
Abstract
The discovery of asunaprevir (1) began with the concept of engaging the small and well-defined S1’ pocket of the hepatitis C virus (HCV) NS3/4A protease that was explored in the context of tripeptide carboxylic acid-based inhibitors. A cyclopropyl-acyl sulfonamide moiety was found to be the optimal element at the P1-P1’ interface enhancing the potency of carboxylic acid-based prototypes by 10- to >100-fold, dependent upon the specific background. Optimization for oral bioavailability identified a 1-substituted isoquinoline-based P2* element that conferred a significant exposure advantage in rats compared to the matched 4-substituted quinoline isomer. BMS-605339 (30) was the first cyclopropyl-acyl sulfonamide derivative advanced into clinical trials that demonstrated dose-related reductions in plasma viral RNA in HCV-infected patients. However, 30 was associated with cardiac events observed in a normal healthy volunteer (NHV) and an HCV-infected patient that led to the suspension of the development program. Using a Langendorff rabbit heart model, a limited structure-cardiac liability relationship was quickly established that led to the discovery of 1. This compound, which differs from 30 only by changes in the substitution pattern of the P2* isoquinoline heterocycle and the addition of a single chlorine atom to the molecular formula, gave a dose-dependent reduction in plasma viral RNA following oral administration to HCV-infected patients without the burden of the cardiac events that had been observed with 30. A small clinical trial of the combination of 1 with the HCV NS5A inhibitor daclatasvir (2) established for the first time that a chronic genotype 1 (GT-1) HCV infection could be cured by therapy with two direct-acting antiviral agents in the absence of exogenous immune-stimulating agents. Development of the combination of 1 and 2 was initially focused on Japan where the patient population is predominantly infected with GT-1b virus, culminating in marketing approval which was granted on July 4, 2014. In order to broaden therapy to include GT-1a infections, a fixed dose triple combination of 1, 2, and the allosteric NS5B inhibitor beclabuvir (3) was developed, approved by the Japanese health authorities for the treatment of HCV GT-1 infection on December 20, 2016 and marketed as Ximency®.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Acyl sulfonamide
- Asunaprevir
- Beclabuvir
- Daclatasvir
- HCV NS3/4A protease inhibitor
- Hepatitis C virus
- Pegylated interferon-α
- Ribavirin
1 Introduction
Asunaprevir (BMS-650032, 1) is a tripeptidic acyl sulfonamide derivative that has been approved in 17 countries for the treatment of chronic hepatitis C virus (HCV) infection and is marketed as Sunvepra® [1,2,3,4,5,6,7]. Asunaprevir (1) is a potent inhibitor of the HCV NS3/4A protease and is approved for clinical use either in combination with the HCV NS5A inhibitor daclatasvir (2, Daklinza®) or as part of a triple therapeutic regimen that includes 2 and the HCV NS5B inhibitor beclabuvir (3), a fixed dose combination marketed as Ximency® [8,9,10,11,12,13,14,15]. The dual combination provides a highly effective treatment for patients infected with hepatitis C genotype 1b (GT-1b) virus, while the triple combination extends therapy to include the GT-1a variant that is prevalent in the United States and Europe [3,4,5,6,7,8,9,10, 15]. Notably, early clinical studies with 1 and 2 provided proof of concept that a chronic HCV infection could be cured with direct-acting antiviral agents (DAAs) in the absence of the exogenous immune stimulation provided by pegylated-interferon-α (PEG-IFN-α), a component of the standard of care therapy for HCV infection prior to the advent of DAAs [16, 17]. This clinical result was described as a “watershed moment in the treatment of HCV” since it provided a clear clinical path to the orally administered, DAA drug regimens that are available today and which are capable of curing a chronic HCV infection after just 8–12 weeks of therapy [18,19,20,21,22,23,24].

The discovery of 1 began with an evaluation of the hexapeptide derivative 4, a compound that had been described in the literature as a modestly potent inhibitor of the HCV NS3/4A enzyme [25,26,27]. An X-ray co-crystal structure of 4 bound to an engineered NS3/4A complex (Fig. 1) provided key insights into the nonbonding interactions governing the ligand-protein complex. Of particular interest were the touchpoints between the terminal carboxylic acid functionality of this product-based enzyme inhibitor and the catalytic triad of the NS3 protease, which were essential for potent enzyme inhibitory activity [27,28,29]. These critical interactions included a salt bridge between the oxygen-centered anion of acid 4 and the imidazole ring of the catalytic histidine (His57) as well as hydrogen-bonding interactions between the carbonyl moiety and the backbone N-H of Gly139 that contributes to the oxyanion hole of the enzyme. As part of an effort to design more potent inhibitors, consideration was given to the introduction of structural motifs that would maintain the key interactions between the carboxylic acid moiety and the catalytic site elements while allowing an extension of functionality into the small and well-defined S1’ sub-pocket [30]. As illustrated in Fig. 2, the S1’ site is a shallow pocket that is contiguous with the oxyanion hole of the enzyme whose boundaries are defined by the side chains of Phe43, Val55, and Gly58. An acyl sulfonamide was selected as a potential structural motif with which to replace the terminal carboxylic acid moiety since it appeared to fulfill the targeted criteria [31,32,33]. Modeling studies provided support for the design concept which preserves an acidic element while projecting the sulfone substituent toward the S1’ pocket. The contemporaneous disclosure of the tripeptidic carboxylic acid-based inhibitors of the NS3/4A protease represented generically by 5 provided a more compelling vehicle with which to explore this design proposal, and an exploratory discovery effort was initiated [1, 25, 26, 34, 35].

Details of the binding interactions of peptide inhibitor 4 bound to an engineered HCV NS3/4A protease complex. (a) View of the hexapeptide 4 bound to HCV NS3/4A. (b) Close-up view of the P1 element of 4 bound to HCV NS3/4A showing His157

Surface of the HCV NS3/4A enzyme illustrating the S1 and S1’ subsites

2 The Discovery of the First Clinical Candidate, BMS-605339
The carboxylic acid 6 was exploited as the vehicle with which to explore the acyl sulfonamide design concept based on its potent GT-1a NS3/4A enzyme inhibition, IC50 = 54 nM, and efficacy in a GT-1b replicon assay where the EC50 value was 550 nM. The prototypical acyl sulfonamide, the methyl derivative 7, exhibited enzyme and replicon inhibitory activities that were comparable to 6, validating the design principle and encouraging further molecular editing (Table 1) [26]. Modeling studies suggested that the cyclopropyl ring deployed in 11 would be the optimal element with which to fill the S1’ pocket and the preparation of this compound was, accordingly, given priority. In vitro evaluation of 11 revealed it to be a potent inhibitor of the GT-1a NS3/4A enzyme with an IC50 value of 1 nM, a 54-fold advantage over 6 that extended to the GT-1b replicon where sub-genomic virus replication was half-maximally inhibited at a concentration of 4 nM. These in vitro potency values met the criteria of <10 nM that we had set as the standard for a clinical candidate for all of our HCV inhibitor programs, and the results fostered additional study of the acyl sulfonamide chemotype. Confirmation that 11 represented the optimal P1’ element came from the systematic analysis of the structure-activity relationship (SAR) studies that are summarized in Table 1. The ethyl homologue 8 offered sixfold increased potency over the methyl prototype 7, while the propyl analogue 9 was twofold more potent than 7. The isopropyl derivative 10 is the ring-opened analogue of 11, and remarkably, the addition of just two hydrogen atoms to 11 is associated with a 20-fold reduction in potency in both the enzyme and cell-based assays, reflecting the precise demands associated with filling the NS3/4A S1’ pocket [26]. Interestingly, methyl substitution of the cyclopropyl ring proximal to the sulfone, as in 12, was tolerated, with only a modest effect on antiviral potency. However, potency decreased progressively in both in vitro assays as the size of the ring was increased, as exemplified by compounds 13–15, although in this series, the phenyl derivative 16 was an order of magnitude more potent than the cyclohexyl analogue 15 [36]. This SAR point was attributed to the flexibility of the Gln41 side chain of the enzyme allowing accommodation of larger P1’ structural elements. Modeling studies suggested that the key drug-target interactions of 11 revolved around the acidic acyl sulfonamide moiety engaging the imidazole of the catalytic His57 via the nitrogen rather than the carbonyl oxygen atom (Fig. 3). This contention was supported by calculations of the electrostatic potential of acyl sulfonamide derivatives and solid-state data for this structural motif [26]. This scenario anticipated that the carbonyl oxygen atom of the acyl sulfonamide would accept a H-bond from the backbone N-H of Gly137 in the oxyanion hole. These predictions were ultimately confirmed by X-ray co-crystal structures of inhibitors with the NS3/4A construct which also revealed that one of the sulfone oxygen atoms engaged in a H-bonding interaction with the side chain OH of the catalytic Ser139, while the other oxygen atom coordinated with the N-H of Gly137, resulting in this N-H engaging in a bifurcated interaction (Fig. 3).


