
Abstract
Sepsis is newly defined as life-threatening organ dysfunction caused by a dysregulated host response to infection. The pathophysiological mechanism of sepsis is highly complex, and the mortality of in-patients suffering from sepsis is more than 10%. Severe unmanaged inflammation and inappropriate immune response characterize sepsis. Anti-inflammation therapies alone are not successful for the reason that disbalance of anti-inflammatory and pro-resolving agents. In the recent researches, the host responses during the course of self-resolving infections are found to have the involvements of specialized pro-resolution mediators (SPMs), namely, lipoxins, resolvins, protectins and maresins. These endogenous lipid metabolites are core signal molecules in the resolution of inflammation, playing a key role in regulating the inflammation and promoting return to homeostasis. Besides, heme oxygenase-1 (HO-1, a sensitive marker for oxidative stress) is also known for upregulation in inflammation profiling. Carbon monoxide, synthesized by HO-1, performs multiple stances of anti-inflammation and pro-resolution along with the SPMs. If the potentially beneficial effects of these mediators would be well evaluated in clinical trials, they present encouraging new hints in managing infectious maladies especially sepsis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
8.1 Introduction
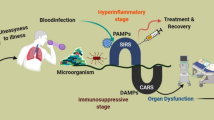
Sepsis is a complicated series of pathophysiological and biochemical disbalance due to the uncontrolled inflammatory response to the invasive pathogens. Its newest definition is defined as organ dysfunction due to infection, which remains a major public health concern that is associated with poor clinical outcomes (in-hospital mortality is more than 10%) and substantial healthcare expenditures [1]. Data showed that the cost of medical care for sepsis patients was more than $20 billion (5.2%) of total US hospital costs in 2011 [2]. Although the accurate incidence of sepsis is unknown, the reported data are increasing [3, 4], and conservative estimates indicate that sepsis is a leading cause of mortality in the US hospitals and critical illness in the world [5, 6].
The inflammatory host response was long considered as a passive process, which is terminated by the clearance of pro-inflammatory mediators. Recently, it is recognized that the initiation and cessation of immune and inflammatory responses is an initiative and symphony process. The pathogenesis of sepsis is intricate. The early stages of sepsis are characterized by excessive generation of inflammatory mediators; however, as sepsis develops into severe chronic sepsis, immunosuppression dominates. This demonstrates that excessive uncontrolled inflammation and inappropriate immune response characterize sepsis [7]. The new lights into sepsis contribute to the explanation of failure in current treatment strategies, including inhibition of the activation phase of the acute inflammatory response to infection (e.g. glucocorticoids, nonsteroidal anti-inflammatory drugs and anti-TNF-α drugs) [8]. Therefore, it’s urgent and beneficial to explore new paradigms in managing sepsis.
As of recent studies relating the host response to a self-resolving infection, it is brought into light that a genus of endogenous bioactive lipid mediators (SPMs) is produced by the innate immune system cells. A process of stereoselective enzyme conversion of essential fatty acids including arachidonic acid (AA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and n-3 docosapentaenoic acid (n-3 DPA) produces these molecules. The synthetases include cyclooxygenase (COX), lipoxygenase (LOX) and cytochrome P-450 monooxygenase. The SPMs can further be subdivided into lipoxins, resolvins, protectins, maresins and immunoresolvents (RvTs, PCTRs and MCTRs) (Fig. 8.1) [9,10,11,12].

SPMs derived from essential fatty acids and their receptors
Through the G-protein-coupled receptor-dependent manner, these SPMs exhibit the fundamental bioactivities in the maintaining of host responses. Hence, the productions of pro-inflammatory cytokines (TNF-α and IL-1β) and chemokines are inhibited, and the downregulation of inflammation-initiating eicosanoids (prostaglandins and leukotrienes) is promoted. This as well regulates the leukocyte infiltration and stimulates the macrophage efferocytosis of apoptotic cells and debris [10, 11]. Recent studies have shown that the SPMs exhibit beneficial impacts at the site and in course of sepsis while maintaining the homeostasis between the abundance of microorganisms and the host. In this chapter, we aim at:
-
(1)
The functions of SPMs in sepsis.
-
(2)
Because aspirin partakes in the synthesis of lipoxins and resolvins, studies related to aspirin-triggered lipid mediators are included [13].
-
(3)
Protective effects of carbon monoxide (CO) on inflammation and its impacts of enhanced productions of pro-resolving lipids [14, 15]. Therefore carbon monoxide and its synthetase HO-1 are summarized.
-
(4)
Since Annexin (A1) is a stress response protein, the current evidence for beneficial pro-resolving effects of this mediator in sepsis is also addressed in this chapter [16].
8.2 Mediators Related to Resolution of Inflammation
8.2.1 Lipoxins
Lipoxins (trihydroxy-tetraene-containing eicosanoids) are derived from the omega-6 polyunsaturated fatty acid arachidonic acid (AA) through sequential reactions involving lipoxygenase enzymes, including 5-lipoxygenase (5-LOX), 12-lipoxygenase (12-LOX) and 15-lipoxygenase type 1 (15-LOX-1). There are two series of lipoxins, lipoxin-A4 (LXA4) and lipoxin-B4 (LXB4) [17]. We mainly summarize the activities of LXA4 in this chapter.
