
Abstract
The PI3K/AKT pathway is one of the most frequently altered in cancer and several promising small-molecule inhibitors of this pathway have entered advanced stages of clinical research. This chapter intends to highlight substantial medicinal chemistry lead identification and optimization efforts that led to the discovery of key isoform- and PIK family-selective PI3K inhibitors. Coverage is given for those disclosed PI3K isoform-selective inhibitors that have entered clinical trials in oncology, regardless of their current status. Where possible based on existing information, a focus is placed on discussion of potential structural rationale for isoform selectivity.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
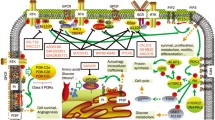
There are four isoforms of phosphatidylinositide-3-kinase (PI3K) referred to as PI3Kα, -β, -δ, and -γ. These Class-I PI3Ks exist as heterodimers between catalytic p110 isoforms (α, β, δ, γ) and regulatory subunits (i.e., p85α, p50α, p85β, p55γ). The Class-I PI3Ks have proven to play a crucial role in signaling networks that promote cell growth and survival [1, 2]. All PI3K isoforms propagate a signaling cascade by phosphorylation of PIP2 (phosphatidylinositol-4,5-bisphosphonate) to PIP3 (phosphatidylinositol (3,4,5)-triphosphate). The lipid messenger PIP3 mediates activation of several downstream protein kinases, including promoting phosphorylation and activation of AKT.
The summation of over 20 years of preclinical and clinical investigation supports the importance of phosphoinositol-3-kinases in oncology. As may be expected based on their cellular function, activation of the PI3K/AKT pathway is a frequent event in human tumors. This activation can be achieved by several mechanisms. Somatic loss of PTEN, upregulation of upstream RTKs, and genetic modification of PI3K itself can lead to pathway dysregulation [3]. PIK3CA (the gene that encodes for p110α) itself is found to be mutated in a high percentage of a number of cancers [3, 4]. For example, in a study of 452 patients with unilateral invasive primary breast cancer, PIK3CA mutations were identified in 151 tumors (33.4%) [5].
Most early PI3K inhibitors that entered clinical investigation for oncology indications were pan-isoform PI3K inhibitors, many of which also inhibited other PIK-family members (PIK: phosphatidylinositol kinases, e.g. mTOR, DNA-PK, see the chapter “Modulation of the DNA Damage-Response by Inhibitors of the Phosphatidylinositol 3-Kinase Related Kinase (PIKK) Family”). The clinical efficacy of these pan-isoform PI3K inhibitors has been described as “modest at best” [6]. Pivotal Ph-II and -III results from pictilisib (GDC-0941) and buparlisib (BKM-120) in mBC are informative as these are perhaps the most advanced pan-isoform PI3K inhibitors in patients.
Both studies are promising and provide proof-of-concept for PI3K inhibition in breast cancer, however, the changes in median progression free survival (mPFS) and overall response rate (ORR) were not as substantial as hoped. In late 2014, the results of pictilisib in combination with fulvestrant vs. fulvestrant plus placebo in patients with AI-resistant ER+ advanced or metastatic breast cancer were reported (168 total patients, “FERGI” trial [7]). In analysis of the full population, addition of pictilisib to fulvestrant was associated with an mPFS improvement from 3.8 to 6.2 mo (HR, 0.77; 95% CI, 0.50–1.19). In 2015, the results of a phase III investigation of buparlisib in combination with fulvestrant vs. fulvestrant plus placebo in patients with HR+/HER- AI-resistant advanced breast cancer were reported (1,147 patients, “BELLE-2” trial [8]). In analysis of the full population, addition of buparlisib to fulvestrant was associated with an mPFS (median progression free survival) improvement from 5.0 to 6.9 mo (HR, 0.78; P < 0.001). The overall response rate (ORR) for the treated vs. control group in the total population was 11.8% vs 7.7%.
One hypothesis for the modest mPFS and ORR for these agents (and others) is the exposure, and thus efficacy, of pan-PI3K inhibitors is limited by toxicity associated with the simultaneous inhibition of multiple PI3K isoforms. Common human adverse events resulting from pan-PI3K inhibition are hyperglycemia, maculopapular rash, gastrointestinal intolerance (anorexia, nausea, vomiting, dyspepsia, severe diarrhea/colitis), and stomatitis [9]. Although not transformative for many patients, clinical efficacy for these pan-inhibitors has been observed and has supported further investigation of clinical PI3K inhibitors.
Despite similar structure and catalytic function, preclinical observations of different mechanisms of activation, tissue localization, and phenotypes resultant from genetic alteration suggest distinct native physiological roles of each PI3K isoform [3]. Thus, a diverse pharmacology/toxicity is likely associated with inhibition of specific isoforms. p110α and p110β are ubiquitously expressed in normal tissues. Correspondingly, homozygous genetic loss of p110α or β in mice results in embryonic lethality [10, 11]. Even heterozygous mice expressing one copy each of kinase dead and wild-type p110α show metabolic defects including reduced body weight, insulin resistance, and increased adiposity [12]. p110δ and p110γ are primarily restricted to leukocytes. Deletion of p110δ and p110γ leads to viable mice, but they have immune deficiencies. For example, p110δ was shown to play a critical role in B cell homeostasis and function [13] and p110γ was shown important in thymocyte development, T-cell activation, and neutrophil migration [14]. p110δ kinase-dead knock-in mice show impaired immune responses and develop inflammatory bowel disease [15].
Whilst predicted to have efficacy in broad tumor types, the above-mentioned preclinical and clinical toxicities associated with pan-PI3K inhibition may limit the therapeutic index of pan-PI3K inhibitors. The development of isoform-selective PI3K inhibitors may thus improve therapeutic outcomes as single agents or in combination. There is great interest in the clinical study of selective PI3Kα inhibitors in cancers with PIK3CA mutations, PI3Kβ inhibitors in tumors with PTEN loss and PI3Kδ in hematological malignancies. Several of these “next generation” isoform-selective PI3K inhibitors are expanding the understanding and re-invigorating the enthusiasm of therapeutic intervention within this pathway. This chapter intends to highlight the significant medicinal chemistry efforts leading to isoform- and PIK family- selective PI3K inhibitors that entered clinical trials in oncology.
2 Structural Biology
Drug discovery programs directed at PI3-kinases have been highly enabled through analysis of ligand-bound and apo X-ray structures of catalytic p110 subunits alone. It is apparent that most drug discovery programs, regardless of targeted isoform selectivity profile, initially relied on the well precedented p110γ co-crystallization system of Walker et al. [16, 17]. Homology models for other isoforms derived from these p110γ structures were also utilized. Reports of X-ray structures for the p110 subunits of the α [18,19,20], β [21], and δ [22] isoforms have been of more recent value for designing and/or rationalizing isoform selective inhibitors.
2.1 Domains of p110 and Interaction with Regulatory Domains
Class IA (PI3Kα, β, δ) and class IB (PI3Kγ) PI3Ks possess multi-domain catalytic p110 subunits. For reference, an X-ray structure and the domain sequence architecture of p110α (PDB-id 4JPS) are displayed in Fig. 1 [23]. Class 1A p110s include an adapter binding domain (ABD), a Ras binding domain (RBD), a C2 domain, a helical domain, and a kinase domain. The class 1B p110γ shares similar structural features, however lacks an ABD at the N-terminus. The kinase domain (magenta Fig. 1) is mechanistically responsible for the primary function of the PI3Ks: ATP-mediated phosphorylation of membrane localized phosphatidylinositol-4,5-bisphosphonate (PIP2) to phosphatidylinositol (3,4,5)-triphosphate (PIP3). The kinase domain is the site of action of all clinical PI3K inhibitors which act ATP-competitive.


Ribbon diagram of the p110α structure (2.2 Å resolution, PDB-id 4JPS) highlighting the different domains of this catalytic subunit [23]. The ATP-binding site of the kinase domain (magenta) occupied by BYL-719 is shown as the magenta solvent-accessible surface. Domain boundaries adapted from Huang et al. [18]
As noted above, PI3Ks exist as heterodimers between catalytic subunits (p110) and regulatory subunits (p85 and splice variants p55, p50). There are multiple mechanisms hypothesized for activating and localizing PI3Ks at the membrane where PIP2 is located. Binding to pRTKs, RAS superfamily members, GPCRs, Gβγ subunits, and plasma membrane have all been proposed as important depending on stimulus and PI3K isoform invoked [24]. The specific manners of activation and localization are structurally dependent on the different domains of p110 isoforms as well as regulatory subunits that are associated as part of the heterodimeric PI3K complex. The regulatory subunits contain SH2 (Src homology 2) domains thought to interact with RTKs in response to RTK auto-phosphorylation generated by ligand stimulus. It is hypothesized that these pRTK (and/or membrane) interactions are a major mechanism to achieve active and localized p110 subunits. In all reported structures of p110 isoforms, the DFG and αC-helix are “in” suggesting a persistent catalytically active state of p110. This may be consistent with the hypothesis that PI3K activity is controlled primarily by co-peptide binding (i.e., pRTKs, Ras) and co-localization at the membrane with PIP2.
Although a complete structure of a heterodimer has not been disclosed, a model for p110/p85 interaction can be built based on aligned overlays of partial structures. Shown in Fig. 2 is an overlay of structures containing full p110α and p110β with partial p85 fragments [19, 21]. From this model, positioning of the inter-, n-terminal, and c-terminal SH2 domains can be approximated. The regulatory p85 makes extensive contacts with different domains of p110; in particular, a significant contact along the spine of the i-SH2. Contacts between the n- and c-SH2 domains are predicted to be mediated by only a few residues. It is believed that the n-terminal and/or c-terminal SH2 domains are substantially displaced upon pRTK binding. It is interesting to note that two hotspot mutations of p110α (H1047R and E545K, shown in red), as well as many others reported in the literature, occur at or near the interface of the n- and c-SH2 domains of p85 [4].