(a) Model of the acyl sulfonamide moiety of 11 bound to the NS3/4A protease enzyme construct. (b) Key interactions between the acyl sulfonamide moiety and the NS3/4A protein. (c) Model of the cyclopropyl moiety bound to the S1’ pocket
With the identification of the cyclopropyl-acyl sulfonamide as the optimal P1’ moiety, this motif was adopted as a key structural element for further studies, stimulating the development of the synthetic methodologies to prepare cyclopropanesulfonamide and its derivatives that are summarized in Scheme 1 [37]. Commercially available 3-chloropropane-1-sulfonyl chloride was reacted with ammonia or tert-butylamine to afford the corresponding sulfonamide, with the primary sulfonamide converted to the N-t-Boc derivative by exposure to Boc anhydride in CH2Cl2 in the presence of Et3N and a catalytic amount of DMAP. Reaction of the tert-butyl or N-Boc sulfonamides with two equivalents of n-BuLi in THF effected an intramolecular alkylative ring closure to afford the anions of N-(tert-butyl)cyclopropanesulfonamide or tert-butyl (cyclopropylsulfonyl)carbamate, respectively, in situ. These products could be quenched with H2O and then exposed to CF3CO2H to deprotect the nitrogen atom to afford cyclopropanesulfonamide. Alternatively, the anions could be treated with an equivalent of n-BuLi in situ to generate dianions that were alkylated with CH3I to afford, after deprotection of the products by exposure to CF3CO2H, the methylated derivative explored in the context of 12. The dianionic species could also be generated directly from N-(tert-butyl)cyclopropanesulfonamide or tert-butyl (cyclopropylsulfonyl)carbamate by treatment with two equivalents of n-BuLi in THF [37].

Synthetic approaches developed to access cyclopropanesulfonamide derivatives
The promising potency of 11 led to an evaluation of its pharmacokinetic (PK) profile in a rat snapshot experiment which captured plasma levels of the compound over a 4-h period and liver levels at the termination of the experiment following oral dosing. After the administration of a dose of 20 mpk of 11, the plasma AUC was 0.73 μM·h, while IV administration at a dose of 5 mpk provided information on clearance (55 mL/min/kg) and half-life (1 h), data that translated into an oral bioavailability of 9% [26]. However, liver levels of 11 at the end of the experiment (4-h post-dose) were 5.8 μM, tenfold higher than the concentration of 0.58 μM measured in plasma at the same time point. In a separate experiment, the liver levels at 8-h post-dose were 0.6 μM, while plasma levels were 0.004 μM, a 150-fold differential. In rat liver microsomes (RLMs), 11 demonstrated a t 1/2 value of 162 min, a level of metabolic stability that indicated that the clearance pathways in vivo were not simply a function of metabolic modification and suggested the potential for the involvement of transporter-mediated liver uptake. The PK profile of 11 in the dog was similar to that observed in the rat, with a t 1/2 value of 170 min in dog LMs, a plasma AUC of 2.0 μM·h and bioavailability of 20% following an oral dose of 3 mpk, while the clearance after IV dosing of the compound was 16 mL/min/kg, translating into a plasma half-life of 0.6 h.
The hepatotropic disposition of 11 in the rat and, particularly, the sustained exposure in the liver were considered to be a favorable attribute since this is the organ where HCV predominantly replicates. In order to expedite in vivo studies as the program evolved, test compounds were dosed intraduodenally (ID) to surgically prepared rats and plasma levels monitored for 4-h post-drug administration, with the liver concentration of a test compound determined at the end of the study. In this phase of the program, structural manipulation of 11 was broad-based in nature and directed toward developing a detailed understanding of SARs associated with the P1, P3, and P4 elements of the molecule. In addition, modifications to the substituted quinoline heterocycle attached to C-4 of the P2 proline residue, a motif that bound to the protease outside of the substrate-binding groove and which was therefore designated as the P2* element, were explored. This approach recognized the importance of the backbone H-bond donors and acceptors in molecular recognition, while the side chains and the P4 moiety established van der Waals contacts with their respective pockets in the enzyme. This SAR survey also acknowledged the high molecular weight of 11 (789 Da) and its potential impact on PK parameters to the extent that pruning elements of the molecule was considered an important part of the design strategy. The data presented in Table 2 summarize the key informative discoveries that emerged from this line of inquiry and which shaped the remainder of the program ultimately leading to the discovery of 1. Truncating the P1 and P3 elements by removal of the vinyl and tert-butyl substituents (17 and 18, respectively) resulted in a greater than tenfold reduction in intrinsic enzyme inhibitory potency that translated into a more severe loss of antiviral activity in the replicon assay. Removal of the t-Boc moiety at P4 (19) also eroded potency significantly, but replacing this element with a methoxycarbonyl (20) was more readily tolerated. However, this reduction in molecular weight resulted in an inferior in vivo profile, with both the plasma and, particularly, the liver exposure significantly reduced after ID dosing compared to 11. Further molecular modification of P4 in the context of the urea 21, reverse carbamate 22, the isopropyl carbamate 23, and the cyclopentyl carbamate 24 led to additional insightful and interesting SAR points, but none provided the kind of in vivo profile that was considered to be promising, with plasma and liver exposures in the 4-h rat experiment less than that observed for 11 [26, 38]. However, modifications to the P2* element proved to be a more promising avenue of study, with the removal of the C-3 phenyl substituent associated with only a modest reduction in both intrinsic and replicon potency, as exemplified by 25. Unfortunately, the reduction in molecular weight was not helpful with respect to improving the in vivo exposure. A more severe truncation of the quinoline heterocycle at P2* to the pyridine ring found in 26 defined the structural requirements at this site for potency which was enhanced by the introduction of the C-3 phenyl substituent in 27, although with no benefit to the in vivo properties. However, configuration of the quinoline moiety as the 2-substituted topology represented in 28
was not only tolerated with some facility in the in vitro assays, but the in vivo profile of this compound in the rat was illuminating, revealing that changes at the P2* moiety could significantly affect PK parameters. The plasma exposure of 28 offered a threefold advantage over that of 11, while the 4-h liver levels were sixfold higher. The installation of a 1-substituted isoquinoline at P2* gave a compound 29
that preserved much of the intrinsic enzyme inhibitory potency of 11 but, more importantly, offered an improved in vivo PK profile, with tenfold higher plasma exposure and almost fourfold higher liver levels at 4 h [26]. The oral bioavailability of 29 in the rat was 20%, with IV clearance measured as 19 mL/min/kg and a plasma t
1/2 of 2.2 h. The introduction of a 6-methoxy substituent to the isoquinoline of 29
gave 30, a compound with improved potency in both assays while further boosting the PK profile in the rat ID experiment. In the rat, the oral bioavailability of 30 was 18%, with IV clearance lower than for 29 at 4.4 mL/min/kg and a longer plasma t
1/2 of 4.4 h. Comparison of the profile of 30 with that of its 4-substituted quinoline isomer 25 provides a remarkable example of the effect of a subtle structural modification on the properties in different biological systems against the structural backdrop of a large molecule (MW = 713) [39]. While the antiviral effects of the two compounds are similar, the PK profiles are quite disparate, with the plasma exposure of 30 almost 700-fold higher than that of 25, while the 4-h liver levels are 100-fold higher, an observation amplified in the comparison with the matched pair of macrocyclic inhibitors 31 and 32 where the AUC and liver levels of the former compound exceed those of the latter by 28,000- and 1,500-fold, respectively [40].


The preliminary in vitro and in vivo profiling of 30, which was designated as BMS-605339, indicated that this was a promising compound, with antiviral potency that met the criteria for advancement. An extensive survey of the SARs associated with substitution of the isoquinoline heterocycle of 30 confirmed its candidacy, with the compounds that provided seminal insights into SAR and PK profiles captured in Table 3. In the X-ray co-crystal structure of 30 with the NS3/4A construct, the isoquinoline ring is positioned over the side chain of Arg155 which forms a salt bridge with Asp168 (Fig. 4). The SARs associated with substitution of the isoquinoline ring of 30 were interpreted in the context of the effect on the electrostatic potential of the heterocycle that modulates a π-cation interaction with Arg155. However, the C-6-substituted nitrile 41 is an outlier, explained by modeling studies that suggested the potential for the electron-rich tip of the nitrile substituent to engage the positively charged center of the guanidine moiety of Arg155 [26]. Hence, the interaction of the isoquinoline ring with the NS3/4A protease and the effect of the C-6 substituent appear to take advantage of two distinct modes of engagement with the enzyme surface and, particularly, Arg155 that is sensitive to the substitution pattern (Table 4).


