
Abstract
The pleura are composed of two layers, parietal and visceral layers, separated by a pleural space. The parietal pleuron is supplied by systemic vessels and drains into the right atrium via the azygos, hemiazygos, and internal mammary veins. The visceral pleuron is supplied by bronchial and pulmonary vessels and drains into the pulmonary veins.
You have full access to this open access chapter, Download chapter PDF
Keywords
- Cystic Fibrosis
- Obstructive Sleep Apnea
- Idiopathic Pulmonary Fibrosis
- Obstructive Sleep Apnea Syndrome
- Cardiac Sarcoidosis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
7.1 Pleural Diseases
The pleura are composed of two layers, parietal and visceral layers, separated by a pleural space. The parietal pleuron is supplied by systemic vessels and drains into the right atrium via the azygos, hemiazygos, and internal mammary veins. The visceral pleuron is supplied by bronchial and pulmonary vessels and drains into the pulmonary veins.
The pleural space normally contains interstitial fluid (1–5 mL) that is cleared by the parietal pleural lymphatic vessels. There is no direct communication between the visceral pleura lymphatics and the pleural space.
The pleura appear normally on radiographs only when the X-ray beam is tangentially set on the film. On radiographs, the pleura appear as fissures and junctional lines. Fissures are made up of two layers of visceral pleura. The normal parietal pleuron is never visualized on posteroanterior (PA) radiographs.
Different pathological conditions affecting the pleura can be diagnosed with confidence by PA chest radiographs alone. This topic discusses the main pathological pleural conditions with their typical radiologic manifestations.
7.1.1 Pleural Effusion
Pleural effusion is a condition characterized by abnormal fluid collection between the parietal and visceral pleura (excess pleural space fluid). The pleural fluid can be water (edematous effusion), blood (hemothorax), pus (empyema), tumor cells (malignant pleural effusion), or lymph (chylothorax).
Pathologically, pleural effusion is divided into serous or exudative according to the protein content after lab analysis. Serous plural effusion contains little protein content (<2.5 g/dL) and usually arises due to systemic disease like cardiac failure, nephrotic syndrome, or liver failure. Exudative pleural effusion contains high protein count (>2.5 g/dL) and usually arises due to inflammatory or infectious process like tuberculosis, malignancy, and acute pancreatitis.
Disruption of the thoracic duct due to lymphoma or a tumor can cause lymphatic blockage and leakage into the pleural space causing chylothorax. Malignant effusion typically results from metastasizing of the malignant cells into the pleural cavity via the parietal pleura lymphatics, and it is often massive.
Bronchopleural fistula is a condition characterized by opening of a bronchus into the pleural space. It can develop occasionally following thoracic surgery, infection, medical intervention, or malignancy. Bronchopleural fistula is seen in 2–3 % of postpneumonectomy cases.
Signs on Chest Radiographs
-
Obliteration of the lateral costophrenic angle with a meniscus like arc at the interface between the fluid and the chest wall in PA radiographs (Meniscus sign) (Fig. 7.1.1).
-
Obliteration of the posterior costophrenic angle in lateral radiographs (Fig. 7.1.1). This angle is more sensitive to plural effusion collection due to gravity effect. Up to 50 mL of fluid is necessary to obliterate the posterior costophrenic angle, and 200 mL is necessary to obliterate the lateral costophrenic angle.
-
Subpulmonic pleural effusion (SPE) is a pleural effusion that occurs below the lungs at the diaphragmatic surface. SPE does not obliterate the costophrenic angle, but it distorts the shape of the diaphragmatic dome, giving the impression of raised hemidiaphragm. You can suspect SPE in the left lung when the space between the gastric bubble and the lower lung margins increases up to 3 cm instead of usual few millimeters. Beside the raised hemidiaphragm, the lung appears to end early on PA radiographs (Fig. 7.1.2).
-
Encysted (loculated) pleural effusion is a localized encysted fluid at the fissures between lobes of the lung. It occurs usually at the right lung’s minor fissure, and it has biconvex contour mimicking a mass (Fig. 7.1.3). Very rarely, a benign form of mesothelioma can grow along the major or minor fissures mimicking encysted pleural effusion, a condition known as pseudotumor.
-
Parapneumonic effusion is an effusion that develops adjacent to pneumonias (empyema). Almost 30 % of patients with pneumonia develop pleural effusion and usually resolve with antibiotic therapy.
-
Mediastinal pleural effusion is a fluid collection around the mediastinum. It is an unusual condition, and when it occurs, it forms silhouette sign along the mediastinal borders causing mediastinal widening. Silhouette sign is a term used to describe any opacity within the chest radiograph that obliterates a mediastinal border.

Posteroanterior (a) and lateral (b) chest radiographs in two different patients with pleural effusion show meniscus sign with right pleural effusion obliterating the lateral costophrenic angle (arrowhead) in (a) and pleural effusion obliterating the posterior costophrenic angle in (b) (arrow)

Posteroanterior chest radiograph of a patient with right subpulmonic pleural effusion (SPE) shows raised hemidiaphragm, and the lung seems to end early (arrowhead)

Posteroanterior (a) and lateral (b) chest radiographs show right-sided encysted pleural effusion (arrowheads)
Signs on US
Pleural effusion appears as anechoic or hypoechoic collection that lies between the echogenic line of the visceral pleura and lung (Fig. 7.1.4).

Transverse ultrasound image shows right-sided pleural effusion (arrowhead). The diaphragm can be visualized as a hyperechoic line separating the right lung base from the liver (arrow)
Signs on CT
-
Serous pleural effusion is visualized as a crescent peripheral area with CT water density. Exudative effusion can be hyperdense.
-
Empyema characteristically demonstrates thickened parietal/visceral pleura (e.g., > 2 mm) with effusion in between (split pleura sign) (Fig. 7.1.5). Enhancement of both pleura occurs in 80–100 % cases after contrast injection. Multiple gas pockets within the empyema may be seen.
-
Bronchopleural fistula occurs when a bronchus opens into the pleural space due to lung parenchymal destruction (e.g., pneumonia with empyema formation). It is seen as pleural effusion with air–fluid level on radiographs or HRCT (Fig. 7.1.6).

Posteroanterior chest radiograph (a) and axial chest CT (b) of a patient with huge left-sided empyema show split pleura sign in (b), with thickened, enhanced pleura with effusion in between (arrowheads)

Axial chest CT shows huge right bronchopulmonary fistula
7.1.1.1 Differential Diagnoses and Related Diseases
-
Meigs’ syndrome is a disease characterized by ascites, pleural effusion, and one of the following ovarian tumors (fibroma, thecoma, granulose cell tumor, or Brenner’s tumor). In contrast, Pseudo-Meigs’ syndrome is defined as ascites, pleural effusion, and ovarian tumor other than the ones mentioned previously. The absence of malignant cells from the ascites or the pleural effusion is mandatory for the diagnosis of Meigs’ syndrome. Typically, the ascites and the pleural effusions resolve after tumor resection. Meigs’ syndrome often occurs in postmenopausal women.
-
Yellow nail syndrome is a rare disease characterized by extremities lymphedema and thickened, slowly growing, yellowish-green nails that are excessively curved from side to side (Fig. 7.1.7). The disease is commonly accompanied by idiopathic pleural effusion, chronic bronchiectasis, chronic sinusitis, and lymphedema of the face. Yellow nail syndrome may be accompanied by rheumatoid arthritis or thyroid disease. The disease is believed to be caused by hypoplasia, atresia, or varicosity of the lymphatics.
Fig. 7.1.7 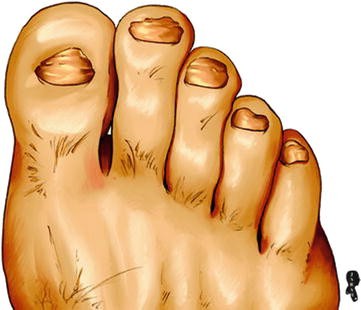
An illustration demonstrates the yellowish-green nails of the yellow nail syndrome