Aligned overlay of X-ray structures of niSH2-p85α/p110α (PDB-id: 4L23) and ciSH2-p85β/p110β (2Y3A) [19, 21]. The p110α and p110β subunits are represented by dark gray and light gray ribbon diagram, respectively. The niSH2-p85α is represented by dark blue ribbon; the ciSH2-p85β is colored magenta. Key residues H1047 and E545 of p110α (highlighted red) likely influence contact with the cSH2 and nSH2 domains of p85α
2.2 Structural Features of the ATP Site and Inhibitors
A number of different chemotypes have been discovered to selectively inhibit PI3-kinases. There are some general features of the ATP-binding site that are typically exploited for potency and/or selectivity. A representation of the key features of the ATP binding site is shown schematically in Fig. 3. The N- and C-lobes of p110 subunits for the ATP pocket and solvent access occurs between the lower hinge and p-loop/kβ3 strand (kβ3 = kinase beta 3, nomenclature adapted from Walker [16]). Three key isoform-conserved residues border the adenine pocket: a hinge valine, a gatekeeper isoleucine, and a tyrosine residue. PI3K inhibitors necessitate a hydrogen bond from this valine hinge residue. Interestingly, as opposed to many other kinase inhibitors, single- rather than dual- interaction with this valine residue is common for PI3K inhibitors. The phosphate binding pocket (“affinity pocket”; Berndt [22]) is lined, as typical across the kinome, with a catalytic lysine and an acidic residue projecting from the αC-helix (aspartate in the case of p110s). Many PI3K inhibitors fill this pocket directly or indirectly (through water) interacting with the tyrosine, lysine, and/or aspartate residues. Many inhibitors also occupy a hydrophobic pocket on the N-lobe side of the ribose pocket. Lipophilic substitution of the ligand here is enclosed by isoform-conserved hydrophobic amino acid side-chains (catalytic lysine Cα-Cγ methylenes, proline, and methionine from the kβ4 strand and an isoleucine from the kβ5 strand; residues not shown). With little exception [25], isoform selective or specific inhibitors primarily take advantage of structural differences in the lower hinge (C-lobe) or kβ3/kβ4 strands (N-lobe, discussed below). Representative in Fig. 3 is a schematic representation of clinical PI3K-inhibitor BYL719 is mapped onto the above description of key binding features. The aminothiazole contacts the hinge, polar and hydrophobic interactions are achieved from the 4-pyridyl and substituents. Isoform selectivity is achieved through interaction with the lower hinge (discussed later).


(a) Schematic representation of key residues and features of the ATP binding site of catalytic p110 subunits (gray) and typical features of ligands (multicolored). (b) Ligand features mapped on to PI3Kα-selective inhibitor BYL719
Representative of all p110 isoforms, the ATP-binding site of p110γ (PDB-id 3DBS) in complex with pan-PI3K inhibitor GDC-0941 (pictilisib) is shown in Fig. 4. The key isoform-conserved residues within the binding site, mentioned above, are highlighted (hinge Val882γ, catalytic Lys833γ, αC-helix Asp841γ, and the DFG motif). A series of β-strands (kβ3–kβ7) forms the N-lobe of the ATP-binding site, as typical for protein kinases. The formal gatekeeper residue in all p110 isoforms is an isoleucine (Ile879γ, not shown). However, due to a change in orientation of the kβ7 strand (kinase beta strand 7) leading to the hinge, Tyr876γ (on the kβ6 strand, conserved across isoforms) acts as a functional gatekeeper. We hypothesize that this change in strand orientation accommodates ligands with non-aromatic, sterically encumbering hinge-binding motifs (i.e., the morpholine of GDC-0941). This may lead to a greater volume for ligand binding and may explain the relative ease of attaining selectivity for PIK family members over the broad kinome by PI3K inhibitors.


X-ray structure of GDC-0941 (blue) bound to p110γ (magenta) [PDB-id: 3DBS] [26]. Key conserved residues across p110 isoforms are labelled with black text; key motifs are highlighted in magenta text.
Despite a high degree of homology among the isoforms, there are notable differences in primary structure and amino acid sidechain mobility within the lower hinge and kβ3/kβ4 strands. Most isoform selective inhibitors reported were designed for (or later rationalized by) exploitation of these particular differences. Key differences in primary structure on the kβ3/kβ4 strands and lower hinge are labelled in Fig. 5 (GDC-0941 in p110γ). For example, a difference in residue Lys809γ (Gln859α, Asp856β, Asn836δ) is believed to key in a mechanism of selectivity for NVP-BYL-719 (selective interaction with Gln859α; Furet [23]). It has been hypothesized that mobility/flexibility of conserved Met804γ and Trp812γ contribute to the selectivity of inhibitors. These differences have been used to rationalize PI3Kβ [27] and PI3Kδ [22] selective inhibitors, as well as those that spare inhibition of PI3Kβ [28].


X-ray structure of GDC-0941 (blue) bound to p110γ (magenta) [PDB-id: 3DBS] [26]. Residues on the kβ3/kβ4 strands and lower hinge that have been hypothesized to aid in isoform selective inhibition are labelled in black text
3 Tabulated Selectivity Data for Isoform Selective PI3K Inhibitors
Inhibitors discussed in this chapter are presented in Table 1. Activity against the different PI3K isoforms is reported as disclosed by the various discovery teams.
4 Medicinal Chemistry: Discoveries Leading to Isoform Selective PI3K Inhibitors
4.1 PI3Kδ Inhibitors
The most efficacious single-agent PI3K inhibitors to date are those of that selectively target PI3Kδ in hematological malignancies, including US FDA approved Zydelig®. Notably, dramatic responses are observed even though genetic alteration of the PI3K pathway in these patients is not present. The compounds block signaling downstream of the B-cell receptor [13], thus targeting dysfunctional leukemic B-cells.
4.1.1 Cal-101 (GS-1101, Idelalisib, Zydelig®), PI3Kδ Inhibitor
Idelalisib (Cal-101, GS-1101, compound 1, Scheme 1), originally discovered by Icos Corporation, entered clinical trials in 2008 and was approved under the trade name Zydelig® by the FDA in 2014 for relapsed CLL, FL, and SLL. Idelalisib is reported to have high isoform selectivity for PI3Kδ over the other Class-1 isoforms [29]. Despite being the most advanced and only approved PI3K inhibitor, very little has been published related to the medicinal chemistry effort that led to this groundbreaking compound.


Chemical structures of IC87114 and idelalisib. Biochemical data from Somoza et al. [29]
The structure of idelalisib is exemplified in a 2005 patent application from Icos Corporation where they disclose a series of quinazolinone inhibitors of PI3Kδ [38]. An earlier structurally related compound, compound 2, was reported by Icos to be a “close analog” of a compound identified from a screen of their diverse chemical matter. It is presumed that this was a lead that resulted in the discovery of idelalisib (1).
The crystal structure of idelalisib in p110δ was recently disclosed [29] confirming the hypothesized binding mode [22]. Most PI3Kδ selective inhibitors possess a unique three dimensional architecture compared to pan-PI3K inhibitors, commonly referred to as “propeller” or “T-shaped.” In a seminal publication containing the first crystal structures of p110δ, Berndt et al. [22] proposed that selectivity for PI3Kδ was achieved for this class of inhibitors through exploitation of a differential conformational flexibility and sequence identity of active-site residues that aren’t important for ATP binding. This conformational mobility can be visualized in Fig. 6 through examination of the induced fit binding of compound 1 to p110δ (pdb id: 4XE0, Somoza et al. [29]) compared to the p110δ apo structure (pdb id: 2WXR, Berdnt et al. [22]). The adenine of compound 1 exists in the expected plane bisecting the N- and C-lobes of p110δ. Consistent with many other PI3Kδ selective inhibitors, this inhibitor appears to interact with the hinge-valine and does not appear to directly make any contacts to conserved residues in the phosphate binding pocket. The quinazolinone is oriented orthogonal to this plane and occupies a region between a tryptophan and methionine residues that are conserved among the isoforms. This occupied pocket doesn’t exist in the apo form of p110δ (or other isoforms) and Berdnt et al. hypothesize this conformational flexibility is more facile in p110δ compared to the other isoforms.


Induced fit binding of “T-shaped” PI3Kδ inhibitors exemplified by idelalisib (1). (a) p110δ in complex with idelalisib (4XE0). (b) p110δ apo structure (pdb id 2WXR). A reorganization of residues on the kβ3 and kβ4 strands (p-loop) is required for binding to idelalisib
This inhibitor is an exciting treatment for patients with relapsed refractory CLL. For example, in a phase III study of idelalisib in combination with rituximab vs. rituximab alone, patients receiving idelalisib had improved overall response rates (81% vs. 13%) and overall survival at 12 months (92% vs. 80%, Furman et al. [39]).
4.1.2 INK-1197 (IPI-145, Duvelisib), PI3Kδ/γ Inhibitor
In 2011, INK-1197 (3) entered a phase I dose escalation in patients with advanced hematological malignancies. The structure of INK-1197 is exemplified in a 2009 application from Intellikine [40] and acknowledged first as the clinical candidate in a 2013 research publication [30]. INK-1197 was renamed as IPI-145 when licensed to Infinity in 2011. Infinity has subsequently partnered with Abbvie to co-develop this inhibitor which recently entered phase III clinical trials in patients with hematological malignancies.
There hasn’t been any information disclosed about the optimization of this compound. The quinazolone of idelalisib (1) is replaced with an isoquinolone (Scheme 2).


Chemical structure and biochemical isoform inhibition data for INK-1197 (3) [30]
The compound 3 has been claimed to be differentiated from idelalisib (1) on the basis of improved activity against the PI3Kγ isoform. Although structurally similar, 3 possesses greater dual activity against PI3Kδ and PI3Kγ. A structure based rationale for this feature has not been reported. However, based on structural similarity, compound 3 very likely adopts an induced fit binding to PI3Kδ akin to compound idelalisib (1).
Given there are distinct roles and expression of PI3Kδ and PI3Kγ isoforms in lymphocyte subset function, it has been hypothesized that activity associated with dual inhibition may differ from that of inhibition of PI3Kδ alone [30, 41, 42]. Clinical results for INK-1197 in hematological malignancies have been compelling but it remains to be seen whether differentiation from idelalisib is possible with this unique selectivity profile.
4.2 PI3Kβ Inhibitors
PI3Kβ inhibitors, such as TGX-221 and AZD6482, appear to have been originally developed with the promise of preventing platelet aggregation and blood clots. More recently, reports of the importance of the PI3Kβ isoform in PTEN-deficient or null cancers [43, 44] has moved some interest into treating this specific population of cancer patients. PTEN (phosphatase and tensin homolog) is a negative regulator of PI3K signaling and catalyzes the dephosphorylation of PIP3 to PIP2 [45]. Indeed, one of the most common observed mechanisms leading to activated PI3K signaling in patients is somatic loss of PTEN through epigenetic or genetic alteration [46].
4.2.1 SAR260301, PI3Kβ
In 2012, SAR260301 (4, Scheme 3) entered phase I dose escalation to determine MTD in patients with advanced solid tumors. SAR260301 is reported to be a selective inhibitor of PI3Kβ, with >20-fold selectivity over the other PI3K isoforms. The Sanofi oncology drug discovery group published a series of manuscripts from 2012 to 2014 detailing a successful optimization campaign [31, 47, 48].


