
Abstract
In this paper, we evaluate 15 methods for gene set analysis in microarray classification problems. We employ four datasets from myeloma research and three types of biological gene sets, encompassing a total of 12 scenarios. Taking a two-step approach, we first identify important genes within gene sets to create summary gene set scores, we then construct predictive models using the gene set scores as predictors. We propose two powerful linear methods in addition to the well-known SuperPC method for calculating scores. By comparing the 15 gene set methods with methods used in individual-gene analysis, we conclude that, overall, the gene set analysis approach provided more accurate predictions than the individual-gene analysis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Gene expression profiling (GEP) via DNA microarrays has been used extensively in cancer research to study disease mechanisms and make predictions of clinical outcomes. A typical microarray data analysis focuses on the selection of individual genes. For example, to identify differentially expressed genes under different conditions, one typically calculates a statistic and p value for each gene, followed by multiple comparison adjustments since normally tens of thousands of genes are measured in a microarray experiment. To select genes for predicting clinical outcomes, one can resort to methods such as semi-supervised principal component analysis (SuperPC) [1], partial least squares [2], Lasso [3], random forest [4], and so on. However, this type of analysis can miss some important genes whose individual contributions to a particular outcome may be moderate but whose combined effects are significant. Another limitation of the individual-gene approach is frequently inconsistent gene findings from similar studies conducted by different institutes [5, 6]. These problems of the individual-gene analysis were discussed in Mootha et al. [7] and Subramanian et al. [8], where they proposed a gene set enrichment analysis (GSEA) idea, incorporating prior biological knowledge into the analysis routine to identify important genes through gene sets. Since then many new statistical methods have been proposed for making inference on associations or predictions at gene set levels instead of individual-gene levels.
A gene set is a group of genes related in certain ways (e.g., they may be from the same pathway or perform similar molecular functions). There are public databases holding such information, for example those with the Gene Ontology (GO) annotations [9] and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways [10]. For differential expression analysis, a gene set method aims to determine via hypothesis testing whether a gene set as a whole is associated with an outcome of interest. Examples include the pioneering GSEA algorithm [8], the Global Test [11], ANCOVA Global Test [12], SAM-GS [13], and GSA [14], to name just a few. For biomarker discovery, i.e. finding genes to build models for diagnostic/prognostic purposes, the idea of incorporating gene set information is to improve both performance and interpretability of resulting models. Tai and Pan [15] proposed a modified linear discriminant analysis (LDA) approach for classification by regularizing the covariance matrix and incorporating correlations among the genes within gene sets. With simulated and real datasets plus information from KEGG pathways, they showed that the new approach performed better than not incorporating the correlations within gene sets. Chen and Wang [16] proposed a two-step procedure: first to create a “super gene score” using SuperPC [1] within each a priori gene set obtained from GO and then to use Lasso or SuperPC again to build a final model based on the super gene scores. With two survival microarray data they demonstrated that their gene set-based models enjoyed improved prediction accuracy and generated more biologically interpretable results. Ma et al. [17] also took a two-step approach, where they first divided genes into clusters by k-means, followed by applying Lasso within each cluster to get refined gene clusters, and then they selected important gene clusters with group Lasso [18]. Luan and Li [19] proposed a group additive regression model to incorporate pathway information and the use of gradient descent boosting for model fitting. With both simulations and a real microarray survival dataset, they showed improved accuracy by their method when compared to not using gene group information.
In this paper, we aim to investigate several score methods in conjunction with trees and random forests for gene set analysis and compare with individual-gene analysis in classification problems. In the individual-gene analysis, neither the gene selection nor the prediction process utilizes any biological information. For the gene set analysis, we first identify important genes within a priori gene sets to create summary gene set scores, and we then use the gene set scores as predictors for constructing predictive models. We explore four myeloma microarray datasets and three types of gene sets, and demonstrate that predictive accuracy depends on both the method and the type of gene sets being investigated. In the next section we first introduce our datasets from myeloma research. We then describe the analysis methods in Sect. 3 and show our results from applying the methods to the myeloma datasets in Sect. 4. Finally in Sect. 5, we conclude with a comparison of our results with findings reported by others.
