
Abstract
The High-performance High-throuhput Virtual Screening Framework (HHVSF) has been developed to accelerate High-Throughput Virtual Screening (HTVS) on high-performance computers. Task management and data management are two core components in HHVSF. Fine-grained computing resources are configured to support serial or threaded applications. Each task gets the input file from database through a preemptive algorithm and the failed tasks can be found and corrected. NoSQL database MongoDB is used as the data repository engine. Data is mobilized between the RAMDISK in computing node and the database. Data analysis is carried out after the computing process, and the results are stored in the database. Among the most popular molecular docking and molecular structure similarity packages, Autodock_vina (ADV) and WEGA were chosen to carry out experiments. Results show that when ADV was used for molecular docking, 10 million molecules were screened and analyzed in 22.31 h with 16000 cores, and the throughput reached up to 1324 molecules per second, averaging 145 molecules per second during the steady-running process. For WEGA, 958 million conformations were screened and analyzed in 34.12 min with 4000 cores, of which throughput reached up to 9448 molecules per second, 6430 molecules per second on average.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
- High-Throughput Virtual Screening
- Drug discovery
- High-Performance Computing
- Molecular docking
- Molecular structure similarity
1 Introduction
Computational methodology has become a significant component in pharmaceutical industry for drug design and discovery [1,2,3,4]. Typically, molecular docking and molecular structure similarity are two frequently used computational approaches. High-Throughput Virtual Screening (HTVS) is known to computationally screen large compound libraries. These libraries contain a huge number of small-molecules varying from tens of thousands to millions, require a high volume of lots-of-small files scenario for a virtual screening campaign. With the development of high-performance computers, the virtual drug screening is accelerating. However, HTVS still faces challenges while a large scale virtual screening application is executed on High-Performance Computing (HPC) resources, such as distributing massive tasks, analyzing lots-of-small molecular structure files, and implementing fault tolerance.
Tools have been developed to accelerate the process of HTVS on HPC resources. Falkon [5] is a lightweight execution framework to enable loosely coupled program to run on peta-scale systems. The benchmark [6] shows that DOCK5 can scale up to 116,000 cores with high efficiency by Falkon. VinaMPI is a MPI version program based on ADV package, which uses a large number of cores to speed-up individual docking tasks. VinaMPI successfully ran on 84,672 cores on Kraken supercomputer and efficiently reduce the total time-to-completion. While all the above works focus on performance and efficiency of distributing tasks, ignoring the whole HTVS process, for instance, robustness, recoverability and result analysis. FireWorks (FWS) [7] is a workflow software for high-throughput calculation running on supercomputer, effectively solve the problem of concurrent task distribution and fault tolerance management, and provide an intuitive graphical interface. However, FWS pays more attention on versatility and usability. DVSDMS [8] is a distributed virtual screening data management system, only focusing on high-throughput docking process in the data management issues. Therefore, the architecture of high-performance computers, as well as the computational characteristics of the application, needs to be considered to design the framework for HTVS on high-performance computers.
In this work, we report a general framework - High-performance High-throughput Virtual Screening Framework (HHVSF) - to enable large-scale, multitasking and small-size input and output (IO) applications to efficiently execute on HPC resources. This framework contains task management and data management systems, which can handle thousands of tasks, manage a large volume of lots-of-small files, and reduce the long processing time for analyzing. The purpose of HHVSF is to provide high computational performance based on portability, availability, serviceability and stability (PASS).
2 Experimental and Computational Details
The framework of HHVSF is comprised of two parts: task management and distributed data management (see Fig. 1). In order to access and store data efficiently and flexibly, the executions of a program are coupled loosely by MongoDB C driver, while the application codes do not need to be modified. The following three subsections document the overall framework of HHVSF, the simulation parameters and the data sets of the experiments are introduced at the end of this section. ADV [9] and WEGA [10] are chosen as typical applications to carry out the experiments, and others can be integrated into the HHVSF in similar way.

