
Abstract
Cell membranes are central to all life on earth. The lipid bilayer composing cell membranes enables discrimination between the cell and its external environment. Far from a passive bystander, the lipid membrane plays an active role in sequestering and accumulating genetic material, proteins, nutrients and ions essential for life. Indeed, cells allocate a significant portion of their energy budget to maintain an unequal distribution of ions across the cell membrane. The unequal distribution of ions across the cell membrane results in an electrochemical potential, or voltage. Rapid changes in this membrane voltage drive the unique physiology of excitable cells like neurons and cardiomyocytes. Despite the central importance of voltage in the brain and body, the full contributions of voltage changes to both physiology and disease remain incompletely characterized due to a lack of methods that can report on membrane potential with high fidelity, high throughput, and minimal invasiveness.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


1 Introduction
Cell membranes are central to all life on earth. The lipid bilayer composing cell membranes enables discrimination between the cell and its external environment. Far from a passive bystander, the lipid membrane plays an active role in sequestering and accumulating genetic material, proteins, nutrients and ions essential for life. Indeed, cells allocate a significant portion of their energy budget to maintain an unequal distribution of ions across the cell membrane. The unequal distribution of ions across the cell membrane results in an electrochemical potential, or voltage. Rapid changes in this membrane voltage drive the unique physiology of excitable cells like neurons and cardiomyocytes. Despite the central importance of voltage in the brain and body, the full contributions of voltage changes to both physiology and disease remain incompletely characterized due to a lack of methods that can report on membrane potential with high fidelity, high throughput, and minimal invasiveness.
Voltage imaging with fluorescent dyes offers a solution to this problem because it combines a direct readout of voltage changes, providing temporal resolution, with an imaging approach that offers spatial resolution. Attempts at voltage imaging with small molecules date back to the 1970s (Braubach et al. 2015; Loew 2015), and it is a measure of the difficulty of the task that voltage imaging has not yet become a wide-spread experimental approach. Rather, surrogates of voltage changes, notably Ca2+ ion fluxes have become the primary means of assessing neuronal and cellular activity. This is due, in part, to the fact that the experimental constraints on Ca2+ imaging are somewhat less stringent than voltage imaging in the context of neurobiology. First, transient fluctuations in intracellular Ca2+ concentration follow time-courses that are 10–100,000 times slower than neuronal action potentials. Second, an individual cell can accumulate >1000-fold more Ca2+ indicators relative to voltage indicators, owing to the requirement that voltage indicators must localize to the plasma membrane (which occupies ~0.1% the volume of the cytosol) in order to retain function. Finally, because the fluorescent signals from Ca2+ indicators emanate from the cytosol, it is relatively straightforward to segregate signals arising from distinct cells. For voltage indicators that localize to the plasma membrane, fluorescent signals from adjoining cells can overlap. These barriers, and others, have hindered the widespread adoption of voltage indicators because, compared to Ca2+ indicators, voltage indicators must do more with less (Kulkarni and Miller 2017).
To address these challenges, we have recently initiated a program to develop fluorescent sensors of membrane potential that circumvent previous limitations of speed, sensitivity and disruptive capacitive load. Our strategy uses small molecule fluorescent voltage sensors that employ photoinduced electron transfer (PeT) (De Silva et al. 1995; Li 2007) through a molecular wire as a voltage-sensing trigger. In the first generation of voltage-sensitive fluorophores, also known as VoltageFluors, or VF dyes, an electron-rich aniline donor is coupled to a fluorescent, fluorescein reporter via a phenylenevinylene molecular wire (Miller et al. 2012). Due to their amphipathic nature, VF dyes inserts into the plasma membrane with the chromophore protruding slightly above the phospholipid layer, on account of the anionic sulfonate. We hypothesized that, at resting membrane potential, which is slightly negative (approximately −60 mV in a typical mammalian neuron), the orientation of the electric field across the membrane accelerates PeT from the aniline donor to the excited chromophore, quenching fluorescence (Fig. 9.1). When a neuron fires an action potential, the membrane potential changes rapidly (<5 ms), depolarizing to approximately +40 mV. During this depolarization, the electric field across the membrane reverses, resulting in a reduction of the rate of PeT and subsequent fluorescence brightening. In this way, the membrane potential modulates PeT, (De Silva et al. 1995; Li 2007) which in turn alters the fluorescence, which can be read out optically using fluorescence microscopy. The first generation dye, VoltageFluor2.1.Cl, or VF2.1.Cl, possesses high voltage sensitivity (approximately 27% ΔF/F per 100 mV), linear and fast (sub-millisecond) fluorescence response to voltage changes, and no detectable increase in membrane capacitance (Miller et al. 2012). VF2.1.Cl (which has now been licensed by ThermoFisher and sold under the tradename “FluoVolt”) has been utilized in a number of contexts, including leech ganglia (Fathiazar et al. 2016; Miller et al. 2012), cultured rat hippocampal neurons (Miller et al. 2012), mouse pancreatic tissue (Dolensek et al. 2013), cochlear tissues (Ceriani and Mammano 2013) and in cardiomyocytes (McKeithan et al. 2017). Below, we outline new chemical directions that have enabled improved sensitivity, voltage imaging in multiple colors, and targeting to defined cells.