Lipoxins exert effects through a G-protein-coupled receptor, formyl peptide receptor-2 (FPR2/ALX). This receptor mainly expresses on the membrane of cells in the immune system along with the resident fibroblasts and epithelial cells [18]. In general, lipoxins are potent inhibitors of inflammatory activity, inducing the resolution of leukocyte activity. They block the response to leukotrienes and chemotactic factors such as leukotriene B4 (LTB4) and platelet-activating factor [19]. These effects inhibit the infiltration of pro-inflammatory cells. The resolution of inflammation involves reduction of neutrophil recruitment, promotion of macrophage migration and augmentation of apoptotic cells efferocytosis. Lipoxins exert immune-modulatory activity as well [17, 18].
LXA4, treated after injury, was proved to limit inhaled LPS-induced lung injury [20]. In other experimental models, 5 h after caecal ligation and puncture (CLP), LXA4 was administered to rats which resulted in decreasing of plasma IL-6, chemokine (C-C motif) ligand 2 (CCL2) and IL-10 and reducing NF-κB activity in peritoneal macrophages. LXA4 further enhanced the phagocytosis of macrophage recruited to the peritoneum and reduced bacterial load in the blood. Hence, LXA4 improved the mortality of this CLP model [21].
Further study showed that flavocoxid, a dual inhibitor of COX-2 and 5-LOX, reduces the expression of NF-κB, COX-2 and 5-LOX which improves survival rate in a murine CLP sepsis model. Plasma IL-10 and LXA4 concentrations are increased, while tumour necrosis factor-α (TNF-α), IL-6, LTB4 and PGE2 are decreased [22].
Neutrophil phagocytosis involving the Fc receptor I (CD64) is increased and enhanced by LXA4. Meanwhile, LXA4 decreases the release of the exotoxin pyocyanin by Pseudomonas aeruginosa, reducing its pathogenicity [23]. Moreover, LXA4 blocks trafficking of neutrophils, inhibits their adhesion and their release of azurophilic granules. And in activated T-cells, LXA4 lowers TNF release [24]. The finding shows that LXA4 not only modulates the host response but also affects bacterial toxicity [23].
A test was run to investigate the role of receptor FPR2/ALX in mediating the protective effects in sepsis using the receptor agonist BML-111. It ameliorates intestinal inflammation in septic rats. The anti-inflammatory cytokine transforming growth factor-β (TGF-β) which is supposed to protect intestinal cells from apoptotic cell death was induced [25]. In a murine model of non-lethal polymicrobial sepsis, FPR2/ALX-deficient animals developed more serious disease and exhibit higher cytokine levels and reduced recruitment of monocytes in peritoneal lavages. Treatment with an FPR2/ALX agonist protected wild type but not the knock-out mice from cardiac dysfunction [26]. These findings prove that LXA4 protect from organ dysfunction, a major cause of mortality in sepsis [27].
As known to all, acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS) are major common complications of severe sepsis [27]. With the inoculation of Klebsiella pneumoniae in mice, pulmonary sepsis occurred and induced LXA4 and FPR2/ALX expression in the lung. Later, the treatment of receptor antagonists and inhibition of 5-lipoxygenase and 15-lipoxygenase in early sepsis (1 h postinfection) even increased leukocyte migration to the infected tissues, and survival rate increased. On the contrary, receptor agonist and LXA4 application consequently worsened early infection and reduced migration of leukocytes. But, 24 h postinfection, LXA4 improved animal survival. Here, this research demonstrates the dual role of LXA4 and highlights the time dependence when targeting the LXA4 pathway in pulmonary infection [28].
In the case of septic patients, all the mediators identified in preclinical studies and tested for the treatment in clinical trials have failed [29]. Pro-resolving lipid analysis in critically ill patients may reveal a novel orientation for treatment and bring in further insights into the pathways playing a role in the pathophysiology of sepsis. While comparing to 27 non-survival septic patients for 28 days of admission to the intensive care unit, LXA4 was significantly reduced in 39 patients that survived, but levels of this lipid were not associated with death [30].
8.2.2 Resolvins
Resolvins are also derived from omega-3 polyunsaturated fatty acids and exist as two series (D and E). E-series resolvins (RvE1 and RvE2) are products of eicosapentaenoic acid (EPA) involving 5-LOX, cytochrome P450 and aspirin-acetylated COX-2 as well. D-series resolvins (RvD1–RvD6) are synthesized from docosahexaenoic acid (DHA) metabolism involving enzymes 5-LOX/15-LOX [13, 31]. The biological activities of resolvins are similar to lipoxins. RvE1 and its analogues are more potent than LXA4 on a molar basis. RvE1 binds to the leukotriene receptor BLT1 and blocks TNF-α-stimulated NF-κB activation at the ChemR23 receptor [32].
In a mouse model of aspiration pneumonia and subsequent involvement of one lobe with E. coli infection, the function of RvE1 in acute lung injury was analysed and found that the RvE1, when injected before the acid injury, reduced pulmonary neutrophil infiltration and enhanced bacterial clearance. This was accompanied by lower levels of inflammatory cytokines and chemokines and marginally improved survival rate [33].
In other two murine models of acute lung injury, RvE1 enhanced cell death of neutrophils arising from the phagocytosis of opsonized E. coli or yeast and is mediated by the leukotriene B4 receptor BLT1. Consequently, RvE1 enhanced the resolution of the established pulmonary inflammation [34]. LXA4, RvE1 and protectin D1 increase levels of the C-C chemokine receptor 5 (CCR5) on apoptotic polymorphonuclear cells (PMNs) and thereby terminate chemokine signalling [35]. RvE1 and 15-epi-lipoxin protect macrophages from oxidative stress-associated apoptotic cell death, and this contributes to the removal of cytotoxic debris and the inflammation resolution [36, 37].
