
Abstract
Shiga toxins and ricin are potent inhibitors of protein synthesis. In addition to causing inhibition of protein synthesis, these toxins activate proinflammatory signaling cascades that may contribute to the severe diseases associated with toxin exposure. Treatment of cells with Shiga toxins and ricin have been shown to activate a number of signaling pathways including those associated with the ribotoxic stress response, Nuclear factor kappa B activation, inflammasome activation, the unfolded protein response, mTOR signaling, hemostasis, and retrograde trafficking. In this chapter, we review our current understanding of these signaling pathways as they pertain to intoxication by Shiga toxins and ricin.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Shiga toxins (Stxs) and ricin are AB toxins consisting of a single A-subunit bound to either 5 B-subunits in the case of Stxs, or 1 B-subunit in the case of ricin. In both toxins, the B-subunits recognize and bind to host cell-surface receptors. For Stxs, these are thought to be primarily neutral glycolipids, namely globotriaosyl ceramide (Gb3 or CD77) or globotetraosyl ceramide (Gb4); for ricin, these are thought to be glycoproteins or glycolipids containing terminal galactose and N-acetylgalactosamine (Gal/GalNac) residues (Baenziger and Fiete 1979; Debray et al. 1981; DeGrandis et al. 1989; Jacewicz et al. 1986; Lindberg et al. 1987; Nicolson and Blaustein 1972; Sandvig and van Deurs 1996; Waddell et al. 1988). The A-subunits of both toxins have N-glycosidase activity which results in depurination of a single adenine (A-4324) located on the alpha-sarcin loop (sarcin/ricin loop) of the 28S ribosomal RNA (Endo et al. 1987; Endo and Tsurugi 1987; Endo et al. 1988). This particular event is discussed elsewhere in this volume.
The B-subunit is essential for host cell binding, endocytic uptake, and retrograde trafficking of the holotoxin following which the toxin follows one or more retrograde trafficking pathways from the early endosome, through the Golgi to the endoplasmic reticulum (ER) (Arab and Lingwood 1998; Girod et al. 1999; Lingwood et al. 1998; Rapak et al. 1997; Sandvig et al. 1992; Sandvig and van Deurs 1996; Sandvig and van Deurs 1999; Sandvig and van Deurs 2000; Walchli et al. 2008; White et al. 1999). In the ER, the A-subunit of Shiga toxin is proteolytically cleaved and undergoes reduction of an intramolecular A-subunit disulfide bond, thereby freeing the enzymatically active portion of the A-subunit from the B-subunits (Garred et al. 1995, 1997; Yu and Haslam 2005). Similarly, once in the ER, reduction of a disulfide bond in ricin allows separation of the A- and B-subunits (Simpson et al. 1999; Spooner et al. 2004). The freed A-subunits are retrotranslocated from the ER to the cytoplasm where the toxins have access to the ribosome (Simpson et al. 1999; Wesche et al. 1999; Yu and Haslam 2005). These events are discussed in more detail elsewhere in this volume. The depurination of the ribosome by Stx and ricin A-subunits is a critical event in activation of host signal transduction pathways resulting in the proinflammatory response (Foster et al. 2000; Foster and Tesh 2002; Iordanov et al. 1997; Lindauer et al. 2010; Smith et al. 2003; Thorpe et al. 1999).
In this chapter, we will primarily discuss the activation of specific host signal transduction pathways by Shiga toxin and ricin that result in host cellular stress responses, and thus may drive the proinflammatory signaling observed in response to intoxication with these agents. We will also discuss effects of the Shiga toxin B-subunit on host signal transduction pathways. Data from animal models and human illness in which Shiga toxin- and ricin-induced proinflammatory responses are observed will be discussed elsewhere in this volume. It should be noted that while some toxin-associated signaling seems to drive uptake and retrograde trafficking of the toxins, other pathways seem to be more directed to activation of inflammatory/apoptotic pathways. However, in some cases, the distinction between trafficking-related and inflammatory/apoptotic signaling is not entirely clear, and will be noted. Finally, the details of toxin-induced effects on apoptosis will be discussed in detail elsewhere in this volume. Here we will simply note when a specific host signal transduction event is also linked to eventual apoptosis.
2 The Ribotoxic Stress Response
Activation of one or more members of the mitogen-activated protein kinase family (MAPK family) in response to Stxs or ricin treatment has been demonstrated in several different cell lines. In general, activation of MAPK signaling begins with the sensing of either mitogenic or stress-related stimuli by cells (Fig. 1). This results in activation of a MAPKinase signaling module, in which one or more MAP3Kinases phosphorylate and activate MAP2Kinases, which subsequently phosphorylate and activate one or more of the MAPK family. The MAPK family is comprised of extracellular-receptor kinases (ERKs), p38, and the jun-N-terminal kinases (JNKs) (reviewed in Kyriakis and Avruch (2001)). The “stress activated protein kinase family” or “SAPKs” is sometimes used to further describe p38 and JNKs.

The MAPKinase signaling module
Activation of MAPKs results in changes in gene regulation at both transcriptional and post-transcriptional levels. Genes that are upregulated by MAPKs include proinflammatory cytokines such as IL-8, GRO-α, IL-1β, and TNF-α, as well as pro-apoptotic genes such as FasL (Jung et al. 2002; Kyriakis and Avruch 2001; Means et al. 2000; Thorpe et al. 1999; Verhaeghe et al. 2007).
As previously discussed, the Shiga toxin and ricin A-subunits are N-glycosidases that specifically depurinate the alpha-sarcin loop of the 28S ribosomal subunit at a single adenine. Although this damage to the ribosome results in inhibition of protein synthesis, a MAPKinase-driven proinflammatory (and eventually pro-apoptotic) signaling cascade is also activated. This signaling cascade was originally termed the ribotoxic stress response (RSR) after Iordanov et al. (1997) observed that treatment of cells with certain protein synthesis inhibitors including ricin, anisomycin, and α-sarcin caused the activation of jun-N-terminal Kinase (JNKs), while other protein synthesis inhibitors, including cycloheximide, emetine, T-2 toxin, pactamycin, and puromycin, did not (Iordanov et al. 1997). Also, treatment of cells with emetine, which arrests the ribosome in a pretranslocation state, prevented subsequent induction of the RSR. This important work demonstrated that initiation of the RSR required actively translating ribosomes at the time of toxic insult. Furthermore, these data suggested that specific interactions and/or damage to the ribosome, versus inhibition of protein synthesis per se, were the triggering events for JNKs activation.
In addition to activation of JNKs, activation of p38 and ERKs can also occur as part of the RSR (Colpoys et al. 2005; Iordanov et al. 1998; Shifrin and Anderson 1999; Zhou et al. 2003). Therefore, the RSR can be defined as the activation of JNKs, p38, and/or ERKs by toxicants that act to disrupt the 28S ribosomal RNA on functional ribosomes. Also, in addition to Stxs and ricin, the list of ribotoxic stressors includes anisomycin, α-sarcin, UV-light, and the trichothecene toxins (Iordanov et al. 1997, 1998; Laskin et al. 2002; Shifrin and Anderson 1999; Thorpe et al. 1999; Zhou et al. 2003). Despite the fact that these agents cause inhibition of global translation, activation of the RSR results in a paradoxical increase in expression of proinflammatory proteins (Cherla et al. 2006; Foster and Tesh 2002; Gonzalez et al. 2006; Thorpe et al. 1999, 2001). Although much of the increase in proinflammatory gene expression is mediated at the transcriptional level, post-transcriptional events may also be important (discussed below in Sect. 4).
The ability of Shiga toxin and ricin to activate the RSR and induce proinflammatory and pro-apoptotic signaling has been demonstrated in vitro in HCT-8, Vero cells, THP-1 cells, human primary airway cells, RAW 264.7 cells, and murine primary macrophages, as well as in vivo in the murine kidney, lung, and intestine (see Table 1 for a description of the cell lines discussed in this review) (Higuchi et al. 2003; Korcheva et al. 2005; Korcheva et al. 2007; Lee et al. 2005; Lindauer et al. 2009; Smith et al. 2003; Thorpe et al. 1999, 2001; Wong et al. 2007a, b; Yoder et al. 2007). Due to the ability of ribotoxic stressors to activate proinflammatory and pro-apoptotic pathways, blockade of the RSR or its downstream effect(s), may constitute a therapeutic strategy to treat illnesses associated with ribotoxic stressors such as hemolytic uremic syndrome (HUS) or ricin-induced acute respiratory distress syndrome (ARDS). Indeed, MAPK pathways have been attractive therapeutic targets for the treatment of other diseases such as cancer, Crohn’s disease, and diabetes (Force et al. 2004; Pratilas and Solit 2010).
