
Abstract
Although comprising only 1% of all brain tumors, optic pathway glioma (OPG) is the most common optic pathway tumor. While many cases are found in young children, 20–40% are found in patients ≥15 years. Neurofibromatosis type 1 (NF1) is a well-known risk factor, with about 15–20% with NF1 developing OPG which tend to be more indolent than the non-NF1 OPG. Most are pilocytic astrocytomas, but other types of low-grade and malignant gliomas have been reported. The role of surgery is to obtain tissue diagnosis or resection in the patient with glioma confined just to the optic nerve and complete visual loss. Chemotherapy consisting of carboplatin and vincristine is a popular treatment approach to delay radiotherapy (RT) in children <10 years and preserve neurocognitive function. In NF1 patients, RT is often avoided because of the higher chance of developing vascular and neoplastic events. RT is the mainstay of treatment for non-NF1 patients who are ≥10 years. The 10-year overall survival and progression-free survival rates range from 80% to 95% and 70% to 85%, respectively, with RT. In addition, stabilization and improvement in vision have been observed in more than 70% of patients receiving RT.
Similar content being viewed by others


Keywords
- Optic pathway glioma
- Hypothalamic chiasmatic glioma
- Radiotherapy
- Surgery
- Chemotherapy
- Observation
- Neurofibromatosis type 1
Learning Objectives
-
To determine the most common histology, risk factors, and clinical presentation associated with optic pathway glioma.
-
To define the roles of observation, surgery, chemotherapy, and radiotherapy in the management of optic pathway glioma.
-
To describe the results of treatment, including survival, progression-free survival, and visual preservation outcomes with the different treatment modalities.
-
To determine the risk of complications with the use of radiotherapy as well as tolerance doses of the surrounding normal organs in the treatment of optic pathway glioma.
Epidemiology
Optic pathway gliomas account for about two thirds of all primary optic pathway tumors [1]. Optic pathway glioma (OPG) is a relatively rare tumor, accounting for 1% of all intracranial tumors . In children, OPG comprises about 5% of all brain tumors. These tumors usually present during the first 7 years of life, with about 90% of cases diagnosed within the first two decades of life [2]. Males and females are equally affected. In adults, OPG can be divided into two groups . The first includes patients who were diagnosed as children and followed through adulthood. The second group includes patients diagnosed with OPG during adulthood.
The optic pathway includes the retina, optic nerve, optic chiasm, optic radiations, and occipital lobe. When OPG is diagnosed, it is often classified as prechiasmatic, chiasmatic, and postchiasmatic. Chiasmatic and postchiasmatic OPGs are more common in sporadic cases, whereas those limited to the optic nerve (prechiasmatic) are more frequently found in children with neurofibromatosis type 1 (NF1). In one study, the most common site of involvement in the NF1 patients was the optic nerve (66%), followed by the chiasm (62%). In the non-NF1 patients, the chiasm was the most common site of involvement (91%), while the optic nerves were involved in only 32% [3].
Risk Factors
OPG is seen in about 15–20% of patients with NF1, an autosomal dominant disorder with an incidence of 1 in 2000–2500 individuals [4, 5]. The main disease manifestations of NF1 include café au lait macules, skinfold freckling (axillary and inguinal regions), Lisch nodules or iris hamartomas, OPGs, and skeletal deformities. In a study from Cincinnati, 18% of 826 NF1 individuals were identified to have OPG with a median age at detection of 3 years. Fifteen percent of OPG patients had a radiologic or clinical progression requiring therapy [6]. Conversely, in series of OPG patients, about 20–58% of patients have been reported to have NF1 [7, 8].
Diagnosis
Clinical Presentation
Many patients with OPG are asymptomatic, particularly those with NF1 [6]. For symptomatic patients, visual loss is the most common complaint. Patients with optic nerve gliomas typically present with unilateral, slow vision loss with a relative afferent pupillary defect, optic disk swelling or atrophy, or strabismus. Ocular pain is uncommon, but proptosis can be a presenting sign. In tumors with chiasmatic involvement, bitemporal field defects can be seen. Intracranial extension of the tumor can result in hypothalamic and endocrine problems. Malignant optic gliomas have been reported in adults and can present as sudden visual loss in 70–80% of patients [9].
