope protein gp120 (Bissantz 2003; Deng et al. 1996). There are 18 chemokine receptors and over 45 known chemokine ligands. The chemokines can be divided into the CC and CXC family, the former contains two cysteine residues adjacent within the protein sequence while in the latter they are separated by a single amino acid. CCR4 is a chemokine receptor expressed on Th2-type CD4+ T cells and has been linked to allergic inflammation diseases such as asthma, atopic dermatitis, and allergic rhinitis. There are two chemokines which bind the CCR4 receptor exclusively, CCL22 and CCL17 (Feng et al. 1996). Inhibition of the two ligands has been shown to reduce the migration of T cells to sites of inflammation, suggesting than any CCR4 antagonist could provide an effective treatment for allergic reactions, specifically in the treatment of asthma. Both anti-CCL17 and anti-CCL22 antibodies have been observed to have efficacy, the property that enables a molecule to impart a pharmacological response, in murine asthma models.
It is possible for the CCR4 receptor to act as an adjuvant due to its expression by regulatory T cells (Tregs) that normally downregulate an immune response (Chvatchko et al. 2000). The Tregs inhibit dendritic cell maturation and thus downregulate expression of the co-stimulatory molecule. A successful CCR4 antagonist would therefore be able to enhance human T cell proliferation in an in vitro immune response model by blocking the Treg proliferation. This suggests that an effective CCR4 antagonist would have the properties of an adjuvant. A combination of virtual screening and experimental validation has been used to identify several potential adjuvants capable of inhibiting the proliferation of Tregs. Small-molecule adjuvant discovery is amenable to techniques used routinely by the pharmaceutical industry. Three-dimensional virtual screening is a fast and effective way of identifying molecules by docking a succession of ligands into a defined binding site (Lieberam and Forster 1999). A large database of small molecules can be screened quickly and efficiently in this way. Using ‘targeted’ libraries containing a specific subset of molecules is often more effective. It is possible to use ‘privileged fragments’ to construct combinatorial libraries, those which are expected to have increased probability of success. A pharmacophore is a specific three-dimensional map of biological properties common to all active confirmations of a set of ligands exhibiting a particular activity that can be used to discover new molecules with similar properties. Several small molecules that have been investigated for adjuvant properties in this way (Schellhammer and Rarey 2004). More recently, molecules that selectively interfere with chemokine-mediated T Cell migration have shown the potential to act as adjuvants by down-regulating the expression of co-stimulatory molecules, limiting T Cell activation. Small-molecule chemokine receptor antagonists have been identified and shown to be effective at blocking chemokine function in vivo (Charoenvit et al. 2004b; Godessart 2005), although to date no compound has reached a phase II clinical trial.
Discussion
In 1900, prime causes of human mortality encompassed influenza, enteritis, diarrhoea, and pneumonia: together accounting for over 30% of fatalities. Conversely, cancer and heart disease were responsible for only 12%. Compare that to the final 25 years of the seventeenth century, when average life expectancy was below 40. Principal causes of death were again infectious disease: smallpox, tuberculosis, malaria, yellow fever, and dysentery, which affected adult and children alike. Seemingly little had changed in the intervening 150 years. Today, the picture is very different, at least in developed countries. Infectious disease accounts for below 2% of deaths. Chronic disease now account for over 60% of deaths in the First World.
To a first approximation, life expectancy has, on average, escalated consistently throughout the last few thousand years. Obviously, a few great epidemic diseases – principally the Black Death – have, on occasion, made not insignificant dents in this inexorable upward progression. Citizens of the Roman Empire enjoyed a mean life expectancy at birth of about 22 years. By the Middle Ages, this had, in Europe at least, increased to be about 33 years. By the middle of the nineteenth century general life expectancy had risen to roughly 43 years. In the early 1900s, mean life spans in more developed countries ranged from 35 to 55. Life expectancy has accelerated over the last hundred years or so and today over 40 countries have an average life expectancy exceeding 70 years. The average life span across the whole human population is somewhat lower, however; it is estimated at 64.8 years – 63.2 years for men and 66.47 years for women.