X-ray co-crystal structure of 30 bound to a HCV NS3/4A protein
The results of antiviral and rat PK screening identified 30 and 33 as compounds of interest for more detailed studies. Full PK profiling of 33 in the rat revealed that while the oral bioavailability was an encouraging 20% following a dose of 15 mpk, the compound could not be detected in plasma 24-h post-dose, although liver levels at this time point were high at 4.2 μM. This observation was concordant with IV PK parameters, with 33 cleared rapidly from plasma but partitioning readily into the liver, where drug concentration was measured as 8.0 μM at 24 h following administration of a 5 mpk dose. This profile was not considered suitable for further progression of 33, and attention was focused on the regioisomer 30 which, although displaying lower oral bioavailability in the rat, exhibited a less hepatotropic disposition in vivo [39]. After dosing of 30 to rats as a solution formulation, the oral bioavailability ranged from 5 to 18%, with plasma exposure of 14.1 μM·h over a 24-h period following a 20 mpk dose [39]. Plasma levels of 24-h post-dose were 8 nM, with the liver concentration almost 100-fold higher at 730 nM. In the rat, 30 is classified as a low clearance compound, with values of 4.7 and 4.4 mL/min/kg after IV administration of doses of 2 and 10 mpk, respectively (Table 4).
In the dog, the plasma clearance of 30 was classified as moderate, with a moderate V ss and a short t 1/2 of 0.6 h following a 1 mpk IV dose. However, the AUC increased in a greater than dose-proportional fashion following a 4 mpk dose of 30, with lower clearance and a longer t 1/2 of 5.1 h, suggesting that the drug elimination pathways were subject to saturation. In PO dosing experiments, the absorption of 30 was rapid at all doses examined, but oral bioavailability increased with exposure, rising from 23% at 1 mpk to 51% at 3 mpk. Similar to observations in the rat, the oral bioavailability of 30 in the dog was lower following administration of a suspension of the drug compared to a solution formulation, indicative of solubility- or dissolution-limited absorption. Liver levels in the dog were considerably higher than in plasma, with ratios ranging from 51 to 64 that remained constant over time (2, 8, and 24 h), consistent with parallel elimination rates from both compartments [39]. While IV clearance was moderate in the cynomolgus monkey, volume of distribution (V ss) was low, and the t 1/2 value was 0.8 h after administration of a 1 mpk dose and 3.7 h following a dose of 3 mpk. The oral bioavailability of 30 in the cynomolgus monkey was poor at just 1%, attributed to rapid metabolism that was reflective of the high turnover observed in cynomolgus monkey LMs.
The HCV NS3 protease and replicon inhibitory spectrum of 30 is summarized in Table 5. In vitro analyses indicated excellent selectivity of 30 for HCV NS3/4A protease with IC50 values >1,000-fold higher for a panel of mammalian serine and cysteine proteases and the phylogenetically closest relative, the GBV-B NS3 protease. Compound 30 was inactive in cell culture replication assays for bovine viral diarrhea virus, canine parainfluenza virus, and HIV-1, and additional liability profiling of 30 confirmed its suitability for advancement into clinical trials [26].
After successfully completing IND toxicology studies, 30 was advanced into clinical trials following a protocol that comprised of a single ascending dose (SAD) study (10, 30, 60, and 120 mg) conducted in normal healthy volunteers (NHVs) and a subsequent SAD study conducted in HCV-infected patients [26]. In the NHV study, plasma levels of 30 increased with dose, although not in a dose-proportional fashion, with a t max of ~1.5 h indicative of relatively rapid absorption. The terminal t 1/2 was 4–8 h, and 30 was detectable in plasma at 24-h post-drug administration. The SAD in patients infected with GT-1 HCV assessed doses of 10, 60, and 120 mg, selected based on the PK parameters determined in NHVs, with the effect of 30 on plasma viral load compared with those of a placebo control. Plasma viral RNA levels were monitored closely for 24 h and at 48, 72 and 144 h post-dose. A dose-dependent reduction in viral load was observed following administration of 30, with the 120 mg dose associated with a mean decline of 1.8 log10 international units per mL (IU/mL) measured at 12-h post-dose [26]. The dose-response data are compiled in Fig. 5.

Mean maximal reductions in plasma HCV levels following the administration of single oral doses of 30 to HCV-infected subjects. Maximal reductions at each dose occurred 12 h after drug administration
3 The Discovery of Asunaprevir (BMS-650032)
While the antiviral efficacy observed with 30 offered considerable promise, enthusiasm was tempered by the occurrence of clinically relevant electrocardiographic changes in one NHV and one HCV-infected patient following a dose of 120 mg of the drug. The effects of 30 on cardiac function were manifested as transient mild bradycardia, PR interval prolongation, and junctional rhythm disturbances that, although asymptomatic in presentation, were considered to represent an unacceptable safety risk for further clinical study, and clinical development of 30 was ended [26].
The C max value of 30 in the NHV who experienced cardiac effects was 113 nM, while in the HCV-infected patient, the C max was 206 nM, and, in each case, the effects were observed at 1-h post-dose. In preclinical studies conducted in conscious telemetrized dogs administered two 15 mpk doses of 30, the observed cardiovascular effects were a reduction in heart rate and prolongation of the PR interval. While these general electrophysiological effects had some similarity with the observations in humans, they occurred at plasma exposures estimated to be approximately 12 μM based on studies conducted in non-telemeterized animals [1]. Since the affinity of 30 for plasma proteins was similar in dogs (98.2%) and humans (99.1%), it was concluded that humans were considerably more sensitive to the CV effects of 30 than preclinical species. In an effort to identify a predictive preclinical model, the CV effects of 30 in rabbits were investigated following IV administration of the drug to anesthetized animals. Heart rate (HR), blood pressure, and sinus node recovery time (SNRT) were monitored following incrementally increasing doses of 1, 3, and 10 mpk of 30, with each dose infused over a 5-min period. A mild 32 beats per minute (bpm) decline in HR was noted in this experiment along with a modest reduction in blood pressure amounting to 14–23 mmHg from the baseline value; however, only the heart rate decline was drug related since changes in blood pressure were also observed in control animals infused with vehicle. The plasma concentration of 30 in this experiment peaked at 27 μM and by 60 min after the last infusion had fallen to 1.65 μM when drug concentration in cardiac tissue was 0.25 μM. The results of this study indicated that an in vivo rabbit evaluation protocol would not be a useful approach to identifying compounds predicted to offer reduced CV effects in humans. Evaluation of 30 in a battery of in vitro cardiac ion channel and receptor screens failed to identify a biochemical target, and so a Langendorff isolated rabbit heart model was investigated in an effort to more precisely understand the effects of 30 on cardiac function [1, 41]. In this model, the isolated rabbit hearts were perfused with Krebs-Henseleit solution containing 30 at concentrations of 0.3, 1, 3, and 10 μM for a period of ~12 min, while HR and SNRT were monitored. Declines in HR were observed at concentrations of 3 μM (−15 bpm) and 10 μM (−23 bpm) of 30 with an increase in SNRT of ≥425 ms, effects that continued to increase during the 10 min washout period following drug perfusion. However, no effects on atrioventricular conduction time or QRS interval were observed. When rabbit hearts were perfused for 60 min at a single concentration of 30, effects on HR and SNRT were observed when drug was perfused at a concentration of 1 μM, but the effects were attenuated at a lower perfusion concentration of 0.3 μM and absent at 0.1 μM. These data suggested that the cardiac effects of 30 were due to a direct action of the drug on the SA node which acts as the cardiac pacemaker. Although the rabbit heart was less sensitive to the effects of 30 than humans, the Langendorff model was adopted as an in vitro liability screen since test compounds could be evaluated at high, protein-free drug concentrations, with a perfusion concentration of 10 μM selected to profile test compounds. However, the Langendorff model is a complex assay with limited throughput, necessitating a careful selection of compounds for evaluation, which sought to develop an understanding of the structure-liability relationships by evaluating systematic structural changes to 30 [1].
The data presented in Table 6 provided key insights into effect of molecular editing on cardiac parameters in the rabbit model. The observation of heart rate and SNRT changes associated with the carboxylic acid analogue 42 that were milder in severity than 30 was considered to absolve the cyclopropyl-acyl sulfonamide moiety as causative. This was important since the value of the cyclopropyl-acyl sulfonamide is clearly exemplified by the 100-fold compromised antiviral activity associated with 42 compared to 30. Compounds with modifications at P3 (43), P4 (44), and P2* (45 and 46) also exhibited significant effects on the measured rabbit heart parameters, results that indicated that identifying a compound containing potent antiviral activity with the targeted PK profile while exhibiting reduced CV effects would require considerable experimentation [1]. This consideration, coupled with the knowledge base that had been generated around the P2* element and its effects on antiviral potency and PK, focused the attention on assessing the effects of isoquinoline substitution pattern on CV parameters, with results for the salient compounds 33, 47–51, and 1 compiled in Table 6. All seven of these compounds are potent GT-1a enzyme inhibitors with EC50 values in the GT-1b replicon that, with the exception of 33 and 47, are ≤10 nM. However, the CV effects in the Langendorff assay show a marked dependence on substitution pattern, with early structure-liability studies implicating the C-6 methoxy substituent as a contributing element. Consistent with this observation, the CV effects were moderated when the methoxy substituent was installed at the 5-position (47) or the 4-position (33) or at both the 4- and 5-position (48) when compared to 30. The PK profile of 33 offered promise, with oral bioavailability of 19%, an AUC of 0.37 μM·h, low clearance (9.8 mL/min/kg), and a t 1/2 of 4.3 h following a 30 mpk dose, while for 47 the oral F was 9%, the AUC was 2.7 μM·h, clearance was low at 6.0 mL/min/kg, and the t 1/2 was 2.2 h. However, 33 and 47 were not progressed because of the modest potency in the GT-1b replicon with EC50 values of 25 and 19 nM, respectively, that exceeded the target of ≤10 nM. The PK profile of 48 was less than optimal, with a short t 1/2 in the rat of 0.3 h. Nevertheless, these results encouraged further structural manipulation which revealed that the combination of 4- and 5-methoxy substitution in 49 was associated with significant effects on HR and SNRT as was the 4-methoxy and 5-chloro combination exemplified by 50. However, the combination of a 4-methoxy substituent with either a 6-chloro (51) or 7-chloro (1) substituent moderated the CV effects, with those of 1 comparable to vehicle in the Langendorff rabbit heart assay. This CV profile was confirmed in a 60-min perfusion protocol at a drug concentration of 10 μM where the maximal reduction of heart rate associated with 1 was 10% and the maximal increase in SNRT was 14%, values that were similar to the vehicle control in this experiment. This result provides a second demonstration of the significant effects of subtle structural modifications on biological properties in the context of a large molecule in this series of HCV NS3/4A protease inhibitor, with the only difference in the molecular formulae between 30 and 1 being the addition of a chlorine atom [1].