Diagnostic Imaging
On magnetic resonance imaging (MRI) of the brain, these lesions are T1-hypointense and T2-hyperintense with typically homogeneous gadolinium enhancement . Larger tumors may present with heterogeneous enhancement and may be associated with hydrocephalus. MRI with fat suppression can be done to visualize the entire optic pathway.
Pathology
Most OPGs are low-grade astrocytomas , of which pilocytic astrocytomas are the predominant subtype. Malignant transformation is unusual. Optic nerve gliomas are typically expansile lesions which transgress the pia; on microscopic examination, the optic nerve is surrounded by neoplastic astrocytes with reactive elements enclosed within the dura mater. Pilomyxoid astrocytomas, fibrillary or diffuse astrocytomas, and gangliogliomas have also been reported but are not as common as pilocytic astrocytomas.
Overall Treatment Strategy
Tissue Diagnosis
Currently, tissue diagnosis is recommended in most tumors that look like OPG. In one study, MRI was shown to have a sensitivity of 83.3% and a specificity of 50% for diagnosing chiasmatic-hypothalamic gliomas [10]. However, most agree that radiologic diagnosis is sufficient in infants with chiasmatic-hypothalamic lesions and in children with NF1 [11].
Observation
In general, the management of OPG can include observation, surgery, chemotherapy, and radiotherapy. Because of its indolent nature, observation of OPG has been performed, especially in children with NF1 where tumors can “wax and wane.” Occasionally, OPGs may undergo spontaneous regression [12]. One of the hardest decisions is to decide when to intervene and treat the patient. Considerations for intervention include functional, typically visual, and neurologic deterioration. In patients with non-NF1 OPG, neuraxis dissemination and diencephalic syndrome may trigger intervention [13].
Surgery
Complete resection of the tumor is recommended for the glioma confined just to the optic nerve, presenting with complete visual loss. For other tumors, a biopsy or surgical debulking have been reported. In one study of 42 children with OPG, 22 debulking procedures were performed. None of the patients had visual deterioration but six had diabetes insipidus (three permanent, three transient). One had transient hemiparesis that improved during rehabilitation. Surgery alone was found to control 13 of 17 tumors. Surgery was able to relieve mass effect and in some cases delay radiotherapy (RT). The authors conclude that surgical debulking of tumor is safe and effective for carefully selected patients [14]. Others have reported improvements in tumor control by surgical debulking while others have not [15,16,17].
Chemotherapy
Chemotherapy is often used in children with OPG who have subtotal resection/biopsy to delay the use of RT. Delay in the delivery of RT can be beneficial in children with low-grade glioma as shown by better neurocognitive outcomes in older children receiving conformal RT [18]. The standard chemotherapy regimen consists of vincristine and carboplatin (“Packer regimen”). Packer et al. reported on 78 children with a mean age of 3 years who had newly diagnosed, progressive low-grade gliomas were treated with combined carboplatin and vincristine chemotherapy. Majority had diencephalic tumors, and the objective response rate was 56%. The 2- and 3-year progression-free survival (PFS) rates were 75% and 68%, respectively [19]. A phase II Pediatric Oncology Group (POG) was conducted to evaluate the activity of carboplatin in children ≤5 years with progressive optic pathway tumors (OPTs). Of the 50 children, 21 had progressive disease (15 during the course of carboplatin therapy and 6 patients progressed after) [20]. In the HIT-LGG 1996 study, 198 patients with a median age of 3.6 years had either a chiasmatic-hypothalamic (n = 144), chiasmatic (n = 34) or hypothalamic tumor (n = 20), and 98 of these children had severe visual impairment as their first symptom. A total of 123 children received vincristine and carboplatin at a median age of 3.7 years, of which 105 had initial complete or partial response or stable disease. The 5-year overall survival was 93% for the entire group of patients; the 5-year PFS for those receiving chemotherapy was 61%, with the 5-year radiotherapy-free survival was 83% [21]. The literature is scarce regarding the use of chemotherapy in adult patients with OPG. A Canadian study of adolescent patients with OPG suggests that chemotherapy be considered for first-line treatment rather than RT for progressive disease. In this particular report, children >10 years had PFS comparable to younger children with first-line chemotherapy, avoiding potential RT-related toxicity [22]. Many institutions, however, consider RT to be the standard treatment for progressive OPG in children greater than 10 years of age and adult patients [23].