Lead compound 5 and SAR260301 (4)
Lead compound 5 was identified following up a high throughput screen of 550 K compounds. Compound 5 is reported to be active against PI3Kβ (IC50 = 42 nM) and to possess variant selectivity over the PI3Kα, δ and γ isoforms (IC50 = 2,138, 118 and >10,000 nM, respectively). Optimization of potency and selectivity for PI3Kβ was achieved through monitoring a biochemical assay as well as pAKT inhibition in a PC3 line (PTEN-deficient prostate cancer cell line). Initial efforts were directed at positional scan of substitution on the pendant aniline, and representative data is shown in Table 2. Small para (fluoro, 6) and meta-substitution (methoxy, 7) gave analogs that were slightly more potent against the PI3Kβ isoform. Ortho substitution (methyl, 9) was detrimental to PI3Kβ potency. Most interestingly, meta and ortho substitution, in general, improved selectivity over PI3Kδ.

Two strategies were reported that called for more drastic changes to the potentially labile aniline amide. Isosteric replacement via a benzimidazole and benzoxazole was well tolerated (Fig. 7; Certal et al. [47]). Representative is benzoxazole 10, which has similar levels of activity and isoform selectivity compared to starting anilide 5. Cyclization of the aniline amide to indoline 11 gave a very potent and isoform selective PI3Kβ inhibitor [31]. The low aqueous solubility of 11 was hypothesized to be a result of strong crystal packing, confirmed by a high melting point of the crystalline solid (285°C). An analysis of the small molecule X-ray structure indicated intermolecular hydrogen bonding interactions between pyridone monomers as well as π-stacking of the indoline moieties. Blocking the intermolecular hydrogen bonds by introduction of an N-methyl within the pyridone resulted in a loss of activity without strong improvement of aqueous solubility. Indoline methyl substitution, however, in analogs 12 and 4 gave a dramatic improvement in solubility. The (S)-methyl enantiomer possessing the greater level of selectivity over the PI3Kδ isoform. Compound 4 was advanced based on its overall combination of activity, selectivity, solubility, permeability (Caco2 Papp = 48 × 10−6 cm/s), and microsomal stability (57% remaining after 20 min incubation with HLM).


Aniline modification through cyclization including benzoxazole 10 and indolines 4, 11 and 12
Compound 4 was shown to inhibit pAKT-S473 formation in vitro in a PC3 cell line with an IC50 = 49 nM. Pathway suppression and tumor growth inhibition were demonstrated in two PTEN-deficient xenograft models, PC3 and UACC-62.
Notably, this group also was able to solve a crystal structure of murine p110β in complex with 4 (Fig. 8) [31]. The binding mode of 4 was in good agreement with that hypothesized throughout the optimization process. The morpholine contacts the hinge-valine (residue 848). The pyrimidone carbonyl engages in a likely water-mediated interaction with Tyr833, whereas the “propeller” shape of the inhibitor directs the indoline under the p-loop in between Trp781 and Met773. Notably, the orientation of the Trp781 and Met773 residues differ substantially compared to the structure of p110β with pan-inhibitor GDC-0941 (pdb id: 2Y3A). The authors propose this flexibility in PI3Kβ and PI3Kδ allows for selectivity over the PI3Kα and PI3Kγ isoforms. They also hypothesize that the differences they observe in PI3Kβ over PI3Kδ activity can be attributed to a difference in primary structure on the kβ3 strand in this area: Lys771β vs. Thr750δ. Indeed, some of the divergent SAR against the PI3Kβ and PI3Kδ isoforms based on changes in the 4-position of the indoline [31] and aniline substitution of compound 5 above may be explainable by this difference in primary structure.


(a) Structure of SAR260301 (4) in p110β (pdb id: 4BFR). (b) Structure of pan-inhibitor GDC-0941 in p110β (pdb id: 2Y3A). Compound 4 occupies a hydrophobic cleft created by reorientation of the Trp781 and Met773 residues relative to the GDC-0941 structure
The results of the phase I dose escalation of 4 were disclosed at the 2015 ASCO annual meeting. A maximum tolerated dose was not reached and no objective responses were observed (3 of 21 patients were PTEN null, Bedard et al. [49]). The authors concluded that the rapid clearance observed for 4 was responsible for a lack of sustained pathway inhibition that was necessary to see anti-tumor activity in preclinical models.
4.2.2 GSK2636771, PI3Kβ
In 2011, GSK2636771 (13) entered a phase I dose escalation in patients with advanced solid tumors with PTEN-deficiencies. The structure and initial clinical data for 13 was disclosed in 2012 [50]; the structure is disclosed in a 2012 patent application (Qu et al. [32], Example 31) and subsequent combination method of use applications (Scheme 4).


TGX-221 (14) and GSK2636771 (13)
Medicinal chemistry was directed at ligand-based modification of TGX-221 (14). A series of publications from GSK detailed a number of different scaffolds possessing similar binding motifs (Scheme 5). Representative chemotypes included imidazopyrimidones (i.e., 15, Lin et al. [27]), triazolopyrimidinones (i.e., 16, Sanchez et al. [51]), pyrazolopyrimidines (i.e., 17, Yu et al. [52]), and thiazolopyrimidinones (i.e., 18, Lin et al. [53]).


Chemotypes published by GSK related to GSK2636771
Representative of all of these scaffolds, optimization to 15 was accomplished through a combination of SAR at the 1- and 2-positions of the imidazopyrimidinone core (Table 3). Dual ortho and meta substitution (R1 = Me, R2 = CF3) was preferred to achieve maximum inhibition of PI3Kβ (compare 19, 20 and 21). Notably, these modifications did not strongly influence the selectivity for PI3Kβ over PI3Kδ (five to eightfold selectivity). Imidazopyrimidinone 2-methyl substitution resulted in a modest improvement in potency against PI3Kβ and larger substitution such as ethyl was tolerated (compound 22).

Scientists at GSK used p110β homology models for structure based design and there aren’t any published crystal structures from within these chemical series. However, binding modes can be assumed based on those observed for TGX-221 and SAR260301 (Fig. 8). All 5,6-bicyclic heteroaromatic scaffolds possess morpholino substitution which presumably interact with the conserved hinge valine. The ring carbonyls likely make a water mediated interaction with the tyrosine in the back pocket. The arene substitution of these “propeller-shaped” inhibitors likely relies on the induced fit movement of the conserved methionine and tryptophan residues (akin to SAR260301).
GSK2636771 itself diverges from what is published in these manuscripts as it has a carboxylate substituent in replacement of the carbonyl present in TGX-221, 15, 16, 17, and 18. The patent application that exemplifies GSK2636771 has many examples where substitution at this position is changed including carboxylic acid derivatives and small aromatic heterocycles. It is likely that the carboxylate substitution results in displacement of a back pocket water molecule and results in direct interaction with the tyrosine and catalytic lysine residues.
Within this patent application, GSK2636771 (13) is reported to inhibit soft-agar growth of PC3 cells (PTEN-null) with an IC50 = 27 nM and promote tumor growth stasis in a PC3-xenograft model at doses ≥10 mg/kg.
Results from the phase I dose escalation were reported at ASCO in 2014 [54]. In this disclosure, 13 was reported to be a potent inhibitor of PI3Kβ (IC50 = 0.89 nM) with >900-fold selectivity over PI3Kα/γ and >10-fold selectivity over PI3Kδ. MTD was determined to be 500 mg and substantial pAKT suppression in surrogate tissue was noted. One patient had a RECIST PR and 13 patients had stable disease (53 patients reported to be treated).
4.2.3 AZD8186, PI3Kβ/δ
In 2013, AZD8186 (23) entered phase I dose escalation including patients with PTEN-deficient/mutated or PIK3CB mutated/amplified advanced solid malignancies tumors classified as PTEN-null or low. AZD8186 is reported to have good biochemical potency against the PI3K-β and -δ isoforms, reduced activity against the PI3K-α isoform and minimal activity against the PI3K-γ isoform. It is commonly referred to as a β/δ inhibitor as it is approximately 200-fold more potent in PTEN-null MDA-MB-468 cells compared to a PIK3CA-mt line, B7H4. Several sites at Astra Zeneca collaborated on a series of publications in 2014 describing medicinal chemistry leading to this unique selectivity profile [33, 55, 56].
AZD8186 (23) was generated by a combination of properties and structure guided-design that started from TGX-221 (14, Scheme 6). A related early compound, AZD6482, originally disclosed by some of the academic researchers that discovered TGX-221 [57], was progressed into clinical trials in healthy volunteers to assess influence on platelet activation. Although well tolerated, this compound was apparently discontinued as a result of significant concentration-dependent increase in plasma insulin as well as relatively short plasma half-life. Subsequently this compound was optimized to oral inhibitor AZD8186.


Chemical structures of TGX-221 (14), AZD6482 (24), and AZD8186 (23)
A crystal structure of compound 24 in PI3Kγ (pdb id: 4URK) indicates a predictable binding mode wherein the morpholine makes a single point interaction with the hinge valine and the 4-carbonyl interacts with the catalytic lysine. This compound adopts a “T-shape” with orthogonal orientation of the anthranilic acid. Compounds of this type likely derive isoform selectivity through the induced fit model discussed above (Fig. 8). Optimization from 24 is reported to have focused on improving the pharmacokinetic profile to allow once a day dosing as well as alterations in the overall isoform selectivity profile. Elimination of the carboxylate improved passive permeability, however, didn’t improve metabolic stability even though addressing an observation of UDP-mediated acyl glucuronide formation in human hepatocytes (compound 25, Scheme 7). The ADME properties of this series were improved through a series of LogD reductions. Amido substitution at the 7-position of the pyridopyrimidinone (i.e., compound 26) gave some improvement in human hepatocyte stability. Although poorly soluble, compound 26 was able to show some efficacy at high doses in nude mice implanted with a PC3 prostate tumor xenograft.


