
Abstract
Antibody-drug conjugates (ADCs) are a therapeutic modality that enables the targeted delivery of highly potent cytotoxic payloads to tumors. This chapter describes the components of the ADCs and discusses the medicinal chemistry principles that guide the design of this class of therapeutics. A description of main classes of drugs and the linkers used to attach to monoclonal antibodies with an emphasis on the design, historical development, and linking strategies is presented. Clinical use of the approved ADCs for the treatment of cancer is briefly described.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
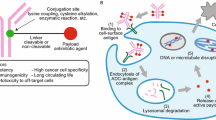
Antibody–drug conjugates (ADCs) can be described as a sum of components that are subject to several processes that lead to the desired outcome of selective delivery of a drug to an entity targeted by the specificity of the antibody. While the concept upon which ADCs are built appears deceptively simple, the elements involved in putting this concept into practice and the interdependencies between these components underscore the complexity of this family of drugs. The components of an ADC are monoclonal antibody, a set of linking sites targeted with bioorthogonal conjugation chemistry, a tether that bridges the linking site to the “prodrug,” the drug releasing moiety (sometimes called a “trigger”) that acts as both a tether and a means for selective release of the active “drug” to the desired tissue compartment and the drug. A set of processes are required for achieving efficacy with an ADC: plasma stability is a desired characteristic for ADCs as all ADCs known to date are administered IV, binding to the antigen situated on the surface of the cell is followed by internalization of the ADC-antigen complex that carries the ADC through the endosomal-lysosomal pathway leading to protein degradation by the action of hydrolases and release of the active drug followed by cytoplasmic and nuclear delivery of the active drug that interacts with the intracellular target. In case of anti-cancer drugs the cell dies and releases the drug into the tumor environment and, depending on the cell membrane permeability of the drug, can affect neighboring cells that do not express the antigen at the levels needed for a pharmacological effect, a process called “bystander effect.” The aim of this account is to describe the design principles of the components of ADCs and capture how those design principles translate into therapeutic outcomes. So far most of the applications of this therapeutic modality have been in Oncology where a highly cytotoxic drug is delivered to tumor cells. In an oncology setting minimizing the innate toxicity of the drug to healthy tissues becomes a critical design criterion.
2 Components of the ADCs
2.1 The Monoclonal Antibody
Antibodies, also called immunoglobulins or Ig, are ~150 kDa proteins that are produced by the immune system in response to the presence of foreign proteins called antigens. In human blood, IgG is the major type of antibody present. The use of antibodies as therapeutics was made possible by the work of Köhler and Millstein [1] who described methods to produce murine hybridomas that led to development of monoclonal antibody (mAb) technology. It took over 20 years to develop cancer targeting monoclonal antibodies with clinical applications. These cancer therapeutics bind to tumor antigens, protein expressed on the surface of the cancer cells, to induce tumor cell death [2]. The efficiency of cancer cell killing by immunotherapies can be enhanced by attachment of cytotoxic drugs to antibodies [3]. As opposed to chemotherapy where the cytotoxic drugs are administered systemically, the concept of ADCs introduced the possibility of targeted cytotoxic killing by virtue of mAb binding to antigens selectively expressed on cancer cells. mAbs are proteins with a defined chemical structure represented by a sequence of amino acids. The chemical structure of antibodies is determinant for the function of these molecules. The key function of the antibody is to preferentially bind to antigens on target cells. These Y-shaped molecules (Fig. 1) present a C2 symmetry with two identical arms held together by disulfide bonds and non-covalent interactions. Each half is made of a large subunit (~50 kDa) called heavy chain (HC) and a smaller subunit (~23 kDa) called light chain (LC). The assembly of two subunits of HCs and entire two LCs forms two variable units are called Fabs (fragments of antigen binding) while the constant unit made of HC is called Fc (fragment crystallizable). In terms of primary protein structure each chain contains a large constant region (C) and a variable (V) region. Within the heavy chain (also named gamma chain) there are 3 constant regions CH1, CH2, and CH3 encompassing positions ~120–446 at the C-terminus and one variable region at the N-terminus of about 120 amino acid residues. The light chain also named (kappa chain) consists of 214 amino acid sequence of which the N-terminus 108 amino acids are variable and the remaining amino acid sequence is constant. Within the variable regions, there are complementarity-determining regions (CDR) that are essential for antigen binding. The tertiary structure of these regions derived from the unique sequence of amino acids leads to large diversity of structure and accounts for the antibody specificity. CH2 region of the IgG contains an amino acid Asp 297 that is the anchor for N-glycans that contain a defined sequence of monosaccharides with various degrees of heterogeneity. The Fc is responsible for the effector function of the mAb by interacting with receptors present on cells of the immune system. The carbohydrate chains are important for Fc receptor recognition. The Fab and the Fc regions are connected by a sequence of amino acids, called the hinge, that allow for ample movement of the distinct regions that is essential for the function of the mAb. The number of amino acids of the hinge region varies between different subclasses of mAbs with the most commonly used IgG1 subclass having 15 amino acids while IgG4 subclass has just two amino acids. IgG1 is most commonly used as a therapeutic agent and it has a total of 32 cysteine residues forming disulfide bridges within the macromolecule. Twenty-four of these form intrachain disulfide bonds. They are in general considered to be less solvent accessible and hence less reactive than the eight cysteines that form interchain disulfides, including two that are located at the hinge region of the antibody. A crystal structure of an IgG was reported at high resolution (PDB: 1hzh) [4].


Components of immunoglobulin G antibody (IgG1 is shown for illustrative purposes)
Early clinical experience with mAbs used murine monoclonal antibodies. It was quickly recognized that murine molecules led to development of immune responses (were immunogenic) that resulted in rapid clearance of the drug and impaired efficacy. The continuous improvements in antibody engineering technologies led to gradual elimination of murine elements in therapeutic mAbs as follows: chimeric format where variable regions of both HC and LC were murine, humanized format that used mouse CDR regions and finally, the currently preferred format for ADCs, fully human antibodies. The half-lives of human mAbs are up to 3 weeks, significantly longer than the murine mAb that could only be detected in the blood for a few days.
2.2 Biorthogonal Conjugation Chemistry
Two factors have shaped the process of attachment a drug to an antibody and those were the requirement that conjugation be performed under aqueous conditions within a narrow range of pH and the constraints imposed by the reactivity of the natural amino acid residues. The amino group in lysine residues and the thiol group in cysteines are functional groups that were widely used for conjugation to a drug by reaction with electrophiles.
A human antibody contains 80–90 lysine residues of which a small number will react selectively without causing changes to the stability of or antigen binding ability of the antibody. Acylation of lysine residue leads to a stable amide bond. The reaction is performed with heterobifunctional N-hydroxysuccinimide-activated ester (NHS esters). Most common reagents for antibody lysine functionalization are N-succinimidyl-4-(2-pyridyldithio)butanoate (SPDB), N-succinimidyl-4-(2-pyridyldithio)pentanoate (SPP), N-succinimidyl 4-methyl-4-(2-pyridyldithio)pentanoate (SMPP), and succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) (Fig. 2). The resulting labelled antibody displays disulfide or maleimide groups, which can react with thiols. While lysine conjugation is a chemically straightforward process, controlling the number and location of modified lysine residue remains a great challenge. The extent of modification of solvent accessible lysine residue will be dependent on the molar equivalents of added heterobifunctional reagent and various other parameters of the conjugation protocol. The product of this process will have a stochastic distribution of reactive residues ranging between 0–10 modified residues with most conjugates having an average number of 3.5–4 molecules per antibody.


Reagents used for functionalization of mAbs
Cysteine conjugation to an antibody requires partial reduction of native interchain disulfides by a mild reducing agent such as 1,4-Dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) followed by modification of the sulfhydryl group by nucleophilic addition to a Michael acceptor or nucleophilic displacement with the thiolate group as a nucleophile. Conjugation to maleimide leads to a stochastic distribution of several species with 0, 2, 4, 6 and 8 functionalized residues with the species bearing 2 and 4 conjugated molecules being predominant. The most common reagent for cysteine conjugation is maleimide and the resulting thiosuccinimide product (Fig. 3) was shown to undergo retro-Michael addition reactions in the presence of thiols [5]. Loss of drug due to retro-Michael deconjugation was recognized as a potential safety liability and several groups have provided evidence that rapid hydrolysis of the resulting thiosuccinimide leads to lower propensity to loss of drug by deconjugation [6].Several approaches were shown to achieve hydrolysis of the succinimide: the use of a proximal basic amine was shown to improve stability of the cysteine conjugates by promoting hydrolysis of the succinimide “maleimido-DPR” [7], promoting hydrolysis by introduction of N-PEG tethers and higher pH [8] or use of phenylmaleimides that have a propensity for hydrolysis once conjugated [9]. Reduced interchain disulfides were targeted with bisalkylating reagents that replace the disulfide bridge with a propylidene-disulfide by an addition-elimination-second addition reaction sequence [10]. This approach allows for generation of ADCs with 1–4 drugs per antibody that are stable in presence of thiol-bearing plasma proteins [11]. A similar approach used dibromomaleimide [12] or dibromopyridazinedione [13] to cross-link the reduced disulfide bonds based on a concept introduced by Smith [14]. This approach presents the potential drawback of incorrectly re-bridged IgG1 mAb where the hinge cysteines form an intra-heavy-chain cross-linked product as opposed to inter-heavy-chain cross-linked product. This drawback was addressed by using a reagent that contains a TCEP-like structural moiety and a re-bridging moiety within the same molecule that allows for reduction and functional re-bridging. Reagent 1 was shown to lead to homogenous ADCs with no observable scrambling of the di-sulfide staple (Fig. 4) [15].


The chemistry of maleimide-cysteine conjugation


Rebridging reagent that contains disulfide bridge reducing moiety
In addition to lysine and cysteine one additional native amino acid – glutamine – was used for conjugation. Conjugation to glutamine uses the enzymatic recognition of a specific amino-acid sequence that contains the amino acid in the primary structure of the antibody. This approach leads to conjugates at a specified site of the antibody thus allowing for modulation of properties of the resulting conjugate. Glutamine conjugation to antibodies utilizes a bacterial transglutaminase (TG) isolated from Streptoverticillium mobaraense [16]. Unlike common transglutaminases that can catalyze the formation of amide bonds between the primary amine of a lysine and the amide group of any glutamine, the bacterial TG can catalyze the formation of an amine bond between any primary amine and the amide group of sequence specific glutamines [17]. A positional scan of antibody constant domains was performed by engineering a glutamine tag (LLQG) into surface accessible regions of an IgG1 antibody and several sites were identified that showed good biophysical properties and a high degree of conjugation [18].
A chemoenzymatic bioconjugation approach to cysteine conjugation used formyl glycine generating enzyme (FGE). FGE recognizes a pentapeptide consensus sequence, CxPxR, and it specifically oxidizes the cysteine in this sequence to an unusual aldehyde-bearing formyl glycine (fGly) [19]. This “tagged” construct is produced recombinantly in cells that coexpress the FGE, which co-translationally converts the cysteine within the tag into an fGly residue, generating an antibody expressed with two aldehyde tags per molecule. The aldehyde functional group serves as a chemical handle for bioorthogonal conjugation. A hydrazino-iso-Pictet-Spengler (HIPS) ligation was used to connect the payload to fGly, resulting in the formation of a stable, covalent C–C bond between the cytotoxin payload and the antibody [20]. This approach was used to generate ADCs that showed the desired activity [21]. Conjugation chemistry with faster kinetics that uses formyl glycine was described recently. The new technology is described as trapped-Knoevenagel ligation using a thiopyrazolone nucleophile (Fig. 5) [22]. One drawback of the formyl glycine approach is the observed formation of the aldehyde hydrate species that can impact the efficiency of conjugation under suboptimal conditions [19].