Iceland heads the most recent 2003 league table with a mean life expectancy 78.7 years, next is Japan (78.4 years), followed by Sweden (77.9 years), then Australia (77.7 years). Next come Israel and Switzerland jointly with 77.6 years, Canada (77.4 years), then Italy (76.9 years), New Zealand and Norway jointly (76.8 years), and then Singapore with a mean life expectancy of 76.7 years. The United Arab Emirates is in tenth place with 76.4 years, followed by Cyprus (76.1 years), and in joint twelfth place Austria and the United Kingdom (76.0 years). Perversely, perhaps, the richest and most economically successful country on Earth, The United States of America, only manages twentieth place (74.6); while the newly emergent tiger economies of China (69.9 years, 41st place) and India (61.8; 77th place), which threaten to dominate the economic and fiscal landscapes of the coming century, come even lower on the list.
The population in many developed countries is now said to be ageing, putting burgeoning pressure on social welfare systems. However, this so-called ageing is only comparative, and comparative to the past. In fact, around 27% of the global population are aged 14 or under; of this, 0.91 billion are male and 0.87 billion are female. Only an estimated 100 million children attend school. The gender imbalance also varies across the world. In Hennan province, the most populous region of China, the balance is skewed 118:100 in favour of boys, probably due to the elective abortion of female foetuses; the average in industrialised nations of the First World is 103–108:100. The global average is 104:100.
65.2% of the world’s population are between the ages of 15 and 64, with a ratio of 2.15 billion men to 2.10 billion women. Worldwide, only 7.4% of people are actually 65 and over, with a balance of men to women of 0.21–0.27 billion. However, when compared to centuries past, this shift is remarkable; one might almost call it a lurch, it has been so rapid. One way in which this shift manifests itself is in the vastly increased longevity of the individual, as well as an increasing proportion of the population reaching old age. This phenomenon is, in part, a by product of the First World’s ever more comfortable, ever more urbanised post-industrial environment. Coupled to decades of better nutrition, medical advances in both treatment regimes and medicines will allow an ever-increasing section of the population to exploit their own genetic predisposition to long life. In North America, it has been estimated that by the middle of the century, those living beyond the age 100 would number over 100,000. On this basis, some have predicted that the human life span will be routinely stretched to 120. However, the total global population of so-called super-centenarian – those living beyond 110 – is roughly 80; while only one in two billion will live beyond 116.
With this growth in life expectancy has come a concomitant growth in the diseases of old age. These include hitherto rare, or poorly understood, neurodegenerative diseases, such as Parkinson’s or Alzheimer’s disease which proportionally affect the old more, cardiovascular diseases, and stroke, the prevalence of which is also increasing.
Patterns of disease have changed over the past hundred years and will change again in the next hundred. Some of these changes will be predictable, others not. Nonetheless, many diseases, have, at least in the west, have been beaten, or seemingly beaten, or, at least, subdued and kept in check. This is due to many factors which have militated against the severity and spread of disease; these include improvements to the way that life is lived – precautionary hygiene, nutrition, water quality, reduced overcrowding, improved living conditions – as well as more significant, interventionary measures, such as quarantining, antibiotic therapy, and, of course, vaccines.
Vaccine design and development is an inherently laborious process, but the programs and techniques outlined here have the potential to simplify the process greatly. The techniques described also have the potential to identify candidate proteins that would be overlooked by conventional experimentation. Reverse vaccinology has in particular proved effective in the discovery of antigenic subunit vaccines that would otherwise remain undiscovered.
It is sometimes difficult for outsiders to assess properly the relative merits of in silico vaccine design compared to mainstream experimental studies. The potential – albeit largely unrealised – is huge, but only if people are willing to take up the technology and use it appropriately. People’s expectations of computational work are often largely unrealistic and highly tendentious. Some expect perfection, and are soon disappointed, rapidly becoming vehement critics. Others are highly critical from the start and are nearly impossible to reconcile with informatic methods. Neither appraisal is correct, however. Informatic methods do not replace, or even seek to replace, experimental work, only to help rationalise experiments, saving time and effort. They are slaves to the data used to generate them. They require a degree of intellectual effort equivalent in scale yet different in kind to that of so-called experimental science. The two disciplines – experimental and informatics – are thus complementary albeit distinct.
