
Abstract
One of the most intriguing and less known aspects of the interaction between viruses and their host is the impact of the viral infection on the heat shock response (HSR). While both a positive and a negative role of different heat shock proteins (HSP) in the control of virus replication has been hypothesized, HSP function during the virus replication cycle is still not well understood. This chapter describes different aspects of the interactions between viruses and heat shock proteins during infection of mammalian cells: the first part focuses on the modulation of the heat shock response by human viral pathogens; the second describes the interactions of HSP and other chaperones with viral components, and their function during different steps of the virus replication cycle; the last part summarizes our knowledge on the effect of hyperthermia and HSR modulators on virus replication.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
Viruses are fascinating micromachines that are able to invade virtually all forms of life. Despite their structural simplicity, viruses have evolved sophisticated tactics to exploit the metabolic machinery of the host cell and to reprogram it for the synthesis of the macromolecular constituents that are required for their multiplication and invasion of new cells. During the last years the study of the elaborate relationship between viruses and their hosts has led to the understanding of how viral pathogens not only hijack the host transcriptional/translational machinery, but are also able to control cellular signaling pathways and transcription factors to their own advantage. A fascinating aspect of the interaction between viruses and their host is the impact of the viral infection on the heat shock response (HSR).
The HSR is considered a fundamental cellular defense mechanism against the deleterious effects of physiological and environmental stress provoking cell damage due to protein misfolding, degradation and insoluble aggregation, including hyperthermia, alterations in the intracellular redox environment, and exposure to different types of chemicals [1]. The stress signal, which is usually triggered by a flux of non-native proteins, results in the activation of heat shock factors (HSF) and in the synthesis of heat shock proteins (HSP), many of which function as molecular chaperones to guide conformational states critical in the synthesis, folding, assembly and disassembly, translocation and degradation of proteins [2]. This poses the question on whether viruses are able to hijack the chaperone machinery necessary for the correct folding of the abundant amount of viral proteins rapidly synthesized in bulk, and for their correct assembly into viral components during different phases of the virus replication cycle. Since HSP are also crucial factors in many signal transduction pathways and the proper regulation of chaperone expression is critical to the regulation of apoptosis [1–3], it is evident that the control of HSP expression would be a powerful tool for the invading virus to manipulate the fate of the host cell.
A different perspective of the relationship between viruses and the HSR comes from studies which investigate the effect of high intracellular levels of HSP induced by hyperthermia or chemical agents on virus replication during acute or persistent infections. Several of these studies describe an antiviral activity of HSR inducers in infected cells. In addition, the heat shock response physiologically induced in vivo by fever is a host defense mechanism known to have a beneficial role during viral infection [4].
Despite the large amount of literature describing the impact of virus infection on the host HSR, the significance of this interaction is still not well understood, and it is not clear whether the outcome may benefit the host or the pathogen. This chapter analyzes different aspects of the interactions between viruses and heat shock proteins during infection of mammalian cells, focusing on the modulation of the HSR by viruses that cause diseases in humans.
3.2 Modulation of the Heat Shock Response During Viral Infection
Different types of DNA and RNA viruses influence the cellular heat shock response (Fig. 3.1). In the case of DNA viruses, a pioneering study by Khandjian and Turler [5] showed that during lytic infection of monkey and mouse cells with simian virus 40 (SV40) or polyoma virus there is a marked increase in the synthesis of two host heat-inducible proteins of 92 and 72 kDa. The main SV40-inducible member of the hsp70 family was subsequently shown to be the hsc70 gene in monkey CV-1 cells [6]. An up-regulation of hsp60, hsc70, and hsp90 and a down-regulation of the small heat shock protein hsp28 were instead shown by Honoré et al. [7] in SV40-transformed keratinocytes, indicating a different response in different types of host and different conditions of infection. In addition to polyomavirus, adenoviruses were among the first DNA viruses shown to increase the expression of HSP genes, and in particular of the hsp70 genes, in human cells. Several studies have shown that the adenoviral gene products E1A and E1B synergistically stimulate hsp70 expression in a cell cycle specific manner [8–13], as described in the next section.

Viruses that induce HSP expression in mammalian cells. Several human viral pathogens belonging to different DNA (top) and RNA (bottom) virus families are able to activate the expression of one or more heat shock proteins. Double-strand (white arrow) and single-strand RNA viruses of positive (dark gray) and negative (light gray) polarity are depicted in the bottom section. See text for details
Numerous reports have shown alteration of the HSR during herpesvirus infection. In the case of herpes simplex virus (HSV), mutants of HSV type 1 (HSV-1) and 2 (HSV-2) induce HSP during infection of chick embryo fibroblasts [14] and human neuroblastoma cells [15] respectively. The presence of abnormal forms of the HSV-1 immediate early polypeptide Vmw175 was found to be the signal for induction of the stress response in chick embryo fibroblasts infected with the HSV mutant tsK [16]. An altered HSV-1 envelope gB glycoprotein that is retained in the ER of mammalian cells, but not the normal viral envelope protein, was also found to transactivate the grp78 promoter [17]. However, the presence of abnormal proteins is not necessary for HSR stimulation by herpesviruses. In fact, lytic infection of BHK cells with several strains of HSV-2 causes intracellular accumulation and translocation to the cell surface of a protein related to the hsp90 family [18]. In addition, the presence of elevated hsp70 mRNA levels was reported in rodent cells early after infection with HSV types 1 and 2 [19]; hsp70 induction was dependent on viral protein synthesis but not on viral DNA replication, suggesting that one or more HSV-encoded protein(s) could be involved in inducing hsp70 expression. This turned out in fact to be the case, as described in the next section. HSR activation was also shown after infection with a different α-herpesvirus, the Varicella Zoster virus (VZV) [20].
Also β- and γ-herpesviruses activate the HSR. The β-herpesvirus HCMV (human cytomegalovirus) was shown to transiently induce hsp70 gene expression in human diploid fibroblasts [21], whereas infection of human B lymphocytes with the γ-herpesvirus EBV (Epstein-Barr virus) induces the expression of both hsp70 and hsp90 proteins, independently of viral protein synthesis [22]. Peripheral blood B cells immortalized in vitro by EBV were also shown to express elevated levels of hsp70 and hsp90 [22]. In this case hsp90, but not hsp70, was found to be localized on the surface of EBV-immortalized lymphoblastoid cell lines. This expression was shown to be important in the stimulation of γδ T cells, suggesting that hsp90 serves as an immune sentinel trigger during acute virus infection, or as an aid in the generation of EBV-specific T cells during acute infection mononucleosis convalescence [23].
Cytoplasmic DNA viruses can also control HSP expression. Jindal and Young reported that infection of human monocyte-macrophages by vaccinia virus, caused a dramatic decrease in the levels of cellular mRNAs, but did not cause a significant reduction in the levels of hsp90 and hsp60 mRNA, rather it led to a substantial increase in hsp70 mRNA levels, indicating an increased resistance of HSP transcription and translation during cytopathic virus infection [24]. Interestingly, HSP expression was shown to be also enhanced during poxvirus infection of mouse ovaries in vivo [25].
In the case of RNA viruses, cytoplasmic replication is the rule with a few exceptions which include influenza viruses. Most RNA viruses do not need to interact directly with the cellular transcriptional machinery, carrying their own either in the form of RNA-dependent RNA polymerase complexes present in the viral capsid (negative-strand RNA viruses) or synthesizing the polymerase soon after infection of the host cell (positive-strand RNA viruses). RNA viruses have evolved different strategies to control the host translational apparatus, and usually provoke a dramatic shut-off of host cell protein synthesis. However, a small number of known cellular proteins are synthesized at increased rates after infection by both positive and negative polarity RNA viruses. The proteins of the interferon system are the most studied example, however induction of stress proteins has also been reported. Starting from the initial observation by Peluso et al. that infection of cultured chick embryo cells by the paramyxoviruses Sendai virus and Simian virus 5 (SV5) stimulated the synthesis of glucose-regulated proteins (GRP) [26, 27], a growing body of literature has described the induction of stress proteins by different types of RNA viruses (Fig. 3.1). In the case of SV5, a fivefold increase in the rate of grp78-BiP transcription and an increase in grp-BiP protein levels were shown in monkey cells. When the individual SV5 polypeptides were expressed from cloned cDNAs, the synthesis of the hemagglutinin-neuraminidase (HN) glycoprotein led to an increase of grp78-BiP accumulation, whereas the fusion (F) glycoprotein or the viral proteins P, V and M had no effect, indicating that the flux of folding-competent HN molecules through the ER of infected cells stimulates grp-BiP synthesis [28]. In a similar manner, the hepatitis C (HCV) virus E2 envelope protein, which is retained in a pre-Golgi compartment in a partially misfolded state bound by grp78, was found to induce overproduction of a number of ER resident chaperones including grp78 and grp94 [29]. Since grp78 is part of the sensing system that detects misfolding in the ER, its interaction with partially folded viral proteins may activate the feedback mechanism of the unfolded protein response (Fig. 3.2).

Modulation of the heat shock response during viral infection. Different viruses interact with the heat shock response at different levels. 1. Virus attachment at cell membrane receptors may activate signal transduction pathways interfering with the HSR. 2. Adenoviruses and poxviruses induce HSF1 nuclear translocation and DNA-binding activity. 3. In some cases, HSR activation may be due to hsp70 sequestering by viral proteins present in an unfolded aggregation-prone state in the nucleus and/or cytoplasm of the host cell, causing HSF freeing and activation. 4. Some DNA virus proteins with promiscuous trans-activating ability directly induce hsp70 gene expression. 5. Adenoviruses specifically promote hsp70 mRNA nuclear export, evading the virus-induced cellular mRNAs export block. 6. Similarities in the mechanism of preferential translation during stress conditions between hsp70 mRNA and viral mRNA may favor host HSP mRNA translation under conditions where cellular protein synthesis is inhibited. 7. Fluxes of folding-competent viral glycoproteins through the ER of infected cells may stimulate GRP synthesis. Viruses known to function at the different levels are indicated. See text for details
While GRP appear to be preferentially induced by RNA viruses, accumulation of HSP was also reported after infection with the paramyxovirus NDV (Newcastle disease virus) [30], and a direct association between in vivo virus infection and HSR induction was shown by Oglesbee and Krakowka in brain tissue from dogs infected with a different member of the Paramyxoviridae family, the morbillivirus CDV (canine distemper virus) [31]. In this case, elevated levels of a 72 kDa HSP were found in CDV-infected astrocytes as compared to non-infected cells. Translocation of hsp70 to the nucleus [32] and to the lipid-raft membranes [33], but not increase in expression, was instead observed after infection with the pneumovirus RSV (Respiratory Syncytial Virus).
Garry et al. reported stress protein induction by the togavirus Sindbis virus and by the rhabdovirus vesicular stomatitis virus (VSV) in chick embryo cells [34]; notably, HSP induction was resistant, relative to the synthesis of most host proteins, to alterations in the intracellular ionic concentration. The capsid proteins of a different member of the Togaviridae family, Semliki Forest virus, was also found to efficiently modulate the stress response of target cells and to confer thermal resistance to HeLa cells [35].
Increased levels of hsp70 were detected in murine neonatal myocardial cells after infection with two different picornaviruses, encephalomyocarditis (EMC) virus and coxsackievirus B-3 (CVB3) [36]. Virus inactivation by ultraviolet irradiation prevented hsp70 induction in these cells. Among the Picornaviridae, polioviruses (PV) are known to cause a dramatic shut-off of the host cell protein cap-dependent translation by proteolytically inactivating the cap-binding protein complex [37]. Poliovirus infection is known to inhibit constitutive [38] as well as heat shock- or prostaglandin-induced [39, 40] hsp70 synthesis early after infection of human cells; however, hsp70 synthesis was found to be more resistant to inhibition than normal host proteins after PV infection. On the other hand, the important studies by Sarnow [41], demonstrating that grp78 translation is increased in poliovirus-infected HeLa cells at a time when cap-dependent translation of cellular mRNA is inhibited, opened new scenarios on how stress protein mRNA may adopt differential translation strategies that, in some cases, have similarities with viral RNA.
Induction of grp78 and grp94 [42], and a transient increase in hsp70 levels [43] was described during infection with rotaviruses, which possess a segmented double strand RNA genome. In this case, the nonstructural glycoprotein NSP4, which represents the viral enterotoxin, was responsible for GRP induction via an unknown mechanism [42]. It should be noted, however, that rotavirus infection was found to prevent induction of hsp70 expression by the cyclopentenone prostanoid PGA1 [44].
