
Abstract
This chapter summarizes recent developments in the design, synthesis, and structure–activity relationship studies of organometallic antimalarials. It begins with a general introduction to malaria and the biology of the parasite Plasmodium falciparum, with a focus on the heme detoxification system. Then, a number of metal complexes from the literature are reported for their antiplasmodial activity. The second half of the chapter deals with the serendipitous discovery of ferroquine, its mechanism(s) of action, and the failure to induce a resistance. Last, but not least, we suggest that the bioorganometallic approach offers the potential for the design of novel therapeutic agents.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
1.1 Malaria: The Burden and the Problems
Malaria is the most common parasitical disease in the world [1]. Half of the World’s population is exposed to this disease caused by a protozoan, Plasmodium, and transmitted by the female Anopheles mosquito. The clinical phase in human infection corresponds to the invasion of red blood cells (RBC) by the merozoïte form of the parasite; this follows a first phase of development of the sporozoïte in the liver after its injection by Anopheles. In the RBC, the parasite develops into a trophozoïte that internalizes and digests its host cell content, it then differentiates into schizont containing multiple merozoïtes which will invade new RBC. Two forms of malaria have a particularly severe prognostic: (1) cerebral malaria (mainly in young children), due to sequestration of mature parasites in the brain, the consequence of adherence of parasitized RBC to endothelium of cerebral capillary vessels, and (2) malaria occurring in pregnant women, due to mass sequestration of infected erythrocytes in placenta.
Among the five species currently recognized to infect humans, Plasmodium falciparum is the most deadly (the others being P. vivax, P. malariae, P. ovale, and P. knowlesi). Among 2.37 billion people are exposed across 87 countries, malaria is the cause of 300–500 million clinical cases per year [2], but even though falciparum malaria is rife in nearly all tropical and subtropical areas of the World, Africa is currently the worst affected, accumulating (WHO 2004 statistics) 70% of total malaria clinical episodes which are the cause of 7% of the total number of deaths, 20% of direct child mortality (one child dies of malaria every 30s) [3], 10% of indirect child mortality [4], and a severe prognosis for mothers and children when infection occurs during the first pregnancy [5, 6]. Of these fatal outcomes, malaria is the cause of many sequelae in the surviving patients, mainly in young children [5], and disability in adults during infectious episodes. The malaria burden, expressed as DALYs (disability-adjusted life year), was ranked fourth out of all diseases for low-income countries in 2004, behind lower respiratory infections, diarrheal diseases, and AIDS [7]. Other heavy consequences of malaria include anemia that can increase child mortality and can markedly reduce power productivity in adults and a deficit in intellectual developments, which has consequences on the future evolution of development and productivity in concerned areas [8]. In lower income countries, malaria can represent 1–3% of Gross Domestic Product (GDP) per capita. As a result, malaria is a poverty impeding factor in developing countries that are affected by this disease [8]. To face such a challenge, the international financing was increased. The Global Fund to Fight AIDS, Tuberculosis and Malaria (GFTAM) and other supports committed US$ 991.5 million for malaria in 2007. However, among the 136 countries supported by this strategy since 2002, 16 of them, representing 710 million people living in stable transmission conditions, received an estimated combined annual support lower than US$ 0.5 per capita [9], which is not enough to fight efficiently malaria.
Vaccination, which would theoretically represent the best way to settle the problem of malaria, remains very far despite the large amount of money devoted to this research area in the last 40 years and the numbers of papers published, even if protection against malaria appears possible, based on the sum of observations collected. International organizations such as Programs for Appropriate Technologies in Health (PATH), the Malaria Vaccine Initiative (MVI), the European Malaria Vaccine Initiative (EMVI), and United States Aid (USAID) are involved in feasibility studies concerning all aspects of malaria vaccination; however, currently there are acute debates concerning acquisition of antimalarial immunity and optimization and design of a malaria vaccine [10].
An efficient fight against malaria mainly includes the control of the mosquito vector by indoor residual spraying, protection against bites by insecticide treated nets, chemoprophylaxis and chemotherapy of stated infection which gave promising results, mainly in low transmission rate areas, but failed to eradicate malaria in regions of intense transmission, even if progress was noticeable in several countries [5, 11–13]. However, the cost of these interventions is very high, representing up to 30% of domestic expenses in low-income countries and must be supported in a large part by international interventions (Multilateral Initiative on Malaria, Roll Back Malaria Project, GFTAM, Medicines for Malaria Venture), which have played an important role in the progress observed so far. Resistance, which spread both in Anopheles and Plasmodium, disproved the efficacy of the global fight program because low cost insecticides and drugs, the most interesting for concerned populations, are less and less efficient. For some insecticides as DDT, economical interests of concurrent products and environmental problems are adding to mosquito resistance problems. As a consequence, eradication programs were abandoned in the 1970s. With the extension of the resistance to chloroquine and some other low cost antimalarials, a resurgence of malaria occurred in many parts of the world, sometimes with devastating local epidemic episodes such as in Sri Lanka and Madagascar. High priority research includes development of new drugs for the treatment of malaria, the development of safe molecules for mass drug administration in chemoprevention, the development of new insecticides for indoor residual spraying and long-lasting insecticide-treated bed nets, the development of vaccines with high transmission-blocking potential, improved access to treatment for poor populations, and cost efficiency of drug combinations [14].
Malaria chemotherapy has been based for a long time on a reduced number of drugs (mainly quinine, chloroquine, antimetabolites, and primaquine). The occurrence of resistance of P. falciparum to several of these antimalarials provoked the search for new active molecules. Various strategies were adopted to find new antimalarials that resulted in the industrial development of mefloquine, halofantrine, artemisinin derivatives, and several antibiotics. The spread of resistance to CQ and sulphadoxine-pyrimethamine and the decrease in susceptibility of the parasite to some of the new drugs increased interest in a new idea concerning the future of malaria chemotherapy, one interesting suggestion being the use of some already known drugs in Artemisinin Combination Therapy (ACT) to delay the occurrence of resistance.
Deciphering the P. falciparum genome gave access to thousands of potential new targets, as 60% of genes identified in the parasite have no equivalent orthologues in any other genome sequenced. But the validation of new targets is a long way away since it requires complex genetic reverse manipulations which can only be performed on a large scale in a few laboratories worldwide. After the validation of a target, the design of specific inhibitors is another time and money consuming step, since it requires a very fine knowledge of the target structure to design specific inhibitors. Moreover, adequate and practical methods to test a large numbers of molecules remain limited [15].
Out of the strategies targeting specific parasite probes, there are many methods used to search for new active molecules, including ethnopharmacology, medicinal chemistry, combinatorial chemistry and chemical libraries screening by high throughput screening, and drug design. These approaches have been the source of numerous potential antimalarials, but few molecules have been successful enough to enter in the queue for clinical trials.
1.2 The Digestive Vacuole of Parasite and Hemoglobin Digestion
When invading the RBC, Plasmodium has to solve multiple problems to successfully achieve its intraerythrocytic cycle: (1) keeping communication and exchanging tools with the extracellular compartment (necessary nutrients not being provided by its host cell); (2) to get the necessary space within the RBC to welcome it, siphoning off from the amino acids necessary for its growth; (3) to detoxify heme liberated during hemoglobin (Hb) digestion by forming hemozoin (also called malarial pigment); (4) to reorganize the surface of the RBC in order to escape to the host immune defenses (Fig.1).

Schematic representation of a malaria parasite
Digestion of Hb and heme detoxication in hemozoin appears to be very critical steps in the intraerythrocytic cycle. By providing necessary amino acids to the parasite [16], the digestion of Hb is crucial for equilibrium of osmotic pressure in the parasitized RBC in order to prevent a premature lysis before the completion of the cell cycle [17]. Such processes also provide space for the parasite to increase in size. If about 70–80% of the host cell Hb is digested by the parasite [18], only about 16% of produced aminoacids are used for protein synthesis [16, 19].
Hb digestion requires specialized structures which are organized by the parasite as soon as it enters the ring stage, but are modulated in importance throughout the cell cycle. Several mechanisms have been proposed for the capture of Hb by the parasite [20–24].
Recently, a primary process, called “big gulp” [21], occurring at the ring stage, has been proposed at the origin of the parasite digestive vacuole. The “big gulp” is different from classical macropinocytosis or phagocytosis, which is actin-dependent, as it is not affected by treatment by Cytochalasin D. Analysis of the evolution of “big gulp” structure suggests that it is at the origin of the food vacuole (containing hemozoin). Indeed, the “big gulp” goes before the food vacuole during the cell cycle, and the two structures cannot be observed simultaneously in the same parasite.
Cytostomal structures can be observed in the parasite shortly after invasion of the host cell [20–24]. The cytostome is an invagination of both parasite and parasitophorous membranes [23]. Currently, there is an active discussion about the role of actin in the formation of this structure [21, 23]. This is due to recent evidence of rab5, a marker associated with endosome formation and transport in eukaryotes [21]. Cytostomes are formed throughout the erythrocytic cell cycle and often many are found in one parasite. Subsequent individualization of small vesicules containing Hb was observed by numerous authors [20–22, 24]. These vesicules appear at the ring stage [20] but their importance in Hb uptake becomes more important at the trophozoïte stage [21]. They are transported to the digestive vacuole and finally fuse with it. This process is actin and rab5-dependent. Digestion occurs in these structures because hemozoin can appear in them even if their fusion with the digestive vacuole is inhibited, indicating that they can acquire the necessary enzymes and factors to degrade Hb and to biocrystallize hematin [21, 24].
A cytostomal tube can be produced by the parasite, starting from a cytostome by elongation of small vesicles described above. Its formation requires actin intervention [21, 23]. Cytostomal tubes were thought to have an important role in Hb uptake by the parasite. Individual digestive vesicles were proposed to either bud off from the terminal portion of the tube or by the individualization of the different portions of the tube itself. Finally, the pigment vesicles were proposed to fuse rapidly to produce a “residual vacuole,” where malarial pigment was supposed to be sequestrated [24]. Recent observations suggest a direct fuse of cytostomal tube with the food vacuole resulting from the evolution of the “big gulp” and not by individualization of vesicles [23], but these results are controversial [21]. Cytostomal tubes appear about 30h after erythrocyte invasion and their global volume seems to be small, limiting their role in Hb uptake by the parasite.
Phagotrophy is a fourth means by which the parasite captures Hb. Many phagosomes appear at the trophozoïte and schizont stages. Unlike typical phagosome found in other eukaryotes, their formation is not actin-dependent and very similar in mechanism to the “big gulp” [21].
The four means of uptake occurred at different times during the erythrocytic cycle. Big gulp might take part in more than 90% of hemoglobin uptake at the ring stage, while small vesicules deriving from cystostomes and phagotrophy might have the prominent role in uptake during the trophozoïte and schizont stages [21]. Doubts remain about this hypothesis and concern the role of cytostomal tubes in uptake which might be more important than proposed [23].
Digestion of Hb engulfed in endocytic structures needs particular conditions which are of importance concerning the action of many antimalarials. Four aspartic proteases (falcipains), three cysteic proteases (plasmepsins), a metalloprotease (falcilysin), a dipeptidylpeptidase I, and probably aminopeptidases are involved in the digestion of Hb [25]. Hb is primarily attacked by plasmepsins and falcipains and cleaved into smaller fragments. These fragments are hydrolyzed by falcilysin in small peptides, and finally aminoacids were liberated by dipeptidylpeptidase and aminopeptidase. Inhibitors of the falcipain and plasmepsins impair or kill the parasite in vitro and in vivo, but their specificity is not strict. Knockouts experiments on several of these enzymes, individually or in combination, demonstrated that in normal culture conditions P. falciparum is able to acquire the important parts of the aminoacids necessary from its environmental medium (isoleucine, which is absent in Hb is entirely taken up from parasitized RBC outside compartment) as a redundant way with Hb degradation. Such experiments also showed that some proteolytic enzymes have an overlapping role in Hb digestion and, for these reasons, plasmepsins do not represent valuable drug targets [26]. It must be noted that if Hb degradation is strictly necessary for parasite survival [26], it is not restricted to the use of produced amino acids for nutrition, but also to make room for itself [16, 19] and to maintain the osmotic pressure required to avoid lysis of the infected RBC [16, 17]. All enzymes involved in Hb digestion are more active at a pH less than 5. Consequently, the DV is an acidic compartment in the parasite and represents a primary interest in the mechanism of action of many schizontocidal antimalarials.
During hemoglobin degradation, free heme, ferriprotoporphyrin-IX or Fe(II)PPIX (Fig.2) is released in the digestive vacuole. The toxicity of heme to the parasite has been demonstrated [27–30]; it is supposed to cause the disruption of metabolic functions by means of peroxidation of membranes and inhibitions of enzymes via the generation of oxidative free radicals [31].