7.1.2 Pneumothorax
Pneumothorax is a condition characterized by the presence of air between the parietal and visceral pleura. There are three types of pneumothoraces:
-
Primary (spontaneous) pneumothorax: this type occurs without a defined cause and mainly seen in young males who are tall, thin, and smokers. Primary pneumothorax is attributed to rupture of subpleuritic blebs at lung apices according to some investigators.
-
Secondary pneumothorax: this type occurs usually after penetrating trauma, ruptured bulla, or an interventional thoracic procedure (e.g., lung mass biopsy).
-
Tension pneumothorax: this type occurs when the air collection within the subpleural space is large enough to push the mediastinum to the other side, interfering with blood circulation within the major vessels.
Up to 40 % of pneumothoraces may not be detected by chest radiographs. CT is 100 % sensitive for detection of pneumothoraces. When pneumothorax opens into the mediastinum, a pneumomediastinum develops. Pneumomediastinum is characterized by the presence of air around the mediastinal structures.
Sign on Radiograph
-
Thin visceral pleural line: it is visible on radiographs. The line is outlined by air with the absence of the peripheral vasculature laterally and lung tissue with possible increased density due to collapse, medially (Fig. 7.1.8). Lung apices are the best sites checked for early detection of pneumothorax.
-
Deep sulcus sign: the costophrenic angle deepens at the site of the pneumothorax (Fig. 7.1.9). It is seen in pneumothorax with large air collection.
-
Tension pneumothorax: it is seen as complete collapse of the lung and shift of the trachea and mediastinum to the contralateral (other side) collapsed lung (Fig. 7.1.10).
-
Pitfall: a skinfold and underlying clothing can mimic a pneumothorax (Fig. 7.1.11). Always correlate the radiological findings with the patient history and current status.
-
Pneumomediastinum: it is detected when the mediastinal structures are surrounded by dark radiolucent line of air (Fig. 7.1.12).

Posteroanterior chest radiograph shows right pneumothorax with lung collapse (arrowhead)

Posteroanterior chest radiograph shows right-sided pneumothorax (arrowhead) with right deep sulcus sign (arrow)

Posteroanterior chest radiograph of a child with right tension pneumothorax shows mediastinal shift toward the left side

Posteroanterior chest radiograph (a) and axial chest CT (b) of a patient with skin flap that appears as right-sided pneumothorax on chest radiograph (arrowheads), while on the HRCT, no pneumothorax is detected

Posteroanterior chest radiograph (a) and axial chest CT (b) of a patient with pneumomediastinum show air around the heart and the mediastinal structures (arrows)
7.1.3 Pleural Calcification
Pleural calcification can be seen following chronic pleural damage. Pleural calcification can be unilateral or bilateral. Unilateral pleural calcification occurs usually as a late complication of empyema, hemothorax, fungal infection, or tuberculosis. Bilateral pleural calcification is commonly caused by asbestosis
Asbestosis is a pathological condition that results from previous exposure to asbestos. Pleural plaques are the commonest manifestation of asbestosis, and they usually develop 20–30 years after the exposure to asbestosis. They are composed of focal areas of parietal pleural thickening with dense hyaline collagen. Mesothelioma is an uncommon primary tumor of the serosal lining of the pleura or peritoneum. Only 5–7 % of patients with asbestosis develop mesothelioma. Mesothelioma has poor prognosis, with survival rate of 12 months. Bronchogenic carcinoma develops in 25 % of cases of asbestosis, and it is the main cause of death.
Signs on Chest Radiographs
-
Pleural plaques are seen as smoothly demarcated, well-defined opacities (in profile) or faint, ill-defined plaques (en face), in a bilateral fashion. Unilateral pleural plaques may be seen in 25 % of cases and usually located on the left side. The plaques usually are <1 cm in thickness and seen parallel to the chest wall.
-
Pleural calcification is seen in 15–25 % of cases after a latency period of 30–40 years (Fig. 7.1.13). Diaphragmatic calcification is pathognomonic finding of asbestosis.
-
Asbestos-related diffuse pleural thickening is a bilateral thickening involving at least 25 % of the chest or 50 % if unilateral, plus pleural thickness >5 mm at any site. The diffuse pleural thickening can affect the visceral layer and the parenchyma below, causing “fluffy fibrous strands.”
-
Round atelectasis is seen as a pleural mass (3–5 cm) that abuts over the pleura with a curvilinear tails entering the mass (comet tail sign). The curvilinear densities are composed of fibrosed vessels, and the mass is commonly visualized at the base of the lungs (Fig. 7.1.14). Air bronchograms within the mass are common.
-
Malignant mesothelioma is visualized as visceral or parietal pleural nodules that are indistinguishable from pleural metastases. The most common manifestation of malignant mesothelioma is a unilateral massive pleural effusion.

Posteroanterior chest radiograph of a patient with asbestosis shows bilateral calcified pleural plaques

Axial chest HRCT illustration demonstrates round atelectasis with comet tail sign (arrowhead)
Signs on HRCT
-
Pleural plaques appear as well-circumscribed areas of pleural thickening separated from the underlying ribs by thin layer of fat. The edges of the pleural plaque are typically thicker than its center.
-
Malignant mesothelioma is visualized as nodular pleural thickening (94 %) that commonly involves the lung bases (50 %) (Fig. 7.1.15). Diaphragmatic involvement (80 %), pleural calcification (20 %), and pleural effusion (80 %) are other common features.

Posteroanterior chest radiograph (a) and axial chest CT (b) of a patient with malignant pleural mesothelioma show nodular thickening of the pleura in the right lung field with extension toward the apex in (a) and (b). Notice the nodular mass that follows the pleural distribution in (b) (arrowhead)
Further Reading
-
Akira M, et al. Asbestosis: high-resolution CT-pathologic correlation. Radiology. 1990;176:389–94.
-
DeCoste SD, et al. Yellow nail syndrome. J Am Acad Dermatol. 1990;22:608–11.
-
Gallardo X, et al. Benign pleural diseases. Eur J Radiol. 2000;34:87–97.
-
Goyal M, et al. Malignant pleural mesothelioma in a 13-yearold girl. Pediatr Radiol. 2000;30:776–8.
-
Nicholas G, et al. Asbestosis and malignancy. AJR. 1967;100:597–602.
-
Nimkin K, et al. Localized pneumothorax with lobar collapse and diffuse obstructive airway disease. Pediatr Radiol. 1995;25:449–51.
-
O’Lone E, et al. Spontaneous pneumothorax in children: when is invasive treatment indicated? Pediatr Pulmonol. 2008;43:41–6.
-
Qureshi NR, et al. Imaging in pleural disease. Clin Chest Med. 2006;27:193–213.
-
Sacco O, et al. Yellow nail Syndrome and bilateral cystic lung disease. Pediatr Pulmonol. 1998;26:429–33.
-
Váuez JL, et al. Pneumomediastinum and pneumothorax as presenting signs in severe Mycoplasma pneumoniae pneumonia. Pediatr Radiol. 2007;37:1286–8.
7.2 Alveolar Lung Diseases
Alveolar lung diseases (ALD) are group of disorders characterized by pathological insult involving mainly the alveoli. The alveoli can be imagined as an empty cup, and alveolar diseases are classified according to the content of this cup. Alveolar diseases are characterized by filling of the alveoli with materials that impede its normal physiological function (ventilation). Alveolar diseases can be localized (focal) or diffuse. Names of the conditions depend upon the content of the material filling the alveoli.
7.2.1 Types of Alveolar Lung Diseases
-
Alveoli filled with serous fluid: cardiogenic and noncardiogenic edema
-
Alveoli filled with blood: pulmonary hemorrhage, commonly due to vasculitis (e.g., Churg–Strauss syndrome)
-
Alveoli filled with pus: pneumonia
-
Alveoli filled with proteins: alveolar proteinosis and amyloidosis
-
Alveoli filled with malignant cells: bronchoalveolar carcinoma
-
Alveoli filled with calcium: alveolar microlithiasis
7.2.1.1 Pulmonary Edema
The alveoli are the main units for respiratory–blood ventilation and oxygenation and normally are full of air on inspiration. You can think of the alveoli as an empty cup, and any pathological condition that fills this cup will form a pathological condition according to the cup content. Pulmonary edema arises due to alveolar filling with serous fluid (water).
Pulmonary edema can be either due to cardiac disease (cardiogenic) or other conditions (noncardiogenic). Most cases of noncardiogenic pulmonary edema are due to acute respiratory distress syndrome (ARDS).
Cardiogenic pulmonary edema is commonly seen with heart failure. It starts as an interstitial edema before it turns into alveolar edema, because the pulmonary veins lie in the interstitium. As the hydrostatic pressure within the veins rises, they leak into the interstitium first and then progress to fill the alveoli. This process is rapid, and only very early edema can be seen as a pure interstitial linear pattern in chest radiographs.
Noncardiogenic pulmonary edema has the same radiographic features as the cardiogenic pulmonary edema, but the causes are different: ARDS, chemical pneumonitis, drug-induced pulmonary edema, and transfusion reaction are the most common causes for noncardiogenic pulmonary edema. ARDS is a situation where an alveolar capillary injury occurs as a result of variety of causes (e.g., sepsis). Chemical pneumonitis is a pulmonary edema that occurs due to inhalation of noxious chemical substance such as ammonia, smoking inhalation, near-drowning situations, and gastric acid aspiration. The mechanism of pulmonary edema is the result of one of the three mechanisms: irritation of the tracheobronchial tree that leads to inflammation and pulmonary edema formation; absorption of the noxious material from the respiratory tract, which can affect the lungs directly by its metabolites; and asphyxiation due to inhalation of high concentration of the noxious material that will displace oxygen from the blood and cause tissue hypoxia. Drug-induced and transfusion reactions pulmonary edema arise due to anaphylactic lupus-like reaction formation. The radiographic picture cannot be differentiated from ARDS unless you have history of drug ingestion or recent transfusion reaction. Classic examples of drugs causing pulmonary edema are heroin, aspirin, and penicillin. Negative pressure pulmonary edema is a term used to describe noncardiogenic edema that arises due to acute airway obstruction (type 1) or after the relief of chronic airway obstruction (type 2).
Signs on Radiograph
-
There is a centrally located, bilateral, symmetrical diffuse alveolar opacities emitting from the helium and spares the periphery (butterfly or batwings sign) (Fig. 7.2.16). Usually, pulmonary edema causes homogenous opacities, but sometimes they can cause nodular or blotchy opacities.
-
Cardiomegaly and signs of congestive heart failure (e.g., congested pulmonary vessels).
-
Kerley lines represent thickening of the interlobar septae. Lung lymphatics and veins run in the interstitium, leakage of the veins (edema), or tumor infiltration of the lymphatics (lymphangitis carcinomatosis) can result in thickening of the interlobar septa, which are called Kerley lines. Kerley A lines are long lines located near the lung hilum and extend obliquely near the bronchoarterial bundle into the peripheries. Kerley B lines are short white lines seen perpendicular to the pleural surface at the lung bases, commonly near the costophrenic angles (Fig. 7.2.17). Kerley C lines are a mixture between the two lines resulting in a reticular pattern.
-
Air-bronchogram sign is a sign seen when the alveoli are filled with fluid and the terminal bronchioles and bronchi are devoid of fluid (filled with air). The bronchioles appear as radiolucent lines within whitish radio-opaque opacities (Fig. 7.2.18). This sign is specific for alveolar disease, but nonspecific for the cause. Pulmonary edema, pulmonary hemorrhage, pneumonia, and alveolar carcinoma all look the same on radiographs. All appears as ALD with air bronchogram. The medical history plays a very important role in differentiating these conditions because the radiographic signs can be nonspecific.
-
In blood diversion, normally, the upper lobe vessels are not visualized on radiographs, and the lower lobe vessels are mildly dilated and visible due to the gravity effect in upright posteroanterior (PA) radiographs. In cases of cardiac diseases and pulmonary hypertension, the upper lobe vessels will be as wide as the lower lobe vessels in upright radiographs. Note that the upper lobe vessels can be seen dilated normally in supine (lying) chest radiographs (e.g., in intensive care unit radiographs).
-
Silhouette sign refers to a patchy, ill-defined radio-opaque shadow that obscures part of the normal mediastinal configuration.