The PK profile of 1 in rats was acceptable, with an oral AUC of 1.0 μM·h following a 20 mpk dose, 12% oral bioavailability, and high liver levels of 15.2 μM 24-h post-dose, reflective of a more hepatotropic disposition than 30 [1, 42]. An IV PK experiment allowed the determination of the in vivo clearance of 1, which was 38 mL/min/kg, although the t 1/2 was reasonable at 4.2 h (Table 7). Although extensive SAR studies were conducted around 1, with variations at P1, P2*, and P4 examined, none of the analogues synthesized offered an overall profile in vitro and in vivo that surpassed that of the prototype, and 1 was examined in further detail in order to determine its suitability as a candidate for development. The PK profiles of 1 in mouse, dog, and cynomolgus monkey are compiled along with the rat data in Table 7 and reveal moderate clearance in all species with the exception of the cynomolgus monkey, a PK profile that reflects the high turnover of 1 in LMs from this species (50 ± 22 pmol/min mg which compares to 4.9 pmol/min mg in RLM, 4.7 pmol/min mg in dog LM, and 5.8 ± 1.8 in HLM). The measured in vitro clearance values correlated reasonably well with the in vivo results in the dog and monkey but underpredicted in vivo clearance in the rat and the dog, although the data were within twofold in the latter species. This was attributed to direct excretion of 1 into the bile, which amounted to 9% of the dose in the rat and 22% in the dog, and suggested the involvement of liver uptake transporters. Incubation of 1 with individual recombinant CYP enzymes revealed significant metabolic turnover only by CYP 3A4, with metabolism in HLM inhibited by ketoconazole and troleandomycin and selective CYP 3A4 inhibitors, although additional contributions to metabolism by CYP 2D6, CYP 2C9, and CYP1A2 could not be definitively ruled out. Further studies with recombinant CYP enzymes indicated that 1 was not an inhibitor of CYP 2D6, CYP 2C9, CYP 2C19, and CYP 1A2, with IC50 values >40 μM, but 1 was a moderate CYP 3A4 inhibitor with IC50 values of 8, 29, and 27 μM when benzyloxyresorufin (BZR), 7-benzyloxy-trifluoromethylcoumarin (BFC), and midazolam were used as substrate probes, respectively.
The permeability of 1 in a parallel artificial membrane permeability assay (PAMPA) was high (>473 nm/s at pH = 5.5 and >492 nm/s at pH = 7.4) predictive of good absorption [1, 43, 44]. The high membrane permeability associated with 1 has been attributed to its inherent conformational flexibility which allows dynamic modulation of the exposure of its polar surface elements in different environments [44]. However, in a bidirectional Caco-2 cell assay, the measured efflux ratio (ER) was 31-fold at a drug concentration of 5 μM and threefold at the higher concentration of 25 μM, indicative of saturation of efflux [43, 44]. Efflux of 1 (5 μM) was reduced significantly in these assays in the presence of inhibitors of P-gp and breast cancer resistance protein (BCRP) (ER = 1.9) and P-gp and multidrug resistance protein 2 (MRP2) (ER = 1.4) but less so in the presence of a BCRP inhibitor alone (ER = 20), suggesting that 1 is a substrate for P-gp and, possibly, MRP2. A role for P-gp in the disposition of 1 in vivo was confirmed with studies in P-gp knockout mice where the oral bioavailability increased to >100% compared to 28% in wild-type animals [43].
The plasma protein binding of 1 was high in rat, dog, and cynomolgus monkey plasma (97.2–98.8%) and comparable to human plasma, 98.8%. The V ss in vivo was higher than total body water in mice and rats but similar to the total body water volume in dog and cynomolgus monkey. The liver to plasma ratios of 1 in mouse, rat, dog, and monkey measured at 8-h post-dose were 82, 555, 293 (7 h), and 248, respectively, high levels that persisted out to 24-h post-dose in the rat (300) and dog (410), indicative of a hepatotropic disposition in all of the preclinical species studied. However, 1 did not distribute effectively to CNS, endocrine, reproductive, or fatty tissues.
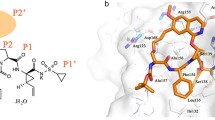
Biochemical profiling indicated that 1 was a potent and competitive inhibitor of recombinant GT-1a and GT-1b HCV NS3/4A proteases with K i values of 0.4 nM and 0.24 nM, respectively, and an X-ray co-crystal structure highlighted the key drug-target interactions (Fig. 6) [1, 43, 45,46,47]. In vitro genotypic and phenotypic analysis of the emergence of resistance in GT-1a and GT-1b replicons in response to selective pressure exerted by 1 identified several substitutions that conferred low (5-fold) to moderate (21-fold) resistance to GT-1a virus in transient-transfection susceptibility assays [45]. The primary mutations were either Arg155Lys or Asp168Gly, which interacts in the NS3 protein to form a salt bridge, part of which makes contact with the isoquinoline P2* moiety, or at Ile170Thr, a residue interacts with the head group of Arg155 and contributes to the pocket around the Arg155-Asp168 salt bridge. While the Ile170Thr mutation suggests a subtle effect on interactions of the isoquinoline P2* heterocycle with the Arg155-Asp168 salt bridge, disruption of the Asp168-Arg155 salt bridge renders the Arg155 side chain mobile, modifying the planarity of the S2* region of NS3/4A and thereby affecting binding of the isoquinoline P2* heterocycle. In addition, comparison of the single crystal conformation to the bound conformation of 1 (Fig. 7) suggests significant preorganization of the ligand in the P2*-P4 region. Mutations occurring in the S2* region would thus be expected to exert an impact on the contact surface with the protein and require disruption of the preorganized conformation of the ligand, with the fold change in resistance reflective of the extent of disruption in the optimal planar S2* site. In GT-1b replicons, the same selection pressures resulted in replicons with a higher level of resistance to 1, with the primary site of mutation mapped to Asp168 and with several substitutions (Asp168Ala/Gly/His/Val/Tyr) observed that conferred high levels (16- to 280-fold) of resistance. The most prevalent GT-1b resistance mutations were Asp168Gly and Asp168Val, and these substitutions had a deleterious impact on replication capacity.

Co-crystal structures of 1 bound to a HCV NS3/4A enzyme construct. (a) Co-crystal structure of 1 bound to a HCV NS3/4A enzyme construct. (b) An overlay of the co-crystal structures of 1 and 30 bound to a HCV NS3/4A enzyme construct

(a) Single crystal structure of 1. (b) Overlay of the single crystal structure of 1 with the protease-bound conformation of 1 (green “C” sticks)
In replicon assays, 1 was a potent inhibitor of GT-1 and GT-4 replication with EC50 values of <5 nM but was less potent toward GT-2 and GT-3 replicons where the EC50 values ranged from 67 to 1,162 nM, data compiled in Table 8. In combination studies, 1 demonstrated additive or synergistic activity when combined with IFN-α, the nucleoside derivative ribavirin (52), the HCV NS5A inhibitor 2, and the allosteric HCV NS5B inhibitor 3.

4 Clinical Studies with Asunaprevir
The clinical development program for 1 began with a double-blind, placebo-controlled sequential SAD study in which doses of 10, 50, 100, 200, 400, 600, and 1,200 mg of 1 were administered as an oral suspension to panels of eight NHVs in order to establish a safety profile and glean insight into plasma pharmacokinetic properties [48,49,50,51]. Adverse events (AEs) were generally mild to moderate and comparable to the placebo controls, with headache and diarrhea as the most common events that occurred in both the drug-treated and placebo groups. Most importantly in this short duration study, a detailed analysis of cardiac safety in patients revealed no evidence of an increased risk of adverse cardiac events, and there were no dose- or time-related trends in the QT interval corrected for heart rate [48]. Although 1 was detected in plasma of NHVs at 30 min post-dose, reflective of facile absorption, the PK profile of the drug was complex in nature. The terminal half-life of 1 ranged from 14 to 20 h, but the apparent oral clearance was high, and plasma levels of the compound declined in a biphasic fashion [49,50,51]. The rapid plasma clearance associated with 1 was consistent with the hepatotropic disposition observed in preclinical species and in subsequent studies evaluating the effect of concomitant administration of a single dose of the OATP inhibitor rifampin; the plasma C max of 1 was 21-fold higher, although there was considerable variability in the values, while the mean AUCinf increased by 15-fold [52]. The plasma exposure of 1 in the context of dose was nonlinear, with incremental increases in exposure that were greater below 200 mg than above this dose (Table 9) [49,50,51]. In addition, the plasma exposure was higher following the dosing of 1 as a solution formulation with the mean C max and AUCinf, 4.4- and 1.8-fold higher, respectively, at the 50 mg dose and 10- and 2.8-fold higher, respectively, at the 200 mg dose. Collectively, these data were indicative of solubility- and dissolution-limited absorption, and when a tablet formulation of 1 was dosed concomitantly with a high-fat meal at a dose of 600 mg, the C max and AUCinf values were 29.6- and 11.5-fold higher, respectively, than administration of the tablet under fasted conditions [49,50,51].
Based on the PK profile in NHVs, doses of 10, 50, 200, and 600 mg of 1 were selected for a double-blind, placebo-controlled sequential SAD study conducted in HCV GT-1-infected patients to assess the effect of compound on plasma HCV levels [48]. The PK parameters of 1 in this patient population were similar to NHVs although plasma exposure appeared to be slightly higher [48,49,50,51]. HCV viral load was followed for 144-h post-drug dosing, and while the virological response to 1 was minimal at the 10 mg dose, mean plasma HCV RNA declined by 0.64 log10, 2.26 log10, and 2.87 log10 IU/mL following administration of single 50, 200, and 600 mg doses of 1, respectively (Fig. 8a). A multiple ascending dose study in which 1 was administered at doses of 200, 400, and 600 mg on a twice daily schedule for 3 days to GT-1 HCV-infected individuals under a double-blind, placebo-controlled protocol confirmed the antiviral effects, with the mean maximal reductions in viral load compiled in Fig. 8b.

