
Abstract
Adult-onset Still’s disease (AOSD) is a rare systemic, autoinflammatory disorder that often presents in adolescence and early adulthood with fever, rash, and polyarthritis. There are significant genetic and clinical similarities with systemic juvenile idiopathic arthritis (sJIA) with a different chronological disease onset. The disease can have many protean characteristics leading to delays in diagnosis. Treatment includes corticosteroids; traditional immunomodulators, such as methotrexate; and targeted biologic treatments that include IL-1 and IL-6 inhibitors.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Adult-onset Still’s disease
- Pathogenesis
- Etiology
- Treatment
- Macrophage activation syndrome (MAS)
- IL-1 inhibitors
Introduction
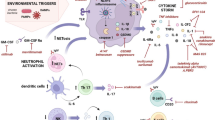
Adult-onset Still’s disease (AOSD) is a rare, idiopathic, systemic, autoinflammatory disorder. Patients can present with the “Still’s triad”, arthritis, evanescent salmon-colored rash, and quotidian or double quotidian fevers, although atypical cases abound. Described as early as 1896 by George Still in children and further characterized in 1971 by Eric Bywaters who an adult onset of symptoms, “Still’s disease” has come to define a disease spectrum with systemic juvenile idiopathic arthritis (SJIA) at one end and AOSD at the other, based on age of symptom onset [1]. Clinical manifestations may include hepatosplenomegaly, lymphadenopathy, serositis, and aseptic meningitis. Severe disease complications include endocarditis, myocarditis, and pericarditis, coagulation abnormalities, and, especially, macrophage activation syndrome (MAS). The precise etiology of AOSD remains unclear; however, activation of the innate immunity via an unknown trigger has been described as the key inciting factor. Establishing the diagnosis of AOSD is often difficult due to the protean nature of the disease; the presence of non-specific symptoms, especially in the early phases of the disease; and the absence of characteristic serological diagnostic markers. Importantly, albeit inconsistent, laboratory findings include neutrophil-predominant leukocytosis, negative rheumatoid factor (RF), and antinuclear antibodies (ANA) as well as frequently elevated serum ferritin levels with a low-glycosylated fraction [2].
AOSD remains a diagnosis of exclusion , since more common illnesses can present with very similar symptoms. Definitive diagnosis should only be made after ruling out infectious, malignant, and other autoimmune and autoinflammatory diseases. Yamaguchi criteria are often used to aid in diagnosis and, especially, classification [3, 4]. Patients frequently have a favorable prognosis with timely diagnosis, unless life-threatening complications such as macrophage activation syndrome (MAS) ensue.
Epidemiology
Prevalence of AOSD is estimated to be less than one case per 100,000 people. AOSD is rare, and hence there are currently no consensus on its incidence and prevalence in different populations. Based on larger reviews from the 1980s, it appears that AOSD occurs worldwide and may affect slightly more women than men. This disease characteristically affects younger people between the age of 16 and 35 years of age [5, 6]. A bimodal peak at ages 15–25 and 36–46 without a sex bias has been described in a retrospective study of 62 patients from western France [7]. However, an epidemiological survey from Japan described that 67% of the cases presented after the age of 35, the majority (65–70%) being women [8]. AOSD affects all ages, and stress has been suggested as an important risk factor for all ages [9]. No familial trend has been reported in the recent literature; however, an association with certain HLA alleles has been observed in certain populations. These HLA subtypes include HLA DR4, B17, B18, B35, DR2, DR5, and DQ1.
Etiopathogenesis
Pathophysiology of AOSD was largely obscure until the recent past. A myriad of factors such as genetics, infectious (bacterial and viral) agents, and environmental factors have been thought to play a causative role. Concurrent elevations of serologic markers of infectious agents have been noted in some patients with Still’s disease. These infectious triggers include EBV; parvovirus B19; CMV; HHV; HIV; Coxsackievirus; mumps; rubella; echovirus; hepatitis A, B, and C viruses; Campylobacter jejuni; Chlamydia pneumoniae; adenovirus; influenza virus; parainfluenza virus; and Mycoplasma [5, 10]. However, to date definite insight in to precise role of infection in AOSD is lacking.
Several associations with distinct HLA alleles and AOSD have been described thus far. Pouchot et al. described a strong association between HLA-B17, B18, B35, and DR2 and AOSD [11]. In a small study of 25 AOSD patients, HLA-Bw35 was associated with disease susceptibility conferring a favorable prognosis [12]. Wouters et al. noted an increased frequency of the HLA-DR4 allele in 29 patients with AOSD compared to normal controls, with the presence of HLA-DRw6 being linked to root joint involvement [13]. An association between a chronic articular form of AOSD and HLA-DRB1*1501 (DR2), DRB1*1201 (DR5), and DQB1*0602 (DQ1) was previously reported, while HLA-DQB1*0602 (DQ1) have been also associated with the systemic form of the disease in Japanese population [14]. Statistics from a Korean report supported an association between HLA-DRB1*12 and DRB1*15 and AOSD, while HLA-DRB1*04 seemed to be protective. Conversely, HLA-DRB1*14 alleles were more frequently present in patients with the monocyclic systemic type of AOSD [15].