Anteroposterior plain chest radiographs in two different patients show bilateral symmetrical pulmonary edema with bat wings appearance in (a), and bilateral, almost symmetrical pulmonary hemorrhage in (b). Notice that without history, you cannot differentiate pulmonary hemorrhage from pulmonary edema based on radiographic presentation alone

Posteroanterior plain chest radiograph shows Kerley B lines (arrowheads)

Posteroanterior plain chest radiographs in two different patients with pneumonia show pneumonic lung patch with air columns within the patchy due to unaffected bronchi in (a) (arrowhead) and pneumonic lung patch with no air-bronchogram sign in (b). Patient (a) presents with airspace pneumonia, whereas patient (b) presents with bronchopneumonia
7.2.2 How to Differentiate Between Cardiogenic Edema from ARDS on Plain Chest Radiographs?
-
ARDS usually has a normal heart size, while cardiogenic pulmonary edema shows signs of heart failure.
-
ARDS usually affects peripheral lung field more than central, whereas cardiogenic edema typically starts from the center to the periphery.
-
ARDS usually has no Kerley B lines.
7.2.2.1 Pneumonia
Pneumonia is a condition characterized by an infectious inflammation of the lung parenchyma and deposition of pus within the alveoli. Pneumonia can be caused by bacteria (e.g., methicillin-resistant Staphylococcus aureus (MRSA)), fungi (e.g., Pneumocystis carinii), and viruses (e.g., Cytomegalovirus (CMV)).
Patients with pneumonia present with dyspnea, purulent sputum, fever, tachycardia, and maybe hemoptysis (e.g., tuberculosis). Complications of pneumonia include lung abscess formation, septicemia, and empyema. Rarely, arthritis and neurological symptoms may be encountered in atypical pneumonias (e.g., Mycoplasma pneumonia).
Pneumonias are divided into “typical pneumonia,” which is caused by Streptococcus pneumoniae (pneumococcus), and “atypical pneumonia,” which is caused by any pathogen that is not pneumococcus. Typical pneumonia is clinically dominated by respiratory symptoms, whereas atypical pneumonia clinically is dominated by symptoms of fever and malaise more than the respiratory symptoms.
7.2.3 Types of Pneumonias
-
Airspace pneumonia (lobar pneumonia): in this type, the infection is confined to a single lobe. There is usually one patch filling the whole affected lobe. This type is seen with pneumococcus, Legionella, Pseudomonas, and primary tuberculosis infection. Lobar pneumonia is characterized by an “air-bronchogram sign.”
-
Bronchopneumonia: this type is characterized by an infection that starts in the bronchioles and small bronchi walls and then spreads to the alveoli. This type is seen with Staphylococcus aureus, Haemophilus influenza, and Mycoplasma pneumonias.
-
Interstitial pneumonia: this type is characterized by an infection that involves the interstitial septa and giving reticular interstitial pattern on chest radiograph. This type can be seen with viral infections like influenza virus and varicella-zoster virus (VZV) and Mycoplasma infections (30 % of cases).
MRSA is a serious infection with antibiotic-resistant staphylococci. MRSA is categorized as community-acquired, nosocomial, and healthcare-associated infection. MRSA is the leading cause of nosocomial and healthcare-associated bloodstream infection, globally. Also, it is responsible for 30–50 % of ventilator-associated pneumonia. MRSA causes metastatic foci of infections in 30 % of cases into the lungs, liver, kidneys, heart valves, and joints. Most community-associated MRSA strains carry the Panton–Valentine leukocidin (PVL) gene, which is rarely found in the hospital-acquired MRSA or the normal strain of S. aureus. PVL toxin is a potent lethal factor to neutrophils, which causes tissue necrosis and severe necrotizing pneumonia. MRSA pneumonia is more frequently associated with sepsis, high-grade fever, hemoptysis, pleural effusion, and death compared to PVL-negative S. aureus. MRSA pneumonia can result in the formation of pulmonary cavitary infiltration due to the development of necrotizing pneumonia. The development of MRSA necrotizing pneumonia should be suspected in a young patient presenting with hypoxia, hemoptysis, and single or multiple cavitary lung lesions.
Viral pneumonias are characterized by several pathologies that include bronchiolitis, tracheobronchitis, and classical pneumonia. Viruses that attack immunocompetent patients include influenza viruses, Epstein–Barr virus, and adenoviruses. Viruses that attack immunocompromised patients include measles virus, VZV, and CMV. Measles virus attacks usually children due to immunosuppression or vaccine failure. VZV pneumonia is a common complication of VZV septicemia in children with a mortality rate of 9–50 %. Up to 90 % of VZV pneumonia cases are seen in patients with lymphoma or immunosuppression. CMV pneumonia is commonly seen in transplant patients and immunocompromised patients. Patients may develop severe necrotizing pneumonia in spite of antiviral therapy.
7.2.3.1 Differential Diagnoses and Related Diseases
Hyperimmunoglobulinemia E syndrome (Job’s syndrome) is a rare condition characterized by marked elevation of serum IgE levels against S. aureus, resulting in decreased production of antistaphylococcus IgG. The patient with this syndrome presents with frequent attacks of S. aureus pneumonia, pustular dermatitis, eczema, and sinusitis. Formation of chronic lung abscesses is a common feature on radiographs.
Signs on Radiograph
-
Radio-opaque patches with an air-bronchogram sign (Fig. 7.2.18).
-
For bulging fissure sign, some infections will increase the volume of the lobe involved, causing the adjacent fissure to bulge (commonly the transverse fissure) (Fig. 7.2.19). This sign is classically seen in Klebsiella pneumonia.
-
In bronchopneumonia, there are multiple patchy infiltrations of the lung with or without segmental lobe atelectasis (if the bronchus is totally obstructed) (Fig. 7.2.18).
-
Interstitial pneumonia shows nonspecific linear or reticular interstitial lung pattern. Correlation with history and laboratory findings is essential to establish the diagnosis.
-
Viral pneumonias can appear as poorly defined nodules (4–10 mm in diameter), with lung hyperinflation due to bronchiolitis.
-
Measles pneumonia shows mix pattern of reticular interstitial pattern with patchy pneumonia (Fig. 7.2.20). Hilar lymphadenopathy may be associated.
-
VZV pneumonia appears as multiple, ill-defined micronodules (5–10 mm) (Fig. 7.2.21). The lesions may calcify persisting as well-defined, randomly scattered, dense pulmonary calcification.
-
CMV pneumonia is commonly seen as a mixed nodular interstitial pattern with ill-defined patchy lung infiltration. The patchy filling is caused pathologically by hemorrhage, neutrophilic and fibrinous exudates, and hyaline membrane formation (Fig. 7.2.22).
-
Chronic pneumonia can lead to fibrosis, traction bronchiectasis, and paracicatricial emphysema (Fig. 7.2.23).
-
In MRSA necrotizing pneumonia, a pneumonic patch or a pulmonary mass with central cavitary lesion can be found. The lesion can be single or multifocal. The same manifestations are observed in HRCT. Differential diagnoses of cavitary lung infiltrations include lung abscess, metastases, pulmonary lymphoma, and Wegener’s granulomatosis.