(a) Mean maximal viral load reduction following oral administration of 1 to GT-1 HIV-infected patients. Maximal viral load reduction was observed at 12-h post-dose following administration of the 10, 50, and 20 mg doses, while the nadir occurred at 24 h following the 600 mg dose of 1. (b) Mean maximal viral load reduction following oral administration of 1 twice daily to GT-1 HIV-infected patients for 3 days
The development of 1 was part of a broader strategy that sought to advance at least two direct-acting antiviral agents into clinical trials that were designed to be used in combination therapy as a means of addressing concerns surrounding the rapid development of resistance to mono-therapeutic drug regimens. Viral RNA polymerases are recognized for their poor proofreading ability which leads to the incorporation of mismatched base pairs and the production of viruses with mutations that can resist drug action and lead to virological failure [53,54,55,56,57,58,59,60]. The frequency with which HCV RNA polymerase introduces mutations and the influence of replication rate on virus diversity has been elegantly analyzed by Perelson and explains the rapid emergence of resistance seen with the first-generation HCV NS3 protease inhibitors [61,62,63,64,65,66,67,68,69]. The HCV NS5A inhibitor 2 progressed into clinical trials contemporaneously with 1, providing the opportunity to conduct a clinical trial in which the potential of the two compounds to treat a chronic HCV infection was examined [17, 18]. The patient population selected for this trial was comprised of patients who had either failed the existing standard of care therapy at the time, PEG-IFN-α, and the nucleoside analogue 52, or who had failed to respond to this therapeutic regimen, so-called null responders [70,71,72]. This patient population was considered to be the most difficult to treat, and a trial against this clinical background was viewed as a stringent evaluation of the potential of DAAs to affect the course of a chronic HCV infection. In this phase IIa clinical trial, a combination of 1 (600 mg BID) and 2 (60 mg QD) was administered to 11 HCV GT-1-infected subjects for 24 weeks. The control arm for this study comprised of 10 GT-1-infected subjects who received the same dosing regimen of 1 and 2 in conjunction with the standard of care therapy of PEG-IFN-α2a and 52 dosed for 24 weeks. The two patient cohorts were matched for viral load and virus genotype, with the majority infected with GT-1a and with 10/11 patients in the dual combination group and 9/10 in the quadruple arm group expressing the IL28B CT or TT genotypes that predict a poor response to peg-IFNα therapy rather than the responsive CC genotype (Table 10) [73,74,75,76]. In the quadruple therapy cohort, all patients had undetectable levels of HCV RNA in plasma at the end of therapy, a circumstance that persisted to week 12 post-therapy (SVR12), data compiled in Table 10. However, one patient had detectable HCV RNA in plasma at week 24 after treatment but was undetectable 35 days later; however, this patient was classified as a failure based on the written clinical protocol. In the direct-acting antiviral cohort, 5/11 subjects had undetectable HCV RNA at the end of therapy with 1 GT-1a-infected subject relapsing post-therapy to leave 4/11 subjects who achieved SVR12 (Table 10). In this cohort, both GT-1b-infected subjects achieved SVR12 and SVR24, while viral breakthrough was restricted to the GT-1a-infected subjects. Those subjects experiencing viral breakthrough had peg-IFNα2a and 52 added to their drug treatment regimen, and while most responded with declines in viral load, the effect was not durable [17].
This study demonstrated for the first time that a chronic HCV infection could be cured with DAA therapy alone in the absence of an exogenous immune stimulant and 52 and was characterized as “a watershed moment” in the development of HCV DAAs [18]. The efficacy of the combination of 1 and 2 in treating GT-1b infections redirected the clinical development program toward Japan where the patient population is comprised predominantly (70%) of subjects infected with this virus genotype. In the phase III clinical trials that formed the basis of marketing approval of the dual combination of 1 and 2 for the treatment of GT-1b infection in Japan, the combination was administered at doses of 100 mg BID and 60 mg QD, respectively, for 24 weeks of therapy [77,78,79,80,81,82,83,84,85]. This regimen was associated with SVR12 rates of 81% in the non-responder population and 87% in those either ineligible for or intolerant of PEG-IFNα therapy. In a multinational clinical trial conducted in a broad-based patient population infected with GT-1b virus, the SVR12 rates were 90% for those naïve to therapy and 82% for those in the ineligible/intolerant group.
Marketing approval of the dual drug combination of 1 and 2 by the Japanese Pharmaceutical and Medical Devices Agency (PMDA) occurred on July 4, 2014 [8]. The licensing of 1 and 2 represented the first approval of a combination of DAAs for the treatment of HCV infection although the combination of 52 and the nucleoside-based HCV NS5B inhibitor prodrug sofosbuvir (53) was approved by the FDA in December of 2013 [86, 87].