Replacement of the anthranilic acid and re-distribution of polarity led to analog 26 (Papp measured in a Caco-2 cell line)
Replacement of the pyridopyrimidinone core by a more polar variant was targeted to further reduce lipophilicity in order to improve solubility and metabolic stability with hopes of maintaining high potency. Although calculated to be more lipophilic, chromenone replacement of the pyridopyrimidinone resulted in a consistent decrease in Log D. A matched-pair analysis over 54 pairs showed an average Log D7.4 decrease of 0.69. Chromenone 23 (AZD8186) has improved metabolic stability in human hepatocytes (Clint < 4 μL/min/10−6 cells) and acceptable permeability (Papp = 8 × 10−6 cm/s). Compound 23 has moderate/high clearance and moderate/low bioavailability in mice and dogs. It was active in PTEN-null breast adenocarcinoma MDA-MB-463 cells (pAKT IC50 = 3 nM) and a cell-line sensitive to PI3Kδ inhibition (Jeco B cell pAKT IC50 = 17 nM). It was not active in a cell line sensitive to PI3Kα inhibition, B7H4 (PIK3CA mutant breast ductal carcinoma, pAKT IC50 = 752 nM). Pathway modulation and tumor growth inhibition was demonstrated in a PC3-xenograft model where ABT co-administration was necessary to achieve tumor stasis at 60 mg/kg of compound 23.
Clinical results for this inhibitor have not been disclosed.
4.3 PI3Kα Inhibitors
Selective inhibition of PI3Kα is anticipated to be desirable in patient populations with solid tumors that have activating PIK3CA (or PIK3R1) mutations. In this patient population, off-target inhibition of PI3Kβ, PI3Kδ, and PI3Kγ may cause unnecessary adverse events related to metabolic function, immune suppression, or activation. Avoiding adverse events related to inhibition of isoforms that do not contribute to efficacy is hypothesized to lead to greater efficacy compared to pan-inhibitors like pictilisib and buparlisib within this PIK3CA mutant population.
4.3.1 GDC0032 (Taselisib), PI3Kα/δ/γ
At the AACR in 2013, researchers at Genentech reported the structure and preclinical activity of GDC0032 (Olivero et al. [58], later given the generic name taselisib). GDC0032 (27) is a pan PI3K inhibitor with the exception that it substantially spares the PI3Kβ isoform. The desired profile at this stage of Genentech’s PI3K effort was to eliminate PI3Kβ activity with the hypothesis that resultant compounds would have decreased metabolic effects in patients. GDC0032 was discovered through a combination of structure and property guided optimization from high-throughput screening hit thienobenzopyran 28 [59]. Throughout optimization, starting from lipophilic lead compound 28, polarity was progressively increased and LLE improved (Scheme 8).


Chemical structures of thienobenzopyran HTS hit 28 and benzoxazepin GDC-0032 (27)
A positional scan of arene substituents of compound 28 identified a modest potency improvement with 2-chloroarene 29 substitution. Thienobenzopyran compounds such as 28 and 29 possess a doubly activated methylene believed likely susceptible to metabolism. In addition, many compounds made were found to be unstable presumably via hydrolytic ring opening at this position. Simple ring expansion to thienobenzoxepin 30 resulted in a ~5-fold increase in biochemical potency and solved chemical stability issues [59]. Metabolic stability determined through in vitro and in vivo experimentation was still poor for this chemical series (i.e., 30, HLM Clhep > 18 mL/min/kg, rat Clp = 60 mL/min/kg). Researchers hypothesized that cleavage of the aniline amide could be leading to instability and the concomitant production of potentially reactive/toxic metabolites. The hypothesis was supported by substantial turnover of most compounds from this series was observed in HLM in the absence of NADPH. The investigators sought to modify amide substitution, as in analogs 31 and 32, but potency was decreased substantially. Presumably this is a result of the unique cis-amide preferred small molecule conformation of N-methyl aniline amides (Scheme 9).


Ring expansion to thienobenzoxepin 30 and attempted aniline amide replacement
Isosteric replacement of the N-methyl amide with 5- and 6-membered heterocycles that would enforce the observed binding conformation was successful (Fig. 9; Staben et al. [60]). This replacement, in turn, allowed replacement of the arene with smaller alkyl substituents. A class effect was noted in this effort: greater aqueous solubility, passive permeability, and HLM stability for alkyl-substituted heterocyles compared to compounds possessing the N-methyl amide. For alkyl triazole 34, higher solubility and permeability improved bioavailability. A large improvement in rodent free clearance and free exposure on oral dosing was observed. A crystal structure of compound 34 in p110γ confirmed polar back pocket interactions of the triazole.


(a) Alkyl triazole 34 had improved ADME properties with maintained potency. (b) Crystal structure of compound 34 in p110γ (pdb id: 4HLE) [60]. The triazole makes polar contacts within the phosphate binding region including accepting an H-bond from catalytic Lys833
Additional efforts focused on improving selectivity over PI3Kβ and structural replacement of the thiophene (Scheme 10). Compound 34 had ~7-fold selectivity for PI3Kα over PI3Kβ and this could be improved through larger substitution at the 8-position of the tricyclic core [59, 61]. For example, compound 35 and 27 possess 8-(4-pyrazolyl) substitution and had 56- and 31-fold selectivity over PI3Kβ, respectively. Five-membered heteroarenes were designed to maintain appropriate bi-aryl dihedral with the 1-isopropyl-1,2,4-triazole. The replacement of the core thiophene with an imidazole gave inhibitors with the highest LLE and superior ADME properties; most notable was an improvement in free clearance that allowed for higher free exposure. This trend is observed across a series of matched pairs looking at direct replacement of the 5-membered ring [34]. This imidazole is a key feature in GDC0032 (compound 27).


Modifications from compound 34 targeted at improving selectivity over PI3Kβ and isosteric replacement of the thiophene
Selectivity over PI3Kβ was followed empirically throughout the optimization process as there was only access to crystal structures of compounds bound to PI3Kγ. Publication of crystal structures of all PI3K isoforms has allowed subsequent evolution of a model for observed selectivity for compounds like 27 and 35 [28]. A conserved tryptophan among the PI3K isoforms is predicted to be in close proximity to the 1-alkylpyrazole substituent. This tryptophan adopts a unique orientation in PI3Kβ which is hypothesized to be driven by steric insult of nearby residues. This conformer would result in greater steric clash with the 1-alkylpyrazole substituent.
Compound 27 inhibits pAKT and proliferation in an MCF7-neo/HER2 cell line at IC50 values equal to 4 and 25 nM, respectively. Maximal suppression of pAKT and tumor growth stasis is observed in an MCF7-neo/HER2-xenograft model at doses ≥5.8 mg/kg.
A notable feature of GDC0032 is its reported mutant selectivity [58, 62, 63]. While most PI3K inhibitors have increased anti-proliferative activity in PIK3CA-mutant cell lines due to the cell lines oncogene addition, GDC0032 uniquely is able to selectively inhibit the PI3K pathway and proliferation in PIK3CA-mutant lines compared to those that are pathway wild-type. In an isogenic matched set containing stable knock-in of wt-PI3Kα, H1047R-PI3Kα and E545K-PI3Kα (SW48 cells), GDC0032 selectively inhibits signaling and growth in the mutant lines. It is anticipated that this feature could improve the ability to provide efficacy in PIK3CA-mutant patients while minimizing wt-PI3Kα driven pharmacological effects.
In a phase I dose escalation that enrolled 34 patients, clinical partial responses were observed in 5 patients treated with doses of GDC-0032 ranging from 3 to 12 mg. GDC-0032 has a notable oral half-life of 40 h. Metabolic partial responses were observed via FDG-PET in 7 of 13 patients assessed [64]. A number of clinical trial are ongoing including “SANDPIPER,” a phase III study to examine the efficacy of taselisib in combination with fulvestrant in patients with ER-positive, HER2-negative mBC enriched for patients that have PIK3CA mutant tumors [65].
4.3.2 CH5132799, PI3Kα/β/γ
In 2011, researchers at Chugai disclosed the structure, in vitro activity, and preclinical pharmacology of clinical PI3Kα/γ selective inhibitor CH5132799 (36, Tanaka et al. [66]). CH5132799 (36) bears a 2-morpholino-dihydropyrrolopyrimidine skeleton that was designed by a ligand based approach combining attributes of PI103 (37) and an early Chiron/Novartis PI3K inhibitor (38) (Scheme 11; Tanaka et al. [66]; Owada et al. [35]). Compound 36 is reported to have a unique profile with 9-fold and 36-fold selectivity for PI3Kα over the PI3Kβ and PI3Kδ isoforms, respectively.


Lead structures used in ligand-based design leading to CH5132799 (36)
An early ligand based combination of PI103 and compound 37 was 2-morpholino-dihydropyrrolopyridine 39. Compound 39 is reported to have a PI3Kα IC50 = 8.6 nM (activity against other isoforms is not reported, Scheme 12). In vitro testing of phenolic compound 39 identified glucuronidation as a potential mechanism for clearance. Incubation of compound 39 with alamethicin-treated human or mouse liver microsomes with added UDPGA resulted in rapid turnover of compound 39 (T1/2 = 11 min, 6.1 min for human and mouse, respectively). The medicinal chemistry team triaged designs of phenolic isosteres using virtual docking in PI3Kγ and compounds were subsequently tested for activity in the PI3Kα biochemical assay. Many common isosteric replacements for phenols such as the indazole in compound 40 (PI3Kα IC50 > 50 μM) were not tolerated. A 5-(2-amino)-pyrimidine replacement of the phenol gave compound 41 with only moderate loss in potency. Notably, this compound was more resistant to glucuronidation and had greater overall metabolic stability when tested in human liver microsomes (HLM).


Isosteric replacement of the phenol of compound 39 to reduce glucuronidation
Further optimization was focused on dihydropyrrolopyrimidinyl 7-substitution to improve ADME properties. Notably, replacing typical heteroaryl substitution as in pyridine 41 with a methanesulfonyl (36, CH5132799, Kawada et al. [67]) reduced HLM clearance and improved bioavailability in mouse. CH5132799 (36) possesses very high selectivity over other PIK-family kinases, such as PI3K C2 α/β, VPS34 and mTOR. This compound has strong anti-proliferative activity in a range of PIK3CA-mutant cancer cells (KPL4 IC50 = 32 nM; T-47D IC50 = 56 nM; ME-180 IC50 = 140 nM). A combination of good PK properties and strong cellular potency in PIK3CA mutant cell lines led to strong efficacy at relatively low dose in a number of reported efficacy studies with xenograft-implanted mice [66]. For example, tumor regression was observed in mice containing a KPL-4 xenograft at doses greater than 1.6 mg/kg.
A crystal structure of CH5132799 has been reported in p110γ (PDB ID: 3APC, Fig. 10). The morpholine is in position to accept a hydrogen-bond from hinge Val882, whilst the aminopyrimidine is in proximity to both donate hydrogen bonds to α-C-helix Asp841 and Asp836 and accept a hydrogen-bond from the catalytic Lys833. Although not observed, there is likely a water molecule that bridges interaction between the remaining pyrimidine nitrogen and functional-gatekeeper Tyr867. This same key pentad of interactions has also been described for other classes of morpholine containing pan-PI3K inhibitors such as GDC-0980 and BKM-120. Given the lack of disclosed isoform selectivity SAR, as well as absence of a p110α crystal structure of CH5132799, hypotheses surrounding the structural basis of isoform selectivity over the PI3Kδ and PI3Kβ isoforms cannot be supported. However, the sulfonamide interestingly is directed toward the vicinity of differences in primary sequence among the isoforms (underneath the kβ3/4 strands and adjacent to the lower hinge).