Hallmark of AOSD involves neutrophil and macrophage activation triggered by the pro-inflammatory cytokine IL-18. Neutrophil (PMN) CD64, a marker of neutrophil activation, has been found to be upregulated in patients with active AOSD. Calprotectin (calcium-binding protein) secreted by neutrophils and macrophages and macrophage migration inhibitory factor (MIF) have been found to be useful markers of disease activity [13]. Intercellular adhesion molecule (ICAM-1) upregulated by IL-18 has been implicated as a useful clinical marker whose expression typically reflects the level of disease activity. Macrophage colony-stimulating factor, a cytokine which orchestrates proliferation and differentiation of macrophages, also appears to play a role in AOSD.
More recently, regulation of cytokine production has been noted in patients with AOSD. A predominance of Th1 subset of cytokines has been seen in peripheral blood and tissues of active untreated AOSD patients. Th1 immune cascade is characterized by elevated secretion of interferon γ (IFNγ), interleukin-2 (IL-2), and tumor necrosis factor α (TNFα) cytokines that direct B cells toward IgG2a production, activate macrophages and natural killer (NK) cells, and promote cell-mediated immunity [10]. When compared with controls, serum levels of IL6, TNFα, and IFNγ were significantly increased in 12 patients with active AOSD [16]. IL18 is a pro-inflammatory cytokine that is overproduced in the acute phase of AOSD and is believed to be the cytokine initiating the inflammatory cascade that includes IFNγ, IL6, and TNFα [17]. Genetic polymorphisms of the human IL18 gene have been described to confer disease susceptibility in a Japanese study [18]. Conversely, in another Japanese study, serum levels of soluble IL2 receptors, IL4, and IL18 correlated with chronic articular AOSD activity, whereas IFNγ and IL8 levels were found to be persistently raised, even in disease remission.
Understanding of the Still’s disease was also enhanced by the description of autoinflammatory syndromes. These disorders are associated with recurrent bouts of inflammation without an instigating antigenic stimulus. Defective interleukin-1 processing , regulation of nuclear factor-B transcription factor, and possible uncharacteristic apoptosis are all anticipated mechanisms that may possibly play a role in the generation and perseverance of an inflammatory cascade. Patients with autoinflammatory syndromes, in particular, the typical hereditary periodic fever syndromes, may share certain genetic traits; MEFV gene mutation associated with familial Mediterranean fever (FMF) and IL-1 hypersecretion was seen with augmented frequency in Turkish children with SJIA. Mutation of perforin and the MUNC13–4 genes have been seen in patients with macrophage activation syndrome (MAS), a known severe, life-threatening complication of AOSD [3]. Mutations in genes encoding the tumor necrosis factor (TNF) receptor and pyrin superfamilies of molecules may result in the endurance of leukocytes that would customarily go through apoptosis [3]. As a result, relatively minor pro-inflammatory triggers may lead to an exaggerated and potentially harmful, inflammatory response.
IL-1b , the pivotal cytokine in AOSD and other autoinflammatory syndromes, activates the thermoregulatory center, resulting in fever; may activate IL-1 receptors on the endothelium, resulting in rash; and can also act on the bone marrow to increase mobilization of granulocyte progenitors and mature neutrophils, resulting in peripheral neutrophilia. IL-1 also causes an increase in platelet production, which results in thrombocytosis, and decreases the response to erythropoietin, causing anemia. IL-1 induces the production of IL-6. Circulating IL-6 stimulates the hepatocytes to synthesize several acute-phase proteins, such as CRP, ferritin, and D-dimer.
Clinical Features
In general, three types of AOSD have been described. The monocyclic pattern, which is the most benign form, is characterized by a single episode of AOSD without recurrence. In the polycyclic pattern, the patient experiences recurrent attacks, although the subsequent AOSD attacks often seem not as severe as the first one. In both the monocyclic and the polycyclic forms, systemic symptoms (rash, fever) are very prominent. The worst prognosis is carried by the chronic articular form, which is thought to be an unfortunate evolution of the polycyclic form. Often, the systemic symptoms are absent or so remotely in the past that the patient may not even remember them and the main morbidity is from a chronic articular polyarthritis that mimics RA.
Common findings of AOSD are fever, arthralgia, rash, and sore throat. Other accompanying symptoms include, but are not limited to, myalgia, pharyngitis, lymphadenopathy, splenomegaly, and serositis. Fever is usually quotidian and often precedes other manifestations. Temperature spikes of >39°C frequently occur and are associated with chills and rigors, joint pain, or rash. AOSD is one of the main causes of pyrexia of unknown origin (PUO). Temperature fluctuations can be dramatic. Fever may persist between spikes in approximately 20% of cases, and complete defervescence is not always a characteristic of the quotidian fevers [11]. High-grade temperatures, more than 39.5° C, can be associated more strongly with monophasic pattern of AOSD [19].