In general, activation of MAPKs in any host cell can occur by many types of stimuli, some involving growth and differentiation (such as growth factors), and others involving responses to various types of stress (Kyriakis and Avruch 2001). Thus, one potential caveat to using MAPKinases as therapeutic targets may be the lack of pathway specificity. Therefore developing inhibitors that target signaling components upstream in the MAPK signaling module such as a specific MAP3kinase might provide a better therapeutic strategy for treating the disease. In the case of illnesses mediated by Stxs and ricin, understanding how damage to the ribosome is specifically detected by the cell and how this information is relayed through to the MAPKinase cascade could prove important in the discovery of novel therapeutic targets that block the RSR. However, the mechanism cells use to detect damage to the 28S rRNA and subsequently transduce this signal through the RSR remains unknown. To date, three upstream effectors of the RSR have been described and are reviewed herein. These include the double-stranded RNA (dsRNA) activated protein kinase (PKR); hematopoietic cell kinase (Hck); and the zipper sterile alpha motif kinase (ZAK) (Fig. 2).

The ribotoxic stress response by Shiga toxin and ricin results in MAPKinase activation which together with NFκB activation promotes expression of proinflammatory and pro-apoptotic genes
Deoxynivalenol (DON) is a trichothecene toxin which when added to macrophage and monocyte-like cells induces the RSR with activation of ERKs, p38, JNKs, production of TNF-α, and induction of apoptosis (Moon and Pestka 2002; Shifrin and Anderson 1999; Yang et al. 2000; Zhou et al. 2003). The exact mechanism by which DON causes damage to the 28S rRNA is not clear, but DON does not have a direct effect on purified mammalian ribosomes in vitro (Li and Pestka 2008). Zhou et al. (2003) demonstrated that DON treatment of RAW 264.7 cells resulted in possible activation of PKR as evidenced by eIF2α phosphorylation, a downstream target of PKR (Zhou et al. 2003). Furthermore, treatment of cells with PKR inhibitors or antisense knockdown of PKR resulted in a decrease of DON-induced MAPKinase activation as well as apoptosis. Knockdown of PKR also blocked apoptosis by the ribotoxic stressor anisomycin and by the protein synthesis inhibitor emetine. Using human monocytic U937 cells, Gray et al. 2008, demonstrated that expression of a dominant-negative PKR blocked Stx1-, ricin-, or DON-induced IL-8, verifying that PKR activation was not specific to DON treatment, but may be activated by other ribotoxic stressors as well (Gray et al. 2008).
Hck, a Src family kinase (SFK) was shown by Zhou et al. (2005) to play a potential role in transduction of the RSR (Zhou et al. 2005). DON treatment of RAW 264.7 cells resulted in phosphorylation of Hck, and treatment of cells with Src kinase inhibitors blocked DON-induced activation of ERKs, p38, and JNKs. Furthermore, siRNA knockdown of Hck decreased both DON-induced TNF-α production and apoptosis, demonstrating that Hck is also a transducer of the DON-induced RSR (Zhou et al. 2005).
Further support for PKR and Hck as upstream mediators of the RSR comes from the findings that both PKR and Hck interact with the 40S rRNA subunit, and interactions between Hck and the 40S subunit disappear upon knocking down PKR expression (Bae et al. 2010). That PKR is a critical player in the macrophage/monocyte RSR is further supported by the findings of Bae et al. 2010, who were able to demonstrate in murine peritoneal macrophages that DON is able to recruit p38 to the ribosome (Bae et al. 2010). Since p38 is not recruited to the ribosome in mice deficient in PKR, PKR activation may be required for DON-induced p38 activation. Because PKR is activated by double-stranded RNA, it has been proposed that damage to the 28 rRNA by ribotoxic stressors such as Stx, ricin, or DON (which ultimately results in cleavage of the 28S rRNA in intact mammalian cells (Li and Pestka 2008)) provides a substrate (presumably double-stranded RNA) that activates PKR. This would result in recruitment and activation of MAPKinases to the ribosome thereby initiating subsequent downstream signaling (Bae and Pestka 2008; Gray et al. 2008).
A third upstream mediator of the RSR is the MAP3Kinase ZAK. ZAK, a mixed lineage kinase, is also known as MRK and MLTK (in humans), or MLK7 (in mice). ZAK was first shown to transduce activation of JNKs and p38 by anisomycin, and was later shown to do the same for Stx2 and ricin (Jandhyala et al. 2008; Wang et al. 2005). Stx2-, ricin-, and anisomycin-induced activation of p38 and JNKs was shown to be blocked in COS-7, Vero, and HCT-8 cells by pretreatment with the ZAK-specific inhibitor 7-[3-fluoro-4-aminophenyl-(4-(2-pyridin-2-yl-5,6-dihydro-4H-pyrrolo[1,2-b]pyrazol-3-yl))]-quinoline also called DHP-2 (IC50 = 17 nM), or by knocking down ZAK with siRNA (COS-7 and Vero cells) (Jandhyala et al. 2008; Wang et al. 2005). DHP-2 pretreatment was also shown to block Stx2 and ricin induction of interleukin-8 expression as well as inhibit caspase-3 activation and some toxin-mediated cell death. Similarly, the chemotherapeutic agents sorafenib (K d for ZAK = 6.3 nM) and nilotinib (K d for ZAK = 3 nM) decreased ricin-induced p38 and JNKs activation in lipopolysaccharide (LPS) primed murine bone marrow-derived macrophages (Lindauer et al. 2010). However, in these cells, ZAK inhibition did not effect ricin-induced activation of the NALP3 inflammasome and subsequent post-translational IL-1β processing (discussed further below). These data support the concept that signaling cascades outside the RSR are also important for the proinflammatory effects of ricin.
As a member of the family of mixed lineage kinases, ZAK has limited homology to other members of this family. There are two isoforms of ZAK, ZAKα and ZAKβ, which are different splice variants of the same gene (Gotoh et al. 2001; Tosti et al. 2004; Wang et al. 2005). Both ZAKα and ZAKβ share an identical N-terminus composing the kinase domain and leucine zipper, but ZAKα has an extended C-terminus containing a sterile alpha motif (Gross et al. 2002). While the respective roles of the two isoforms in ribotoxic stress signaling are currently not known, ZAKβ may become phosphorylated following treatment with the antineoplastic agent and ribotoxic stressor doxorubicin, as suggested by the presence of an extra band of higher molecular weight on western blots (Sauter et al. 2010). ZAK activation of p38 and JNKs seems to be fairly specific for the RSR, as other stimuli including TLR 5 signalling, IL-1β, and TNF-α do not seem to signal through ZAK (Jandhyala et al. 2010; Wang et al. 2005). Therefore ZAK may constitute a therapeutic target for blocking or reducing Stx- and ricin-induced inflammation and apoptosis.
In summary, Stx- and ricin-induced RSR is mediated by the A-subunit catalytic activity, but the exact sequence of molecular events that occur following intoxication by either of these agents resulting in activation of the MAPKinase module remains somewhat unclear. Evidence suggests that the RSR is one of the essential host responses that promote proinflammatory signaling following intoxication with Stx and ricin.
3 Superinduction
Superinduction occurs when a protein synthesis inhibitor is added along with a stimulus, resulting in massive over-accumulation of mRNA transcripts of certain primary response genes, including cytokines, at concentrations much larger than that seen following addition of stimulus alone. The superinduction phenomenon has been observed with a number of different protein synthesis inhibitors having different mechanisms of action; 28S rRNA damage does not appear to be required. The exact mechanism(s) by which protein synthesis inhibitors cause these effects remains unknown, and multiple mechanisms may contribute. We have implicated ZAK activation in the Stx2 superinduction of flagellin-induced IL-8 (Jandhyala et al. 2010).
There is substantial data supporting the idea that Shiga toxins and ricin can superinduce proinflammatory cytokines which may be associated with the pathogenic process that occurs in response to these agents (Cherla et al. 2006; Lindauer et al. 2010; Pestka and Zhou 2006; Thorpe et al. 1999, 2001). Indeed, some data exists from animal models in which both ricin and Stx were co-administered with LPS, resulting in enhanced toxicity (Fu et al. 2004; Keepers et al. 2006; Korcheva et al. 2005; Taylor et al. 1999). Superinduction of some LPS-responsive genes by the toxins may be one of the contributory mechanisms in these models.