X-ray structure of 36 in p110γ. The pyrimidine makes polar contacts with residues in the phosphate binding region. The sulfonamide is directed toward the solvent front
Data from a first-in-human study of CH5132799 was disclosed in 2014 [68]. Thirty-eight patients were treated in the dose escalation and an MTD of 48 mg BID was determined. A metabolic partial response was observed in five of seven patients assessed at MTD. Preliminary clinical activity was observed but no RECIST partial or complete responses were recorded.
4.3.3 AZD8835, PI3Kα/δ
In 2015 and 2016, researchers at Astra Zeneca disclosed the structure, in vitro activity and preclinical pharmacology of the clinical PI3K-α/δ inhibitor AZD8835 (compound 42, Barlaam et al. [69]; Hudson et al. [36]). AZD8835 (42, Scheme 13) is reported to be a single digit nM inhibitor of PI3Kα and δ while substantially sparing the β and γ isoforms.


Early lead compound 43 and clinical PI3K α/δ inhibitor AZD8835
Early lead compound 43 identified by Astra Zeneca researchers possessed good potency against PI3Kα and good levels of selectivity over PI3Kβ. However, containing a common hinge-binding aminopyrazine, these compounds were not particularly selective over the broad kinome [70].
Crystal structures of these ligands in PI3K isoforms have not been published, but the authors report docking studies in PI3Kα and a hypothesized binding mode. The aminopyrazine making a donor–acceptor interaction with the hinge (Val851 and Glu849), the pyrazine 3-substitutent directed toward the phosphate-binding region and the 5-substituent directed toward the solvent front in between the lower hinge and β3/4 strands. The authors hypothesize the 5-membered heteroarene is within proximity to make a single-point interaction with Gln859 (PI3Kα) which they believe provides their observed selectivity over PI3Kβ for the entire series.
The team pursued two strategies to improve selectivity over non-lipid kinases while maintaining a level of isoform selectivity over the PI3Kβ isoform: modification of the 5-membered heteroarene (pyrazole) and replacement of the benzotriazole. The former proved effective (Scheme 14). While early lead compound 43 inhibits 13 of 76 tested kinases at >80% when tested at 1 μM concentration, compound 44 (pyrazole replaced with a 1,2,4-triazole) is significantly more selective, inhibiting only 3 of 76 kinases at >60%. The authors provide a number of potential explanations for the improved kinase selectivity resultant from this 5-membered heterocycle modification. While designed to electronically repel with the hinge carbonyl residues in off-target kinases, the authors hypothesize insult of residues on the p-loop of off-target kinases and selective π-stacking with the PI3K-conserved tryptophan (Trp780, PI3Kα) may also contribute to this desirable property.


Kinase selectivity of compounds 43 and 44
AZD8835 (compound 42) was subsequently identified through simultaneous optimization potency and HLM stability within a related subseries of aminopyrazines containing a 1,3,4-oxadiazole at the 3-position (Table 4). Piperidine amide (R1), Triazole 1-subsitution (R2), and oxadiazole 5-substitution were explored. The authors noted larger triazole substituents, such as ethyl (46), improved potency (compare 45 to 46, Table 4). Amongst disclosed examples, human liver microsome stability seemed to roughly correlate with measured Log D7.4. Piperidine amide substitution was modified to decrease Log D and resulted in stable compound 42.

The reported biochemical isoform selectivity for AZD8835 has been supported by additional pharmacological assessment in cell based assays [36, 69]. This molecule is very active in cells sensitive to PI3Kα inhibition (i.e., BT7H4 pAKT IC50 = 57 nM) and PI3Kδ (i.e., Jeko-1 pAKT IC50 = 49 nM), but less active in PI3Kβ-sensitive PTEN null lines (i.e., MDA-MB-468) or PI3Kγ-sensitive lines (i.e. RAW264 cells). AZD8835 was shown to have good activity in PIK3CA-mutant xenograft models, such as BT474 where 93% TGI was observed dosing AZD8835 at 25 mg/kg BID. The authors also explored intermittent dose scheduling as a single agent or in combination [36].
AZD8835 entered a phase I clinical trial in patients with advanced solid tumors in 2014. Results from clinical studies have not been presented to date.
4.3.4 BYL-719 (NVP-BYL-719, Alpelisib), PI3Kα
In 2013, researchers at Novartis disclosed the structure and first-in-human clinical data PI3Kα-specific inhibitor BYL-719 (47, Gonzalez-Angulo et al. [71]). BYL-719 was generated from a medicinal chemistry campaign that spanned multiple Novartis sites starting from screening compound acylaminothiazole 48 (Scheme 15). The Novartis aminothiazoles, such as 42, were the first selective PI3Kα inhibitors reported and they were the first to publish a structure-based hypothesis for achieving PI3Kα selectivity (for a similar hypothesis, see also Heffron et al. [72]).


Optimization of aminothiazole 43 led to BYL-719 (42)
In a 2012 publication [73], Bruce et al. disclose a parallel chemistry optimization to make isoform selective tool compounds starting with non-selective screening hit 48. A reduction in TPSA and donor count was realized by replacement of the primary sulfonamide with a methyl sulfone, N-pyrazole, or methyl ketone resulting in analogs with improved cell potency as exemplified by compound 49 (Scheme 16). This analog has moderate potency against PI3Kα, but little selectivity over the δ or γ isoforms. Compound 49 had an IC50 of 0.32 μM in a neutrophil/B-cell respiratory burst assay (fMLP = f-Met-Leu-Phe).


Representative early inhibitor compound 44 and focused combinatorial library strategy
Novartis researchers hypothesized that modification near the solvent front of ligands such as 49 (binding mode discussed later) would alter isoform selectivity as this was a region of sequence diversity among the isoforms. They took a combinatorial approach to test this hypothesis. A library of 80 amines was reacted with aminothiazoles 50 to generate ureas 51 (Scheme 16). Trends in isoform selectivity were realized and the process repeated with more focused amine sets. An outcome of this effort was PI3Kα selective compound 52 (Scheme 17). Compound 52 is a single-digit nanomolar inhibitor of PI3Kα with >44-fold selectivity over the other isoforms. Also, it isn’t active against other PIK-family kinases like mTOR, DNA-PK, or Vps34. Quite notably this diversity approach resulted in selective inhibitors of not only PI3Kα but also the δ and γ isoforms [73].


Optimization of back pocket substitution leading to compound 47
An improvement in physical properties of compound 52 was made by removing of the 4-N-pyrazolobenzene and replacing with substituted pyridine and pyrimidine derivatives. The pyridyl 2-substitution occupies the hydrophobic side of the ribose pocket of p110α and these analogs maintained strong inhibition of PI3Kα and high selectivity (Scheme 17 [23]). Metabolite ID of compound 53 leant evidence to potential hydroxylation of one of the tert-butyl methyl groups. Replacement of one of the methyls with a trifluoromethyl gave BYL-719 (47). Compared to 53, 47 had lower turnover in rat liver microsomes and somewhat lower rat clearance in vivo. Compound 47 was tested in cell assays to confirm isoform selectivity. Compound 47 inhibited pAKT with IC50 values of 0.074 uM, 2.2 uM and 1.2 uM in Rat1 cells containing transfected Myr-p110α, β and δ, respectively (Myr = myristoylated).
The aminothiazole core both accepts and donates a hydrogen bond to hinge-residue Val851 (Fig. 11). Notably, there aren’t the typical significant polar interactions with back-pocket residues Asp810 or Lys802 observed for most PI3-kinase inhibitors. The trifluoromethyl substituent resides in a hydrophobic region of the ribose pocket and potentially interacts with Lys802 via nonclassical hydrogen bonds. Novartis researchers nicely rationalize that selective PI3Kα inhibition is accomplished through an isoform-specific interaction between the primary amide and Gln859. This residue deviates from glutamine in PI3Kβ (aspartate), in PI3Kδ (asparagine), and in PI3Kg (lysine). At the time of this review, this is the only reported structure-based hypothesis that rationalizes PI3Kα selectivity (similar to Heffron et al. [72]). In support of this hypothesis, a crystal structure of BYL-719 in PI3Kα details the proximity of the key primary amide to Gln859 (Fig. 11). A series of three hydrogen bonds are noted: the primary amide of BYL-719 accepts and donates a hydrogen bond to Gln859 as well as accepts a hydrogen bond from a backbone N-H from Ser854 of the lower hinge.


(a) X-ray structure of 47 in p110α (pdb id: 4JPS) [23]. (b) Expansion and schematic highlighting interactions of the primary amide with Gln859α
This structure-based rationale is backed-up by data for closely related analogs from these optimization disclosures (Table 5). Compound 54 containing the (S)-pyrrolidine carboxamide moiety present in BYL-719 shows good inhibition of the PI3Kα isoform and corresponding selectivity over PI3Kβ, δ and γ. When the carboxamide is completely absent as in pyrrolidine 55 a large drop in activity and isoform selectivity is observed. Similarly, secondary amide (S)-pyrrolidine N-methyl-carboxamide 56 is quite inactive and non-selective. Even inversion of the stereochemistry eliminated PI3Kα inhibition resulting in non-selective high micromolar inhibitor 57. It is indeed striking that this single residue at the solvent front of PI3Kα can be harnessed to achieve such high selectivity.