Anteroposterior plain chest radiograph of a bedridden patient shows right upper lobe pneumonia with bulging of the transverse fissure (arrowhead)

Posteroanterior plain chest radiograph of a 5-year-old child with measles presenting with dyspnea shows ill-defined patchy pneumonia in the upper zone of the left lung (arrowhead)

Posteroanterior plain chest radiograph of a patient with varicella-zoster virus (VZV) pneumonia shows diffuse micronodular interstitial lung pattern bilaterally

Posteroanterior plain chest radiograph of a patient with cytomegalovirus (CMV) pneumonia after heart transplant shows mixed patchy lung infiltration with micronodular interstitial lung pattern

Posteroanterior plain chest radiograph of a patient with mycoplasma pneumonia shows right upper lobe fibrosis with honeycombing due to traction bronchiectasis (arrow) and paracicatricial emphysema (arrowheads)
Further Reading
-
Anuradha G. Methicillin-resistant staphylococcus aureus bacteremia and pneumonia. Dis Mon. 2008;54:787–92.
-
Chuang YC, et al. Negative pressure pulmonary edema: report of three cases and review of the literature. Eur Arch Otorhinolaryngol. 2007;264:1113–6.
-
Connolly B, et al. Bronchial artery aneurysm in hyperimmunoglobulinemia E syndrome. Pediatr Radiol. 1994;24:592–3.
-
Corriere MD, et al. MRSA: an evolving pathogen. Dis Mon. 2008;54:751–5.
-
Decker CF. Pathogenesis of MRSA infection. Dis Mon. 2008;54:774–9.
-
Ebert MD, et al. Necrotizing pneumonia caused by community-acquired methicillin-resistant Staphylococcus aureus: an increasing cause of “mayhem in the lung”. Emerg Radiol. 2009;16:159–62.
-
Fujinaga S, et al. Pulmonary edema in a boy with biopsy-proven poststreptococcal glomerulonephritis without urinary abnormalities. Pediatr Nephrol. 2007;22:154–5.
-
Gattinoni L, et al. The role of CT-scan studies for the diagnosis and therapy of acute respiratory distress syndrome. Clin Chest Med. 2006;27:559–70.
-
Kawamata M, et al. Acute pulmonary edema associated with transfusion of packed red blood cells. Intensive Care Med. 1995;21:443–6.
-
Kim EA, et al. Viral pneumonias in adults: radiologic and pathologic findings. Radiographics. 2002;22:S137–49.
7.3 Atelectasis (Lung Collapse)
Atelectasis is a condition characterized by lung collapse, which can be subtotal (25–50 % collapse) or total (100 % collapse).
Atelectasis can result due to air resorption (resorptive atelectasis), lung compression (compression atelectasis), or loss of the surfactant in acute respiratory distress syndrome (ARDS) and hyaline membrane disease (respiratory distress syndrome) in preterm infants (microatelectases). Pulmonary atelectasis is a recognized complication of general anesthesia.
Pulmonary surfactant is secreted by pneumocytes type II, which is composed of phospholipids and proteins. The surfactant stabilizes the lung by reducing the surface tension at the air–liquid interface in the alveoli. Therefore, deficiency of pulmonary surfactant could result in collapse of the alveolar spaces.
Patients with atelectasis commonly present with dyspnea, tachypnea, cough, and pleuritic chest pain on inspiration. Hypoxemia may result from atelectasis due to reduced ventilation–perfusion equilibrium.
7.3.1 Types of Pulmonary Atelectases
-
Resorptive atelectasis can arise due to intrinsic obstruction (e.g., mucus plug) or extrinsic obstruction (e.g., hilar lymphadenopathy). Brock’s syndrome is a term used to describe right middle lobe (RML) atelectasis by enlarged hilar lymphadenopathy compressing the right main bronchi.
-
Passive atelectasis results when the natural tendency of lung tissue to collapse due to elastic recoil goes unstopped. This condition can be seen in atelectasis due to pneumothorax.
-
Compressive atelectasis is a variant of passive atelectasis and occurs when a space-occupying lesion abuts the lung causing atelectasis (e.g., massive pleural effusion).
-
Cicatrization atelectasis is seen with fibrosis, where the scar tissue contracts and collapses the alveoli.
-
Adhesion atelectasis occurs due to surfactant deficiency, which is classically seen in hyaline membrane disease in infants and ARDS and pulmonary embolism in adults.
-
Plate atelectasis is composed of sheets of horizontal tissue collapse, which is commonly located 1–3 cm above the diaphragm. This type is commonly seen in conditions which impede normal respiration (e.g., inflammatory conditions in the chest or abdomen).
-
Congenital atelectasis is seen in newborn infants due to failure to aerate the lung after pregnancy.
-
Round atelectasis is seen in asbestosis, and it is characterized by atelectasis of parenchymal tissues near the pleura. It is best diagnosed by CT, which will show bronchovascular marks entering the mass (comet tail sign).
-
Segmental atelectasis is an uncommon type of atelectasis characterized by an entire lung segment collapse. It is highly suggestive of a tumor blocking the bronchial feeding of that segment.
Signs on Chest Radiograph
-
Diaphragmatic elevation due to reduced lung volume.
-
Shift of the right horizontal fissure upward due to upper lobe collapse (Fig. 7.3.24).
-
RML and left lower lobe (LLL) atelectases are located behind the heart. They can be seen as dense radio-opaque triangles overlying the heart shadow (Figs. 7.3.25 and 7.3.26). They can be easily missed if the atelectasis is examined in posteroanterior view only; lateral views are advised if RML or LLL atelectases are suspected.
-
Left upper lobe (LUL) atelectasis is generally seen as increase in lung density on posteroanterior (PA) view. This is explained by the fact that the LUL collapses anteriorly. Lateral view is shown clearly as an anterior mediastinal radio-opaque shadow representing the collapsed lobe (Fig. 7.3.27).
-
Right upper lobe (RUL) atelectasis is seen as homogenous opacity located at the right upper lung zone and bounded inferiorly by the transverse fissure (Fig. 7.3.28).
-
Shift of the trachea and the mediastinum toward the collapse.
-
For spine sign, normally the lower vertebrae on lateral view are less dense than the upper vertebra. The upper vertebrae appear denser due to the arm and axilla shadow overlying them. With progressive atelectasis of the lower lobes, the lobes will move more posteromedially, making the lower vertebra appears as dense as the upper vertebrae (Fig. 7.3.26).
-
Golden S sign is seen when the RUL is collapsed due to hilar mass blocking the right main bronchus (e.g., in Brock’s syndrome).
-
Plate atelectasis is detected on radiographs as linear horizontal radio-opaque lines commonly located 1–3 cm above the diaphragm (Fig. 7.3.29).