4 Initiation of Translation
There is some data to suggest that host translation initiation pathways may be activated in response to Shiga toxins. There are three ways by which translation initiation can be effected. First, activation of Mnk1 by ERKs and/or p38 results in phosphorylation of eukaryotic translation initiation factor 4E (eIF4E). When phosphorylated, eIF4E promotes initiation through enhanced recognition of mRNAs with complex 5′ capping, preferentially recruiting these mRNAs to the ribosome. Second, activation of FRAP/mTOR can result in hyperphosphorylation of 4E-binding protein 1 (4E-BP1). When hypo- or unphosphorylated, 4E-BP1 has decreased affinity for eIF4E, resulting in increased activity of eIF4E. Finally, FRAP/mTOR activation results in phosphorylation of S6 Kinase 1, which promotes translation initiation of mRNA species that have a 5′ oligopyrimidine tract. 5′ oligopyrimidine tracts are frequently found in mRNAs encoding proteins required for ribosome biogenesis. Activation of all three pathways by Stx1 have been demonstrated in both HCT-8 and differentiated THP-1 cells (Cherla et al. 2006, 2009; Colpoys et al. 2005), and may act to maintain translation despite ribosomal intoxication (Colpoys et al. 2005) and/or allow for translation of specific cytokines (Cherla et al. 2006, 2009). Thus, host cells appear to be capable of modulating translational initiation in response to these toxins, perhaps with some host benefit and/or contribution to pathogenicity.
5 Nuclear factor-kappa B Signaling
Nuclear factor-kappa B (NF-κB) signaling is important for the regulation of a variety of genes including those associated with hemostasis, inflammation, and immunity (Hayden and Ghosh 2011; Kollader et al. 2010; Wiggins et al. 2010). “NF-κB” is a term used collectively to describe transcription factors comprised of one (homo-dimer) or two (hetero-dimer) of the five RelA/NF-κB proteins RelA (p65), RelB, c-Rel, NF-κB1 (p50), or NF-κB2 (p52) (Hauf and Chakraborty 2003; Hayden and Ghosh 2011). NF-κB in its inactive form resides in the cytoplasm where it binds to a member of the IκB family of proteins IκBα, IκBβ, or IκBε. Activation of NF-κB occurs by phosphorylation of IκB, which leads to the subsequent trafficking of phospho-IκB to the proteosome for degradation. This allows the liberated NF-κB, with its now exposed nuclear localization motif, to translocate to the nucleus. Once in the nucleus, NF-κB activates the transcription of genes with promoters containing an NF-κB binding site. Many immediate early response genes, and proinflammatory genes have NF-κB binding sites in their promoters.
Stx1 and Stx2 have been shown to modulate NF-κB signaling in a number of different cell lines including differentiated THP-1 cells (e.g. macrophage-like), human peripheral blood monocytes, Vero cells, murine podocytes, T84 cells, human umbilical vein endothelial cells (HUVECs), and human glomerular endothelial cells (Cameron et al. 2002; Morigi et al. 2006; Sakiri et al. 1998; Zanchi et al. 2008; Zoja et al. 2002). It is difficult to identify a consensus role for Stxs in NF-κB signaling, as in some systems, Stxs are associated with activation of NF-κB, and in others, they are associated with inhibition of NF-κB. Thus, we will review the relevant data that describes the effects of Stxs on NF-κB.
The first demonstration of NF-κB signaling in response to Stx treatment employed differentiated THP-1 cells and human peripheral blood monocytes (Sakiri et al. 1998). Sakiri et al. showed that Stx1/NF-κB signaling primarily involves p65 and p50 with maximum nuclear translocation occurring by 2 h post-toxin treatment. Similarly, p65 and p50 appeared to represent the NF-κB family members involved in Stx signaling in Vero cells and HUVECs (Cameron et al. 2002; Zoja et al. 2002).
However, Gobert et al. (2007) have shown that Stx may inhibit NF-κB activation by other agonists, since treatment of T84 cells with an enterohemorrhagic E. coli (EHEC) strain in which the Stx genes had been deleted induced greater NF-κB activation than did the isogenic strain expressing both Stx1 and Stx2. In this study, the activation of NF-κB by the Stx-negative EHEC strain was shown to occur via PI3Kinase/Akt signaling suggesting that Stx treatment may inhibit effectors associated with this pathway. Because T84 cells do not express the Stx receptor Gb3, signaling by Stx may occur in a different manner than is seen in Gb3 positive cells.
Wong et al. (2007a, b) have shown that ricin-induced expression of proinflammatory genes in human primary airway cells is dependent on activation of NF-κB and in a manner independent of TNF-α, thereby suggesting a direct response to ricin (Wong et al. 2007b). Optimal activation of NF-κB occurred 6 h after treatment with 100 ng/ml ricin. Knockdown of NF-κB by siRNA was shown to result in decreased mRNA for CXCL1, CCL2, IL-8, IL-1β, TNF-α, but not IL-6, supporting the role for NF-κB in the ricin-induced activation of certain cytokines. Similarly, lung tissue harvested from mice 48 h after intratracheal instillation of ricin revealed nuclear localization of NF-κB (Wong et al. 2007a).
The mechanism by which NF-κB is activated following treatment with ricin is not known; however, it is possible that the RSR is contributory to its activation. Activation of MAP3Ks, including ZAK, have been shown to result in NFκB activation (Liu et al. 2000; Malinin et al. 1997), and activation of NF-κB by the ribotoxic stressor DON has been shown to occur subsequent to MAPK activation (Zhou et al. 2003), suggesting that MAPK activation is upstream to that of NF-κB. Consistent with Stx- and ricin-induced NF-κB activation being downstream to MAPK activation, we have shown that inhibition of ZAK results in decreased expression of the CXC chemokine IL-8 (Jandhyala et al. 2008), which is regulated at the transcriptional level by both AP-1 and NF-κB (Jung et al. 2002; Mukaida et al. 1994). However, we did not specifically assess the role of NF-κB activation in this response, and we have observed enhanced stabilization of multiple CXC chemokine mRNAs following Stx treatment (Thorpe et al. 2001). Similarly, by treating U937 cells with inhibitors of PKR or by expressing a dominant- negative PKR in U937 cells, Gray et al. 2008 were able to decrease Stx1-, ricin-, and DON-induced IL-8 message (Gray et al. 2008). While this inhibition of IL-8 could result from decreased AP-1 activation or post-transcriptional events such as mRNA destabilization, Gray et al. 2008 also demonstrated that following DON treatment, U937 cells expressing the dominant-negative PKR had decreased NF-κB binding activity, and treatment of U937 cells with SB203580, a p38 inhibitor, resulted in decreased DON-induced NF-κB binding activity. Together these studies suggest that NF-κB activation by DON and possibly Stx and ricin, may occur at least in part from RSR-induced MAPK activation. However, further studies using Stx and ricin are needed to confirm that NF-κB activation is indeed associated with the RSR.
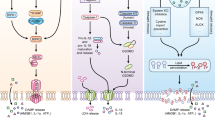
6 Ricin Activation of IL-1β Through the NALP3 Inflamasome
Inflamasomes (reviewed in Lamkanfi 2011) are multiprotein complexes that are activated as part of an innate immune response to stimuli associated with pathogen-associated molecular patterns (PAMPs) such as LPS and flagellin and danger-associated molecular patterns (DAMPs) such as uric acid and ATP (Lamkanfi 2011). Although it is not completely understood how PAMPs and/or DAMPs transduce inflammasome activation, upon detection of these stimuli, the inflammasome complex is assembled by a member of the NALP protein family of NOD-like receptors (NLR), such as NLRP1, NLRP3, AIM2, and the adaptor protein ASC that forms a scaffold connecting the NLR with caspase-1 (Fernandes-Alnemri et al. 2009; Tschopp et al. 2003). Inflammasomes act by promoting “proximity-induced autoactivation” of caspase-1, best known for its role in proteolytic activation of IL-1β and IL-18 from their inactive pro-forms (Lamkanfi 2011).
Ricin’s proinflammatory and lethal effects, including neutrophil recruitment, have been shown to be strongly suppressed in mice deficient in expression of IL-1α/β (Lindauer et al. 2009). However, in this same study, the co-administration of ricin plus IL-1β restored the ability of ricin to elicit pulmonary inflammation and neutrophil recruitment in IL-1α/β-deficient mice. By contrast, lipopolysaccharide (LPS), which plays an important role in the development and progression of chronic respiratory disease including asthma (Liu 2004), requires neither IL-1R nor IL-18R or caspase-1 to mediate a pulmonary inflammatory response, including neutrophil recruitment and vascular leak (Togbe et al. 2006). Administration of aerosolized ricin to macrophage-depleted mice resulted in reduced expression of proinflammatory transcripts, reduced accumulation of neutrophils, and decreased microvascular barrier permeability, indicating that macrophages are required for ricin to mediate inflammatory responses in the lungs (Lindauer et al. 2009). Taken together, the evidence demonstrates that IL-1 plays a key role in mediating proinflammatory responses signaled by ricin in the airways, and that macrophages are required for mediating these proinflammatory responses.