(a) Chemical and (b) molecular structure of heme
Detoxification of the heme is achieved by hemozoin formation via a biomineralization process [32] and not via a polymerization process as previously envisaged. First, the iron II in Fe(II)PPIX is oxidized to iron III in Fe(III)PPIX or hematin (Fig.3)

Oxidation of heme in hematin
These molecules of Fe(III)PPIX are then linked into dimers through reciprocal iron-carboxylate bonds to one of the propionic side chains of each porphyrin, and these dimers form chains linked by hydrogen bonds (Fig.4, [33, 34]).

Chemical structures of hemozoin
A crystal is formed and its characteristics have been recently defined and discussed concerning the mechanism of action of antimalarials interacting with hemozoin formation [35, 36]. With the mechanism of biomineralization, the low soluble hemozoin is thus removed from the biological environment of the parasite.
However, if some doubts are remaining on how β-hematin is formed in the malaria parasite, recent results have shed light on the mechanisms involved. Among the three proposed hypotheses to explain catalysis or nucleation of the hematin biocrystallization, the role of histidin rich protein 2 (HRP2) [37] is now called into question based on: (1) the localization of the protein and the capability of HRP2 knockout parasites to form pigment [38] and (2) the fact that organisms lacking HRP2 are still able to produce a pigment similar to hemozoin [39]. There is now clear evidence that the formation of hemozoin occurs in a hydrophobic environment within or at the surface of lipids in Plasmodium and in other organisms producing hemozoin [40–42]. Experimental works have confirmed this hypothesis [43–47], and that the activity of some antimalarials is in clear relation with an interaction with parasite lipids [48]. The intervention of specific enzymes in biocrystallization of hemozoin has been largely discussed since the observation of heme polymerase activity in parasite extracts [49, 50]. The difficulty of purifying a protein showing such activity, the demonstration that biocrystallization could occur in absence of protein [51], and the discovery of the role of lipids have diverted interest for this hypothesis. Recently, the gene of a heme detoxification protein (HDP) was characterized. Both the recombinant protein and the authentic protein purified by immunoprecipitation were shown to interact with heme with high affinity (more than four times that of HRP2) and to catalyze very efficiently the biocrystallization in β-hematin. The catalytic activity of the protein is optimal at pH 5.2 or lower. After its synthesis, the protein seems to be exported into the infected RBC cytoplasm and is internalized again at the same time as Hb by the parasite. Knockout experiments showed that in P. falciparum, the hdp locus could not be mutated and authors concluded from the results obtained that the gene was certainly critical for parasite survival [52]. This protein appears well conserved within the Plasmodium genus [53]. The fraction of total hemozoin related to HDP activity is still under discussion.
1.3 Drug Resistance
Resistance to CQ was first reported in Columbia and Thailand in the early 1960s and has now spread worldwide. It was only in 1987 that a capital observation concerning CQ resistance was done. The Krogstad’s group [54] showed that: (1) CQ-resistant parasites accumulated significantly less CQ than sensitive one; (2) this decrease in accumulation was due to a more rapid release of CQ in resistant parasites (40–50 times); (3) this accelerated efflux was slowered or inhibited by calcium channel blockers such as verapamil (Fig.5), dilthiazem, or TMB-8 and by vinblastin and daunomycin whereas a calcium ionophore such as A23187 was not active; (4) none of these drugs affected the CQ accumulation or efflux in sensitive parasites; (5) reversion phenomenon was dependent of the acid pH of the digestive vacuole. Authors pointed out the similarity between their observations and the reversion of resistance in multidrug-resistant cancer cells which is affected in the same manner as verapamil.

Chemical structure of verapamil
In fact, at this time, two main research methods were developed independently to search the origin of CQ resistance in P. falciparum.
The route of multidrug resistance led to the discovery and characterization of pfmdr1 gene and then to other transporters [55]. If pfmdr1 was proposed to be at origin of CQ resistance, first by gene amplification [56, 57] then by polymorphim of the gene [58], field observations did not confirm this hypothesis. Nevertheless, it was shown that pfmdr1 amplification and polymorphism were involved, possibly in association with other genetic factors [59], in the modulation of susceptibility to several antimalarials (mefloquine, halofantrine, quinine, and artemisinin) in fields studies, and in transfected parasites [60–64]. Recent studies using transfection of Xenopus leavis oocyte provided direct evidence of the transport of antimalarials by pgh1, the pfmdr1 gene product [65], conducting to a new interpretation relation between pgh1 and CQ transport [66].
The other route resulted from a genetic cross between CQ-susceptible and a CQ-resistant clones of P. falciparum and of the genetic analysis of progeny [67]. Results showed that: (1) resistance was linked to accelerated efflux of CQ outside the parasite and susceptible to verapamil; (2) resistance level was independent from phenotype and copy number of pfmdr1; (3) the locus for resistance was situated on chromosome 7 and not on chromosome 5 where pfmdr1 was found. Starting from these results, Wellems et al. completely dissected the 400kb area suspected to contain chr 7 and finally selected a 47.5kb area containing 13 genes, which were analyzed. The first gene (cg2) postulated to be at the origin of CQ resistance [68] was not confirmed in many epidemiological studies and by transfection experiments [69]. The discovery of a 14th gene (pfcrt) coding for a protein localized in the digestive vacuole membrane and carrying specific mutations in CQ-resistant parasites provided one key capital of CQ resistance [70]. The role of PfCRT protein mutations in CQ resistance was rapidly confirmed on field isolates [71, 72].
Mutations of PfCRT have been described in all CQ-resistant P. falciparum isolates. All CQ-resistant isolates share a common mutation at position K76T, but this mutation is never present in the gene of resistant parasites alone, in which at least 19 other points of mutations have been found [73]. Two major haplotypes were found, and several studies showed that the level of CQ resistance was associated with haplotypes [73, 74].
The mechanism by which PfCRT confers resistance to chloroquine is still under discussion [75]. The first aspect concerns the nature of the protein: whether PfCRT, postulated to possess ten transmembrane helices, is considered as a member of the drug-metabolite transporter family of proteins [55, 76–78]. Some authors discuss about the channel nature of the protein [79, 80]. It has been suggested that the change of the charged lysine to uncharged threonine affects the electrostatic interaction with the diprotonated CQ [81]. In the mutated forms, the absence of electrostatic interactions allows the drug to cross the channel (Fig.6). This results in the efflux of the drug out of the DV [81]. Note here that the reduced accumulation can be partially reversed by verapamil, a lipophilic compound (Fig.5).

Schematic representation native and mutant (K76T) PfCRT. Postulated mechanism of the efflux of charged CQ reversed by verapamil
Other experiments have shown that the mutation K76T is not the only factor to modulate the action of PfCRT. The Cooper group [82] demonstrated that mutations in transmembrane domains 1, 4, and 9 altered susceptibility to CQ, quinine, and quinidine. Photoaffinity labeling of PfCRT with a novel perfluorophenylazido CQ [83] provided new information concerning interaction of the drug with the protein. Mutations conferring CQ resistance increased the CQ-associated H+ leak in the parasite digestive vacuole [84]. Recent observations have shown that PfCRT may exert its protective role even in erythrocytic stages when hemoglobin digestion and production of pigment is not very active, as in rings and late schizonts [85]. On the other hand, we should keep in mind that CQ resistance has a biological cost for the parasite. In some geographical areas, the abandonment of CQ use results in resurgence of susceptible phenotypes [86, 87]. As PfCRT is considered by some authors as a metabolite/drug transporter, it can be expected that mutations in such a protein can modify the physiology of parasite. Comparison of transcriptomes of clones carrying wild type PfCRT, with clones carrying a single mutation in the gene showed that the mRNA level from 45 genes was modified compared with the wild type parent. Most of the up-regulated genes are involved in invasion, cell growth and development, signal transduction, and transport activities. Some genes encode proteins involved in transport and/or regulation of cytoplasmic or compartmental pH [88].
Current knowledge is in favor of intervention of more than one gene in modulation of CQ resistance, even if PfCRT appears to play the main role. It seems to be difficult to simultaneously target several genes to overcome CQ resistance, especially when some of these factors remain to be precisely identified. However, the reversal of Q resistance, by verapamil and related drugs, is a common characteristic of all CQ-resistant strains of P. falciparum and open ways for future therapeutics [89, 90].
2 Metal Complexes as Antimalarials: An Overview
Due to the success of cis-platin in anticancer therapy, medicinal chemists have rapidly begun to evaluate the utility of metal complexes as antimalarials [91–94]. We will not report all of the bioinorganic compounds that have been synthesized and tested. In the following, we will just focus on the most representative agents. For example, we will not review the metal chelators.
In order to exploit the antimalarial activity of CQ, coordination of CQ by different metals has been explored. The triphenylphosphinegold(I)–CQ complex (Fig.7) was found to have a ninefold higher activity in vitro than CQ against FcB1 and FcB2 Colombian CQ-resistant strains of P. falciparum. The complexation of Au to CQ also increased the in vivo susceptibility of P. berghei to CQ [95].