Extending the therapeutic utility of 1 and 2 to include the treatment of HCV GT-1a infections required the addition of a third agent, the NS5B inhibitor beclabuvir (3), which was ultimately developed as a fixed dose combination comprising of 200 mg of 1, 30 mg of 2, and 75 mg of 3 administered on a BID schedule for 12 weeks [88,89,90,91,92,93]. In an international phase III clinical study designated the UNITY 1 trial that was conducted in 415 patients infected with HCV GT-1 who were non-cirrhotic, 91% of the patients achieved SVR12. This patient population in this trial comprised of both treatment-naive and treatment-experienced patients with SVR12 achieved by 92% in the former and 89% in the latter groups with virologic failure occurring in 8% of the patients. In a phase III study conducted in 202 GT-1-infected patients with compensated cirrhosis who resided in the United States, Canada, France, and Australia (UNITY-2), the SVR12 rates were 93% and 87% for patients in the treatment-naïve and treatment-experienced groups, respectively. In this study, the addition of 52 to the therapeutic regimen improved SVR12 rates to 98% for those naïve to treatment and 93% in those who were treatment-experienced. In the UNITY 3 phase III clinical trial that was conducted in Japanese patients infected with GT-1 HCV, SVR12 rates were ≥95% in both the larger treatment-naive (n = 152) and the interferon-experienced (n = 65) groups following 12 weeks of therapy. In this study, SVR12 rates were similar for this with cirrhosis and those aged ≥65 years. These studies contributed to the approval of the fixed dose combination of 1, 2, and 3 for marketing as Ximency® in Japan on December 20, 2016. Interestingly, a short duration intensification study in which HCV-infected patients were treated with a combination of 1, 2, 3, and 53 for 4 or 6 weeks failed to effect high rates of cure despite 96% of the patients achieving undetectable HCV RNA levels in plasma at the end of therapy [94].
5 Conclusion
The discovery of 1 relied upon an approach rooted in structure-based drug design that identified the beneficial effects of the cyclopropyl-acyl sulfonamide moiety as the optimal P1-P1’ moiety in a series of tripeptide-based NS3/4A inhibitors. This structural element conferred a significant enhancement in intrinsic enzyme inhibitory activity compared to carboxylic acid-based prototypes that translated effectively to potent antiviral effects in HCV replicons, which had just become available at the time that 11 was first synthesized. The value of the cyclopropyl-acyl sulfonamide in NS3/4A inhibitor drug design is underscored by the widespread adoption of this moiety as a prominent structural element that is present in all of the currently marketed HCV NS3/4A protease inhibitors. The development of the tripeptide-based inhibitors leading to 1 presented several challenges that were associated with exploring drug space beyond the rule-of-5 parameters that were beginning to be widely embraced at the time that 11 was discovered [95,96,97,98,99,100,101]. While identifying compounds with targeted PK profiles proved to be less arduous than the rule-of-5 parameters might have predicted, the observations of cardiac side effects in phase I clinical trials with 30 was a major disappointment and a significant hurdle to surmount. However, despite the low throughput associated with the Langendorff isolated rabbit heart assay used to triage compounds for CV effects, by the time that the decision was made to end the clinical development of 30, the team had been able to identify 1 by finessing the salient structure-liability relationships after screening approximately 20 compounds. The clinical trial assessing a combination of 1 with the NS5A inhibitor 2, the discovery of which relied upon the diametrically opposite strategy of phenotypic screening and optimization, in infected HCV patients proved to be of significance to the field [11, 16, 17]. The results of this small clinical study, which demonstrated for the first time that a chronic infection could be cured by DAAs alone, subtended the advent of the well-tolerated small molecule drug combinations that have been widely approved. These DAA combinations are very effective therapeutics for the treatment of chronic HCV infections, effecting high rates of cure after just 8–12 weeks of therapy, a sharp contrast to the 48 weeks of therapy that had to be endured with PEG-IFN-α-based regimens.
References
Scola PM, Sun L-Q, Wang AX, Chen J, Sin N, Venables BL, Sit S-Y, Chen Y, Cocuzza A, Bilder DM, D'Andrea SV, Zheng B, Hewawasam P, Tu Y, Friborg J, Falk P, Hernandez D, Levine S, Chen C, Yu F, Sheaffer AK, Zhai G, Barry D, Knipe JO, Han Y-H, Schartman R, Donoso M, Mosure K, Sinz MW, Zvyaga T, Good AC, Rajamani R, Kish K, Tredup J, Klei HE, Gao Q, Mueller L, Colonno RJ, Grasela DM, Adams SP, Loy J, Levesque PC, Sun H, Shi H, Sun L, Warner W, Li D, Zhu J, Meanwell NA, McPhee F (2015) The discovery of asunaprevir (BMS-650032), an orally efficacious NS3 protease inhibitor for the treatment of hepatitis C virus infection. J Med Chem 57:1730–1752
Meanwell NA (2016) 2015 Philip S. Portoghese medicinal chemistry lectureship. Curing hepatitis C virus infection with direct-acting antiviral agents: the arc of a medicinal chemistry triumph. J Med Chem 59:7311–7351
Reviriego C (2012) Asunaprevir. Drugs Future 37:247–254
Gentile I, Buonomo AR, Zappulo E, Minei G, Morisco F, Borrelli F, Coppola N, Borgia G (2014) Asunaprevir, a protease inhibitor for the treatment of hepatitis C infection. Ther Clin Risk Manag 10:493–504
Yang SS, Kao JH (2015) Asunaprevir-containing regimens for the treatment of hepatitis C virus infection. Expert Rev Gastroenterol Hepatol 9:9–20
Akamatsu N, Sugawara Y, Kokudo N (2015) Asunaprevir (BMS-650032) for the treatment of hepatitis C virus. Expert Rev Anti-Infect Ther 13:1307–1317
Eley T, Garimella T, Li W, Bertz RJ (2017) Asunaprevir: an HCV protease inhibitor with preferential liver distribution. Clin Pharmacol Drug Dev 6:195–200
Poole RM (2014) Daclatasvir plus asunaprevir: first global approval. Drugs 74:1559–1571
Zeuli JD, Adie SK, Rizza SA, Temesgen Z (2015) Asunaprevir plus daclatasvir for the treatment of chronic hepatitis C virus infection. Drugs Today 51:629–643
Hayes CN, Imamura M, Chayama K (2017) The practical management of chronic hepatitis C infection in Japan – dual therapy of daclatasvir plus asunaprevir. Expert Rev Gastroenterol Hepatol 11:103–113
Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun J-H, O'Boyle II DR, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG (2010) Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100
Belema M, Nguyen VN, Bachand C, Deon DH, Goodrich JT, James CA, Lavoie R, Lopez OD, Martel A, Romine JL, Ruediger EH, Snyder LB, St. Laurent DR, Yang F, Zhu J, Wong HS, Langley DR, Adams SP, Cantor GH, Chimalakonda A, Fura A, Johnson BM, Knipe JO, Parker DD, Santone KS, Fridell RA, Lemm JA, O’Boyle II DR, Colonno RJ, Gao M, Meanwell NA, Hamann LG (2014) Hepatitis C virus NS5A replication complex inhibitors: the discovery of daclatasvir. J Med Chem 57:2013–2032
Belema M, Meanwell NA (2014) Discovery of daclatasvir, a pan-genotypic hepatitis C virus NS5A replication complex inhibitor with potent clinical effect. J Med Chem 57:5057–5071
Gentles RG, Ding M, Bender JA, Bergstrom CP, Grant-Young K, Hewawasam P, Hudyma T, Martin S, Nickel A, Regueiro-Ren A, Tu Y, Yang Z, Yeung K-S, Zheng X, Chao S, Sun J-H, Beno BR, Camac DM, Chang C-H, Gao M, Morin PE, Sheriff S, Tredup J, Wan J, Witmer MR, Xie D, Hanumegowda U, Knipe J, Mosure K, Santone KS, Parker DD, Zhuo X, Lemm J, Liu M, Pelosi L, Rigat K, Voss S, Wang Y, Wang Y-K, Colonno RJ, Gao M, Roberts SB, Gao Q, Ng A, Meanwell NA, Kadow JF (2014) Discovery and preclinical characterization of the cyclopropylindolobenzazepine BMS-791325, a potent allosteric inhibitor of the hepatitis C virus NS5B polymerase. J Med Chem 57:1855–1879
Esposito I, Marciano S, Trinks J (2018) Pharmacokinetic and pharmacodynamic evaluation of daclatasvir, asunaprevir plus beclabuvir as a fixed-dose co-formulation for the treatment of hepatitis C. Expert Opin Drug Metab Toxicol 14:649–657
Lok AS, Gardiner DF, Lawitz E, Martorell C, Everson GT, Ghalib R, Reindollar R, Rustgi V, McPhee F, Wind-Rotolo M, Persson A, Zhu K, Dimitrova DI, Eley T, Guo T, Grasela DM, Pasquinelli C (2012) Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 366:216–224
Lok AS, Gardiner DF, Hézode C, Lawitz EJ, Bourlière M, Everson GT, Marcellin P, Rodriguez-Torres M, Pol S, Serfaty L, Eley T, Huang S-P, Li J, Wind-Rotolo M, Yu F, McPhee F, Grasela DM, Pasquinelli C (2014) Randomized trial of daclatasvir and asunaprevir with or without PegIFN/RBV for hepatitis C virus genotype 1 null responders. J Hepatol 60:490–499
Chung RT (2012) A watershed moment in the treatment of hepatitis C. N Engl J Med 366:273–275
Chayama K, Imamura M, Hayes CN (2016) Hepatitis C virus treatment updated. A new era of all-oral HCV treatment. Adv Dig Med 3:153–160
Zhang X (2016) Direct anti-HCV agents. Acta Pharm Sin B 6:26–31
Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, Sulkowski MS (2017) Oral direct-acting agent therapy for hepatitis C virus infection. A systematic review. Ann Intern Med 166:637–648
Geddawy A, Ibrahim YF, Elbahie NM, Ibrahim MA (2017) Direct acting anti-hepatitis C virus drugs: clinical pharmacology and future direction. J Transl Int Med 5:8–17
Li G, De Clercq E (2017) Current therapy for chronic hepatitis C: the role of direct-acting antivirals. Antivir Res 142:83–122
Spengler U (2018) Direct antiviral agents (DAAs) – a new age in the treatment of hepatitis C virus infection. Pharmacol Ther 183:118–126
Campbell JA, Good A (2002) World Patent Application WO-2002/060926 A2, 8 Aug 2002
Scola PM, Wang A, Good AC, Sun L, Combrink KD, Campbell JA, Chen J, Tu Y, Sin N, Venables BL, Sit SY, Chen Y, Cocuzza A, Bilder DM, D’Andrea S, Zheng B, Hewawasam P, Ding M, Thuring J, Hernandez D, Yu F, Falk P, Zhai G, Sheaffer AK, Chen C, Lee MS, Barry D, Knipe JO, Han YH, Jenkins S, Gesenberg C, Sinz MW, Santone KS, Zvyaga T, Rajamani R, Klei HE, Colonno RJ, Grasela DM, Hughes E, Chien C, Adams S, Levesque PC, Li D, Zhu J, Meanwell NA, McPhee F (2014) Discovery and early clinical evaluation of BMS-605339, a potent and orally efficacious tripeptidic acylsulfonamide NS3 protease inhibitor for the treatment of hepatitis C virus infection. J Med Chem 57:1708–1729
Ingallinella P, Altamura S, Bianchi E, Taliani M, Ingenito R, Cortese R, De Francesco R, Steinkuhler C, Pessi A (1998) Potent peptide inhibitors of human hepatitis C virus NS3 protease are obtained by optimizing the cleavage products. Biochemistry 37:8906–8914
Llinàs-Brunet M, Bailey M, Fazal G, Goulet S, Halmos T, Laplante S, Maurice R, Poirier M, Poupart M-A, Thibeault D, Wernic D, Lamarre D (1998) Peptide-based inhibitors of the hepatitis C virus serine protease. Bioorg Med Chem Lett 8:1713–1718
Steinkühler C, Biasiol G, Brunetti M, Urbani A, Koch U, Cortese R, Pessi A, De Francesco R (1998) Product inhibition of the hepatitis C virus NS3 protease. Biochemistry 37:8899–8905
Imperiali B, Abeles RH (1987) Extended binding inhibitors of chymotrypsin that interact with leaving group subsites S1′-S3′. Biochemistry 26:4474–4477
Meanwell NA (2011) Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem 54:2529–2591
Heidler P, Link A (2005) N-Acyl-N-alkyl-sulfonamide anchors derived from Kenner’s safety-catch linker: powerful tools in bioorganic and medicinal chemistry. Bioorg Med Chem 13:585–599
Ammazzalorso A, De Filippis B, Giampietro L, Amoroso R (2017) N-acylsulfonamides: synthetic routes and biological potential in medicinal chemistry. Chem Biol Drug Des 90:1094–1105
Llinas-Brunet M, Bailey MD, Cameron D, Faucher A-M, Ghiro E, Goudreau N, Halmos T, Poupart M-A, Rancourt J, Tsantrizos YS, Wernic DM, Simoneau B (2000) Preparation of hepatitis C inhibitory tripeptides. World Patent Application WO 2000/0009543. 24 Feb 2000
Llinas-Brunet M, Bailey M, Cameron D, Ghiro E, Goudreau N, Poupart M-A, Rancourt J, Tsantrizos YS (2000) Hepatitis C inhibitor peptides. World Patent application WO 2000/0009558, 24 Feb 2000
Gunaydin H, Bartberger MD (2016) Stacking with no planarity? ACS Med Chem Lett 7:341–344
Li J, Smith D, Wong HS, Campbell JA, Meanwell NA, Scola PM (2006) A facile synthesis of 1-substituted cyclopropylsulfonamides. Synlett 2006:725–728
Venables BL, Sin N, Wang XA, Sun L-Q, Hernandez D, Sheaffer A, Lee M, Franco D, Friborg J, Yu F, Knipe J, Sandquist J, Good AC, Rajamani R, Mcphee F, Meanwell NA, Scola PM (2018) P3-P4 Ureas and reverse carbamates as potent HCV NS3 serine protease inhibitors: effective transposition of the P4 hydrogen bond donor. Bioorg Med Chem Lett 28:1853–1859
Jenkins S, Scola P, McPhee F, Gesenberg C, Sinz M, Arora V, Pilcher G, Santone K (2014) Preclinical pharmacokinetics and in vitro metabolism of BMS-605339: a novel HCV NS3 protease inhibitor. J Pharm Sci 103:1891–1902
Scola P, Sun L-Q, Gillis, E, Bowsher M, Chen J, Wang X, Sit SY, Chen Y, Zheng Z, D'Andrea S, Sin N, Venables B, Mull E, Chen Q, Kandhasamy S, Pulicharla N, Vishwakrishnan S, Reddy S, Trivedi R, Sinha S, Sivaprasad S, Rao A, Desai S, Ghosh K, Rajamani R, Friborg J, Levine S, Chen C, Falk P, Jenkins S, Kramer M, Haskell R, Johnson K, Loy J, Levesque P, Zhu J, Cockett M, Meanwell N, McPhee F (2015) Discovery of a second generation, pan genotype NS3/4A protease inhibitor (BMS-986144) for the treatment of hepatitis C. In: Proceedings of 250th national ACS meeting and exposition, Boston, MA, 16–20 Aug 2015. MEDI-309
Langendorff O (1895) Untersuchungen am uberleberden saugerleizen. Pflugers Arch Gesamte Physiol 61:291–332
Mosure KW, Knipe JO, Browning M, Arora V, Shu Y-Z, Phillip T, McPhee F, Scola P, Balakrishnan A, Soars MG, Santone K, Sinz M (2015) Preclinical pharmacokinetics and in vitro metabolism of asunaprevir (BMS-650032), a potent hepatitis C virus NS3 protease inhibitor. J Pharm Sci 104:2813–2823
McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun L-Q, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P (2012) Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob Agents Chemother 56:5387–5396
Rossi Sebastiano M, Doak BC, Backlund M, Poongavanam V, Over B, Ermondi G, Caron G, Matsson P, Kihlberg J (2018) Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the rule of 5. J Med Chem 61:4189–4202
McPhee F, Friborg J, Levine S, Chen C, Falk P, Yu F, Hernandez D, Lee MS, Chaniewski S, Sheaffer AK, Pasquinelli C (2012) Resistance analysis of the hepatitis C virus NS3 protease inhibitor asunaprevir. Antimicrob Agents Chemother 56:3670–3681
Ali A, Aydin C, Gildemeister R, Romano KP, Cao H, Ozen A, Soumana D, Newton A, Petropoulos CJ, Huang W, Schiffer CA (2013) Evaluating the role of macrocycles in the susceptibility of heptatits C virus NS3/4A protease inhibitors to drug resistance. ACS Chem Biol 8:1469–1478
Soumana DI, Ali A, Schiffer CA (2014) Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS Chem Biol 9:2485–2490
Pasquinelli C, McPhee F, Eley T, Villegas C, Sandy K, Sheridan P, Persson A, Huang S-P, Hernandez D, Sheaffer AK, Scola P, Marbury T, Lawitz E, Goldwater R, Rodriguez-Torres M, DeMicco M, Wright D, Charlton M, Kraft WK, Lopez-Talavera J-C, Grasela DM (2012) Single- and multiple-ascending-dose studies of the NS3 protease inhibitor asunaprevir in subjects with or without chronic hepatitis C. Antimicrob Agents Chemother 56:1838–1844
Eley T, He B, Huang S-P, Li W, Pasquinelli C, Rodrigues AD, Grasela DM, Bertz RJ (2013) Pharmacokinetics of the NS3 protease inhibitor, asunaprevir (ASV, BMS-650032), in phase I studies in subjects with or without chronic hepatitis C. Clin Pharmacol Drugs Dev 2:316–327
Eley T, Garimella T, Li W, Bertz RJ (2015) Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug–drug interactions. Clin Pharmacokinet 54:1305–1222
Gong J, Eley T, He B, Arora V, Philip T, Jiang H, Easter J, Humphreys WG, Iyer RA, Li W (2016) Characterization of ADME properties of [14C]asunaprevir (BMS-650032) in humans. Xenobiotica 46:52–64
Eley T, Han Y-H, Huang S-P, He B, Li W, Bedford W, Stonier M, Gardiner D, Sims K, Rodrigues AD, Bertz RJ (2015) Organic anion transporting polypeptide-mediated transport of, and inhibition by asunaprevir, an inhibitor of hepatitis C virus NS3 protease. Clin Pharmacol Ther 97:159–166
Steinhauer DA, Domingo E, Holland JJ (1992) Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 122:281–288
Elena SF, Sanjuán R (2005) Adaptive value of high mutation rates of RNA viruses: separating causes from consequences. J Virol 79:11555–11558
Barr JN, Fearns R (2010) How RNA viruses maintain their genome integrity. J Gen Virol 91:1373–1387
Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R (2010) Viral mutation rates. J Virol 84:9733–9748
Graci JD, Gnädig NF, Galarrage JE, Castro C, Vignuzzu M, Cameron CE (2012) Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J Virol 86:2869–2873
Campagnola G, McDonald S, Beaucourt S, Vignuzzi M, Peersen OB (2015) Structure-function relationships underlying the replication fidelity of viral RNA-dependent RNA polymerases. J Virol 8:275–286
Shu B, Gong P (2016) Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc Natl Acad Sci U S A 113:4005–4014
Venkataraman S, Prasad BVLS, Selvarajan R (2018) RNA dependent RNA polymerases: insights from structure, function and evolution. Viruses 10:76. https://doi.org/10.3390/v10020076
Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS (1998) Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy. Science 282:103–107
Perelson AS, Herrmann E, Micol F, Zeuzem S (2005) New kinetic models for the hepatitis C virus. Hepatology 42:749–754
Shudo E, Ribeiro RM, Perelson AS (2009) Modeling HCV kinetics under therapy using PK and PD information. Expert Opin Drug Metab Toxicol 5:321–332
Rong L, Perelson AS (2010) Treatment of hepatitis C virus infection with interferon and small molecule direct antivirals: viral kinetics and modeling. Crit Rev Immunol 30:131–148
Ribeiro RM, Li H, Wang S, Stoddard MB, Learn GH, Korber BT, Bhattacharya T, Guedj J, Parrish EH, Hahn BH, Shaw GM, Perelson AS (2012) Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog 8(8):e1002881. https://doi.org/10.1371/journal.ppat.1002881