Skin rash associated with AOSD is a salmon-pink colored, maculopapular eruption that tends to accompany or, more frequently, be exacerbated by fever. Rash usually presents centrally at the trunk and the adjacent extremities (arms/thighs). Histopathology of the rash often reveals non-specific findings including dermal edema and mild perivascular inflammation in the superficial dermis with lymphocytes and histiocytes. Complement deposits (C3) have been described with immunofluorescence. Arthralgias and myalgias are common manifestations of AOSD. Most commonly involved joints include the wrists, ankles, knees, elbows, proximal interphalangeal joints, and shoulders. These manifestations can evolve into more severe and potentially destructive polyarthritis that can mimic other systemic inflammatory arthritides, such as rheumatoid arthritis [20]. Myalgia can be debilitating and often associated with fever spikes. The muscle involvement, when severe, may be accompanied by an elevation of serum creatinine kinase and aldolase concentrations. However, muscle biopsy and electromyographic (EMG) studies are typically normal. Sterile pharyngitis manifesting as throat pain can occasionally precede the development of fever or rash by weeks, or even months, as a prodromal symptom and can often reoccur with disease relapses.
Hepatomegaly and modest elevation of serum hepatic aminotransferases and alkaline phosphatase are not uncommon in patients with AOSD. Several cases of fulminant liver failure in association with AOSD have been described and may be associated with overexpression of IL-18 [11]. Myocarditis, pericarditis, and pleural effusions have also been observed in AOSD patients, and they seem to respond to anti-inflammatory treatment. Uncommonly, some patients may develop severe interstitial lung disease and some progress to acute respiratory distress syndrome (ARDS). Enlarged, symmetrical, cervical nodes are seen in about one half of patients with AOSD. Lymphadenopathy is often accompanied by fever, leukocytosis creating diagnostic confusion with lymphoma. Lymph node biopsy typically shows intense, paracortical immunoblastic hyperplasia [21]. Splenomegaly is also seen in up to one third of patients.
Macrophage activation syndrome (MAS) or reactive hemophagocytic syndrome (RHS) is a life-threatening complication of AOSD. Mortality rate ranges between 10% and 22% [22,23,24,25], and an incidence of 12–14% has been noted in two recent series, a rate higher than other rheumatic diseases [26, 27]. It is categorized by an uncontrollable activation of the reticuloendothelial system within the bone marrow, reticuloendothelial system, and central nervous system, with successive phagocytosis of hematopoietic cells by tissue macrophages (histiocytes) [25, 28]. Patients developing MAS present with acute high fever, lymphadenopathy, and hepatosplenomegaly. Laboratory findings include pancytopenia, elevated ferritin levels, triglycerides, and liver enzymes, often accompanied by normal erythrocyte sedimentation rate (ESR). The most commonly implicated triggers include infections, medications, and disease flares [29,30,31]. Patients with MAS have a decreased ability to eliminate antigen stimulation, thereby inducing T cell activation and proliferation resulting in cytokine secretion (interferon-gamma and granulocyte macrophage colony-stimulating factor) and macrophage hyperactivation. The end result is an uncontrollable increase in cytokines, specifically TNFα, interleukin-1, and interleukin-6 production resulting in severe systemic inflammatory reaction, i.e., “cytokine storm” [25]. There is also a suggestion that certain therapeutic agents, such as nonsteroidal anti-inflammatory drugs, methotrexate, sulfasalazine, penicillamine, and lately TNF-α, IL-1, and IL-6 inhibitors may be capable of provoking MAS, often complicating their therapeutic use [27]. In theory, these therapies may create a state of immunodeficiency resulting in the reactivation of latent viruses (Epstein-Barr virus or Cytomegalovirus) which in turn can stimulate MAS. The counter-argument would be that anti-inflammatory medications may not be able to prevent the development of MAS, at least in the dosages used in AOSD or SJIA.
Early suspicion of MAS is most commonly raised by the detection of subtle laboratory changes, whereas clinical symptoms may be delayed. A recent international effort to identify candidate markers using an expert consensus process identified nine criteria that included a falling platelet count, hyperferritinemia, evidence of macrophage hemophagocytosis in the bone marrow, increased liver enzymes, falling leukocyte count, a persistent continuous fever ≥38 °C, a falling erythrocyte sedimentation rate (ESR), hypofibrinogenemia, and hypertriglyceridemia [32]. Hemophagocytosis, seen in bone marrow aspiration and biopsy, establishes the diagnosis, even though hemophagocytosis could be seen more frequently in biopsies from the liver, spleen, and/or lymph nodes. Bone marrow aspiration is considered the gold standard and is usually required in atypical cases, causing a diagnostic dilemma. There is significant overlap between AOSD and MAS, and these two conditions are often thought to be anchoring the same disease spectrum, with AOSD representing the milder form.