The hardware and relevant operations in HHVSF.
2.1 Task Management
The followings are mainly considered in the task management system: two-level task scheduling, preemptive scheduling algorithm for worker and failed tasks recovery.
2.1.1 Task Scheduling
HTVS employs massive computing resources to support a large number of independent computing tasks. Because most molecular docking and molecular structure similarity tools, for instance, ADV, Gold [11], Glide [12], FlexX [13] and WEGA, are serial or threaded codes, these computing tasks are typical fine-grained Many-Task Computing (MTC) [6]. Such MTC tasks cannot take full advantage of the static scheduling solution with coarse scheduling granularity, while most traditional large-scale HPC resources are configured with coarse scheduling granularity under a control of batch queuing system such as Simple Linux Utility for Resource Management (SLURM) [14], Portable Batch System (PBS)/Torque [15], and Sun Grid Engine (SGE) [16].
Multi-level scheduling method can effectively solve the different application requirements for scheduling granularity, while maintaining the unified management of computing resources. The first level scheduler applies for a number of resources to the second level for task distribution. The second level scheduler can refine the computing resources and then distributes the tasks. HTCondor [17] is chosen to be the second level scheduler to dispatch tasks. HTCondor is full-featured batch workload management system for coordinating a large number of independent serial or parallel jobs in High-Throughput Computing (HTC) environment. We configure the HTCondor with one core per slot to provide more flexible task scheduling.
2.1.2 Preemptive Scheduling Algorithm
Molecular docking and molecular structure similarity are typical MTC applications, while maintaining millions of tasks by HTCondor to screen a large database with millions of ligands or conformers is still a touch work. Thus, we transform MTC into HTC by wrapping ADV or WEGA program with MongoDB C driver (version 1.4.2) as a worker. Each worker accesses database preemptively to get input files until all data is traversed. MongoDB provides atomic operation with “inc” to ensure data security when multitudinous workers start concurrently, so that each worker can get unique job. After the worker obtains data from the database, the data is written to a file and stored on the local file system implemented in RAMDISK. The kernel function’s computational procedure is shown in the Fig. 2.

The pseudo code of vina_wrapper and wega_wrapper.
2.1.3 Fault Tolerance
Fault tolerance in this context can be simplified as the ability to automatically restart a task when the original run fails. When the HTVS scales to millions of tasks or run a long-time task, it is easy to get failures for bad input parameters, such as computing node fault, IO blocking, and network latency. There are two ways to consider fault tolerance in this case, one is monitoring the job status during the running by job management system, another is making a successful or failed tag on each task after the task is finished. HTCondor provides checkpoint mechanism in the standard universe by using condor_compile to relink the execution with the HTCondor libraries, while those coupled program vina_wrapper and wega_wrapper, containing system calls like system(), cannot provide check pointing services with HTCondor. As a result, we choose the second method. When a worker calls the execution of ADV or WEGA successfully, a tag that represents the task status will insert into the corresponding document in MongoDB database. After the job is finished, it needs to check the document’s failed tag and then restart the failed jobs.
2.2 Data Management
Data storage, data relocation and data analysis are the bottlenecks when a virtual screening is scaled up to handle millions of tasks on thousands of cores. Such scattered lots-of-small files can overburden the shared file system with abundant IO operations if the plain files are accessed and stored directly. Database offers an attractive solution to both the storage and the data querying. In our framework, we avoid using shared file system by replacing it with the combination of MongoDB and local RAMDISK. The IO files are stored in MongoDB, while they are cached in the local RAMDISK during computation. The following three subsections describe the details on the data storage, data relocation and data analysis in HHVSF.
2.2.1 NoSQL Database for Storage
Chemical databases are the critical components of HTVS, which provide the basic information to build knowledge-based models for discovering and designing drug. Such as, PubChem [18], ZINC [19], ChEMBL [20], ChemDB [21], contain millions of compounds, provide shape data, physical properties, biological activities, and other information for pharmaceutical evaluations. Currently, many molecular docking programs and molecular structure similarity algorithms read the input and store the output in plain text files, which is not suitable for management when data grow up rapidly. Maintaining and analyzing such data are difficult.
MongoDB [22] is used as the data repository engine, which is a high performance, high availability, automatic scaling, open source NoSQL (Not Only SQL) database. This architecture is suitable for sparse and document-like data storage. By using MongoDB “index”, molecules can be queried and ranked easily. In addition, MongoDB uses “sharding” (a method for distributing data across multiple machines) to support deployments with large data sets in high throughput manner, enhancing the computational performance by balancing query loading as the database growing. Finally, MongDB accepts big data up to 16 MB. MongDB is employed for WEGA to access the big conformation SDF input file.
2.2.2 Data Relocation
ADV and WEGA involve in processing large sized plain text files. Without modifying their source codes, the programs have to process huge number of small molecular structure files by moving on the shared file disks when screening a large-scale compound library. Hence, the RAMDISK in computing nodes are used to temporarily store the IO files needed by the applications (see Fig. 3). The RAMDISK provides high-speed, low-latency IO operations for handling lots-of-small files, while the high storage capacity of shared file disk is still fully occupied to store the resulting data. By relocating data between MongDB and RAMDISK, the IO pressure for shared file storage is effectively mitigated.