HIPS conjugation and Trapped-Knoevenagel ligation
Glycoengineering was used for generation of site specific conjugates by either metabolic engineering or post-translational remodeling of native glycan located at Asn-297 conserved position in the Fc domain of the antibody. The objective of glycoengineering is modification of the glycan in a manner that a functional group becomes available for biorthogonal conjugation. Metabolic engineering was achieved by substituting the fucose residue with a functionalized fucose residue that had a reactive handle for conjugation. A thiofucose residue was incorporated in the mAb during expression in CHO cells in medium that contained the modified fucose. The resulting antibody was used to generate conjugates where maleimide-bearing payloads were conjugated at the thiofucose site. The efficiency of the process did not exceed 70% thus limiting the utility of this particular conjugation technique [23]. Early efforts around post translational remodeling of native glycans have used a chemical oxidation step of the carbohydrate [24] or enzymatic introduction of sialic acid [25] to generate aldehyde functional group for oxime ligation. Engineering the active site of glycosyltransferases has provided tools for introducing chemically modified sugar substrates [26]. Native IgGs were converted to a homogenous G0f glycoform population by using β-1,4-galactosidase from Streptococcus pneumoniae. The resulting antibody was modified with a synthetic galactose bearing a chemical handle at the C2 position by using mutant β4Gal-T1-Y289L [27]. A keto- or an azido-group can be introduced in a site specific manner to generate an antibody with four functional groups. Further refinement of this technique was presented recently where an endoglycosidase was used to trim the antibody glycan to the fucosylated Acetylglucosamine (GlcNAc). Transfer of N-azidoacetylgalactosamine (GalNaz) in presence of β4Gal-T1-Y289L led to efficient installation of two azide groups onto the antibody [28].
Recent advances in development of methodologies for the genetic incorporation of unnatural amino acids into proteins [29] opened the possibility of site-selective modification of antibodies [30]. The side chains of unnatural amino acids provide novel functional groups for biorthogonal chemistry thus enabling generation of stable conjugates. The two main functional groups introduced using genetic incorporation of non-natural amino acids are the keto- and azido- that showed utility for conjugation by oxime ligation and the Huisgen alkyne-azide cycloaddition (“click”) reaction, respectively. The incorporation of p-acetylphenylalanine into trastuzumab generated an antibody with ~2 acetyl groups that were used for conjugation with alkoxyamine to generate the oxime at pH 4.5 with >95% efficiency [31]. N6-((2-azidoethoxy)carbonyl)-l-lysine was efficiently incorporated into trastuzumab and generated antibodies with ~2 azide groups that were efficiently conjugated by both strain promoted alkyl azide cycloaddition (SPAAC) and Cu(I) alkyl azide cycloaddition (CuAAC) to generate homogenous ADCs [32].
2.3 The Linking Sites
Conjugation to antibodies is governed by two parameters: the position of the amino acid in the protein sequence that carries the conjugated moiety and the number of amino acid residues that are functionalized during the conjugation process known as the drug-to-antibody ratio (DAR). Both these parameters impact the in vivo behavior of ADCs. Stochastic conjugation to Lys or native cysteines generates a heterogeneous mixture of molecular species where the sites of conjugation are random and the drug-to-antibody ratio is defined as an average [33, 34]. Each of the molecular species of such a mixture would display a distinct behavior in vivo as a consequence of different pharmacokinetics [35]. Discovery of solvent exposed engineered Cys IgGs was driven by the need to introduce a thiol-labelling site that did not adversely affect the structure or function of the antibody [36]. Following expression in mammalian cells mutant cysteines were capped with either a Cys or glutathione. Efforts to find an uncapping reagent that did not affect the native disulfide bonds were partially successful when alkylation with iodoacetate of a serine to cysteine mutant at the position 442 in the Fc domain was attempted [37]. These early discoveries set the stage for the development of antibodies containing engineered reactive cysteine residues at specific sites in antibodies that allow for drugs to be conjugated with defined stoichiometry without disruption of interchain disulfide bonds (termed THIOMABS) [38, 39]. These ADCs were produced by global reduction of blocked cysteine residues and interchain disulfides, subsequent oxidation in the presence of CuSO4 or dehydroascorbic acid to regenerate the interchain disulfide bonds, and then conjugation of the reactive cysteine thiol to maleimide reagents. This method generated site specifically modified ADCs with a homogenous distribution consisting of 92.1% species with two drugs/antibody. Importantly, the THIOMAB conjugate displayed a larger therapeutic index, as it was tolerated at much higher doses in animals and displayed better in vivo activity. The higher therapeutic index correlated with a higher in vivo stability of the THIOMAB when compared to equivalent stochastic ADCs. The authors hypothesized that engineered cysteines may be in relatively “protected” sites that resist proteolytic attack in circulation and they observed that the accessibility of a cysteine residue varies depending on the position of the mutated amino acid in the antibody protein sequence [39]. In a subsequent study the THIOMAB team explored whether the conjugation site could modulate the stability of a cysteine-maleimide adducts. The Genentech team prepared three THIOMABs of trastuzumab in which the mutant cysteine residues were positioned at sites that differed by their local structural environment. One site was chosen based on high solvent accessibility, while the other two were relatively buried sites but one was located in a positively charged environment while the other was in a relatively neutral environment. The in vitro potency of the three ADCs was comparable for all sites but the serum stability showed significant differences in the stability of the ADCs: the solvent-accessible conjugate underwent rapid thiol exchange with serum albumin, the conjugate at the positively charged site showed succinimide hydrolysis, resulting in improved stability, and the conjugate at the neutral site exhibited intermediate behavior between the other two ADCs. Serum stability results correlated well with in vivo efficacy, pharmacokinetic properties, and toxicity. That study demonstrated for the first time that the structural and chemical dynamics of the conjugation site can be exploited to design optimal protein conjugates for therapeutic applications [6]. A team at Seattle Genetics that were exploring conjugation of a highly hydrophobic drug pyrrolobenzodiazepine (PBD, see below) was faced with the challenge of finding a solution to the issues associated with stochastic conjugation of the maleimide-bearing PBD that led to formation of high percentage of aggregate. Scanning various conjugation sites with solvent accessible Cys mutants led to the discovery of position S239C on the HC where they conjugated the hydrophobic payload with only 1.6% aggregate. Thus they concluded that the recombinant construct provided superior ADCs compared to hinge disulfide conjugates in terms of ADC uniformity and aggregation levels [40].
2.4 The Drug
The choice of effective cytotoxic drugs for ADCs is governed by the relatively small number of antigen molecules on the cancer cell surface to which the antigen can bind (~104–106 receptors/cell) and by the efficiency of the internalization of cell-surface bound antigen–antibody complex and intracellular processing to release the active drug. Provided the intracellular delivery and release are efficient, the number of cytotoxic drug delivered to the individual cell by the ADC needs to be well above the number of cytotoxic agent molecules required to kill a cell. Thus cytotoxic drugs with potency in the pM range are needed. The early ADCs used drugs that were cancer chemotherapeutics as is the case with vinblastine analogs or doxorubicin. Following conjugation to an antibody both these drugs showed diminished potency as ADCs. This observation was attributed to the different modes of cellular uptake of the conjugated vs. non-conjugated drug. For cell membrane permeable drugs, the free diffusion can lead to high concentration of the drug inside the cell dependent on the dose of the administered drug. In addition to normal tissue target-mediated uptake that can be minimized by judicious choice of tumor-specific target, non-specific uptake by pinocytosis and plasma degradation of the ADC in circulation means that only ~1% of the administered ADC reaches the desired tumor tissue [41]. From a medicinal chemistry perspective, the hydrophobicity of the drug and its impact upon aqueous solubility could present a significant challenge. Also medicinal chemists need to develop structure activity relationships that reveal sites on the cytotoxic drug that tolerate a linker and possess the required reactivity for attachment to an antibody as well as enable the release of the drug inside the cancer cell. All cytotoxic drugs that have been reported as ADC therapeutics are derived from naturally occurring molecules and have very large molecular surfaces that appear to be a common feature for all highly potent cytotoxic molecules. Consequently, the structural complexity of the cytotoxic drugs for ADCs presents a major challenge for development of such molecules. The intracellular targets of cytotoxic drugs are quite limited and two main mechanisms have been targeted successfully with ADCs, i.e. tubulin and DNA.
2.4.1 Antimitotic Drugs
Disruption of microtubule dynamics impairs the ability of mitotic spindles to assemble and alter the architecture of the cytoskeleton, causing cell death [42]. Due to their mechanism of action, antimitotic agents are particularly cytotoxic to cancer cells that divide faster than non-cancerous cells. However, normal tissues that contain rapidly dividing cells such as cells lining the digestive tract, hair follicles, and bone marrow can also be killed, causing undesired toxicity.
2.4.1.1 Maytansines
Maytansine 2 (Fig. 6) is a benzoansamacrolide that was first isolated from the bark of the Ethiopian shrub Maytenus ovatus [43]. Maytansine binds to tubulin and causes disruption of microtubule dynamics resulting in cell death by apoptosis. While being widely cytotoxic, maytansine was shown to be particularly highly toxic against breast cancer cell lines, i.e. SK-BR3 and MCF-7 with an IC50 in the 30–40 pM range [44]. Structure–activity relationship studies on maytansine showed that several pharmacophores were required to maintain in vitro cytotoxicity. The carbinolamide at C9 and the ester side chain at C3 were required for biological activity, but the structure of this side chain could be varied without loss of potency (Fig. 5) [45]. Since the C3 position was amenable to modification, it was chosen for the incorporation of new ester side chains bearing a functional group to enable linkage to antibodies. Maytansine can be synthesized from ansamitocins that are obtained as a mixture of esters by fermentation of the microorganism Actinosynnema pretiosum. Ester hydrolysis in ansamitocins was shown to be challenging due to propensity for elimination of the C3 ester under basic conditions [46]. The ester group in the ansamitocins 3 (Fig. 6) was efficiently cleaved using the mild reducing agent lithium trimethoxyaluminum hydride, under controlled temperature (−30 to −40°C), to give maytansinol 4 in good yields [44]. A stable acetal intermediate 5 that can be stored was synthesized when the pH was adjusted to neutral following the reaction. The stable intermediate generated the desired maytansinol under specific conditions (Fig. 6). With maytansinol in hand, a thiol-based linking was pursued for linking to mAbs functionalized with thiol-reactive groups (SPBD, SPP or SMPP) resulting in disulfide linkages or thiosuccinimide (maleimide). The disulfide linkage in the ADC was hypothesized to be stable in circulation due to low concentration of thiol-bearing species in the blood while still able to release the drug in the reducing environment of the cytoplasm (where concentration of glutathione is 1–10 mM). A series of drug-maytansinoids DM1, DM3, and DM4 that were capped with a thiomethyl were made by coupling a series of disulfide-bearing acids (Fig. 7) to maytansinol in presence of a coupling agent (DCC, EDC, etc.) and a Lewis acid (ZnCl2). All resulting maytansinoid disulfides showed cytotoxicity as least as potent as maytansine [44]. These compounds were reduced with a mild agent, as is the case with dithiothreitol (DTT) prior to conjugation to functionalized mAbs.