Laboratory and Radiographic Findings
AOSD, unlike other systemic rheumatic diseases driven by adaptive immunity, is not typically associated with rheumatoid factor (RF) or antinuclear antibody (ANA) positivity, although several cases have been published with low-level positivity of these autoantibodies. This has been considered in various sets of classification criteria . Elevated ESR is a common finding in most patients [11, 33]. C-reactive protein (CRP) may also be found to be raised. Other laboratory abnormalities include leukocytosis, thrombocytosis, and anemia which often accompany increased disease activity. Pancytopenia is often an alarming sign of coexisting or developing MAS and necessitates prompt intervention. Abnormal coagulation testing can rarely be seen and, in extreme cases, may develop into full-blown disseminated intravascular coagulation (DIC) which can be fatal [11]. Abnormal liver and biliary function tests (increase in lactic dehydrogenase, aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transferase, and bilirubin) can also be seen in up to 75% of patients and often accompany fever and exacerbation of arthritis. Mild periportal inflammation with monocyte infiltration may be seen on liver biopsy.
Serum ferritin has gained attention as both a diagnostic test for AOSD and a marker of disease activity (biomarker). In AOSD, ferritin is an acute phase reactant involved in inflammation, is linked to initiation of histiocyte-macrophage system and/or augmented release from damaged hepatocytes, and is not related to iron metabolism and storage. Ferritin production is driven by cytokines such as IL1β, IL18, TNFα, and IL6 [2]. Serum ferritin levels as high as 1000 ng/ml, five times the upper limits of normal, have been described in these patients. Levels ranging from 4000 ng/ml to 30,000 ng/ml are not uncommon. Fautrel et al. evaluated the validity of hyperferritinemia as a diagnostic tool in a retrospective analysis looking at 49 patients with Still’s disease [2]. A fivefold increase in serum ferritin had 80% sensitivity and 41% specificity. Similar results were seen in a Japanese study57 with 82% sensitivity and 46% specificity [4]. Serum ferritin levels are seen in other diseases such as hemochromatosis, Gaucher’s disease, infections (sepsis, HIV), and malignancies (leukemia, lymphomas). Serum ferritin levels correlate with disease activity and often normalize when the disease flare subsides. However, the absence of elevated serum ferritin does not exclude active AOSD. Some patients with active AOSD do not have elevated serum ferritin levels at all, or the rise of serum ferritin may lag behind the symptom presentation.
Glycosylated fraction is a more specific diagnostic marker than ferritin, albeit not commercially available in the USA. In healthy subjects, 50–80% of ferritin is glycosylated. Saturation of glycosylation mechanisms results in a drop in glycosylated fraction to drop to 20–50%. This phenomenon is particularly prevalent in AOSD. Glycosylated ferritin often remains elevated for months after the disease goes into remission and hence cannot be used to monitor disease activity or response to treatment. Improved diagnostic tests are clearly desirable, and new immunological tests, such as IL18, may prove useful in the near future for diagnosis as well as monitoring disease activity and response to treatment [17]. Currently available tests: complete blood count and differential, ESR, CRP, ANA, and RF (both negative), liver function tests (LFTs) and albumin, ferritin, and glycosylated ferritin (if available) are of practical use to most clinicians.
Radiographs are often of limited significance in the early phase of the disease. Images are either normal or show soft tissue swelling, joint effusion, or mild periarticular demineralization. Bone scan and gadolinium-enhanced magnetic resonance imaging were assessed in small series and may prove to be more sensitive imaging modalities for early diagnosis and successful treatment in follow-up. In one study, distinctive pattern of intercarpal and carpometacarpal joint space narrowing was seen in 41% of the subjects (bilateral in 69%) that led to pericapitate ankylosis in 25% of the cases [11]. Other investigators have also reported a tendency for distal interphalangeal, intertarsal, and cervical zygapophyseal ankylosis. Patients who have the chronic articular disease pattern can present with joint erosions making the differential diagnosis from RA problematic, especially in the absence of systemic signs and symptoms.
Diagnosis
The differential diagnosis of AOSD is extensive, especially at presentation, when the systemic symptoms predominate. Conditions such as infections, neoplasms, and autoimmune disorders should be ruled out before the diagnosis of AOSD can be confidently made. Viral syndromes (e.g., rubella, cytomegalovirus, Epstein-Barr virus, mumps, Coxsackievirus, adenovirus) can be excluded if the symptoms persist for more than 3 months and serologies are negative. Neoplastic disorders that can mimic AOSD include leukemia and lymphoma, such as angioimmunoblastic lymphoma. However, the clinical presentation can differ substantially, with atypical rashes and/or isolated lymph node enlargement. At times bone marrow and/or lymph node biopsy may be needed to differentiate these entities. Common mimickers of AOSD include reactive arthritis and the other spondyloarthropathies, hemophagocytic syndrome, dermatomyositis, Kikuchi’s syndrome, Sweet’s syndrome, granulomatous disorders, and the vasculitides. Other differentials of AOSD are the periodic fever syndromes and, in particular, familial Mediterranean fever, hyper IgD syndrome (HIDS), and TNF receptor-associated periodic syndrome (TRAPS). Patients with familial Mediterranean fever often present with acute, self-limited episodes of fever accompanied by signs of peritonitis, pleuritis, synovitis, and erysipelas-like erythema. The disease commonly starts in childhood or early adolescence. A significant family history, clinical presentation, and response to colchicine can aid in making the correct diagnosis. This can be verified, in many cases, with genetic analysis for the MEFV gene. TRAPS and HIDS commonly start in childhood and also have strong familial distributions. However, adult-onset cases of those rare syndromes do exist, and the genetic tests that are commercially available can be helpful in excluding them, especially when atypical presentations are present.