Finally, retroviruses also affect the cellular stress response. Stimulation of GRP synthesis was initially shown in cells transformed by Rous sarcoma virus [27], while expression of hsp70 was detected on the surface of cells infected with human T cell leukemia virus type I (HTLV-I); this last observation suggested that HSP immunity plays a role in the pathogenesis of HTLV-I infection [45]. Increased levels of hsp70 synthesis were also reported by D’Onofrio et al. [46] in human Molt-4 leukemic T-cells after exposure to HTLV-I in a cell-to-cell transmission model. In addition to hsp70, hsp90 was shown to be overexpressed in HTLV-1-infected cell lines as well as in primary cells derived from patients with adult T-cell leukemia/lymphoma (ATL), a peripheral T-lymphocytic malignancy caused by HTLV-1 [47]. Hsp90 is involved in the stabilization and conformational maturation of many signaling proteins [3]. In these cells hsp90 plays a critical role in the folding and conformational maturation of proteins required for cell survival, including Cdk4, Cdk6 and survivin, and is required for the activity of cellular kinases such as IKK, Akt and PDK1 which are essential for the induction and maintenance of T cell proliferation and transformation by HTLV-1. In fact, degradation of client proteins via inhibition of hsp90 by the geldanamycin derivative 17-AAG leads to impaired signaling and cell apoptosis [47].
In the case of the human immunodeficiency virus (HIV), an increase in hsp70 synthesis and nuclear translocation was observed early after infection of permissive CD4+ cells [48–50]. The fact that exposure to heat-inactivated HIV-1 or to purified recombinant gp120 HIV-1 envelope glycoprotein resulted in hsp70 induction indicated that HIV-1 replication is not required for hsp70 synthesis and intracellular translocation, and that the simple interaction of the virus with the CD4 receptor on the cell membrane delivers the signal activating these events (see next section). Increased hsp70 expression was also shown in human lymphoma cells chronically infected with HIV [51], as well as in lymphocytes from HIV-infected subjects [52]. In addition to hsp70, increased levels of hsp40 [53, 54] and hsp27 [55] were also reported in HIV-infected cells. In particular, HIV-1 Vpr protein, which has been implicated in host–virus interactions at multiple levels, was shown to induce HSF-dependent hsp27 expression, and this effect was associated with an antagonistic effect of the small heat shock protein on the virus replication cycle [56].
The results described above clearly indicate that the entry of a virus into a eukaryotic cell initiates a cascade of events, which may result in switching on HSP gene expression. This is not surprising. The questions to be answered are: (1) how do viruses activate the HSR, and (2) which is the functional significance of stress protein induction during viral infection?
3.3 How Do Viruses Activate the Heat Shock Response?
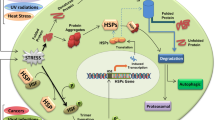
The heat shock response is regulated at the transcriptional level by the activities of a family of heat shock transcription factors (HSF) [2]. Of the three human HSF genes, HSF1, -2, and -4, HSF1 is the best characterized and essential for the heat shock response. In non-stress conditions HSF1 is located in the cytoplasm of mammalian cells in a negatively regulated state as an inert monomer, associated with chaperones and co-chaperones, including hsp70 and hsp90. As a consequence of the appearance of unfolded proteins and release of interacting chaperones, HSF1 DNA-binding activity is de-repressed and monomers oligomerize to a trimeric state, translocate to the nucleus, are inducibly phoshorylated and bind to specific sequence elements referred to as HSE (heat shock elements), located within the hsp gene promoters, activating transcription of heat shock genes and synthesis of HSP.
In the case of viral infection, depending on the type of virus, HSP induction may be the indirect result of rapid accumulation of large amount of viral proteins present in an unfolded aggregation-prone state in the nucleus and the cytoplasm of the host cell, which would result in hsp70 sequestering and freeing HSF for heat shock gene transcription (Fig. 3.2). Several viruses, however, activate the HSR in a specific manner, and can act at different levels on the cascade of events leading to HSP expression and accumulation.
Some viruses may interfere with stress signal transduction pathways upstream of HSF, independently of the presence of unfolded proteins inside infected cells. For example, EBV-induced HSP synthesis is dependent on virus attachment to the host cell membrane, but independent of viral protein synthesis, and involves virus-induced trans-membrane Ca2+ fluxes, indicating that HSR activation occurs trough the interference of the virus with cellular signal transduction pathways [22].
In the case of retroviruses, the HIV envelope protein Env (gp120), which binds to the CD4 receptor and, together with the fusion protein gp41 mediates entry of the virus into the host cell, has been shown to play a role in HSR activation. In fact, the simple interaction of the virus with the CD4 receptor on the cell membrane was found to deliver the signal activating HSP expression [48]. Triggering of hsp70 expression by HIV–Env interaction with the host cell receptor in the absence of viral replication was demonstrated by a proteomic approach after co-culturing effector adherent cells that stably express Env with nonadherent target blood mononuclear cells [57].
Surprisingly, despite the large number of studies demonstrating virus-triggered HSP expression, there is limited evidence of direct HSF activation during viral infection. These include two DNA viruses: HSF activation by adenovirus infection was demonstrated by gel retardation assay early after infection of HeLa cells [11]; more recently, induction of HSF DNA-binding activity, phosphorylation and nuclear translocation was shown in human blood monocyte derived macrophages at late stage of infection with vaccinia virus, leading to increased levels of hsp70 and hsp90 mRNA, and accumulation of hsp70 protein [58]. As far as we know, there is no evidence of induction of HSF DNA-binding activity by RNA viruses.
On the other hand, activation of HSF1 would be predicted to trigger the expression of several heat shock genes; instead, in several instances, selective transcription of specific heat shock genes has been demonstrated, suggesting that individual viral components may interact directly with the host transcriptional machinery. This is well documented for some DNA viruses encoding immediate-early proteins with promiscuous trans-activating capabilities (Fig. 3.3). An example is the SV40 large T-antigen that has been reported to induce hsp70 gene expression by binding to the general transcription initiation factors, the TATA-binding protein (TBP) and TFIIA, and selectively stabilizing the transcription pre-initiation complex at the hsp70 promoter TATA-element [59, 60]. In the case of herpesviruses, the HSV-1 ICP4, EBV EBNA3A and the HCMV IE2 proteins were found to selectively induce hsp70 transcription [61–66]. The HCMV IE2 protein is able to transactivate the human hsp70 promoter by a TATA box-dependent mechanism: IE2 can interact directly with the TATA-binding protein TBP via IE2 C-terminal regions which are important for trans-regulation [61, 64]. Furthermore, it was found that IE2 is able to overcome Dr1 (down-regulator of transcription 1)-mediated repression of hsp70 promoter in vivo, and can interact with Dr1 in vitro and in vivo. This suggested that IE2-induced transcriptional activation of the hsp70 promoter is due to the ability of IE2 to alleviate Dr1-mediated repression by disruption of the inhibitory complex of Dr1 with TBP [65]. It should be noted that EBNA3A was also found to upregulate the expression of the cochaperones hsp40 and Bag-3 [66].

Modulation of HSP70 gene transcription by viral products. Schematic representation of the HSP70 promoter. Heat shock binding elements (HSE), CCAAT-box promoter proximal elements (CCAAT) and TATA-box elements (TATA) are indicated. Viral proteins, including E1A of Adenovirus, large T antigen (T ag) of Simian virus 40 (SV40), ICP4 of herpes simplex virus type 1 (HSV-1), IE2 of human cytomegalovirus (HCMV), and EBNA3 (EBV nuclear antigen 3) of Epstein-Barr virus (EBV), are able to modulate HSP70 gene transcription by direct interaction with different components of the basal transcription apparatus. See text for details. TFIIs: general transcription factors II; CBF: CCAAT-binding factor
Also in the case of adenoviruses, stimulation of hsp70 expression is mediated through an interaction of the viral early protein E1A with the CCAAT-box binding factor [67, 68]. As for HCMV, E1A-mediated disruption of the inhibitory complex Dr1 with the TATA-binding protein was reported [69]. In the case of the avian adenovirus CELO, the viral product Gam-1 was found to be responsible for relocalization and increase in intracellular levels of hsp70 and hsp40 [70]. In addition to stimulating its transcription, adenoviruses specifically promote hsp70 mRNA nuclear export by interaction with the early E1B protein, thus evading the virus-induced cellular mRNAs export block [71, 72], and revealing different levels of complexity of virus-mediated regulation of hsp70 expression.
Finally, it should be noted that similarities in the mechanism of preferential translation during stress conditions between hsp70 mRNA and viral mRNA have been reported. This is an extremely interesting and as yet poorly understood level of interaction between viruses and their host. One example is represented by the described similarities between the 5ʹ noncoding region of human adenovirus late mRNAs (the tripartite leader) and the human hsp70 mRNA for selective translation, promoting ribosome shunting through the use of conserved sequences that are complementary to the stem of the 3ʹ hairpin of 18S rRNA during conditions in which cap-dependent protein synthesis is blocked [73]. It has been known for several years that both viral and heat shock mRNAs can be translated under stress conditions in which cellular protein synthesis is inhibited. This implies that since several viruses cause a shut-off of cap-dependent protein synthesis and hijack the host translational machinery for their exclusive use, they may favor host HSP mRNA translation at the same time. Whether this is an advantage or a disadvantage for the invading pathogen remains to be established and it represents a central question in the interaction of the virus with its host.
One important consideration drawn from the findings described above is that, with rare exceptions, viruses may have evolved different strategies to increase the level of selected HSP without apparently inducing a “canonical” heat shock response where HSF DNA-binding activity and phosphorylation is needed. This interpretation would favor the hypothesis that virus-induced HSP could be actively involved in the regulation of the virus life cycle, rather than reflecting a nonspecific stress situation of the host cell.
3.4 Which Is the Functional Significance of HSP Induction During Viral Infection?
Viruses, independently of their amazing variation and complexities, in order to accomplish their basic mission, multiplication, need to complete a series of common tasks. These which start with the recognition and binding to host receptors and co-receptors, followed by the entry into the cell by endocytosis or membrane fusion, disassembly of the viral capsid and protective structures, and release of the viral genome into the cytoplasm or the nucleus where transcription and replication occur. Then large quantities of newly manufactured viral proteins begin to accumulate, many of which undergo complex steps of maturation and processing, before packaging the viral genome and assembling into capsidic structures. Some viruses, like herpesviruses, also produce scaffolding proteins that are involved in virion assembly, but are subsequently disposed of and do not form part of the mature virion. Enveloped viruses also need to utilize the cellular trafficking apparatus for transporting and inserting into cell membranes viral glycoproteins that direct formation of the envelope and the exit of mature virions from the host cell.
There is a large amount of literature that describes a remarkable number of possible functions of HSP during every step of the viral life cycle. Rather than attempting to summarize all of the relevant literature, we will describe some examples for each stage of the replication of important human pathogens, focusing on the best understood models for each step.
3.4.1 Virus Entry
The first step in infection of a target cell consists of binding of the virion to the host surface, an event dependent on the recognition of specific receptors and co-receptors, which therefore represent important determinants of virus tissue tropism and pathogenesis. Although cytosolic HSP do not contain leader peptides enabling membrane localization, hsc70 and hsp70 are found on the surface of different types of cells [74]. In particular hsp70 has been detected in detergent-soluble microdomains enriched in sphingolipids [75] and was shown to directly interact with the lipid phosphatidylserine in plasma membranes of tumor cells [76]. In addition to the well known implication of this phenomenon in immunological responses [77], surface-exposed hsp70 and hsc70 proteins have been involved in virus entry into cells. The best studied example of this HSP function is rotavirus infection.
Rotaviruses are non-enveloped double-stranded RNA viruses, which represent the leading cause of severe gastroenteritis in infants and young children word-wide [78]. Although they have a preferential cell tropism for the mature enterocytes of the villi of the small intestine, rotaviruses can infect different types of tissues, in a complex multistep process in which different domains of the virus surface proteins interact with different cell surface molecules acting as attachment and entry receptors, including sialic acid (SA), several integrins and hsc70. The initial contact of the virus with the cell plasma membrane is through an SA-containing receptor, via the VP8 domain of viral surface protein VP4. This initial binding allows the virus to interact with integrin α2β1, and subsequently with integrins αvβ3, αxβ2 and hsc70 present in cell membrane lipid microdomains [79] (Fig. 3.4a). The viral interaction with hsc70 was shown to occur through the C-terminal domain of the viral structural protein VP5 and the peptide binding-domain of hsc70 [80, 81]. The fact that incubation of host cells with antibodies to hsc70, as well as preincubation of the virus particles with a recombinant human hsc70 protein, selectively inhibited rotavirus infectivity, without affecting virus attachment to the cell membrane, indicates that rotaviruses interact with hsc70 after the initial binding to other cell surface molecules, and suggested that hsc70 may play an active role in the rotavirus entry process at a post-attachment step [82]. It has been speculated that the chaperone activity of hsc70 could have a pivotal role to help conformational rearrangements of the virus outer layer proteins VP4 and VP7 taking place during virus particle attachment and entry into cells [82]. The interaction of rotavirus with hsc70 may, however, be more complex. The fact that double layered rotavirus particles (DPLs) were recently shown to bind to hsc70 through a different rotavirus structural protein, VP6, indicated a different level of interaction [83] and have suggested a fundamental role of HSP for rotavirus infectivity not only during entry, but also at a post-entry level, in rotavirus uncoating, similarly to hsc70-mediated outer capsid disassembly of reovirus [84].