Maytansinoids SAR and semisynthetic approach to Maytansinol


Synthesis of maytansinoid DM family of drugs
2.4.1.2 Auristatins
Following 15 years of work isolating and characterizing a series of cytotoxic peptides extracted from the sea hare Dolabella auricularia, Pettit discovered Dolastatin 10 (Fig. 8) that was at the time the most potent cytotoxic molecule ever known [47] with a potency of 50–300 pM against most cancer cell lines tested. The discovery and characterization of the molecule was an extraordinary achievement as it required extensive fractionation and purification followed by characterization by mass spectrometry and NMR. The definitive structure of Dolastatin 10 was established following the publication of the absolute configuration and the total synthesis of the molecule [48]. Dolastatin 10 and its derivatives have been shown to inhibit tubulin-dependent GTP binding, cause noncompetitive inhibition of vincristine binding to tubulin, and inhibit microtubule dynamics resulting in cell cycle arrest and apoptosis [49] that was confirmed by an X-ray crystal structure of a synthetic analog – soblidotin – bound to tubulin [50]. Following the discovery of Dolastatin 10, a significant effort was dedicated to making analogs in a quest for compounds with chemotherapeutic utility. The analogs of Dolastatin 10 were named “auristatins” and Pettit described Auristatin Phe and Auristatin E (Fig. 8) both with slightly attenuated cytotoxicity (200–400 pM) by replacing the C-terminal Dolaphenine with a Phe methyl ester and norephedrine moiety, respectively [51]. While several of the auristatin analogs were tested in the clinic, none of them progressed due to unacceptable side effects at doses that did not achieve efficacy. An important discovery was made that the N-terminal end of the molecule tolerates a secondary amine resulting in monomethyl auristatin D (MMAD) (Fig. 8), which opened the door to attaching a linker to these molecules [52]. The significance of this discovery was recognized in 2003 when scientists at Seattle Genetics developed monomethyl auristatin E (MMAE) (Fig. 8) as a hybrid of Pettit’s Auristatin E and MMAD [53]. This allowed, for the first time, the linking of an auristatin to a monoclonal antibody. The Seattle Genetics team later developed monomethyl auristatin F (MMAF) (Fig. 8). MMAF showed weak cytotoxic activity and was initially overlooked as a potential drug for ADCs. Working on the hypothesis that the lack of activity was due to poor cell membrane permeability (that was supported by the observation that an Auristatin Phe analog of MMAF showed activity) they envisaged that delivering MMAF intracellularly could overcome this drawback. This work led to discovery of the first auristatin non-cleavable payload mcMMAF [54]. Recently a team at Pfizer used crystallography data for auristatin analogs bound at the α,β-tubulin interface to design analogs where the dolavaline in Dolastatin 10 was replaced with a variety of amino acids. This work led to the discovery of PF-06380101 (Fig. 8) that is being progressed into the clinic as a drug for ADCs [55]. Research targeting new auristatins for ADCs is very active and is expected to continue in the future [56].


Development of synthetic drugs derived from natural product Dolastatin 10
2.4.1.3 Tubulysins
Tubulysins were isolated from myxobacteria and were described as a family of tetrapeptides that displayed antimitotic activity in a series of cancer cell lines including multidrug resistant cell lines [57]. Tubulysin D was reported to display 10–500 times higher cytotoxicity than paclitaxel and vinblastine [58] while the family of natural tubulysins A–I (Fig. 9) showed IC50 values in the range of 0.3–8.4 nM in human cervix carcinoma, multidrug-resistant cell line KB-V1 [59]. Structurally, the tubulysins (Fig. 9) consist of an (R)-N-methylpipecolic acid (Mep), a natural amino acid l-Ile, a tubuvaline (Tuv) containing a thiazole and two potentially labile acyloxymethyl and acetate ester groups and two defined chiral centers and a tubuphenylalanine (Tup) (R2 = H) or tubuphenyltyrosine (Tut) (R2 = OH) each displaying two defined chiral centers (Fig. 9). Simplified synthetic analogs of natural tubulysins were described where the acyloxymethyl group was replaced with a methyl group [60,61,62]. The cellular potency of the resulting derivative N14-desacetoxytubulysin H (Fig. 9) was shown to be little affected by this structural alteration (L929 IC50 = 0.34 nM; SW-480 IC50 = 0.02 nM, KB-3-1 IC50 = 0.19 nM) [61]. Linking tubulysin requires introduction of a reactive group that comes at a cost for potency. Endocyte developed a folate conjugate that contained a derivative of Tubulysin B (R1 = butyrate, R2 = OH), i.e. Tubulysin B hydrazide that was shown to be ~3-fold less potent than the corresponding acid [63]. Introducing an aniline in the molecule of Tubulysin (R2 = NH2) leads to substantial drop-off in potency, however the potency is regained by modification of the N-substituent of tubuvaline (Tuv) as illustrated by MMETA that was developed by MedImmune/Astrazeneca. MMETA [64] (Fig. 9) showed consistent sub-nM potency in a wide range of tumor cell lines (DU 145 IC50 = 0.2 nM; MDA-MB-361 IC50 = 0.05 nM, NCI-N87 IC50 = 0.04 nM)) [65]. The aniline was used to generate payloads that were successfully conjugated to antibodies.


Tubulysin natural product family and synthetic analogs used as drugs
2.4.2 DNA Targeting Drugs
2.4.2.1 Calicheamicin
The calicheamicins are a family of antibiotics isolated as fermentation products of the bacterium Micromonospora echinospora ssp. calichensis [66]. At the time of their discovery they were considered the most potent tumor agent ever known with a potency 1,000 times higher than adriamycin [67]. Calicheamicin γ1I, the most well-known member of this family (Fig. 10), contains two functional regions: a complex bicyclic enediyne unit with a methyl trisulfide moiety that can induce DNA double-strand scission and an extended sugar residue, which possesses a fully substituted benzene ring that has the ability to bind in a sequence-specific manner to DNA minor grove [67]. The reactive functional region of calicheamicins displays a fascinating mechanism of action (Fig. 10). The enediyne warhead positioned within DNA double helix can be attacked by a nucleophile (e.g., glutathione) at the methyl trisulfide thus releasing a free thiol. The free thiol triggers a cascade of events starting with attack of the α,β-unsaturated ketone followed by a Bergman cycloaromatization reaction leading to a 1,4-benzene diradical. The diradical thus formed abstracts hydrogen atoms from both strands of the duplex DNA which in the presence of oxygen leads to cleavage of DNA double strands and subsequent cell death. The high potency makes Calicheamicin γ1I an ideal candidate for an ADC drug and finding a linking modality was pursued soon after its discovery. Acetylation of secondary amine in Calicheamicin γ1I led to a less toxic 5.6 nM in MX-1 breast carcinoma cells pM derivative N-Acetyl-calicheamicin γ1I6 that was further functionalized to a dimethylhydrazide (DMH) 7 that was used to generate conjugates (Fig. 11) [68].


Natural calicheamicin and mechanism of DNA double strand breaking


Calicheamicin drugs used in ADCs
2.4.2.2 Duocarmycins
The duocarmycin natural product (+)-CC-1065 was isolated from Streptomyces zelensis as a potent antitumor antibiotic with a potency of IC90 = 56 pM [69]. The cytotoxic activity of this natural product was explained by its ability to alkylate adenine in DNA in a sequence specific manner. In case of (+)-CC-1065, the non-covalent DNA binding region (Fig. 12) positions the DNA alkylating region of the molecule into the minor groove where it alkylates the N3 of adenine via the cyclopropane moiety in spirocyclic cyclopropapyrrolo-indole (CPI) unit [70]. (+)-CC-1065 was not progressed into the clinic due to observed delayed hepatotoxicity in preclinical animal studies [71]. A series of analogs were made in an attempt to find molecules with properties suitable for clinical development. The discovery of cyclopropabenzindole (CBI) unit and the corresponding chloromethyl substituent as a precursor to the cyclopropyl ring constituted a breakthrough. This discovery addressed the relatively poor stability of the CPI unit in serum and opened the door for pro-drugging this highly reactive structural feature [72]. The chloromethyl substituent in O-pro-drugged-(S)-1-(chloromethyl)-2,3-dihydro-1H-benzo[e]indol-5-ol generates CBI following the enzymatic removal of the phenol-capping group and a Winstein cyclization (Fig. 13). This approach presented an opportunity for linking this potent warhead to an antibody and also for introduction of much needed water solubilizing groups for this chemotype.


Structure of CC-1065 and mechanism of DNA alkylation


CBI mechanism of formation following enzymatic cleavage
Two duocarmycin containing ADCs have been progressed into the clinic. MED-A used the pro-drugging site to introduce a solubilizing moiety and an aniline in the DNA-binding region for linking to the antibody [73] and seco-DUBA where the DNA-binding region was modified to introduce hydrophilic residue that could promote solubility while the linking was done through the DNA-alkylating unit phenol (Fig. 14). Seco-DUBA showed IC50 = 90–430 pM potency in a range of cancer cell lines [74]. An interesting detoxification mechanism was described for DUBA, the active species following removing of the phenol capping group, where the cleavage of the amide bond between the alkylating region and the binding region in plasma leads to weakly cytotoxic molecules. This degradation was shown to be species dependent with only 2.6% of the DUBA left in rat plasma after 180 min and 68% remaining in human plasma [74].