Several classification criteria have been developed from retrospective data available on AOSD. One such study attempted to validate these classification criteria: Yamaguchi’s criteria were found to be the most sensitive (93.5%), followed by Cush’s (80.6%) and Calabro’s (80.6%) criteria (Table 19.1) [4, 34]. Of late, a French group has proposed a new set of criteria which takes into consideration the two new disease markers: serum ferritin and its glycosylated fraction (Table 19.2). This set provided a sensitivity of 80.6% and a specificity of 98.5%, which remains to be authenticated in a different population before becoming widely accepted [35].
Treatment
Therapeutic goals include controlling physical signs and symptoms of inflammation, which is typically associated with the improvement in laboratory parameters. Decisions about optimal therapy are influenced by the severity and chronicity of the disease and involve taking into consideration the adverse effect profile of the treatments, both long and short term, and the clinical response to initial therapies. The primary goal of therapy not only involves achieving control of acute symptoms but also preventing end-organ damage, including joint injury and major organ complications. The initial choice of therapy depends upon disease severity and extent of organ involvement. There has been a lack of head-to-head randomized controlled trials comparing different therapeutic modalities given the rarity of disease. Treatment options constantly keep evolving as more insight is gained into the pathogenesis of AOSD. Support for the use of biologics as first-line therapy in severe disease is based upon the published experience in AOSD and, especially, more extensive evidence supporting use of these agents in children with SJIA , since recent evidence supports that SJIA and AOSD are different chronological disease onsets of the same “Still’s disease.”
Mild-to-Moderate Disease
Patients with mild-to-moderate disease may present with fevers and rash, as well as with arthralgias or mild arthritis.
NSAIDs
While NSAIDs , such as naproxen or ibuprofen, were the first medications to be used in AOSD, their role today is a very limited one. Their use can be justified either as adjunct treatment or as monotherapy in mild, monocyclic cases for symptomatic relief. In addition to adverse effects that are commonly associated with NSAIDs, an association between the use of NSAIDs and macrophage activation syndrome (MAS) has been described, as well [11]. Patients responsive to NSAIDs alone, who remain asymptomatic for at least a month, could have NSAID doses gradually reduced.
Glucocorticoids
Glucocorticoids can be started immediately, as first-line treatment, or when NSAIDs are insufficient to control signs and symptoms of the disease. Glucocorticoid dose can be gradually tapered once disease activity is controlled. Oral prednisone is typically initiated at a dose of 0.5-1 mg/kg per day, depending on the severity of disease. Intravenous, high-dose steroids are reserved for those with refractory disease [2]. Approximately 70% of patients respond to glucocorticoids alone or to glucocorticoids used after a trial of NSAIDs [36]. In addition to systemic steroids, those with one or two inflamed joints despite systemic therapy may benefit from intraarticular glucocorticoid injections.
DMARDs
Disease-modifying antirheumatic drugs (DMARDs) are typically used in the event of inadequate response to corticosteroids or as steroid-sparing medications. Methotrexate remains the first-line steroid-sparing agent in AOSD and is also a useful agent to treat Still’s arthritis. Methotrexate can result in complete remission in up to 70% of patients and is effective in corticosteroid weaning [37]. Sulfasalazine is contraindicated because of lack of efficacy and association with MAS development.
Moderate-to-Severe Disease
Internal organ damage and debilitating joint symptoms with radiographic are characteristic of moderate-to-severe disease. Severe disease is defined as refractory disease, especially when terminal organ and/or life-threatening involvement exists, such as MAS, severe hepatic injury, cardiac involvement with/or without tamponade, or disseminated intravascular coagulation (DIC). Biologic therapies such as IL-1 inhibitors (anakinra, canakinumab) and IL-6 receptor antagonists (tocilizumab) are used as first line or second line for SJIA often without any prior use of corticosteroids or traditional DMARDs and that practice may be justified in moderate-to-severe AOSD as well. Small series have reported some efficacy of IVIG when used early in the course of AOSD [38, 39].
IL-1 Inhibitors
Target biologic agents have been historically reserved for refractory AOSD (Fig. 19.1) [40]. These agents include a recombinant antagonist of the IL-1 receptor (IL-1Ra, anakinra), a human monoclonal antibody directed against IL-1β (canakinumab), and a soluble IL-1 trap fusion protein (rilonacept).