Anteroposterior plain chest radiograph of a bedridden patient shows mediastinal shift toward the left side due to collapse of the left lung

Posteroanterior (a) and lateral (b) plain chest radiographs show right middle lobe (RML) atelectasis (arrowheads)

Posteroanterior (a) and lateral (b) plain chest radiographs show left lower lobe (LLL) atelectasis (arrowheads). Notice that the lower thoracic vertebrae appear denser than the upper thoracic vertebrae due to the shadow of the atelectatic lobe overlying them (spine sign)

Posteroanterior (a) and lateral (b) plain chest radiographs show left upper lobe (LUL) atelectasis. Notice the high-density left lung field in (a), which is explained by atelectasis of the LUL anteriorly in (b) (arrowheads)

Posteroanterior plain chest radiographs show right upper lobe (RUL) atelectasis bounded inferiorly by the transverse fissure (arrowheads)

Posteroanterior plain chest radiograph shows plate atelectasis as thick radio-opaque shadow at the right costophrenic angle
Further Reading
-
Glay J, et al. Unusual pattern of left lower lobe atelectasis. Radiology. 1981;141:331–3.
-
Sargent MA, et al. Atelectasis on pediatric chest CT: comparison of sedation techniques. Pediatr Radiol. 1999;29:509–13.
-
Tsai KL, et al. Pulmonary atelectasis: a frequent alternative diagnosis in patients undergoing CT-PA for suspected pulmonary embolism. Emerg Radiol. 2004;10:282–6.
-
Westcott JL, et al. Plate atelectasis. Radiology. 1985;155:1–9.
-
Zhao Y, et al. Atelectasis: an unusual and late complication of lung transplant. Clin Transplant. 2002;16:233–9.
7.4 Sarcoidosis
Sarcoidosis, also known as Boeck’s sarcoid, is a multisystemic granulomatous disorder characterized by the formation of multiple epithelioid granulomas within more than one system. Sarcoidosis belongs to a large family of granulomatous disorders, which includes tuberculosis, leprosy, Langerhans cell histiocytosis, and more. All members of the granulomatous disease are characterized by the formation of granulomas within the body system.
Granuloma is a specific kind of chronic inflammation, and it is a term used to describe a nodular chronic inflammation that occurs in foci (granules), with collection of macrophages called epithelioid cells. Epithelioid cells are macrophages with abundant cytoplasm that is similar to the cytoplasm of epithelial cells. When multiple epithelioid cells fuse together, they form a bigger macrophage known as giant cell. Epithelioid cells define granulomatous inflammation. On histological specimens, granulomas show endarteritis obliterans, fibrosis, and chronic inflammatory cells (epithelioid cells). Granuloma can be due to an infection (e.g., tuberculosis) or due to an inorganic foreign body (e.g., silicosis).
Langerhans cells are characteristically found within the sarcoid granuloma, which develops by the fusion of epithelioid cells, resulting in a modified macrophage with nuclei arranged in an arc-like pattern. Langerhans cells secrete lysozyme, collagenase, calcitriol, angiotensin-converting enzyme (ACE), and varied cytokines.
No body tissue is spared from sarcoidosis. There are two forms of the disease, an acute form and chronic form. Acute sarcoidosis responds well to steroids with frequent spontaneous recovery. Moreover, it is characterized by serum elevation of ACE in two thirds of patients and abnormal calcium metabolism (high serum calcium levels). In contrast, the chronic sarcoidosis is persistent, and the serum levels of calcium and ACE are often normal.
Sarcoidosis is often seen between 20 and 40 years of age. However, juvenile form (pediatric sarcoidosis) with a smaller age peak at 13–15 years has been reported to occur rarely.
Sarcoidosis manifestations are seen in almost any part of the body. The definite diagnosis is based on histopathology examination. Radiological investigations play an important role in monitoring the therapy and the disease progression.
7.4.1 Pulmonary Sarcoidosis
The pulmonary system is involved in up to 90 % of patients with sarcoidosis. Patients with sarcoidosis are classically young females presenting with nonspecific symptoms of a systemic disease (e.g., malaise). In pulmonary sarcoidosis, dyspnea and cough are common, whereas hemoptysis (coughing blood) is rare.
7.4.1.1 Sarcoidosis Has Five Radiological Grades on Plain Chest Radiograph
-
Grade 0: Normal chest radiograph.
-
Grade 1: There are clear lung fields with bilateral hilar lymphadenopathy (85 % of cases). It is usually identified by accident, and the patient is asymptomatic (Fig. 7.4.30).
Fig. 7.4.30 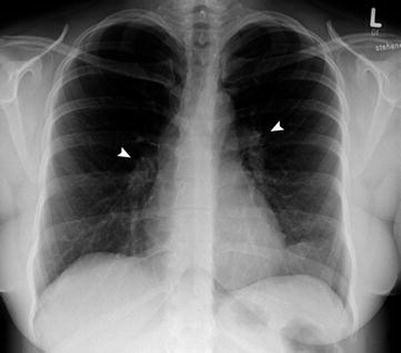
Posteroanterior chest radiograph of a female patient with grade 2 pulmonary sarcoidosis shows bilateral hilar lymphadenopathy (arrowheads) with clear lung fields
-
Grade 2: There is reticulonodular interstitial pattern with hilar lymphadenopathy. The areas affected are usually located in the upper lobes.
-
Grade 3: There is reticulonodular interstitial pattern without hilar lymphadenopathy (Fig. 7.4.31).
Fig. 7.4.31 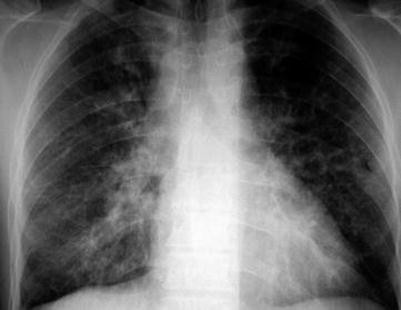
Posteroanterior chest radiograph of a patient with grade 3 pulmonary sarcoidosis shows bilateral diffuse reticular interstitial pattern due to lung fibrosis
-
Grade 4: There is pulmonary parenchymal scarring and fibrosis (Fig. 7.4.32).
Fig. 7.4.32 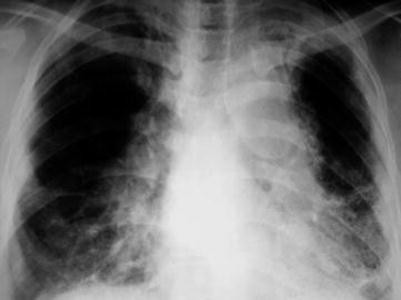
Posteroanterior chest radiograph of a patient with chronic grade 4 pulmonary sarcoidosis shows bilateral lung fibrosis distorting the heart silhouette (shaggy heart appearance)



Signs on HRCT
-
Sarcoid granulomas are typically distributed along the lymphatic vessels within the interstitium. Due to this fact, miliary nodules plus linear thickening of the interlobar septa can be found in a similar fashion to the interstitial disease seen in lymphangitis carcinomatosis and lymphoproliferative diseases.
-
Bilateral hilar lymphadenopathy observed in grade 2 and 4. Punctuate, stippled, or egg shell calcification patterns may be seen.
-
Occasionally, multiple granulomas may aggregate to form a mass-like nodule within the lungs that mimics metastasis (Fig. 7.4.33). Lung nodule is a lesion <3 cm in diameter, whereas lung mass is a lesion >3 cm in diameter.
-
Necrotizing sarcoid granulomatosis is a rare variant of sarcoid characterized by the formation of cavitating granulomas.
-
Hilar lymphadenopathy with eggshell calcification and bilateral upper lobe fibrosis are typical findings in pulmonary silicosis. Sarcoidosis may mimic silicosis when it produces the same set of radiographic manifestations on plain chest radiograph.