D-galactosamine-sensitized mouse endotoxin shock mode was also tested for the effects of RvD1, which counteracted the induction of high-mobility group box-1 (HMGB1) and pro-inflammatory cytokines. Hepatocyte apoptosis was suppressed, and also neutrophil immigration to the peritoneum was reduced by the effects of RvD1 [38].
In a mouse model of intraperitoneal E. coli peritonitis, RvD5 enhanced phagocytosis of bacteria compared to the control group. RvD1 had a similar but smaller effect. Both RvD1 and RvD5 reduced significantly titre of viable bacteria in peritoneal exudates and blood and lowered degree of hypothermia as well. Plasma levels of pro-inflammatory cytokines (TNF-α and IL-1β) were reduced by RvD1 and RvD5. Interestingly, it was found that RvD1 enhanced the antimicrobial effect of ciprofloxacin in resolving E. coli peritonitis and increased survival rate in this model [39].
Administration of resolvin D2 could improve outcomes of burn-related sepsis by regulating PMN chemotaxis. In a rat model of burn-related sepsis, RvD2 restored the chemotaxis of PMN to almost normal level. Furthermore, when burned rats received intravenous LPS 9 days after their burn injury, with intravenous pretreated RvD2, the survival of rats improved significantly. Similarly, RvD2 pretreatment increased survival, following caecal ligation after burn injury [40].
RvD1 injected after CLP model of sepsis increased bacterial clearance and mice survival. The numbers of peritoneal neutrophils were decreased, while CD3 T-lymphocytes apoptosis in thymus got significant improvement [41]. When AT-RvD1 was administered 1 h after the toxin in a LPS-induced acute kidney injury mouse model, renal function was improved. Lower expression of adhesion molecules, less activation of NF-κB and reduced infiltration of neutrophils have been reported as well [42].
Previous studies have shown that IV administration of RvD2 on a CLP sepsis model exhibits the following protective pro-resolution effects and increases survival rate: (1) reduce viable aerobic bacterial load in peritoneal exudates and blood; (2) reduce PMN migration into the peritoneum; (3) reduce plasma levels of IL-10 and IL-17; (4) reduce pro-inflammatory cytokine (IL-6, IL-1β, IL-23 and TNF-α) levels in plasma and peritoneum; (5) reduce concentrations of the pro-inflammatory lipids PGE2 and LTB4; while (6) increase clearance of bacteria by phagocytes in inguinal lymph nodes and in vitro; (7) enhance phagocytosis of E. coli by human PMN and also increase intracellular production of reactive oxygen species; and (8) increase survival as a result [43].
Inflammatory pain is mainly caused due to the activation of transient receptor potential subtype vanilloid 1 (TRPV1) and TRP ankyryn 1 (TRPA1). RvD2 is a potent inhibitor of both channels in primary sensory neurons, while RvE1 inhibits TRPV1 and RvD1 inhibits TRPA1, respectively. Hence, these lipids contribute to pain-relieving activities [44].
The potential therapeutic uses of exogenous resolvins are currently under investigation. In a recent phase 2 clinical trial involving patients with dry eye syndrome, an RvE1 analogue significantly improved signs and symptoms of corneal inflammation. This is the first trial to show the clinical efficacy of the novel class of resolvins therapeutics that stimulate resolution rather than inhibit inflammatory mediators [14].
8.2.3 Protectins
Protectins are also omega-3 polyunsaturated fatty acid derivatives, generated from docosahexaenoic acid (DHA) through 12-LOX/15-LOX-mediated pathways. Neuroprotectin D1 (NPD1) and protectin D1 (PD1) are included [45].
In a severe human influenza model, H1N1-A virus was introduced through the intratracheal route in mice, and PD1 levels were found to be reduced, suggesting that endogenous production was suppressed by the virus. When other influenza A virus strains were tested, the reduction of PD1 was found to inversely correlate with the virulence of the virus strain used. In the same model of severe human influenza, intravenous administration of PD1 reduced in vitro replication of the H1N1 influenza A virus and increased survival. Besides, When PD1 was administered in combination with the antiviral agent peramivir, a dramatic increase in survival was found compared to use of peramivir alone [46].
Protectin DX (PDX, an isomer of protectin D1) also exhibits protective effects of anti-inflammation and pro-resolution. In a CLP sepsis mice model, PDX increased overall survival rate and attenuated multiple organ injury. In addition, PDX reduced pro-inflammatory cytokines and bacterial load 24 h after CLP. Moreover, PDX promoted phagocytosis of peritoneal macrophages and increased the percentage of M2 macrophages in peritoneum of septic mice [47].
In a recent report encompassing 22 sepsis patients, it was shown that plasma levels of inflammation-initiating mediators including PGF2α and LTB4 and pro-resolving mediators, including RvE1, RvD5 and 17R-PD1, were significantly higher in non-survivors than in surviving sepsis subjects. Further analysis revealed increased respiratory failure in non-survivors. These results indicate that peripheral blood lipid mediator profiles (RvE1, RvD5 and 17R-PD1) in sepsis correlate with survival and ARDS development, thus suggesting plausible novel biomarkers and biological targets for critical illness [48].