Thirty-six patients with advanced solid tumors carrying a somatic mutation of PIK3CA were dosed in the first-in-human study up to 450 mg/day [71]. The MTD for once daily dosing was determined to be 400 mg/day. Four confirmed partial responses were reported. At the time of writing, Alpelisib continues on in late stage clinical trials, recently initiating a pivotal PhIII trial in ER+ metastatic breast cancer in combination with letrozole. The PI3K community urgently awaits the results of this trial of the most advanced pure PI3Kα inhibitor.
4.3.5 INK1117 (MLN1117), PI3Kα
In 2011, researchers at Intellikine announced the discovery of INK1117 and entry in to phase I clinical trials. INK1117 was later renamed MLN1117 after Intellikine was acquired by Millenium (Takeda) in 2011. The structure of INK1117 has not been directly announced and there has not been a report of medicinal chemistry optimization leading to its discovery.
To the author’s knowledge, the reported PI3Kα isoform selectivity for INK1117 is the highest for any disclosed PI3K inhibitor. It is believed that INK1117 is exemplified in a 2011 patent application primarily disclosing a series of 5,6 and 6,5 heterobicyclic PI3K inhibitors [74]. “Compound 54” (58) from this patent application (Scheme 18) was latter the sole exemplification in a latter Intellikine patent application claiming a method for polymorph synthesis (58 referred to as Formula 1, Martin et al. [75]). It was also the sole exemplification in a latter Millenium patent application focused on pharmaceutical combinations comprising inhibitors of PI3Ks (58 referred to as “compound A,” Zoren et al. [76]). Within this application, clinical AE data for 35 patients treated with 58 was reported. Thus, 58 is a hypothesized but unconfirmed structure of INK1117 (MLN1117).


Reported data and hypothesized structure (compound 58) of INK1117
Although details have not been reported, INK1117 is reported to have high isoform selectivity as a result of structure based design. Similar imidazopyridines, such as compound 59, are known to bind to PI3K in a fashion where they accept (and in some cases donate) a hydrogen bond from the hinge valine whilst projecting 6-substitution to the polar back pocket that contains the catalytic lysine and αC-helix aspartate (pdb ID: 4KZC, Fig. 12). It is likely that compound 52 also binds through in a similar orientation, accepting a hydrogen bond from Val882α and projecting the aminobenzoxazole substituent to achieve hydrogen-bonding interactions with Lys802α and Asp810α. These hypothesized interactions are with residues conserved among the PI3K isoforms. The 3-amido-substitution of the imidazopyridine core is hypothesized to extend toward the solvent front between the lower hinge and β3/β4 strands. Although this is a region that has differences among the isoforms, it is difficult to propose a rationale for the striking isoform selectivity reported. Modelling into the active site suggests a potential single point interaction between the morpholine oxygen and α-specific Gln859.