Indication for Irradiation
Because most of the OPG patients will survive for a long time, minimizing the sequelae of treatment is paramount in the management of this disease. As such, RT is considered to be the standard treatment for progressive OPG in adult patients, while for children <10 years old, most would consider chemotherapy to be the standard therapy. In our institution, children >10 years requiring treatment are treated with RT although there is no consensus in the literature [11, 22,23,24,25].
Target Volume Delineation and Radiotherapy Dose Prescription
The type of OPG often dictates what imaging study to use for target delineation . In pilocytic astrocytoma, T1 enhancement is often seen and is utilized for gross tumor volume (GTV) delineation. For other low-grade gliomas, tumors are often non-enhancing, and FLAIR abnormalities are used for GTV delineation. The safety margin beyond the GTV have ranged from 0 to 10 mm for the clinical target volume (CTV). Most would use a 3 mm margin around the CTV to create the planning target volume (PTV). At St. Jude Children’s Research Hospital, a 10 mm margin was added to the GTV to create the CTV, while the Children’s Oncology Group ACNS0221 employed a 5 mm margin for the CTV [26]. A previous study did not find a difference between 5 or 10 mm safety margin for the CTV [27]. At the Dana-Farber Cancer Institute where stereotactic RT was used, the GTV and CTV are essentially the same, with a 2 mm expansion for the PTV [28]. Typically dose ranges from 45 to 50.4 Gy for pilocytic astrocytomas and 50.4 to 54 Gy for non-pilocytic low-grade gliomas.
For the rare malignant glioma of the optic pathway, both the contrast-enhanced T1 and FLAIR images are employed to create the GTV and CTV. In our institution the FLAIR abnormality is typically covered by the 50 Gy isodose line, while the tumor gross disease with T1 enhancement gets the higher dose, typically 54 Gy because of the tolerance of the optic nerve and chiasm.
Organs at Risk and Tolerance Doses
Table 15.1 lists the normal tissue constraints that are used by the author for treatment of optic pathway tumors.
Radiation Toxicity: Acute and Late
The acute effects of radiation can include erythema and itching of irradiated skin, partial alopecia, fatigue, and conjunctivitis. Late effects and possible complications of radiotherapy may include cataract, cognitive dysfunction, endocrine dysfunction, Moyamoya disease, dryness of the eye, and secondary malignancy [29]. Blindness is uncommon and <5% when the D max or maximum dose to the optic nerves and chiasm is less than 54 Gy and retina is <50 Gy. In patients with NF1, secondary tumors and Moyamoya disease are more frequent with the use of RT [30, 31].
Protons may be able to decrease the chance of developing a secondary neoplasm as integral dose will be less [32]. It may also minimize the low-dose region in the hippocampus and hypothalamus that one gets with the use of photons. In one study, protons resulted in reduced doses to the contralateral optic nerve, optic chiasm, pituitary gland, and bilateral temporal and frontal lobes compared to photons [33].
Radiotherapy Outcomes
Tumor Control and Survival
Table 15.2 lists multiple single-institution studies using RT as the primary treatment for OPG. In some of the studies, about 20–40% of the patients are adolescents or adult patients. The 5- and 10-year overall survival rates range from 90% to 100% and 79% to 94%, while the 10-year PFS rates range from 69% to 100% [8, 34,35,36,37,38]. These results compare favorably to patients treated with primary chemotherapy where the 5-year PFS rates are in the range of 30–40% [11]. As a result many children treated with primary chemotherapy require more than one chemotherapy regimen. Despite these results, younger children <10 years old with progressive OPG are typically treated with chemotherapy first because of the possible side effects of RT on cognition and endocrine function [39]. In NF1 patients, secondary tumors and vascular problems are heightened by the use of RT, so chemotherapy is usually the first-line treatment for progressive OPG [30, 31].