8.2.4 Maresins
Maresins are a new family of anti-inflammatory and pro-resolving lipid mediators derived from docosahexaenoic acid (DHA) by macrophages via human 12-lipoxygenase (12-LOX). The first member of this family, termed as maresin 1 (MaR1), exhibits potent phagocyte-directed actions that include inhibition of neutrophil infiltration and stimulation of macrophage efferocytosis by dihydroxyl products in this pathway [10, 49, 50]. In a murine model of ARDS, lipid mediator metabololipidomics discovered that MaR1 was temporally generated and regulated in vivo. Early intravascular MaR1 was organ-protective, and MaR1 production was dependent on platelet-neutrophil interactions leading to reduced lung neutrophils, oedema, tissue hypoxia and inflammatory mediators [51].
Pro-resolution effects are activated to terminate inflammation as soon as the inflammatory response initiates. In a peritonitis model, MaR1 was one of the first SPMs upregulated in the peritoneum during self-resolving infections. Acting as a partial agonist/antagonist to the LTB4 receptor (BLT1), MaR1 supressed the activity of LTB4 so as to promote the uptake and clearance of apoptotic cells and bacteria [52]. Interestingly, levels of MaR1 and LTB4 reached a maximum level in the early stages of the inflammatory response, suggesting that early MaR1 production impacts leukocyte infiltration to the inflammatory site [39, 52].
In a LPS-induced ALI mice model, high dose of MaR1 exhibited protective activities by mitigating patho-histological changes, attenuating pulmonary oedema and restoring oxygenation. Besides, high-dose MaR1 inhibited the increasement of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6) and chemokines, while anti-inflammatory cytokine IL-10 was upregulated. Moreover, MaR1 lowered LPS-induced neutrophil adhesions and suppressed the expression of intercellular adhesion molecule (ICAM)-1, P-selection and CD24 [53, 54]. Another study reported that MaR1 can maintain the permeability of lung epithelial cells by upregulating the expression of claudin-1 and ZO-1 in LPS-induced ALI [55].
In a CLP sepsis mice model, it was found that MaR1 markedly mitigated the levels of pro-inflammatory cytokines (TNF-α, IL-1β and IL-6). Intervention of MaR1 lowered the LPS level in serum and enhanced the bacterial clearance. Furthermore, MaR1 attenuated lung injury and decreased serum level of alanine transaminase (ALT), aspartate transaminase (AST), creatinine (Cre) and blood urea nitrogen (BUN) in this sepsis model. Consequently, the survival rate was improved. Inhibition of NF-κB activation pathway by MaR1 is the possible protective mechanism [56].
Maresin 2 (MaR2) was identified later. MaR2 exhibits similar potency to MaR1 in limiting PMN recruitment but has an apparent optimal concentration 2–3 log orders lower than MaR1 in enhancing human macrophage phagocytosis of zymosan. MaR2 also enhanced human macrophage uptake of apoptotic PMN but was less potent than MaR1 [57].
8.2.5 Aspirin-Triggered Lipid Mediators
Aspirin is a classic anti-platelet anti-inflammatory agent. Acetylating COX-2 facilitates aspirin to induce a shift from the synthesis of pro-inflammatory to pro-resolving lipid mediators termed as aspirin-triggered lipoxins (AT-LX) and aspirin-triggered resolvins (AT-Rv) [13, 58]. AT-LX and AT-Rv share the pro-resolution effects of LXA4 and RvD1, respectively, and act by the same intracellular pathways [59].
The aspirin-triggered lipoxin, also known as 15-epi-LXA4 (AT-LXA4), was found to be increased with administration of aspirin in both in vitro and in vivo models of infection; with infection alone increase in 15-epi-LXA4 levels was observed [60]. In murine models of sepsis and ARDS, aspirin increased the survival rate effectively [61]. Low dosage of aspirin administered 30 min prior to the endotoxin model of sepsis improved the survival and reduced the levels of thromboxane and prostaglandins derived from arachidonic acid [62]. Another research indicated that low-dose aspirin, however, did not reduce cytokine and prostaglandin levels while enhancing 15-epi-LXA4 synthesis [63]. In an E. coli sepsis model, 15-epi-LXA4 injected 24 h after the injury lowered PMN number in broncho-alveolar lavage by stimulating the apoptosis [64].
Several observational clinical studies have confirmed the benefits of aspirin in those who took it prior to medical consult in sepsis patients. In a group of 1149 critically ill patients, 25% of them with preclinical aspirin use had a decreased risk of developing to ARDS and tended to a lower mortality [65]. A different study with 5523 patients suffering from systemic inflammatory response syndrome or sepsis suggested that a lower mortality was found in 2082 patients when given aspirin within 24 h after diagnosis [66]. Similarly, in a study comprising 1005 patients with community-acquired pneumonia, 100 mg/day intake of aspirin was associated with a lower mortality rate within 30 days [67]. Hence, a clinical trial indicates that the aspirin and essential fatty acid supplemented in healthy human volunteers increase the endogenous production of pro-resolving mediators including 17-R-PD1 and augment bacterial clearance of leukocytes [68].
However, a multicentre study with 3855 patients did not pinpoint a significant association between preclinical aspirin therapy and progression to ARDS [69]. A prospective observational study with 972 patients indicated that preclinical aspirin therapy was neither associated with the development of organ failure nor shock nor 90-day mortality up to 90 days after hospitalization [70].