X-ray structure of 59 in p110γ and schematic representation of the hypothesized binding mode of compound 58
It is interesting that inhibitors from the 2011 patent application that are unlikely to encroach regions of differences in primary structure possess qualitative differences in isoform selectivity. For example, structurally related quinolone 60 (example 205, Ren et al. [74]) is reported to also have very high selectivity for PI3Kα.
MLN1117 is currently in phase I clinical trials and the results from the first-in-human dose escalation and regimen frequency study in patients with advanced solid malignancies were reported in 2015 [37]. At the time of report, 76 patients had enrolled and 4 confirmed partial responses (RECIST) were observed.
5 Conclusion and Perspective
The past 15+ years of research directed at the discovery of small-molecule inhibitors of PI3Ks and 9 years of clinical research has resulted in great advancements in our understanding of the therapeutic potential of these targets. Early on, a majority of pharmaceutical companies focused on the discovery and development of pan-PI3K or multi-PIK family inhibitors for solid tumors. Although some would argue the modest clinical success of these pan-inhibitors has not justified the enormous resource investment, clinical results have supported proof-of-efficacy and provided hypotheses for maximizing therapeutic benefits of PI3K inhibition. Pan-PI3K inhibitors may not provide sufficient efficacy/safety in all situations and there is a theoretical advantage to isoform-selective inhibition of PI3K. Inhibition of PI3K isoforms that don’t contribute strongly to efficacy within a given population can lead to undesirable pharmacology/toxicity. Indeed, the extent and duration of the oncogenic driver, and subsequent clinical efficacy, could be limited by toxicity driven by a lack of selectivity. Optimism remains in the form of isoform selective inhibitors. Clinical success in treating solid tumors with PI3K inhibitors will likely be a result of maximizing therapeutic index through this selectivity for oncogenic drivers in diagnostically driven patient populations and appropriate combination therapies.
Pharmaceutical research teams have now provided the tools to test these hypotheses. The outstanding, transformative clinical efficacy of PI3Kδ-specific inhibitors such as Idelalisib against hematological malignancies has proven the validity of isoform-specific inhibition. Early clinical efficacy of PI3Kβ and PI3Kα isoform selective inhibitors observed in first-in-human dose escalation studies has been promising. Ongoing, pivotal, clinical trials in combination with SOC agents will determine if there is advantage to PI3Kα-selective or PI3Kβ-selective inhibitors in patients with solid tumors over their pan-PI3K predecessors.
Abbreviations
- AE:
-
Adverse event
- AI:
-
Aromatase inhibitor
- AKT:
-
Protein kinase B
- AUC:
-
Area under the curve
- CaCo:
-
Heterogeneous human epithelial colorectal adenocarcinoma cells
- CI:
-
Confidence interval
- Clhep:
-
Predicted hepatic clearance
- Clint:
-
Intrinsic clearance
- CLL:
-
Chronic lymphocytic leukemia
- Clp:
-
Plasma clearance
- DNA-PK:
-
DNA-dependent protein kinase
- ER:
-
Estrogen receptor
- FDG-PET:
-
Fluorodeoxyglucose positron emission tomography
- FL:
-
Follicular lymphoma
- HEP:
-
Hepatocytes
- HER2:
-
Receptor tyrosine protein kinase ErbB2
- HR:
-
Hazard ratio
- HR+:
-
Hormone receptor positive
- HTS:
-
High throughput screen
- LLE:
-
Lipophilic ligand efficiency
- LM:
-
Liver microsomes (H, human; R, rat; M, mouse)
- MDCK:
-
Madin-Darby canine kidney cells
- mPFS:
-
Median progression free survival
- MTD:
-
Maximum tolerated dose
- mTOR:
-
Mechanistic target of rapamycin
- ORR:
-
Overall response rate
- pAKT:
-
Phosphorylated protein kinase B
- Papp:
-
Apparent passive permeability
- PI3K:
-
Phosphatidylinositide-3-kinase
- PIK:
-
Phosphoinositide kinases
- PIP2:
-
Phosphatidylinositol-4,5-bisphosphonate
- PIP3:
-
Phosphatidylinositol (3,4,5)-triphosphate
- p-loop:
-
Phosphate-binding loop (glycine-rich loop)
- PR:
-
Partial response
- pRTK:
-
Phosphorylated receptor tyrosine protein kinase
- PTEN:
-
Phosphatase and tensin homologue
- Ras:
-
Rat sarcoma
- RTK:
-
Receptor tyrosine protein kinase
- SH2:
-
Src homology domain
- SLL:
-
Small lymphocytic lymphoma
- TPSA:
-
Topological polar surface area
- UDP:
-
Uridine diphosphate
- UDGPA:
-
Uridine 5′-diphosphoglucuronic acid
- VPS34:
-
Vacuolar sorting protein-34
References
Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD (2001) Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis and cancer. Annu Rev Cell Dev Biol 17:615–675
Engleman JA, Luo J, Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619
Thorpe LM, Yuzugullu H, Zhao JJ (2015) PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 15:7–24
Samuels Y, Wang Z, Bardelli A (2004) High frequency of mutations of the PIK3CA gene in human cancer. Science:304–554
Cizkova M, Susini A, Vacher S, Cizeron-Clairac G, Andrieu C, Driouch K, Fourme E, Lidereau R, Bieche I (2012) PIK3CA mutation impact on survival in breast cancer patients and in ERα, PR and ERBB2-based subgroups. Breast Cancer Res 14:R28
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10:143–153
Krop I, Johnston S, Mayer IA, Dickler M, Ganju V, Forero-Torres A, Melichar B, Morales S, de Boer R, Gendreau S, Dernck M, Lackner M, Spoerke J, Yeh R, Levy G, Ng V, O’Brien C, Savage H, Xiao Y, Wilson T, Lee SC, Petrakova K, Vallentin S, Yardley D, Ellis M, Piccart M, Perez EA, Winer E, Schmid P (2014) The FERGI phase II study of the PI3K inhibitor pictilisib (GDC-0941) plus fulvestrant vs fulvestrant plus placebo in patients with ER+, aromatase inhibitor (AI)-resistant advanced or metastatic breast cancer – Part 1. Results. In: San Antonio Breast Cancer Society annual meeting 2014, Abstract S2-02
Baselga J, Im S, Iwata H, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng L, Hurvits S, Masuda N, Cortés J, De Laurentiis M, Arteaga CL, Jiang Z, Jonat W, Hachemi S, Le Maouhaer S, Di Tomaso E, Urban P, Massacesi C, Campone M (2015) PIK3CA status in circulating tumor DNA (ctDNA) predicts efficacy of buparlisib (BUP) plus fulvestrant (FULV) in postmenopausal women with endocrine-resistant HR+/HER2− advanced breast cancer (BC): first results from the randomized, phase III BELLE-2 trial. In: San Antonia Breast Cancer Society annual meeting 2015, Abstract S6-01
Chia S, Gandhi S, Joy AA, Edwards S, Gorr M, Hopkins S, Kondejewski J, Ayoub JP, Califaretti N, Rayson D, Dent SF (2015) Novel agents and associated toxicities of inhibitors of the PI3K/AKT/mTOR pathway for the treatment of breast cancer. Curr Oncol 22:33–48
Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL (1999) Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110α subunit of PI3K-kinase. J Biol Chem 274:10963–10968
Bi L, Okabe I, Bernard DJ, Nussbaum RL (2002) Early embryonic lethality in mice deficient in the p110β catalytic subunit of PI3-kinase. Mamm Genome 13:169–172
Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, Sancho S, Smith AJH, Withers DJ, Vanhaesebroeck B (2006) Critical role for the p110α phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature 441:366–370
Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, Humphries LA, Rawlings D, Reynolds H, Vigorito E, Turner M (2002) A crucial role for the p110δ subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med 196:753–763
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287:1040–1046
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJH, Vanhaesebroeck B (2002) Impaired B and T cell antigen receptor signaling in p110δ PI 3-kinase mutant mice. Science 297:1031–1034
Walker EH, Perisic O, Ried C, Stephens L, Williams RL (1999) Structural insights into phosphoinositide 3-kinase catalysis and signaling. Nature 402:313–320
Walker EH, Pacold ME, Perisic O, Stephans L, Hawkins PT, Wymann MP, Williams RL (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6:909–919
Huang C-H, Mandelker D, Schmidt-Kittler O, Samules Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabeli SB, Amzel LM (2007) The structure of human p110α/p85α complex elucidates the effects of oncogenic PI3Kα mutations. Science 318:1744–1748
Zhao Y, Zhang X, Lu S, Peng Y, Wang X, Guo C, Zhang J, Luo Y, Shen Q, Ding J, Meng L, Zhang J (2014) Crystal structures of PI3Kα complexed with PI103 and its derivatives: new directions for inhibitors design. ACS Med Chem Lett 5:138–142
Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM (2009) A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A 106:16996–17001
Zhang X, Vadas O, Perisic O, Anderson KE, Clark J, Hawkins PT, Stephens LR, Williams RL (2011) Structure of lipid kinase p110β/p85β elucidates and unusual SH2-domain-mediated inhibitory mechanism. Mol Cell 41:567–578
Berndt A, Miller S, Williams O, Lee DD, Housman BT, Pacold JI, Gorrec F, Hon W-C, Liu Y, Rommel C, Gaillard P, Ruckle T, Schwarz MK, Shokat KM, Shaw JP, Williams RL (2010) The p110 delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors. Nat Chem Biol 6:117–124
Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, Hamon J, Fabbro D, Caravatti G (2013) Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett 23:3741–3748. PDB-id 4JPS. The p85 iSH2 domain and small isothiocyanate fragment present in this structure have been removed for presentation clarity
Cully M, You H, Levine AJ, Mak TW (2006) Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 6:184–192
Safina BS et al. (2012) Discovery of novel PI3-kinase-delta specific inhibitors for the treatment of rheumatoid arthritis: taming CYP3A4 time-dependent inhibition. J Med Chem 55:5887–5900
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJA, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ (2008) The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 51:5522–5532
Lin H, Shulz MJ, Xie R, Zeng J, Luengo JI, Squire MD, Tedesco R, Qu J, Erhard K, Mack JF, Raha K, Plant R, Rominger CM, Ariazi JL, Sherk CS, Schaber MD, McSurdy-Freed J, Spengler MD, Davis CB, Hardwicke MA, Rivero RA (2012) Rational design, synthesis and SAR of a novel thiazolopyrimidinone series of selective PI3K-beta inhibitors. ACS Med Chem Lett 3:524–529
Staben ST, Ndubaku C, Blaquiere N, Belvin M, Bull RJ, Dudley D, Edgar K, Gray D, Heald R, Heffron TP, Jones GE, Jones M, Kolesnikov A, Lee L, Lesnick J, Lewis C, Murray J, McLean NJ, Nonomiya J, Olivero AG, Ord R, Pang J, Price S, Prior W, Rouge L, Salphati L, Sampath D, Wallin J, Wang L, Wei B, Wiesmann C, Wu P (2013) Discovery of thiazolobenzoxepin PI3-kinase inhibitors that spare the PI3-kinase B isoform. Bioorg Med Chem Lett 23:2606–2613
Somoza JR, Koditek D, Villasenor AG, Novikov N, Wong MH, Liclican A, Xing W, Lagpacan L, Wang R, Schultz BE, Papalia GA, Samuel D, Lad L, McGrath ME (2015) Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase δ. J Biol Chem 290:8439–8446
Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, Pink MM, Proctor JL, Lussier J, Martin CM, Hoyt JG, Tillotson B, Murphy EL, Lim AR, Thomas BD, MacDougall JR, Ren P, Liu Y, Li L, Jenssen KA, Fritz CC, Dunbar JL, Porter JR, Rommel C, Palombella VJ, Changelian PS, Kutok JL (2013) PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol 20:1364–1374
Certal V, Carry J, Halley F, Virone-Oddos A, Thompson F, Filoche-Rommé B, El-Ahmad Y, Karlsson A, Charrier V, Delorme C, Rak A, Abecassis P, Amara C, Vincent L, Bonnevaux H, Nicolas J, Mathieu M, Bertrand T, Marguette J, Michot N, Benard T, Perrin M, Lemaitre O, Guerif S, Perron S, Monget S, Gruss-Leleu F, Doerflinger G, Guizani H, Brollo M, Delbarre L, Bertin L, Richepin P, Loyau V, Garcia-Echeverria C, Lengauer C, Schio L (2014) Discovery and optimization of pyrimdone indoline amide PI3Kβ inhibitors for the treatment of Phosphatase and Tensin Homologue (PTEN)-deficient cancers. J Med Chem 57:903–920
Qu J, Rivero R, Sanchez R, Tedesco R. Benzimidazole derivatives as PI3 kinase inhibitors. WO2012047538
Barlaam B, Cosulich S, Degorce S, Fitzek M, Green S, Hancox U, Labert-van der Brempt C, Lohmann J, Maudet M, Morgantin R, Pasquet M, Péru A, Plé P, Saleh T, Vautier M, Walker M, Ward L, Warin N (2015) Discovery of (R)-8-(1-(3,5-difluorophenylamino)ethyl)-N,N-dimethyl-2-morpholinio-4-oxo-4H-chromen-6-carboxamide (AZD8186): a potent and selective inhibitor of PI3Kβ and PI3Kδ for the treatment of PTEN-deficient cancers. J Med Chem 58:943–962
Ndubaku C, Heffron TP, Staben ST, Baumgardner M, Blaquiere N, Bradley E, Bull R, Do S, Dotson J, Dudley D, Edgar KA, Friedman LS, Goldsmith R, Heald RA, Kolesnikov A, Lee L, Lewis C, Nannini M, Nonomiya J, Pang J, Price S, Prior W, Salphati L, Sideris S, Wallin JJ, Wang L, Wei B, Sampath D, Olivero AG (2013) Discovery of 2-{3-[2-(1-isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): a β-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor. J Med Chem 56:4597–4610