CBI-based duocarmycin drugs progression to the clinic
2.4.2.3 Pyrrolobenzodiazepine Dimers
Anthramycin, the first member of this family of antibiotics, was isolated as the active constituent from the fermentation broth of Streptomyces refuineus var. thermotolerans [75]. Many analogs have been discovered or synthesized since then (Fig. 15). The key analogs that led to warheads currently used in ADCs are Tomaymycin [76] and Sibiromycin [77]. The key features of pyrrolobenzodiazepines (PBDs) are the presence of a substituted phenyl ring A, the diazepine ring B, a nitrogen containing five-membered heterocycle C containing various levels of unsaturation and a specific (S)-configuration at the B-C ring junction, which is essential for the interaction with the DNA minor groove (Fig. 15). PBDs are believed to owe their biological activity to their ability to react at the electrophilic N10–C11 imine with the amino group in C2 position of a guanine residue within the minor groove (Fig. 16) [78]. PBD monomers have shown nM toxicity against cancer cell lines and have limited applications for ADCs. A seminal discovery was made when a 1,3-propane-diether-linked dimer was shown to have much improved cytotoxicity due to its ability to bind to minor groove DNA over a span of six base pairs. The dimer 8 (DSB-120) showed up to 600 times improved activity over the monomer [79]. Further improvements in potency were made by introduction of unsaturation at C2 that led to the discovery of 9 (SJG-136 and SG2000), which was progressed into the clinic for hematological malignancies [80, 81]. Low pM in vitro potency was achieved by making the tether between the two PBD monomers longer, i.e.1,5-pentane-diether as illustrated by the discovery of 10 (SG2057) that showed 212 pM potencies in cancer cell lines [82]. In addition, highly potent analogs of PBD dimers were obtained when an aryl group was introduced at the C2-position; compounds 11 (SG2202) and its water soluble analog 13 (SG2285) showed 1.3–53.5 pM activity in a panel of cancer cell lines [83]. Compound 11 presented the opportunity to introduce a functional group for linking to an antibody when it was shown that an aniline at C-2 as in 12 (SG1882) showed 0.1–1 nM activity in a panel of lymphoma and renal cell carcinoma cell line panel [40].


Naturally occurring pyrrolobenzodiazepines


Mechanism of DNA alkylation with PBDs
An interesting linking strategy was used for the warhead in compound 14 (D6.5) where the N-10 position was used to introduce the linker [84] (Fig. 17).


Pyrrolobenzodiazepine dimers that were progressed into the clinic
2.4.3 Anthracyclines
Doxorubicin was used in early ADCs in which the molecule was linked to the antibody via a hydrazone formed with the ketone functional group (Fig. 18). In a randomized trial, chimeric BR96 antibody was conjugated to doxorubicin and the resulting ADC BR96-doxorubicin was advanced to a Phase II human clinical trial in metastatic breast cancer. The trial failed to show meaningful therapeutic benefit [85]. Clinical use of doxorubicin analogs is limited by their dose-related cardiotoxicity. Compound 15 (Nemorubicin, MMDX) (Fig. 18) was a synthetic analog of doxorubicin that showed reduced toxicity of 400–800 nM against doxorubicin resistant cancer cell lines [86]. During preclinical studies involving incubation of the drug with NADPH-supplemented rat liver microsomes a highly toxic metabolite was identified compound 16 (PNU-159682) (Fig. 18). This metabolite was shown to be 3,000-fold more cytotoxic than the parent 15. The cytotoxicity against cultured human cancer cell lines was 0.07–0.58 nM/L [87]. The mechanism of action of 16 is intercalation between CG base pairs with a much higher binding affinity than 15 [88]. Compound 16 was linked to antibodies via the hydroxyl group (Fig. 18).


Anthracycline drugs for antibody drug conjugates
2.4.4 RNA Polymerase Inhibitors
2.4.4.1 α-Amanitin
α-Amanitin 17 is a bicyclic octapeptide isolated from Amanita mushrooms and responsible for their extreme toxicity (Fig. 19). Amanitin binds with high specificity and low nM affinity near the catalytic active site of RNA polymerase II, as shown by its high resolution structure bound to the enzyme [89]. α-Amanitin “freezes” the enzyme in a conformation that prevents nucleotide incorporation and stops translocation of the transcript [90, 91]. Despite its low cell membrane permeability, α-amanitin is hepatotoxic by a mechanism that involves the organic anion-transporting polypeptide (OATP3), which acts as an uptake transporter in human hepatic cells [92]. α-Amanitin was conjugated to proteins in the 1980s by using the phenol as the linking functional group or a diazo-linker with uncertain structure [93]. It was shown that once conjugated to antibodies via the 4-hydroxy-l-allothreonine 18 (Fig. 19), the anti-epithelial cell adhesion molecule (EpCAM) targeting amanitin ADCs showed 105 enhancement of cytotoxicity as an ADC targeting an antigen expressed on the Colo205 cancer cell lines [94]. This observation was later attributed to the fact that suppression of RNA polymerase IIA with amanitin led to selective inhibition of proliferation, survival and tumorigenic potential of colorectal cancer cells with hemizygous TP53 loss in a p53-independent manner [95]. Introducing a linking reactive group or a phenyl-ether 19 (Fig. 19) led to potent anti-PSMA targeting ADCs that showed 17–620 pM potency in PSMA expressing cells in vitro. The corresponding cleavable and non-cleavable conjugates showed tumor stasis in CWR-22rv1 mouse model at an iv dose of 150 μg/kg with respect to toxin [96].


Structure of natural α-amanitin and derivatives for linked drugs
2.4.4.2 SN-38 and the Camptothecins
Camptothecin (CPT) was first isolated from the Camptotheca acuminata tree in 1966 (Fig. 20) [97]. Preclinical studies revealed that CPT had remarkable activity against leukemia cell lines. The low aqueous solubility of CPT led to its use as a sodium salt of the open cycle lactone end of the molecule, which leads to loss of activity. In efforts to improve the aqueous solubility of analogs, the derivatives Topotecan and Irinotecan were developed. The potent antitumor activity of CPT was attributed to selective inhibition of DNA topoisomerase I, thus impairing DNA replication and resulting in the apoptotic cell death of tumor cells [98]. The X-ray crystal structure of a ternary complex formed between DNA, topoisomerase I and Topotecan showed the only H-bond interaction between the drug and the enzyme was with the 20-hydroxy-group of Topotecan and an important water interaction was observed with the phenol [99]. This discovery provided an explanation for much lower potency for Irinotecan, a result of inefficient carboxylesterase-catalyzed hydrolysis of the prodrug to its active metabolite in patients. The active metabolite of Irinotecan (SN38) was plagued by low aqueous solubility despite higher activity than CPT. However, SN-38 was successfully conjugated to an antibody via its hydroxy group at C20 [100]. Compound 20 (Dxd, DX-8951) was described as an inhibitor of Topoisomerase I with higher potency than SN-38. The warhead was incorporated into an ADC by linking with the hydroxyacetate via an aminomethylene moiety [101]. A trastuzumab conjugate with compound 20 showed activity in breast cancer patients [102].


Structure of camptothecin and derivatives used as cytotoxic drugs and as linked drugs
2.5 The Tether
The tether is defined as the region of the linker that connects the drug to a reactive group that accomplishes the conjugation to the antibody as described in Sect. 2.2. The tether must be stable in plasma for the period between the IV administration and cellular internalization and processing thus addressing the primary drug loss mechanism from the ADC (Fig. 21). The secondary source of drug loss is the stability of the whole conjugate in plasma. The attachment of the linked drug negatively impacts the pharmacokinetics of the ADC, thus leading to release of drug by virtue of antibody plasma metabolism (Fig. 21) and the tether can minimize the negative impact of drug conjugation on pharmacokinetics by counterbalancing the hydrophobicity of the drug. In addition to modulation of the overall stability of the ADC, the tether can provide at least one intracellular specific mechanism of release of the active species. Based on these considerations the tether plays a critical role in achieving true targeted delivery of the drug by harnessing the long half-lives of mAbs and utilizing tumor and cancer cell specific cytotoxic drug release. Two classes of tethers have been utilized in generation of ADCs: non-cleavable and cleavable tethers.


Representative pharmacokinetic profiles of molecular components of ADCs
2.5.1 Non-cleavable Tethers
A non-cleavable tether does not contain a tumor or cancer cell-specific release unit, called a “trigger.” For a medicinal chemist, the use of a non-cleavable tether poses the challenge of finding a linking site on the cytotoxic drug where attachment of the tether will not impact cytotoxicity. Additionally, following cellular catabolism, the non-cleavable tether ADCs generates a species that contains a residual charged amino acid residue. In vitro assessment of the cytotoxicity of such species is not always trivial due to the potential low cell membrane permeability thus making the evaluation of linked drugs with non-cleavable tethers challenging. The particular nature of non-cleavable tethers has two apparently conflicting consequences: on one hand the stable linkage translates to higher plasma stability and reduced toxicity as shown by higher Maximum Tolerated Dose (MTD) in vivo. On the other hand, in tumors with heterogeneous expression of the antigen, the efficacy can be negatively affected by the reduced cell membrane permeability of the active species, leading to reduced ability of the resulting drug to kill neighboring cells.
Two prominent examples of non-cleavable tether linked warheads are MCC-DM-1 and mc-MMAF (maleimidocaproyl-MMAF). The lysosomal catabolites of ADCs containing these linked drugs are DM1-MCC-Lys 21 and MMAF-mc-Cys 22, respectively (Fig. 22). As the catabolism of ADCs was shown to be lysosomal [103] and the target of these drugs is located in the cytoplasm, the exact mechanism of lysosomal escape was unknown. Recent data suggest that protein transporters imbedded in the lysosomal membrane could be responsible for the exit of these highly charged molecules from the lysosomal compartment into the cytoplasm. Moreover, the study showed that transporters have the ability to selectively transport species with certain linked drug structure thus providing evidence that development of non-cleavable tethers could face higher hurdles than is the case for cleavable analogs [104].