Involvement of heat shock proteins in different phases of virus replication. Heat shock proteins have been shown to function at many levels during different stages of the viral life cycle. Some examples of HSP function during entry (a, Rotavirus), uncoating (b, Adenovirus), viral gene transcription (c, Human Immunodeficiency virus type 1, HIV-1), viral genome replication (d, Hepatitis B virus, HBV) and viral capsid assembly (e, Polyomavirus) are shown. Details are described in the text
Apart of rotaviruses, HSP have been involved in cell tropism and entry of other human viral pathogens, including Japanese encephalitis virus, Dengue virus and the human retrovirus HTLV-1 [85–87]. In the case of HTLV-1, cell-surface exposed hsc70, which has been shown to bind the HTLV-1 glycoprotein gp46, appears to be essential for cell-to-cell interactions between HTLV-1-harboring cells and target cells leading to syncytium formation and allowing direct cell-to-cell transfer of the virus [86, 87]. Finally, in addition to hsc70/hsp70, cell-surface exposed ER stress protein grp78/BiP has also been shown to interact with viruses, and in particular with coxsackievirus A9 (CAV-9), a nonenveloped enterovirus of the Picornaviridae family. Interestingly, both the attachment and cell entry of CAV-9 can be prevented by BiP-specific antibodies [88].
3.4.2 Uncoating
After penetration into the target cell, the viral genome, which is packaged in protective nucleocapsid structures, has to be released in a form able to be transcribed, translated and replicated. Depending on the type of virus, uncoating may occur simultaneously with the internalization of the pathogen, or it may take place only after a complex series of membrane fusion and transport steps. The molecular mechanism of uncoating is still not well understood for several viruses. Since hsp70 chaperones are known to be involved in the disassembly of oligomeric protein structures, including the uncoating of clathrin vesicles [89, 90] which are utilized by many viruses for internalization into the host, it is feasible that the hsp70 chaperone machinery may participate in the uncoating of virion particles.
In fact, an involvement of chaperones and cochaperones in virus uncoating has been shown during infection with adenoviruses (AdV). Adenoviruses, nonenveloped DNA viruses responsible for common respiratory and gastrointestinal infections, present a highly complex icosahedral capsidic structure composed by 11 proteins organized in “hexon” and “penton base” building blocks, and 12 fibers projecting from the vertices of the icosahedron. AdV enter by endocytosis and soon after the release of the virion particles from endocytic vesicles into the cytoplasm, hsp70 and hsc70 can be found attached to the hexon protein [91], while hsp70 and its co-chaperone Bag-3 interact with the penton protein [92]. The intact capsid is transported to the nuclear pore complex using the cell nuclear localization signal (NLS)-dependent nuclear import machinery and docks with the nuclear pore by interaction of the hexon protein with components of the pore complex [93]. The viral DNA, without the capsid, is subsequently translocated into the nucleus in an hsp70-dependent process (Fig. 3.4b). It is hypothesized that the capsid, which is too large to pass through the nuclear pore, is disassembled in an hsp70-dependent manner allowing the viral DNA to enter the nucleus [93]. However, the fact that in a reconstituted import assay, containing hsp70 with the hexon import factors, the viral DNA could not be transferred into the nucleus, suggested that hsp70 cochaperones such as J-domain proteins or Bag-domain proteins may be necessary for coat disassembly and genome nuclear transfer. Thus, similarly to the uncoating of clathrin-coated vesicles where the clathrin-associated J-domain protein auxilin targets hsc70 to clathrin for the multiple ATPase cycles-requiring uncoating reaction [94], cochaperones may be needed for targeting hsp70 to the AdV capsid, and for assisting in the disassembly process.
In addition to AdV, hsp70 chaperones were shown to efficiently disassemble polyoma- and papillomavirus-like particles and virions in energy-dependent reactions in vitro [95], supporting a more general role for cell chaperones in virus capsidic structure disassembly.
3.4.3 Viral Gene Expression
Animal viruses have evolved a remarkable range of strategies for regulating the expression of their genes in order to optimize the use of the host cell resources. For transcription of their genome, RNA viruses utilize viral-encoded RNA polymerases and/or polymerase complexes, which are translated directly from the genomic RNA immediately after entry into the host in the case of positive-strand RNA viruses, or are carried in the viral capsid strictly associated to the genome in the case negative-strand RNA viruses. DNA viruses, with the exception of poxviruses, transcribe their genes in the nucleus and exploit the cellular transcription machinery engaging host cell initiation and elongation factors associated with specialized viral proteins. Since some of the required host factors interact with components of the hsp70 system, the chaperone network also intervenes in this phase of the viral life cycle. One example is represented by the hsp70 cochaperone Bag-1, which was shown to interact physically with several transcription initiation factors in vitro and to stimulate general transcription activity in an hsp70-dependent manner [96–98]. The M isoform of Bag-1 was shown to strongly enhance the activity of the HCMV promoter under unstressed conditions; this effect was mediated by the dual activity of the protein, i.e. binding to the hsp70/hsc70 chaperones through the C-terminal BAG domain, and to the HCMV promoter element through the N-terminal DNA-binding motif [99]. It has been hypothesized that Bag-1 M may function by activating or stabilizing host transcription factors through hsp70/hsc70 chaperone interactions [99]. Bag-1 was also identified as a nuclear factor activating human polyomavirus JCV early and late promoters, mimicking transcription factors [100]; however, the molecular mechanisms responsible for transcriptional activation are still unclear.
Among RNA viruses, retroviruses, in addition to viral proteins, also have to depend on the host transcriptional machinery for viral gene expression. A unique feature that distinguishes HIV and lentiviruses from other retroviruses is the ability to productively infect nondividing cells, by actively transporting the viral preintegration complex (PIC) into the nucleus without breakdown of the nuclear envelope during cell division [101]. Nuclear import is regulated by different PIC components, comprising the central DNA flap, and several viral proteins, including Vpr for HIV type 1 (HIV-1) and Vpx for HIV type 2 (HIV-2). Interestingly, hsp70 has been suggested to play a role in HIV-1 PIC nuclear import also in the absence of Vpr [102], whereas it has been recently shown that the cochaperone hsp40/DNAJB6 specifically enhances the nuclear localization of Vpx and modulates the targeting of HIV-2 PICs to the nuclei of infected cells [103]. After nuclear import and integration into the host genome, the efficient transcription of the full-length proviral DNA depends on the viral transactivator protein Tat, that stimulates the processivity of host RNA polymerase II, and its specificity for the HIV RNA stem-loop structure TAR located at the 5ʹ end of the nascent viral transcripts [101]. This activation is mediated by the human positive transcription elongation factor P-TEFb, which interacts with Tat and phosphorylates the C-terminal domain of RNA polymerase II. The catalytic subunit of the P-TEFb complex is the cyclin-dependent protein kinase Cdk9, which has been shown to form a heterodimer with cyclin T1 and, in addition, to interact with hsp70 and the kinase-specific chaperone complex hsp90/Cdc37 to form two separate chaperone Cdk9 complexes (Fig. 3.4c). These two complexes act sequentially to facilitate Cdk9 folding/stabilization and the production of the mature Cdk9/cyclin T1 P-TEFb complex [104]. In addition to hsp70 and hsp90, hsp40 was also implicated in the enhancement of HIV-1 gene expression, but in this case transcriptional stimulation is mediated by the viral protein Nef [53].
Interestingly, HSP do not interact only with cellular factors involved in transcriptional regulation. For example, hsp90 has been shown to interact with the viral RNA polymerase in the case of influenza virus. Influenza A virus genome consists of eight segmented, single-stranded RNAs of negative polarity, each one associated with the nucleocapsid protein (NP) and the viral RNA polymerase complex consisting of three subunits, PB2, PB1 and PA, to form the ribonucleoprotein complexes (vRNP) [105]. Since the viral polymerase is not able to synthesize a cap structure, host-derived oligo RNA containing cap structures are necessary for initiation of transcription. The host capped RNA is recognized by the carboxyl-terminal region of the PB2 subunit, and cleaved by the PB1 subunit 10–15 bases downstream from the 5ʹ end; the capped RNA fragment then serves as a primer for viral mRNA synthesis catalyzed by PB1 [106]. It has been shown that hsp90 is re-localized to the cell nucleus after viral infection, and interacts with the viral RNA polymerase PB2 subunit through the amino-terminal chaperone domain and the middle region containing a highly acidic domain. Hsp90 chaperone activity appears not be responsible for the stimulatory activity that in fact resides in the acidic middle region, but not in the amino-terminal chaperone domain [107]. It was speculated that hsp90 acts by facilitating the association of RNA-free RNA polymerase to template RNA and/or stabilize the RNA polymerase during its translocation between templates. It was suggested that hsp90 might be considered a host factor for the influenza virus RNA polymerase. More recently it has been shown that hsp90 is also involved in the nuclear import and assembly of influenza virus RNA polymerase subunits [108]. Interestingly, the chaperone activity of hsp70 did not mimic the hsp90 stimulatory activity in influenza virus transcription, whereas hsp70 was shown to increase the viral polymerase activity of canine distemper virus (CDV), a member of the Paramyxoviridae family, by a reversible and direct interaction with the viral core particle [109]. In this case, a stimulatory effect of hsp70, but not hsc70, on the viral polymerase activity was shown in CDV nucleocapsid isolated from infected cells. Hsp70 was also found to interact in an ATP-dependent manner with the polymerase of a human member of the Paramyxoviridae family, the respiratory syncytial virus, in host cell lipid-rafts [33].
All together, these results indicate that chaperones and cochaperones may be involved in viral gene expression at the level of transcription initiation and elongation, and may act by direct interaction with host factors, as well as viral polymerases.
3.4.4 Genome Replication
A critical role for chaperones in viral genome replication was first demonstrated more than 30 years ago by the pioneering studies of Costa Georgopoulos in bacteriophage λ DNA replication [110]. Subsequent studies on animal viruses have highlighted the role of the cellular protein folding machinery in viral genome replication, reflecting the requirement for selected viral proteins to undergo essential conformational changes in order to carry out their functions. One model in which viral genome replication has been studied extensively is the hepatitis B virus (HBV). In addition to its importance in human pathology (worldwide, there are approximately 350–400 million persons infected with chronic hepatitis B), HBV also represents a distinct entity in the world of viruses with its extremely compact 3.2 kb circular, not covalently closed, DNA genome, which has the unusual property of being partly double-stranded, with a single-stranded region of variable length [111]. These peculiar features arise from the unique mechanism of generation of the DNA genome via reverse transcription. As in all hepdnaviruses, a pregenomic RNA (pgRNA) that also acts as mRNA for the capsid protein and the viral reverse transcriptase, termed P protein, is packaged into viral capsids where is reverse-transcribed into progeny DNA genomes. Both pregenomic RNA encapsidation and initiation of DNA synthesis are triggered by the binding of P to the ɛ 5ʹ-proximal RNA stem-loop of the pgRNA. After binding a “priming reaction” occurs, in which a tyr-residue on the “terminal protein” (TP) domain of the reverse transcriptase serves for the protein-primed synthesis of a ɛ-encoded and covalently TP-linked short (3–4 nt) DNA oligonucleotide. After translocation of the complex to a 3ʹ proximal RNA element, the oligo is extended into the complete minus-strand DNA; P-driven plus-strand DNA synthesis then yields the circular incomplete DNA genome present in mature virions [111].
It is well established that the priming reaction strictly requires prior activation of the reverse transcriptase by host chaperones, including interactions with hsp40, hsp70, hsp90 and Hop in an ATP-dependent reaction. Following the initial observation that hsp90 is associated with the reverse transcriptase of the duck HBV [112], mechanistic studies initially in cell-free rabbit reticulocyte systems and more recently in in vitro reconstitution of priming-active P complexes entirely from purified components [113] have allowed the role of the individual chaperones that are required to induce functionally crucial structural rearrangements in both the P protein and the RNA to be dissected. The following model of action of the chaperone machine for HBV replication has been recently proposed [111]. In the inactive state of P, the ɛ-RNA binding pocket is inaccessible; hsp40/hsp70 activation promotes the structural changes in the reverse transcriptase required for binding the ɛ-RNA (Fig. 3.4d). Complexes containing primer-competent ɛ-RNA undergo induced-fit rearrangements in the RNA and the protein enabling them to initiate DNA synthesis. However, maintaining a steady-state level of the active P form, which is metastable and decays in the inactive state within minutes, requires constant re-activation by the hsp40/hsp70 system and a continuous supply of ATP. In the presence of substrate, the weak hsp70 ATPase activity is stimulated up to 1,000 fold by hsp40, explaining the important role of this co-chaperone in this system. Since the ADP-ATP exchange on hsp70 represents a rate-limiting step in the priming reaction, the presence of Bag-1, an established nucleotide exchange factor of hsp70, markedly stimulates activation by increasing the steady-state concentration of the activated metastable P form. Whether in addition to the hsp70 system, the presence of hsp90 and the hsp70-hsp90 organizing protein Hop is essential for P activation has been controversial, though it is generally accepted that the presence of hsp90, as well as the co-chaperone p23, highly enhances P activity [113–118]. It should be noted that a negative role of hsp40 in the regulation of HBV replication through destabilization of the viral X protein has been also recently suggested [119]. In addition to the hsp70 and hsp90 systems, hsp60 was found to be important for HBV replication in vivo, presumably through activation of the reverse transcriptase before encapsidation of P into HBV core particles [120–122]. Finally hsp70 and hsp90 (but not hsp60) are found incorporated into the released virions supporting the hypothesis that these chaperones may be needed both to stabilize the reverse transcriptase in the extracellular particle and to allow immediate reactivation after entry into host cells.