Visual Acuity
Table 15.2 lists the visual outcomes of patients treated with RT for OPG. In general, about one third of patients had improvement in vision while another one half had stabilization [8, 34,35,36,37,38]. The results for visual outcome after primary chemotherapy appear to be worse compared to primary RT. In a study of 20 optic glioma patients treated with conformal RT, patients who received chemotherapy prior to RT had worse visual acuity compared to those treated with primary RT [40]. In another OPG study, older children treated with RT had better visual acuity compared to younger children treated with primary chemotherapy [41]. Published studies on childhood low-grade gliomas have not shown that chemotherapy improves vision in the majority of children with OPG [42].
Follow-Up: Radiographic Assessment
For patients with OPG being observed for NF1, MRI of the brain is typically done every 3 months during the first year and less frequently (every 6 months then every year) thereafter. These tumors tend to wax and wane and are more indolent compared to the non-NF1 OPG.
For patients receiving RT for OPG, we typically follow these patients with a MRI of the brain and visual examination every 4 months for the first 2 years and then every 6 months for the next 3 years and yearly thereafter.
Case Presentation
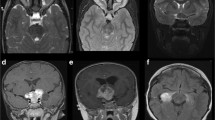
The patient is a 19-year-old male who initially was diagnosed with a hypothalamic-chiasmatic optic pathway glioma after a biopsy when he was 9 years old. At presentation he had right eye blindness, and he was subsequently treated with 80 weeks of vincristine and carboplatin and had stable disease until when he was 18 years old when he was noticed to have progression. Representative MRI slices are shown in Fig. 15.1a–c. The patient was treated using protons to a dose of 50.4 cobalt Gy equivalent (CGE). The CTV was 0.5 cm beyond the GTV, respecting anatomic barriers (Fig. 15.1d–f).

Proton therapy in a 19-year-old patient with optic pathway glioma. (a–c) Coronal, sagittal, and axial magnetic resonance imaging (MRI) slices of a patient with progressive pilocytic astrocytoma of the right hypothalamic optic pathway. (d–f) Axial, coronal, and sagittal slices showing treatment plan with protons. The prescribed dose was 50.4 cobalt Gy equivalent. The gross tumor volume (GTV) is outlined in maroon, while the clinical target volume (CTV) is outlined in mustard
Summary
Most optic pathway gliomas (OPGs) are pilocytic astrocytomas, but other types of low-grade and malignant gliomas have been reported. The role of surgery is to obtain tissue diagnosis or resection in the patient with glioma confined just to the optic nerve and complete visual loss. Chemotherapy consisting of carboplatin and vincristine is a popular treatment approach to delay radiotherapy (RT) in children <10 years and preserve neurocognitive function. In patients with neurofibromatosis, type 1, RT is often avoided because of the higher chance of developing Moyamoya syndrome and secondary neoplasms. RT is the mainstay of treatment for non-NF1 patients who are ≥10 years. The 10-year overall survival and progression-free survival rates range from 80% to 95% and 70% to 85%, respectively, with RT. In addition, stabilization and improvement in vision have been observed in more than 70% of patients receiving RT.
Self-Assessment Questions
-
1.
What proportion of neurofibromatosis type 1 patients will have optic pathway glioma?
-
A.
<1%
-
B.
15%
-
C.
50%
-
D.
85%
-
A.
-
2.
What is the most likely histology for an optic pathway glioma?
-
A.
Ganglioglioma
-
B.
Pleomorphic xanthoastrocytoma
-
C.
Pilocytic astrocytoma
-
D.
Subependymal giant cell astrocytoma
-
A.
-
3.
What would be the most appropriate treatment for an optic nerve glioma in an eye with complete visual loss?
-
A.
Stereotactic radiosurgery
-
B.
Carboplatin and vincristine chemotherapy
-
C.
Intensity modulated radiation therapy
-
D.
Surgical resection
-
A.
-
4.
At 10 years after radiotherapy, what is the expected local control for an optic pathway glioma?
-
A.
20%
-
B.
40%
-
C.
60%
-
D.
80%
-
A.
-
5.
After radiotherapy for an optic pathway glioma, what is the expected proportion of patients with stable or improved vision?