Linked drugs with non-cleavable tethers
MCC-DM-1 contains the thiosuccinimide moiety, which was shown to lead to drug loss following exchange with thiol-containing plasma protein, i.e. albumin. Two truly non-cleavable linkers that lacked the reversible thioether succinimide connection between the drug and antibody were shown to possess superior efficacy and stability relative to MCC-DM1. These two linkers are May-mc 23 and May-MPA 24 and the corresponding lysosomal released drugs are shown in Fig. 22 [105].
2.5.2 Cleavable Tethers
Cleavable tethers contain a spacer and a structural feature designed to release the drug inside the cell called a “trigger.” The spacer is usually a short carbon chain for most ADCs but for some is a series of polyethylene glycol units, which are designed to reduce the logP of the linked drug during the conjugation process and minimize the impact of drug lipophilicity upon the pharmacokinetics of the ADC. The trigger is a tool that exploits changes that occur in vesicles along the endosomal, lysosomal and cytoplasmic pathway following receptor-mediated endocytosis [106]. The changes that have been exploited by trigger designs are: (1) gradual drop in pH from physiological range at the cell surface of 7.2–7.4 to 5.5–6 within the endosome and to ~5.0 in lysosome; (2) the activation of protein digesting enzymes at the lower pH of the lysosome; and (3) the increase in concentrations of reducing co-factors such as glutathione and cysteine and activation of enzymes that can reduce disulfide bonds [107].
Early examples of pH sensitive triggers are shown in Fig. 23. The linker in gemtuzumab ozogamicin (Mylotarg) showed good stability at pH 7.4 (94%) and complete hydrolysis at pH 4.5 (97%) following an investigation of a range of structural analogs. This linker, called AcBut, showed the best stability at pH resembling plasma yet can be efficiently cleaved in the acidic environment of the lysosome. AcBut led to ADC that showed potency 100,000-fold higher toward antigen-positive HL-60 cells than when conjugated to a non-targeted antibody [108]. One additional example of an acid sensitive trigger is AEVB (Fig. 23), which contains Auristatin E linked at the alcohol moiety as an ester to a ketone containing acid. The ketone is linked to the antibody via a pH sensitive hydrazone. The linkage was relatively stable at pH 7.2 (t1/2 >60 h) but labile at pH 5.0 (t1/2 3 h). Once conjugated, AEVB was released non-enzymatically at pH 5 (t1/2 4.4 h) more rapidly than at pH 7.2 (t1/2 183 h). A human plasma stability study showed that after 4 days ~30% hydrazine was hydrolyzed. In mouse plasma ~40% of drug was cleaved at the ester site after 4 days incubation [53].


Low pH cleavable hydrazone-linked drugs
The most successful trigger has been the peptidase-cleavable peptide pioneered following discoveries made at Bristol Myers Squibb in the late 1990s. Dubowchik and his colleagues hypothesized that replacing the hydrazone trigger with a cathepsin B cleavable peptide should lead to tethers that are stable in circulation, due to absence of the protease in extracellular space, yet labile in the lysosomal compartment, thus releasing the drug intracellularly [109]. The study showed that cathepsin B–mediated release of doxorubicin from constructs containing various dipeptide triggers was observed only when a spacer was introduced between the bulky doxorubicin and the dipeptide. The spacer chosen was para-amino-benzyl (PAB) that presented the unique feature of self-immolation at pH ~5. An illustration of the dipeptide linker design (Fig. 24) uses mc-VC-MMAE. The dipeptide used in this tether is Valine-Citrulline flanked by a maleimidocaproyl spacer at the N-terminus and by the PAB spacer at the C-terminus. Upon cleavage of the PAB-Cit amide bond, a MMAE-PAB carbamate is released. This species can be identified transiently but it decomposes by a 1,6-elimination with the release of unstable MMAE carbamic acid, which spontaneously eliminates carbon dioxide to generate the active drug. In a detailed comparison of peptide and hydrazone cleavable tethers, scientists brought strong evidence that peptide tethers were superior in almost all aspects when linking auristatins to antibodies [53]. In addition to the Val-Cit peptide trigger, several groups have employed a Val-Ala linker as the one described in SC16LD6.5, in which the tether for pyrolobenzodiazepine dimer D6.5 contains a Val-Ala-PAB trigger and a PEG8, which lowers the hydrophobicity of the whole payload (Fig. 25) [84].


Dipeptide trigger design for linked MMAE


Val-Ala dipeptide trigger in a PBD dimer linked drug
A highly complex linker design was reported for duocarmycin in SYD985 [74]. The main challenge with duocarmycin derived ADCs is linking via a phenol group. Design criteria employed for linking seco-DUBA were: introducing a tether that lowers lipophilicity, as is the case with PEGs and alcohols, use of a self-immolative 1,5-cyclization moiety and use of a valine-citrulline-PAB cathepsin B-sensitive trigger. Unlike the Val-Cit-PAB alone, for which a mildly acidic environment is needed to facilitate enzymatic cleavage, the tether in SYD985 requires a pH of greater than 6 for efficient release of the DUBA active species. This requirement is met by the transport of the prodrug carbamate from the lysosomal compartment to the cytoplasm where the higher pH triggers the release of DUBA (Fig. 26) [74]. This additional step could present challenges as it was shown that transporter proteins could play a critical role [103] and structural characteristics of the tether may impact the efficiency of crossing intracellular membranes. The tether in NMS249 utilizes a similar self-immolative 1,5-cyclization unit that connects the protease cleavable trigger to a primary alcohol. This tether is more hydrophobic and contains no PEG to attach the maleimide. This design was driven by the hydrophilic nature of the PNU-159682 parent drug and the presence of a primary alcohol as the linking handle (Fig. 27) [110]. The Val-Cit-PAB motif adds considerable hydrophobicity to tethers and ultimately to ADCs.


Design of phenol-linked dipeptide-trigger duocarmycin in SYD985


Design for linking a primary alcohol with a dipeptide trigger anthracycline linked drug in NMS249
Unlike small molecule drugs mAb-based drugs owe their long half lives in circulation, among other factors, to their peptidic structures and optimal lipophilicity. Structural modifications of mAb that add hydrophobicity to the molecule have been shown to reduce the half-life of these constructs in circulation. Hydrophobic interaction chromatography (HIC) is an analytical tool that allows for evaluation of “hydrophobic penalty” paid by ADCs following linking drugs to antibodies. This penalty is evidenced by a longer retention time in HIC when compared to unconjugated mAb and has a significant impact upon pharmacokinetics of the conjugate in vivo [35, 111]. To illustrate the impact of hydrophobicity on pharmacokinetics, a tether that contained a hydrophilic moiety (AT-glu-Dpr-(mDpr)) in which the phenylalanine of MMAF was replaced with threonine and linked directly to a hydrophilic cleavable peptide via a hydrolysable maleimide tether (see earlier) was designed. The resulting ADC was compared to the mc-MMAF and mc-VC-PAB-MMAF in the HIC assay (Fig. 28). As mentioned previously, for a given ADC, the number of drugs loaded to a mAb is defined as drug to antibody ratio (DAR). In the context of hydrophobic penalty, the DAR refers to the number of molecular entities attached to the mAb that contain the active drug that contributes to added hydrophobicity to the ADC. In the study above, the hydrophilic tether ADC showed a retention time similar to unconjugated antibody even at a high DAR 8. The observed plasma pharmacokinetics with DAR 8 mirrored the HIC hydrophobicity profiles. Moreover, the in vivo efficacy for an ADC with a DAR of 4 with mc-VC-PAB tether was significantly higher than an analogous ADC with a DAR of 8. By contrast, the efficacy of a DAR 8 hydrophilic AT-glu-Dpr-(mDpr) tether ADC was much higher than DAR 8 with mc-VC-PAB tether at one quarter of the dose [111].


Structure of linked drugs used to evaluate the hydrophilic linked auristatins
The PAB spacer in peptide triggers was omitted in cases where a phenyl group is part of the warhead. The peptide trigger was attached directly to the warhead via an aniline. The PBD dimer 12 (SG1882) (Fig. 17) was linked to an antibody via a tether that contained the Val-Ala-mc. Compound 25 (Fig. 29) was conjugated at position HC S239C to yield the ADC (see Sect. 3 for discussion) [40]. Another example of a minimalistic linker is mc-Lys-MMETA 26 where a Lys was linked directly to the warhead to yield highly potent ADCs following conjugation (Fig. 29).


Linked drugs that do not use a spacer for dipeptide trigger
β-Glucuronidase is an enzyme that was shown to be highly abundant in breast tumor tissue and peritumoral space [112]. β-Glucuronidase can cleave the glycosidic bond between glucuronic acid and a hydroxyl-bearing substrate. In the context of ADCs a β-glucuronide trigger was coupled to a tri-substituted self-immolative tether that acts a linking point for antibody attachment and as a drug release device analogous to the previously described PAB. This type of linker was used to link MMAF to CD30 and CD70 targeting antibodies where the resulting ADCs showed pM potency in antigen positive cancer cell lines (Fig. 30) [113]. The ability of this tether to generate increase hydrophilicity was exploited when hydrophobic Duocarmycin analogs were successfully conjugated to anti-CD30 and anti-CD70 antibodies to yield potent ADCs [114]. A recent extension of the glucuronide linking technology used a quaternary ammonium linker (Fig. 31) instead of a carbamate to link the self-immolative moiety to the drug. This approach introduces one additional hydrophilic element to the tether thus leading to highly hydrophilic ADCs, which have improved pharmacokinetics. This new technology was used to link Auristatin E, the drug in GlucQ-AE, and N14-desacetoxy-tubulysin H, the drug in GlucQTub, to anti-CD30 and anti-CD70 antibodies with a DAR of 4 that showed potent cytotoxicity in an antigen specific cytotoxicity assay (Fig. 31) [115].


Glucuronide as a trigger for linked MMAF


Quaternary ammonium linker combined with glucuronide trigger
The higher concentration of glutathione (0.5–10 mmol/L) and relatively low extracellular concentration (2–10 μm/L in plasma) [116] was exploited by tether design for ADCs. In addition to hydrolysis of the hydrazine, the calicheamicin conjugates require the reduction of a disulfide bond for activity (see above). The development of a plasma stable disulfide tether that could efficiently release the drug in the intracellular compartment was critical for the development of the maytansinoid drugs DM1 and DM4 with cleavable linkers (Fig. 32). A series of huC242 ADCs 27a–f were prepared with the intent to explore the effect of steric hindrance adjacent to the disulfide bond upon the ADC characteristics. All ADCs were highly active (low pM potency) and the activity was antigen expression status specific [117]. The conjugates were incubated with DTT at pH 6.5 to determine the relative loss of drug and it was shown that higher hindrance leads to much slower drug release. The higher stability of the disulfide translated to improved pharmacokinetics as defined by the half-life but the tolerability in vivo was not affected by hindrance (Fig. 32). In comparison to non-cleavable linker DM1-SMCC-tether, the cleavable disulfide ADCs were less well tolerated in mice. The disulfide tethers however showed increased bystander activity (as defined above). A bystander kill assay, in which the ADCs were incubated with antigen negative cells and increasing number of antigen positive cells, was carried out for ADCs 27c and 27b in Fig. 33 along with the non-cleavable DM1-SMCC. As long as enough antigen positive cells were present, the cleavable linker containing ADC was able to kill antigen negative cells, with ADC 27b showing much higher efficiency than 27c while the non-cleavable tether was ineffective. An understanding of this behavior was achieved using a radiolabeled maytansinoid conjugated to huC242 by using SPP (27c in Fig. 33) and SPDB (27b in Fig. 33) tethers for DM1 and DM4, along with the non-cleavable DM1-SMCC. The study analyzed the products of cell catabolism for all ADCs. All ADCs yielded Lys-bearing catabolites, indicating full degradation of the antibody in the lysosome. The non-cleavable tether was not further degraded but the cleavable tethers yielded the reduced drugs DM1 and DM4 following disulfide reduction. Interestingly the S-methylated analogs of the two drugs were observed as well with that being the major species for DM4 while for DM1only small amounts of methylated analog were observed. This indicated that DM4 was efficiently methylated by endogenous S-methyl transferases. This finding, associated with the enhanced cell membrane permeating ability of the S-Me analogs, explained the observed differential activity against non-antigen expressing cells (Fig. 29) [118].