General scheme for voltage sensing. At hyperpolarizing potentials, alignment between membrane potential and electron transfer accelerates electron transfer, quenching fluorescence. At depolarized potential, the rate of electron transfer is slowed, resulting in enhanced fluorescence. (Adapted with permission from Kulkarni and Miller, Biochemistry 2017, 56, 5171. Copyright 2017 American Chemical Society)
2 Rational Design of VoltageFluor Dyes
While VF dyes have enabled voltage imaging in a variety of biological preparations, we hoped to increase the performance of VF dyes through tuning the redox properties of both the aniline donor and fluorescent reporter. The voltage sensitivity of VF dyes can be improved in this way, providing indicators with sensitivities approaching 50% ΔF/F per 100 mV (Woodford et al. 2015). Seeking a more general solution to improved performance, we also systematically investigated the orientation of VF dyes in cell membranes using a combination of computation and experiment (Kulkarni et al. 2017b). We hypothesized that a perfect perpendicular orientation of VF dyes with the plane of membrane should yield indicators with the maximum theoretical sensitivity. We used molecular dynamics (MD) simulations to calculate the tilt angle, or degree of displacement away from membrane normal and found that a typical VF dye containing a single sulfonate group shows significant variability and an approximate 35° departure from membrane normal (Fig. 9.2a and b). The calculations hinted at a structure—containing a symmetrical sulfonation pattern—that would resolve this rather large tilt angle, reducing it to 19°. Based on a theoretical model of voltage sensing, we hypothesized this change in angle would result in an approximate 16% increase in voltage sensitivity—a large improvement in a field where sensitivities typically fall below 20% ΔF/F per 100 mV.

Disulfonated VoltageFluors have better membrane orientation than monosulfonated VoltageFluors. (a) Molecular dynamics modeling of disulfoVoltageFluor in a POPC lipid bilayer demonstrated a slight deviation in angle from the normal. (b) DisulfoVF, or dsVF, has a small average angle of deviation as well as a smaller standard deviation, suggesting that it is more rigid in the membrane than msVF. (c) Confocal fluorescence image of dsVF in rat hippocampal neurons with structure of VF dyes overlaid. (Adapted with permission from Kulkarni, Yin, Pourmandi, James, Adil, Schaffer, Wang and Miller, ACS Chem. Biol. 2017, 12, 407. Copyright 2016 American Chemical Society)
We then confirmed this computational result by devising a new route to doubly sulfonated VF dyes. We synthesized nine new dyes, enabling six pairwise, experimental comparisons of monosulfoVF (msVF) and disulfoVF (dsVF) dyes in living cells to complement our computational study. In all cases, we saw increased voltage sensitivity for the dsVF dye variants relative to their msVF counterparts, with an average increase of 19%, which is in close agreement to the value predicted by MD simulation. This result not only validates our computational model of VF dyes in a lipid membrane, but has provided the most sensitive VF to date, dsVF2.2(OMe). Cl (Fig. 9.2c), with a voltage sensitivity of 63% ΔF/F per 100 mV. We showed that with this increased sensitivity, dsVF2.2(OMe). Cl can readily monitor spontaneous activity in mid-brain dopaminergic neurons derived from human pluripotent stem cells. Over the course of differentiation, the stem cell-derived neurons began to demonstrate the characteristic pacemaker potential and activity of dopaminergic neurons (Rodrigues et al. 2017). Together, these results suggest that this particular modification – introduction of a second, complementary sulfonate – may be a general solution for improving the voltage sensitivity of VF-type dyes. More broadly, efforts to improve the orientation of the molecular wire voltage sensing domain with the electric field across the membrane represents a general strategy for improving voltage sensitivity.
3 Voltage Imaging with Red-Shifted Dyes
While synthetic modifications to fluorescein-based VFs have greatly improved their performance, they are inherently limited by their blue/green excitation and emission profiles, which overlap spectrally with optical tools such as GFP and ChannelRhodopsin2 (ChR2). VF dyes also rely on a synthetically challenging sulfonic acid for proper membrane targeting and orientation. With these limitations in mind, we aimed to develop a voltage sensing scaffold that would retain the high sensitivity of the VF dye series, expand the spectrum of colors available for voltage sensing, and circumvent the inclusion of the sulfonate that makes synthetic efforts challenging.
Toward this end we developed a new class of VF dyes based on tetramethylrhodamine (TMR), dubbed Rhodamine Voltage Reporters, or RhoVRs (Deal et al. 2016). RhoVRs were synthesized in isomerically pure form from 4′- and 5′-bromo TMRs. The bromo TMR derivatives were themselves generated from isomerically pure bromo phthalides, as we found separating the regioisomers at the phthalide stage (Mirabdolbaghi and Dudding 2012) much simpler than at the rhodamine stage. Pd-catalyzed Heck couplings were used to attach aniline-substituted styrenes, generating the carboxy-functionalized TMR voltage reporters in good yields (41–55%). Initial studies with carboxy-functionalized RhoVRs revealed their tendency to localize to internal membranes of cells (Fig. 9.3b). We hypothesized this was due to the formation of a neutral spirocycle which could freely pass through the cell membrane. This problem was surmounted by the formation of an N-methylglycine-derived tertiary amide that both prevented spirolactonization and retained a negatively charged carboxylate group. Confocal microscopy confirmed that this strategy led to membrane staining identical to that seen with sulfonate-containing VFs (Fig. 9.3g–i).