References
Andersen PH et al (2006) Prediction of residues in discontinuous B Cell epitopes using protein 3D structures. Protein Sci 15:2558–2567
Babiuk LA et al (2000) Nucleic acid vaccines: research tool or commercial reality. Vet Immunol Immunopathol 76:1–23
Babiuk LA et al (2003) Induction of immune responses by DNA vaccines in large animals. Vaccine 21:649–658
Bateman A et al (2000) The Pfam protein families database. Nucleic Acids Res 28:263–266
Bissantz C (2003) Conformational changes of G protein-coupled receptors during their activation by agonist binding. J Recept Signal Transduct Res 23:123–153
Blythe MJ, Flower DR (2004) Benchmarking B Cell epitope prediction: underperformance of existing methods. Protein Sci 14:246–248
Booth JS et al (2007) Innate immune responses induced by classes of CpG oligodeoxynucleotides in ovine lymph node and blood mononuclear cells. Vet Immunol Immunopathol 115(1–2):24–34
Bulashevska A, Eils R (2006) Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinformatics. 7:298
Charoenvit Y, Goel N, Whelan M, Rosenthal KS, Zimmerman DH (2004a) CEL-1000 – a peptide with adjuvant activity for Th1 immune responses. Vaccine 22(19):2368–2373
Charoenvit Y et al (2004b) A small peptide (CEL-1000) derived from the beta-chain of the human major histocompatibility complex class II molecule induces complete protection against malaria in an antigen-independent manner. Antimicrob Agents Chemother 48:2455–2463
Christopoulos A, Kenakin TG (2002) protein-coupled receptor allosterism and complexing. Pharmacol Rev 54:323–374
Chvatchko Y, Hoogewerf AJ, Meyer A, Alouani S, Juillard P, Buser R, Conquet F, Proudfoot AE, Wells TN, Power CA (2000) A key role for CC chemokine receptor 4 in lipopolysaccharide-induced endotoxic shock. J Exp Med 191:1755–1764
Davies MN et al (2003) A novel predictive technique for the MHC class II peptide-binding interaction. Mol Med 9:220–225
Davies MN et al (2006) Statistical deconvolution of enthalpic energetic contributions to MHC-peptide binding affinity. BMC Struct Biol 6:5–17
De Groot AS (2006) Immunomics: discovering new targets for vaccines and therapeutics. Drug Discov Today 11:203–209
de Lisle GW, Wards BJ, Buddle BM, Collins DM (2005) The efficacy of live tuberculosis vaccines after presensitization with Mycobacterium avium. Tuberculosis (Edinb) 85(1–2):73–79
Delcher AL et al (1999) Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27:4636–4641
Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR (1996) Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666
Donnes P, Elofsson A (2002) Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics 3:25
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:4
Doytchinova IA et al (2005) Towards the chemometric dissection of peptide-HLA-A*0201 binding affinity: comparison of local and global QSAR models. J Comput Aided Mol Des 19:203–212
Ebo DG, Bridts CH, Stevens WJ (2008) IgE-mediated large local reaction from recombinant hepatitis B vaccine. Allergy 63(4):483–484
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2(4):953–971
Falquet L et al (2002) The PROSITE database. Nucleic Acids Res 30:235–238
Feng Y, Broder CC, Kennedy PE, Berger EA (1996) HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877
Gether U, Asmar F, Meinild AK, Rasmussen SG (2002) Structural basis for activation of G-protein-coupled receptors. Pharmacol Toxicol 91:304–312
Girard MP, Osmanov SK, Kieny MP (2006) A review of vaccine research and development: the human immunodeficiency virus (HIV). Vaccine 24(19):4062–4081
Godessart N (2005) Chemokine receptors: attractive targets for drug discovery. Ann N Y Acad Sci 1051:647–657
Greenbaum JA, Andersen PH, Blythe M, Bui HH, Cachau RE, Crowe J, Davies MN et al (2007) Towards a consensus on datasets and evaluation metrics for developing B Cell epitope prediction tools. J Mol Recognit 20(2):75–82
Gupta RK (1998) Aluminum compounds as vaccine adjuvants. Adv Drug Deliv Rev 32:155–172