Chemical structure of the gold CQ complex
To the contrary, coordination of CQ by the organometallic moiety Rh(COD)Cl (Fig.8) did not significantly alter the in vitro antimalarial activity of CQ. In in vivo tests, equivalent concentrations of free CQ reduced the parasitemia by 55%, while for the complex CQRh(COD)Cl the reduction reached 73% without any sign of acute toxicity being observed up to 30 days after treatment [96].

Chemical structure of the rhodium CQ complex
Schiff-base phenolate metal complexes (Fig.9) have been shown to inhibit both HB3 CQ-susceptible and FCR3 CQ-resistant parasites and correlate in a free metal-independent manner with their ability to inhibit hemozoin formation in vitro [97].

Chemical structure of the schiff-base phenolate iron complex
More recently, the synthesis of an amine–phenol complex of gallium(III) was described (Fig.10). The antimalarial activities were in the micromolar range against the HB3 CQ-susceptible and Dd2 CQ-resistant strains. The authors proposed that the cytotoxic targeting properties of these kinds of metal complexes lie in the spatial orientation of substituents on the peripheral part of the molecule [98].

Chemical structure of the amine–phenol gallium complex
2.1 Ferrocenic Molecules with Antimalarial Properties
The synthesis of ferrocenyl sugars as exemplified by Fig.11 and their antimalarial activities have been reported. Whereas the organic parent compound, an ellagitannin derivative, trideca-O-methyl-α-pedunculagin, displayed no antimalarial activity, the diferrocenyl glucoside showed inhibitory activity similar to that of quinine against the FCR3 CQ-susceptible strain [99].

Chemical structure of the ferrocenyl sugar
A series of ferrocenyl chalcones as exemplified by the two compounds in Fig.12 were evaluated for in vitro antimalarial activity against the K1 CQ-resistant strain. Disappointing low activities in the micromolar range were reported [100].

Chemical structures of selected ferrocenyl chalcones
The results showed that the location of ferrocene influenced the ease of oxidation of iron II and the polarity of the carbonyl linkage. These parameters were found to influence antiplasmodial activity. Better antimalarial activities were obtained when the ferrocene moiety is adjacent to the carbonyl group [101].
In order to exploit the cationic redox behavior of methylene blue as antimalarial agent, new ferrocene benzimidazolium conjugates (Fig.13) have been designed, synthesized, and tested against the P. falciparum malarial parasite strain NF54. Compound with a thiophene moiety as R group showed IC50=40nM similar to that of CQ (IC50=20nM) and artemeter (IC50=25nM). Unfortunately, no data about their in vivo activity is available [102].

Chemical structures of methylene blue and ferrocenic benzimidazolium salts. R is an (hetero)aromatic ring
2.2 Ferrocene Conjugates with Antimalarial Drugs Other Than Chloroquine
2.2.1 Artemisinin
Introduction of a ferrocene core in the chemical structure of artemesinin (Fig.14) did not enhance antimalarial activity compared with artemesinin (or dihydroartemisinin) alone.

Chemical structures of artemisinin and the ferrocenyl conjugates
Whatever the chosen linker (ether, ester, amine, …) between both fragments, no improvement of the antimalarial activity was noted. It was therefore concluded that incorporation of the ferrocene moiety into an artemesinin skeleton did not improve its activity [103, 104].
2.2.2 Atovaquone
A similar strategy based on the coupling of active fragments led to the design of two new series of atovaquone conjugates (Fig.15). The compounds were tested on both Toxoplasma gondii and P. falciparum [105]. The very lipophilic derivatives with an aliphatic chain of 6–8 carbon atoms were active on atovaquone-resistant ATO T. gondii clone. Only antimalarial activities in the micromolar range were reported against the 3D7 CQ-susceptible strain and Dd2 CQ-resistant strain. Whereas 1,4-naphthoquinone derivatives, such as atovaquone, were reported to interfere with the oxidation of dihydroorotate, no data for these derivatives is available.

Chemical structures of atovaquone and the ferrocenyl conjugates
2.2.3 Mefloquine and Quinine
Exploiting the structure-based strategy, the quinuclidinyl and the piperidinyl side chains of quinine (QN) and mefloquine (MF) were, respectively, substituted with a ferrocene moiety while maintaining a basic amino group (Fig.16). In vitro, lower activities than the parent compounds were reported [106]. In acidic aqueous solution, these ferrocenyl analogs seemed to be unstable, leading to the formation of the presumably inactive carbeniums.

Chemical structures of quinine and mefloquine, and their ferrocenyl conjugates. The hypothetical carbenium is presented on the right
2.2.4 Ferrocenyl Pyrrolo[1,2-a]quinoxaline Derivatives
Inspired by the bioorganometallic approach, two series of ferrocenyl pyrrolo[1,2-a]quinoxalines were designed and prepared (Fig.17). The derivatives were tested in vitro against three different strains of P. falciparum: F32, FcB1, and PFB [107]. The best results (IC50 between 30 and 70nM in comparison to CQ IC50=225nM) were observed in the first series with a bis(3-aminopropyl)piperazine as a linker. These compounds were tested for their ability to inhibit β-hematin formation. For all but one case, the derivatives did not interfere with hemozoin formation suggesting a different mechanism of action compared with the classical 4-aminoquinoline family. Their toxicity being too high, the development was stopped.

Chemical structures of the ferrocenyl pyrrolo[1,2-a]quinoxaline derivatives
2.2.5 Ciprofloxacin
Quinolones and fluoroquinolones are widely used against bacteria and mycobacteria [108]. Recently, it has been shown that, similarly to T. gondii, at pharmalogical concentrations, ciprofloxacin (Fig.18) acts on P. falciparum [109]. Among all the (fluoro)quinolones active on bacteria and tested against P. falciparum strains (3D7 or NF54-R), none of them showed an activity better than 1µg/mL (the most active proved to be grepafloxacin, gatifloxacin, and moxifloxacin) [110]. Moreover, several quinolones proved to be active in vitro against CQ-susceptible (D6) and CQ-resistant (Dd2) P. falciparum strains [111, 112].

Chemical structures of ciprofloxacin and its ferrocenyl conjugates. Doxycycline is the reference compound
Curiously, although hundreds of analogs of ciprofloxacin have been synthesized and structure–activity relationship has been analyzed, the bioorganometallic strategy has not been previously envisaged. Therefore, new ciprofloxacin derivatives bearing a ferrocenyl substituent at position N(1) or at C(7) of the quinolone ring were designed (Fig.18).
A lead compound bearing a ferrocenyl moiety at position N(1) was identified. This derivative is more active than Ciprofloxacin and Doxycycline. The activity is remarkably constant regardless of the level of resistance to CQ of the strains. Contrary to other antibiotics, no “delayed-death” effect was noted. Isobologram analysis showed that this compound exerts an antagonist effect with the main quinoline-containing antimalarials. In vitro results have to be confirmed in vivo to check the bioavailability of the molecule and its potential interest as a new antimalarial [113].
2.3 Ferrocene Conjugates with Chloroquine
First, CQ was associated with the ferrocenecarboxylic acid by a weak salt bridge interaction (Fig.19). Indeed, the ferrocene moiety may independently potentiate the activity of CQ by enhancing oxidative stress. Regrettably, this hypothesis was proven wrong. In vitro against the SGE2 CQ-susceptible and the FCM17 CQ-resistant strains, this salt was even less active than CQ diphosphate alone, suggesting an antagonist effect between both parts [114].