Evidence above does not make a solid statement of conclusion on exact beneficial prognoses of aspirin therapy in sepsis. Further studies are required to evaluate a potential protective effect of aspirin in sepsis patients.
8.2.6 Novel Families of SPMs
8.2.6.1 RvTs
A novel family of pro-resolving mediators termed 13 series resolvins (RvTs) were identified recently in the very early stages (≤4 h) of self-resolving E. coli infections model. These RvTs mediators include RvT1, RvT2, RvT3 and RvT4, which are derived from n-3 docosapentaenoic acid (n-3 DPA) through sequential reactions involving COX-2/5-LOX in the crosstalk of vascular endothelia and neutrophils. Each of these molecules activates the host immunity in a dose-dependent manner for bacterial clearance and counter-regulates the production of pro-inflammatory molecules including endothelin-1, plasminogen activator inhibitor-1 and inflammatory eicosanoids. Moreover, concentrations of RvTs in peripheral blood increased rapidly after exercise (a self-resolving inflammatory state) in healthy volunteers and were significantly higher in patients with sepsis than in healthy subjects. These results indicate that RvTs biosynthesis occurs in a coordinated manner in correlation with acute activation of the immune response. Therefore, inability to form these pathways leads to delayed resolution responses and an impaired ability to clear bacterial infections [71].
8.2.6.2 PCTRs
Recent studies suggest that the formation of the immunoresolvent protectin conjugate in tissue regenerations (PCTRs: PCTR1, PCTR2 and PCTR3) by group 3 innate lymphoid cells (ILC-3), which in turn regulates peritoneal macrophage responses to bacterial infections, is promoted by the vagus nerve [72]. PCTRs are derived from DHA through 15-LOX-mediated pathways in leukocytes. Since PCTRs actively promote the termination of bacterial infections by stimulating the uptake and killing the bacteria as well as the repair and regeneration of damaged tissues, these mediators are immunoresolvents [73]. PCTRs parallelly promote a macrophage phenotype shift, downregulating the production of pro-inflammatory cytokines including TNF-α and IL-8 and increase the production of regenerative molecules including TGF-β [74]. Any form of damage to the vagus reflex dysregulates PCTRs formation and macrophage phenotype leading to an impaired ability of the recruited leukocytes to efficiently clear pathogens causing the delayed resolution of inflammatory infections [72].
8.2.6.3 MCTRs
Further researches into mediators formed during the late phases of resolution of E. coli infections pointed out a huge group of molecules that are peptide-lipid conjugates. These molecules termed as maresin conjugates in tissue regeneration (MCTRs: MCTR1, MCTR2 and MCTR3) are produced by 14-lipoxygenation of DHA through 12-LOX-mediated pathways in human macrophages. MCTRs exhibit some potential benefits in regulating bacterial phagocytosis, promoting tissue repair and regeneration [74, 75].
Both PCTRs and MCTRs were found in inflammatory exudates and spleens from infected mice as well as in human plasma, serum and spleens. The levels of these mediators were increased during the later stages of infectious inflammation in mice [73, 74].
8.2.7 Carbon Monoxide and Heme Oxygenase
Carbon monoxide (CO) enhances the immune cell function. CO is produced by the constitutively expressed heme oxygenase-2 (HO)-2, and by HO-1, which is upregulated upon cellular stress. HO-1 catalyses the degradation of heme to biliverdin, CO and iron (which binds to ferritin), all acting as anti-oxidative and anti-inflammatory agents [76]. Endogenously formed CO acts as a signalling molecule and induces antioxidant genes [76, 77]. CO downregulates inflammatory prostaglandins and thromboxanes. On the other hand, it enhances the expression of lipoxygenases which are key synthetases of SPMs [15]. Resolvins and lipoxins in turn upregulate HO-1 in macrophages demonstrating mutual amplification of these two pro-resolving pathways [15]. CO contributes to active pathways on the elimination of microorganisms, promote killing of bacteria and enhance their clearance by macrophages in a protective manner [78]. Furthermore, pro-inflammatory cytokines including TLR2, -4, -5 and -9 are repressed, while anti-inflammatory cytokines such as IL-10 are initiated by CO in macrophages [79]. Enhanced endocytosis of apoptotic cells by efferocytosis is also known [15].
In murine sepsis model, higher mortality and hepatic necrosis in HO-1-deficient mice group were found pointing out the protective function of this enzyme [80]. Hepatoprotective effects were reported with increasing levels of CO in endotoxic rats [81]. Hepatic accumulation of PMN, expression of the intercellular adhesion molecule-1 (ICAM-1) and activation of NF-κB in murine polymicrobial sepsis are reduced by CO. In endotoxin-activated human umbilical vein, the endothelial cells cocultured with CO, the production of reactive oxygen species (ROS), nitric oxide (NO), activation of NF-κB, induction of inducible NO synthase and ICAM-1 and PMN adhesion were reduced [82].
In CLP mice model, levels of circulating inflammatory cytokines and the number of bacteria in blood and organs were decreased by the inhalation of CO which resulted in the increased survival rate of the mice [83]. CO inhalation, 2 h prior to initiation of peritonitis, lowered the number of infiltrating PMN. Monocyte numbers remained unchanged, while the clearance of microbial pathogens and dead PMN significantly raised [15]. Inflammation resolved nearly two times faster than CO nonexposed group. Lipid mediators derived from arachidonic acid, LTB4 and PGE were low in the early stage of inflammation and pro-resolving lipids including RvD1, RvE2 and maresin, were markedly high [15].