The involvement of chaperone systems in genome replication of animal viruses has also been shown in DNA viruses that do not require reverse transcription, including human papillomaviruses [123, 124], herpes simplex virus type 1 [125], and Varicella Zoster virus [126]. In particular, in the case of herpes simplex, HSV-1 DNA polymerase was identified as a putative client protein of the hsp90 chaperone system [127]. The general picture emerging from the literature is that different chaperones, and in particular the hsp70/hsp40 system, appear to play an important role in the initiation of viral DNA replication by facilitating the assembly of initiation complexes.
3.4.5 Assembly of Viral Components and Virion Morphogenesis
Following the observation that prokaryotic chaperones are essential for bacteriophage assembly [128], HSP involvement in assisting folding of viral structural proteins and nucleocapsid organization has been shown during morphogenesis of several plant viruses [129, 130], as well as animal viruses, including adenoviruses [131], poliovirus, coxsackievirus [38, 132] and reovirus [133]. Given the requirements for virion quality control and for architecture regulation of structural proteins intrinsically poised for assembly, it is not surprising that viruses hijack the cellular chaperone machinery also during the final stages of their replication. However, a mechanistic role for chaperones in controlling the quality and intracellular location of viral capsid assembly has been described only in a limited number of models, among which are polyomaviruses. Polyomaviruses are small, nonenveloped, icosahedral DNA viruses that include two human pathogens, BK and JC viruses, and the simian SV40 virus, which is one of the most studied models of DNA virus replication and tumorigenesis. Polyomavirus capsids are composed of 72 pentamers of the major viral protein VP1, while one of the minor proteins, either VP2 or VP3, binds in the central 5-fold cavity of each VP1 pentamer [134]. The C-terminal domain of each VP1 monomer connects to a neighboring pentamer to form the principal interpentamer contacts, which are stabilized by calcium ions. Like most DNA viruses, polyomavirus capsid proteins are synthesized in the cytosol, whereas assembly of virions occurs only in the nucleus and it is rigorously controlled so that virions of uniform size are formed. During viral infection the cognate hsc70 was shown to bind VP1 post-translationally and to colocalize with VP1 in the nucleus [135]. Following this observation, Chromy et al. have shown that, after expression of recombinant VP1 in Escherichia coli, the prokaryotic hsp70 chaperone DnaK copurified with VP1 C-terminal domain [136]. In the presence of ATP, the hsp70 chaperone system, including DnaK, DnaJ and GrpE, was able to assemble VP1 into uniform capsid structures without requiring calcium. Chaperone-mediated in vitro assembly of virion-like particles was similarly catalyzed by the mammalian hsc70 protein in an ATP-dependent process, in combination with the J-domain function of the SV40 large T-antigen protein (Fig. 3.4e). These observations, together with the previous characterization of the colocalization of hsc70 with VP1 into the nucleus of infected cells, support a role for the hsp70 chaperone system in the in vivo regulation of virion assembly.
In addition to the role of HSP in the formation of viral capsids, it is well documented that several proteins of animal viruses, including hepatitis B virus [137], HIV, VSV, influenza A and Sindbis viruses, interact with the ER resident hsp70 chaperone BiP/grp78 during maturation (reviewed in ref [138]); however, description of the role of glucose-regulated proteins and ER stress response in viral replication is beyond the scope of this chapter.
Finally, hsp70 has been identified as one of the minor virion components in several negative-strand RNA viruses, including Newcastle disease virus, influenza A virus and vesicular stomatitis virus, and was found to be selectively incorporated into rabies virions [139]. Hsp70 family members were also found to be incorporated into HIV-1 particles: in particular, hsp70 was found to interact with the HIV-1 Gag polyprotein to which it remains stably associated in HIV-1 virion cores [140].
All together, these observations point to an important role of the cellular chaperone machinery in assisting viral monomer folding, multimer assembly and virus maturation. The function of heat shock proteins in viral morphogenesis is discussed in depth in a different chapter of this book.
3.5 Effect of Hyperthermia and HSR Modulators on Virus Replication
The findings described in the previous sections present convincing evidence that several heat shock proteins are involved at many levels in different phases of the viral replication cycle. The importance of the heat shock response in the control of viral infections has been recently emphasized by the outburst of studies on the antiviral effect of hsp90 inhibitors.
3.5.1 Hsp90 Inhibitors and Virus Replication
Hsp90 is an abundant molecular chaperone which binds to a specific set of client proteins, including intracellular receptors and signal transduction proteins, facilitating their folding and stability [141]. Hsp90 activity can be inhibited by drugs that compete for its N-terminal ATP-binding site, which include natural products (geldanamycin, radicicol) and synthetic compounds such as 17-allylamino-demethoxygeldanamycin (17-AAG) [142]. The antiviral activity of hsp90 inhibitors has been described in a wide variety of experimental models against RNA and DNA viruses, among which picornaviruses [132], vaccinia virus [143], hepatitis B [113] and C [144–146] viruses, herpesviruses [126, 127, 147, 148], and several negative strand RNA viruses, including influenza and parainfluenza viruses, rhabdoviruses and bunyaviruses [149, 150]. In most cases the antiviral activity has been ascribed to destabilization and/or mislocation of important non-structural or structural viral proteins following hsp90 release. One example of a structural viral protein destabilized by hsp90 inhibitors is the large multidomain capsid protein P1 of picornaviruses. Picornavirus replication was found to be critically dependent on the function of the hsp90 chaperone machinery; following hsp90 inhibition, P1 misfolds and is targeted for proteasomal degradation, while the other viral proteins remain unaffected [132]. On the other hand, in the case of herpes simplex viruses, geldanamycin was shown to inhibit HSV-1 replication by impairing nuclear transport and increasing proteasome-dependent degradation of the viral DNA polymerase [127]. Similarly, during influenza virus infection hsp90 inhibition results in preventing nuclear import and decreasing the half-life of the viral polymerase subunits, leading to a reduction in viral ribonucleoprotein complexes [150]. Geldanamycin was also found to inhibit hepatitis C RNA replication in a human hepatoma cell line harboring an HCV RNA replicon by disrupting a complex formed by hsp90 with FKBP8, a member of the FK506-binding proteins, and the HCV nonstructural protein NS5A, a membrane-anchored phosphoprotein containing a zinc metal-binding motif within the N-terminal domain which is essential for viral replication [145]. In addition to NS5A, hsp90 was found to be associated also with the HCV NS2/3 protease, and treatment with hsp90 inhibitors can prevent NS2/3 cleavage, hindering the maturation of the polyprotein that encodes the enzymes essential for HCV replication [144].
These data appear to be in contrast with the vast amount of literature describing inhibition of virus replication under conditions where heat shock protein synthesis is enhanced (see next sections). This contradiction, however, is only apparent if it is considered that geldanamycin and other hsp90 inhibitors can activate an HSF1-dependent heat shock response and their administration leads to increased levels of hsp70 as well as hsp90 itself [151, 152]. As an example, inhibition of hepadnavirus replication by hsp90 inhibitors required concentrations of geldanamycin analogs, which induced a strong heat shock response, causing massive upregulation of hsp70 and hsp90 [113].
3.5.2 Hyperthermia in Viral Infection
One condition in which HSP expression is increased in vivo is fever. Many viral diseases cause the development of fever in the host and, while hyperthermia has been implicated in the reactivation of viral replication in latent/chronic infection, the possibility that fever could be beneficial during acute viral infections has been often suggested (reviewed in ref [153]). Exposure of mammalian cells to elevated temperatures during primary virus infection appears to inhibit virus replication in vitro and in vivo. When animals infected with herpesvirus, rabies virus, group B coxsackievirus and other viruses were kept at elevated temperatures, viral infection was reduced in comparison to animals kept at normal temperature [154, 155]. Survival of newborn pups after inoculation with canine herpesvirus was prolonged and viral growth was greatly reduced when animals were kept in an environment that elevated their body temperature to 38.6–39.5°C [156], whereas whole body hyperthermia (30 min at 40°C on day 5 and 12 after infection) decreased virus titers in the spleen of mice infected with the polycythaemia-inducing strain of Friend virus [157]. A similar correlation between body temperature and virus titers was shown during influenza A virus in ferrets [158]. In humans, local hyperthermia (20–30 min at 43°C) has been found to improve the course of the disease in patients with natural and experimental common colds [159], whereas whole-body hyperthermia at 42°C to inhibits HIV-1 transcription in AIDS patients [160].
There is a large amount of literature that describes the antiviral effect of hyperthermia on different viral pathogens also in vitro. Exposure to febrile temperature (39.5°C) was shown to strongly suppress adenovirus replication in normal human bronchial epithelial cells and lung fibroblasts [161]. During primary infection in cultured cells continuous hyperthermia at 3–4°C above the physiological range can block the replication of several RNA and DNA viruses, including the paramyxoviruses measles and Sendai, influenza virus, poliovirus, herpes simplex viruses and poliomaviruses [162–165].
In addition to continuous exposure to higher temperature, short hyperthermic treatment (HT) has also been shown to suppress the replication of different viruses, including togavirus [166], paramyxovirus [167], rhabdovirus [168], rhinovirus [169], and retroviruses [170]. In the case of the retrovirus HIV, while several studies have suggested that heat shock could activate the transcription of HIV-1 long terminal repeat (LTR) in vitro [171, 172], short hyperthermic treatment was shown to be extremely effective in reducing viral titers in lymphoblastoid cells during acute infection with HIV-1 [170]. Moreover, acutely HIV-infected cells were more susceptible to killing by heat than uninfected cells. Because of the increased heat sensitivity of HIV-1 infected cells and of the relatively rapid inactivation of HIV at 42–45°C [173], hyperthermic treatment was suggested to be potentially beneficial in HIV-1 infection [160, 174, 175].
Studies from our laboratory have shown that short (20 min) hyperthermic treatment, if applied during specific stages of the virus cycle, is extremely effective in blocking the replication of the rhabdovirus VSV during primary infection [168]. In this model, no effect on virus replication was found when heat shock was applied soon after virus entry into host cells, or at late times of infection when the virus has taken control of the cellular protein synthesis machinery and HSP synthesis is impaired, indicating that the antiviral effect is not due to a general change in membrane fluidity, cell metabolism or stability of virus proteins or RNA. If hyperthermic treatment was applied at the time of genomic amplification (2–4 h after virus infection) when virus proteins start to be synthesized, virus yield was reduced to less than 5% of control. In a similar manner, short (20 min) exposure to hyperthermic treatment, when applied at specific stages of the virus replication cycle, strongly suppressed the replication of human rhinoviruses (HRV), the major etiologic agent of the common cold [169]. These results suggested that synthesis of heat shock proteins induced at specific stages of the virus cycle could interfere with virus replication. The possibility that HSP could play a role in the control of rhabdovirus and rhinovirus replication was reinforced by the fact that the antiviral effect was mimicked by different chemical inducers of hsp70 [168, 169].
The mechanism underlying the antiviral effect of hyperthermia is complex and still far from being elucidated. Different aspects of the heat shock response are probably involved, depending on the type of virus and the environmental conditions. Exposure to heat activates different signaling pathways above or independent of HSF; it could be speculated that some of these signals could hinder the virus replicative machinery or could interfere with distinct cellular signaling pathways utilized by some pathogens for progression of their replication cycle. One example is the interaction of the HSR with the signaling pathway leading to activation of nuclear factor-κB (NF-κB). Our studies on the antiviral activity of cyclopentenone prostanoids led to the discovery of a cross-talk between HSF1 and NF-κB [176, 177]. We demonstrated that different chemical inducers of the heat shock response including different cyclopentenone prostanoids, sodium arsenite, serine protease inhibitors, as well as heat shock itself, prevent NF-κB activation triggered by cytokines, mitogens or viruses [176–181]. Following this initial observation, it is now well established that heat stress results in inhibition of NF-κB activity, though the mechanism of this interaction is not yet clearly understood. Since some viruses, including herpesviruses and HIV-1, are dependent on NF-κB for enhancing viral gene expression [182], inhibition of the NF-κB pathway by heat could be, at least in part, responsible for the antiviral activity of hyperthermia during infection with these viruses.
A different type of interaction is the one involving the interferon response. Heat shock was found to induce the expression of 2ʹ, 5ʹ-oligoadenylate synthetase, which is usually induced by interferon (IFN) -α and -β and constitutes one of the pathways of IFN antiviral activity by inhibiting protein synthesis through the activation of a latent endoribonuclease [183]. In addition, the hsp90 chaperone complex was found to regulate the activity of two important components of the interferon pathway: the interferon regulatory factor 3 (IRF3), a critical transcriptional regulator of antiviral immune response mediating the expression of type I IFN and other IFN-stimulated genes [184], and the double-stranded RNA-dependent eIF2-α kinase PKR [185].