The template, chloroquine 1, and the ferrocene conjugates 2–12. R is an alkyl or a ferrocenylmethyl group, n varying from 2 to 6
2.3.1 Quinoline Ring Substitutions
With the same idea in mind, i.e., limiting the number of reaction steps to reduce the costs of production, direct condensation of the ferrocenylmethyl (Fem) moiety on the endocyclic nitrogen of the CQ afforded the quaternary ammonium salt (Fig.19). Nevertheless, such modification of CQ abolished the activity of the parent molecule on both Dd2 CQ-resistant and HB3 susceptible P. falciparum strains [115]. We can hypothesize that the charged species should not be able: (1) to cross the membrane; (2) to stack over the ferriprotoporphyrin ring due to unfavorable electrostatic interaction and/or steric hindrance.
Against the same laboratory strains, a low antimalarial activity was also observed with compound where the quinoline cycle is substituted at the C-3 position by Fem (Fig.19). Here again, the bulky ferrocenyl group should sterically hinder the stacking interaction between the quinoline ring and heme and/or Fem should modify the charge density distribution on the quinoline cycle [115].
2.3.2 Lateral Side Chain Modifications
Particular attention was devoted to studying the impact of the introduction of the ferrocenyl moiety into the lateral side chain of CQ, implicated in DV targeting. Moreover, the length of the side chain and the distance between the two exocyclic nitrogen atoms may both affect resistance against 4-aminoquinolines by P. falciparum [116, 117]. 4-aminoquinolines with shorter (two or three carbon atoms) or longer side chains (10 or 12 carbon atoms) than CQ are more active against CQ-resistant P. falciparum. It has been suggested that these molecules had an N···N spacing which is less suited for binding with the putative CQ transporter and are therefore less efficiently extruded from the food vacuole.
A series of CQ analogs (Fig.19) characterized by the presence of the ferrocenyl group attached to the terminal basic nitrogen atom of the CQ lateral chain were synthesized and tested [118–120]. Whereas most analogs were found to be more active than CQ, they did not present an optimal activity compared with others. For compounds with a benzylcarbamoyl substituent (R), the chain length (n) made no significant difference to the efficacy in D10 CQ-susceptible strain, but a decrease in efficacy with an increase in the length of the methylene spacer was noted in the K1 CQ-resistant strain [120]. Compounds with the Fem group at the end of the side chain showed an activity which decreased rapidly with the level of CQ-resistance among the P. falciparum clones tested. Clearly, a cross resistance could be postulated to emerge very quickly. There was no significant correlation between either the liphophilic character or the in vitro inhibition of hemozoin formation and IC50 values among these CQ analogs [118, 119]. Bis-ferrocenyl conjugates led to erratic activities and impossibility to measure precise IC50 values, mainly due both to stability and solubility problems in culture medium [118, 119]. A decrease of the efficacy was observed between the linear and branched propylamino chain derivatives (Fig.16). Introduction of methyl groups in the side chain was not favorable to the antimalarial activity [118, 119].
2.3.3 N-N Spacer Modifications
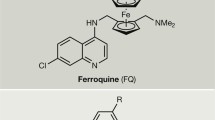
Incorporation of the ferrocene core in the lateral side chain of CQ between the two amine atoms has led to the design of Ferroquine (FQ, SR97193). This compound was shown to be extremely active against both CQ-sensitive and CQ-resistant P. falciparum. FQ was also the most active derivative in vivo and was considered as a lead compound early on (More details in Sect.3) [121]. As 1,2-unsymmetrically substituted ferrocenes are chiral molecules, an effort was made to design an achiral version of these derivatives. The easiest solution was to move the second substituent to the other cyclopentadienyl cycle. These achiral 1,1′-substituted ferrocene analogs (Fig.19) were much more active than CQ but exhibited four times lower activity against the K1 CQ-resistant strain than against the D10 CQ sensitive strain [122]. Unfortunately, no in vivo data were available for the comparison of the substitution patterns: 1,2 vs. 1,1′.
2.3.4 Bisquinolines
In the search for quinoline compounds that evade the resistance problem, bisquinolines were found very promising being active against CQ-resistant strains [123]. The ferrocenyl bisquinoline (Fig.19) remained more efficient on the Dd2 CQ-resistant strain than CQ although this compound was less active on the HB3 CQ-sensitive strain, than FQ [124].
3 Ferroquine: A New Candidate Antimalarial Drug
The presence of the ferrocene moiety within the lateral chain of CQ is the main condition required to retain a strong antimalarial activity on CQ-resistant P. falciparum. FQ was rapidly identified as a lead compound with sufficiently potent in vitro and in vivo activities to meet candidate nomination requirements.
3.1 The Chemistry of Ferroquine
The synthesis of FQ (Fig.20) is simple and quite economical, which renders FQ attractive for the development of an antimalarial drug intended for use in areas, concerned by malaria, that are mostly overlaying with low-income countries. FQ was obtained starting from the commercially available N,N-dimethyl-1-ferrocenylmethanamine. The ferrocenic aldehyde results from a C–C bond formation, a two-step sequence involving metallation with tertio-butyllithium and a reaction with DMF. This step has been previously studied and the 1,2 orientation of the two substituents of the cyclopentadienyl has been unambiguously established [125]. The aldehyde is converted to the corresponding oxime, which is then reduced to the primary amine. The SNAr reaction between the amine and 4,7-dichloroquinoline leads to the desired FQ [121].

Chemical synthesis of ferroquine
FQ possesses planar chirality due to its 1,2-unsymmetrically substituted ferrocene moiety (Fig.21). In the laboratory, a synthetic method was developed to provide separate pure enantiomers of FQ [126]. The approach included a biocatalytic kinetic resolution using Candida rugosa lipase (C.r.l.). The key step (Fig.22) is enantioselective acetylation of primary hydroxy group leading to the formation of enantiomerically enriched acetate. The remaining untransformed alcohol was isolated in an extremely high enantiomeric excess (ee>98%). The deacylation of the enriched acetate was undertaken by means of a transesterification process catalyzed also by C.r.l. This reaction provides the other alcohol with high enantiomeric purity (ee>98%). Finally, both amino alcohols were oxidized to the corresponding carboxaldehydes and then engaged in the total synthesis of (1′R)-FQ and (1′S)-FQ.

Ferroquine enantiomers

Key steps of the synthesis of FQ enantiomers by enzymatic resolution
Note here that for (semi)industrial scale production, both enantiomers were obtained starting from the racemic mixture by preparative chiral chromatography.
Activity of pure enantiomers was compared with the racemate in vitro and in vivo. In vitro, the FQ enantiomers and the racemate were found to be equally active against the CQ-susceptible and CQ-resistant P. falciparum strains HB3 and Dd2. In vivo, both enantiomers were slightly less active than the racemic mixture against CQ sensitive and CQ resistant P. vinckei vinckei, suggesting an additive or a synergetic effect between both enantiomers. Moreover, (1′R)-FQ displayed a slightly improved curative effect than (1′S)-FQ suggesting minor differences in pharmokinetics properties. Actually, the in vitro cytotoxicities of (1′R)-FQ and (1′S)-FQ or the racemate appeared similar in the L5178Y cell proliferative assay. As no critical adverse affect was observed during Phase I and IIa of clinical trials with racemate, the enantiomeric mixture was adopted in further Phase IIb trials in association with artesunate [127].
The apparent partition coefficients (log D) of CQ and FQ were measured at vacuolar (5.2) and cytosolic (7.4) pHs (Table1, [128]). At cytosolic pH, FQ was over 100-fold more lipophilic than CQ, whereas the difference in lipophilicity is only slight at acidic DV pH (about threefold).
In terms of basicity, pK as of the two drugs indicate that FQ is less basic than CQ (Table1, [129]). Crystal structure of FQ (Fig.23) shows the presence of a strong internal hydrogen bond between the anilino (N11) group and the tertiary amino N(24) [128]. This, together with the electron donating properties of the ferrocene moiety, should explain the decreased pK a values.

Crystal structure of neutral ferroquine (CCDC 262108). The dashed line indicates the intramolecular hydrogen bond
In the absence of special transport mechanisms (which is not proven, see further in the text), the vacuolar accumulation ratios (VAR) could be predicted from a derivation of the Hendelson–Hasselbach equation using the calculated log D values. Indeed, for a diacidic base, the log D is a function of the acidity constants (pK a) and the partition coefficient (log P):
So, when using this formula, FQ (VAR=6,402) would not be expected to accumulate in the DV as much as CQ (VAR=19,521). Nevertheless, in view of the high degree of variability of the methods (the pK a were determined in a dioxane–water mixture and the log P in octanol–water mixture), some errors could be included in the results.
Instead of the calculated log D values, the experimental log D values can also be exploited, and the VAR were calculated as follows:
Using this formula, FQ is expected to accumulate 50-times more than CQ in the parasitic DV. However, both mathematical models could be subject to suspicion of bias as special transport mechanism(s) could not be excluded and are even strongly suspected. Indeed, the binding to free heme should contribute to uptake [130, 131].
The debate of the “real” concentration of FQ in the DV compared with CQ is still open. Experimental data will be urgently needed to validate the correct mathematical models.
3.2 Ferroquine Derivatives
In order to study if structural modifications of FQ might improve its antimalarial activity, several derivatives were prepared and tested.
Tertiary amines (Fig.24, R1=R2=C2H5 or R1=C2H5 and R2=CH3) showed strong antimalarial activity, especially against the CQ-resistant Dd2 and W2 strains [118]. These compounds were two- to tenfold more active than CQ and as active as FQ. Secondary amines (R1=H and R2=CH3 or C2H5 or C(CH3)3) also possess antimalarial activity comparable to that of FQ. All these compounds exhibited better inhibitory activity against the Dd2 strain than CQ itself [132].

Chemical structures of the analogs of ferroquine modified on the basic amino group
These structural modifications allow us to conclude that the in vitro antimalarial activity was not disturbed by slight modifications in the lateral basic side chain. To the contrary, when a second ferrocenyl group was introduced on the terminal nitrogen atom, the efficacy of compounds was markedly attenuated [118].
Three FQ derivatives (Fig.25) with R=H, CH3, or C2H5, closely mimicking the antimalarial drug hydroxychloroquine (HCQ), have been prepared [119]. These compounds differed from FQ in their side chains on the basic nitrogen atom N(25). Introduction of a hydroxyl group provides the expected reduction of cytotoxic effects compared with FQ. Moreover, these metallocenic compounds inhibited in vitro growth of P. falciparum far better than CQ. The best results were obtained when R was an ethyl substituent. The high potent antimalarial activity of this compound was confirmed on 25 Cambodian field isolates. This derivative showed almost the same level of activity as that of FQ. As expected, a high correlation (r 2=0.7129) with the IC50 of FQ was noted within the isolates tested. This suggests that the two compounds have a similar mode of action and/or uptake by the parasite.

Chemical structures of hydroxyferroqines. R is a hydrogen or an alkyl group
Oxidative metabolization of tertiary amines like CQ or FQ occurs via the cytochrome P-450 system (see Sect. 3.4 for more details). The main metabolites are generated by side-chain de-alkylation leading first to the monodesalkyl and then to the didesalkyl derivatives. A similar oxidative pathway should mediate the de-alkylation of hydroxyferroquines (R=CH3 or C2H5) as shown for FQ. The loss of the ethanol fragment generates the secondary amine. The other C–N cleavage generates the active mono-N-desmethyl-(dMFQ) and mono-N-desethyl-FQ (dEFQ), respectively. By comparison with HCQ and FQ, it is tempting to suggest that during a clinical use of hydroxyferroquines (R=CH3 or C2H5), formation of these active metabolites may occur and participate in the global activity of the parent products.
Moreover, this new class of bioorganometallic compounds exerts antiviral effects with some selectivity toward SARS-CoV infection. These new drugs may offer an interesting alternative for Asia where SARS originated and malaria has remained endemic.
A chimeric ligand approach (Fig.26) was used to combine the properties of FQ and those of thiosemicarbazones (TSC) [133].