-
A.
20%
-
B.
40%
-
C.
60%
-
D.
80%
-
A.
Answers
-
1.
B
-
2.
C
-
3.
D
-
4.
D
-
5.
C
References
Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994;38(5):427–52.
Binning MJ, Liu JK, Kestle JR, et al. Optic pathway gliomas: a review. Neurosurg Focus. 2007;23(5):E2.
Kornreich L, Blaser S, Schwarz M, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR Am J Neuroradiol. 2001;22(10):1963–9.
King A, Listernick R, Charrow J, et al. Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet A. 2003;122a(2):95–9.
Listernick R, Louis DN, Packer RJ, et al. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol. 1997;41(2):143–9.
Prada CE, Hufnagel RB, Hummel TR, et al. The use of magnetic resonance imaging screening for optic pathway gliomas in children with neurofibromatosis type 1. J Pediatr. 2015;167(4):851–6.e1.
Nicolin G, Parkin P, Mabbott D, et al. Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer. 2009;53(7):1231–7.
Khafaga Y, Hassounah M, Kandil A, et al. Optic gliomas: a retrospective analysis of 50 cases. Int J Radiat Oncol Biol Phys. 2003;56(3):807–12.
Hartel PH, Rosen C, Larzo C, et al. Malignant optic nerve glioma (glioblastoma multiforme): a case report and literature review. W V Med J. 2006;102(4):29–31.
Bommakanti K, Panigrahi M, Yarlagadda R, et al. Optic chiasmatic-hypothalamic gliomas: is tissue diagnosis essential? Neurol India. 2010;58(6):833–40.
Fried I, Tabori U, Tihan T, et al. Optic pathway gliomas: a review. CNS Oncol. 2013;2(2):143–59.
Parsa CF, Hoyt CS, Lesser RL, et al. Spontaneous regression of optic gliomas: thirteen cases documented by serial neuroimaging. Arch Ophthalmol. 2001;119(4):516–29.
Addy DP, Hudson FP. Diencephalic syndrome of infantile emaciation. Analysis of literature and report of further 3 cases. Arch Dis Child. 1972;47(253):338–43.
Goodden J, Pizer B, Pettorini B, et al. The role of surgery in optic pathway/hypothalamic gliomas in children. J Neurosurg Pediatr. 2014;13(1):1–12.
Hoffman HJ, Soloniuk DS, Humphreys RP, et al. Management and outcome of low-grade astrocytomas of the midline in children: a retrospective review. Neurosurgery. 1993;33(6):964–71.
Steinbok P, Hentschel S, Almqvist P, et al. Management of optic chiasmatic/hypothalamic astrocytomas in children. Can J Neurol Sci. 2002;29(2):132–8.
Wisoff JH, Abbott R, Epstein F. Surgical management of exophytic chiasmatic-hypothalamic tumors of childhood. J Neurosurg. 1990;73(5):661–7.
Merchant TE, Conklin HM, Wu S, et al. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol. 2009;27(22):3691–7.
Packer RJ, Ater J, Allen J, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86(5):747–54.
Mahoney DH Jr, Cohen ME, Friedman HS, et al. Carboplatin is effective therapy for young children with progressive optic pathway tumors: a Pediatric Oncology Group phase II study. Neuro Oncol. 2000;2(4):213–20.
Gnekow AK, Kortmann RD, Pietsch T, et al. Low grade chiasmatic-hypothalamic glioma-carboplatin and vincristin chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy — report from the multicenter treatment study for children and adolescents with a low grade glioma — HIT-LGG 1996 — of the Society of Pediatric Oncology and Hematology (GPOH). Klin Padiatr. 2004;216(6):331–42.
Chong AL, Pole JD, Scheinemann K, et al. Optic pathway gliomas in adolescence—time to challenge treatment choices? Neuro Oncol. 2013;15(3):391–400.
Jahraus CD, Tarbell NJ. Optic pathway gliomas. Pediatr Blood Cancer. 2006;46(5):586–96.
Shofty B, Mauda-Havakuk M, Weizman L, et al. The effect of chemotherapy on optic pathway gliomas and their sub-components: a volumetric MR analysis study. Pediatr Blood Cancer. 2015;62(8):1353–9.