Because dysregulated release of IL-1β can be detrimental, IL-1β is kept under stringent control by the requirement for two distinct signals. The first signal, mediated by the activation of NF-κB, is usually conveyed in macrophages by activation of TLRs and induces the expression of the 35 kDa proprotein form IL-1β (pro-IL-1β). The second signal induces the processing of the pro-IL-1β protein into the mature 17 kDa IL-1β by the inflammasome (Mariathasan and Monack 2007; Martinon et al. 2002; Ogura et al. 2006; Yu and Finlay 2008). The IL-1β-converting enzyme (ICE), better known as caspase-1, is required for this cleavage (Burns et al. 2003). Activation of inflammatory caspases in the inflammasome complex is an essential step for the processing and maturation of IL-1β in response to microbial stress or “danger signals” (Martinon and Tschopp 2005).
In LPS-primed murine bone marrow-derived macrophages, ricin was shown to mediate the processing of pro-IL-1β and the release of IL-1β by activating the NLRP3 inflammasome (Lindauer et al. 2010). Ricin failed to induce the expression of pro-IL-1β in unprimed macrophages, demonstrating the requirement for prior exposure of macrophages to an NF-κB-activating agent such as LPS. The proteasome inhibitors bortezomib and MG-132 blocked ricin-induced release of IL-1β from macrophages, suggesting that ricin-induced inhibition of translation may foster the disappearance of labile protein(s) that normally suppress inflammasome formation. Consistent with this hypothesis, inhibition of protein synthesis by a variety of translation inhibitors appears to potently activate the NLRP3 inflammasome (B. Magun, unpublished), suggesting that the ribotoxic effects of ricin and potentially other inhaled toxins may contribute to inflammatory lung disease through inflammasome activation. Figure 3 describes a model that implicates ricin in mediating two distinct signals that lead to expression and processing of IL-1β in macrophages. The first signal leads to the activation of ZAK and subsequent expression of pro-IL-1β. The second signal, inhibition of translation, leads to the activation of the NLRP3 inflammasome and the subsequent release of IL-1β.

Ricin induction of IL-1β results from the activation of MAPKinase and NFκB pathways which promote expression of Pro-IL-1β. Pro-IL-1β is converted to mature IL-1β via activation of the Nalp3 inflammasome
Currently, mechanisms describing how the inflammasome is activated by ricin are lacking. The involvement of inflammasome activation in models of Shiga toxin-associated disease have not been described. However, in a mouse model of HUS, both LPS and Stx are required in order to initiate thrombotic microangiopathy (Keepers et al. 2006). It is tempting to speculate on whether synergistic effects of Stx and LPS contribute to activation of the inflammasome in this model.
7 ER-Stress
Another stress response that may contribute to inflammation/pro-apoptotic signaling during Stx and ricin intoxication is the unfolded protein response (UPR). The UPR is activated when unfolded and/or misfolded proteins accumulate in the ER (reviewed in (Kim et al. 2008; Tsai and Weissman 2010)). The UPR acts by halting global protein translation to limit delivery of nascent proteins to the ER for folding, and preferentially reprograms transcription and translation pathways involved in restoring ER function. This also includes activation of transcription factors that regulate these pathways. In the event that ER homeostasis cannot be regained, apoptotic signaling cascades are then activated. The master regulator of ER homeostasis, BiP (or GRP78), is an ER chaperone that is central to activation of the UPR. ER-stress has been associated with a variety of pathologies including atherosclerosis, neurodegenerative disorders such as prion disease and amyotrophic lateral sclerosis, and diabetes (Hosoi and Ozawa 2009; Kim et al. 2008; Lhotak et al. 2011; Sharma et al. 2006; Zhang and Kaufman 2008).
During ER-stress, the UPR is mediated through the activation of one or more of three effector pathways: PKR-like ER kinase (PERK) pathway, inositol-requiring enzyme 1 (IRE-1) pathway, and activating transcription factor 6 (ATF-6) pathway (Kim et al. 2008; Tsai and Weissman 2010). PERK, IRE-1, and ATF-6 are membrane spanning proteins, the luminal portions of which are thought to interact with BiP. During ER-stress it is believed that BiP is recruited away from these effectors by excess unfolded proteins, an event that triggers their activation (Fig. 4). Phosphorylation and subsequent activation of PERK results in the phosphorylation and inactivation of eIF2α. Inactivation of eIF2α results in translational “reprogramming”, including cessation of global mRNA translation, while preferentially allowing translation of certain transcripts involved in ER recovery, such as that of the transcription factor ATF-4. ATF-4 regulates the promoters of UPR associated genes including that of GRP78 and CHOP the latter of which is important for inducing ER-stress-associated apoptosis. Activation of the second major effector, IRE-1, results in its splicing of the mRNA for X-box binding protein 1 (XBP1). XBP1 protein up-regulates transcription of ER-chaperones and p58 IPK. IRE-1 can also activate the MAPKs p38 and JNKs through a pathway involving TRAF2 and the MAP3K ASK1. Upon activation, the third UPR effector, ATF-6, is trafficked to the Gogli apparatus, modified by proteolytic cleavage, and subsequently translocated to the nucleus to activate the transcription of genes including CHOP and XBP1, BiP, PDI, and GRP94.

The unfolded protein response
Stx1 treatment of the monocyte-like cell line THP-1 has been shown to result in ER-stress with activation of all three UPR effectors PERK, IRE-1, and ATF-6 (Lee et al. 2008). In this study, CHOP mRNA was up-regulated, and accompanied by Ca2+ influx from the ER to the cytosol. This latter event was thought to possibly result in the calpain-dependent activation of caspase-3 and caspase-8, which was detected following Stx1 treatment. Interestingly activation of the UPR was not able to be assigned categorically to either Stx “A” or “B” subunit activity. Catalytically deficient toxin (Stx1AE167Q, R170L) was unable to induce activation of PERK and ATF-6. However, IRE-1 activation, Ca2+ influx, and partial XBP1 splicing effects were retained. These data suggest that at least some aspects of Stx1- mediated ER-stress may be dependent on A-subunit catalytic activity, but signaling from both the A-subunit and the B-subunit appears to contribute. These data would support a complex model, in which multiple Stx activities (such as toxin trafficking, ER-to-cytoplasmic A-subunit translocation, and intoxication) contribute to the overall activation of the UPR.
As part of a study primarily assessing the effects of Stxs on apoptosis in human brain microvascular endothelial cells, Fujii et al. (2008) demonstrated that both CHOP mRNA and ATF-4 mRNA were upregulated at 19 h following treatment with Stx2. Both Stx1 and Stx2 were shown to upregulate CHOP message while Stx1R170L (a Stx1 mutant with approximately 9000-fold decrease in A-subunit activity) did not. Therefore, StxA-subunit catalytic activity may be required for these events. This is consistent with the aforementioned observations by Lee et al. (2008) that suggested Stx A-subunit activity was essential for PERK and ATF-6 activation.
Ricin and three trichothecene toxins including DON (all ribotoxic stressors) have also been shown to induce ER-stress (Horrix et al. 2011; Shi et al. 2009). In one of these studies, treatment of murine peritoneal-derived macrophages with DON resulted in increased concentrations of IRE1, ATF6 and XBP1 mRNA, and an increase in spliced XBP1 message (Shi et al. 2009). It should be noted that unlike Stxs and ricin, DON, being a small molecule of 296.3 Da, does not have a binding subunit. Interestingly, although BiP is generally upregulated during ER-stress, in this study it was shown to be proteolytically degraded in response to DON treatment. Also, RNAi knockdown of BiP expression caused an increase in IL-6, suggesting that DON-induced BiP degradation might promote an IL-6 response in these cells. Interestingly ricin was also shown to induce BiP degradation. Together these results suggests that DON- and ricin-induced ER-stress might trigger IL-6 production through BiP degradation.
In a more recent study by Horrix et al. (2011), ricin treatment of the human adenocarcinoma cell lines MDA-MB-231 and HCT116 was shown to result in the UPR with eIF2α phosphorylation and ATF-6 activation (Horrix et al. 2011). However, unlike the effects of Stx noted by Lee et al. (2008), IRE1-dependent XBP-1 mRNA splicing was lacking (Lee et al. 2008).