2 Datasets
All GEP datasets used in this investigation were from the Myeloma Institute (MI) at the University of Arkansas for Medical Sciences (UAMS). Multiple myeloma (MM) is a cancer of plasma cells in the bone marrow, with symptoms such as elevated calcium, renal failure, anemia, and bone lesions (the so-called CRAB symptoms). Normal plasma cells produce many immunoglobulins (antibodies) that the body needs to identify and fight pathogens such as bacteria and viruses. With MM, abnormal plasma cells from a single clone accumulate and eventually crowd out normal plasma cells, causing the body to produce only one type of immunoglobulin. It is not clear what causes MM, but it is characterized by genetic abnormalities such as gene mutations and translocations. For example, deletions of chromosome 17p and P53 gene mutations have been linked to poor clinical outcomes in numerous MM studies. Typically prior to developing MM, abnormal plasma cells accumulate in the body and the patient undergoes an asymptomatic phase, comprising monoclonal gammopathy of uncertain significance (MGUS) and smoldering multiple myeloma (SMM). Compared to MGUS, SMM has more abnormal plasma cells in the bone marrow and higher levels of monoclonal immunoglobulin (M-protein) in the serum. Both MGUS and SMM patients lack the CRAB symptoms that define MM. However, MGUS patients have an approximately 1% risk per year of developing MM [20]. Among patients with SMM, about 10% annually will progress to MM within 5 years, and after the 5-year mark the progression rate is similar to MGUS [21].
In previous work, based on an earlier Affymetrix platform with ~12,000 genes, we identified differentially expressed genes that could distinguish in plasma cells between normal and MM and between normal and MGUS [22]. An interesting finding at the time was a lack of ability of the models to discriminate between MGUS and MM at the gene expression level. Based on the newer platform U133Plus2, and more samples, we aimed to do a more refined analysis in this investigation, specifically to identify signature genes and build predictive models to distinguish between (1) normal and MGUS, (2) MGUS and SMM, (3) SMM and MM, and (4) P53 deletion and no deletion in MM. The MM patients in this study were enrolled in a series of Total Therapy (TT) clinical trials, with the MGUS/SMM patients in two observational clinical trials (SWOG S0120 and MI M0120). P53 deletion was determined at baseline by interphase fluorescence in situ hybridization (iFISH). For GEP, purified plasma cells (PC) by CD138 expression were obtained from normal healthy subjects and the MM (MGUS/SMM) patients prior to therapy (at registration of the observational trials). Microarray raw intensity values were preprocessed and normalized using the MAS5 algorithm provided by the manufacturer, and the normalized data also went through batch effect checking and corrections [23].
3 Methods
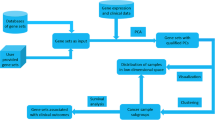
Table 1 gives the sample sizes in each dataset. To ensure data quality, we first implemented the following steps prior to analysis:
-
1.
Use the genes with current annotations from Affymetrix.
-
2.
Take the median if a gene is represented by more than one probe set.
-
3.
Keep only those genes whose raw intensity values are >128 in at least 80% of the samples to avoid any resolution problems that may be encountered by low microarray intensity values.
Since applying the above procedure to each GEP dataset separately produced similar sets of genes, for simplicity we applied it to all the data combined to obtain a total of 9624 genes before analysis.
3.1 What Gene Sets to Use?
There are different types and sources of biological gene sets. The Molecular Signatures Database (MSigDB) [24] on the Broad Institute website is one of the largest and most popular repositories. We downloaded three types of gene sets from MSigDB: those associated with the GO biological processes (BP), the hallmark gene sets, and the positional gene sets. Each gene set groups certain genes together that share a particular biological property. GO BP gene sets contain genes associated with biological processes, each of which is made up of many chemical reactions or events leading to chemical transformations. However, the GO BP gene sets are a broad category and do not necessarily comprise co-regulated genes. On the other hand, the hallmark gene sets represent well-defined biological states or processes and contain genes with coordinate expression [25]. The positional gene sets group genes by chromosome and cytogenetic band. Such gene sets are helpful in identifying effects related to chromosome abnormalities.