Axial thoracic lung-window HRCT illustration demonstrates a pulmonary mass that is composed of multiple aggregated sarcoid granulomas. Although it is a difficult diagnosis to confirm without biopsy, the presence of air bronchogram or areas of normal tissue lines within the mass (arrowhead) can differentiate this rare lesion from bronchogenic carcinoma
7.4.2 Hepatic, Splenic, and Gastric Sarcoidosis
Hepatic sarcoidosis is seen in 5–15 % of patients with high ACE levels (acute disease). There are multiple hepatic granulomas that can be easily mistaken on CT and MRI for metastasis or lymphoma. Simultaneous involvement of the spleen favors the diagnosis of sarcoidosis and lymphoma.
Splenic sarcoidosis classically affects the white bulb and the arterial circulation. The spleen is affected in 5–14 % of patients with sarcoidosis. Patients may suffer from symptoms of hypersplenism, anemia, thrombocytopenia, and leukopenia.
Gastric sarcoidosis is the most common feature of gastrointestinal involvement of sarcoidosis. It often involves the antrum. Massive retroperitoneal lymphadenopathy may be rarely encountered in sarcoidosis.
Signs on CT
-
Hepatic sarcoidosis is seen as multiple hypodense lesions with irregular shapes on liver contrast-enhanced images.
-
Splenic sarcoidosis is seen on contrast-enhanced images as multiple, irregularly diffuse, hypodense lesions within the spleen representing granulomas (Fig. 7.4.34). Hepatic lesions may be noticed in the same scan (50 % of cases). The same lesions are seen hypoechoic on US and hypointense on T1W and T2W images on MRI compared to the background.
-
Gastric sarcoidosis features range from ulceration mimicking peptic ulcer to mucosal thickening mimicking Menetrier disease. Diagnosis requires endoscopic biopsy to confirm the epithelioid granuloma.

Abdominal nonenhanced CT illustration demonstrates multiple hypodense lesions within the spleen representing splenic sarcoid granulomas
7.4.3 Dermatological Sarcoidosis
Skin lesions are seen in up to 25 % of patients with sarcoidosis. Skin lesions in sarcoidosis are divided into reactive and specific lesions. Reactive sarcoidosis skin lesions do not contain granuloma formation histologically (e.g., erythema nodosum). In contrast, specific sarcoidosis skin lesions are characterized by noncaseating granuloma formation (e.g., Darier–Roussy nodules).
In the acute reactive sarcoidosis, erythema nodosum is the most common finding, and it is seen as multiple patchy red lesions found over the shin, often in a bilateral fashion. Systemic manifestations like fever, malaise, and polyarthralgia occur in about 50 % of patients with erythema nodosum.
The chronic reactive sarcoidosis, on the other hand, is characterized by a specific lesion called “lupus pernio.” Lupus pernio is a specific skin lesion in sarcoidosis characterized by dusky-red plaques formation on the nose, ears, lips, and face. Lupus pernio is classically seen in women with chronic sarcoidosis and extensive pulmonary infiltration, anterior uveitis, and bone lesions. The nose lesion is typically red to purple in color and seen on the tip of the nose, causing bulbous appearance (Fig. 7.4.35). The nose lesion infiltrates the mucosa and may destroy the underlying nasal bone.

An illustration demonstrates lupus pernio on the ala of the nose
In black patients, maculopapular eruptions are the most common skin manifestations of sarcoidosis. Darier–Roussy nodules are small painless subcutaneous nodules that arise within the dermis and the epidermis. They represent noncaseating granulomas.
7.4.3.1 Differential Diagnoses and Related Diseases
Löfgren syndrome is a disease characterized by the combination of arthralgia, bilateral hilar lymphadenopathy, and erythema nodosum in a patient with sarcoidosis.
7.4.4 Cardiac Sarcoidosis
Sarcoidosis affects the heart in the form of patchy infiltration of the myocardium by granulomas causing fibrosis and scarring. Patients with cardiac sarcoidosis are at risk of sudden cardiac death due to ventricular arrhythmias or conduction block. Most patients are asymptomatic, with only 5 % of cardiac sarcoidosis patients being symptomatic. Cor pulmonale may arise secondary to pulmonary hypertension as a consequence of pulmonary fibrosis.
Signs on Cardiac MRI
-
The protocol of cardiac sarcoidosis should include T1W pre- and postcontrast images (there are multiple areas of contrast enhancement due to noncaseating granulomas), T2-STIR (to show edema or scar formation as low intensity areas), and CE-IR images to assess global function.
-
Inflammatory changes show myocardial high T2 signal intensity lesions (Fig. 7.4.36), with postcontrast enhancement and myocardial thickening. Postinflammatory changes include myocardial high T2 signal intensity lesions, with no contrast enhancement.

Short-axis dark-blood cardiac T2W MR-illustration demonstrates multiple high signal intensity lesions within the myocardium due to granuloma formation in a patient with sarcoidosis
7.4.5 Neurosarcoid
Involvement of the central nervous system by sarcoidosis (neurosarcoid) is noticed in <10 % of patients. There are three patterns of involvement: meningeal, parenchymal, and vascular.
In the brain, neurosarcoid has an affinity to involve the base of the brain and the cranial nerves. It commonly affects the hypothalamus, pons, meninges, spinal cord, basal ganglia, and cranial nerves (optic, facial, and vestibulocochlear). Neurosarcoid is the most common cause of bilateral facial nerve paralysis. Leptomeningeal thickening in the form of aseptic meningitis is commonly seen in neurosarcoid. When the lesion affects the hypothalamus, it leads to abnormal water balance and disturbance of thirst mechanism (sarcoid diabetes insipidus). Neurosarcoid can present in the absence of systemic sarcoidosis in 3 % of cases.
7.4.5.1 Differential Diagnoses and Related Diseases
Klein–Levin syndrome is a disease that arises due to hypothalamic or medial thalamic lesions characterized by episodes of compulsive eating (bulimia), hypersexuality in adolescent males, and hypersomnolence. Patients with hypersomnolence sleep an excessive amount of time at night, take long naps during the day, and generally feel drowsy and distracted when awake. Each episode lasts days to weeks with a symptom-free interval of 3–6 months between attacks. Klein–Levin syndrome is reported to occur rarely due to neurosarcoidosis.
Signs on MRI
-
Within the brain parenchyma, multiple or solitary brain lesions with a ringlike appearance may be seen on T2W and FLAIR images.
-
Thickening and enhancement of the meninges of postcontrast images are a classic finding in neurosarcoid in the area of the sellar diaphragm and the spinal cord (Fig. 7.4.37). Inflammation of basal meninges can lead to interference with cerebrospinal fluid (CSF) flow or aqueduct involvement leading to obstructive hydrocephalus.
-
Cranial nerve neuritis is seen as enhancement of the nerves like the facial or the vestibulocochlear within the internal auditory canal on T1W postcontrast images (Fig. 7.4.38).
-
When diabetes insipidus is present, thickening of the infundibulum and the optic chiasm with isointense T1/T2 high signal intensities and homogenous contrast enhancement is typically observed.
-
Intramedullary spinal cord lesions on T2W images with enhancement after contrast injection may be found representing neurosarcoid granuloma.
-
In Klein–Levin syndrome, hypothalamic T2 high signal lesions with leptomeningeal enhancement on postcontrast injection images may be seen.

Sequential axial T1W postcontrast brain images show nodular thickening and enhancement of the leptomeninges (arrowheads) in a patient with sarcoidosis (neurosarcoid)

Axial cerebellopontine angle T1W postcontrast MR-illustration demonstrates enhancement of the right facial nerve due to neuritis (the labyrinthine segment, the geniculate ganglion, and the proximal tympanic segment)
7.4.6 Musculoskeletal Sarcoidosis
The musculoskeletal system in sarcoidosis present in the form of arthritis (40 %), bony lesions, and muscular lesions. The musculoskeletal manifestations are commonly seen in chronic sarcoidosis, not in the acute form.
Sarcoid arthritis is migratory polyarthritis that involves usually the ankles and the knees, followed by the wrists and the interphalangeal joints. Early sarcoid arthropathy occurs in the first 6 months of symptoms, and it involves migratory polyarthritis (>4 joints). The second form occurs after 6 months or more and is characterized by oligoarthritis (2–3 joints) and inflammation of fingers or toes (dactylitis). Tenosynovitis may occur occasionally, causing a sausage-like finger similar to that seen in psoriatic arthritis.
Bony lesions in sarcoidosis are seen in 5–10 % of patients. They present as extensive bony erosions or cystic-like osteolytic lesions typically seen in the phalanges in the hands and feet (osteitis multiplex cystica). The same type of lesions can be seen in tuberculosis and classically known as osteitis tuberculosa multiplex cystica. Uncommonly, calvarial sarcoidosis may manifest as an expansile bony lesion.
Muscular sarcoidosis often presents as a nodular mass within the muscle due to granuloma formation.
Signs on Plain Radiographs
-
In the phalanges, a sharply demarcated cystic-like lesion is often found in skeletal sarcoidosis (osteitis multiplex cystica) (Fig. 7.4.39). Swelling of the affected finger with soft tissue mass found around the lesion is characteristic.
-
Sarcoid arthritis involving the hands often shows periarticular soft tissue swelling plus punched-out lesions of the phalanges.