Ricin has also been shown to inhibit the ER-stress response when expressed in yeast (Parikh et al. 2008). Parikh et al. show that expressing ricin in yeast such that it is translocated into the ER results in suppression of HAC1 mRNA splicing, an event dependent on the yeast homolog of IRE1, IREp. Ricin mutants that lacked N-glycosidase activity were shown not to inhibit the UPR, suggesting the involvement of the ricin active site. Cell survival during ER-stress is thought to be sustained by prolonging IRE1 activation, which normally terminates around 8 h of continuous ER-stress (Lin et al. 2007). Therefore it is feasible that inhibition of IRE1 by ricin may also promote toxicity in yeast. As noted above, IRE1-mediated XBP-1 splicing was also absent following ricin treatment of human breast and colon cell lines (Horrix et al. 2011). However, it should be noted that unlike mammalian cells, yeast are not able to be intoxicated with Stx or ricin via the B-subunit. Thus, toxin A-subunit-encoding constructs with appropriate ER delivery signals must be used. Furthermore, unlike mammals, yeast do not have known PERK and ATF-6 homologs, thus limiting the ability of using yeast to study mammalian ER-stress events.
Together, these data suggest that ER-stress may play a role in ricin-and Stx-induced apoptosis, and that activation of the UPR is largely mediated by the A-subunits of Stx and ricin. However, the precise mechanisms by which Stx and ricin induce ER-stress are currently not understood. It is possible that following inactivation by Stx or ricin, ribosomes that are engaged in active translation of secreted proteins at the cytosolic surface of the ER remain attached to the incompletely translated nascent peptides at the cytosolic domain of the Sec61 translocon, thereby clogging the secretion machinery of the ER. In an attempt to remediate this situation, intoxicated cells may activate the UPR. However, it is also possible that B-subunit accumulation in the ER, or the partial unfolding of Stx or ricin A-subunits prior to cytosolic translocation, may effect ER-stress. Further studies are required to elucidate the exact cause(s) of Stx- and ricin-mediated ER-stress.
8 Signaling Associated with the B-Subunit of Stxs and/or Stx Uptake and Trafficking
B-subunits of both Stxs and ricin are required for binding and intracellular trafficking of toxin, and several studies have demonstrated that the Stx B-subunit may induce a variety of host-cell responses. These include cytoskeletal remodeling, endocytosis of toxin, retrograde trafficking of toxin, stimulation of von Willebrand factor (vWF) secretion, activation of apoptotic cascades, and possibly Toll-like receptor-4 (TLR4) signaling (Fischer et al. 2007; Huang et al. 2010; Lauvrak et al. 2006; Mangeney et al. 1993; Takenouchi et al. 2004; Walchli et al. 2008). These studies have been performed in several different systems and are summarized in Table 2. We will not discuss data supporting a possible effect(s) of B-subunit on pro-apoptotic pathways here, as this is discussed elsewhere in this volume.
In general, interactions between StxB-subunit and receptor(s) on lipid rafts may induce signaling events as early as 2.5–15 min following toxin treatment (Falvo et al. 2000; Katagiri et al. 1999; Lauvrak et al. 2006; Taga et al. 1997; Torgersen et al. 2007; Walchli et al. 2008). These signaling events have been shown to result in the activation of tyrosine kinase signaling, p38 stress-activated protein kinase signaling (p38 SAPK), protein kinase C signaling (PKC), and Ca2+ signaling. Activation of these pathways seems to be important not only for toxin uptake and trafficking, but may also have more direct pathological consequences.
Treatment of cells with Stx has been shown to result in the activation of tyrosine kinases including Syk, and the SFK Yes and Lyn (Katagiri et al. 1999; Lauvrak et al. 2006; Malyukova et al. 2009; Mori et al. 2000). Activation of these tyrosine kinases appear to be associated with binding and uptake of the toxin since activation occurs within 15 min of toxin exposure and precedes inhibition of protein synthesis by A-subunit activity.
Early work by Katagiri et al. (1999), was important in demonstrating that Gb3 associated with lipid rafts were important for early Stx1-mediated signaling through the Yes tyrosine kinase; more detailed information has followed. In the human renal tubular cell line ACHN the Stx receptor Gb3 can be found associated with the SFK Yes in detergent-insoluble microdomains (DIM) or lipid rafts (Katagiri et al. 1999). Treatment of these cells with Stx1 results in transient tyrosine phosphorylation of several DIM proteins, and this occurs with similar kinetics to the activation and autophosphorylation of Yes. In addition to activating Yes, treatment with Stx1 results in recruitment of Yes to the lipid rafts. (However, during or following activation, Yes then becomes detergent soluble). Yes activation is transient: maximal activation of Yes occurs within 10 min post-Stx1 treatment, and diminishes by 30–60 min. Similarly, tyrosine phosphorylation of DIM proteins in response to Stx1 treatment occurs by 10 min and basal phosphorylation levels are regained by 30 min.
Two other tyrosine kinases, Lyn and Syk were shown to be promptly activated in Burkitt’s lymphoma Ramos cells (BL-cells) following treatment with Stx1 (Mori et al. 2000). As with Yes, Lyn was localized in lipid rafts containing Gb3, and became detergent soluble following toxin treatment. Unlike Yes, Lyn does not immunoprecipitate with Gb3 (Mori et al. 2000). Stx1 treatment of BL-cells also resulted in complex formation between Syk and Lyn. It is possible that Syk and/or Lyn activation may also play a role in endocytosis and trafficking of Stxs as Syk has been shown to be a regulator of clathrin-dependent endocytosis in HeLa cells (Lauvrak et al. 2006).
Stx was the first lipid binding ligand shown to use a clathrin-dependent mechanism to promote its own uptake (Sandvig et al. 1989). Although Stxs can be taken up by clathrin-dependent and clathrin-independent pathways, clathrin pit-associated endocytosis appears to predominate in Stx-sensitive cells, and clathrin-dependent endocytosis is required for retrograde Golgi transport (Lauvrak et al. 2004; Saint-Pol et al. 2004). Sandvig et al. (1989) demonstrated that Stx could promote its own uptake in HeLa cells, and Lauvrak et al. (2006) were able to show that this, at least in part, was due to Stx activation of Syk and the promotion of complex formation between Syk and clathrin heavy chain (CHC) (Lauvrak et al. 2006; Sandvig et al. 1989; Utskarpen et al. 2010). Syk appears to regulate Stx uptake by phosphorylating CHC protein, thereby promoting endocytosis, and B-subunit alone is sufficient for Syk actiavation (Lauvrak et al. 2006).
In addition to promoting endocytosis via Syk, Stx may also promote its own endocytosis and subsequent retrograde trafficking by activating and recruiting p38 to the early endosome. Walchli et al. (2008) demonstrated in HeLa cells that treatment with p38-specific inhibitors or siRNA resulted in reduced sulfation of a modified Stx1B-subunit, signifying that p38 played a role in Stx transport to the Golgi (Walchli et al. 2008). Treatment of HeLa cells with Stx1B-subunit resulted in detectable p38 phosphorylation in as early as 1 min and peaked within 10-15 min. In HEp-2 cells, Walchli et al. 2008 did not see translocation of p38 to the nucleus, but instead p38 appeared to be recruited to endosomal fractions. Importantly, modifications of intracellular Ca2+ appeared to be necessary for p38 activation by the Stx B-subunit suggesting that Ca2+ signaling is upstream of Stx-induced p38 activation. This was also supported by the findings that inhibition of p38 by SB203580 does not effect Stx inhibition of histamine-induced Ca2+ oscillations. Finally, inhibition of p38 or siRNA knockdown of p38 had no effect on ricin trafficking to the Golgi, implying that ricin uses a different pathway.
Protein kinase C (PKC) has been shown to be important for Stx trafficking by regulating transport of Stx from the endosome to the Golgi apparatus. Torgersen et al. (2007) demonstrated that inhibiton of PKCδ but not PKCα resulted in accumulation of toxin in the endosome (Torgersen et al. 2007). Phosphorylation of PKCδ following treatment with Stx1B-subunit was detectable at 5 min post-treatment, and maximum PKCδ phosphorylation was attained by 20 min. These observations were also made using Stx1B-subunit alone, suggesting that activation of PKCδ was B-subunit dependent. Supporting a role for Stx in PKC activation, Foster et al. (2000) observed Stx1-mediated activation of PKC in differentiated macrophage-like THP-1 cells. However, studies undertaking PKC isoform determination, or assignment to catalytic activity were not performed.