Chimeras of FQ and TSC. The dashed circles indicate the merged groups
Indeed, derivatives of TSC have shown potent antimalarial activities [134, 135]. Due to their intrinsic metal (e.g., iron) chelating properties, the mechanism of action of TSCs is believed to result from the generation of reactive oxygen radicals [136]. To the contrary, the presence of the ferrocene moiety within the lateral chain is the main condition required to retain a strong antimalarial activity on CQ-resistant P. falciparum.
As can be seen in Fig.27, a covalent binding between both active fragments was envisaged by merging the amino groups. In order to compare the contribution of each fragment, analogs without the ferrocenic moiety and analogs without the 4-aminoquinoline moiety were also synthesized. Chimeras of TSC and FQ were the most active derivatives against four different strains of P. falciparum. Nevertheless, the corresponding purely organic derivatives showed comparable potency. Contrary to previous results, introduction of the ferrocene moiety did not increase antimalarial activity. Here again, we noticed no significant difference in the activity of ferrocenyl compounds between CQ-susceptible and CQ-resistant parasites.

Proposed metabolic pathway of ferroquine in human hepatic models. Main metabolites are in the dashed line box
3.3 Specific Pharmacology
The first tests performed in vitro on P. falciparum clones or acclimated isolates [114, 121, 129] showed that the molecule had an activity equivalent to that of CQ on CQ susceptible parasites and was as efficient on CQ resistant parasites with similar IC50 for both parasites phenotypes. These results were confirmed on other P. falciparum clones which were tested in other laboratories [122, 137].
This preliminary data incited us to explore in vivo activity of FQ on murine malaria models. Subcutaneous administration of FQ to mice infected by P. berghei N, according to the “four-days blood schizonticidal test” [138], showed that drug IC50 and IC90 values were, respectively, 1.22 and 1.95mg/kg/day as against 1.39 and 2.7mg/kg/day for CQ [129]. More than IC50 tests, curative tests showed the efficacy of CQ, but also the limits of some strains for in vivo tests (unpublished results). We conducted the curative test as the “four-days blood schizonticidal test,” but in place of parasitemia determination at day 4, mice are monitored for survival throughout a period of 2 months after the end of treatment, with regular controls to verify if mortality is related to parasite infection [126, 129]. Experiments showed that the curative dose for CQ on the P. berghei N strain was 50mg/kg/day (to be compared with the IC90 reported above). Under the same conditions a daily dose of 8.39mg/kg/day of FQ cured 14/15 mice. Similar results were obtained with P. yoelii NS strain, with no curative effect of CQ at 60mg/kg/day vs. 14/30 mice cured with 10mg/kg/day of FQ, 3/3 cured at 15mg/kg/day, and 5/5 cured at 20mg/kg/day (unpublished results). In all these experiments, the last surviving parasites were observed in immature RBC (reticulocytes). The affinity of P. berghei and some P. yoelii strains for immature RBC is well known [139]. To get round this problem, we selected a strain of P. vinckei vinckei which did not invade immature red cells [129]. In these conditions, FQ cured mice equally from CQ-susceptible or CQ resistant P. vinckei strains at a dose of 8.4mg/kg/day whilst CQ cured the CQ sensitive line at 31mg/kg/day and was not curative for CQ resistant line at a subtoxic dose of 58.9mg/kg/day. One promising result was that FQ was equally curative given subcutaneously or orally, which was an indication of its good bioavailability [126, 129].
In parallel, extensive studies on P. falciparum field isolates in Gabon [140–142], Senegal [143], Cambodia [118, 119, 144], and the Thailand Burmese border [145] corroborated the efficacy of FQ on the parasite whatever its resistance level to chloroquine or to other commonly used antimalarials: mefloquine, quinine, halofantrine, and artemisinin derivatives [146, 147]. The cross reactivity observed in some studies with CQ was limited and it was demonstrated that it was caused by differences in initial parasitemia among isolates at the start of the assays [141]. Independance of susceptibility of P. falciparum with phenotypic variation of pfcrt gene, responsible for CQ resistance, could be suspected from these results, but this was demonstrated at the molecular level on Cambodia isolates [148] and extended further on other genes currently involved in resistance to aminoquinoline antimalarials [89, 90].
Assays were done to select a Plasmodium clone resistant to FQ. All attempts on P. falciparum failed. Parasites appearing after 35 days pressure were unable to develop, even in absence of the drug in the medium [148]. Experiments were conducted in vivo on a murine strain of P. yoelii NS and resulted in a multiresistant phenotype (CQ, mefloquine and FQ) with a very low pathogenicity. The strain was able to develop only in immature erythrocytes and maximum parasitemia was about 2%. Mice were very often able to cure themselves from parasites, either under pressure of FQ or in absence of drug. Moreover, FQ resistance was not fixed genetically, in opposition with CQ resistance [146]. Just removing drug pressure resulted in a rapid loss of resistance to FQ, which was already observed on an artemisinin-resistant phenotype of the same species [149]. When the strain under FQ pressure was cryopreserved, it had lost its resistance by the time the mouse was inoculated again. In these two conditions the strain remained resistant to CQ. Analysis of pycrt and pymdr1 did not show mutation compared with the wild type line in critical regions known for phenotypic resistance [146]. This result posed questions not only concerning the potential mechanism of resistance to FQ in Plasmodium, but also on the origin of CQ resistance in P. yoelii. Recent results obtained on P. berghei and concerning the translocation of the gene of a multidrug resistance-associated protein (mrp gene) from chromosome 13/14 to chromosome 8 in the chloroquine resistant strain RC might be an interesting lead for comprehension of mechanisms involved in FQ resistance [150].
In conclusion, all specific pharmacology studies showed that FQ was very active on all strains of P. falciparum currently resistant to already used antimalarials. The susceptibility to the drug is not dependant to resistance phenotypes already determined. Experiments to induce resistance in vitro or in vivo show clearly that the fit cost of FQ resistance is extremely high for the parasite and disproves its pathogenicity. Nevertheless, one may keep in mind that resistance or decreased susceptibility occurred for all antimalarials currently used. Such resistance could emerge if FQ were used extensively in the future. One way to decrease or to overcome the risk is to use FQ in an ACT [127], with artesunate, as it is planned during its phase II of clinical assays. Out of all cautions taken to delay the occurrence of resistance, experimental studies remain to be performed to select a clone resistant to FQ. The molecular analysis of such resistance might provide useful tools for monitoring the risk of decreased susceptibility of parasite in area when FQ is used.
3.4 Metabolism, ADME, and Toxicology
In humans, CQ is metabolized in the liver, mostly by oxidation via the cytochrome P-450 enzyme system, leading to N-desethylchloroquine and then didesethylchloroquine [151]. Successive dealkylation of the side chain ultimately produces the 7-chloro-4-aminoquinoline. In comparison to CQ, the metabolism of FQ was examined in different in vitro animal and human hepatic models. FQ is also metabolized via a major dealkylation pathway into the mono-N-desmethyl FQ (dMFQ) and then into di-N,N-desmethyl FQ (dEFQ). Other minor metabolic pathways were also identified (Fig.27). Cytochrome P-450 isoforms 2C9, 2C19, and 3A4 and, possibly in some patients, isoform 2D6 are mainly involved in FQ oxidation [148].
Interestingly, dMFQ remains as active as FQ on the CQ-susceptible strain 3D7 and less active than FQ but much more active than chloroquine on CQ-resistant strain W2. The other metabolites were less effective than FQ (eight- to tenfold according to their IC50 values), and their IC90 (>>100nM) values show that they would not be efficient in parasite elimination [148]. Previously, dMFQ was found as active as FQ on CQ-susceptible strain (HB3) and on a moderate CQ-resistant strain (Dd2). dEFQ was found only twofold less active than ferroquine on Dd2 [132].
As these two metabolites are present in significant concentration in blood after administration of FQ, they should be involved in the global antimalarial activity of FQ.
FQ exhibited a good transepithelial transport in a Caco/TC7 monolayer intestinal epithelium model, independent of pH, and not influenced by pgp inhibitors. This indicates a good oral absorption, confirming the experimental results already obtained during in vivo curative assays on mice using an oral way administration [126, 129]. Moreover, the drug shows a low transfer rate through the blood brain barrier in an in vitro model of transendothelial transport across bovine capillary endothelial cells. Both by intravenous and oral administration, FQ showed an extensive plasma clearance, a large volume of distribution, and a large bioavailability (60%). Plasma concentrations are high in a range of 3–8h after oral administration and decrease slowly during the first 24h. A large distribution was observed in liver and in the brain. These properties are close to those already observed for CQ. Concerning the erythrocytic/plasma distribution, the three models tested (rat, macaque monkey, and human) converged toward a high level of accumulation of FQ in RBC (68–85%), close or even better than CQ itself.
In a 14-day test on rats, using an equivalent dose of CQ which caused 100% mortality, FQ caused only about 7% mortality. The liver, and to a lesser extent the kidney, appeared to be the two main target organs. Phase I and IIa of clinical experiments confirmed the good tolerability of FQ and its lower toxicity compared with CQ.
Concerning embryotoxicity and mutagenicity, FQ was found negative on the Frog Embryo Teratogenesis Assay-Xenopus (FETAX) test [152]. FQ and its enantiomers, solubilized in aqueous solutions, were negative in the Ames test [153]. FQ responded negatively in the micronucleus in vitro and in vivo assays conducted under GLP Standards. Interestingly, in the same kind of experiments, CQ was found to be weakly mutagenic and genotoxic [154].
In conclusion, FQ showed a good pharmacological profile after oral absorption, and experimental and clinical phase I assays showed a very good tolerance to FQ given single dose, even at high dosage. These results, associated with the activity of FQ and its first metabolite against CQ-resistant P. falciparum, are a good omen for the future development of FQ as a new antimalarial, confirmed by current passing of the drug in Clinical Phase IIb.
3.5 A Brief Industrial Development Story
-
1994: First chemical synthesis of FQ in the Laboratory of Catalysis of Lille (now Catalysis and Solid State Chemistry, UMR CNRS 8181), ENSCL, University of Sciences and Technologies, Lille, France.
-
1995: French and International Patents of FQ and derivatives.
-
1994–1995: First tests of FQ on Gabon acclimated isolates of P. falciparum by Pascal Millet.
-
1996: First tests on laboratory clones of P. falciparum and on rodent malaria in vivo by Daniel Dive.
-
1997: First contacts with a pharmaceutical industry (Pierre Fabre Medicament, PFM), which asks key questions about the molecule for development.
-
1997–2001: A crystallized form of FQ is obtained.
-
Synthesis of FQ enantiomers by C. Biot. Specific pharmacology on P. falciparum and on rodent malaria in vivo by Daniel Dive.
-
-
2001: PFM stops its activities regarding malaria.
-
New contacts with another pharmaceutical group (Sanofi Aventis).
FQ is licensed by SA. Preliminary development.
-
-
2001–2004: Tests of FQ on field isolates in Gabon, Senegal, Cambodia.
-
06/2003: Development of FQ by SA.
-
2004: Beginning of Phase I clinical trials (first in humans).
-
2005: First administration to human in Gabon.
-
2006: FQ is an international nonproprietary name.
-
2006: Patent on association between FQ and an artemisinine derivative for treating malaria (SA).
-
2007: Beginning of Phase II clinical trials.
-
2008: Comparative safety and activity study with FQ/Artesunate vs. Amodiaquine/Artesunate in African adult patients.
4 Mechanism(s) of Action of Ferroquine
The antimalarial activity of FQ was initially compared with that of the purely organic CQ in an effort to understand how the presence of the ferrocene contributes to the antiplasmodial property. Over the years, the mechanism of CQ has been (and is still) the subject of many discussions and arguments. Nevertheless, there is strong evidence that the action of CQ is linked to its localization in the DV of the parasite and to the inhibition of the formation of hemozoin ([37], see [155–157]).
4.1 Inhibition of Hemozoin Formation
FQ formed a complex with hematin in solution with a stoichiometry of 1:1 (log K=4.95±0.05) [128]. Moreover, in the presence of FQ, hematin is no more converted into β-hematin and a dose-dependent inhibition of β-hematin formation was obtained. The IC50 of FQ was 0.8, whereas the IC50 of CQ was 1.9. This clearly shows that FQ is a strong inhibitor of β-hematin formation and even more potent than CQ [128]. The molecular electrostatic potential (MEP) surfaces have been computed at the DFT-B3LYP level of theory for neutral and diprotonated FQ and CQ. In both cases, FQ and CQ show considerable similarity in the quinoline area. Since this part of the molecule is thought to directly interact with hematin by stacking interaction, a similar mode of interaction between these active drugs (FQ or CQ) and hematin was suggested [128].
4.2 Specific Drug Targeting
However, both the basicity and lipophilicity of FQ are significantly different to that of CQ. The lipophilicity of FQ and CQ is similar when protonated at the putative food vacuole pH of 5.2 (log D=−0.77 and −1.2, respectively), but differ markedly at pH 7.4 (log D=2.95 and 0.85, respectively). In addition, the pK a values of FQ are lower (pK a1=8.19 and pK a2=6.99) than those of CQ (10.03 and 7.94, respectively). As a free base, FQ is more than 100-fold more lipophilic than CQ; this difference may explain the remarkable activity of FQ compared with CQ. Indeed, FQ may target the lipid site of hemozoin formation more efficiently, and thus more powerfully inhibit the process of hemozoin crystallization. To validate or disprove this theory, incubating parasites with [14C] FQ [158] and localizing it by electron microscope autoradiography and subcellular fractionation is envisaged [37].To avoid the use of radioactive material, an alternative method will be to use an electron-dense tracer as ruthenoquine (the ruthenium analog of FQ) to study its uptake by ultrastructural studies of treated parasites.
4.3 A Critical Intramolecular Hydrogen Bond
The solid-state structure of neutral FQ is stabilized by a strong intramolecular hydrogen bond (d=2.173Å) between the anilino N(11) and the tertiary amino N(24) (see Chap. 3.1, [128]). When in solution with a low dielectric constant (chloroform, k=5.5) such as the lipid environment, NMR data show that the spatial structure of FQ is much the same as in the crystal [159]. This peculiar shape leads to an increase of its hydrophobicity and results in the rejection of the bulky ferrocenyl moiety toward the outside [157].
Combined spectroscopic and structural investigations revealed that the extended structure of FQ in polar solvent changes to a more compact conformation via an additional intramolecular hydrogen bond in apolar solvent (Biot et al. 2008). So, the role of this noncovalent interaction on the antimalarial activity was questioned.
To this end, an analog (FQ-Me) bearing a methyl group instead of a hydrogen atom on the anilino N(11) was synthesized (Fig.28). This compound shared similar physicochemical properties with FQ and was also able to inhibit the β-hematin formation (Table2). Nevertheless, this structural modification led to significant reduction in activity against CQ-susceptible and CQ-resistant strains (Table2). The presence of a hydrogen bonding interaction in the lateral side chain of FQ should contribute to the antiplasmodial activity [157]. These results are in accordance with previous observations where FQ analogs (a) and hydroxyferroquines (b) including an intramolecular H-bond in their side chain were shown much more active than molecules lacking this noncovalent interaction.