3.2 Approach for Gene Set Analysis
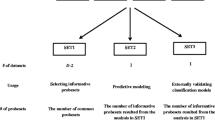
Our general approach for gene set analysis is a two-step procedure: (1) within each a priori gene set create a summary gene set score after gene selection, and (2) construct a predictive model based on the resulting gene set scores. Both Chen and Wang [16] and Ma et al. [17] pointed out that typically not all members of a gene set will participate in a biological process, or be relevant to the outcome of interest, and not doing gene selection within gene sets could result in inferior prediction accuracy. Thus we carry out variable selection twice, first to select important genes within each gene set to calculate a summary gene set score (step 1), and then to select important gene sets based on the gene set scores and build a final predictive model (step 2).
3.3 Variable Selection and Model Building
We investigated several linear and nonlinear methods for variable selection and model building. The linear methods included the Lasso and three univariate score methods, and the nonlinear methods included decision trees and random forests.
Lasso is a multivariate regression technique [3] that has become popular and essential in genomic data analysis. By shrinking regression coefficients using an \( L_{1} \) penalty term in the likelihood function for a logistic regression model, the regression coefficients for some genes become exactly zero, thus enabling variable selection. Classification will be done according to the estimated probabilities from the resulting sparse model with the shrunken coefficients. We implemented Lasso via the R package glmnet [26].
The idea of univariate score methods is to first rank genes by univariate analysis (e.g., doing a t-test for each gene in a two-class problem) and then create a score by a linear combination of the top ranking genes. There are many variants of this method and we investigated three in this paper. In a two-class problem, let \( x_{i} \) and \( t_{i} \) denote the expression level and the two-sample t-statistic for gene \( i \), respectively. The first score is based on a regularized compound covariate , where the t-statistics are shrunken towards 0 by soft-thresholding. We denote it by ccscore, that is,
where \( p \) is the total number of genes, \( \left( x \right)_{\text{ + }} \text{ = }x \) if \( x\text{ > }0 \) and 0 otherwise, and \( 0 \le \Delta { \le }max_{i} \text{(}\left| {t_{i} } \right|\text{)} \) is a tuning parameter to be determined by cross-validation . The non-regularized version of the compound covariate method is also a popular choice for constructing scores, which was originally proposed by Tukey [27] and discussed in Huang and Pan [28] for classification problems with microarray data. The second score is one that, instead of using the t-statistics from univariate analysis, only the signs of the t-statistics are used, followed by dividing by the total number of selected genes. We refer to it as “score”, that is,
where \( S\text{ = }\left\{ {i\text{:}\left| {t_{i} } \right|{ \ge }\Delta } \right\} \), \( \left| S \right| \) = number of genes in \( S \), and \( \Delta \) is a tuning parameter determined by cross-validation. Originally we employed a similar method to develop the robust GEP70 model for risk stratification for MM patients undergoing standard therapy [29]; we then modified it to its current form in (2). The third score is an extension of SuperPC [1], originally developed for time-to-event data and shown to perform well in gene set analysis [16]. It takes the top ranking genes and calculates their first principal component as a score. We denote it here by pcscore, that is,
where \( S = \left\{ {i\text{:}\left| {t_{i} } \right| \ge \Delta } \right\} \), \( b_{i} \) are loadings from the first principal component of the genes selected in \( S \), and \( \Delta \) is a tuning parameter determined by cross-validation . For all the aforementioned score methods, they were first created as continuous variables, and we then dichotomized them, balancing both sensitivity and specificity to create 2-group classification rules.
There is a rich literature concerning the development of predictive models using decision trees and random forests and their applications in genomic data analysis (e.g., see [4, 30,31,32,33,34]). A decision tree model based on recursive partitioning has the advantage of easy interpretation. In a random forest model, many decision trees are built by utilizing bootstrap samples and results from each tree are aggregated by majority voting to make final predictions. By building each tree to the fullest, the method is able to achieve low bias , and by aggregating results from many trees it can also achieve low variance. Importantly, a random forest considers only a random subset of the variables at each split. Doing so allows it to (1) produce less similar bootstrap samples and trees and therefore low variance at the end, and (2) identify a diverse set of important variables associated with the outcome of interest even when there is multicollinearity in the data. We implemented decision trees and random forests via the R packages rpart and randomForest.