The flowchart of the data relocation.
2.2.3 Data Analysis
For virtual screening using molecular docking and molecular structure similarity approaches, scores and molecular similarities have to be calculated before ranking the molecules in a large sized compound library. In consideration of high-performance computing systems with shared file storage, it is necessary to avoid IO overloading problems which are caused by a great number of small files. Thus, it is not wise to analyze the output files on the shared storage disk. When the computations are accomplished in the RAMDISK, the output files are analyzed and the compounds in the library are ranked based upon scores or similarities. This protocol minimizes the IO stress when the number of small files increases dramatically.
2.3 Simulation Parameters and Data Sets
2.3.1 ADV
About twenty million ligands with mol2 format file were obtained from ZINC database FTP server (http://zinc.docking.org/db/bysubset/6/). The pymongo (version 3.2.1) Python library was used for database operations. A python script was developed to insert mol2 files into MongoDB. MGLTools (version 1.5.6) was used to convert mol2 files into pdbqt file for docking. We prepared five different sized data sets (from zinc_ligand_1~5), as shown in Table 2. All data sets are sorted by heavy atom number arranged in ascending order. After finishing molecular docking, the result pdbqt file format was converted to mol file format by Open Babel package [23] (version 2.4.0).
The protein target is a crystal structure of the alpha subunit of glycyl tRNA synthetase (PDB codes: 5F5 W). The (x, y, z) coordinates (in Å) for the center of the docking site is (−94.666, 51.401, 8.991), and the side of the cubic box is (14, 18, 12). The argument of num_modes is set to 1.
2.3.2 WEGA
A SDF file containing about twenty million molecules was obtained from ZINC database FTP server. Approximately 958 million conformers were generated from the SDF file using the CAESAR algorithm [24] in discovery studio (version 3.5) [25] for shape-feature similarity calculation. In order to take advantage of the 16 MB storage space in MongoDB, the conformer files were split into smaller files which occupied 15 MB for each file, and then inserted into the database. Table 2 gives two data sets for WEGA (zinc_conformer_1 and zinc_conformer_2).
The query molecule is 4-amino-1-[4,5-dihydroxy-3-(2-hydroxyethyl)-1-cyclopent-2-enyl]-pyrimidin-2-one (ZINC ID: ZINC03834084). The method for molecular overlay is set to 2 (combing the shape similarity and pharmacophore similarity). Each SDF file corresponds up to 100 similar molecules. The Table 1 shows the detailed information of the data sets which are used throughout the article.
All tests run on Tianhe-2 (MilkyWay-2) supercomputer, which consists of 16,000 computing nodes connected via the TH Express-2 interconnect. Each computing node is equipped with two Intel Xeon E5-2692 CPUs (12-core, 2.2 GHz), and configured with 64 GB memory. The storage subsystem contains 64 storage servers with a total capacity of 12.4 PB. The LUSTRE storage architecture is used as a site-wide global file system.
3 Results and Discussion
3.1 Load Balance
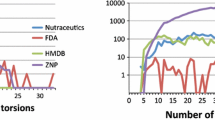
Time for screening a compound ranges from minutes to hours depending on the complexity of the molecular structure. Reports [9, 26] indicate that the compound complexity, for instance, number of active torsions or number of heavy atoms, dominates the computing time of molecular docking. In order to determine the relation between the number of heavy atoms computing time and the computing complexity, the zinc_lignad_5 data set was chosen to record the number of heavy atom in a ligand and computing time, as depicted in Fig. 4. The number of heavy atoms presents a linear relationship with time (logarithmic form), which indicates more heavy atoms in a small molecule requires longer computing time. For zinc_ligand_2~5 data sets are scaled down from zinc_ligand_1 by a certain percentage, the other data sets will also benefit from this approach. Based on this information, the zinc_ligand_4 data set was tested on 8,000 cores. Figure 5a and b demonstrate that the average computing time per worker is reduced by 8.83 s when load balancing protocol was used.

The number of heavy atoms in a compound (x-axis), the computing time (logarithmic form) of a molecular docking (y-axis). The results are based upon zinc_ligand_5 data set.