Chemical synthesis of the methyl FQ derivative (FQ-Me). (a) t-BuLi, Et2O; (b) DMF; (c) CH3NH2, THF; (d) NaBH4, MeOH; (e) 4,7-dichloroquinoline, K2CO3, NEt3, NMP, 135°C
The flip/flop H-bond between the open conformation of the diprotonated FQ and the folded conformation of the neutral FQ should contribute to the transport from water to the hydrophobic membranes (Fig.29).

Proposed flip/flop H-bond that may help transport of FQ through the hydrophobic membranes
The hydrophobic ferrocene moiety should establish favorable van der Waals interactions with lipid structures involved at the interface with aqueous content of the DV, positioning FQ in the same catalytic site as hematin [157]. This preferential location of FQ should cause inhibition of formation of hemozoin more efficiently than CQ does.
Moreover, tight binding interactions between FQ and lipids suggest that the concentration of free FQ in water is decreased and FQ is no longer a substrate for the efflux transporter(s) associated with CQ resistance. More biophysical and biological approaches will be necessary to refine this hypothesis and are currently being developed by our groups.
4.4 Production of Reactive Oxygen Species
In the DV of the parasite, free heme is rapidly converted to hematin, i.e., iron II is oxidized in iron III. During this process, electrons liberated promote the formation of reactive oxygen species (ROS) such as superoxide anion radicals and hydrogen peroxide [160]. ROS can cause cellular damage. Hydrogen peroxide may also be used for the peroxidative degradation of heme. In this context, the influence of H2O2 on the redox behavior of FQ and implications for antimalarial activity was questioned.
Under the specific conditions (acidic and oxidizing) mimicking the parasite DV, FQ shows a reversible one-electron redox reaction [161]. This leads to the formation of ferriquinium and generation of hydroxyl radicals (Fig.30), with kinetics which are relevant for an antimalarial activity on P. falciparum.

Production of hydroxyl radicals by ferroquine
CQ has been shown to induce ROS production in astroglial cells via a signaling pathway involving NFκ-B [162, 163]. Nevertheless, in the RBC devoid of a nucleus, such a pathway cannot occur. Additional spin trapping experiments demonstrate that CQ is not able to produce OH• radicals by itself in the presence of H2O2 at the acidic pH of the parasite DV. On the contrary, using the same experimental conditions, hydroxyl radicals are produced by FQ at micromolar concentration [161]. Whereas the production of OH• is weak, it should be sufficient to induce significant damage.
FQ might thus strike the parasite not only via direct inhibition of hemozoin formation but also by production of lethal hydroxyl radicals. These radicals are known to be particularly aggressive toward unsaturated fatty acids present in membrane phospholipids and to promote an extensive chain reaction of their peroxidation products [164]. The alteration or the destruction of these structures should then influence heme detoxification processes and could result in a complementary harmful effect on the parasite. Further experiments will be necessary to provide answers to these hypotheses.
In conclusion, the activity of FQ is due to more than one route (Fig.31). Due to its physicochemical properties, FQ could specifically target the lipid site of hemozoin formation. Its mechanism of action should be in part similar to that of CQ, based on the inhibition of hemozoin formation. Upon the specific (acidic and oxidizing) DV conditions, production of radical oxygen species (ROS) by FQ should be sufficient to promote significant damage on membranes of the parasite DV. The strong activity of FQ on CQ-resistant clones and isolates of P. falciparum suggests a fundamental difference in interaction with resistance mechanisms of the parasite.