Plain radiograph of the index finger of a patient with chronic sarcoidosis shows multiple osteolytic cystic lesions located within the terminal phalanges (osteitis multiplex cystica)
Signs on MRI
Muscular sarcoidosis presents as a muscular heterogeneous mass with hypointense center in all sequences representing fibrosis. Peripheral enhancement may be seen due to active disease process.
7.4.7 Head and Neck Sarcoidosis
Ocular manifestations of sarcoidosis occur in up to 80 % of patients in the form of bilateral uveitis and lacrimal duct inflammation. However, any structure of the eye may be involved. Conjunctival lesions are the second most common lesions seen in ophthalmic sarcoidosis after anterior uveitis. Keratoconjunctivitis sicca may occur in 5 % of cases when lacrimal gland infiltration occurs.
Parotid gland involvement in a bilateral fashion can be seen in up to 6 % of patients. The features resemble the parotid symptoms observed in Sjögren’s syndrome and lymphoma.
In up to 30 % of cases, patients with sarcoidosis present with cervical, nontender, movable lymphadenopathy commonly located in the posterior triangle.
Hoarseness of voice may rarely arise in patients with sarcoidosis due to vocal cord thickening and granulomas formation. It is a rare manifestation affecting 1–3 % of patients.
Paranasal sarcoidosis may occur, especially with lupus pernio. It has an affinity to involve the mucosa of the inferior turbinate and the nasal septum, causing mucosal thickening and nasal septal destruction.
7.4.7.1 Differential Diagnoses and Related Diseases
Heerfordt syndrome is a disease that occurs in a patient with sarcoidosis characterized by the triad of fever and anterior uveitis, bilateral parotid enlargement, and facial nerve palsy.
Signs on CT and MRI
-
Bilateral enlargement of the lacrimal glands with contrast enhancement is commonly seen in ophthalmic sarcoidosis.
-
Bilateral parotid enlargement, with high signal T2 intensity, and enhancement on postcontrast images are seen in cases of parotid involvement.
-
Inferior turbinate destruction with nasal septum erosion is seen in paranasal sinus CT (Fig. 7.4.40).

Coronal paranasal sinuses CT illustration demonstrates right inferior turbinate destruction (arrowhead) with nasal septum erosions due to sarcoidosis
7.4.8 Genitourinary Sarcoidosis
Renal sarcoidosis manifestations are related to nephrocalcinosis due to hypercalcemia or granuloma formation within the cortex and the medulla (interstitial nephritis). Scrotal sarcoidosis is uncommon but can present in the form of bilateral epididymitis.
Signs on Scrotal US
Epididymitis is seen as enlarged heterogeneous epididymis with marked increased signal flow on color Doppler and power Doppler due to hyperemia.
Signs on CT
-
Interstitial nephritis is seen on postcontrast images as striated nephrogram, usually on both kidneys.
-
Rarely, renal sarcoidosis may present with bilateral hypodense tumorlike nodules on contrast-enhanced images that may be mistaken for lymphoma.
Signs on MRI
Epididymitis is seen as bilaterally enlarged epididymis with high signal intensity on T2W images, with contrast enhancement in postgadolinium injection.
Further Reading
-
Afshar A, et al. Sarcoidosis: a rare cause of Kleine-Levine-Critchley syndrome. Sarcoidosis Vasc Diffuse Lung Dis. 2008;25:60–3.
-
Burov EA, et al. Morpheaform sarcoidosis: report of three cases. J Am Acad Dermatol. 1998;39:345–8.
-
Cummings MM, et al. Sarcoidosis. Dis Mon. 1960;6:1–40.
-
Farman J, et al. Gastric sarcoidosis. Abdom Imaging. 1997;22:248–52.
-
Fodor D, et al. Dactylitis and bone lesions at the onset of sarcoidosis: a case report. Pol Arch Med Wewn. 2008;118:774–7.
-
Geraint JD, et al. Descriptive definition and historic aspects of sarcoidosis. Clin Chest Med. 1997a;18:663–79.
-
Geraint JD, et al. Descriptive definition and historic aspects of sarcoidosis. Clin Chest Med. 1997b;18:663–79.
-
Henry DA, et al. Multiple imaging evaluation of sarcoidosis. Radiographics. 1986;6:75–95.
-
Koyama T, et al. Radiologic manifestations of sarcoidosis in various organs. Radiographics. 2004;24:87–104.
-
Kuhlman JE, et al. The computed tomographic spectrum of thoracic sarcoidosis. Radiographics. 1989;9:449–66.
-
Moore SL, et al. Musculoskeletal sarcoidosis: spectrum of appearances at MR imaging. Radiographics. 2003;23:1389–99.
-
Pattishall EN, et al. Sarcoidosis in children. Pediatr Pulmonol. 1996;22:195–203.
-
Poyanli A, et al. Vertebral sarcoidosis: imaging findings. Eur Radiol. 2000;10:92–4.
-
Rosell A, et al. Lupus pernio with involvement of nasal cavity and maxillary sinus. ORL. 1998;60:236–9.
-
Sharma OP. Sarcoidosis. Dis Mon. 1990;36:474–535.
-
Spilberg I, et al. The arthritis in sarcoidosis. Arthritis Rheum. 1969;12:126–36.
-
Tamme T, et al. Sarcoidosis (Heerfordt syndrome): a case report. Stomatologija Baltic Dent Maxillofac J. 2007;9:61–4.
-
Turkish M, et al. Osteitis tuberculosa multiplex cystica: its treatment with streptomycin and promizole. J Pediatr. 1949;35:625–9.
-
Warshauer DM. Splenic sarcoidosis. Semin Ultrasound CT MRI. 2007;28:21–7.
-
Yanardağ H, et al. Bone cysts in sarcoidosis: what is their clinical significance? Rheumatol Int. 2004;24:294–6.
7.5 Emphysema
Emphysema is a chronic obstructive airway disease characterized by an abnormal, irreversible, permanent enlargement of the air spaces distal to the terminal bronchioles, associated with destruction of the alveolar walls, and without obvious fibrosis.
The mechanism of emphysema is mainly mediated by the proteolytic enzymes (proteases) of the neutrophils and macrophages. The proteolytic enzymes dissolve the alveolar walls, creating holes that facilitate air leak from one alveolus to another, compromising gas exchange and trapping air within the acini. Normally, there are few small physiological holes between the alveoli that connect two adjacent alveoli together (pores of Kohn). In emphysema, the holes between the alveoli are numerous and much bigger than the normal Kohn’s pores, resulting in reducing the surface area for gas exchange.
The enzyme α-1 antitrypsin is a proteinase inhibitor that counteracts the effect of the proteolytic enzymes produced by neutrophils and macrophages. Emphysema results from imbalance between the proteolytic enzymes (proteases) production and α-1 antitrypsin (antiproteases).
The first emphysema mechanism arises due to increased alveolar infiltration by neutrophils and macrophages, with increased proteolytic enzymes’ production that exceeds the capacity of the normal circulating α-1 antitrypsin levels to counteract. This scenario is classically seen in emphysema due to cigarette smoking. The other mechanism of emphysema is seen due to congenital α-1 antitrypsin deficiency disease, where emphysema is produced with normal quantities of proteolytic enzymes.
Pathologically, emphysema is divided according to the level of alveolar destruction and the air trapping pattern within the secondary lobule (e.g., central or peripheral). Four major types of emphysema are described:
-
Centrilobular emphysema: this type starts at the center of the secondary lobule (centrilobular), and it results from the destruction of the alveoli around the proximal respiratory lobule. This type is typically seen in chronic cigarette smokers, and it affects predominantly the upper lung lobes. The emphysematous spaces may coalesce into a lager bulla, which is defined as sharply demarcated area of air collection >1 cm in diameter and with a wall less than 1 mm in thickness (Fig. 7.5.41).
Fig. 7.5.41 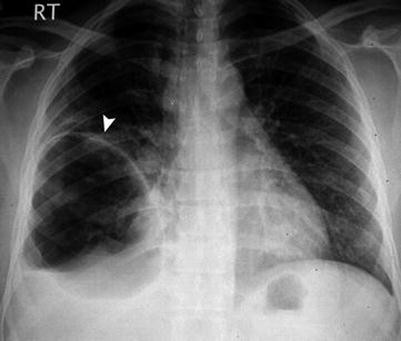
Posteroanterior plain chest radiograph shows large right lower zone bulla (arrowhead)
-
Panlobular (panacinar) emphysema: this type of emphysema is diffuse and involves the whole secondary lobule. This type is classically seen in nonsmoker patients with congenital α-1 antitrypsin deficiency disease and in Swyer–James syndrome (unilateral hyperinflated lung with pulmonary vasculature atresia, and it may be accompanied by bronchiectasis). Panlobular emphysema can be seen in conjunction with centrilobular emphysema in chronic smokers. Panlobular emphysema involves mainly the lower lung lobes.
-
Paraseptal (distal lobular) emphysema: this type is seen as air trapping at the periphery of the secondary lobule, especially adjacent to connective tissue septa. This type is typically seen at the periphery, at the subpleural spaces, and along the fissures and pleural reflections. It plays an important role in the development of spontaneous pneumothoraces.
-
Irregular (paracicatricial) emphysema: this type is an air collection that occurs in an area of massive fibrosis (scar tissue). It is commonly found in the upper lobes in an area of old tuberculosis fibrosis.