Quantification of RhoVR 1 and rhodamine cellular localization. The fluorescence intensity of rhodamine signal was measured by measuring the pixel intensity across a line segment spanning individual HEK cells. Images are shown for RhoVR 1 (g-i), compound 1 (free carboxylate, no sarcosine, a–c), and compound 2(t-Bu ester of RhoVR 1, d–f). Plots (c), (f), and (i) show the normalized fluorescence intensity for the rhodamine derivative (magenta) vs. the normalized widths of the white line segments in the fluorescence images on the left. Thick traces represent the average of five to six cells for each condition. Light traces represent individual intensity profiles. (Adapted with permission from Deal, Kulkarni, Al-Abdullatif, and Miller, J. Am. Chem. Soc. 2016, 138, 9085. Copyright 2016 American Chemical Society)
Four RhoVR derivatives were synthesized to screen for maximal voltage sensitivity. All RhoVRs displayed absorption profiles centered at 564–565 nm (ε = 70,000 to 87,000 M−1 cm−1) and emission profiles centered at 586–588 nm (Φ = 0.89–9.2%). The voltage sensitivity of each RhoVR was assessed in HEK cells using patch-clamp electrophysiology, which we found ranged from 3 to 47% ΔF/F per 100 mV. The most sensitive of the RhoVRs, which we called RhoVR 1, bore a 5′-methoxy substituted aniline (Fig. 9.4a–c). Interestingly, we found that RhoVRs derived from the 5′-substituted rhodamines were both more voltage-sensitive and brighter in cells than the 4′- equivalents. While we are still currently investigating the nature of this improvement. We speculate that the 5′ substitution pattern of RhoVR 1 improves orientation of the molecular wire with respect to the membrane, enhancing voltage sensitivity.

RhoVR 1, a rhodamine-based voltage reporter. (a) Structure of RhoVR 1. (b) Plot of the fractional change in fluorescence vs time for 100 ms hyper- and depolarizing steps (±100 mV in 20 mV increments) from a holding potential of −60 mV for single HEK cells under whole-cell voltage-clamp mode. (c) Plot of % ΔF/F vs final membrane potential summarizing data from nine separate cells, revealing a voltage sensitivity of approximately 47% per 100 mV. Error bars are ±SEM. (d–g) Simultaneous two-color imaging of voltage and Ca2+ in hippocampal neurons using RhoVR 1 and GCaMP6s. (d) DIC image of neurons expressing GCaMP6s and stained with RhoVR 1. (e) Fluorescence image showing membrane localization of RhoVR 1 fluorescence from neurons in (d). (f) Fluorescence image of neurons in (d) showing GCaMP6s fluorescence. Scale bar is 20 μm. Simultaneous two-color imaging of voltage and Ca2+ in hippocampal neurons using RhoVR 1 and GCaMP6s. (g) The green trace shows the relative change in fluorescence from Ca2+-sensitive GCaMP6s, while the magenta trace depicts relative fluorescence changes in RhoVR 1 fluorescence from neuron 1 in (d). (Adapted with permission from Deal, Kulkarni, Al-Abdullatif, and Miller, J. Am. Chem. Soc. 2016, 138, 9085. Copyright 2016 American Chemical Society)
Given the high sensitivity and brightness of RhoVR 1 in HEK cells, we evaluated this dye in cultured rat hippocampal neurons. We found that bath-applying RhoVR 1 to neurons enabled the detection of spontaneously firing action potentials with an average ΔF/F of 15% with good SNR. We then showed that we could perform simultaneous two-color imaging using RhoVR 1 to record action potentials and GCaMP6s (a GFP-based Ca2+ indicator) to record the subsequent Ca2+ transients (Fig. 9.4d–g). From these experiments it was clear that monitoring Vm directly via RhoVR 1 enabled resolution and precise timing of spikes occurring in quick succession from multiple cells, a feat which would be impossible using traditional approaches such as Ca2+ imaging or single-cell electrophysiology.
In summary, we have developed a new class of voltage-sensitive dyes based on tetramethyl rhodamine. The best performing of these voltage indicators, RhoVR 1, displays an excitation and emission profile greater than 550 nm, good photostability and a voltage sensitivity of 47% ΔF/F per 100 mV. The development of RhoVRs revealed ortho-tertiary amides can be used in place of sulfonates to achieve membrane localization (Fig. 9.3), greatly simplifying the synthetic route to long-wavelength voltage sensors. We also revealed the importance of the substitution pattern of the molecular wire, with 5′- substituted RhoVRs outperforming the typical 4′- substitution pattern utilized in our previous voltage sensors and many other PeT-based analyte sensors. Current work with RhoVRs is now focused on utilizing the ortho-tertiary amide strategy to generate genetically targetable RhoVRs which will facilitate their use in vivo.
4 Far-Red Voltage Imaging for High Sensitivity
The biggest challenges in designing voltage-sensitive dyes (VSDs) for the measurement of neuronal action potentials (APs) are born from the fast timescale in which APs occur. Combining high speed imaging with the relatively small pool of dye molecules in the cell membrane leads to photon-starved conditions. To overcome these restrictions, a VF dye which generates signals with low noise and large changes in fluorescence would greatly improve AP detection. We hypothesized that signal-to-noise ratios (SNR) could be improved by red-shifting the VF fluorophore, where less overlap with blue/green spectrum cell autofluorescence should decrease baseline noise levels. Red-shifted VFs are also excited by longer wavelength light which causes less tissue damage and scatters less in tissue preparations. Furthermore, far-red sensors provide even larger spectral separation from useful green-spectrum optical tools and could therefore be used in conjunction with GFP, robust Ca2+ sensors like Oregon Green BAPTA or the GCaMP family, and optogenetic tools like ChannelRhodopsin2 (ChR2).