Proposed step-up therapeutic strategies for AOSD
Anakinra is particularly efficient in the rapid relief of the systemic symptoms. Several retrospective case series and one open-labeled prospective randomized trial have evaluated the use of anakinra in AOSD patients [41, 42]. Anakinra is approved as a daily subcutaneous injection (100 mg) in RA. In AOSD, higher dose may be necessary, since anakinra has a very short half-life (4–6 h). While anakinra does not work in all AOSD cases, for the ones it does work, its onset of action is very rapid. In many cases, resolutions of systemic symptoms and normalization of inflammatory markers were reported within 2 weeks of anakinra use, allowing for rapid tapering of corticosteroids, if they were ever used in the first place. However, relapses are not uncommon with the cessation of these agents. In certain patients, gradual reduction in dose has enabled the weaning of anakinra. Daily anakinra injections are often complicated by frequent injection site reactions. If response to anakinra is incomplete or tolerability issues ensue, rilonacept and/or canakinumab can be considered. They have longer half-lives and can be administered at greater intervals, once or twice weekly for rilonacept and every 4–8 weeks for canakinumab, respectively. Additional supplemental data is available in the setting of SJIA. Swart and associates reviewed data pertaining to 140 children with SJIA treated with anakinra [43]. Systemic symptoms resolved in 98% of the patients, and fatigue and well-being improved in 93% of cases. Arthritis improved in 66% of the cases in time. Absolute disease remission was mostly detected in patients with systemic symptoms, less arthritis, and a shorter duration of disease. Similar findings were corroborated in a large, multicenter, randomized, placebo controlled trial by Quartier et al. [44]. Sample population included 24 patients with a systemic-onset JIA for duration of more than 6 months and steroid dependency. In 1 month, anakinra was effective in 8 out of 12 patients (versus one in the placebo group) who reached the modified American College of Rheumatology (ACR) Pediatric 30 score. After 2 months, majority of patients (9/10) who had been switched to anakinra were also responders. Nigrovic and associates studied 46 patients who received anakinra as first-line treatment. Rapid resolution of systemic symptoms was observed in about 95% of cases, along with a supplementary preventive effect on refractory arthritis in almost 90% of the patients. Based on these results, it was postulated that there could be a benefit for IL-1 blockade therapy in initial phase of the disease (i.e., within 6 months after onset) [45]. These findings were also further reinforced by Vaster et al. in 2014 [46]. In a prospective series of 20 patients who received anakinra as first-line therapy, 85% of the patients showed an American College of Rheumatology Pediatric 90 score response or had inactive disease within 3 months. Overall, 75% of the patients treated with anakinra achieved remission. These results clearly indicate that IL-1 blockade has an early place in the treatment strategy .
IL-6 Inhibitors
Tocilizumab, humanized monoclonal antibody directed against IL-6 receptor , is used in refractory AOSD. This agent has been studied with randomized placebo-controlled trials in SJIA patients but not yet in AOSD. Effects of tocilizumab have been described to persist for ≥6 months after its discontinuation. Tocilizumab appears to have a marked corticosteroid-sparing effect and has a good safety and tolerance profile [47]. Most of the early data was with tocilizumab administered intravenously (IV) at a dosage of 5–8 mg/kg body weight every 2–4 weeks. Nevertheless, larger randomized studies are still needed to further determine the optimal therapeutic scheme for tocilizumab, i.e., optimal dosing, interval, and duration of treatment for both the intravenous but also the subcutaneous route of administration.
TNF-Inhibitors
Although those agents were the first biologics to be used in refractory AOSD, these agents are no longer recommended as first-line treatment in AOSD. Anti-TNF-α agents, in particular infliximab, etanercept, and adalimumab, have been used to treat refractory AOSD, but data on adalimumab are limited to a few cases [48]. Although complete resolution of symptoms has been observed, efficacy of the TNF-inhibitors has been mostly limited to Still’s arthritis. Efficacy was better with the monoclonal antibodies, as compared to the soluble receptor, and switching from one agent to another had no additional effect [19]. Moreover, in one published series, two patients who were started on etanercept and adalimumab developed MAS that could be allied with the initiation of therapy [49, 50]. Overall, TNF-α blockers should be considered for the treatment of chronic polyarticular disease, after the use of IL-1 and/or IL-6 inhibitors.
NSAIDs, Corticosteroids, and Traditional DMARDs
Non-refractory disease comprises of monocyclic and polycyclic AOSD. NSAIDs can be used in monocyclic course of AOSD without major systemic or articular involvement. Preferred NSAID is high-dose indomethacin (150–200 mg/day). Corticosteroids should be started promptly once the diagnosis is confirmed. Usually, corticosteroid therapy starts at a dosage of 0.5–1 mg/kg/day. Pulse dose methylprednisolone is used if there is severe visceral involvement or there is suspicion of MAS complicating the clinical presentation. Higher dosages seem to be more efficient in controlling the disease and lessening the number of relapses. Tapering of corticosteroids can start after 4–6 weeks, when symptoms have resolved, and biological parameters have returned to baseline. Methotrexate (MTX) could be considered early for its steroid-sparing effect. Typically, methotrexate (7.5–20 mg/week) enables complete remission of the disease (70%) and limits frequent corticosteroid use. Blood count and renal and hepatic function should be monitored before initiation of methotrexate and then at monthly intervals. Alternative DMARDs may be use in the event of methotrexate failure; however, more recent literature suggests better results with targeted biologic treatment.