In addition to the effect on signal transduction pathways, distinct mechanisms for the antiviral activity of hyperthermia against different viral pathogens have been proposed. The use of a mutant HSF1, which is transcriptionally active in the absence of heat stress, has shown that HSF can suppress HIV-1 LTR-driven transcription by a mechanism involving interaction with Sp1 elements in the viral promoter [186]. In the case of influenza virus, the suppression of virus production has been attributed to dissociation of the matrix protein M1 from viral ribonucleoprotein complexes (vRNP), which disrupts the nuclear export of vRNP [187]. On the other hand, hyperthermic treatment during acute infection with herpes simplex viruses resulted in repressing virus replication by inhibiting late viral protein synthesis [15, 16]. Selective inhibition of viral protein synthesis was also detected during SV40 poliomavirus infection [163]; the fact that the antiviral effect of hyperthermia was mimicked by chemical inducers of heat shock proteins suggested that inhibition of SV40 replication is an HSP-mediated response.
3.5.3 Induction of Heat Shock Proteins by Antiviral Agents
Several molecules with antiviral activity have been shown to directly induce HSP synthesis or to be able to modulate HSP expression after induction by hyperthermia or other agents.
Among the antiviral molecules able to enhance HSP expression triggered by hyperthermia or chemical stimulation we can include interferon and some non-steroidal anti-inflammatory drugs (NSAIDs). Treatment with type I interferon, even though did not induce HSP synthesis at 37°C, was shown to potentiate the transcription and translation of HSP mRNA after a mild heat shock (39.5–40.5°C) in human [188] and murine cells [189]. IFN was shown to regulate HSP expression at two levels, by enhancing the transcription rate of the heat-shock genes, and by increasing the stability of mRNA coding for HSPs. In addition, the IFN-regulated kinase PKR was found to be essential for efficient activation of the heat shock response and stabilization of hsp70 mRNA [190]. Results from our laboratory showed that human natural IFN-α, also enhanced prostaglandin-induced hsp70 synthesis in cells infected with vesicular stomatitis virus [191]. The increase in hsp70 synthesis was accompanied by an enhanced antiviral activity. Among NSAIDs, indomethacin which has been shown to possess antiviral activity against several DNA and RNA viruses, including the SARS coronavirus [192], is also known to potentiate and prolong HSF1-mediated HSP expression induced by heat shock and to lower the temperature threshold of HSF1 activation [193, 194].
An interesting class of HSP inducers with antiviral activity are the cyclopentenone prostaglandins (cyPG). Starting from the initial discovery in a paramyxovirus infection model [195], it is well established that cyPG possess a potent antiviral activity against a wide variety of DNA and RNA viruses in vitro and in vivo (reviewed in ref [167, 196]). Antiviral prostaglandins share the ability to function as signal for HSF1-dependent induction of HSP synthesis in non-stressing circumstances [197–199]. In the case of SV paramyxovirus, treatment with cyPG in the early phase of infection causes a selective block of viral protein expression, which persists for as long as hsp70 is synthesized by the host cell [200]. Similar results were obtained during infection with the rhabdovirus VSV [201]. A role for hsp70 as the cellular mediator interfering with SV and VSV protein synthesis was hypothesized based on the following observations: i, chemical hsp70 inducers as diverse as sodium arsenite, cadmium, azetidine, 2-cyclopenten-1-one, the serine protease inhibitor 3,4-dichloroisocoumarin, as well as heat shock, mimic the antiviral activity of prostaglandins and cause a selective inhibition of viral protein synthesis associated with HSP expression in infected cells; ii, cyPG have no effect on viral protein synthesis in murine cells which lack the ability to synthesize hsp70 in response to cyPG, and in human cells in which hsp70 translation is impaired by poliovirus infection [167, 199–203]. These data suggest that high levels of HSP antagonize virus protein synthesis in the early phase of acute infection. The mechanism by which HSP can interfere with viral protein synthesis remains to be elucidated. At high concentrations HSP could directly interact with the nascent viral polypeptides, causing a translational block. This possibility is suggested by the studies of Schlesinger et al., showing that the hsp70 presence during in vitro translation of Sindbis virus mRNA interferes with normal polypeptide synthesis [204]. Alternatively, HSP and virus messages, both of which can be translated in conditions where cellular protein synthesis is impaired (i.e. elevated cytoplasmic ionic concentrations, absence of functional eukaryotic initiation factor 4F), could possess similar mechanisms for preferential translation, and could then compete with each other. This hypothesis is supported by the previously described similarities between the tripartite leader of human adenovirus late mRNAs and the human hsp70 mRNA for selective translation, promoting ribosome shunting during conditions in which cap-dependent protein synthesis is blocked [73]. However, other mechanisms involving alterations of the translational machinery itself, including HSR-induced changes in phosphorylation of initiation and/or elongation factors could participate in the antiviral response.
3.6 Concluding Remarks
The picture emerging from the large amount of literature describing the interactions between heat shock proteins and viruses, accumulated in the last years, clearly shows that viruses exploit the host cell chaperone machinery and, in some cases, reprogram it and maximize its efficiency to achieve optimal conditions for their multiplication. It should be pointed out that, with rare exceptions, HSP induction by viruses appears not to be the result of a “canonical” HSF-mediated heat shock response, but rather the effect of virus-controlled transcriptional/translational switches, sometime involving individual viral products.
Due to the many functions of heat shock proteins and to the many levels of interactions described above, the relationship between viruses and HSP appears to be a complex and multifaceted phenomenon, which depends on the type of virus as well as on the environmental conditions. We have described some examples of HSP function during entry, disassembly, gene expression, genome replication, and morphogenesis of important human viral pathogens, indicating that heat shock proteins are potentially involved in all phases of the viral life cycle. HSP are also involved in other important aspects of viral infection, including the control of survival/apoptosis switches and transformation of the host cell (reviewed in ref [205]), and modulation of the immune-response (reviewed in ref [77]), which are not described here being beyond the scope of this chapter.
It should be considered that the studies of the interaction between HSP and viruses during infection of cultured cells may not reflect accurately the events of a viral infection in vivo where other conditions, which include changes in body temperature, inflammation, oxidative stress or increased local concentrations of cytokines and arachidonate cascade products, may influence HSP expression. The large amount of evidence that pharmacologically- or environmentally-induced alteration of HSP levels leads to inhibition of virus replication appears to be in contrast to the well recognized role of heat shock proteins in different stages of viral replication. However, since most of the described functions of heat shock proteins in the virus replication cycle depend upon a delicate balance of chaperones and co-chaperones with metastable forms of viral proteins, it is possible that this equilibrium is disrupted under conditions leading to an abrupt change in the intracellular level of selected HSP. In this case, stress conditions would be unfavorable to the invading virus.
Given the remarkable differences in the replicative strategies utilized by different viruses and the complexity of host-virus interactions, it is evident that we are only starting to understand the influence of the heat shock response during viral infection. A better comprehension of the functional role of heat shock proteins in virus replication may lead to novel strategies to exploit the HSR for therapeutic gain in the treatment of viral diseases.
References
Morimoto, R. I., and Santoro, M. G. (1998) Stress-inducible responses and heat shock proteins: New pharmacologic targets for cytoprotection. Nature Biotechnol 16, 833–838.
Pirkkala, L., Nykänen, P., and Sistonen, L. (2001) Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 15, 1118–1131.
Young, J. C., and Hartl, F. U. (2002) Chaperones and transcriptional regulation by nuclear receptors. Nat Struct Biol 9, 640–642.
Hasday, J. D., and Singh, I. S. (2000) Fever and the heat shock response: distinct, partially overlapping processes. Cell Stress Chaperones 5, 471–480.
Khandjian, E. W., and Türler, H. (1983) Simian virus 40 and polyoma virus induce the synthesis of heat shock proteins in permissive cells. Mol Cell Biol 3, 1–8.
Sainis, I., Angelidis, C., Pagoulatos, G., and Lazaridis, I. (1994) The hsc70 gene which is slightly induced by heat is the main virus inducible member of the hsp70 gene family. FEBS Lett 335, 282–286.
Honoré, B., Rasmussen, H. H., Celis, A., Leffers, H., Madsen, P., and Celis, J. E. (1994) The molecular chaperones HSP28, GRP78, endoplasmin, and calnexin exhibit strikingly different levels in quiescent keratinocytes as compared to their proliferating normal and transformed counterparts: cDNA cloning and expression of calnexin. Electrophoresis 15, 482–490.
Kao, H. T., Capasso, O., Heintz, N., and Nevins, J. R. (1985) Cell cycle control of the human HSP70 gene: implications for the role of a cellular E1A-like function. Mol Cell Biol 5, 628–633.
Wu, B. J., Hurst, H. C., Jones, N. C., and Morimoto, R. I. (1986) The E1A 13S product of adenovirus 5 activates transcription of the cellular human HSP70 gene. Mol Cell Biol 6, 2994–2999.
Herrmann, C. H., Dery, C. V., and Mathews, M. B. (1987) Transactivation of host and viral genes by the adenovirus E1B 19 K tumor antigen. Oncogene 2, 25–35.
Phillips, B., Abravaya, K., and Morimoto, R. I. (1991) Analysis of the specificity and mechanism of transcriptional activation of the human hsp70 gene during infection by DNA viruses. J Virol 65, 5680–5692.
Milarski, K. L., and Morimoto, R. I. (1986) Expression of human HSP70 during the synthetic phase of the cell cycle. Proc Natl Acad Sci USA 83, 9517–9521.
Simon, M. C., Kitchener, K., Kao, H. T., Hichey, E., Weber, L., Voellmy, R., Heintz, N., and Nevins, J. R. (1987) Selective induction of human heat shock gene transcription by the adenovirus E1A gene products, including the 12S E1A product. Mol Cell Biol 7, 2884–2890.
Notarianni, E. L., and Preston, C. M. (1982) Activation of cellular stress protein genes by herpes simplex virus temperature-sensitive mutants which overproduce immediate early polypeptides. Virology 123, 113–122.
Yura, Y., Terashima, K., Iga, H., Kondo, Y., Yanagawa, T., Yoshida, H., Hayashi, Y., and Sato, M. (1987) Macromolecular synthesis at the early stage of herpes simplex type 2 (HSV 2) latency in a human neuroblastoma cell line IMR-32: repression of late viral polypeptide synthesis and accumulation of cellular heat-shock proteins. Arch Virol 96, 17–28.
Russel, J., Stow, E. C., Stow, N. D., and Preston, C. M. (1987) Abnormal forms of the herpes simplex virus immediate early polypeptide Vmw175 induce the cellular stress response. J Gen Virol 68, 2397–2406.
Wooden, S. K., Li, L., Navarro, D., Qadri, I., Pereira, L., and Lee, A. S. (1991) Transactivation of the grp78 promoter by malfolded proteins, glycosylation block, and calcium ionophore is mediated through a proximal region containing a CCAAT motif which interacts with CTF/NF-I. Mol Cell Biol 11, 5612–5623.
La Thangue, N. B., and Latchman, D. S. (1988) A cellular protein related to heat shock protein 90 accumulates during herpes simplex virus infection and is overexpressed in transformed cells. Exp Cell Res 178, 169–179.
Kobayashi, K., Ohgitani, E., Tanaka, Y., Kita, M., and Imanishi, J. (1994) Herpes simplex virus-induced expression of 70KDa heat shock protein (HSP70) requires early protein synthesis but not viral DNA replication. Microbiol Immunol 38, 321–325.
Ohgitani, E., Kobayashi, K., Takeshita, K., and Imanishi, J. (1998) Induced expression and localization to nuclear inclusion bodies of hsp70 in varicella-zoster virus-infected human diploid fibroblasts. Microbiol Immunol 42, 755–760.
Santomenna, L. D., and Colberg-Poley, A. M. (1990) Induction of cellular hsp70 expression by human cytomegalovirus. J Virol 64, 2033–2040.
Cheung, R. K., and Dosch, H. M. (1993) The growth transformation of human B cells involves superinduction of hsp70 and hsp90. Virology 193, 700–708.
Kotsiopriftis, M., Tanner, J. E., and Alfieri C. (2005) Heat shock protein 90 expression in Epstein-Barr virus-infected B cells promotes γδ T-cell proliferation in vitro. J Virol 79, 7255–7261.
Jindal, S., and Young, R.A. (1992) Vaccinia virus infection induces a stress response that leads to association of Hsp70 with viral proteins. J Virol 66, 5357–5362.
Sedger, L., and Ruby, J. (1994) Heat shock response to vaccinia virus infection. J Virol 68, 4685–4689.
Peluso, R. W., Lamb, R. A., and Choppin, P. W. (1977) Polypeptide synthesis in simian virus 5-infected cells. J Virol 23, 177–187.
Peluso, R. W., Lamb, R. A., and Choppin, P. W. (1978) Infection with paramyxoviruses stimulates synthesis of cellular polypeptides that are also stimulated in cells transformed by Rous sarcoma virus or deprived of glucose. Proc Natl Acad Sci USA 75, 6120–6124.