Guzmán E, Romeu A, Garcia-Vallve S (2008) Completely sequenced genomes of pathogenic bacteria: a review. Enferm Infecc Microbiol Clin 26(2):88–98
Harish N, Gupta R, Agarwal P, Scaria V, Pillai B (2006) DyNAVacS: an integrative tool for optimized DNA vaccine design. Nucleic Acids Res 34(Web Server issue):W264–W266
Hedrick JA, Zlotnik A (1996) Chemokines and lymphocyte biology. Curr Opin Immunol 8:343–347
Henry I, Sharp PM (2007) Predicting gene expression level from codon usage bias. Mol Biol Evol 24(1):10–12
Hung CF, Ma B, Monie A, Tsen SW, Wu TC (2008) Therapeutic human papillomavirus vaccines: current clinical trials and future directions. Expert Opin Biol Ther 8(4):421–439
Iellem A, Colantonio L, Bhakta S, Sozzani S, Mantovani A, Sinigaglia F, D’Ambrosio D (2001) Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J Exp Med 194:847–854
Jardetzky TS et al (1996) Crystallographic analysis of endogenous peptides associated with HLADR1 suggests a common, polyproline II-like conformation for bound peptides. Proc Natl Acad Sci USA 93:734–738
Klinman DM et al (1997) Contribution of CpG motifs to the immunogenicity of DNA vaccines. J Immunol 158:3635–3639
Kulkarni-Kale U, Bhosle S, Kolaskar AS (2005) CEP: a conformational epitope prediction server. Nucleic Acids Res 33(Web Server issue):W168–W171
Lee AH et al (1997) Comparison of various expression plasmids for the induction of immune response by DNA immunization. Mol Cells 7:495–501
Lieberam I, Forster I (1999) The murine beta-chemokine TARC is expressed by subsets of dendritic cells and attracts primed CD4+ T cells. Eur J Immunol 29:2684–2694
Liu W et al (2006) Quantitative prediction of mouse class I MHC peptide binding affinity using support vector machine regression (SVR) models. BMC Bioinformatics 7:182
Locati M, Murphy PM (1999) Chemokines and chemokine receptors: biology and clinical relevance in inflammation and AIDS. Annu Rev Med 50:425–440
Luster AD (1998) Chemokines – chemotactic cytokines that mediate inflammation. N Engl J Med 338:436–445
Maione D et al (2005) Identification of a universal Group B streptococcus vaccine by multiple genome screen. Science 309:148–150
McMurry J et al (2005) Analyzing Mycobacterium tuberculosis proteomes for candidate vaccine epitopes. Tuberculosis (Edinb) 85:95–105
Merino EF, Fernandez-Becerra C, Durham AM, Ferreira JE, Tumilasci VF, d’Arc-Neves J, da Silva-Nunes M, Ferreira MU, Wickramarachchi T, Udagama-Randeniya P, Handunnetti SM, Del Portillo HA (2006) Multi-character population study of the vir subtelomeric multigene superfamily of Plasmodium vivax, a major human malaria parasite. Mol Biochem Parasitol 149(1):10–16
Mok BW, Ribacke U, Winter G, Yip BH, Tan CS, Fernandez V, Chen Q, Nilsson P, Wahlgren M (2007) Comparative transcriptomal analysis of isogenic Plasmodium falciparum clones of distinct antigenic and adhesive phenotypes. Mol Biochem Parasitol 151(2):184–192
Montigiani S et al (2002) Genomic approach for analysis of surface proteins in Chlamydia pneumoniae. Infect Immun 70:368–379
Noguchi H et al (2002) Hidden Markov model-based prediction of antigenic peptides that interact with MHC class II molecules. J Biosci Bioeng 94:264–270
O’Hagan DT, MacKichan ML, Singh M (2001) Recent developments in adjuvants for vaccines against infectious diseases. Biomol Eng 18(3):69–85
Ou HY et al (2004) GS-Finder: a program to find bacterial gene start sites with a self-training method. Int J Biochem Cell Biol 36:535–544
Palma-Carlos AG, Santos AS, Branco-Ferreira M, Pregal AL, Palma-Carlos ML, Bruno ME, Falagiani P, Riva G (2006) Clinical efficacy and safety of preseasonal sublingual immunotherapy with grass pollen carbamylated allergoid in rhinitic patients. A double-blind, placebo-controlled study. Allergol Immunopathol (Madr) 34(5):194–198
Pizza M et al (2000a) Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 287:1816–1820