Proposed structure–activity relationships for FQ
5 Conclusion
The contribution of organometallic chemistry in the development of new antimalarials is promising in its potential uses, but so far the outcome remains limited. The attempts to insert an organometallic moiety in already known antimalarials were based on the idea to improve their activity or to overcome the resistance of the parasite to the parent drug. Results remained, for most of them, within expectations, except for CQ analogs and the serendipitous discovery of FQ. The extensive structure–activity studies, realized on the ferrocenic CQ conjugates, provided important information about the role of the ferrocene core in antimalarial activity. The position of the ferrocenyl moiety in the lateral side chain of CQ was probed with precision. The results formed the basis for new ideas and quinine, melfoquine, and artemisinin were derivatised. The influence of the ferrocenyl moietyl on the lipophilicity is really important when considering that an antimalarial drug has to cross at least three membranes before reaching the parasite cytoplasm and five to six membranes to reach the inner part of the parasite mitochondria or the plastid. The fine analysis of FQ properties led to the underlining of the possible specific role of the ferrocene core in the antimarial activity via the production of hydroxyl radicals. The presence of a specific intramolecular hydrogen bond in the molecule seems to be an important factor for its activity. It was postulated that this weak interaction confers to FQ a potency to interact with membrane lipids and to be specifically present at the site of action when it can be the most efficient. The insertion of the ferrocene moiety in the weak base CQ undoubtedly increased its ability to target the parasite digestive vacuole and possibly enables the drug to escape the resistance mechanisms.
FQ (in phase IIb of clinical trials) represents the first organometallic antimalarial currently in the “pipe-line” of promising drugs. Moreover, the pursuit of studies regarding the mechanism of action of FQ and the possible mechanisms of resistance appears promising not only for chemotherapy of malaria, but also as the basis for new concept in drug design.
References
Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI (2005) Nature 434:214
WHO (2004a) http://www.who.int/healthinfo/global_burden_disease/estimates_regional/en/index.html
Alexander K, Rowe KA, Steketee RW (2007) Am J Trop Med Hyg 77:48
Greenwood BM, Fidock DA, Kyle DE, Kappe SHI, Alonso PL, Collins FH, Duffy PE (2008) J Clin Invest 118:1266
Roca-Feltrer A, Carneiro I, Armstrong Schellenberg JRM (2008) Trop Med Int Health 13:771
WHO (2004b) http://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/index.html
Teklehaimanot A, Mejia P (2008) Ann N Y Acad Sci 1136:32
Snow RW, Guerra CA, Mutheu JJ, Hay SI (2008) PLoS Med 5:e142
Plebanski M, Locke E, Kazura JW, Coppel RL (2008) Trends Parasitol 24:532
Bhattarai A et al (2007) PLoS Med 4:e309
Ceesay SJ, Casals-Pascual C, Erskine J, Anya SE, Duah NO, Fulford AJ, Sesay SS, Abubakar I, Dunyo S, Sey O, Palmer A, Fofana M, Corrah T, Bojang KA, Whittle HC, Greenwood BM, Conway DJ (2008) Lancet 372:1545
Noor AM, Mutheu JJ, Tatem AJ, Hay SI, Snow RW (2009) Lancet 373:58
Greenwood BM (2008) Trends Parasitol 24:449
Fidock DA, Eastman RT, Ward SA, Meshnick SR (2008) Trends Parasitol 24:537
Krugliak M, Zhang J, Ginsburg H (2002) Mol Biochem Parasitol 119:249
Esposito A, Tiffert T, Mauritz JM, Schlachter S, Bannister LH, Kaminski CF, Lew VL (2008) PLoS One 3:e3780
Francis SE, Sullivan DJ, Goldberg DE (1997) Annu Rev Microbiol 51:97
Charet P, Prensier G, Slomianny C (1983) Comp Biochem Physiol B 75:347
Bannister LH, Hopkins JM, Margos G, Dluzewski AR, Mitchell GH (2004) Microsc Microanal 10:551
Elliott DA, McIntosh MT, Hosgood HD 3rd, Chen S, Zhang G, Baevova P, Joiner KA (2008) Proc Natl Acad Sci USA 105:2463
Langreth SG, Jensen JB, Reese RT, Trager W (1978) J Protozool 25:443
Lazarus MD, Schneider TG, Taraschi TF (2008) J Cell Sci 121:1937
Slomianny C (1990) Blood Cells 16:369
Goldberg DE (2005) Curr Top Microbiol Immunol 295:275
Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE (2006) Proc Natl Acad Sci USA 103:8840
Chou AC, Fitch CD (1981) J Clin Invest 68:672
Fitch CD, Chevli R, Banyal HS, Phillips G, Pfaller MA, Krogstad DJ (1982) Antimicrob Agents Chemother 21:819
Har-El R, Marva E, Chevion M, Golenser J (1993) Free Radic Res Commun 18:279
Orjih A, Banyal HS, Chevli R, Fitch CD (1981) Science 214:667
Atamna H, Ginsburg H (1993) Mol Biochem Parasitol 61:231
Hempelmann E, Egan TJ (2002) Trends Parasitol 18:11
Bohle DS, Kosar AD, Stephens PW (2002) Acta Crystallogr D Biol Crystallogr 58:1752
Pagola S, Stephens PW, Bohle DS, Kosar AD, Madsen SK (2000) Nature 404:307
Solomonov I, Osipova M, Feldman Y, Baehtz C, Kjaer K, Robinson IK, Webster GT, McNaughton D, Wood BR, Weissbuch I, Leiserowitz L (2007) J Am Chem Soc 129:2615
Weissbuch I, Leiserowitz L (2008) Chem Rev 108:4899
Sullivan DJ, Gluzman IY, Goldberg (1996) Science 271:219
Papalexis V, Siomos MA, Campanale N, Guo XG, Kocak G, Foley M, Tilley L (2001) Mol Biochem Parasitol 115:77
Chen MM, Shi L, Sullivan DJ Jr (2001) Mol Biochem Parasitol 113:1
Jackson KE, Klonis N, Ferguson DJP, Adisa A, Dogovski C, Tilley L (2004) Mol Microbiol 54:109
Oliveira MF, Kycia SW, Gomez A, Kosar AJ, Bohle DS, Hempelmann E, Menezes D, Vannier-Santos MA, Oliveira PL, Ferreira ST (2005) FEBS Lett 579:6010
Pisciotta JM, Coppens I, Tripathi AK, Scholl PF, Shuman J, Bajad S, Shulaev V, Sullivan DJ Jr (2007) Biochem J 402:197
Egan TJ, Chen JY, de Villiers KA, Mabotha TE, Naidoo KJ, Ncokazi KK, Langford SJ, McNaughton D, Pandiancherri S, Wood BR (2006) FEBS Lett 580:5105
Egan TJ (2008) J Inorg Biochem 102:1288
Egan TJ (2008) Mol Biochem Parasitol 157:127
Fitch CD, Cai GZ, Chen YF, Shoemaker JD (1999) Biochim Biophys Acta 1454:31
Pisciotta JM, Sullivan D (2008) Parasitol Int 57:89
Hartwig CL, Rosenthal AS, D’Angelo J, Griffin CE, Posner GH, Cooper RA (2009) Biochem Pharmacol 7:322
Chou AC, Fitch CD (1992) Life Sci 51:2073
Slater AF, Cerami A (1992) Nature 355:167
Dorn A, Stoffel R, Matile H, Bubendorf A, Ridley RG (1995) Nature 374:269
Jani D, Nagarkatti R, Beatty W, Angel R, Slebodnick C, Andersen J, Kumar S, Rathore D (2008) PLoS Pathog 4:e1000053
Vinayak S, Rathore D, Kariuki S, Slutsker L, Shi YP, Villegas L, Escalante AA, Udhayakumar V (2009) Infect Genet Evol 9:286
Krogstad DJ, Gluzman IY, Kyle DE, Oduola AM, Martin SK, Milhous WK, Schlesinger PH (1987) Science 238:1283
Valderramos SG, Fidock DA (2006) Trends Pharmacol Sci 27:594
Foote SJ, Thompson JK, Cowman AF, Kemp DJ (1989) Cell 57:921
Wilson CM, Serrano AE, Wasley A, Bogenschutz MP, Shankar AH, Wirth DF (1989) Science 244:1184
Foote SJ, Kyle DE, Martin RK, Oduola AM, Forsyth K, Kemp DJ, Cowman AF (1990) Nature 345:255
Duraisingh MT, Refour P (2005) Mol Microbiol 57:874
Cowman AF, Galatis D, Thompson JK (1994) Proc Natl Acad Sci USA 91:1143
Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF (2000) Nature 403:906
Sidhu AB, Valderramos SG, Fidock DA (2005) Mol Microbiol 57:913
Sidhu AB, Uhlemann AC, Valderramos SG, Valderramos JC, Krishna S, Fidock DA (2006) J Infect Dis 194:528
Wilson CM, Volkman SK, Thaithong S, Martin RK, Kyle DE, Milhous WK, Wirth DF (1993) Mol Biochem Parasitol 57:151
Sanchez CP, Rotmann A, Stein WD, Lanzer M (2008) Mol Microbiol 70:786
Saliba KJ, Lehane AM, Kirk K (2008) Mol Microbiol 70:775
Wellems TE, Panton LJ, Gluzman IY, do Rosario VE, Gwadz RW, Walker-Jonah A, Krogstad DJ (1990) Nature 345:253
Su X, Kirkman LA, Fujioka H, Wellems TE (1997) Cell 91:593
Fidock DA, Nomura T, Cooper RA, Su X, Talley AK, Wellems TE (2000) Mol Biochem Parasitol 110:1
Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LM, Sidhu AB, Naudé B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE (2000) Mol Cell 6:861
Djimdé A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourté Y, Dicko A, Su XZ, Nomura T, Fidock DA, Wellems TE, Plowe CV, Coulibaly D (2001) N Engl J Med 344:257
Wellems TE (2002) Science 298:124
Jiang H, Joy DA, Furuya T, Su XZ (2006) J Postgrad Med 52:271
Durrand V, Berry A, Sem R, Glaziou P, Beaudou J, Fandeur T (2004) Mol Biochem Parasitol 136:273
Ekland EH, Fidock DA (2007) Curr Opin Microbiol 10:363
Howard EM, Zhang H, Roepe PD (2002) J Membr Biol 190:1
Martin RE, Kirk K (2004) Mol Biol Evol 21:1938
Tran CV, Saier MH Jr (2004) Microbiology 150:1
Sanchez CP, Stein WD, Lanzer M (2007) Trends Parasitol 23:332
Sanchez CP, Rohrbach P, McLean JE, Fidock DA, Stein WD, Lanzer M (2007) Mol Microbiol 64:407