Maytansine disulfide linkers stability studies


Disulfide trigger intracellular processing cleavables vs. non-cleavable
3 Processes
3.1 Antigen Binding and Internalization
At a cellular level, multiple factors contribute to efficient targeting of the cancer cells and subsequent tumor destruction: the antigen copy number expressed on the surface of the cancer cell (as mentioned above), the affinity of ADC for the antigen, the rate and efficiency of internalization of the ADC-antigen complex, the rate of recycling of the complex back to the cell surface and the efficiency of trafficking to the appropriate compartment for drug release and ultimately achieving the interaction with the intracellular target leading to cell death all play critical roles in the success of targeted cancer therapy. Most of these characteristics are antibody target dependent and judicious target selection will have a significant impact on the clinical success of the ADC [119]. However, the efficiency of internalization of the ADC could be induced by conjugation of the drug to the non-internalizing mAbs. Several reports showed that upon binding of unconjugated mAb to the antigen, the antibody/antigen complex remains on the surface while conjugation to drugs such as auristatins L49 mAb [120] or anti-CD-20 [121] led to molecules that efficiently internalize once bound to cell surface antigen. Whether this applies to any drug conjugated to non-internalizing mAbs still remains to be demonstrated.
3.2 ADC Intracellular Processing
Trafficking and intracellular fate of ADCs following internalization has been the object of several detailed studies [118, 122, 123]. Anti-CD30 conjugates bearing the protease cleavable tether mc-VC-MMAE and the non-cleavable mc-MMAF were generated and were shown to display comparable binding and internalization rates with both ADCs showing efficient and antigen-dependent cancer cell killing. Immunofluorescence microscopy showed that the ADC localized to lysosomes following 16 h incubation in L540cy cells. When inhibitors of trafficking (ammonium chloride) were used prior to addition of ADCs to the cells the total intracellular level of ADC was significantly diminished while flow cytometry and fluorescence microscopy showed accumulation of ADC at the surface of the cell. Inhibitors of cathepsin B-mediate proteolysis significantly enhanced the intracellular levels of ADCs. The lysosomal metabolism of the ADC caused the released of the drug was probed by using inhibitors of cysteine proteases that showed subdued cytotoxicity when incubated with the cancer cell pretreated with inhibitor. Interestingly, when the inhibitors were added to the cells hours after the ADC treatment there was minimal effect upon cytotoxicity [123].
4 Clinical Experience
4.1 Hematological Cancers
4.1.1 Gemtuzumab Ozogamicin
Gemtuzumab ozogamicin (Mylotarg) was approved by the FDA in 2000 and was the first ADC to reach the market. It is a humanized IgG4 mAb directed against CD33, a surface antigen present in 85–90% of acute myeloid leukemia (AML), conjugated by random Lys conjugation to N-Ac-γ1I dimethyhydrazide (DMH) (Fig. 11) with an average of DAR4. It was prescribed as a monotherapy in patients over the age of 60 with AML who were not candidates for cytotoxic chemotherapy. Mylotarg was withdrawn in 2010 after a phase III study showed no clinical benefit and a higher risk of fatal adverse events [124].
4.1.2 Brentuximab Vedotin
Brentuximab vedotin (Adcetris) is composed of a mouse IgG1 anti-CD30 mAb conjugated to mc-VC-MMAE via maleimides to native cysteine residues with an average DAR of 4. Adcetris binds to the cell surface CD30 with high affinity (3 nM) and has high potency against CD30+ Hodgkin lymphoma and ALCL tumor cells in vitro (IC50 values of 3–50 pM) [125]. The clinical MTD of Adcetris was 1.8 mg/kg every 3 weeks. At this dose objective responses were obtained, including complete responses (4 of 12 patients) and partial responses (2 of 12 patients) in relapsed CD30-positive lymphomas [126] in a first Phase I study. In a second Phase I trial of brentuximab vedotin carried out to test its effects, when administered weekly the dosing range was 0.4–1.4 mg/kg, the MTD was 1.2 mg/kg and the overall response rate (ORR) was 59%, with 34% complete responses [127]. Adcetris was approved for the treatment of patients with relapsed or refractory CD30+ Hodgkin lymphoma following autologous stem cell transplant (ASCT) or patients not eligible for ASCT who have failed at least two other chemotherapy treatments. Adcetris has also been approved for patients with anaplastic large cell lymphoma (ALCL) as a second line following a phase II study where 86% of patients showed an overall response rate and 54% complete responses [128]. The most common adverse reactions were peripheral sensory neuropathy, neutropenia, fatigue, nausea, and thrombocytopenia.
4.2 Solid Tumors
4.2.1 Ado-Trastuzumab Emtansine
Ado-trastuzumab Emtansine (T-DM1, Kadcyla) [129] is composed of trastuzumab, an anti-Her2 humanized IgG1 antibody random conjugated via Lys to non-cleavable SMCC-DM1 with an average DAR of 3.5. In vitro potency of trastuzumab-MCC-DM1 in a panel of human breast cancer cell lines expressing Her2 was IC50 4–15 ng/mL [129]. The clinical MTD was 3.6 mg/kg every 3 weeks with a t½ of 3.5 days [130]. In early 2013, T-DM1 was approved as a new therapy for patients with HER2-positive, late-stage (metastatic) breast cancer. Currently, T-DM1 is the only ADC approved for treating metastatic breast cancer that overexpresses the HER2 antigen [131, 132].
5 Future Directions and Perspective
Therapeutic ADCs for cancer treatment are experiencing encouraging success with the approval of Adcetris and Kadcyla and promising results with several clinical candidates in advanced clinical trials [133, 134]. The recent advances in linker development led to a breakthrough in the ability to achieve good clinical efficacy with limited toxicity. The two parameters that define the therapeutic index of ADCs are Minimum Effective dose (MED) and MTD and while ADCs present certain advantages over chemotherapy with regard to systemic toxicity still most current ADCs are dosed at or close to the maximum tolerated dose. The most encouraging aspect of ADC development is that targeted therapy, with very few examples, has delivered on its promise of showing efficacy against tumors defined by a particular target. The main challenge for next generation of ADCs is to improve their tolerability. While on-target toxicity can be addressed by a careful choice of target antigen, the off-target toxicity can be addressed with medicinal chemistry. Highly potent drugs have contributed to the success of the current generation of ADCs but all have shown significant dose-limiting toxicities [135]. Generation of homogenous ADCs is a reality with the establishment of site-specific conjugation technology used by ADCs currently in the clinic. This technology, along with the advances in control of the hydrophobicity of linked drugs should lead to significant improvement in pharmacokinetics to a point where the half-lives of the ADCs should closely resemble the ones for the parent mAbs. The small fraction of current ADCs reaching the tumor [41] presents an excellent opportunity for improving the therapeutic index as just a small improvement in efficient targeting could have a great impact on both the MED and MTD. The non-specific uptake of ADC via target-independent internalization, i.e. pinocytosis, was recognized as an area of intervention [136]. Exploiting the genetic differences between tumor and normal cells is in its infancy and medicinal chemists and cell biologists are faced with a great opportunity to harness these yet unknown devices that will enhance the ability to kill cancer cells with future ADCs. Uncovering tumor-specific mechanisms of release will require development of new triggers or combination of triggers within the same tether or linked drug. The suggested dependence of lysosomal escape upon the structure of the linked drug gave us a glimpse into the types of opportunities available to ADC medicinal chemists [104]. From a structural perspective, the interaction of the linked drugs with the antibody is far from being understood. Developing an understanding of these interactions could provide design principles for medicinal chemists when building linked drugs for mAbs. Such an advance could be achieved by making use of molecular modelling or, ideally, by obtaining crystal structures of ADCs.
The efficacy of ADCs is dependent on whether the intracellularly released active species is a substrate of multidrug resistance (MDR) proteins. Most of the ADCs in clinical trials use a small set of cytotoxic warheads: auristatin and maytansinoids. Resistance, innate or acquired, to these drugs could lead to loss of clinical efficacy [137]. It was shown that the chemical structure of the tether can enable the active species to evade efflux mechanisms. Medicinal chemists will always be challenged to discover new chemical warheads that are not efflux substrates, while still displaying the required potency for ADCs. The interaction of medicinal chemists with protein engineers, oncologists, cell biologists, and clinicians will result in better ADCs. These interactions present excellent opportunities to demonstrate that medicinal chemistry is a central science that can provide innovative solutions to the problems currently faced by this fascinating field of scientific endeavor.
References
Köhler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256:495–497
Scott AM, Wolchok JD, Old L (2012) Antibody therapy of cancer. Nat Rev Cancer 12:278–287
Schrama D, Reiseld RA, Becker JC (2006) Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 5:147–159
Saphire EO, Parren PW, Pantophlet R, et al. (2001) Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 293:1155–1159
Alley SC, Benjamin DR, Jeffrey SC, et al. (2008) Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem 19(3):759–765
Shen BQ, Xu K, Liu L, Raab H, et al. (2012) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 30(2):184–189
Lyon RP, Setter JR, Bovee TD, et al. (2014) Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat Biotechnol 32(10):1059–1063
Tumey LN, Charati M, He T, et al. (2014) Mild method for succinimide hydrolysis on ADCs: impact on ADC potency, stability, exposure, and efficacy. Bioconjug Chem 25:1871–1880