As discussed elsewhere in this volume, Shiga toxins are associated with the thrombotic microangiopathic disease HUS. Thrombotic thrombocytopenia purpura has many of the same signs as HUS, but is generally associated with a deficiency in ADAMTS13, a protease responsible for cleaving unusually large vWF multimers. Recently, data has emerged linking B-subunit activity with vWF secretion from endothelial cells. B-subunits alone from Stx1 or Stx2 were able to stimulate secretion of vWF in HUVECs and promote platelet adhesion in human glomerular microvascular endothelial cells (HGMECs) (Huang et al. 2010). In addition, this induction of vWF secretion occurred within 5 min of toxin (B-subunit) treatment, and required cholesterol-rich lipid rafts, but not clathrin. Because Stx2 is more often associated with diarrhea-associated HUS (D+HUS), it was particularly interesting that in this study, Stx2B was found to be a more potent inducer of vWF in HUVECs than Stx1B. Comparisons of Stx2B treatment of TTP-prone, ADAMS13 −/− mice with that of heterozygous ADAMS13 +/− litter mates showed that ADAMS13 −/− mice developed reticulocytosis and fragmented red cells. In addition, 5 of the 13 Stx2B-treated ADAMS13 −/− mice developed thrombocytopenia and/or anemia. These data suggest that StxB-subunits might promote endothelial cell activation by inducing vWF secretion and promoting platelet adhesion. One caveat to these findings is the observation that unlike TTP, thrombi from patients with D+HUS tend to be rich in fibrin, while containing little or no vWF (Tsai et al. 2001).
As with Stxs, the study of ricin has been important in defining and understanding retrograde trafficking. However, unlike Stxs, there is limited data on the contribution(s) of ricin B-subunit to host signal transduction events. There is clearly a role for further investigation of the possible effect(s) of ricin B-subunit on activation of host signal transduction pathway(s) that influence both its toxicity and pathogenic effect(s). These may be, and probably are, very different from effects of StxB-subunit on host cells. It is known that depletion of sphingosine appears to promote ricin trafficking, and depletion of cholesterol inhibits it (Grimmer et al. 2006). This suggests that cholesterol may play a role in endocytosis, and that lipid raft association of ricin receptors may actually impede ricin uptake. The observations that Gb3 located in lipid rafts are required for some of the signal transduction events associated with Stxs suggest that Gb3 in these cell types is “wired” to signaling networks in a way that has not been described for ricin.
9 Concluding Remarks
During the process of intoxication, Stxs and ricin activate a variety of signaling pathways. While much headway has been made in identifying which pathways are activated, and to which cellular outputs they contribute (i.e. inflammation, apoptosis, trafficking), two large gaps in our knowledge remain. The first of these relates to the key signaling events that contribute to morbidity in Stx- and ricin- mediated disease. Filling this gap will involve understanding which outputs are the most necessary for promoting damage to the host. For example, are there particular cytokine responses that if blocked will prevent or help alleviate disease? Alternatively, is it apoptosis, or inflammation, that drives morbidity? In the case of ricin, the development of good animal models has helped elucidate the role of IL-1 in promoting ARDS. However, in the case of Stx, the “connect” between pro-inflammatory and/or pro-apoptotic signaling with microangiopathic disease is still very much undetermined. With the development of new mouse models to study Stx-induced HUS, it will be interesting to see how toxin-mediated cell signaling influences disease progression.
Since protein synthesis inhibition does not seem to be the underlying signal driving the ribotoxic stress response, the second major gap in our knowledge concerning Stx and ricin involves understanding how cells recognize damage to the 28S rRNA subunit. Is there a specific sensor(s), and how does it work? Although a fundamental biological question, the process of answering this question is likely to provide us with new therapeutic targets in addition to providing insight into the greater architecture of the ribosome. In the way that Stx and ricin has help us understand several aspects of retrograde trafficking, the further study of these toxins and the cellular events they initiate are bound to enlighten our understanding of stress signaling in the cell.
References
Arab S, Lingwood CA (1998) Intracellular targeting of the endoplasmic reticulum/nuclear envelope by retrograde transport may determine cell hypersensitivity to verotoxin via globotriaosyl ceramide fatty acid isoform traffic. J Cell Physiol 177:646–660
Bae HK, Pestka JJ (2008) Deoxynivalenol induces p38 interaction with the ribosome in monocytes and macrophages. Toxicol Sci 105:59–66
Bae H, Gray JS, Li M et al (2010) Hematopoietic cell kinase associates with the 40s ribosomal subunit and mediates the ribotoxic stress response to deoxynivalenol in mononuclear phagocytes. Toxicol Sci 115:444–452
Baenziger JU, Fiete D (1979) Structural determinants of ricinus communis agglutinin and toxin specificity for oligosaccharides. J Biol Chem 254:9795–9799
Burns K, Martinon F, Tschopp J (2003) New insights into the mechanism of IL-1beta maturation. Curr Opin Immunol 15:26–30
Cameron P, Bingham D, Paul A et al (2002) Essential role for verotoxin in sustained stress-activated protein kinase and nuclear factor kappa b signaling, stimulated by Escherichia coli o157:H7 in vero cells. Infect Immun 70:5370–5380
Cherla RP, Lee SY, Mees PL et al (2006) Shiga toxin 1-induced cytokine production is mediated by map kinase pathways and translation initiation factor eif4e in the macrophage-like thp-1 cell line. J Leukoc Biol 79:397–407
Cherla RP, Lee SY, Mulder RA et al (2009) Shiga toxin 1-induced proinflammatory cytokine production is regulated by the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin signaling pathway. Infect Immun 77:3919–3931
Colpoys WE, Cochran BH, Carducci TM et al (2005) Shiga toxins activate translational regulation pathways in intestinal epithelial cells. Cell Signal 17:891–899
Debray H, Decout D, Strecker G et al (1981) Specificity of twelve lectins towards oligosaccharides and glycopeptides related to n-glycosylproteins. Eur J Biochem 117:41–55
DeGrandis S, Law H, Brunton J et al (1989) Globotetraosylceramide is recognized by the pig edema disease toxin. J Biol Chem 264:12520–12525
Endo Y, Tsurugi K (1987) RNA n-glycosidase activity of ricin a-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem 262:8128–8130
Endo Y, Mitsui K, Motizuki M et al (1987) The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 s ribosomal RNA caused by the toxins. J Biol Chem 262:5908–5912
Endo Y, Tsurugi K, Yutsudo T et al (1988) Site of action of a vero toxin (vt2) from Escherichia coli o157:H7 and of shiga toxin on eukaryotic ribosomes. RNA n-glycosidase activity of the toxins. Eur J Biochem 171:45–50
Falvo JV, Uglialoro AM, Brinkman BM et al (2000) Stimulus-specific assembly of enhancer complexes on the tumor necrosis factor alpha gene promoter. Mol Cell Biol 20:2239–2247
Fernandes-Alnemri T, Yu JW, Datta P et al (2009) Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513
Fischer H, Ellstrom P, Ekstrom K et al (2007) Ceramide as a tlr4 agonist; a putative signalling intermediate between sphingolipid receptors for microbial ligands and tlr4. Cell Microbiol 9:1239–1251
Force T, Kuida K, Namchuk M et al (2004) Inhibitors of protein kinase signaling pathways: emerging therapies for cardiovascular disease. Circulation 109:1196–1205
Foster GH, Tesh VL (2002) Shiga toxin 1-induced activation of c-jun nh(2)-terminal kinase and p38 in the human monocytic cell line thp-1: possible involvement in the production of tnf-alpha. J Leukoc Biol 71:107–114
Foster GH, Armstrong CS, Sakiri R et al (2000) Shiga toxin-induced tumor necrosis factor alpha expression: requirement for toxin enzymatic activity and monocyte protein kinase c and protein tyrosine kinases. Infect Immun 68:5183–5189
Fu XJ, Iijima K, Nozu K et al (2004) Role of p38 map kinase pathway in a toxin-induced model of hemolytic uremic syndrome. Pediatr Nephrol 19:844–852
Fujii J, Wood K, Matsuda F et al (2008) Shiga toxin 2 causes apoptosis in human brain microvascular endothelial cells via c/ebp homologous protein. Infect Immun 76:3679–3689
Garred O, van Deurs B, Sandvig K (1995) Furin-induced cleavage and activation of shiga toxin. J Biol Chem 270:10817–10821