Treatments Under Development
IL-18 Inhibition/Tadekinig Alpha
An open-label, multicenter, phase II study of subcutaneous Tadekinig alfa (IL-18BP) in patients with AOSD was recently published and showed promise. Tadekinig alfa is the drug name for recombinant human interleukin-18-binding protein (IL-18BP). This study was based on the principle that high levels of IL-18 were noted during active flares of AOSD. Ten patients were assigned to receive 80 mg tadekinig alfa, and 13 patients received the 160 mg dose. At week 3, 5 of 10 patients receiving 80 mg and 6 of 12 patients receiving 160 mg achieved the predefined response criteria. The agent was overall well tolerated with the exception of one case of optic neuropathy [51].
Summary
It is becoming exceedingly evident that AOSD patients fall into two distinct subsets, i.e., those presenting with systemic manifestations and those presenting with prominent articular manifestations. In addition, these findings are also reinforced by molecular evidence, cytokine profiles, clinical course, and response to therapy. Predictors for a prominent articular pattern include female sex, proximal arthritis at disease onset, thrombocytosis, and corticosteroid dependency, whereas high fever, transaminitis, or elevated acute phase reactants are more likely to be connected with a systemic pattern of AOSD [14, 52]. Alternative evidence to identify the systemic subtype of AOSD are the following: thrombocytopenia, RHL, and hyperferritinemia. IL-18, interferon-γ, IL-10, and IL-4 are typically associated with systemic AOSD, whereas IL-6, IL-17, and IL-23 are associated with arthritic AOSD [53]. This dichotomy aids in management as patients fall into one of the two categories and should benefit from different therapies.
Patients with systemic symptom predominant AOSD often benefit from systemic corticosteroid therapy. From traditional DMARDs, methotrexate has been studied the most and may continue to have a role as steroid-sparing and in the treatment of chronic articular disease. IL-1 antagonists should be considered first line in severe or refractory AOSD, either alone or in initial combination with systemic corticosteroids when necessary. Regularly reported side effects with anakinra include injection site reactions. Longer acting IL-1 inhibitors (rilonacept or canakinumab) may play a role in refractory disease or when tolerability issues exist with anakinra. Tocilizumab, the IL-6 receptor antagonist, has shown efficacy in both systemic and articular disease predominant AOSD, even in cases where IL-1 inhibition has been unsuccessful. In contrast to IL-1 and IL-6 inhibitors, anti-TNF-α agents typically have less sustained effect on systemic symptoms, but they may have a limited role for the chronic inflammatory polyarthritis.
In an effort to standardize therapeutic management and evaluate comparative efficacy in an observational setting, the Childhood Arthritis and Rheumatology Research Alliance has developed four consensus treatment plans for SJIA [54]. This includes glucocorticoid plan, a methotrexate plan, an anakinra plan, and a tocilizumab plan. Since no guidelines are available yet in AOSD, this consensus may act as a rough guide for its treatment, as it coincides with the published clinical experience. New therapeutic agents are being developed for AOSD, and future randomized, controlled trials will fill the knowledge gap that currently exists.
References
Still GF. On a form of chronic joint disease in children. Med Chir Trans. 1897;80:47–60. 9.
Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22(5):773–92.
Efthimiou P, et al. Adult onset Still’s disease and autoinflammation. Int J Inflam. 2012;2012:964751.
Yamaguchi M, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19(3):424–30.
van de Putte LB, Wouters JM. Adult-onset Still’s disease. Baillieres Clin Rheumatol. 1991;5(2):263–75.
Ohta A, et al. Adult Still’s disease: review of 228 cases from the literature. J Rheumatol. 1987;14(6):1139–46.
Magadur-Joly G, et al. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54(7):587–90.
Wakai K, et al. Estimated prevalence and incidence of adult Still’s disease: findings by a nationwide epidemiological survey in Japan. J Epidemiol. 1997;7(4):221–5.
Sampalis JS, et al. Risk factors for adult Still’s disease. J Rheumatol. 1996;23(12):2049–54.
Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis. 2006;65(5):564–72.
Pouchot J, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70(2):118–36.
Terkeltaub R, et al. HLA—Bw35 and prognosis in adult Still’s disease. Arthritis Rheum. 1981;24(12):1469–72.
Wouters JMGW, Reekers P, van de Putte LBA. Adult-onset still’s disease. Disease course and HLA associations. Arthritis Rheum. 1986;29(3):415–8.
Fujii T, et al. Cytokine and immunogenetic profiles in Japanese patients with adult Still’s disease. Association with chronic articular disease. Rheumatology (Oxford). 2001;40(12):1398–404.
Joung CI, et al. Association between the HLA-DRB1 gene and clinical features of systemic sclerosis in Korea. Scand J Rheumatol. 2006;35(1):39–43.
Hoshino T, et al. Elevated serum interleukin 6, interferon-gamma, and tumor necrosis factor-alpha levels in patients with adult Still's disease. J Rheumatol. 1998;25(2):396–8.