Christie RJ, Fleming R, Bezabeh B, et al. (2019) Stabilization of cysteine-linked antibody drug conjugates with N-aryl maleimides. J Control Release 220(Pt B):660–670
Badescu G, Bryant P, Bird M, et al. (2014) Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug Chem 25(6):1124–1136
Bryant P, Pabst M, Badescu G, et al. (2015) In vitro and in vivo evaluation of cysteine rebridged trastuzumab–MMAE antibody drug conjugates with defined drug-to-antibody ratios. Mol Pharm 12(6):1872–1879
Behrens CR, Ha EH, Chinn LL, et al. (2015) Antibody–Drug Conjugates (ADCs) derived from interchain cysteine cross-linking demonstrate improved homogeneity and other pharmacological properties over conventional heterogeneous ADCs. Mol Pharm 12(11):3986–3998
Maruani A, Smith ME, Miranda E, et al. (2015) A plug-and-play approach to antibody-based therapeutics via a chemoselective dual click strategy. Nat Commun 6:6645
Smith ME, Schumacher FF, Ryan CP, et al. (2010) Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J Am Chem Soc 132(6):1960–1965
Lee MTW, Maruani A, Baker JR, et al. (2016) Enabling the controlled assembly of antibody conjugates with a loading of two modules without antibody engineering. Chem Sci 7:799–802
Kashiwagi T, Yokoyama K, Ishikawa K, et al. (2002) Crystal structure of microbial transglutaminase from Streptoverticillium mobaraense. J Biol Chem 277(46):44252–44260
Jeger S, Zimmermann K, Blanc A, et al. (2010) Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed Engl 49(51):9995–9997
Strop P, Liu SH, Dorywalska M, et al. (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 20(2):161–167
Rabuka D, Rush JS, deHart GW, et al. (2012) Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat Protoc 7(6):1052–1067
Agarwal P, Kudirka R, Albers AE, et al. (2013) Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug Chem 24:846–851
Drake PM, Albers AE, Baker J, et al. (2014) Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug Chem 25:1331–1341
Kudirka R, Barfield RM, McFarland J, et al. (2015) Generating site-specifically modified proteins via a versatile and stable nucleophilic carbon ligation. Chem Biol 22(2):293–298
Okeley NM, Toki BE, Zhang X, et al. (2013) Metabolic engineering of monoclonal antibody carbohydrates for antibody−drug conjugation. Bioconjug Chem 24(10):1650–1655
Hamann PR, Hinman LM, Beyer CF, et al. (2005) An anti-MUC1 antibody-calicheamicin conjugate for treatment of solid tumors. Choice of linker and overcoming drug resistance. Bioconjug Chem 16:346–353
Zhou Q, Stefano JE, Manning C, et al. (2014) Site-specific antibody-drug conjugation through glycoengineering. Bioconjug Chem 25:510–520
Ramakrishnan B, Qasba PK (2002) Structure-based design of β1,4-galactosyltransferase I (β4Gal-T1) with equally efficient N-acetylgalactosaminyltransferase activity: point mutation broadens β4Gal-T1 donor specificity. J Biol Chem 277(23):20833–20839
Ramakrishnan B, Boeggeman E, Pasek M, Qasba PK (2011) Bioconjugation using mutant glycosyltransferases for the site-specific labeling of biomolecules with sugars carrying chemical handles. Methods Mol Biol 751:281–296
van Geel R, Wijdeven MA, Heesbeen R, et al. (2015) Chemoenzymatic conjugation of toxic payloads to the globally conserved N-glycan of native mAbs provides homogeneous and highly efficacious antibody−drug conjugates. Bioconjug Chem 26(11):2233–2242
Dumas A, Lukas L, Christopher D, et al. (2015) Designing logical codon reassignment – expanding the chemistry in biology. Chem Sci 6:50–69
Hallam TJ, Wold E, Wahl A, Smider VV (2015) Antibody conjugates with unnatural amino acids. Mol Pharm 12(6):1848–1862
Axup JY, Bajjuri KM, Ritland M, et al. (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A 109(40):16101–16106
VanBrunt MP, Shanebeck K, Caldwell Z, et al. (2015) Genetically encoded azide containing amino acid in mammalian cells enables site-specific antibody−drug conjugates using click cycloaddition chemistry. Bioconjug Chem 26(11):2249–2260
Wang L, Amphlett G, Blättler WA, et al. (2005) Structural characterization of the maytansinoid−monoclonal antibody immunoconjugate, huN901−DM1, by mass spectrometry. Protein Sci 14:2436–2446
Sun MM, Beam KS, Cerveny CG, et al. (2005) Reduction−alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjug Chem 16:1282–1290
Hamblett KJ, Senter PD, Chace DF, et al. (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 10(20):7063–7070
Lyons A, King DJ, Owens RJ, et al. (1990) Site-specific attachment to recombinant antibodies via introduced surface cysteine residues. Protein Eng 3(8):703–708
Stimmel JB, Merrill BM, Kuyper LF, et al. (2000) Site-specific Conjugation on Serine→Cysteine Variant Monoclonal Antibodies. J Biol Chem 275(39):30445–30450
Junutula JR, Raab H, Clark S, et al. (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26(8):925–932
Junutula JR, Bhakta S, Raab H, et al. (2008) Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J Immunol Methods 332(1–2):41–52
Jeffrey SC, Burke PJ, Lyon RP, et al. (2013) A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjug Chem 24:1256–1263
Teicher BA, Chari RV (2011) Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res 17(20):6389–6397
Dumontet C, Jordan MA (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9(10):790–803
Kupchan SM, Komoda Y, Court WA, et al. (1972) Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J Am Chem Soc 94(4):1354–1356
Widdison WC, Wilhelm SD, Cavanagh EE, et al. (2006) Semisynthetic maytansine analogues for the targeted treatment of cancer. J Med Chem 49(14):4392–4408
Cassady JM, Chan KK, Floss HG, Leistner E (2004) Recent developments in the maytansinoid antitumor agents. Chem Pharm Bull (Tokyo) 52(1):1–26
Kupchan SM, Sneden AT, Branfman AR, et al. (1978) Structural requirements for antileukemic activity among the naturally occurring and semisynthetic maytansinoids. J Med Chem 21(1):31–37
Pettit GR, Kamano Y, Herald CL, et al. (1987) The isolation and structure of a remarkable marine animal antineoplastic constituent: dolastatin 10. J Am Chem Soc 109(22):6883–6885
Pettit GR, Singh SB, Hogan F, et al. (1989) Antineoplastic agents. Part 189. The absolute configuration and synthesis of natural (−)-dolastatin 10. J Am Chem Soc 111(14):5463–5465
Bai R, Pettit GR, Hamel E, et al. (1990) Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem Pharmacol 39(12):1941–1949
Cormier A, Marchand M, Ravelli RB, et al. (2008) Structural insight into the inhibition of tubulin by vinca domain peptide ligands. EMBO Rep 9(11):1101–1106
Pettit GR, Srirangam JK, Barkoczy J, et al. (1998) Antineoplastic agents 365. Dolastatin 10 SAR probes. Anticancer Drug Des 13(4):243–277
Miyazaki K, Kobayashi M, Natsume T, et al. (1995) Synthesis and antitumor activity of novel dolastatin 10 analogs. Chem Pharm Bull (Tokyo) 43(10):1706–1718
Doronina SO, Toki BE, Torgov MY, et al. (2003) Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat Biotechnol 21(7):778–784
Doronina SO, Mendelsohn BA, Bovee TD, et al. (2006) Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug Chem 17(1):114–124
Maderna A, Doroski M, Subramanyam C, et al. (2014) Discovery of cytotoxic dolastatin-10 analogs with N terminal modifications. J Med Chem 57:10527–10543
Maderna A, Leverett CA (2015) Recent advances in the development of new auristatins: structural modifications and application in antibody drug conjugates. Mol Pharm 12(6):1798–1812
Sasse F, Steinmetz H, Heil J, et al. (2000) Tubulysins, new cytostatic peptides from myxobacteria acting on microtubuli. Production, isolation, physico-chemical and biological properties. J Antibiot (Tokyo) 53(9):879–885
Höfle G, Glaser N, Leibold T, et al. (2003) Semisynthesis and degradation of the tubulin inhibitors epothilone and tubulysin. Pure Appl Chem 75(2–3):167–178
Steinmetz H, Glaser N, Herdtweck E, et al. (2004) Isolation, crystal and solution structure determination, and biosynthesis of tubulysins – powerful inhibitors of tubulin polymerization from myxobacteria. Angew Chem Int Ed Engl 43(37):4888–4892
Wipf P, Wang Z (2007) Total synthesis of N14-desacetoxytubulysin H. Org Lett 9(8):1605–1607
Patterson AW, Peltier HM, Sasse F, Ellman JA (2007) Design, synthesis, and biological properties of highly potent tubulysin D analogs. Chemistry 13(34):9534–9541
Patterson AW, Peltier HM, Ellman JA (2008) Expedient synthesis of N-methyl tubulysin analogues with high cytotoxicity. J Org Chem 73(12):4362–4369
Leamon CP, Reddy JA, Vetzel M, et al. (2008) Folate targeting enables durable and specific antitumor responses from a therapeutically null tubulysin B analogue. Cancer Res 68(23):9839–9844
Li J, Perry SR, Muniz-Medina V, et al. (2016) A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 29(1):117–129
Toader D, Harper J et al (2015) Discovery of tubulysin payloads for antibody drug conjugates with potent in vitro activity and in vivo efficacy in solid tumor models. Presented at molecular targets and cancer therapeutics, Boston, MA, 5–9 Nov 2015. Mol Cancer Ther 14(12 Suppl 2):Abstract nr B170
Lee MD, Ellestad GA, Borders DB (1991) Calicheamicins: discovery, structure, chemistry, and interaction with DNA. Acc Chem Res 24:235–243
Zein N, Sinha AM, McGahren WJ, Ellestad GA (1988) Calicheamicin γ1I: an antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 240(4856):1198–1201
Hinman LM, Hamann PR, Wallace R, et al. (1993) Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res 53(14):3336–3342
Hanka LJ, Dietz A, Gerpheide SA, et al. (1978) CC-1065 (NSC-298223), a new antitumor antibiotic. Production, in vitro biological activity, microbiological assays and taxonomy of the producing microorganism. J Antibiot (Tokyo) 31(12):1211–1217
Boger DL, Johnson DS (1995) CC-1065 and the duocarmycins: unraveling the keys to a new class of naturally derived DNA alkylating agents. Proc Natl Acad Sci U S A 92(9):3642–3649
McGovren JP, Clarke GL, Pratt EA, DeKoning TF (1984) Preliminary toxicity studies with the DNA-binding antibiotic, CC-1065. J Antibiot (Tokyo) 37(1):63–70
Boger DL, Ishizaki T, Kitos PA, Suntornwat O (1990) Synthesis of N-(tert-butyloxycarbonyl)-CBI, CBI, CBI-CDPI1, and CBI-CDPI2: enhanced functional analogs of CC-1065 incorporating the 1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one (CBI) left-hand subunit. J Org Chem 55(23):5823–5832