The clinical patients have been observed for the protective roles of CO and HO-1. Patients with lower respiratory tract infection indeed had higher CO in their breath, and levels descended in those patients recovering from disease after antibiotic treatment [84]. When compared to 30 healthy neonates, CO in plasma of 7 neonates with sepsis was significantly increased [85]. When 36 patients with severe sepsis or septic shock were compared to 21 patients without sepsis, arterial blood CO and HO-1 protein levels in monocytes were ascended. These two molecules were positively related to survival [86]. Exhaled CO is higher in severely ill patients compared to the control group [87]. In mechanically ventilated patients with severe sepsis or septic shock, exhaled CO was nearly threefold higher compared to controls and declined along with therapy, and high amount of CO in exhaled air on the first day during treatment was associated with better outcome [88]. CO is reported to have beneficial impacts in different patient groups; therefore, inhalation of CO would be a potential policy guide for the treatment of sepsis [77].
8.2.8 Annexin A1
Annexin A1 is a 37 kDa monomeric protein, existing abundantly in some pivotal cell types of the innate and adaptive immune systems and the neuroendocrine system. The synthesis and release of Annexin A1 is regulated by glucocorticoids (GCs) and expresses as a stress response protein through binding to formyl peptide family receptors (FPRs) [89].
An increasing number of experimental evidence in present days indicate that Annexin A1 is crucial to many of the acute actions of GCs in several systems relevant to the stress response, including the innate and adaptive host immune systems [89, 90] and the HPA axis [91]. In Annexin A1-null mice models of inflammation, treating with anti-Annexin A1 antibodies or antisense constructs of Annexin A1, have confirmed that Annexin A1 exhibits as an endogenous regulator of anti-inflammatory and pro-resolving as well as a mediator of GC action [89, 90]. Annexin A1, binding to FPR in the innate immune system, exerts general suppressive activities including the reduction of the amounts of pro-inflammatory eicosanoids molecules generated and the release of histamine and preformed cytokines from mast cells. In contrast, ANX-A1 may induce the release of pro-resolving mediators, such as IL-10. Furthermore, Annexin A1 highly downregulates PMN migration into inflammatory sites and accelerates their apoptosis, while the migration of monocytes into inflammatory sites is promoted by Annexin A1 which eventually transform the microenvironment into a resolving procedure [92]. In an in vivo experiment, the absence of Annexin A1 or its major receptor (FPR2) amplifies greatly the duration and intensity of acute and chronic inflammation. Consequently, the acute anti-inflammatory actions of GCs are greatly reduced or even abrogated. Conversely, human recombinant Annexin A1 and peptides derived from the N-terminal domain can rescue this phenotype and exert high anti-inflammatory and pro-resolution effects in many models of inflammation [90, 92].
8.3 Summary and Prospect
Sepsis is a severe organ dysfunction syndrome due to uncontrolled infection with a high mortality risk. Despite of inspiring results in preclinical models, none of the presumptive therapeutic agents tested so far succeed in clinical trials. Excessive uncontrolled inflammation and inappropriate immune responses are both characteristic features of sepsis that complicate identification of suitable drug targets. Inhibition of inflammation may delay the resolution because of an improper induction of anti-inflammatory and pro-resolving pathways. Overcoming sepsis, there is still a long way to run.
An accumulating evidence decodes the role that endogenous resolutions triggered by SPMs demonstrate the ensured internal homeostasis in host body by activating the ultimate cardinal signs of resolution that include pathogen and cellular debris lavage, analgesia and restoration of organ functions. Latest studies point out that monitoring the concentrations of these SPMs in infected patients may provide a better understanding of the inflammation-resolution procedures and hence reflect a positive outcome [48]. Yet, requiring further prospective studies to confirm these observations.
The points and the hints that are mentioned in this review indicate that SPMs act as potential biomarkers and, more importantly, hold a great promise as novel therapeutics in inflammatory diseases. Latest studies reveal that the majority of end-stage fatal sepsis patients are immunosuppressed rather than hyperinflammation [93]. Given, the potential anti-inflammatory and pro-resolution benefits of these SPMs (including CO, HO-1 and Annexin A1) mentioned above, endogenous or exogenous supplement of these mediators analogues or natural extracts may be a new paradigm in treating sepsis.
References
Singer M. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–10.
Torio CM. National inpatient hospital costs: the most expensive conditions by payer, 2011. Statistical Brief #160. Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. 2013. http://www.ncbi.nlm.nih.gov/books/NBK169005/.
Iwashyna TJ. Population burden of long-term survivorship after severe sepsis in older Americans. J Am Geriatr Soc. 2012;60(6):1070–7.
Gaieski DF. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med. 2013;41(5):1167–74.
Vincent J, ICON Investigators. Assessment of the worldwide burden of critical illness: the Intensive Care Over Nations (ICON) audit. Lancet Respir Med. 2014;2(5):380–6.
Fleischmann C, International Forum of Acute Care Trialists. Assessment of global incidence and mortality of hospital-treated sepsis: current estimates and limitations. Am J Respir Crit Care Med. 2016;193(3):259–72.
Das UN. Is sepsis a pro-resolution deficiency disorder? Med Hypotheses. 2013;80(3):297–9.