Silicon-rhodamines (Fu et al. 2008) are particularly desirable red-shifted fluorophores because they display photostable, far-red to NIR fluorescence that can be readily modulated by PeT (Koide et al. 2011). To synthesize a red-shifted VSD, a novel sulfonated silicon-rhodamine called Berkeley Red was first synthesized, which was combined with a phenylenevinylene molecular wire with electron-donating groups to generate Berkeley Red Sensor of Transmembrane potential 1 (BeRST 1, “burst”). As expected from the properties of Berkeley Red and other Si-Rhodamines, photophysical characterization of BeRST 1 showed excitation and emission profiles centered at 658 and 683 nm, respectively. BeRST 1 also showed excellent photostability, with a bleaching half-life of approximately 5 minutes under intense illumination conditions. By comparison, VF2.1.Cl had a bleaching half-life of <60 s under identical illumination intensities. Sensor photostability is highly advantageous, recommending the use of BeRST 1 for stable, relatively long-term imaging of neuronal activity (Huang et al. 2015).
The voltage sensitivity of BeRST 1 was tested after loading it into HEK cell membranes by applying voltage steps across the membrane via whole-cell patch-clamp electrophysiology. BeRST 1 has a voltage sensitivity of approximately 24% ΔF/F per 100 mV, comparable to VF2.1.Cl, and is linear over a physiologically relevant range spanning ±100 mV. Despite a relatively modest voltage sensitivity, BeRST 1 produced action potential traces during extracellular stimulation of dissociated neurons with very high signal-to-noise ratios (SNR; > 60:1). High SNR recordings permitted AP detection in single trials in dissociated hippocampal neurons, meaning that spontaneous neuronal activity could be recorded in multiple neurons simultaneously. By observing spontaneous AP patterns in neurons, it will be possible to study the nascent properties of neuronal firing and neuronal connectivity.
Finally, we showed that BeRST 1 is highly compatible for two-color functional imaging with GFP-based sensors. Using extracellular stimulation of dissociated hippocampal neurons, we could read-out separate BeRST 1 and GCaMP6s signals, examining the differences in the voltage and calcium readouts of AP firing at different frequencies. These experiments pave the way for investigations into interactions between voltage and calcium signaling under a variety of conditions. It was also possible to pair voltage recording with ChR2 where APs triggered by short pulses of cyan light could be detected in ChR2-expressing cells by reading out changes in BeRST 1 fluorescence (Fig. 9.5). This coupling shows the utility of BeRST 1 and ChR2 for non-invasive all-optical electrophysiological experiments.

Using BeRST 1 and ChR2 to perturb network activity. Cultured rat hippocampal (c) neurons transfected with (a) ChR2-YFP and stained with (b) BeRST 1 were stimulated with 475 nm light (80 mW/cm2, 5 ms, 5 Hz, cyan bars) during two separate 3 s periods to evoke activity in the ChR2-YFP-expressing cell. Scale bar is 20 μm. (d) Schematic representation of neurons from DIC image in panel (c), color-coded to match the corresponding traces in (e–g). The blue ChR2(+) cell is depicted making possible connections to other neurons in the field of view. Optical records of BeRST 1 responses were acquired at 500 Hz with an sCMOS camera during (e) an optical recording session and (f) subsequent trial, separated by approximately 30 s (double hash). Numbers and colors of traces refer to specific neurons in panels (a–d). Red boxes indicate areas of the trace that have been magnified for clarity in panel (g). Dotted gray lines are provided in panel (g) to help visually estimate the spike timing of BeRST 1-stained neurons. (Reprinted with permission from Huang, Walker, and Miller, J. Am. Chem. Soc. 2015, 137, 10767)
In summary, BeRST 1 represents a new class of VSDs incorporating a far red/near infrared sulfonated Si-Rhodamine fluorophore. BeRST 1 is bright and highly photostable, making it ideal for imaging on longer timescales. BeRST 1 displays good voltage sensitivity and excellent signal-to-noise, making it advantageous for the detection of APs in single trials. Furthermore, BeRST 1 is ideally suited for multicolor imaging with GFP, Ca2+ indicators like GCaMP, as well as optogenetic tools like ChR2. Use of BeRST 1 alongside ChR2 permits non-invasive recording and control of AP firing.
5 Accessing Two-Photon Infrared Excitation for Imaging in Thick Brain Tissue
Observing neuronal activity in thick tissue preparations remains a challenge in part due to the relative lack of chromophores optimized for imaging under infrared excitation. The popularity of two-photon imaging for imaging intact brains and brain slices made a VoltageFluor optimized for two-photon absorption an attractive chemical target. Rhodols, a group of xanthene chromophores that are intermediates between fluoresceins and rhodamines (Whitaker et al. 1992), are well-suited for imaging applications (Dodani et al. 2014), due in part to their several-fold higher two-photon excitation cross sections than their fluorescein counterparts (Poronik et al. 2013). Rhodols are also relatively photostable compared to fluorescein-based dyes, potentially enabling long-term optical recordings of neurons in a variety of biological sample preparations. We hypothesized that rhodol-based VoltageFluors could serve as a complement to electrode-based techniques for monitoring voltage in neurons with two-photon imaging – facilitating recordings with high spatial resolution and the ability to record from several different neurons simultaneously. To this end, we designed and synthesized a rhodol-based voltage reporter optimized for two-photon microscopy and characterized its one- and two-photon photophysical properties (Kulkarni et al. 2017a).