Watowich, S. S., Morimoto, R. I., and Lamb, R. A. (1991) Flux of the paramyxovirus hemagglutinin-neuraminidase glycoprotein through the endoplasmic reticulum activates transcription of the GRP78-BiP gene. J Virol 65, 3590–3597.
Liberman, E., Fong, Y. L., Selby, M. J., Choo, Q. L., Cousens, L., Houghton, M., and Yen, T. S. B. (1999) Activation of the grp78 and grp94 promoters by Hepatitis C virus E2 envelope protein. J Virol 73, 3718–3722.
Collins, P. L., and Hightower, L. (1982) Newcastle Disease Virus stimulates the cellular accumulation of stress (heat shock) mRNAs and proteins. J Virol 44, 703–707.
Oglesbee, M., and Krakowka, S. (1993) Cellular stress response induces selective intranuclear trafficking and accumulation of morbillivirus major core protein. Lab Invest 68, 109–117.
Brasier, A. R., Spratt, H., Wu, Z., Boldogh, I., Zhang, Y., Garofalo, R. P., Casola, A., Pashmi, J., Haag, A., Luxon, B., and Kurosky, A. (2004) Nuclear heat shock response and novel nuclear domain 10 reorganization in Respiratory Syncytial virus-infected A549 cells identified by high-resolution two-dimensional gel electrophoresis. J Virol 78, 11461–11476.
Brown, G., Rixon, H. W., Steel, J., McDonald, T. P, Pitt, A. R., Graham, S., and Sugrue, R. J. (2005) Evidence for an association between heat shock protein 70 and the respiratory syncytial virus polymerase complex within lipid-raft membranes during virus infection. Virology 338, 69–80.
Garry, R. F., Ulug, E. T. and Bose, H. R. (1983) Induction of stress proteins in Sindbis virus- and vesicular stomatitis virus-infected cells. Virology 129, 319–332.
Michel, M. R., Favre, D., Studer, E., Arrigo, A., and Kempf, C. (1994) Modulation of thermoprotection and translational thermotolerance induced by Semliki Forest virus capsid protein. Eur J Biochem 223, 791–797.
Huber, S. A. (1992) Heat-shock protein induction in adriamycin and picornavirus-infected cardiocytes. Lab Invest 67, 218–224.
Racaniello, V. R. (2007) Picornaviridae : The viruses and their replication. In Fields Virology (5th ed) D. M. Knipe, and P. M. Howley, eds. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, pp. 795–838.
Macejak, D. G., and Sarnow, P. (1992) Association of heat shock protein 70 with enterovirus capsid precursor P1 in infected human cells. J Virol 66, 1520–1527.
Muñoz, A., Alonso, M. A., and Carrasco, L. (1984) Synthesis of heat shock proteins in HeLa cells: inhibition by virus infection. Virology 137, 150–159.
Conti, C., Mastromarino, P., Tomao, P., De Marco, A., Pica, F., and Santoro, M. G. (1996) Inhibition of poliovirus replication by prostaglandins A and J in human cells. Antimicrob Agents Chemother 40, 367–372.
Sarnow, P. (1989) Translation of glucose-regulated protein 78/immunoglobulin heavy-chain binding protein mRNA is increased in poliovirus-infected cells at a time when cap-dependent translation of cellular mRNAs is inhibited. Proc Natl Acad Sci USA 86, 5795–5799.
Xu, A., Bellamy, A. R., and Taylor, J. A. (1998) BiP (GRP78) and endoplasmin (GRP94) are induced following rotavirus infection and bind transiently to an endoplasmic reticulum-localized virion component. J Virol 72, 9865–9872.
Broquet, A. H., Lenoir, C., Gardet, A., Sapin, C., Chwetzoff, S., Jouniaux, A. M., Lopez, S., Trugnan, G., Bachelet, M., and Thomas, G. (2007) Hsp70 negatively controls rotavirus protein bioavailability in Caco-2 cells infected by the rotavirus RF strain. J Virol 81, 1297–1304.
Superti, F., Amici, C., Tinari, A., Donelli, G., and Santoro, M. G. (1998) Inhibition of Rotavirus replication by prostaglandin A: Evidence for a block of virus maturation. J Infect Dis 178, 564–568.
Chouchane, L., Bowers, F. S., Sawasdikosol, S., Simpson, R. M., and Kindt, T. J. (1994) Heat-shock proteins expressed on the surface of human T cell leukemia virus type I-infected cell lines induce autoantibodies in rabbits. J Infect Dis 169, 253–259.
D‘Onofrio, C., Puglianiello, A., Amici, C., Faraoni, I., Lanzilli, G., and Bonmassar, E. (1995) HSP70 production and inhibition of cell proliferation in MOLT-4 T-cell after cell-to-cell transmission of HTLV-1: effect of PGA1. Leukemia Res 19, 345–356.
Kawakami, H., Tomita, M., Okudaira, T., Ishikawa, C., Matsuda, T., Tanaka, Y., Nakazato, T., Taira, N., Ohshiro, K., and Mori, N. (2007) Inhibition of heat shock protein-90 modulates multiple functions required for survival of human T-cell leukemia virus type I-infected T-cell lines and adult T-cell leukemia cells. Int J Cancer 120, 1811–1820.
Furlini, G., Vignoli, M., Re, M. C., Gibellini, D., Ramazzotti, E., Zauli, G., and La Placa, M. (1994) Human immunodeficiency virus type 1 interaction with the membrane of CD4+ cells induces the synthesis and nuclear translocation of 70 K heat shock protein. J Gen Virol 75, 193–199.
Wainberg, Z., Oliveira, M., Lerner, S., Tao, Y., and Brenner, B. G. (1997) Modulation of stress protein (hsp27 and hsp70) expression in CD4+ lymphocytic cells following acute infection with Human Immunodeficiency virus type-1. Virology 233, 364–373.
Iordanskiy, S., Zhao, Y., Di Marzio, P., Agostini, I., Dubrovsky, L., and Bukrinsky, M. (2004) Heat-shock protein 70 exerts opposing effects on Vpr-dependent and Vpr-independent HIV-1 replication in macrophages. Blood 104, 1867–1872.
Di Cesare, S., Poccia, F., Mastino, A., and Colizzi, V. (1992) Surface expressed heat-shock proteins by stressed or human immunodeficiency virus (HIV)-infected lymphoid cells represent the target for antibody-dependent cellular cytotoxicity. Immunol 76, 341–343.
Agnew, L. L., Kelly, M., Howard, J., Jeganathan, S., Batterham, M., Ffrench, R. A., Gold, J., and Watson K. (2003) Altered lymphocyte heat shock protein 70 expression in patients with HIV disease. AIDS 17, 1985–1988.
Kumar, M., and Mitra, D. (2005) Heat shock protein 40 is necessary for Human Immunodeficiency virus-1 Nef-mediated enhancement of viral gene expression and replication. J Biol Chem 280, 40041–40050.
Solis, M., Wilkinson, P., Romieu, R., Hernandez, E., Wainberg, M. A., and Hiscott, J. (2006) Gene expression profiling of the host response to HIV-1 B, C, or A/E infection in monocyte-derived dendritic cells. Virology 352, 86–99.
Brenner, B. G., and Wainberg, M. A. (1999) Heat shock protein-based therapeutic strategies against Human Immunodeficiency Virus type I infection. Infect Dis Obstet Gynecol 7, 80–90.
Liang, D., Benko, Z., Agbottah, E., Bukrinsky, M., and Zhao, R. Y. (2007) Anti-Vpr activities of heat shock protein 27. Mol Med 13, 229–239.
Molina, L., Grimaldi, M., Robert-Hebmann, V., Espert, L., Varbanov, M., Devaux, C., Granier, C., and Biard-Piechaczyk, M. (2007) Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics 7, 3116–3130.
Kowalczyk, A., Guzik, K., Slezak, K., Dziedzic, J., and Rokita, H. (2005) Heat shock protein and heat shock factor 1 expression and localization in vaccinia virus infected human monocyte derived macrophages. J Inflamm 2, 12–21.
Kingston, R. E., Cowie, A., Morimoto, R. I., and Gwinn, K. A. (1986) Binding of polyomavirus large T antigen to the human hsp70 promoter is not required for trans activation. Mol Cell Biol 6, 3180–3190.
Simon, M. C., Fisch, T. M., Benecke, B. J, Nevins, J. R., and Heintz, N. (1988) Definition of multiple, functionally distinct TATA elements, one of which is a target in the hsp70 promoter for E1A regulation. Cell 52, 723–729.
Caswell, R., Hagemeier, C., Chiou, C. J., Hayward, G., Kouzarides, T., and Sinclair, J. (1993) The human cytomegalovirus 86 K immediate early (IE) 2 protein requires the basic region of the TATA-box binding protein (TBP) for binding, and interacts with TBP and transcription factor TFIIB via regions of IE2 required for transcriptional regulation. J Gen Virol 74, 2691–2698.
Colberg-Poley, A. M., Santomenna, L. D., Harlow, P. P., Benfield, P. A., and Tenney, D. J. (1992) Human cytomegalovirus US3 and UL36–38 immediate-early proteins regulate gene expression. J Virol 66, 95–105.
Furnari, B. A., Poma, E., Kowalik, T. F., Huong, S. M., and Huang, E. S. (1993) Human Cytomegalovirus Immediate-Early Gene 2 protein interacts with itself and with several novel cellular proteins. J Virol 67, 4981–4991.
Hagemeier, C., Caswell, R., Hayhurst, G., Sinclair, J., and Kouzarides, T. (1994) Functional interaction between the HCMV IE2 transactivator and the retinoblastoma protein. EMBO J 13, 2897–2903.
Caswell, R., Bryant, L., and Sinclair, J. (1996) Human Cytomegalovirus Immediate-Early 2 (IE2) protein can transactivate the human hsp70 promoter by alleviation of Dr1-mediated repression. J Virol 70, 4028–4037.
Young, P. Anderton, E., Paschos, K., White, R., and Allday, M. J. (2008) Epstein–Barr virus nuclear antigen (EBNA) 3A induces the expression of and interacts with a subset of chaperones and co-chaperones. J Gen Virol 89, 866–877.
Agoff, S. N., and Wu, B. (1994) CBF mediates adenovirus Ela trans-activation by interaction at the C-terminal promoter targeting domain of conserved region 3. Oncogene 9, 3707–3711.
Lum, L. S., Hsu, S., Vaewhongs, M., and Wu, B. (1992) The hsp70 gene CCAAT-binding factor mediates transcriptional activation by the adenovirus E1a protein. Mol Cell Biol 12, 2599–2605.
Kraus, V. B., Inostroza, J. A., Yeung, K., Reinberg, D., and Nevins, J. R. (1994) Interaction of the Dr1 inhibitory factor with the TATA binding protein is disrupted by adenovirus E1A. Proc Natl Acad Sci USA 91, 6279–6282.
Glotzer, J. B., Saltik, M., Chiocca, S., Michou, A. I., Moseley, P., and Cotten, M. (2000) Activation of heat-shock response by an adenovirus is essential for virus replication. Nature 407, 207–211.
Moore, M., Schaack, J., Baim, S. B., Morimoto, R. I., and Shenk, T. (1987) Induced heat shock mRNAs escape the nucleocytoplasmic transport block in adenovirus-infected HeLa cells. Mol Cell Biol 7, 4505–4512.
Yang, U. C., Huang, W., and Flint, S. J. (1996) mRNA export correlates with activation of transcription in human subgroup C adenovirus-infected cells. J Virol 70, 4071–4080.
Yueh, A., and Schneider, R. J. (2000) Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev 14, 414–421.
Multhoff, G., and Hightower, L. E. (1996) Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones 1, 167–176.
Broquet, A. H., Thomas, G., Masliah, J., Trugnan, G., and Bachelet, M. (2003) Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J Biol Chem 278, 21601–21606.
Arispe, N., Doh, M., Simakova, O., Kurganov, B., and De Maio, A. (2004) Hsc70 and Hsp70 interact with phosphatidylserine on the surface of PC12 cells resulting in a decrease of viability. FASEB J 18, 1636–1645.
Pockley, A. G., Muthana, M., and Calderwood, S. K. (2008) The dual immunoregulatory roles of stress proteins. Trends Biochem Sci 33, 71–79.
Estes, M. K., and Kapikian, A. Z. (2007) Rotaviruses. In Fields Virology (5th ed) D. M. Knipe, and P. M. Howley, eds. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, pp. 1917–1974.
López, S., and Arias, C. F. (2004) Multistep entry of rotavirus into cells: a Versaillesque dance. Trends Microbiol 12, 271–278.
Zaráte, S., Cuadras, M. A., Espinosa, R., Romero, P., Juárez, K. O., Camacho-Nuez, M., Arias, C. F., and López, S. (2003) Interaction of rotaviruses with Hsc70 during cell entry is mediated by VP5. J Virol 77, 7254–7260.
Pérez-Vargas, J., Romero, P., López, S., and Arias, C. F. (2006) The peptide-binding and ATPase domains of recombinant hsc70 are required to interact with rotavirus and reduce its infectivity. J Virol 80, 3322–3331.