Bray PG, Martin RE, Tilley L, Ward SA, Kirk K, Fidock DA (2005) Mol Microbiol 56:323
Cooper RA, Lane KD, Deng B, Mu J, Patel JJ, Wellems TE, Su X, Ferdig MT (2007) Mol Microbiol 63:270
Lekostaj JK, Natarajan JK, Paguio MF, Wolf C, Roepe PD (2008) Biochemistry 47:10394
Lehane AM, Kirk K (2008) Antimicrob Agents Chemother 52:4374
Gligorijevic B, Purdy K, Elliott DA, Cooper RA, Roepe PD (2008) Mol Biochem Parasitol 159:7
Chen N, Gao Q, Wang S, Wang G, Gatton M, Cheng Q (2008) Antimicrob Agents Chemother 52:345
Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, Taylor TE, Plowe CV (2006) N Engl J Med 355:1959
Jiang H, Patel JJ, Yi M, Mu J, Ding J, Stephens R, Cooper RA, Ferdig MT, Su XZ (2008) PLoS One 3:e2484
Henry M, Alibert S, Rogier C, Barbe J, Pradines B (2008) Curr Top Med Chem 8:563
Henry M, Briolant S, Fontaine A, Mosnier J, Baret E, Amalvict R, Fusaï T, Fraisse L, Rogier C, Pradines B (2008) Antimicrob Agents Chemother 52:2755
Navarro M, Vasquez F, Sanchez-Delgado RA, Perz H, Sinou V, Schrevel J (2004) J Med Chem 47:5204
Sanchez-Delgado RA, Anzelotti A (2004) Mini Rev Med Chem 4:23
Sanchez-Delgado RA, Anzelotti A, Suarez L (2004) Met Ions Biol Syst 41:379
Sharma V, Piwnica-Worms D (1999) Chem Rev 99:2545
Navarro M, Perez H, Sanchez-Delgado RA (1997) J Med Chem 40:1937
Sanchez-Delgado RA, Navarro M, Perez H, Urbina JA (1996) J Med Chem 39:1095
Goldberg DE, Sharma V, Oksman A, Gluzman IY, Wellems TE, Piwnica-Worms D (1997) J Biol Chem 272:6567
Ocheskey JA, Harpstrite SE, Oksman A, Goldberg DE, Sharma V (2005) Chem Commun 12:1622–1624
Itoh T, Shirakami S, Ishida N, Yamashita Y, Yoshida T, Kim HS, Wataya Y (2000) Bioorg Med Chem Lett 10:1657
Wu X, Wilairat P, Go ML (2002) Bioorg Med Chem Lett 12:2299
Wu X, Tiekink ER, Kostetski I, Kocherginsky N, Tan AL, Khoo SB, Wilairat P, Go ML (2006) Eur J Pharm Sci 27:175
Howarth J, Hanlon K (2001) Tetrahedron Lett 42:751
Delhaes L, Biot C, Berry L, Maciejewski LA, Camus D, Brocard JS, Dive D (2000) Bioorg Med Chem 8:2739
Paitayatat S, Tarnchompoo B, Thebtaranonth Y, Yuthavong Y (1997) J Med Chem 40:633
Baramee A, Coppin A, Mortuaire M, Pelinski L, Tomavo S, Brocard J (2006) Bioorg Med Chem 14:1294
Biot C, Delhaes L, Maciejewski LA, Mortuaire M, Camus D, Dive D, Brocard JS (2000) Eur J Med Chem 35:707
Guillon J, Moreau S, Mouray S, Sinou V, Forfar I, Fabre SB, Desplat V, Millet P, Parzy D, Jarry C, Grellier P (2008) Bioorg Med Chem 16:9133
Fichera ME, Roos DS (1997) Nature 390:407
Dahl EL, Rosenthal PJ (2007) Antimicrob Agents Chemother 51:3485
Anquetin G, Greiner J, Vierling P (2005) Curr Drug Targets Infect Disord 5:227
Saleh A, Friesen J, Baumesteir S, Gross U, Bohne W (2007) Antimicrob Agents Chemother 51:1217
Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK (2008) Exp Parasitol 118:487
Dubar F, Anquetin G, Pradines B, Khalife J, Dive D, Biot C (2009) J Med Chem 52:7954–7957
Domarle O, Blampain G, Agnaniet H, Nzadiyabi T, Lebibi J, Brocard J, Maciejewski L, Biot C, Georges AJ, Millet P (1998) Antimicrob Agents Chemother 42:540
Biot C (1998) Ph.D. thesis, University of Lille1, Villeneuve-d’Ascq, France
De D, Krogstad FM, Cogswell FB, Krogstad DJ (1996) Am J Trop Med Hyg 55:579
De D, Krogstad FM, Byers LD, Krogstad DJ (1998) J Med Chem 41:4918
Biot C, Daher W, Chavain N, Fandeur T, Khalife J, Dive D, De Clercq E (2006) J Med Chem 49:2845
Biot C, Daher W, Ndiaye CM, Melnyk P, Pradines B, Chavain N, Pellet A, Fraisse L, Pelinski L, Jarry C, Brocard J, Khalife J, Forfar-Bares I, Dive D (2006) J Med Chem 49:4707
Chibale K, Moss JR, Blackie M, van Schalkwyk D, Smith PJ (2000) Tetrahedron Lett 41:6231
Biot C, Glorian G, Maciejewski L, Brocard J, Domarle O, Blampain G, Millet P, Georges AJ, Abessolo H, Dive D, Lebibi J (1997) J Med Chem 40:3715
Beagley P, Blackie MAL, Chibale K, Clarkson C, Meijboom R, Moss JR, Smith PJ, Su H (2003) Dalton Trans 15:3046
Raynes K (1999) Int J Parasitol 29:367
Biot C, Dessolin J, Ricard I, Dive D (2004) J Organomet Chem 689:4678
Picart-Goetgheluck S, Delacroix O, Maciejewski L, Brocard J (2000) Synthesis 10:1421
Delhaes L, Biot C, Berry L, Delcourt P, Maciejewski LA, Camus D, Brocard JS, Dive D (2002) Chembiochem 3:101
Fraisse L, Ter-Minassian D (2006) International application PCT/FR2006/000842
Biot C, Taramelli D, Forfar-Bares I, Maciejewski L, Boyce M, Nowogrocki G, Brocard JS, Basilico N, Olliaro P, Egan EJ (2005) Mol Pharm 2:185
Delhaes L, Abessolo H, Biot C, Berry L, Delcourt P, Maciejewski L, Brocard J, Camus D, Dive D (2001) Parasitol Res 87:239
Bray PG, Janneh O, Raynes KJ, Mungthin M, Ginsburg H, Ward SA (1999) J Cell Biol 145:363
Chou AC, Chevli R, Fitch CD (1980) Biochemistry 19:1543
Biot C, Delhaes L, N’Diaye CM, Maciejewski L, Camus D, Dive D, Brocard J (1999) Bioorg Med Chem 7:2843
Biot C, Pradines B, Sergeant MH, Gut J, Rosenthal PJ, Chibale K (2007) Bioorg Med Chem Lett 17:6434
Klayman DL, Bartosevich JF, Griffin TS, Mason CJ, Scovil JP (1979) J Med Chem 22:855
Scovill JP, Klayman DL, Lambros C, Childs GE, Notsch JD (1984) J Med Chem 27:87
Ames JR, Ryan MD, Klayman DL, Kovacic P (1985) J Free Radic Biol Med 1:353
Beagley P, Blackie MAL, Chibale K, Clarkson C, Moss JR, Smith PJ (2002) Dalton Trans 23:4426
Peters W (1975) Ann Trop Med Parasitol 69:155
Peters W (1987) In: Peters W (ed) Chemotherapy and drug resistance in malaria, vol 1. Academic Press, London, pp 437–455
Atteke C, Ndong JM, Aubouy A, Maciejewski L, Brocard J, Lebibi J, Deloron P (2003) J Antimicrob Chemother 51:1021
Kreidenweiss A, Kremsner PG, Dietz K, Mordmüller B (2006) Am J Trop Med Hyg 75:1178
Pradines B, Fusai T, Daries W, Laloge V, Rogier C, Millet P, Panconi E, Kombila M, Parzy D (2001) J Antimicrob Chemother 48:179
Pradines B, Tall A, Rogier C, Spiegel A, Mosnier J, Marrama L, Fusai T, Millet P, Panconi E, Trape JF, Parzy D (2002) Trop Med Int Health 7:265
Chim P, Lim P, Sem R, Nhem S, Maciejewski L, Fandeur T (2004) Ann Trop Med Parasitol 98:419
Barends M, Jaidee A, Khaohirun N, Singhasivanon P, Nosten F (2007) Malar J 6:81
Dive D, Biot C (2008) ChemMedChem 3:383
Stepnicka P (2008) Ferrocenes: ligands, materials and biomolecules. Wiley, Chicester, pp 611–616
Daher WE, Pelinski L, Klieber S, Sadoun F, Meunier V, Bourrie M, Biot C, Guillou F, Fabre G, Brocard J, Fraisse L, Maffrand JP, Khalife J, Dive D (2006) Drug Metab Dispos 34:667
Walker DJ, Pitsch JL, Peng MM, Robinson BL, Peters W, Bhisutthibhan J, Meshnick SR (2000) Antimicrob Agents Chemother 44:344
González-Pons M, Szeto AC, González-Méndez R, Serrano AE (2009) Malar J 8:1
Projean D, Baune B, Farinotti R, Flinois JP, Beaune P, Taburet AM, Ducharme J (2003) Drug Metab Dispos 31:748
Bantle JA, Burton DT, Dawson DA, Dumont JN, Finch RA, Fort DJ, Linder G, Rayburn JR, Buchwalter D, Maurice MA et al (1994) J Appl Toxicol 14:213
Maron D, Ames BN (1983) Mutat Res 113:173
Chatterjee T, Muhkopadhyay A, Khan KA, Giri AK (1998) Mutagenesis 13:619
Gibaud S, Jaouen G (2010) Arsenic – based drugs: from Fowler’s solution to modern anticancer chemotherapy. In: Jaouen G, Metzler-Nolte N (eds) Topics in Organometallic Chemistry. Springer, Heidelberg
Gardella F, Assi S, Simon F, Bogreau H, Eggelte T, Ba F, Foumane V, Henry MC, Kientega PT, Basco L, Trape JF, Lalou R, Martelloni M, Desbordes M, Baragatti M, Briolant S, Almeras L, Pradines B, Fusai T, Rogier C (2008) Malar J 7:124
Dubar F, Khalife J, Dive D, Biot C (2008) Molecules 13:2900
McNeill A, Murrell V, Taylor K (2004) J Label Comp Radiopharm 47:1019
Biot C, Chavain N, Dubar F, Pradines B, Trivelli X, Brocard J, Forfar I, Dive D (2009) J Organomet Chem 694:845
Kannan R, Kumar K., Sahal D, Kukreti S, Chauhan VS (2005) Biochem J 385:409
Chavain N, Vezin V, Dive D, Touati N, Paul J-F, Buisine E, Biot C (2008) Mol Pharmaceutics 5:710
Park J, Kwon D, Choi C, Oh JW, Benveniste EN (2003) J Neuro-chem 84:1266
Park J, Choi K, Jeong E, Kwon D, Benveniste EN, Choi C (2004) Glia 47:9
Cherubini A, Ruggiero C, Polidori MC, Mecocci P (2005) Free Radic Biol Med 39:841
Acknowledgments
We would like to thank Prof. Gérard Jaouen and Prof. Nils Metzler-Nolte for their kind support. We thank Faustine Dubar for the schematic representation of the malaria parasite. Melissa Ladyman is acknowledged for proof-reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Biot, C., Dive, D. (2010). Bioorganometallic Chemistry and Malaria. In: Jaouen, G., Metzler-Nolte, N. (eds) Medicinal Organometallic Chemistry. Topics in Organometallic Chemistry, vol 32. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-13185-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-642-13185-1_7
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-13184-4
Online ISBN: 978-3-642-13185-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)