For the individual-gene analysis, we used methods such as the Lasso, score, ccscore, pcscore, trees, and random forests. For the gene set analysis, to maintain focus we considered only various (instead of all) combinations of the methods from individual-gene analysis. As genes within a biological gene set are more likely to be co-regulated or co-expressed, we restricted to linear methods in step 1 (within gene sets), while in step 2 (between gene sets) we explored both linear and nonlinear methods. There were a total of 15 combinations in the gene set analysis we considered. We denote each combined methodology by using a period between the names of the methods used in the two steps. For example, suppose in step 1 we chose the score method to select genes while in step 2 trees were employed; we would refer to the combined method by score.tree. Tables 2 and 3 list all the methods and their notations for both the gene set and individual-gene analysis.
3.4 Cross-Validation to Determine Tuning Parameter
For the univariate score methods described above, we employed 10-fold cross-validation to select appropriate values for the tuning parameter \( \Delta \) and to achieve variable selection. The search range for \( \Delta \) is normally between 0 and \( max_{i} \text{(}\left| {t_{i} } \right|\text{)} \) as suggested in (1–3), which can be a big range. To reduce computational burden, we restricted our search within the range of 1000 most significant genes when doing the cross-validation. For example, if the absolute values of the t statistic in the top 1000 genes vary between 4.5 and 5.6, we would assess each value from 4.5 to 5.6, with an increment of 0.1 in search of an optimal threshold for \( \Delta \). We used error rate as the performance measure in the cross-validation .
3.5 Model Comparison
Each of the four datasets was split into training and test sets (Table 1), and we only report error rates from the test sets as a guide to compare performance of the different methods. All model building steps were performed in the training sets, including gene selection or shrinkage parameter estimation with cross-validation.
4 Results
There are currently a total of 825 GO BP, 50 hallmark, and 326 positional gene sets on the Broad website that we downloaded. Due to the fact that we had previously performed a gene filtering step, we were left with fewer numbers of gene sets (736 GO BP, 50 hallmark, and 278 positional) as well as fewer genes within the gene sets when we applied these gene sets to our datasets. We also focused on gene sets containing at least 5 genes. Table 4 gives a summary of the number of genes in the gene sets of our datasets. Both the GO BP and positional categories have a small percent of gene sets with a large number of genes in them. However, if we look at the median number of genes within gene sets, the hallmark gene sets have the largest number (86) followed by the GO BP gene sets (54.76) and the positional gene sets (25.9).
4.1 Methods Comparison
Table 5 shows the test set error rates achieved in the individual-gene analysis for each disease comparison. To compare the methods, we ranked them by their averaged error rates (AER) over all the disease comparisons—lower AER is better. Overall, ccscore and score ranked as the top two classifiers in the individual-gene analysis with AER being 0.16 and 0.17 respectively, followed by Lasso (AER = 0.19), random forest (AER = 0.20), and pcscore (AER = 0.20), and the tree method ranked the lowest (AER = 0.22). Note that the AER were rounded to the 2nd decimal point while the rankings were calculated using all decimal points.
For the gene set methods, Table 6 gives the test set error rates for each disease comparison/type of gene sets combination (a total of 12 scenarios). Note that both the method and type of gene sets affected the error rates for each disease comparison. We ranked the methods by their averaged error rates (AER) across all 12 scenarios. It turned out that lasso.lasso and all the methods that employed trees in step 2 of the gene set analysis were low performers. However, Lasso performed well in conjunction with random forests. When not combined with trees in the 2nd step, the ccscore-related methods consistently ranked at the top followed by the pcscore- and score-related methods, although the differences among them were small (≤0.02) by the AER measure. More often than not, random forests were good choices when combined with the score methods or Lasso.