Ohwada J, Ebiike H, Kawada H, Tsukazaki M, Nakamura M, Miyazaki T, Morikami K, Yoshinari K, Yoshida M, Kondoh O, Kuramoto S, Ogawa K, Aoki Y, Shimma N (2011) Discovery and biological activity of a novel class I PI3K inhibitor, CH5132799. Bioorg Med Chem Lett 21:1767–1772
Hudson K, Hancox UJ, Trigwell C, McEwen R, Polanska UM, Nikolaou M, Gutierrez PM, Avivar-Valderas A, Delpuech O, Dudley P, Hanson L, Ellston R, Jones A, Cuberbatch M, Cosulich SC, Ward L, Cruzalegui F, Green S (2016) Intermittent high-dose scheduling of AZD8835, a novel selective inhibitor of PI3Kα and PI3Kδ, demonstrates treatment strategies for PIK3CA-dependent breast cancers. Mol Cancer Ther. doi:10.1158/1535-7163.MCT-15-0687
Juric D, De Bono JS, LoRusso P, Nemunaitis JJ, Heath EI, Kwak EL, Macarulla T, Geuna E, Luken MJM, Patel C, Kuida K, Sankoh S, Zohren F, Shou Y, Tabernero J (2015) First-in-human, phase I, dose-escalation study of selective PI3Kα isoform inhibitor MLN1117 in patients (pts) with advanced solid malignancies. In: ASCO annual meeting 2015, Abstract 2501
Fowler KW, Huang D, Kesicki EA, Ooi HC, Oliver AR, Ruan F, Treiberg J, Puri KD. Quinazolinones as inhibitors of human phosphatidylinositol 3-kinase delta. WO2005113556
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Path FRC, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn (2014) Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 370:997–1007
Ren P, Liu Y, Wilson TE, Li L, Chan K, Rommel C. Substituted isoquinolin-1(2H)-ones, and methods of use thereof. US20090312319
Okkenhaug K (2013) Two birds with one stone: dual p110δ and p110γ inhibition. Chem Biol 20:1309–1310
Balakrishnan K, Peluso M, Fu M, Rosin NY, Burger JA, Wierda WG, Keating MJ, Faia K, O’Brien S, Kutok JL, Gandhi V (2015) The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia 29:1811–1822
Edgar KA, Wallin JJ, Berry M, Lee LB, Prior WW, Sampath D, Friedman LS, Belvin M (2010) Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res 70:1164–1172
Wee S, Wiederschain D, Maira S-M, Loo A, Miller C, de Beaumont R, Stegmeir F, Yao Y-M, Lengauer C (2008) PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 105:13057–13062
Song MS, Salamena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13:283–296
Parsons R (2004) Human cancer, PTEN and the PI-3 kinase pathway. Semin Cell Dev Biol 15:171–176
Certal V, Halley F, Virone-Oddos A, Thompson F, Filoche-Rommé B, El-Ahmad Y, Carry J, Delorme C, Karlsson A, Abecassis P, Vincent L, Bonnevaux H, Nicolas J, Morales R, Michot N, Vade I, Louboutin A, Perron S, Doerlinger G, Tric B, Monget S, Lengauer C, Schio L (2012) Preparation and optimization of new 4-(morpholin-4-yl)-(6-oxo-1-6-dihydropyrimidin-2-yl)amide derivatives as PI3Kβ inhibitors. Bioorg Med Chem Lett 23:6381–6384
Certal V, Halley F, Virone-Oddos A, Delorme C, Karlsson A, Rak A, Thompson F, Filoche-Rommé B, El-Ahmad Y, Carry J, Abecassis P, Lejeune P, Vincent L, Bonnevaux H, Nicolas J, Bertrand T, Marquette J, Michot N, Benard T, Below P, Vade T, Chatreaux F, Lebourg G, Pilorge F, Angouillant-Boniface O, Louboutin A, Langauer C, Schio L (2012) Discovery and optimization of new benzimidazole- and benzoxazole-pyrimidone selective PI3Kβ inhibitors for the treatment of phosphatase and TENsin homology (PTEN)-deficient cancers. J Med Chem 55:4788–4805
Bedard PL, Davies MA, Kopetz S, Flaherty KT, Shapiro G, Luke JJ, Spreafico A, Wu B, Gomex C, Cartot-Cotton S, Mazuir F, Micallef S, Demers B, Juric D, Margaret P (2015) First-in-human phase I trial of the PI3Kb-selective inhibitor SAR260301 in patients with advanced solid tumors. In: ASCO annual meeting 2015, Abstract 2564
Blackman SC, Gainer SD, Suttle BB, Skordos KW, Greshock JD, Motwani M, Roadcap LT, Hardwicke MA, Wooster RF (2012) Abstract 1752: a phase I/IIa, first time in human, open-label dose-escalation study of GSK2636771 in subjects with advanced solid tumors with PTEN deficiency. In: AACR 103rd annual meeting 2012
Sanchez RM, Erhard K, Hardwicke MA, Lin H, McSurdy-Freed J, Plant R, Raha K, Rominger CM, Schaber MD, Spengler MD, Moore ML, Yu H, Luengo JI, Tedesco R, Rivero RA (2012) Synthesis and structure-activity relationships of 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones as novel series of potent β isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg Med Chem Lett 22:3198–3202
Yu H, Moore ML, Erhard K, Hardwicke MA, Lin H, Luengo JI, McSurdy-Freed J, Plant R, Qu J, Raha K, Rominger CM, Schaber MD, Spengler MD, Rivero RA (2013) [3a,4]-dihydropyrazolo[1,5a]pyrimidines: novel, potent, and selective phophatidylinositol-3-kinase β inhibitors. ACS Med Chem Lett 4:230–234
Lin H, Erhard K, Hardwicke AA, Luengo JI, Mack JF, McSurdy-Freed J, Plant R, Raha K, Rominger CM, Sanchez RM, Schaber MD, Shulz MJ, Spengler MD, Tedesco R, Xsie R, Zeng JJ, Rivero RA (2012) Synthesis and structure-activity relationships of imidazo[1,2-a]pyrimidin-5-(1H)-ones as a novel series of beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg Med Chem Lett 22:2230–2234
Arkenau H-T, Mateo J, Lemech CR, Infante JR, Burris HA, Bang Y-J, Eder JP, Herbst RS, Sharma S, Chung HC, Decordova S, Swales KE, Garrett MD, Loftiss JI, Durante M, Russo MW, Suttle BB, Motwani M, Kumar R, De Bono JS (2014) A phase I/II, first-in-human dose-escalation study of GSK2636771 in patients (pts) with PTEN-deficient advanced tumors. In: 2014 ASCO annual meeting, Abstract 2514
Barlaam B, Cosulich S, Degorce S, Fitzek M, Giordanetto F, Green S, Inghardt T, Hennequin L, Hancox U, Lambert-van der Brempt C, Morgentin R, Pass S, Plé P, Saleh T, Ward L (2014) Discovery of 9-(1-anilinoethyl)-2-morpholino-4-oxo-pyrido[1,2-a]pyrimidine-7-carboxamides as PI3Kβ/δ inhibitors for the treatment of PTEN-deficient tumours. Bioorg Med Chem Lett 24:3928–3935
Giordanetto F, Barlaam B, Berglund S, Edman K, Karlsson O, Lindberg J, Nylander S, Inghardt T (2014) Discovery of 9-(1-phenoxyethyl)-2-morpholino-4-oxo-pyrido[1,1-a]pyrimidine-7-carboxamides as oral PI3Kβ inhibitors, useful as antiplatelet agents. Bioorg Med Chem Lett 24:2936–3943
Kim S, Mangin P, Dangelmaier C, Lillian R, Jackson SP, Daniel JL, Kunapuli SP (2009) Role of phosphoinositide 3-kinase β in glycoprotein VI-mediated Akt activation in platelets. J Biol Chem 284:33763–33772
Olivero A et al (2013) Discovery of GDC-0032: a beta-sparing PI3K inhibitor active against PIK3CA mutant tumors. In: AACR 2013, Washington, 6–10 Apr 2013, Abstract DDT02-01
Staben ST, Siu M, Goldsmith R, Olivero AG, Do S, Burdick DJ, Heffron TP, Dotson J, Sutherlin DP, Zhu B-Y, Tsui V, Le H, Lee L, Lesnick J, Lewis C, Murray JM, Nonomiya J, Pang J, Prior WW, Salphati L, Rouge L, Sampath D, Sideris S, Wiesmann C, Wu P (2011) Structure-based design of thienobenzoxepin inhibitors of PI3K. Bioorg Med Chem Lett 21:4054–4058
Staben ST, Blaquiere N, Tsui V, Kolesnikov A, Do S, Bradley EK, Dotson J, Goldsmith R, Heffron TP, Lesnick J, Lewis C, Murray J, Nonomiya J, Olivero AG, Pang J, Rouge L, Salphati L, Wei B, Wiesmann C, Wu P (2013) Cis-amide isosteric replacement in thienobenzoxepin inhibitors of PI3-kinase. Bioorg Med Chem Lett 23:897–901
Heffron TP, Wei B, Olivero A, Staben ST, Tsui V, Do S, Dotson J, Folkes A, Goldsmith P, Goldsmith P, Gunzner J, Lesnick J, Lewis C, Mathieu S, Nonomiya J, Shuttleworth S, Sutherlin DP, Wan NC, Wang S, Wiesmann C, Zhu B-Y (2011) Rational design of phosphoinositide 3-kinase alpha inhibitors that exhibit selectivity over the phosphoinositide 3-kinase beta isoform. J Med Chem 54:7815–7833
Belvin M, Friedman L, Sampath D, Wallin J. Mutant selectivity and combinations of a phosphoinositide 3 kinase inhibitor compound and chemotherapeutic agents for the treatment of cancer. WO2013182668
Edgar KA, Nannini M, Hong R, Eigenbrot C, Schmidt S, Young A, Sampath D, Wallin JJ, Friedman LS (2015) Characterization of the enhanced potency of PI3K inhibitor taselisib (GDC0032) in PI3K mutant cell lines and models. In: AACR 106th annual meeting 2015, Abstract 2672
Juric D, Krop I, Ramanathan RK, Xiao J, Sanabria S, Wilson TR, Choi Y, Parmar H, Hsu J, Baselga J, Von Hoff DD (2013) Abstract LB-64: GDC-0032, a beta isoform-sparing PI3K inhibitor: results of a first-in-human phase Ia dose escalation study. In: AACR annual meeting 2013
Baselga J, Cortes J, De Laurenitis M, Diéras V, Harbeck N, Im Y, Jacot W, Krop IE, Verma S, Wilson TR, Lin R, Schimmoller F, Hsu JY (2015) SANDPIPER: phase III study of the PI3-kinase (PI3K) inhibitor taselisib (GDC-0032) plus fulvestrant in patients (pts) with estrogen receptor (ER)-positive, HER2-negative locally advanced or metastatic breast cancer (BC) enriched for pts with PIK3CA mutant tumors. In: ASCO annual meeting 2015, Abstract TPS629
Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O, Aoki Y (2011) The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res 17:3272–3281
Kawada H, Ebiike H, Tsukazaki M, Nakamura M, Morikami K, Yoshinari K, Yoshida M, Ogawa K, Shimma N, Tsukuda T, Ohwada J (2013) Lead optimization of a dihydropyrrolopyrimidine inhibitor against phosphoinositide 3-kinase (PI3K) to improve the phenol glucuronic acid conjugation. Bioorg Med Chem Lett 23:673–678
Blagden S, Omlin A, Josephs D, Stavraka C, Zivi A, Pinato DJ, Anthoney A, Decordova S, Swales K, Riisnaes R, Pope L, Noguchi K, Shiokawa R, Inatani M, Prince J, Jones K, Twelves C, Spicer J, Banerji U (2014) First-in-human study of CH5132799, an oral class I PI3K inhibitor, studying toxicity, pharmacokinetics and pharmacodynamics in patients with metastatic cancer. Clin Cancer Res 20:5908–5917
Barlaam B, Cosulich S, Delouvrié B, Ellston R, Fitzek M, Germain H, Green S, Hancox U, Harris CS, Hudson K, Labert-van der Brempt C, Lebraud H, Magnien F, Lamorlette M, Le Griffon A, Morgentin R, Ouvry G, Page K, Pasquet G, Polanska U, Ruston L, Saleh T, Vautier M, Ward L (2015) Discovery of 1-(4-(5-(5-amino-6-(5-tert-butyl-1,3,4-oxadiazol-2-yl) pyrazin-2-yl)-1-ethyl-1,2,4-triazol-3-yl)piperidin-1-yl)-3- hydroxypropan-1-one (AZD8835): a potent and selective inhibitor of PI3Kα and PI3Kδ for the treatment of cancers. Bioorg Med Chem Lett 25:5155–5162
Barlaam B, Cosulich S, Fitzek M, Green S, Harris CS, Hudson K, Lambert-van der Brempt C, Ouvry G, Page K, Ruston L, Ward L, Delouvrié B (2015) Design of selective PI3Kα inhibitors starting from a promiscuous pan kinase scaffold. Bioorg Med Chem Lett 25:2679–2685
Gonzalez-Angulo AM, Juric D, Argilés G, Schellens JHM, Burris HA, Berlin J, Middleton MR, Shuler MH, Van Geel R, Helgason T, Bootle D, Boehm M, Goggin TK, Demanse D, Quadt C, Baselga J (2013) Safety, pharmacokinetics, and preliminary activity of the α-specific PI3K inhibitor BYL719: results from the first-in-human study. In: ASCO annual meeting 2013, Abstract 2531
Heffron TP, Heald RA, Ndubaku C, Wei B, Augistin M, Do S, Edgar K, Eigenrot C, Friedman L, Gancia E, Jackson PS, Jones G, Kolesnikov A, Lee LB, Lesnick JD, Lewis C, McLean N, Mörtl M, Nonomiya J, Pang J, Price S, Prior WW, Salphati L, Sideris S, Staben ST, Steinbacher S, Tsui V, Wallin J, Sampath D, Olivero AG (2016) The rational design of selective benzoxazepin inhibitors of the α-isoform of phosphoinositide 3-kinase culminating in the identification of (S)-2-((2-(1-isopropyl-1H-1,2,4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxepin-9-yl)oxy)propanamide (GDC-0326). J Med Chem 59:985–1002
Bruce I, Akhlaq M, Bloomfield GC, Budd E, Cox B, Cuenoud B, Finan P, Gedeck P, Hatto J, Hayler JF, Head D, Keller T, Kirman L, Leblanc C, Le Grand D, McCarthy C, O’Connor D, Owen C, Oza MS, Pilgrim G, Press NE, Sviridenko L, Whitehead L (2012) Development of isoform selective PI3-kinase inhibitors as pharmacological tools for elucidating the PI3K pathway. Bioorg Med Chem Lett 22:5445–5550
Ren P, Liu Y, Li L, Chan K, Wilson TE, Cambell SF. Heterocyclic compounds and uses thereof. WO 2011022439. Only qualitative PI3K isoform inhibition data is reported in this patent application for select compounds
Martin M, Worrall CP, Gancedo SD, Ren P. Kinase inhibitor polymorphs. WO2013071272
Zohren F, Patel C. Enhanced treatment regimens using PI3Kα inhibitors. WO2015051193
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Staben, S.T. (2017). Isoform Selective PI3K Inhibitors for Treating Cancer. In: Waring, M.J. (eds) Cancer II. Topics in Medicinal Chemistry, vol 28. Springer, Cham. https://doi.org/10.1007/7355_2016_27
Download citation
DOI: https://doi.org/10.1007/7355_2016_27
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75924-1
Online ISBN: 978-3-319-75926-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)