(a) The computing time of each worker without load balancing. The red line is the average computing time per task. (b) The computing time per worker with load balance. The red line is the average computing time per task. (Color figure online)
3.2 Throughput with Data
The MongoDB monitors the status of a running instance. When a worker starts, a “connection” operation is activated by MongoDB’s server. After the computing task is accomplished, the resulting data (score, structural conformation, and running status) will be inserted into MongoDB’s collection. Figure 6 shows the “connection” and “insert” operations of MongoDB’s server every second with vina_wrapper during the whole computing period, the points of inverted triangle clearly reveal the three stages of running tasks: startup, steady-running and finish. The total time during the startup was 1,396 s to start 16,000 workers, averaging 11 tasks per second. The points of rhombus become higher gradually as time progresses, reaching up to 1,324 molecules per second and averaging 145 molecules per second. Table 2 gives the results for other data sets. As for WEGA, Fig. 7 shows that the data throughput can reach up to 9,448 molecules per second, averaging 6,430 molecules per second, indicating a high performance and a high data throughput.

The number of “insert” operation and “connection” operation in MongoDB’s server when running ADV application. The zinc_ligand_3 data set was used to run on 16,000 cores.

The number of “insert” operation and “connection” operation in MongoDB’s server when running WEGA application. The zinc_conformer_1 data set was used to run on 4,000 cores.
3.3 Scalability
To test scalability, we perform the experiments of speedup and parallel efficiency with zinc_ligand_4 data set and zinc_ligand_3 data set. Figure 8a shows zinc_ligand_4 data set can be scaled to 8,000 cores with parallel efficiency of 0.84, and the zinc_ligand_4 data set can be scaled to 16,000 cores with parallel efficiency of 0.83 (see Fig. 8b). It is shown that the parallel efficiency decreases sharply when computing resource is scaled up to more than 8,000 cores. This is because more cores represent more workers, and thus, more time will be cost by HTCondor to start those workers.