4.2 Gene Set Analysis Versus Individual-Gene Analysis
The question is: did the gene set analysis improve prediction accuracy over the individual-gene analysis? We compared the two types of analysis by calculating differences in error rates. For example, suppose in individual-gene analysis we used the Lasso, then we would compare it with those gene set methods that employed Lasso in 2nd step of the gene set analysis such as ccscore.lasso, score.lasso, pcscore.lasso, and lasso.lasso. By doing such comparisons, one can gauge whether step 1 of the gene set analysis is necessary—without step 1 the gene set analysis just reduces to individual-gene analysis. Table 7 lists reductions in error rate by using gene set analysis compared to individual-gene analysis in all such comparisons. Note that each gene set method was applied for each disease comparison three times, each time utilizing a different kind of gene sets (either GO BP, hallmark, or positional), while each individual-gene method was applied only once for each disease comparison. Thus when calculating the differences in error rate, we replicated those error rates of the individual-gene methods three times. We can see in Table 7 that both the method and the type of gene sets affected whether there was any improvement in performance by doing gene set analysis, where improvement was measured by reduction in error rate. We highlighted those scenarios when the reductions in error rate by doing gene set analysis were somewhat meaningful (≥0.04), although 0.04 is an arbitrary choice. The fact that there are both positive and negative values in Table 7 indicates that sometimes individual-gene analysis was better than gene set analysis in terms of prediction accuracy. Averaged reductions in error rate were also calculated for each gene set method in comparison to an appropriate individual-gene method (last column of Table 7). By this measure, all 15 gene set methods except two produced more accurate models with reduced error rates. Figure 1 provides a visualization of the reductions in error rate. Despite the variations, overall across all the methods, gene set analysis reduced error rates by 0.02 on average, and 25% of the time by at least 0.05.

Boxplots of the reductions in test set error rate (shown in Table 7) by using each of 15 gene set methods compared to corresponding individual-gene methods
4.3 Disease Comparisons
Tables 5 and 6 is also a good summary of the overall prediction error rates for the disease comparisons. To have a focused discussion here, we consider only the ccscore and ccscore.lasso methods in this subsection and the next, as respectively they were among the top methods used in the individual-gene and gene set analysis. It appears that both the individual-gene and gene set methods were able to classify SMM versus MM and P53 deletion versus no deletion very well with error rates varying between 0.06 and 0.10. Clinically, SMM is characterized by a higher percentage of abnormal plasma cells in the bone marrow and higher levels of M-protein in the serum than MGUS. Thus being able to classify SMM versus MM implies being able to discriminate between MGUS and MM as well. It turned out that our hypothesis was right when we went to verify it—the test set error rate for discriminating between MGUS and MM by an individual-gene analysis with the ccscore was 0.099. This is contradictory to the findings reported in Hardin et al. [22], where all the models failed to classify MGUS versus MM. A couple of factors could be the cause here. First, the newer microarray platform U133Plus2 covers the whole genome more comprehensively than the older platform, so there is a better chance to detect differentiable genes and therefore create more powerful models. Second, we had more MGUS samples in this investigation: 73 compared to 21 in Hardin et al. [22], while the MM samples in both investigations were plenty (174 in ours and 218 in Hardin et al.). Nonetheless, the difficult case for us was to discriminate between MGUS and SMM, as the error rates for this classification were between 0.3 and 0.37 for the individual-gene and gene set analysis. This indicates that at the molecular level MGUS and SMM are different for the most part, yet they share certain genetic features that make them less indistinguishable. Also interesting were the error rates for classifying MGUS versus normal varied between 0.16 and 0.21.
Taken together, these data seem to suggest that in terms of gene expression levels SMM is very different from MM, while MGUS is somewhere between normal and SMM, but more similar to SMM. At this point careful interpretation of the results is warranted. When using CD138 expression to isolate plasma cells (PC) before GEP—a standard procedure routinely performed at the Myeloma Institute, the MGUS/SMM PC samples were infiltrated with normal PC, while the MM PC samples were largely abnormal. Consequently, some of the differentially expressed genes we identified between MGUS/SMM PC and MM PC samples might be due to differences in the amount of normal PC in the samples rather than due to disease differences. This problem was less when comparing normal versus MGUS and MGUS versus SMM PC samples, as they were more comparable in terms of the amount of normal PC in the samples.
4.4 Gene Lists and Gene Selection
We provide a list of genes and gene sets identified for each disease comparison by the ccscore and ccsore.lasso methods from the individual-gene and gene set analysis respectively (for the same reason described in the last subsection) (Tables 8, 9, 10, and 11). For the ccscore.lasso gene set analysis, we chose the gene set that gave the best result for each disease comparison (2nd row in Table 6). Furthermore, we summarized the total number of genes identified by the two types of analysis. In all except the comparison of P53 deletion versus no deletion, more genes were selected by ccscore.lasso than ccscore, with comparatively few overlapping genes (Table 12).