Lee CR. Resolvin infectious inflammation by targeting the host response. N Engl J Med. 2015;373(22):2183–5.
Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101.
Serhan CN. Protectins and maresins: new pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta. 2015;1851(4):397–413.
Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. 2017;31(4):1273–88.
Dalli J. Does promoting resolution instead of inhibiting inflammation represent the new paradigm in treating infections? Mol Aspects Med. 2017;58:12–20. pii: S0098-2997(17)30021–3.
Serhan CN. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196:1025–37.
Shinohara M. Novel endogenous proresolving molecules: essential fatty acid-derived and gaseous mediators in the resolution of inflammation. J Atheroscler Thromb. 2016;23(6):655–64.
Chiang N. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J Immunol. 2013;190:6378–88.
Gobbetti T. Annexin A1 and resolution of inflammation: tissue repairing properties and signalling signature. Biol Chem. 2016;397:981–93.
Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids. 2005;73:141–62.
Serhan CN. Novel anti-inflammatory--pro-resolving mediators and their receptors. Curr Top Med Chem. 2011;11(6):629–47.
Psychogios N. The human serum metabolome. PLoS One. 2011;6(2):e16957.
Jin SW. Posttreatment with aspirin-triggered lipoxin A4 analog attenuates lipopolysaccharide-induced acute lung injury in mice: the role of heme oxygenase-1. Anesth Analg. 2007;104:369–77.
Walker J. Lipoxin A4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock. 2011;36:410–6.
Bitto A. Flavocoxid, a dual inhibitor of COX-2 and 5-LOX of natural origin, attenuates the inflammatory response and protects mice from sepsis. Crit Care. 2012;16:R32.
Wu B. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J. 2016;30:2400–10.
Ariel A. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J Immunol. 2003;170:6266–72.
Liu H. Effect of BML-111 on the intestinal mucosal barrier in sepsis and its mechanism of action. Mol Med Rep. 2015;12:3101–6.
Gobbetti T. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc Natl Acad Sci U S A. 2014;111:18685–90.
Khadaroo RG. ARDS and the multiple organ dysfunction syndrome: common mechanisms of a common systemic process. Crit Care Clin. 2002;18:127–41.
Sordi R. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int Immunopharmacol. 2013;7:283–92.
Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20:195–203.
Tsai WH. Plasma levels in sepsis patients of annexin A1, lipoxin A4, macrophage inflammatory protein-3a, and neutrophil gelatinase-associated lipocalin. J Chin Med Assoc. 2013;76:486–90.
Serhan CN. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111:5922–43.
Aoki H. Protective effect of resolvin E1 on the development of asthmatic airway inflammation. Biochem Biophys Res Commun. 2010;400(1):128–33.
Seki H. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol. 2010;184:836–43.
El Kebir D. Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109:14983–8.
Ariel A. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7:1209–16.
Lee HN. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem Pharmacol. 2013;86:759–69.
Prieto P. Activation of autophagy in macrophages by pro-resolving lipid mediators. Autophagy. 2015;11:1729–44.
Murakami T. Suppressive action of resolvin D1 on the production and release of septic mediators in D-galactosamine-sensitized endotoxin shock mice. Exp Ther Med. 2011;2:57–61.
Chiang N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature. 2012;484:524–8.
Kurihara T. Resolvin D2 restores neutrophil directionality and improves survival after burns. FASEB J. 2013;27:2270–81.
Chen F. Resolvin D1 improves survival in experimental sepsis through reducing bacterial load and preventing excessive activation of inflammatory response. Eur J Clin Microbiol Infect Dis. 2014;33:457–64.
Chen J. Aspirin-triggered resolvin D1 down-regulates inflammatory responses and protects against endotoxin-induced acute kidney injury. Toxicol Appl Pharmacol. 2014;277:118–23.
Spite M. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–91.
Park CK. Resolvin D2 is a potent endogenous inhibitor fortransient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. J Neurosci. 2011;31:18433–8.
Serhan CN. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. 2006;176:1848–59.
Morita M. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 2013;153:112–25.
Xia HF. Protectin DX increases survival in a mouse model of sepsis by ameliorating inflammation and modulating macrophage phenotype. Sci Rep. 2017;7:99.
Dalli J. Human sepsis eicosanoid and proresolving lipid mediator temporal profiles: correlations with survival and clinical outcomes. Crit Care Med. 2017;45(1):58–68.
Serhan CN. Maresins: novel macrophage mediators with potent anti-inflammatory and proresolving actions. J Exp Med. 2009;206:15–23.
Sasaki K. Total synthesis and bioactivities of two proposed structures of maresin. Chem Asian J. 2011;6(2):534–43.
Abdulnour RE. Maresin 1 biosynthesis during platelet–neutrophil interactions is organ-protective. Proc Natl Acad Sci U S A. 2014;111(46):16526–31.
Colas RA. Identification and actions of the maresin 1 metabolome in infectious inflammation. J Immunol. 2016;197(11):4444–52.
Gong J. Maresin 1 mitigates LPS-induced acute lung injury in mice. Br J Pharmacol. 2014;171(14):3539–50.
Gong J. Maresin 1 prevents lipopolysaccharide-induced neutrophil survival and accelerates resolution of acute lung injury. Shock. 2015;44(4):371–80.
Chen L. Maresin 1 maintains the permeability of lung epithelial cells in vitro and in vivo. Inflammation. 2016;39(6):1981–9.