Guerrero, C. A., Bouyssounade, D., Zárate, S., Isa, P., López, T., Espinosa, R., Romero, P., Méndez, E., López, S., and Arias, C. F. (2002) Heat shock cognate protein 70 is involved in rotavirus cell entry. J Virol 76, 4096–4102.
Gualtero, D. F., Guzmán, F., Acosta, O., and Guerrero, C. A. (2007) Amino acid domains 280–297 of VP6 and 531–554 of VP4 are implicated in heat shock cognate protein hsc70-mediated rotavirus infection. Arch Virol 152, 2183–2196.
Ivanovic, T., Agosto, M. A., Chandran, K., and Nibert, M. L. (2007) A role for molecular chaperone Hsc70 in reovirus outer capsid disassembly. J Biol Chem 282, 12210–12219.
Ren, J., Ding, T., Zhang, W., Song, J., and Ma, W. (2007) Does Japanese encephalitis virus share the same cellular receptor with other mosquito-borne flaviviruses on the C6/36 mosquito cells? Virol J 4, 83–89.
Fang, D., Haraguchi, Y., Jinno, A., Soda, Y., Shimizu, N., and Hoshino, H. (1999) Heat shock cognate protein 70 is a cell fusion-enhancing factor but not an entry factor for human T-cell lymphotropic virus type I. Biochem Biophys Res Commun 261, 357–363.
Sagara, Y., Ishida, C., Inoue, Y., Shiraki, H., and Maeda, Y. (1998) 71-kilodalton heat shock cognate protein acts as a cellular receptor for syncytium formation induced by human T-cell lymphotropic virus type 1. J Virol 72, 535–541.
Triantafilou, K., Fradelizi, D., Wilson, K., and Triantafilou, M. (2002) GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J Virol 76, 633–643.
Greene, L. E., and Eisenberg, E. (1990) Dissociation of clathrin from coated vesicles by the uncoating ATPase. J Biol Chem 265, 6682–6687.
Ungewickell, E. (1985) The 70-kd mammalian heat shock proteins are structurally and functionally related to the uncoating protein that releases clathrin triskelia from coated vesicles. EMBO J 4, 3385–3391.
Niewiarowska, J., D’Halluin, J. C., and Belin, M. T. (1992) Adenovirus capsid proteins interact with HSP70 proteins after penetration in human or rodent cells. Exp Cell Res 201, 408–416.
Chroboczek, J., Gout, E., Favier, A. L., and Galinier, R. (2003) Novel partner proteins of adenovirus penton. Curr Top Microbiol Immunol 272, 37–55.
Saphire, A. C., Guan, T., Schirmer, E. C., Nemerow, G. R., and Gerace, L. (2000) Nuclear import of adenovirus DNA in vitro involves the nuclear protein import pathway and hsc70. J Biol Chem 275, 4298–4304.
Rapoport, I., Boll, W., Yu, A., Böcking, T., and Kirchhausen, T. (2008) A motif in the clathrin heavy chain required for the Hsc70/auxilin uncoating reaction. Mol Biol Cell 19, 405–413.
Chromy, L. R., Oltman, A., Estes, P. A., and Garcea, R. L. (2006) Chaperone-mediated in vitro disassembly of polyoma- and papillomaviruses. J Virol 80, 5086–5091.
Zeiner, M., Niyaz, Y., and Gehring, U. (1999) The hsp70-associating protein Hap46 binds to DNA and stimulates transcription. Proc Natl Acad Sci USA 96, 10194–10199.
Niyaz, Y., Zeiner, M., and Gehring, U. (2001) Transcriptional activation by the human Hsp70-associating protein Hap50. J Cell Sci 114, 1839–1845.
Niyaz, Y., Frenz, I., Petersen, G., and Gehring, U. (2003) Transcriptional stimulation by the DNA binding protein Hap46/BAG-1 M involves hsp70/hsc70 molecular chaperones. Nucleic Acids Res 31, 2209–2216.
Takahashi, N., Sasaki, R., Takahashi, J., Takayama, S., Reed, J. C., and Andoh, T. (2001) BAG-1 M, an isoform of Bcl-2-interacting protein BAG-1, enhances gene expression driven by CMV promoter. Biochem Biophys Res Commun 286, 807–814.
Devireddy, L. R., Kumar, K. U., Pater, M. M., and Pater, A. (2000) BAG-1, a novel Bcl-2-interacting protein, activates expression of human JC virus. J Gen Virol 81, 351–357.
Freed, E. O., and Martin, M. A. (2007) HIVs and their replication. In Fields Virology (5th ed) D. M. Knipe, and P. M. Howley, eds. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, pp. 2107–2186.
Agostini, I., Popov, S., Li, J., Dubrovsky, L., Hao, T., and Bukrinsky, M. (2000) Heat-shock protein 70 can replace viral protein R of HIV-1 during nuclear import of the viral preintegration complex. Exp Cell Res 259, 398–403.
Cheng, X., Belshan, M., and Ratner, L. (2008) Hsp40 facilitates nuclear import of the human immunodeficiency virus type 2 Vpx-mediated preintegration complex. J Virol 82, 1229–1237.
O’Keeffe, B., Fong, Y., Chen, D., Zhou, S., and Zhou, Q. (2000) Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J Biol Chem 275, 279–287.
Palese, P., and Shaw, M. (2007) Orthomyxoviridae: the viruses and their replication. In Fields Virology (5th ed) D. M. Knipe, and P. M. Howley, eds. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, pp. 1647–1689.
Engelhardt, O. G., and Fodor, E. (2006) Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol 16, 329–345.
Momose, F., Naito, T., Yano, K., Sugimoto, S., Morikawa, Y., and Nagata, K. (2002) Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem 277, 45306–45314.
Naito, T., Momose, F., Kawaguchi, A., and Nagata, K. (2007) Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 81, 1339–1349.
Oglesbee, M. J., Liu, Z., Kenney, H., and Brooks, C. L. (1996) The highly inducible member of the 70 kDa family of heat shock proteins increases canine distemper virus polymerase activity. J Gen Virol 77, 2125–2135.
Georgopoulos, C. P. (1977) A new bacterial gene (groPC) which affects lambda DNA replication. Mol Gen Genet 151, 35–39.
Beck, J., and Nassal, M. (2007) Hepatitis B virus replication. World J Gastroenterol 13, 48–64.
Hu, J., and Seeger, C. (1996) Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci USA 93, 1060–1064.
Stahl, M., Retzlaff, M., Nassal, M., and Beck, J. (2007) Chaperone activation of the hepadnaviral reverse transcriptase for template RNA binding is established by the Hsp70 and stimulated by the Hsp90 system. Nucleic Acids Res 35, 6124–6136.
Hu, J., Toft, D. O., and Seeger, C. (1997) Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J 16, 59–68.
Hu, J., and Anselmo, D. (2000) In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J Virol 74, 11447–11455.
Cho, G., Park, S. G., and Jung, G. (2000) Localization of HSP90 binding sites in the human hepatitis B virus polymerase. Biochem Biophys Res Commun 269, 191–196.
Park, S.G., Rho, J.K., and Jung, G. (2002) Hsp90 makes the human HBV Pol competent for in vitro priming rather than maintaining the human HBV Pol/pregenomic RNA complex. Arch Biochem Biophys 401, 99–107.
Hu, J., Flores, D., Toft, D., Wang, X., and Nguyen, D. (2004) Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78, 13122–13131.
Sohn, S. Y., Kim, J. H., Baek, K. W., Ryu, W. S., and Ahn, B. Y. (2006) Turnover of hepatitis B virus X protein is facilitated by Hdj1, a human Hsp40/DnaJ protein. Biochem Biophys Res Commun 347, 764–768.
Park, S. G., and Jung, G. (2001) Human hepatitis B virus polymerase interacts with the molecular chaperonin Hsp60. J Virol 75, 6962–6968.
Park, S. G., Lim, S. O., and Jung, G. (2002) Binding site analysis of human HBV pol for molecular chaperonin hsp60. Virology 298, 116–123.
Park, S. G., Lee, S. M., and Jung, G. (2003) Antisense oligodeoxynucleotides targeted against molecular chaperonin Hsp60 block human hepatitis B virus replication. J Biol Chem 278, 39851–39857.
Liu, J. S., Kuo, S. R., Makhov, A. M., Cyr, D. M., Griffith, J. D., Broker, T. R., and Chow, L. T. (1998) Human Hsp70 and Hsp40 chaperone proteins facilitate human papillomavirus-11 E1 protein binding to the origin and stimulate cell-free DNA replication. J Biol Chem 273, 30704–30712.
Lin, B. Y., Makhov, A. M., Griffith, J. D., Broker, T. R., and Chow, L. T. (2002) Chaperone proteins abrogate inhibition of the human papillomavirus (HPV) E1 replicative helicase by the HPV E2 protein. Mol Cell Biol 22, 6592–6604.
Tanguy Le Gac, N., and Boehmer, P. E. (2002) Activation of the herpes simplex virus type-1 origin-binding protein (UL9) by heat shock proteins. J Biol Chem 277, 5660–5666.
Kyratsous, C. A., and Silverstein, S. J. (2007) BAG3, a host cochaperone, facilitates varicella-zoster virus replication. J Virol 81, 7491–7503.
Burch, A. D., and Weller, S. K. (2005) Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol 79, 10740–10749.
Georgopoulos, C.P., and Hohn, B. (1978) Identification of a host protein necessary for bacteriophage morphogenesis (the groE gene product). Proc Natl Acad Sci USA 75,131–135.
Napuli, A. J., Falk, B. W., and Dolja, V. V. (2000) Interaction between HSP70 homolog and filamentous virions of the Beet yellows virus. Virology 274, 232–239.
Satyanarayana, T., Gowda, S., Mawassi, M., Albiach-Marti, M. R., Ayllón, M. A., Robertson, C., Garnsey, S. M., and Dawson, W. O. (2000) Closterovirus encoded HSP70 homolog and p61 in addition to both coat proteins function in efficient virion assembly. Virology 278, 253–265.
Macejak, D. G., and Luftig, R. B. (1991) Association of hsp70 with the adenovirus type 5 fiber protein in infected HEp-2 cells. Virology 180, 120–125.
Geller, R., Vignuzzi, M., Andino, R., and Frydman, J. (2007) Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev 21, 195–205.
Leone, G., Coffey, M. C., Gilmore, R., Duncan, R., Maybaum, L., and Lee, P. W. K. (1996) C-terminal trimerization, but not N-terminal trimerization, of the reovirus cell attachment protein is a posttranslational and Hsp70/ATP-dependent process. J Biol Chem 271, 8466–8471.
Imperiale, M. J., and Major, E. O. (2007) Polyomaviruses. In Fields Virology (5th ed) D. M. Knipe, and P. M. Howley, eds. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, pp. 2263–2298.
Cripe, T. P., Delos, S. E., Estes, P. A., and Garcea, R. L. (1995) In vivo and in vitro association of hsc70 with polyomavirus capsid proteins. J Virol 69, 7807–7813.
Chromy, L. R., Pipas, J. M., and Garcea, R. L. (2003) Chaperone-mediated in vitro assembly of Polyomavirus capsids. Proc Natl Acad Sci USA 100, 10477–10482.
Lambert, C., and Prange, R. (2003) Chaperone action in the posttranslational topological reorientation of the hepatitis B virus large envelope protein: Implications for translocational regulation. Proc Natl Acad Sci USA 100, 5199–5204.
He, B. (2006) Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ 13, 393–403.
Sagara, J., and Kawai, A. (1992) Identification of heat shock protein 70 in the rabies virion. Virology 190, 845–848.
Gurer, C., Cimarelli, A., and Luban, J. (2002) Specific incorporation of heat shock protein 70 family members into primate lentiviral virions. J Virol 76, 4666–4670.
Whitesell, L., and Lindquist, S. L. (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5, 761–772.
Wandinger, S. K., Richter, K., and Buchner, J. (2008) The Hsp90 chaperone machinery. J Biol Chem 283, 18473–18477.
Hung, J. J., Chung, C. S., and Chang, W. (2002) Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J Virol 76, 1379–1390.
Waxman, L., Whitney, M., Pollok, B. A., Kuo, L. C., and Darke, P. L. (2001) Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci USA 98, 13931–13935.
Okamoto, T., Nishimura, Y., Ichimura, T., Suzuki, K., Miyamura, T., Suzuki, T., Moriishi, K., and Matsuura, Y. (2006) Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J 25, 5015–5025.
Nakagawa, S., Umehara, T., Matsuda, C., Kuge, S., Sudoh, M., and Kohara, M. (2007) Hsp90 inhibitors suppress HCV replication in replicon cells and humanized liver mice. Biochem Biophys Res Commun 353, 882–888.
Li, Y. H., Tao, P. Z., Liu, Y. Z., and Jiang, J. D. (2004) Geldanamycin, a ligand of heat shock protein 90, inhibits the replication of herpes simplex virus type 1 in vitro. Antimicrob Agents Chemother 48, 867–872.