Rhodol VoltageFluor-5, or RVF5, features a dichlororhodol xanthene chromophore, similar to the parent VF dye, VF2.1.Cl, but photobleaches four-fold more slowly and possess a four-fold higher two-photon excitation cross section relative to its parent dye. RVF5 demonstrates robust voltage sensitivity in HEK cells (28% ΔF/F per 100 mV) under one- and two-photon illumination. RVF5 can also be used to detect spontaneous and evoked activity in cultured neuron preparations (signal to noise = 10:1, ΔF/F = 11% per action potential). Hippocampal neurons stained with RVF5 show no difference in action potential kinetics relative to neurons without RVF5, as measured by whole-cell patch-clamp electrophysiology. Additionally, action potentials recorded optically with RVF5 perfectly track with electrophysiologically recorded signals.
RVF5 enables dissection of epileptic-like activity in cultured neurons. We made optical recordings of RVF5 in neurons deficient in the protein tuberous sclerosis complex 1 (Tsc1). Fluorescence imaging revealed that these neurons exhibit increased spontaneous activity relative to wild-type neurons, consistent with multi-electrode array recording data (Bateup et al. 2013). Providing a powerful complement these electrode-based measures, the optical recordings showed that the proportion of active neurons in the network increased with the loss of Tsc1. RVF5 can also be used to characterize the firing activity of human stem-cell derived neurons as they mature. Finally, the high two-photon cross section of RVF5 enables imaging of neuronal voltage changes in acutely prepared mouse brain slices using two-photon microscopy. Bath applied RVF5 can be used to detect chemically evoked hippocampal spiking activity in mouse brain slices, validating RVF5 as a tool for performing voltage imaging in thick brain tissue samples (Fig. 9.6). Next generations of two-photon VoltageFluor dyes will be targeted towards imaging voltage changes in live animals to provide complementary information to classical extracellular electrode-based recordings.

Two-photon voltage imaging with RVF5 in mouse brain slices. Transmitted light images of mouse hippocampal brain slice stained with 10 μM RVF5 in oxygenated ACSF show (a) the entire hippocampus and (b) a zoomed region of CA1. (c) Fluorescence signals from RVF5 (10 μM, oxygenated ACSF) from the same region in panel (b) show membrane-localized staining. (d) and (e) A zoomed-in region from panel (c) shows RVF5 fluorescence primarily in cellular membranes and excluded from the cytosol and (e) nuclear-localized mCherry-Cre indicating Tsc1 KO neurons. Functional imaging was performed by creating a 8 × 64 pixel region over an area of CA1 and imaging at ~200 Hz for 20–40 s, first in the absence of glutamate (panel f) and then following addition of glutamate to the perfusate (panel g (+)Glu). Responses were recorded from neurons in the sham injected (control, “WT”, black traces) and mCherry-Cre injected (“Tsc1 KO”, red traces) hemispheres. Scale bars are 100 μm for panel a and 20 μm for panels (b–e). Fluorescence traces are single-trial ΔF/F values from single pixels and are uncorrected for photobleaching
6 Targeting VoltageFluor Dyes to Specific Cells
Amphipathic VoltageFluor dyes stain tissue indiscriminately (Fig. 9.6c), making it difficult to resolve signals from individual cells and placing important constraints on in vivo and ex vivo experiments, where it is often desirable to image from sparsely labeled neurons, or a genetically defined sub-population therein. To overcome these issues we designed a small-molecule, photoactivatable optical sensor of transmembrane potential, or SPOT2.1.Cl, a dimly fluorescent, caged derivative of VF2.1.Cl which could release the functional parent dye upon irradiation with near-UV light, permitting optical recording from arbitrarily defined cells of interest (Grenier et al. 2015).
SPOT2.1.Cl is accessible in a single step from VF2.1.Cl through alkylation of the phenolic oxygen with 2-nitro-4,5-dimethoxybenzyl bromide. The quantum yield of SPOT2.1.Cl (Φ = 0.002) is 28 times lower than that of VF2.1.Cl (Φ = 0.057). Although the quantum yield of uncaging is low (Φ = 0.007) HPLC analysis of photolysis products indicated that VF2.1.Cl was cleanly released after irradiation. In HEK cells, we observed a maximal 12 ± 1.2 fold increase in fluorescence intensity after illumination at 390 nm. Fluorescence recordings indicated that voltage-sensitivity was maintained after triggered release of VF2.1.Cl – we measured a response of 17% ΔF/F per 100 mV, 77% of the response obtained from VF2.1.Cl under identical recording conditions. In cultured embryonic rat hippocampal neurons we measured a ΔF/F of 9 ± 0.2% in response to evoked action potentials, without a measurable change in action potential full width at half maximum. As an assessment of the effects on UV-irradiation and nitrobenzyl photochemistry on cell health we measured the electrophysiological parameters of HEK cells in whole-cell clamp before and after SPOT2.1.Cl uncaging and found no evidence that the membranes of target cells were compromised.