Chatterjee A, Guedj J, Perelson AS (2012) Mathematical modeling of HCV infection: what can it teach us in the era of direct-acting antiviral agents? Antivir Ther 17:1171–1182
Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Müh U, Welker M, Wincheringer D, Zhou Y, Chu H-M, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD (2007) Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777
Rong L, Dahari H, Ribeiro RM, Perelson AS (2010) Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med 2:1–9
Susser S, Welsch C, Wang Y, Zettler M, Domingues FS, Karey U, Hughes E, Ralston R, Tong X, Herrmann E, Zeuzem S, Sarrazin C (2009) Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 50:1709–1718
Hilgenfeldt EG, Schlachterman A, Firpi RJ (2015) Hepatitis C: treatment of difficult to treat patients. World J Hepatol 7:1953–1963
Suzuki H, Kakizaki S, Horiguchi N, Ichikawa T, Sato K, Takagi H, Mori M (2010) Clinical characteristics of null responders to Peg-IFNα2b/ribavirin therapy for chronic hepatitis C. World J Hepatol 2:401–405
Palumbo E (2011) Pegylated interferon and ribavirin treatment for hepatitis C virus infection. Ther Adv Chronic Dis 2:39–45
Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchison JG, Goldstein DB (2009) Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401
Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Lorena Abate M, Bassendine M, Spengler U, Dore GJ, Powell E, Riordan S, Sheridan D, Smedile A, Fragomeli V, Müller T, Bahlo M, Stewart GJ, Booth DR, George J, Hepatitis C Study (2009) IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat Genet 41:1100–1104
Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, Nakagawa M, Korenaga M, Hino K, Hige S, Ito Y, Mita E, Tanaka E, Mochida S, Murawaki Y, Honda M, Sakai A, Hiasa Y, Nishiguchi S, Koike A, Sakaida I, Imamura M, Ito K, Yano K, Masaki N, Sugauchi F, Izumi N, Tokunaga K, Mizokami M (2009) Genome-wide association of IL28B with response to pegylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat Genet 41:1105–1109
Asselah T, Estrabaud E, Bieche I, Lapalus M, De Muynck S, Vidaud M, Saadoun D, Soumelis V, Marcellin P (2010) Hepatitis C: viral and host factors associated with non-response to pegylated interferon plus ribavirin. Liver Int 30:1259–1269
Chayama K, Takahashi S, Toyota J, Karino Y, Ikeda K, Ishikawa H, Watanabe H, McPhee F, Hughes E, Kumada H (2012) Dual therapy with the nonstructural protein 5A inhibitor, daclatasvir, and the nonstructural protein 3 protease inhibitor, asunaprevir, in hepatitis C virus genotype 1b-infected null responders. Hepatology 55:742–748
Suzuki Y, Ikeda K, Suzuki F, Toyota J, Karino Y, Chayama K, Kawakami Y, Ishikawa H, Watanabe J, Hu W, Eley T, McPhee F, Hughes E, Kumada H (2013) Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol 58:655–662
Kumada H, Suzuki Y, Ikeda K, Toyota J, Karino Y, Chayama K, Kawakami Y, Ido A, Yamamoto K, Takaguchi K, Izumi N, Koike K, Takehara T, Kawada N, Sata M, Miyagoshi H, Eley T, McPhee F, Damokosh A, Ishikawa H, Hughes E (2014) Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology 59:2083–2091
McPhee F, Suzuki Y, Toyota J, Karino Y, Chayama K, Kawakami Y, Yu ML, Ahn SH, Ishikawa H, Bhore R, Zhou N, Hernandez D, Mendez P, Kumada H (2015) High sustained virologic response to daclatasvir plus asunaprevir in elderly and cirrhotic patients with hepatitis C virus genotype 1b without baseline NS5A polymorphisms. Adv Ther 32:637–649
Kanda T, Yasui S, Nakamura M, Suzuki E, Arai M, Haga Y, Sasaki R, Wu S, Nakamoto S, Imazeki F, Yokosuka O (2016) Daclatasvir plus asunaprevir treatment for real-world HCV genotype 1-infected patients in Japan. Int J Med Sci 13:418–423
Kumada H, Suzuki F, Suzuki Y, Toyota J, Karino Y, Chayama K, Kawakami Y, Fujiyama S, Ito T, Itoh Y, Tamura E, Ueki T, Ishikawa H, Hu W, McPhee F, Linaberry M, Hughes E (2016) Randomized comparison of daclatasvir+asunaprevir versus telaprevir+peginterferon/ribavirin in Japanese hepatitis C virus patients. J Gastroenterol Hepatol 31:14–22
Akuta N, Sezaki H, Suzuki F, Kawamura Y, Hosaka T, Kobayashi M, Kobayashi M, Saitoh S, Suzuki Y, Arase Y, Ikeda K, Kumada H (2017) Favorable efficacy of daclatasvir plus asunaprevir in treatment of elderly Japanese patients infected with HCV genotype 1b aged 70 and older. J Med Virol 89:91–98
Toyoda H, Kumada T, Tada T, Shimada N, Takaguchi K, Senoh T, Tsuji K, Tachi Y, Hiraoka A, Ishikawa T, Shima T, Okanoue T (2017) Efficacy and tolerability of an IFN-free regimen with DCV/ASV for elderly patients infected with HCV genotype 1B. J Hepatol 66:521–527
Sezaki H, Suzuki F, Hosaka T, Akuta N, Fujiyama S, Kawamura Y, Kobayashi M, Suzuki Y, Saitoh S, Arase Y, Ikeda K, Kobayashi M, Kumada H (2017) The efficacy and safety of dual oral therapy with daclatasvir and asunaprevir for genotype 1b in Japanese real-life settings. Liver Int 37:1325–1333
Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM (2013) Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 368:34–44
Keating GM (2014) Sofosbuvir: a review of its use in patients with chronic hepatitis C. Drugs 74:1127–1146
Poordad F, Sievert W, Mollison L, Bennett M, Tse E, Brau N, Levin J, Sepe T, Lee SS, Angus P, Conway B, Pol S, Boyer N, Bronowicki J-P, Jacobson I, Muir AJ, Reddy KR, Tam E, Ortiz-Lasanta G, de Ledinghen V, Sulkowski M, Boparai N, McPhee F, Hughes E, Swenson ES, Yin PD, UNITY-1 Study Group (2013) Fixed-dose combination therapy with daclatasvir, asunaprevir, and beclabuvir for noncirrhotic patients with HCV genotype 1 infection. J Am Med Assoc 313:1728–1735
Muir AJ, Poordad F, Lalezari J, Everson G, Dore GJ, Herring R, Sheikh A, Kwo P, Hézode C, Pockros PJ, Tran A, Yozviak J, Reau N, Ramji A, Stuart K, Thompson AJ, Vierling J, Freilich B, Cooper J, Ghesquiere W, Yang R, McPhee F, Hughes EA, Swenson ES, Yin PD (2015) Daclatasvir in combination with asunaprevir and beclabuvir for hepatitis C virus genotype 1 infection with compensated cirrhosis. J Am Med Assoc 313:1736–1744
Hassanein T, Sims KD, Bennett M, Gitlin N, Lawitz E, Nguyen T, Webster L, Younossi Z, Schwartz H, Thuluvath PJ, Zhou H, Rege B, McPhee F, Zhou N, Wind-Rotolo M, Chung E, Griffies A, Grasela DM, Gardiner DF (2015) A randomized trial of daclatasvir in combination with asunaprevir and beclabuvir in patients with chronic hepatitis C virus genotype 4 infection. J Hepatol 62:1204–1206
Toyota J, Karino Y, Suzuki F, Ikeda F, Ido A, Tanaka K, Takaguchi K, Naganuma A, Tomita E, Chayama K, Fujiyama S, Inada Y, Yoshiji H, Watanabe H, Ishikawa H, Hu W, McPhee F, Linaberry M, Yin PD, Swenson ES, Kumada H (2017) Daclatasvir/asunaprevir/beclabuvir fixed-dose combination in Japanese patients with HCV genotype 1 infection. J Gastroenterol 52:385–395
Kao JH, Yu ML, Peng CY, Heo J, Chu CJ, Chang TT, Lee YJ, Hu TH, Yoon KT, Paik SW, Lim YS, Ahn SH, Isakov V, McPhee F, Hu W, Swenson ES, Yin PD, Treitel M (2017) Daclatasvir/asunaprevir/beclabuvir, all-oral, fixed-dose combination for patients with chronic hepatitis C virus genotype 1. J Gastroenterol Hepatol 32:1998–2005
Ahmed AM, Doheim MF, Mattar OM, Sherif NA, Truong DH, Le Hoa PT, Hirayama K, Huy NT (2017) Beclabuvir in combination with asunaprevir and daclatasvir for hepatitis C virus genotype 1 infection: a systematic review and meta-analysis. J Med Virol 90:907–918
Sulkowski MS, Flamm S, Kayali Z, Lawitz EJ, Kwo P, McPhee F, Torbeyns A, Hughes EA, Swenson ES, Yin PD, Linaberry M (2017) Short-duration treatment for chronic hepatitis C virus with daclatasvir, asunaprevir, beclabuvir and sofosbuvir (FOURward study). Liver Int 37:836–842
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249
Meanwell NA (2011) Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol 24:1420–1456
Meanwell NA (2016) Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem Res Toxicol 29:564–616
Doak BC, Over B, Giordanetto F, Kihlberg J (2014) Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol 21:1115–1142
Doak BC, Zheng J, Dobritzsch D, Kihlberg J (2016) How beyond rule of 5 drugs and clinical candidates bind to their targets. J Med Chem 59:2312–2327
Zhao H (2011) Lead optimization in the nondrug-like space. Drug Discov Today 16:158–163
Compliance with Ethical Standards
Conflict of Interest: Nicholas A. Meanwell is an employee of Bristol-Myers Squibb and holds stock in the company.
Ethical Approval: This article does not contain any preclinical studies with animals or clinical trials with human participants that were performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Meanwell, N.A., Rajamani, R., Scola, P.M., Sun, Q. (2019). The Discovery and Early Clinical Evaluation of the HCV NS3/4A Protease Inhibitor Asunaprevir (BMS-650032). In: Sofia, M. (eds) HCV: The Journey from Discovery to a Cure. Topics in Medicinal Chemistry, vol 31. Springer, Cham. https://doi.org/10.1007/7355_2018_58
Download citation
DOI: https://doi.org/10.1007/7355_2018_58
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28206-6
Online ISBN: 978-3-030-28207-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)