4.5 Computing Time
We recorded computing time for all the methods in the individual-gene analysis (Table 13). The evaluations were conducted on a laptop using 64-bit Windows 7 and running on a 4-core 3 GHz CPU with 8 GB of memory. For all the methods except Lasso, we started with all 9624 genes. For random forests, however, we included another filtering step to consider only the top 1500 differentially expressed genes prior to model selection. Our experience is that random forests can be very slow without pre-filtering. As shown in Table 13, computing time increases as sample size increases. The ccscore/score/pcscore finished in decent amounts of time (a couple of minutes) but Lasso was no doubt the fastest algorithm in all cases.
5 Conclusions and Discussion
In this paper, we evaluated 15 methods for gene set analysis in classification problems using four GEP myeloma datasets and three types of biological gene sets, encompassing a total of 12 scenarios. By comparing the 15 methods with individual-gene methods, we conclude that, overall, the gene set analysis provided more accurate models than the individual-gene analysis. Within a biologically defined gene set, genes are more likely to be co-regulated or co-expressed. We propose to use linear methods such as the ccscore, score, and pcscore (an extension of the SuperPC [1]) for calculating gene set scores before constructing final predictive models.
Our overall results after averaging across different datasets/gene sets are comparable to those reported by other authors. For example, Ma et al. [17] proposed using Lasso within gene clusters to first select important genes before applying group Lasso [18] on the refined gene clusters. In four microarray datasets, they demonstrated either equal or better performance of their method than using regular Lasso in individual-gene analysis. In our study, lasso.lasso, ccscore.lasso, score.lasso, and pcscore.lasso are similar to their approach. Although both are two-step procedures, in their 1st step they created refined gene clusters rather than summary genet set scores. As shown in Tables 5 and 6, when compared to regular Lasso, the ccscore.lasso, score.lasso, and pcscore.lasso each had a reduction in error rate between 0.01 and 0.02, although lasso.lasso had a 0.01 increase in error rate.
Our general approach resembles with that of Chen and Wang [16]. In the 1st step they created “super gene scores” with SuperPC [1], and in the 2nd step they employed either Lasso or again SuperPC using the super gene scores as predictors. With two microarray survival datasets they demonstrated the superiority of their methods when compared to not using gene set information. Our pcscore is essentially an extension of the SuperPC for binary outcomes, and therefore our pcscore.pcscore and pcscore.lasso directly correspond to their methods except that they focused on survival prediction instead of classification . When comparing to only using pcscore or lasso in individual-gene analysis, we saw an averaged reduction of at least 0.02 in error rate for pcscore.pcscore and pcscore.lasso (Tables 5 and 6), confirming comparability of our results with theirs.
Additionally, Tai and Pan [15] proposed a modified LDA approach to incorporate pathway information. With both simulated and real datasets, their method was shown to perform better than not incorporating pathway information (e.g., when compared to PAM [35], which considers genes as independent). Genes are naturally not independent from each other, therefore the improvements by their method were reasonable and expected. Importantly, our ccscore/score/pcscore methods already draw strength by combining correlated genes. As Park et al. [36] have shown, averaging genes within gene clusters can improve prediction accuracy. Our score method is essentially an extension of the averaged gene expression method to account for genes with both positive and negative correlations with the outcome. Although it is beyond the scope of this paper, it would be interesting to apply their approach and PAM on myeloma datasets in future research. Further investigations on the genes identified to examine whether the gene set analysis could provide more coherent biological insights into the myeloma disease mechanisms would be another avenue of research.
References
Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004;2(4):e108.
Nguyen DV, Rocke DM. Tumor classification by partial least squares using microarray gene expression data. Bioinformatics. 2002;18(1):39–50.
Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc Ser B (Methodol). 1996;1:267–88.
Díaz-Uriarte R, De Andres SA. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006;7(1):1.
Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21(2):171–8.
Solé X, Bonifaci N, López-Bigas N, Berenguer A, Hernández P, Reina O, Maxwell CA, Aguilar H, Urruticoechea A, de Sanjosé S, Comellas F. Biological convergence of cancer signatures. PLoS ONE. 2009;4(2):e4544.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–50.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(suppl 1):D355–60.
Goeman JJ, Van De Geer SA, De Kort F, Van Houwelingen HC. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20(1):93–9.