(a) Speedup (right triangle) and parallel efficiency (read dot) of molecular docking experiment on zinc_ligand_4 data set. (b) Speedup (block dot) and parallel efficiency (upper triangle) of molecular docking experiment on zinc_ligand_3 data set.
3.4 Fault Tolerance
Fault tolerance management can avoid task failures due to external environments, for instances, compute node fault, network blocking, IO latency, etc. Table 3 gives the information of failed tasks on different data sets. The zinc_ligand_2 data set has a high failure rate than others due to ten million ligands containing more ligands with high molecular weight which are not suitable for docking space of the protein (PDB code: 5W5F). In addition, longer calculations can lead to higher failures. The zinc_conformer_1 data set has fewer files (95712 SDF files in total) and less computing time, as a result, produces a low failure rate.
4 Conclusions
HHVSF includes task management and relocating data management, and supports the high-throughput applications of large-scale, multitasking and small sized IO files running on HPC resources. There are two types of virtual drug screening applications: (1) computation-intensive applications (such as molecular docking), and (2) data-intensive applications (such as molecular structure similarity based virtual screening campaigns). With HHVSF, two types of applications can run on Tianhe-2 supercomputer with high performance. Testing results show that when use ADV for molecular docking, the protein target (PDB code: 5W5F) was used to screen nearly half of compounds from the ZINC database within one day on 16,000 cores. For WEGA, 958 million conformations were screened by using about a half hour on 4,000 cores. The ranked ligands or conformers can be accessed in milliseconds by specifying the “sort” method from the database. Meanwhile, the IO pressure of shared file storage affected by lots-of-small files in HPC resources can be mitigated. Thus, HHVSF can significantly accelerate HTVS campaigns on HPC resources.
References
Manglik, A., Lin, H., Aryal, D.K., Mccorvy, J.D., Dengler, D., Corder, G., Levit, A., Kling, R.C., Bernat, V., Hübner, H.: Structure-based discovery of opioid analgesics with reduced side effects. Nature 537(7619), 1 (2016)
Rodrigues, T., Reker, D., Schneider, P., Schneider, G.: Counting on natural products for drug design. Nat. Chem. 8(6), 531–541 (2016)
Hao, G.F., Wang, F., Li, H., Zhu, X.L., Yang, W.C., Huang, L.S., Wu, J.W., Berry, E.A., Yang, G.F.: Computational discovery of picomolar Q(o) site inhibitors of cytochrome bc1 complex. J. Am. Chem. Soc. 134(27), 11168–11176 (2012)
Forli, S., Huey, R., Pique, M.E., Sanner, M.F., Goodsell, D.S., Olson, A.J.: Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 11(5), 905 (2016)
Raicu, I.: Falkon: a Fast and Light-weight tasK executiON framework, p. 43 (2007)
Raicu, I., Zhao, Z., Wilde, M., Foster, I., Beckman, P., Iskra, K., Clifford, B.: Toward loosely coupled programming on petascale systems, pp. 1–12 (2008)
Jain, A., Ong, S.P., Chen, W., Medasani, B., Qu, X., Kocher, M., Brafman, M., Petretto, G., Rignanese, G.M., Hautier, G.: FireWorks: a dynamic workflow system designed for high-throughput applications. Concurr. Comput. Pract. Exp. 27(17), 5037–5059 (2015)
Zhou, T., Caflisch, A.: Data management system for distributed virtual screening. J. Chem. Inf. Model. 49(1), 145–152 (2009)
Trott, O., Olson, A.J.: AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31(2), 455–461 (2010)
Yan, X., Li, J., Liu, Z., Zheng, M., Ge, H., Xu, J.: Enhancing molecular shape comparison by weighted gaussian functions. J. Chem. Inf. Model. 53(8), 1967–1978 (2013)
Jones, G., Willett, P., Glen, R.C., Leach, A.R., Taylor, R.: Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267(3), 727–748 (1997)
Friesner, R.A., Banks, J.L., Murphy, R.B., Halgren, T.A., Klicic, J.J., Mainz, D.T., Repasky, M.P., Knoll, E.H., Shelley, M., Perry, J.K.: Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47(7), 1739–1749 (2004)
Rarey, M., Kramer, B., Lengauer, T., Klebe, G.: A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 261(3), 470–489 (1996)
Yoo, A.B., Jette, M.A., Grondona, M.: SLURM: simple linux utility for resource management. In: Feitelson, D., Rudolph, L., Schwiegelshohn, U. (eds.) JSSPP 2003. LNCS, vol. 2862, pp. 44–60. Springer, Heidelberg (2003). https://doi.org/10.1007/10968987_3
Bode, B., Halstead, D.M., Kendall, R., Lei, Z., Jackson, D.: The Portable batch scheduler and the Maui Scheduler on Linux Clusters (2000)
Gentzsch, W.: Sun Grid Engine: Towards Creating a Compute Power Grid, pp. 35–36 (2001)
Thain, D., Tannenbaum, T., Livny, M.: Distributed computing in practice: the Condor experience: research articles. Concurr. Comput. Pract. Exp. 17(2–4), 323–356 (2010)
Wang, Y., Xiao, J., Suzek, T.O., Jian, Z., Wang, J., Bryant, S.H.: PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 37(Web Server issue), W623 (2009)
Irwin, J.J., Sterling, T., Mysinger, M.M., Bolstad, E.S., Coleman, R.G.: ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52(7), 1757–1768 (2012)
Gaulton, A., Bellis, L.J., Bento, A.P., Chambers, J., Hersey, A., Light, Y., Mcglinchey, S., Michalovich, D., Allazikani, B.: ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 40(Database issue), D1100 (2012)
Chen, J., Swamidass, S.J., Dou, Y., Bruand, J., Baldi, P.: ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics 21(22), 4133–4139 (2005)
Banker, K.: MongoDB in Action. Manning Publications Co., Greenwich (2011)
O’Boyle, N.M., Banck, M., James, C.A., Morley, C., Vandermeersch, T., Hutchison, G.R.: Open babel: an open chemical toolbox. J. Cheminform. 3(1), 1–14 (2011)
Li, J., Ehlers, T., Sutter, J., Varma-O’Brien, S., Kirchmair, J.: CAESAR: a new conformer generation algorithm based on recursive buildup and local rotational symmetry consideration. J. Chem. Inf. Model. 47(5), 1923–1932 (2007)
Visualizer, D.S.: Release 3.5. Accelrys Inc., San Diego (2012)
Jaghoori, M.M., Bleijlevens, B., Olabarriaga, S.D.: 1001 ways to run AutoDock Vina for virtual screening. J. Comput. Aided Mol. Des. 30(3), 1–13 (2016)
Acknowledgments
We would like to thank Prof. Xin Yan for permission to use WEGA program to test. Helpful discussions with Guixin Guo, Lin Li, Wen Wan and technical assistance by HTCondor Team (University of Wisconsin-Madison) are gratefully acknowledged. This work was performed by the auspices of the NSFC (U1611261), GD Frontier & Key Techn, Innovation Program (2015B010109004).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made. The images or other third party material in this book are included in the book's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the book's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2018 The Author(s)
About this paper

Cite this paper
Chen, P., Yan, X., Li, J., Du, Y., Xu, J. (2018). HHVSF: A Framework to Accelerate Drug-Based High-Throughput Virtual Screening on High-Performance Computers. In: Yokota, R., Wu, W. (eds) Supercomputing Frontiers. SCFA 2018. Lecture Notes in Computer Science(), vol 10776. Springer, Cham. https://doi.org/10.1007/978-3-319-69953-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-69953-0_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-69952-3
Online ISBN: 978-3-319-69953-0
eBook Packages: Computer ScienceComputer Science (R0)