We next demonstrated our ability to uncage SPOT2.1.Cl with high spatial precision. Using either confocal microscopy or an iris diaphragm on a widefield microscope, we were able to release VF2.1.Cl with single cell precision in both HEK cells and cultured neurons. By illuminating over a small region of a single HEK cell we determined that released VF2.1.Cl diffuses laterally through cell membranes at an appreciable rate, gradually filling out the entire plasma membrane without a substantial increase in fluorescence in adjacent cells. To achieve our objective of selective recording from genetically defined populations of cells we envisioned using an expressed fluorescent protein as a fiduciary marker which could define regions for photoactivation. We sparsely transfected cultured neurons with a plasmid encoding mCherry, uncaged SPOT2.1.Cl in these cells, and recorded action potentials from target neurons (Fig. 9.7).

SPOT2.1.Cl uncaging in single neurons. Dissociated, cultured rat neurons (a) were transiently transfected with CAAX-mCherry for use as a fiducial marker for illumination. Panels (a) and (b) show DIC and mCherry fluorescence images, respectively. Cells were loaded with 500 nM SPOT2.1.Cl and then photoactivated (390 nm, 10 s, 22.3 W/cm2) over a region defined by the somatic staining of the mCherry signal (white arrow). (c) Following photoactivation, VF fluorescence appears to be membrane-localized. (d–f) EMCCD image of indicated cell, showing mCherry fluorescence. VF dye fluorescence from the mCherry-positive neuron, (e) prior to SPOT photoactivation and (f) immediately after. (g) Field stimulation of the SPOT-stained neuron at 5 Hz produced a train of optically recorded action potentials, which were captured at 500 Hz with an EMCCD camera. Scale bars are 20 μm for panels (a–c) and 10 μm for panels (d–f). (Reprinted with permission from Grenier, Walker, Miller, J. Am. Chem. Soc. 2015, 137, 10894. Copyright 2015 American Chemical Society)
Retrograde tracers of neuronal connectivity have proven to be valuable tools for studying brain physiology. We envisioned that SPOT2.1.Cl could serve as a dual use, functional tracer. We first used a modified fluorescence recovery after photobleaching assay over a monolayer of HEK cells to determine that while VF2.1.Cl does have some ability to travel across cellular membranes, it does so at the same rate as the canonical tracer dye DiO. In a proof-of-principle experiment we next released VF2.1.Cl in the processes of cultured neurons, allowing the dye to back-fill the cell body of a neuron outside the illuminated region, and recorded optical action potentials from that cell. Further development of photoactivatable VoltageFluors will require the use of photocages with high 2-photon cross-sections to enable photoactivation with single-cell resolution in tissue slice.
Based on the success of the photoactivation approach for staining single cells, we envisioned a similar strategy could be employed for enzymatic activation of voltage-sensitive dye, providing contrast without the need for a separate photoactivation step (Liu et al. 2017). To achieve targeting, the parent VF dye is chemically modified to be minimally fluorescent and must be enzymatically activated prior to imaging (Fig. 9.8a). We appended a bulky methylcyclopropyl ester (Tian et al. 2012), a moiety cleaved only by pig liver esterase (PLE), to VF2.1.Cl to prepare two dyes VF-EX1 and VF-EX2. While VF-EX1 is masked only at the phenol, VF-EX2 is esterified at both phenol and sulfonic acid positions. Compared to the parent dye, both VF-EX1 and VF-EX2 showed diminished fluorescence, with 19- and 6.7-fold decreases in quantum yield, respectively. Robust fluorescence turn-on was observed upon enzyme treatment, confirming that both dyes are PLE substrates in vitro. Kinetic studies were also performed and revealed that VF-EX2 is a better substrate than VF-EX1, as evidenced by the larger kcat/KM value for VF-EX2 (1.3 × 106 M−1 s−1) versus VF-EX1 (2.1 × 105 M−1 s−1).

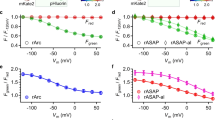
VoltageFluor targeted by esterase expression (VF-EX), a fluorogenic voltage reporter targeted to cells of interest. (a) Scheme of neuron-specific targeting with ester/esterase pairs (components are not to scale). Widefield fluorescence microscopy of cultured hippocampal neurons (b–e) stained with VF-EX2 (1 μM) show membrane-associated fluorescence only in cells expressing cell-surface PLE. (b) Epifluorescence images showing VoltageFluor-associated fluorescence. (c) Fluorescence image of nuclear-localized mCherry indicating PLE expression. (d) Transmitted light image of neurons. (e) Merged imaging showing overlay of both VF and mCherry fluorescence. Scale bar is 20 μm. ΔF/F vs. time plots for action potentials evoked and optically recorded with either VF-EX2/PLE (f), ASAP 1 (g), or Ace2N (h). (i–j) Quantification of ΔF/F (i) and signal-to-noise (SNR, j). (Data are mean ± S.E.M Adapted with permission from Liu, Grenier, Hong, Muller, and Miller, J. Am. Chem. Soc. 2017, 139, 17334. Copyright 2017 American Chemical Society)
We next engineered a cell-surface PLE by removing a ER retention signal from the native sequence and adding a secretion signal derived from immunoglobulin K (IgK) and a membrane targeting sequence (a transmembrane domain from platelet-derived growth factor receptor) or a glycophosphatidyl inositol anchor signal). These modifications resulted in clear membrane localization of the enzyme on the cell surface in both HEK cells and neurons as determined by immunostaining. Bath application of the dyes exhibited selective staining in PLE-expressing HEK cells, with a 7- and 17-fold turn-on for VF-EX1 and VF-EX2 respectively. The activated dyes displayed high and linear voltage sensitivities around 20% ΔF/F per 100 mV, comparable to the value of 27% measured for VF2.1.Cl. By using neuron specific promoters, Synapsin (pan-neuronal) or CamKIIα (excitatory neuron), we expressed PLE selectively in neurons of interest. VF-EX dyes provided enhanced contrast in transfected neurons (Fig. 9.8b–e) and were able to record field-stimulation electrode-evoked action potentials as well as spontaneous spiking events in single trials. VF-EX2 was selected for further applications due to its greater brightness.