Stieber VW. Radiation therapy for visual pathway tumors. J Neuroophthalmol. 2008;28(3):222–30.
Merchant TE, Kun LE, Wu S, et al. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J Clin Oncol. 2009;27(22):3598–604.
Paulino AC, Mazloom A, Terashima K, et al. Intensity-modulated radiotherapy (IMRT) in pediatric low-grade glioma. Cancer. 2013;119(14):2654–9.
Marcus KJ, Goumnerova L, Billett AL, et al. Stereotactic radiotherapy for localized low-grade gliomas in children: final results of a prospective trial. Int J Radiat Oncol Biol Phys. 2005;61(2):374–9.
Jeganathan VS, Wirth A, MacManus MP. Ocular risks from orbital and periorbital radiation therapy: a critical review. Int J Radiat Oncol Biol Phys. 2011;79(3):650–9.
Desai SS, Paulino AC, Mai WY, et al. Radiation-induced moyamoya syndrome. Int J Radiat Oncol Biol Phys. 2006;65(4):1222–7.
Sharif S, Ferner R, Birch JM, et al. Second primary tumors in neurofibromatosis 1 patients treated for optic glioma: substantial risks after radiotherapy. J Clin Oncol. 2006;24(16):2570–5.
Sethi RV, Shih HA, Yeap BY, et al. Second nonocular tumors among survivors of retinoblastoma treated with contemporary photon and proton radiotherapy. Cancer. 2014;120(1):126–33.
Fuss M, Hug EB, Schaefer RA, et al. Proton radiation therapy (PRT) for pediatric optic pathway gliomas: comparison with 3D planned conventional photons and a standard photon technique. Int J Radiat Oncol Biol Phys. 1999;45(5):1117–26.
Tao ML, Barnes PD, Billett AL, et al. Childhood optic chiasm gliomas: radiographic response following radiotherapy and long-term clinical outcome. Int J Radiat Oncol Biol Phys. 1997;39(3):579–87.
Horwich A, Bloom HJ. Optic gliomas: radiation therapy and prognosis. Int J Radiat Oncol Biol Phys. 1985;11(6):1067–79.
Erkal HS, Serin M, Cakmak A. Management of optic pathway and chiasmatic-hypothalamic gliomas in children with radiation therapy. Radiother Oncol. 1997;45(1):11–5.
Grabenbauer GG, Schuchardt U, Buchfelder M, et al. Radiation therapy of optico-hypothalamic gliomas (OHG)—radiographic response, vision and late toxicity. Radiother Oncol. 2000;54(3):239–45.
Combs SE, Schulz-Ertner D, Moschos D, et al. Fractionated stereotactic radiotherapy of optic pathway gliomas: tolerance and long-term outcome. Int J Radiat Oncol Biol Phys. 2005;62(3):814–9.
Lacaze E, Kieffer V, Streri A, et al. Neuropsychological outcome in children with optic pathway tumours when first-line treatment is chemotherapy. Br J Cancer. 2003;89(11):2038–44.
Awdeh RM, Kiehna EN, Drewry RD, et al. Visual outcomes in pediatric optic pathway glioma after conformal radiation therapy. Int J Radiat Oncol Biol Phys. 2012;84(1):46–51.
Campagna M, Opocher E, Viscardi E, et al. Optic pathway glioma: long-term visual outcome in children without neurofibromatosis type-1. Pediatr Blood Cancer. 2010;55(6):1083–8.
Moreno L, Bautista F, Ashley S, et al. Does chemotherapy affect the visual outcome in children with optic pathway glioma? A systematic review of the evidence. Eur J Cancer. 2010;46(12):2253–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Paulino, A.C. (2018). Optic Pathway Gliomas. In: Chang, E., Brown, P., Lo, S., Sahgal, A., Suh, J. (eds) Adult CNS Radiation Oncology. Springer, Cham. https://doi.org/10.1007/978-3-319-42878-9_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-42878-9_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-42877-2
Online ISBN: 978-3-319-42878-9
eBook Packages: MedicineMedicine (R0)