Li RD. Maresin 1 mitigates inflammatory response and protects mice from sepsis. Mediators Inflamm. 2016;2016:3798465.
Deng B. Maresin biosynthesis and identification of maresin 2, a new anti-inflammatory and pro-resolving mediator from human macrophages. PLoS One. 2014;9(7):e102362.
Spite M. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res. 2010;107:1170–84.
Rogerio AP. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol. 2012;189:1983–91.
Alfredo MB. Protective role of acetylsalicylic acid in experimental Trypanosoma cruzi infection: evidence of a 15-epi-lipoxin A4-mediated effect. PLoS Negl Trop Dis. 2013;7:e2173.
Toner P. Aspirin as a potential treatment in sepsis or acute respiratory distress syndrome. Crit Care. 2015;19:374.
Halushka PV. Studies on the beneficial effects of aspirin in endotoxic shock: relationship to inhibition of arachidonic acid metabolism. Am J Med. 1983;74:91–6.
Morris T. Effects of low-dose aspirin on acute inflammatory responses in humans. J Immunol. 2009;183:2089–96.
El Kebir D. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. 2009;180:311–9.
Chen W. Prehospital aspirin use is associated with reduced risk of acute respiratory distress syndrome in critically ill patients: a propensity-adjusted analysis. Crit Care Med. 2015;43:801–7.
Eisen DP. Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med. 2012;40:1761–7.
Falcone M. Lower mortality rate in elderly patients with community-onset pneumonia on treatment with aspirin. J Am Heart Assoc. 2015;4:e001595.
Colas RA. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. Am J Physiol Cell Physiol. 2014;307(1):C39–54.
Kor DJ. Association of prehospitalization aspirin therapy and acute lung injury: results of a multicenter international observational study of at-risk patients. Crit Care Med. 2011;39:2393–400.
Wiewel MA. Chronic antiplatelet therapy is not associated with alterations in the presentation, outcome, or host response biomarkers during sepsis: a propensity-matched analysis. Intensive Care Med. 2016;42:352–60.
Dalli J. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med. 2015;21(9):1071–5.
Dalli J. Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity. 2017;46(1):92–105.
Dalli J. Novel proresolving and tissue-regenerative resolvin and protectinsulfido-conjugated pathways. FASEB J. 2015;29(5):2120–36.
Ramon S. The protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am J Pathol. 2016;186(4):962–73.
Dalli J. Maresin conjugates in tissue regeneration biosynthesis enzymes in human macrophages. Proc Natl Acad Sci U S A. 2016;113(43):12232–7.
Sacerdoti D. EETs and HO-1 cross-talk. Prostaglandins Other Lipid Mediat. 2016;125:65–79.
Nakahira K. Carbon monoxide in the treatment of sepsis. Am J Physiol Lung Cell Mol Physiol. 2015;309:1387–93.
Wegiel B. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Investig. 2014;124:4926–40.
Nakahira K. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203:2377–89.
Poss KD. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U S A. 1997;94:10925–30.
Kyokane T. Carbon monoxide from heme catabolism protects against hepatobiliary dysfunction in endotoxin-treated rat liver. Gastroenterology. 2001;120:1227–40.
Cepinskas G. Carbon monoxide liberated from carbon monoxide-releasing molecule CORM-2 attenuates inflammation in the liver of septic mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:184–91.
Lee S. Carbon monoxide confers protection in sepsis by enhancing beclin 1-dependent autophagy and phagocytosis. Antioxid Redox Signal. 2014;20:432–42.
Biernacki WA. Exhaled carbon monoxide in patients with lower respiratory tract infection. Respir Med. 2001;95:1003–5.
Shi Y. Plasma carbon monoxide levels in term newborn infants with sepsis. Biol Neonate. 2000;78:230–2.
Takaki S. Beneficial effects of the heme oxygenase-1/carbon monoxide system in patients with severe sepsis/septic shock. Intensive Care Med. 2010;36:42–8.
Morimatsu H. Increased heme catabolism in critically ill patients: correlation among exhaled carbon monoxide, arterial carboxyhemoglobin, and serum bilirubin IX alpha concentrations. Am J Physiol Lung Cell Mol Physiol. 2006;290:114–9.
Zegdi R. Increased endogenous carbon monoxide production in severe sepsis. Intensive Care Med. 2002;28:793–6.
D’Acquisto F. Annexin-A1: a pivotal regulator of the innate and adaptive immune systems. Br J Pharmacol. 2008;155(2):152–69.
Perretti M. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9(1):62–70.
Buckingham JC. Annexin 1, glucocorticoids, and the neuroendocrine-immune interface. Ann N Y Acad Sci. 2006;1088:396–409.
Perretti M. Annexin I is stored within gelatinase granules of human neutrophil and mobilized on the cell surface upon adhesion but not phagocytosis. Cell Biol Int. 2000;24(3):163–74.
Hotchkiss RS. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–74.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Liu, Y., Hao, Y., Bhandari, S., Jin, S. (2019). Pro-resolution of Inflammation: New Hints to Manage Sepsis?. In: Fu, X., Liu, L. (eds) Severe Trauma and Sepsis. Springer, Singapore. https://doi.org/10.1007/978-981-13-3353-8_8
Download citation
DOI: https://doi.org/10.1007/978-981-13-3353-8_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3352-1
Online ISBN: 978-981-13-3353-8
eBook Packages: MedicineMedicine (R0)