Mansmann U, Meister R. Testing differential gene expression in functional groups Goeman’s global test versus an ANCOVA approach. Methods Inf Med. 2005;44(3):449–53.
Dinu I, Potter JD, Mueller T, Liu Q, Adewale AJ, Jhangri GS, Einecke G, Famulski KS, Halloran P, Yasui Y. Improving GSEA for analysis of biologic pathways for differential gene expression across a binary phenotype. COBRA Prepr Ser. 2007; 16.
Efron B, Tibshirani R. On testing the significance of sets of genes. Ann Appl Stat. 2007;1:107–29.
Tai F, Pan W. Incorporating prior knowledge of gene functional groups into regularized discriminant analysis of microarray data. Bioinformatics. 2007;23(23):3170–7.
Chen X, Wang L. Integrating biological knowledge with gene expression profiles for survival prediction of cancer. J Comput Biol. 2009;16(2):265–78.
Ma S, Song X, Huang J. Supervised group Lasso with applications to microarray data analysis. BMC Bioinform. 2007;8(1):1.
Meier L, Van De Geer S, Bühlmann P. The group lasso for logistic regression. J R Stat Soc Ser B (Stat Methodol). 2008;70(1):53–71.
Luan Y, Li H. Group additive regression models for genomic data analysis. Biostatistics. 2008;9(1):100–13.
Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, Melton LJ III. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–9.
Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, Larson DR, Plevak MF, Jelinek DF, Fonseca R, Melton LJ III. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–90.
Hardin J, Waddell M, Page CD, Zhan F, Barlogie B, Shaughnessy J, Crowley JJ. Evaluation of multiple models to distinguish closely related forms of disease using DNA microarray data: an application to multiple myeloma. Stat Appl Genet Mol Biol. 2004;3(1):1–21.
Stein CK, Qu P, Epstein J, Buros A, Rosenthal A, Crowley J, Morgan G, Barlogie B. Removing batch effects from purified plasma cell gene expression microarrays with modified ComBat. BMC Bioinform. 2015;16(1):1.
Liberzo A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40.
Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection. Cell Syst. 2015;1(6):417–25.
Glmnet vignette. http://www.stanford.edu/~hastie/glmnet/glmnet_alpha.html.
Tukey JW. Tightening the clinical trial. Control Clin Trials. 1993;14(4):266–85.
Huang X, Pan W. Linear regression and two-class classification with gene expression data. Bioinformatics. 2003;19(16):2072–8.
Shaughnessy JD, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, Stewart JP, Kordsmeier B, Randolph C, Williams DR, Xiao Y. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–84.
Breiman L, Friedman J, Stone CJ, Olshen RA. Classification and regression trees. Boca Raton: CRC Press; 1984.
Breiman L. Random forests. Mach Learn. 2001;45(1):5–32.
Cutler A, Cutler DR, Stevens JR. Random forest. In: Machine learning. 2011. http://www.researchgate.net/publication/236952762.
Hastie T, Tibshirani R, Friedman J. The elements of statistical learning: data mining, inference, and prediction. 2nd ed. Springer series in statistics. 2011.
Boulesteix AL, Janitza S, Kruppa J, König IR. Overview of random forest methodology and practical guidance with emphasis on computational biology and bioinformatics. Wiley Interdiscip Rev Data Min Knowl Discov. 2012;2(6):493–507.
Tibshirani R, Hastie T, Narasimhan B, Chu G. Class prediction by nearest shrunken centroids, with applications to DNA microarrays. Stat Sci. 2003;1:104–17.
Park MY, Hastie T, Tibshirani R. Averaged gene expressions for regression. Biostatistics. 2007;8(2):212–27.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Qu, P., Tian, E., Barlogie, B., Morgan, G., Crowley, J. (2017). An Evaluation of Gene Set Analysis for Biomarker Discovery with Applications to Myeloma Research. In: Matsui, S., Crowley, J. (eds) Frontiers of Biostatistical Methods and Applications in Clinical Oncology. Springer, Singapore. https://doi.org/10.1007/978-981-10-0126-0_25
Download citation
DOI: https://doi.org/10.1007/978-981-10-0126-0_25
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-0124-6
Online ISBN: 978-981-10-0126-0
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)