Basha, W., Kitagawa, R., Uhara, M., Imazu, H., Uechi, K., and Tanaka, J. (2005) Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir Chem Chemother 16, 135–146.
Connor, J. H., McKenzie, M. O., Parks, G. D., and Lyles, D. S. (2007) Antiviral activity and RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology 362, 109–119.
Chase, G., Deng, T., Fodor, E., Leung, B. W., Mayer, D., Schwemmle, M., and Brownlee, G. (2008) Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 377, 431–439.
Bagatell, R., Paine-Murrieta, G. D., Taylor, C. W., Pulcini, E. J., Akinaga, S., Benjamin, I. J., and Whitesell, L. (2000) Induction of a Heat Shock Factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res 6, 3312–3318.
Shen, H. Y., He, J. C., Wang, Y., Huang, Q. Y., and Chen, J. F. (2005) Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem 280, 39962–39969.
Santoro, M. G. (1996) Viral infection. In Stress-inducible cellular responses, U. Feige, R. I. Morimoto, I. Yahara, and B. S. Polla, eds. Birkhauser Verlag, Basel, Switzerland, pp. 337–359.
Teisner, B., and Haahr, S. (1974) Poikilothermia and susceptibility of suckling mice to Coxsackie B1 virus. Nature 274, 568–569.
Roberts, N. Jr. (1979) Temperature and host defense. Microbiol Rev 43, 241–259.
Carmichael, L. E., Barnes, F. D., and Percy, D. H. (1969) Temperature as a factor in resistance of young puppies to canine herpesvirus. J Infect Dis 120, 669–678.
Lu, L., Shen, R. N., Zhou, S. Z., Wu, B., Kim, Y. J., Lin, Z. H., Ruscetti, S., Ralph, P., and Broxmeyer, H. E. (1991) Efficacy of recombinant human macrophage colony-stimulating factor in combination with whole-body hyperthermia in the treatment of mice infected with the polycythemia-inducing strain of the Friend virus complex. Exp Hematol 19, 804–809.
Husseini, R. H., Sweet, C., Collie, M. H., and Smith, H. (1982) Elevation of nasal viral levels by suppression of fever in ferrets infected with influenza viruses of differing virulence. J Infect Dis 145, 520–524.
Tyrrell, D., Barrow, I., and Arthus, J. (1989) Local hyperthermia benefits natural and experimental common colds. Br Med J 298, 1280–1283.
Steinhart, C. R., Ash, S. R., Gingrich, C., Sapir, D., Keeling, G. N., and Yatvin, M. B. (1996) Effect of whole-body hyperthermia on AIDS patients with Kaposi’s sarcoma: a pilot study. J Acquir Immune Defic Syndr Hum Retrovirol 11, 271–281.
Thorne, S. H., Brooks, G., Lee, Y. L., Au, T., Eng, L. F., and Reid, T. (2005) Effects of febrile temperature on adenoviral infection and replication: implications for viral therapy of cancer. J Virol 79, 581–591.
Bennett, I. L., and Nicastri, A. (1960) Fever as a mechanism of resistance. Bacteriol Rev 24, 16–34.
Angelidis, C. E., Lazaridis, I., and Pagoulatos, G. N. (1988) Specific inhibition of simian virus 40 protein synthesis by heat and arsenite treatment. Eur J Biochem 172, 27–34.
Panasiak, W., Oraczewska, A., and Luczak, M. (1990) Influence of hyperthermia on experimental viral infections in vitro. In Consensus on Hyperthermia, H. I. Bicher, J. R. McLaren, and G. M. Pigliucci, eds. Plenum Press, New York, pp. 471–475.
Ishida, Y., Hiraki, A., Hirayama, E., Koga, Y., and Kim, J. (2002) Temperature-sensitive viral infection: inhibition of hemagglutinating virus of Japan (Sendai virus) infection at 41 degrees. Intervirology 45, 125–135.
da Costa Carvalho, M. G., and Fournier, M. V. (1991) Effect of heat shock on gene expression of Aedes albopictus cells infected with Mayaro virus. Res Virol 142, 25–31.
Santoro, M. G. (1997) Antiviral activity of cyclopentenone prostanoids. Trends Microbiol 5, 276–281.
De Marco, A., and Santoro, M. G. (1993) Antiviral effect of short hyperthermic treatment at specific stages of vesicular stomatitis virus replication cycle. J Gen Virol 74, 1685–1690.
Conti, C., De Marco, A., Mastromarino, P., Tomao, P., and Santoro, M. G. (1999) Antiviral effect of hyperthermic treatment in rhinovirus infection. Antimicrob Agents Chemother 43, 822–829.
Wong, G. H. W., McHugh, T., Weber, R., and Goeddel, D. V. (1991) Tumor necrosis factor α selectively sensitizes human immunodeficiency virus-infected cells to heat and radiation. Proc Natl Acad Sci USA 88, 4372–4376.
Stanley, S. K., Bressler, P. B., Poli, G., and Fauci, A. S. (1990) Heat shock induction of HIV production from chronically infected promonocytic and T cell lines. J Immunol 145, 1120–1126.
Hashimoto, K., Baba, M., Gohnai, K., Sato, M., and Shigeta, S. (1996) Heat shock induces HIV-1 replication in chronically infected promyelocyte cell line OM10.1. Arch Virol 141, 439–447.
Gordon, L. M., Jensen, F. C., Curtain, C. C., Mobley, P. M., and Aloia, R. C. (1988) Thermotropic lipid phase separation in the human immunodeficiency virus. Biochim Biophys Acta 943, 331–342.
Yatvin, M. B., Stowell, M. H., and Steinhart, C. R. (1993) Shedding light on the use of heat to treat HIV infections. Oncology 50, 380–389.
Pennypacker, C., Perelson, A. S., Nys, N., Nelson, G., and Sessler, D. J. (1995) Localized or systemic in vivo heat inactivation of human immunodeficiency virus (HIV): a mathematical analysis. J Acquir Immune Defic Syndr Hum Retrovirol 8, 321–329.
Rossi, A., Elia, G., and Santoro, M. G. (1997) Inhibition of nuclear factor-kappaB by prostaglandin A1: an effect associated with heat shock transcription factor activation. Proc Natl Acad Sci USA 94, 746–750.
Rossi, A., Kapahi, P., Natoli, G., Takahashi, T., Chen, Y., Karin, M., and Santoro, M. G. (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 403, 103–108.
Santoro, M. G. (2000) Heat shock factors and the control of the stress response. Biochem Pharmacol 59, 55–63.
Rossi, A., Elia, G., and Santoro, M. G. (1998) Activation of the heat shock factor 1 by serine protease inhibitors. An effect associated with nuclear factor-kappaB inhibition. J Biol Chem 273, 16446–16452.
Amici, C., Belardo, G., Rossi, A., and Santoro, M. G. (2001) Activation of IkappaB kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J Biol Chem 276, 28759–28766.
Bernasconi, D., Amici, C., La Frazia, S., Ianaro, A., and Santoro, M. G. (2005) The IkappaB kinase is a key factor in triggering influenza A virus-induced inflammatory cytokine production in airway epithelial cells. J Biol Chem 280, 24127–24134.
Santoro, M. G., Rossi, A., and Amici, C. (2003) NF-kappaB and virus infection: who controls whom. EMBO J 22, 2552–2560.
Chousterman, S., Chelbi-Alix, M. K., and Thang, M. N. (1987) 2ʹ,5ʹ-oligoadenylate synthetase expression is induced in response to heat shock. J Biol Chem 262, 4806–4811.
Yang, K., Shi, H., Qi, R., Sun, S., Tang, Y., Zhang, B., and Wang, C. (2006) Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol Biol Cell 17, 1461–1471.
Donzé, O., Abbas-Terki, T., and Picard, D. (2001) The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J 20, 3771–3780.
Ignatenko, N. A., and Gerner, E. W. (2003) Regulation of the HIV1 long terminal repeat by mutant heat shock factor. Exp Cell Res 288, 1–8.
Sakaguchi, A., Hirayama, E., Hiraki, A., Ishida, Y., and Kim, J. (2003) Nuclear export of influenza viral ribonucleoprotein is temperature-dependently inhibited by dissociation of viral matrix protein. Virology 306, 244–253.
Chang, C. C., Konno, S., and Wu, J. M. (1991) Enhanced expression of heat shock protein and mRNA synthesis by type I interferon in human HL-60 leukemic cells. Biochem Int 24, 369–377.
Dubois, M. F., Mezger, V., Morange, M., Ferrieux, C., Lebon, P., and Bensaude, O. (1988) Regulation of the heat shock response by interferon in mouse L cells. J Cell Physiol 137, 102–109.
Zhao, M., Tang, D., Lechpammer, S., Hoffman, A., Asea, A., Stevenson, M. A., and Calderwood, S. K. (2002) Double-stranded RNA-dependent protein kinase (pkr) is essential for thermotolerance, accumulation of HSP70, and stabilization of ARE-containing HSP70 mRNA during stress. J Biol Chem 277, 44539–44547.
Pica, F., Rossi, A., Santirocco, N., Palamara, A., Garaci, E., and Santoro, M. G. (1996) Effect of combined αIFN and prostaglandin A1 treatment on vesicular stomatitis virus replication and heat shock protein synthesis in epithelial cells. Antiviral Res 29, 187–198.
Amici, C., Di Caro, A., Ciucci, A., Chiappa, L., Castilletti, C., Martella, V., Decaro, N., Buonavoglia, C., Capobianchi, M. R., and Santoro, M. G. (2006) Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir Ther 11, 1021–1030.
Amici, C., Rossi, A., and Santoro, M. G. (1995) Aspirin enhances thermotolerance in human erythroleukemic cells: an effect associated with the modulation of the heat shock response. Cancer Res 55, 4452–4457.
Lee, B. S., Chen, J., Angelidis, C., Jurivich, D. A., and Morimoto, R. I. (1995) Pharmacological modulation of heat shock factor 1 by antiinflammatory drugs results in protection against stress-induced cellular damage. Proc Natl Acad Sci USA 92, 7207–7211.
Santoro, M. G., Benedetto, A., Carruba, G., Garaci, E., and Jaffe, B. M. (1980) Prostaglandin A compounds as antiviral agents. Science 209, 1032–1034.
Santoro, M. G., and Roberts, S. M. (1999) Search for novel cytoprotective and antiviral prostanoids. Drug News Perspect 12, 395–400.
Santoro, M. G., Garaci, E., and Amici, C. (1989) Prostaglandins with antiproliferative activity induce the synthesis of a heat shock protein in human cells. Proc Natl Acad Sci USA 86, 8407–8411.
Amici, C., Sistonen, L., Santoro, M. G., and Morimoto, R. I. (1992) Anti-proliferative prostaglandins activate heat shock transcription factor. Proc Natl Acad Sci USA 89, 6227–6231.
Santoro, M. G. (1994) Heat shock proteins and virus replication: hsp70s as mediators of the antiviral effects of prostaglandins. Experientia 50, 1039–1047.
Amici, C., Giorgi, C., Rossi, A., and Santoro, M. G. (1994) Selective inhibition of virus protein synthesis by prostaglandin A1 a translational block associated with HSP70 synthesis. J Virol 68, 6890–6899.
Pica, F., De Marco, A., De Cesare, F., and Santoro, M. G. (1993) Inhibition of vesicular stomatitis virus replication by Δ12-prostaglandin J2 is regulated at two separate levels and is associated with induction of stress protein synthesis. Antiviral Res 20, 193–208.
Rossi, A., Elia, G., and Santoro, M. G. (1996) 2-Cyclopenten-1-one, a new inducer of heat shock protein 70 with antiviral activity. J Biol Chem 271, 32192–32196.
Santoro, M. G., Jaffe, B. M., and Esteban, M. (1983) Prostaglandin A inhibits the replication of vesicular stomatitis virus: effect on virus glycoprotein. J Gen Virol 64, 2797–2801.
Schlesinger, M. J., Ryan, C., Sadis, S., and Hightower, L. E. (1991) In vitro inhibition of nascent polypeptide formation by HSP70 proteins. In Heat Shock Proteins, B. Maresca, and S. Lindquist, eds. Springer-Verlag, Berlin, pp. 111–117.
Mayer, M. P. (2005) Recruitment of Hsp70 chaperones: a crucial part of viral survival strategies. Rev Physiol Biochem Pharmacol 153, 1–46.
Acknowledgements
This work was supported by the Italian Ministry of Public Health (ISS, ACC Oncology Project), and the Italian Ministry of University and Scientific Research (FIRB and PRIN Projects).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Netherlands
About this chapter
Cite this chapter
Santoro, M.G., Amici, C., Rossi, A. (2009). Role of Heat Shock Proteins in Viral Infection. In: Pockley, A., Calderwood, S., Santoro, M. (eds) Prokaryotic and Eukaryotic Heat Shock Proteins in Infectious Disease. Heat Shock Proteins, vol 4. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-2976-8_3
Download citation
DOI: https://doi.org/10.1007/978-90-481-2976-8_3
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-2975-1
Online ISBN: 978-90-481-2976-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)