Pizza M et al (2000b) Whole genome sequencing to identify vaccine candidates against serogroup B meningococcus. Science 287:1816–1820
Ponomarenko JV, Bourne PE (2007) Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct Biol 7:64
Rammensee H et al (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219
Rey S, Acab M, Gardy JL, Laird MR, deFays K, Lambert C, Brinkman FS (2005) PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res 33(Database issue):D164–D168
Rombel IT et al (2003) ORF-FINDER: a vector for high-throughput gene identification. Gene 282:33–41
Ross BC et al (2001) Identification of vaccine candidate antigens from a genomic analysis of Porphyromonas gingivalis. Vaccine 19:4135–4142
Salomon J, Flower DR (2006) Predicting Class II MHC-Peptide binding: a kernel based approach using similarity scores. BMC Bioinformatics 7:501
Schellhammer I, Rarey M (2004) FlexX-Scan: fast, structure-based virtual screening. Proteins 57(3):504–517
Schijns VE (2003) Mechanisms of vaccine adjuvant activity: initiation and regulation of immune responses by vaccine adjuvants. Vaccine 21:829–831
Schreiber A, Humbert M, Benz A, Dietrich U (2005) 3D-Epitope-Explorer (3DEX): localization of conformational epitopes within three-dimensional structures of proteins. J Comput Chem 26(9):879–887
Servant F et al (2002) ProDom: automated clustering of homologous domains. Brief Bioinform 3:246–251
Singh M, O’Hagan DT (2002) Recent advances in vaccine adjuvants. Pharm Res 19:715–728
Singh M, Srivastava I (2003) Advances in vaccine adjuvants for infectious diseases. Curr HIV Res 1:309–320
Stills HF Jr (2005) Adjuvants and antibody production: dispelling the myths associated with Freund’s complete and other adjuvants. ILAR J 46:280–293
Tettelin H et al (2000) Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science 287:1809–1815
Tongchusak S, Brusic V, Chaiyaroj SC (2008) Promiscuous T cell epitope prediction of Candida albicans secretory aspartyl proteinase family of proteins. Infect Genet Evol 8(4):467–473
Uchijima M et al (1998) Optimization of codon usage of plasmid DNA vaccine is required for the effective MHC class I-restricted T Cell responses against an intracellular bacterium. J Immunol 161:5594–5599
Vekemans J, Ballou WR (2008) Plasmodium falciparum malaria vaccines in development. Expert Rev Vaccines 7(2):223–240
Vivona S, Bernante F, Filippini F.(2006) NERVE: new enhanced reverse vaccinology environment. BMC Biotechnol 6:35
Wan S et al (2004) Large-scale molecular dynamics simulations of HLA-A*0201 complexed with a tumor-specific antigenic peptide: can the alpha3 and beta2m domains be neglected? J Comput Chem 25:1803–1813
Wan J et al (2006) SVRMHC prediction server for MHC-binding peptides. BMC Bioinformatics 7:463
Winnenburg R, Urban M, Beacham A, Baldwin TK, Holland S, Lindeberg M, Hansen H, Rawlings C, Hammond-Kosack KE, Köhler J (2008) PHI-base update: additions to the pathogen host interaction database. Nucleic Acids Res 36(Database issue):D572–D576
Wizemann TM et al (2001) Use of a whole genome approach to identify vaccine molecules affording protection against Streptococcus pneumoniae infection. Infect Immun 69:1593–1598
Xu ZL et al (2001) Optimization of transcriptional regulatory elements for constructing plasmid vectors. Gene 272:149–156
Yang J, Chen L, Sun L, Yu J, Jin Q (2008) VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res 36(Database issue):D539–D542
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Davies, M.N., Flower, D.R. (2009). Computational Vaccinology. In: Flower, D., Davies, M., Ranganathan, S. (eds) Bioinformatics for Immunomics. Immunomics Reviews:, vol 3. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-0540-6_1
Download citation
DOI: https://doi.org/10.1007/978-1-4419-0540-6_1
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-0539-0
Online ISBN: 978-1-4419-0540-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)