Wang H, Rangan VS, Sung MC, et al. (2016) Pharmacokinetic characterization of BMS-936561, an anti-CD70 antibody-drug conjugate, in preclinical animal species and prediction of its pharmacokinetics in humans. Biopharm Drug Dispos 37(2):93–106
Elgersma RC, Coumans RGE, Huijbregts T, et al. (2015) Design, synthesis, and evaluation of linker-duocarmycin payloads: toward selection of HER2-targeting antibody-drug conjugate SYD985. Mol Pharm 12(6):1813–1835
Leimgruber W, Batcho AD, Schenker F (1965) The structure of anthramycin. J Am Chem Soc 87:5793–5795
Tozuka Z, Yazawa H, Murata M, Takaya T (1983) Studies on tomaymycin. III. Syntheses and antitumor activity of tomaymycin analogs. J Antibiot (Tokyo) 36(12):1699–1708
Brazhnikova MG, Konstantinova NV, Mesentsev AS (1972) Sibiromycin. Isolation and characterization. J Antibiot (Tokyo) 25(11):668–673
Hurley LH, Thurston DE (1984) Pyrrolo(l,4)benzodiazepine antitumor antibiotics: chemistry, interaction with DNA, and biological implications. Pharm Res 1(2):52–59
Subhas Bose D, Thompson AS, Ching J, et al. (1992) Rational design of a highly efficient irreversible DNA interstrand cross-linking agent based on the pyrrolobenzodiazepine ring system. J Am Chem Soc 114:4939–4941
Hartley JA, Spanswick VJ, Brooks N, et al. (2004) SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: Part 1: Cellular pharmacology, in vitro and initial in vivo antitumor activity. Cancer Res 64(18):6693–6699
Hartley JA, Hamaguchi A, Coffils M, et al. (2010) SG2285, a novel C2-aryl-substituted pyrrolobenzodiazepine dimer prodrug that cross-links DNA and exerts highly potent antitumor activity. Cancer Res 70(17):6849–6858
Hartley JA, Hamaguchi A, Suggitt M, et al. (2010) DNA interstrand cross-linking and in vivo antitumor activity of the extended pyrrolo[2,1-c][1,4]benzodiazepine dimer SG2057. Investig New Drugs 30(3):950–958
Howard PW, Chen Z, Gregson SJ, et al. (2009) Synthesis of a novel C2/C2′-aryl-substituted pyrrolo[2,1-c][1,4]benzodiazepine dimer prodrug with improved water solubility and reduced DNA reaction rate. Bioorg Med Chem Lett 19(22):6463–6466
Saunders LR, Bankovich AJ, Anderson WC, et al. (2015) A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med 7(302):302ra136
Tolcher AW, Sugarman S, Gelmon KA, et al. (1999) Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J Clin Oncol 17(2):478–484
Bakker M, Renes J, Groenhuijzen A, et al. (1997) Mechanisms for high methoxymorpholino doxorubicin cytotoxicity in doxorubicin-resistant tumor cell lines. Int J Cancer 73(3):362–366
Quintieri L, Geroni C, Fantin M, et al. (2005) Formation and antitumor activity of PNU-159682, a major metabolite of nemorubicin in human liver microsomes. Clin Cancer Res 11(4):1608–1617
Mazzini S, Scaglioni L, Mondelli R, et al. (2012) The interaction of nemorubicin metabolite PNU-159682 with DNA fragments d(CGTACG)(2), d(CGATCG)(2) and d(CGCGCG)(2) shows a strong but reversible binding to G:C base pairs. Bioorg Med Chem 20(24):6979–6988
Bushnell DA, Cramer P, Kornberg RD (2002) RNA polymerase II cocrystal at 2.8 A resolution. Proc Natl Acad Sci U S A 99:1218–1222
Kaplan CD, Larsson KM, Kornberg RD (2008) The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Mol Cell 30:547–556
Brueckner F, Cramer P (2008) Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat Struct Mol Biol 15:811–818
Letschert K, Faulstich H, Keller D, Keppler D (2006) Molecular characterization and inhibition of amanitin uptake into human hepatocytes. Toxicol Sci 91:140–149
Faulstich H, Fiume L (1985) Protein conjugates of fungal toxins. Methods Enzymol 112:225–237
Moldenhauer G, Salnikov AV, Lüttgau S, et al. (2012) Therapeutic potential of amanitin-conjugated anti-epithelial cell adhesion molecule monoclonal antibody against pancreatic carcinoma. J Natl Cancer Inst 104(8):622–634
Liu Y, Zhang X, Han C, et al. (2015) TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature 520(7549):697–701
Hechler T, Kulke M, Mueller C et al (2014) Amanitin-based antibody-drug conjugates targeting the prostate-specific membrane antigen. Poster presented at annual meeting of the American Association for Cancer Research, San Diego, CA, 5–9 Apr 2014. Cancer Res 74(19 Suppl):Abstract nr 664
Wall ME, Wani MC, Cook CE, et al. (1966) Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminate. J Am Chem Soc 88:3888–3890
Hsiang YH, Hertzberg R, Hecht S, Liu LF (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260:14873–14878
Staker BL, Hjerrild K, Feese MD, et al. (2002) The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A 99(24):15387–15392
Moon SJ, Govindan SV, Cardillo TM, et al. (2008) Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem 51:6916–6926
Ogitani Y, Yamaguchi J, Ishii C et al (2015) DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a potent anti-tumor efficacy with differentiation from T-DM1 in preclinical studies. Poster presented at international conference: molecular targets and cancer therapeutics, Boston, MA, 5–9 Nov 2015. Mol Cancer Ther 14(12 Suppl 2):Abstract nr A145
Tamura K, Shitara K, Naito Y, et al. (2016) Single agent activity of DS-8201a, a HER2-targeting antibody-drug conjugate, in breast cancer patients previously treated with T-DM1: phase 1 dose escalation. Ann Oncol 27(suppl 6):LBA17
Rock BM, Tometsko ME, Patel SK, et al. (2015) Intracellular catabolism of an antibody drug conjugates with a noncleavable linker. Drug Metab Dispos 43:1341–1344
Hamblett KJ, Jacob AP, Gurgel JL, et al. (2015) SLC46A3 is required to transport catabolites of noncleavable antibody maytansine conjugates from the lysosome to the cytoplasm. Cancer Res 75(24):5329–5340
Pillow TH, Tien J, Parsons-Reponte KL, et al. (2014) Site-specific trastuzumab maytansinoid antibody-drug conjugates with improved therapeutic activity through linker and antibody engineering. J Med Chem 57:7890–7899
Dubowchik GM, Walker MA (1999) Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharmacol Ther 83:67–123
Saito G, Swanson JA, Lee KD (2003) Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities. Adv Drug Deliv Rev 55:199–215
Hamann PR, Hinman LM, Hollander I, et al. (2002) Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem 13(1):47–58
Dubowchik GM, Firestone RA, Padilla L, et al. (2002). Bioconjug Chem 13(4):855–869
Yu SF, Zheng B, Go M, et al. (2015) A novel anti-CD22 anthracycline-based antibody-drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Clin Cancer Res 21(14):3298–3306
Lyon RP, Bovee TD, Doronina SO, et al. (2015) Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol 33(7):733–735
Albin N, Massaad L, Toussaint C, et al. (1993) Main drug metabolizing enzyme systems in human breast tumors and peritumoral tissues. Cancer Res 53:3541–3546
Jeffrey SC, Andreyka JB, Bernhardt SX, et al. (2006) Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjug Chem 17:831–840
Jeffrey SC, Nguyen MT, Moser RF, et al. (2007) Minor groove binder antibody conjugates employing a water soluble beta-glucuronide linker. Bioorg Med Chem Lett 17:2278–2280
Burke PJ, Hamilton JZ, Pires TA, et al. (2016) Development of novel quaternary ammonium linkers for antibody-drug conjugates. Mol Cancer Ther 15(5):938–945
Wu G, Fang YZ, Yang S, et al. (2004) Glutathione metabolism and its implications for health. J Nutr 134(3):489–492
Kellogg BA, Garrett L, Kovtun Y, et al. (2011) Disulfide-linked antibody-maytansinoid conjugates: optimization of in vivo activity by varying the steric hindrance at carbon atoms adjacent to the disulfide linkage. Bioconjug Chem 22(4):717–727
Erickson HK, Park PU, Widdison WC, et al. (2006) Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res 66:4426–4433
Sadekar S, Figueroa I, Tabrizi M (2015) Antibody drug conjugates: application of quantitative pharmacology in modality design and target selection. AAPS J 17(4):828–836
Smith LM, Nesterova A, Alley SC, et al. (2006) Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol Cancer Ther 5(6):1474–1482
Law CL, Cerveny CG, Gordon KA, et al. (2004) Efficient elimination of B-lineage lymphomas by anti-CD20-auristatin conjugates. Clin Cancer Res 10:7842–7851
Okeley NM, Miyamoto JB, Zhang X, et al. (2010) Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res 16(3):888–897
Sutherland MS, Sanderson RJ, Gordon KA, et al. (2006) Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J Biol Chem 281(15):10540–10547
Petersdorf SH, Kopecky KJ, Slovak M, et al. (2013) A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 121(24):4854–4860
Senter PD, Sievers EL (2012) The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol 30(7):631–637
Younes A, Bartlett NL, Leonard JP, et al. (2010) Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363:1812–1821
Fanale MA, Forero-Torres A, Rosenblatt JD, et al. (2012) A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res 18:248–255
Pro B, Advani R, Brice P, et al. (2012) Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 30(18):2190–2196
Lambert JM, Chari RV (2014) Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J Med Chem 57:6949–6964
Krop IE, Beeram M, Modi S, et al. (2010) Phase I study of trastuzumab−DM1, an HER2 antibody−drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol 28:2698–2704
LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX (2011) Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res 17(20):6437–6447
Verma S, Miles D, Gianni L, et al. (2012) Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med 367(19):1783–1791
Diamantis N, Banerji U (2016) Antibody-drug conjugates – an emerging class of cancer treatment. Br J Cancer 114(4):362–367
Smaglo BG, Aldeghaither D, Weiner LM (2014) The development of immunoconjugates for targeted cancer therapy. Nat Rev Clin Oncol 11(11):637–648
Hinrichs MJ, Dixit R (2015) Antibody drug conjugates: nonclinical safety considerations. AAPS J 17(5):1055–1064
Polakis P (2016) Antibody drug conjugates for cancer therapy. Pharmacol Rev 68(1):3–19
Shefet-Carasso L, Benhar I (2015) Antibody-targeted drugs and drug resistance-challenges and solutions. Drug Resist Updat 18:36–46
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Toader, D. (2017). Antibody–Drug Conjugates. In: Waring, M.J. (eds) Cancer II. Topics in Medicinal Chemistry, vol 28. Springer, Cham. https://doi.org/10.1007/7355_2017_29
Download citation
DOI: https://doi.org/10.1007/7355_2017_29
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75924-1
Online ISBN: 978-3-319-75926-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)