Garred O, Dubinina E, Polesskaya A et al (1997) Role of the disulfide bond in shiga toxin a-chain for toxin entry into cells. J Biol Chem 272:11414–11419
Girod A, Storrie B, Simpson JC et al (1999) Evidence for a cop-i-independent transport route from the golgi complex to the endoplasmic reticulum. Nat Cell Biol 1:423–430
Gobert AP, Vareille M, Glasser AL et al (2007) Shiga toxin produced by enterohemorrhagic Escherichia coli inhibits pi3k/nf-kappa b signaling pathway in globotriaosylceramide-3-negative human intestinal epithelial cells. J Immunol 178:8168–8174
Gonzalez TV, Farrant SA, Mantis NJ (2006) Ricin induces IL-8 secretion from human monocyte/macrophages by activating the p38 map kinase pathway. Mol Immunol 43:1920–1923
Gotoh I, Adachi M, Nishida E (2001) Identification and characterization of a novel map kinase kinase kinase, mltk. J Biol Chem 276:4276–4286
Gray JS, Bae HK, Li JC et al (2008) Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, shiga toxin 1, and ricin in monocytes. Toxicol Sci 105:322–330
Grimmer S, Spilsberg B, Hanada K et al (2006) Depletion of sphingolipids facilitates endosome to golgi transport of ricin. Traffic 7:1243–1253
Gross EA, Callow MG, Waldbaum L et al (2002) Mrk, a mixed lineage kinase-related molecule that plays a role in gamma-radiation-induced cell cycle arrest. J Biol Chem 277:13873–13882
Hauf N, Chakraborty T (2003) Suppression of nf-kappa b activation and proinflammatory cytokine expression by shiga toxin-producing Escherichia coli. J Immunol 170:2074–2082
Hayden MS, Ghosh S (2011) Nf-kappa b in immunobiology. Cell Res 21:223–244
Higuchi S, Tamura T, Oda T (2003) Cross-talk between the pathways leading to the induction of apoptosis and the secretion of tumor necrosis factor-alpha in ricin-treated raw 264.7 cells. J Biochem 134:927–933
Horrix C, Raviv Z, Flescher E et al (2011) Plant ribosome-inactivating proteins type ii induce the unfolded protein response in human cancer cells. Cell Mol Life Sci 68:1269–1281
Hosoi T, Ozawa K (2009) Endoplasmic reticulum stress in disease: mechanisms and therapeutic opportunities. Clin Sci (Lond) 118:19–29
Huang J, Motto DG, Bundle DR et al (2010) Shiga toxin b subunits induce vwf secretion by human endothelial cells and thrombotic microangiopathy in adamts13-deficient mice. Blood 116:3653–3659
Iordanov MS, Pribnow D, Magun JL et al (1997) Ribotoxic stress response: activation of the stress-activated protein kinase jnk1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28s rRNA. Mol Cell Biol 17:3373–3381
Iordanov MS, Pribnow D, Magun JL et al (1998) Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J Biol Chem 273:15794–15803
Jacewicz M, Clausen H, Nudelman E et al (1986) Pathogenesis of shigella diarrhea. Xi. Isolation of a shigella toxin-binding glycolipid from rabbit jejunum and hela cells and its identification as globotriaosylceramide. J Exp Med 163:1391–1404
Jandhyala DM, Ahluwalia A, Obrig T et al (2008) Zak: a map3kinase that transduces shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol 10:1468–1477
Jandhyala DM, Rogers TJ, Kane A et al (2010) Shiga toxin 2 and flagellin from shiga-toxigenic Escherichia coli superinduce interleukin-8 through synergistic effects on host stress-activated protein kinase activation. Infect Immun 78:2984–2994
Jung YD, Fan F, McConkey DJ et al (2002) Role of p38 mapk, ap-1, and nf-kappa b in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells. Cytokine 18:206–213
Katagiri YU, Mori T, Nakajima H et al (1999) Activation of src family kinase yes induced by shiga toxin binding to globotriaosyl ceramide (gb3/cd77) in low density, detergent-insoluble microdomains. J Biol Chem 274:35278–35282
Keepers TR, Psotka MA, Gross LK et al (2006) A murine model of hus: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol 17:3404–3414
Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7:1013–1030
Kollander R, Solovey A, Milbauer LC et al (2010) Nuclear factor-kappa b (nf kappa b) component p50 in blood mononuclear cells regulates endothelial tissue factor expression in sickle transgenic mice: implications for the coagulopathy of sickle cell disease. Transl Res 155:170–177
Korcheva V, Wong J, Corless C et al (2005) Administration of ricin induces a severe inflammatory response via nonredundant stimulation of erk, jnk, and p38 mapk and provides a mouse model of hemolytic uremic syndrome. Am J Pathol 166:323–339
Korcheva V, Wong J, Lindauer M et al (2007) Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol Immunol 44:2761–2771
Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81:807–869
Lamkanfi M (2011) Emerging inflammasome effector mechanisms. Nat Rev Immunol 11:213–220
Laskin JD, Heck DE, Laskin DL (2002) The ribotoxic stress response as a potential mechanism for map kinase activation in xenobiotic toxicity. Toxicol Sci 69:289–291
Lauvrak SU, Torgersen ML, Sandvig K (2004) Efficient endosome-to-golgi transport of shiga toxin is dependent on dynamin and clathrin. J Cell Sci 117:2321–2331
Lauvrak SU, Walchli S, Iversen TG et al (2006) Shiga toxin regulates its entry in a syk-dependent manner. Mol Biol Cell 17:1096–1109
Lee SY, Cherla RP, Caliskan I et al (2005) Shiga toxin 1 induces apoptosis in the human myelogenous leukemia cell line thp-1 by a caspase-8-dependent, tumor necrosis factor receptor-independent mechanism. Infect Immun 73:5115–5126
Lee SY, Lee MS, Cherla RP et al (2008) Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol 10:770–780
Lhotak S, Zhou J, Austin RC (2011) Immunohistochemical detection of the unfolded protein response in atherosclerotic plaques. Meth Enzymol 489:23–46
Li M, Pestka JJ (2008) Comparative induction of 28s ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and t-2 toxin in the macrophage. Toxicol Sci 105:67–78
Lin JH, Li H, Yasumura D et al (2007) Ire1 signaling affects cell fate during the unfolded protein response. Science 318:944–949
Lindauer ML, Wong J, Iwakura Y et al (2009) Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J Immunol 183:1419–1426
Lindauer M, Wong J, Magun B (2010) Ricin toxin activates the nalp3 inflammasome. Toxins 2:1500–1514
Lindberg AA, Brown JE, Stromberg N et al (1987) Identification of the carbohydrate receptor for shiga toxin produced by shigella dysenteriae type 1. J Biol Chem 262:1779–1785
Lingwood CA, Khine AA, Arab S (1998) Globotriaosyl ceramide (gb3) expression in human tumour cells: intracellular trafficking defines a new retrograde transport pathway from the cell surface to the nucleus, which correlates with sensitivity to verotoxin. Acta Biochim Pol 45:351–359
Liu AH (2004) Something old, something new: indoor endotoxin, allergens and asthma. Paediatr Respir Rev 5(Suppl A):S65–S71
Liu TC, Huang CJ, Chu YC et al (2000) Cloning and expression of zak, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-alpha motif. Biochem Biophys Res Commun 274:811–816
Malinin NL, Boldin MP, Kovalenko AV et al (1997) Map3k-related kinase involved in nf-kappa b induction by tnf, cd95 and IL-1. Nature 385:540–544
Malyukova I, Murray KF, Zhu C et al (2009) Macropinocytosis in shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am J Physiol Gastrointest Liver Physiol 296:G78–G92
Mangeney M, Lingwood CA, Taga S et al (1993) Apoptosis induced in burkitt’s lymphoma cells via gb3/cd77, a glycolipid antigen. Cancer Res 53:5314–5319
Mariathasan S, Monack DM (2007) Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7:31–40
Martinon F, Tschopp J (2005) Nlrs join tlrs as innate sensors of pathogens. Trends Immunol 26:447–454
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of pro IL-beta. Mol Cell 10:417–426
Means TK, Pavlovich RP, Roca D et al (2000) Activation of tnf-alpha transcription utilizes distinct map kinase pathways in different macrophage populations. J Leukoc Biol 67:885–893
Moon Y, Pestka JJ (2002) Vomitoxin-induced cyclooxygenase-2 gene expression in macrophages mediated by activation of erk and p38 but not jnk mitogen-activated protein kinases. Toxicol Sci 69:373–382
Mori T, Kiyokawa N, Katagiri YU et al (2000) Globotriaosyl ceramide (cd77/gb3) in the glycolipid-enriched membrane domain participates in b-cell receptor-mediated apoptosis by regulating lyn kinase activity in human b cells. Exp Hematol 28:1260–1268
Morigi M, Buelli S, Zanchi C et al (2006) Shigatoxin-induced endothelin-1 expression in cultured podocytes autocrinally mediates actin remodeling. Am J Pathol 169:1965–1975
Mukaida N, Okamoto S, Ishikawa Y et al (1994) Molecular mechanism of interleukin-8 gene expression. J Leukoc Biol 56:554–558
Nicolson GL, Blaustein J (1972) The interaction of ricinus communis agglutinin with normal and tumor cell surfaces. Biochim Biophys Acta 266:543–547