Kawashima M, et al. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001;44(3):550–60.
Sugiura T, et al. Association between adult-onset Still’s disease and interleukin-18 gene polymorphisms. Genes Immun. 2002;3(7):394–9.
Gerfaud-Valentin M, et al. Adult-onset still disease: manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine (Baltimore). 2014;93(2):91–9.
Elkon KB, et al. Adult-onset Still’s disease. Twenty-year follow up and further studies of patients with active disease. Arthritis Rheum. 1982;25(6):647–54.
Valente RM, Banks PM, Conn DL. Characterization of lymph node histology in adult onset Still’s disease. J Rheumatol. 1989;16(3):349–54.
Silva CA, et al. Macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. J Pediatr (Rio J). 2004;80(6):517–22.
Ramanan AV, Schneider R. Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003;30(2):401–3.
Sawar H, Espinoza LR, Gedalia A. Macrophage activation syndrome and etanercept in children with systemic juvenile rheumatoid arthritis. J Rheumatol. 2004;31(3):623. author reply 623-4.
Tristano AG. Macrophage activation syndrome: a frequent but under-diagnosed complication associated with rheumatic diseases. Med Sci Monit. 2008;14(3):RA27–36.
Arlet JB, et al. Reactive haemophagocytic syndrome in adult onset Still’s disease: report of 6 patients and review of the literature. Ann Rheum Dis. 2006;65:1596.
Singh S, Samant R, Joshi VR. Adult onset Still’s disease: a study of 14 cases. Clin Rheumatol. 2008;27(1):35–9.
Durand M, et al. Macrophage activation syndrome treated with anakinra. J Rheumatol. 2010;37(4):879–80.
Arlet JB, et al. Reactive haemophagocytic syndrome in adult-onset Still’s disease: a report of six patients and a review of the literature. Ann Rheum Dis. 2006;65(12):1596–601.
Ravelli A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146(5):598–604.
Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421–6.
Davi S, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38(4):764–8.
Wouters JM, van de Putte LB. Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med. 1986;61(235):1055–65.
Cush JJ. Adult-onset Still’s disease. Bull Rheum Dis. 2000;49(6):1–4.
Fautrel B, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002;81(3):194–200.
Franchini S, et al. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthritis Rheum. 2010;62(8):2530–5.
Fautrel B, et al. Corticosteroid sparing effect of low dose methotrexate treatment in adult Still’s disease. J Rheumatol. 1999;26(2):373–8.
Vignes S, et al. Intravenous immunoglobulin in adult Still’s disease refractory to non-steroidal anti-inflammatory drugs. Clin Exp Rheumatol. 1998;16(3):295–8.
Permal S, et al. Treatment of Still disease in adults with intravenous immunoglobulins. Rev Med Interne. 1995;16(4):250–4.
Kadavath S, Efthimiou P. Adult-onset Still’s disease-pathogenesis, clinical manifestations, and new treatment options. Ann Med. 2015;47(1):6–14.
Lequerre T, et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: preliminary experience in France. Ann Rheum Dis. 2008;67(3):302–8.
Fitzgerald AA, et al. Rapid responses to anakinra in patients with refractory adult-onset Still’s disease. Arthritis Rheum. 2005;52(6):1794–803.
Swart JF, et al. The efficacy and safety of interleukin-1-receptor antagonist anakinra in the treatment of systemic juvenile idiopathic arthritis. Expert Opin Biol Ther. 2010;10(12):1743–52.
Quartier P, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70(5):747–54.
Nigrovic PA, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–55.
Vastert SJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol. 2014;66(4):1034–43.
De Benedetti F, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–95.
Benucci M, et al. Adalimumab (anti-TNF-alpha) therapy to improve the clinical course of adult-onset Still’s disease: the first case report. Clin Exp Rheumatol. 2005;23(5):733.
Kaneko K, et al. Exacerbation of adult-onset Still’s disease, possibly related to elevation of serum tumor necrosis factor-alpha after etanercept administration. Int J Rheum Dis. 2010;13(4):e67–9.
Agarwal S, et al. A rare trigger for macrophage activation syndrome. Rheumatol Int. 2011;31(3):405–7.
Gabay C, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann Rheum Dis. 2018;77:840–47.
Ichida H, et al. Clinical manifestations of adult-onset Still’s disease presenting with erosive arthritis: association with low levels of ferritin and Interleukin-18. Arthritis Care Res (Hoboken). 2014;66(4):642–6.
Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61(2):345–8.
DeWitt EM, et al. Consensus treatment plans for new-onset systemic Juvenile idiopathic arthritis. Arthritis Care Res. 2012;64(7):1001–10.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Efthimiou, P., Yadlapati, S. (2019). Adult-Onset Still’s Disease. In: Efthimiou, P. (eds) Auto-Inflammatory Syndromes. Springer, Cham. https://doi.org/10.1007/978-3-319-96929-9_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-96929-9_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-96928-2
Online ISBN: 978-3-319-96929-9
eBook Packages: MedicineMedicine (R0)