Our dye displays improved SNR in cultured neurons relative to purely genetically-encoded voltage indicators (GEVIs) including ASAP1 and Ace2N (Fig. 9.8f–j). This is due in part to the improved membrane localization of uncaged VF-EX dyes. Whereas a significant fraction of expressed fluorescent GEVIs remains in the cytosol and internal compartments, contributing to unresponsive background fluorescence, the genetically-encoded component PLE is non-fluorescent and the dye only localizes to, but does not cross, the cell membrane. By targeting VF-EX2 to excitatory neurons, we could interrogate the neuromodulatory effects of serotonin (5-HT), an important neuromodulator. Treatment of hippocampal cultures with 5-HT showed decreased spiking rates in excitatory neurons while washout of 5-HT resulted in a recovery of neuronal spiking. Using pharmacological blockade of 5-HT receptors, we identified 5-HT1A as the receptor responsible for the inhibitory effect of 5-HT on neuronal activity. Current work focuses on extending this fluorogenic approach to other VF dyes with different chromophores and improved sensitivity as well as applying these probes for in vivo and ex vivo imaging.
7 Conclusion/Summary
Voltage imaging remains an outstanding challenge. Methods that can faithfully report on changes in membrane potential in a non-invasive, high-throughput fashion will be critical in dissecting the roles that membrane potentials play in biology. Optical imaging of voltage, whether through small molecule fluorescent dye approaches described here, or through other creative solutions (Abdelfattah et al. 2016; Akemann et al. 2013; Gong et al. 2015; Hochbaum et al. 2014; Jin et al. 2012; Kralj et al. 2011a, b; Piatkevich et al. 2018), must address problems of low voltage sensitivity, low brightness, poor membrane localization, and/or poor localization to cells of interest (Kulkarni and Miller 2017). In a related fashion, new modalities to enable high-speed optical sampling (~kHz) from deep brain regions, with high spatial resolution (μm) across large regions (mm) are required to fully realize the potential of voltage imaging.
References
Abdelfattah AS et al (2016) A bright and fast red fluorescent protein voltage indicator that reports neuronal activity in organotypic brain slices. J Neurosci 36:2458–2472. https://doi.org/10.1523/JNEUROSCI.3484-15.2016
Akemann W, Sasaki M, Mutoh H, Imamura T, Honkura N, Knopfel T (2013) Two-photon voltage imaging using a genetically encoded voltage indicator. Sci Rep 3:2231. https://doi.org/10.1038/srep02231
Bateup HS, Johnson CA, Denefrio CL, Saulnier JL, Kornacker K, Sabatini BL (2013) Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78:510–522. https://doi.org/10.1016/j.neuron.2013.03.017
Braubach O, Cohen LB, Choi Y (2015) Historical overview and general methods of membrane potential imaging. In: Canepari M, Zecevic D, Bernus O (eds) Membrane potential imaging in the nervous system and heart. Springer, Cham, pp 3–26. https://doi.org/10.1007/978-3-319-17641-3_1
Ceriani F, Mammano F (2013) A rapid and sensitive assay of intercellular coupling by voltage imaging of gap junction networks. Cell Commun Signal 11:78. https://doi.org/10.1186/1478-811X-11-78
De Silva AP, Gunaratne HQN, Habibjiwan JL, Mccoy CP, Rice TE, Soumillion JP (1995) New fluorescent model compounds for the study of photoinduced electron-transfer - the influence of a molecular electric-field in the excited-state. Angew Chem Int Edit 34:1728–1731. https://doi.org/10.1002/anie.199517281
Deal PE, Kulkarni RU, Al-Abdullatif SH, Miller EW (2016) Isomerically pure tetramethylrhodamine voltage reporters. J Am Chem Soc 138:9085–9088. https://doi.org/10.1021/jacs.6b05672
Dodani SC et al (2014) Copper is an endogenous modulator of neural circuit spontaneous activity. Proc Natl Acad Sci 111:16280–16285. https://doi.org/10.1073/pnas.1409796111
Dolensek J, Stozer A, Skelin Klemen M, Miller EW, Slak Rupnik M (2013) The relationship between membrane potential and calcium dynamics in glucose-stimulated beta cell syncytium in acute mouse pancreas tissue slices. PLoS One 8:e82374. https://doi.org/10.1371/journal.pone.0082374
Fathiazar E, Anemuller J, Kretzberg J (2016) Statistical identification of stimulus-activated network nodes in multi-neuron voltage-sensitive dye optical recordings. Conf Proc IEEE Eng Med Biol Soc 2016:3899–3903. https://doi.org/10.1109/EMBC.2016.7591580
Fu MY, Xiao Y, Qian XH, Zhao DF, Xu YF (2008) A design concept of long-wavelength fluorescent analogs of rhodamine dyes: replacement of oxygen with silicon atom. Chem Commun:1780–1782. https://doi.org/10.1039/B718544h
Gong Y, Huang C, Li JZ, Grewe BF, Zhang Y, Eismann S, Schnitzer MJ (2015) High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 350:1361–1366. https://doi.org/10.1126/science.aab0810