Ogura Y, Sutterwala FS, Flavell RA (2006) The inflammasome: first line of the immune response to cell stress. Cell 126:659–662
Parikh BA, Tortora A, Li XP et al (2008) Ricin inhibits activation of the unfolded protein response by preventing splicing of the hac1 mRNA. J Biol Chem 283:6145–6153
Pestka J, Zhou HR (2006) Toll-like receptor priming sensitizes macrophages to proinflammatory cytokine gene induction by deoxynivalenol and other toxicants. Toxicol Sci 92:445–455
Pratilas CA, Solit DB (2010) Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin Cancer Res 16:3329–3334
Rapak A, Falnes PO, Olsnes S (1997) Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc Natl Acad Sci USA 94:3783–3788
Saint-Pol A, Yelamos B, Amessou M et al (2004) Clathrin adaptor epsinr is required for retrograde sorting on early endosomal membranes. Dev Cell 6:525–538
Sakiri R, Ramegowda B, Tesh VL (1998) Shiga toxin type 1 activates tumor necrosis factor-alpha gene transcription and nuclear translocation of the transcriptional activators nuclear factor-kappa b and activator protein-1. Blood 92:558–566
Sandvig K, van Deurs B (1996) Endocytosis, intracellular transport, and cytotoxic action of shiga toxin and ricin. Physiol Rev 76:949–966
Sandvig K, van Deurs B (1999) Endocytosis and intracellular transport of ricin: recent discoveries. FEBS Lett 452:67–70
Sandvig K, van Deurs B (2000) Entry of ricin and shiga toxin into cells: molecular mechanisms and medical perspectives. EMBO J 19:5943–5950
Sandvig K, Olsnes S, Brown JE et al (1989) Endocytosis from coated pits of shiga toxin: a glycolipid-binding protein from shigella dysenteriae 1. J Cell Biol 108:1331–1343
Sandvig K, Garred O, Prydz K et al (1992) Retrograde transport of endocytosed shiga toxin to the endoplasmic reticulum. Nature 358:510–512
Sauter KA, Magun EA, Iordanov MS et al (2010) Zak is required for doxorubicin, a novel ribotoxic stressor, to induce sapk activation and apoptosis in hacat cells. Cancer Biol Ther 10:258–266
Sharma P, Senthilkumar RD, Brahmachari V et al (2006) Mining literature for a comprehensive pathway analysis: a case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis 5:1
Shi Y, Porter K, Parameswaran N et al (2009) Role of grp78/bip degradation and er stress in deoxynivalenol-induced interleukin-6 upregulation in the macrophage. Toxicol Sci 109:247–255
Shifrin VI, Anderson P (1999) Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-jun n-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J Biol Chem 274:13985–13992
Simpson JC, Roberts LM, Romisch K et al (1999) Ricin a chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett 459:80–84
Smith WE, Kane AV, Campbell ST et al (2003) Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and jnk activation and induction of apoptosis in intestinal epithelial cells. Infect Immun 71:1497–1504
Spooner RA, Watson PD, Marsden CJ et al (2004) Protein disulphide-isomerase reduces ricin to its a and b chains in the endoplasmic reticulum. Biochem J 383:285–293
Taga S, Carlier K, Mishal Z et al (1997) Intracellular signaling events in cd77-mediated apoptosis of burkitt’s lymphoma cells. Blood 90:2757–2767
Takenouchi H, Kiyokawa N, Taguchi T et al (2004) Shiga toxin binding to globotriaosyl ceramide induces intracellular signals that mediate cytoskeleton remodeling in human renal carcinoma-derived cells. J Cell Sci 117:3911–3922
Taylor CM, Williams JM, Lote CJ et al (1999) A laboratory model of toxin-induced hemolytic uremic syndrome. Kidney Int 55:1367–1374
Thorpe CM, Hurley BP, Lincicome LL et al (1999) Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immun 67:5985–5993
Thorpe CM, Smith WE, Hurley BP et al (2001) Shiga toxins induce, superinduce, and stabilize a variety of c-x-c chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect Immun 69:6140–6147
Togbe D, Schnyder-Candrian S, Schnyder B et al (2006) Tlr4 gene dosage contributes to endotoxin-induced acute respiratory inflammation. J Leukoc Biol 80:451–457
Torgersen ML, Walchli S, Grimmer S et al (2007) Protein kinase cdelta is activated by shiga toxin and regulates its transport. J Biol Chem 282:16317–16328
Tosti E, Waldbaum L, Warshaw G et al (2004) The stress kinase mrk contributes to regulation of DNA damage checkpoints through a p38gamma-independent pathway. J Biol Chem 279:47652–47660
Tsai YC, Weissman AM (2010) The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer 1:764–778
Tsai HM, Chandler WL, Sarode R et al (2001) Von willebrand factor and von willebrand factor-cleaving metalloprotease activity in Escherichia coli o157: H7-associated hemolytic uremic syndrome. Pediatr Res 49:653–659
Tschopp J, Martinon F, Burns K (2003) Nalps: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4:95–104
Utskarpen A, Massol R, van Deurs B et al (2010) Shiga toxin increases formation of clathrin-coated pits through syk kinase. PLoS One 5:e10944
Verhaeghe C, Remouchamps C, Hennuy B et al (2007) Role of ikk and erk pathways in intrinsic inflammation of cystic fibrosis airways. Biochem Pharmacol 73:1982–1994
Waddell T, Head S, Petric M et al (1988) Globotriosyl ceramide is specifically recognized by the Escherichia coli verocytotoxin 2. Biochem Biophys Res Commun 152:674–679
Walchli S, Skanland SS, Gregers TF et al (2008) The mitogen-activated protein kinase p38 links shiga toxin-dependent signaling and trafficking. Mol Biol Cell 19:95–104
Wang X, Mader MM, Toth JE et al (2005) Complete inhibition of anisomycin and uv radiation but not cytokine induced jnk and p38 activation by an aryl-substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J Biol Chem 280:19298–19305
Wesche J, Rapak A, Olsnes S (1999) Dependence of ricin toxicity on translocation of the toxin a-chain from the endoplasmic reticulum to the cytosol. J Biol Chem 274:34443–34449
White J, Johannes L, Mallard F et al (1999) Rab6 coordinates a novel golgi to er retrograde transport pathway in live cells. J Cell Biol 147:743–760
Wiggins JE, Patel SR, Shedden KA et al (2010) Nf-kappa b promotes inflammation, coagulation, and fibrosis in the aging glomerulus. J Am Soc Nephrol 21:587–597
Wong J, Korcheva V, Jacoby DB et al (2007a) Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am J Pathol 170:1497–1510
Wong J, Korcheva V, Jacoby DB et al (2007b) Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and nf-kappa b. Am J Physiol Lung Cell Mol Physiol 293:L1385–L1394
Yang GH, Jarvis BB, Chung YJ et al (2000) Apoptosis induction by the satratoxins and other trichothecene mycotoxins: relationship to erk, p38 mapk, and sapk/jnk activation. Toxicol Appl Pharmacol 164:149–160
Yoder JM, Aslam RU, Mantis NJ (2007) Evidence for widespread epithelial damage and coincident production of monocyte chemotactic protein 1 in a murine model of intestinal ricin intoxication. Infect Immun 75:1745–1750
Yu HB, Finlay BB (2008) The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe 4:198–208
Yu M, Haslam DB (2005) Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone hedj/erdj3. Infect Immun 73:2524–2532
Zanchi C, Zoja C, Morigi M et al (2008) Fractalkine and cx3cr1 mediate leukocyte capture by endothelium in response to shiga toxin. J Immunol 181:1460–1469
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462
Zhou HR, Islam Z, Pestka JJ (2003a) Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol Sci 72:130–142
Zhou HR, Lau AS, Pestka JJ (2003b) Role of double-stranded RNA-activated protein kinase r (pkr) in deoxynivalenol-induced ribotoxic stress response. Toxicol Sci 74:335–344
Zhou HR, Jia Q, Pestka JJ (2005) Ribotoxic stress response to the trichothecene deoxynivalenol in the macrophage involves the src family kinase hck. Toxicol Sci 85:916–926
Zoja C, Angioletti S, Donadelli R et al (2002) Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via nf-kappa b dependent up-regulation of IL-8 and mcp-1. Kidney Int 62:846–856
Acknowledgments
We would like to thank the National Institutes of Health, Bethesda, MD, USA for its support of our work through the following grants: AI-59509 (C.M.T), AI0883360O1A1 (C.M.T and D.M.J), and AI1059335 (B.E.M).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Jandhyala, D.M., Thorpe, C.M., Magun, B. (2011). Ricin and Shiga Toxins: Effects on Host Cell Signal Transduction. In: Mantis, N. (eds) Ricin and Shiga Toxins. Current Topics in Microbiology and Immunology, vol 357. Springer, Berlin, Heidelberg. https://doi.org/10.1007/82_2011_181
Download citation
DOI: https://doi.org/10.1007/82_2011_181
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-27469-5
Online ISBN: 978-3-642-27470-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)