Grenier V, Walker AS, Miller EW (2015) A small-molecule photoactivatable optical sensor of transmembrane potential. J Am Chem Soc 137:10894–10897. https://doi.org/10.1021/jacs.5b05538
Hochbaum DR et al (2014) All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat Methods 11:825–833. https://doi.org/10.1038/nmeth.3000
Huang YL, Walker AS, Miller EW (2015) A photostable silicon rhodamine platform for optical voltage sensing. J Am Chem Soc 137:10767–10776. https://doi.org/10.1021/jacs.5b06644
Jin L, Han Z, Platisa J, Wooltorton JR, Cohen LB, Pieribone VA (2012) Single action potentials and subthreshold electrical events imaged in neurons with a fluorescent protein voltage probe. Neuron 75:779–785. https://doi.org/10.1016/j.neuron.2012.06.040
Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T (2011) Evolution of group 14 rhodamines as platforms for near-infrared fluorescence probes utilizing photoinduced electron transfer. ACS Chem Biol 6:600–608. https://doi.org/10.1021/cb1002416
Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D, Cohen AE (2011a) Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat Methods 9:90–95. https://doi.org/10.1038/nmeth.1782
Kralj JM, Hochbaum DR, Douglass AD, Cohen AE (2011b) Electrical spiking in Escherichia coli probed with a fluorescent voltage-indicating protein. Science 333:345–348. https://doi.org/10.1126/science.1204763
Kulkarni RU, Miller EW (2017) Voltage imaging: pitfalls and potential. Biochemistry 56:5171–5177. https://doi.org/10.1021/acs.biochem.7b00490
Kulkarni RU, Kramer DJ, Pourmandi N, Karbasi K, Bateup HS, Miller EW (2017a) Voltage-sensitive rhodol with enhanced two-photon brightness. Proc Natl Acad Sci U S A 114:2813–2818. https://doi.org/10.1073/pnas.1610791114
Kulkarni RU et al (2017b) A rationally designed, general strategy for membrane orientation of photoinduced electron transfer-based voltage-sensitive dyes. ACS Chem Biol 12:407–413. https://doi.org/10.1021/acschembio.6b00981
Li LS (2007) Fluorescence probes for membrane potentials based on mesoscopic electron transfer. Nano Lett 7:2981–2986. https://doi.org/10.1021/nl071163p
Liu P, Grenier V, Hong W, Muller VR, Miller EW (2017) Fluorogenic targeting of voltage-sensitive dyes to neurons. J Am Chem Soc 139:17334–17340. https://doi.org/10.1021/jacs.7b07047
Loew LM (2015) Design and use of organic voltage sensitive dyes. In: Canepari M, Zecevic D, Bernus O (eds) Membrane potential imaging in the nervous system and heart, Advances in experimental medicine and biology, vol 859. Springer, Berlin, pp 27–53. https://doi.org/10.1007/978-3-319-17641-3_2
McKeithan WL et al (2017) An automated platform for assessment of congenital and drug-induced arrhythmia with hiPSC-derived cardiomyocytes. Front Physiol 8:766. https://doi.org/10.3389/fphys.2017.00766
Miller EW, Lin JY, Frady EP, Steinbach PA, Kristan WB Jr, Tsien RY (2012) Optically monitoring voltage in neurons by photo-induced electron transfer through molecular wires. Proc Natl Acad Sci U S A 109:2114–2119. https://doi.org/10.1073/pnas.1120694109
Mirabdolbaghi R, Dudding T (2012) An indium-mediated allylative/transesterification DFT-directed approach to chiral C(3)-functionalized phthalides. Org Lett 14:3748–3751. https://doi.org/10.1021/ol301566f
Piatkevich KD et al (2018) A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat Chem Biol. https://doi.org/10.1038/s41589-018-0004-9
Poronik YM, Clermont G, Blanchard-Desce M, Gryko DT (2013) Nonlinear optical chemosensor for sodium ion based on rhodol chromophore. J Org Chem 78:11721–11732. https://doi.org/10.1021/jo401653t
Rodrigues GMC et al (2017) Defined and scalable differentiation of human oligodendrocyte precursors from pluripotent stem cells in a 3D culture system. Stem Cell Rep 8:1770–1783. https://doi.org/10.1016/j.stemcr.2017.04.027
Tian L et al (2012) Selective esterase-ester pair for targeting small molecules with cellular specificity. Proc Natl Acad Sci U S A 109:4756–4761. https://doi.org/10.1073/pnas.1111943109
Whitaker JE, Haugland RP, Ryan D, Hewitt PC, Prendergast FG (1992) Fluorescent rhodol derivatives: versatile, photostable labels and tracers. Anal Biochem 207:267–279
Woodford CR et al (2015) Improved PeT molecules for optically sensing voltage in neurons. J Am Chem Soc 137:1817–1824. https://doi.org/10.1021/ja510602z
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this paper

Cite this paper
Deal, P.E., Grenier, V., Kulkarni, R.U., Liu, P., Walker, A.S., Miller, E.W. (2020). Making Life Visible: Fluorescent Indicators to Probe Membrane Potential. In: Toyama, Y., Miyawaki, A., Nakamura, M., Jinzaki, M. (eds) Make Life Visible. Springer, Singapore. https://doi.org/10.1007/978-981-13-7908-6_9
Download citation
DOI: https://doi.org/10.1007/978-981-13-7908-6_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-7907-9
Online ISBN: 978-981-13-7908-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)