Other types of emphysema include:
-
Emphysema due to old age: it occurs due to the loss of lung volume (atrophy). It is panacinal type without airways obstruction.
-
Compensatory emphysema (postpneumonectomy syndrome): this type occurs when a lung lobe collapses or has been removed. The other lung will expand to occupy the space of lung deficiency. There is no airway obstruction with this type.
-
Giant bullous emphysema (vanishing lung syndrome): it is airways destruction due to extensive alveolar atrophy due to avascular necrosis of the lung parenchyma, resulting in hyperinflation of the affected lung. It is most commonly seen in young men with bilateral upper lobes bullae. It is a panlobular type affecting the upper lobes mainly and commonly present in individuals in their 40s. Up to 20 % of patients have congenital α-1 antitrypsin deficiency disease.
-
Bronchial atresia emphysema: this type arises due to developmental bronchial atresia. The segment with the atrophic bronchus receives its aeration by the “collateral air-drift mechanism” via “pores of Kohn” and “canals of Lambert.” It is panacinar type and usually affects the left upper lobe.
-
Foreign body emphysema: it is lung hyperinflation due to an obstructed bronchus. It’s a reversible airway obstruction.
-
Subcutaneous (surgical) emphysema: it is defined as collection of air at the level of subcutaneous tissues superficial to the deep fascia that covers the skeletal muscle plane. This type is commonly seen after trauma to the trachea or the esophagus in car accidents, stab wounds, or gunshot wounds. It can also be seen in intensive patients on a positive airway pressure ventilator. Air-leak syndrome is a term used to describe generalized thoracic air leak that includes subcutaneous emphysema, pneumomediastinum, and pneumopericardium, with or without pneumothorax.
Signs on Chest Radiographs
-
Lung hyperinflation, which is detected as posterior rib counts >10 ribs, anterior rib count >7 ribs, and increased intercostals spaces distance.
-
Prominent hilar vessels with disappearance of the peripheral vessels.
-
Increase in the retrosternal trans-radiant area size on lateral radiographs, which is the area behind the sternum where the two lungs come in contact. This space is usually up to 3 cm deep. An increase in this area above 3 cm might indicate emphysema (Fig. 7.5.42).
-
Deep sulcus sign: the costophrenic angle deepens due to lung hyperinflation (Fig. 7.5.43).
-
Flattening of the diaphragm with barrel (funnel-shaped) chest configuration (Fig. 7.5.44).
-
Bulla is visualized as a hyperlucent area surrounded by a thin wall.
-
Compensatory emphysema: a hyperinflated lung with a part herniating into the other side of the chest to compensate an area of lung deficiency or atelectasis (Fig. 7.5.43).
-
Vanishing lung syndrome: bilateral upper zones giant bullae (Fig. 7.5.45).
-
Foreign body emphysema: unilateral hyperinflated lung usually with radio-opaque structure located at the areas of the main bronchi or trachea.
-
Subcutaneous emphysema: radiolucent air is visualized under the skin and around the muscles (Fig. 7.5.46).

Lateral plain chest radiograph of a patient with congenital α-1 antitrypsin deficiency disease shows increased retrosternal space due to emphysema (arrowheads)

Posteroanterior plain chest radiograph of a patient with postpneumonectomy of the right lower lung lobe shows compensatory emphysema of the left lung with deep sulcus sign (arrowhead)

Posteroanterior plain chest radiograph (a) and coronal chest HRCT (b) of two patients with congenital α-1 antitrypsin deficiency disease shows flattened diaphragm in (a) (arrowheads), and bilateral panlobular and centrilobular emphysema in (b)

Posteroanterior plain chest radiograph of a patient with vanishing lung syndrome shows bilateral giant upper lobes bullae (arrowheads)

Anteroposterior plain chest radiograph of a patient with subcutaneous emphysema shows air that surrounds the pectoralis muscle fibers bilaterally
Signs on HRCT and Conventional CT
-
Centrilobular emphysema appears as focal, oval, or round areas of low attenuation up to 1 cm in diameter, within a homogenous background of lung parenchyma (Fig. 7.5.47), and not associated with fibrosis. It has a characteristic of upper lung zone predominance.
-
Panlobular emphysema appears as large, uniform low-attenuation areas with characteristic lower lung zone predominance (Fig. 7.5.47).
-
Paraseptal emphysema appears as multiple small areas of low attenuation located typically at lung peripheries with subpleural location (Fig. 7.5.47). It has thin walls and should not be confused with honeycombing, which is characterized by thick wall bronchiectasis, with signs of fibrosis and architectural distortion.
-
Irregular emphysema appears as air bullae trapped within areas of fibrosis.
-
Congenital α-1 antitrypsin deficiency disease is typically associated with signs of liver cirrhosis, with risks of developing hepatocellular carcinoma. Patients are typically in their 40s presenting with dyspnea with signs of hepatic dysfunction.
-
Air-leak syndrome is detected as generalized subcutaneous emphysema, pneumomediastinum, parenchymal emphysema, and pneumopericardium, with or without pneumothorax (Fig. 7.5.48).

Axial thoracic HRCT illustration demonstrates types of emphysema on HRCT: (1) centrilobular, (2) paraseptal, and (3) panlobular

Axial thoracic HRCT of a female patient with air-leak syndrome shows right panlobular emphysema, subcutaneous emphysema affecting the thoracic wall and breasts bilaterally, and mild pneumopericardium
Further Reading
-
Bergin C, et al. The secondary pulmonary lobule: normal and abnormal CT appearance. AJR Am J Roentgenol. 1988;151:21–5.
-
Kazerooni EA, et al. Imaging of emphysema and lung volume reduction surgery. Radiographics. 1997;17:1023–36.
-
Marti de Gracia M, et al. Subcutaneous emphysema: diagnostic clue in the emergency room. Emerg Radiol. 2009;16:343–8. doi:10.1007/s10140–009–0794-x.
-
Stern EJ, et al. CT of the lung in patients with pulmonary emphysema: diagnosis, quantification, and correlation with pathologic and physiologic findings. AJR Am J Roentgenol. 1994;162:791–8.
-
Thurlbeck WM, et al. Radiographic appearance of chest in emphysema. AJR Am J Roentgenol. 1978;130:429–40.
-
Thurlbeck WM, et al. Emphysema: definition, imaging, and quantification. AJR Am J Roentgenol. 1994;163:1017–25.
-
Yamanoha A, et al. Air-leak syndrome associated with bronchiolitis obliterans after allogeneic peripheral blood stem cell transplantation. Int J Hematol. 2007;85:95–6.
7.6 Idiopathic Interstitial Pneumonias
Idiopathic interstitial pneumonias (IIPs) are a group of diseases characterized by parenchymal lung fibrosis. IIPs are classified by the American Thoracic Society (ATS) and the European Respiratory Society (ERS) into seven disease entities: idiopathic pulmonary fibrosis, nonspecific interstitial pneumonia (NSIP), cryptogenic organizing pneumonia (COP), respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), desquamative interstitial pneumonia (DIP), lymphoid interstitial pneumonia (LIP), and acute interstitial pneumonia (AIP).
Although the ATS-ERS classification differentiates between the subtypes of IIPs based on histopathology findings, radiology can help in the diagnosis assessment based on the computed tomography findings of each disease. Lung extension can be characteristic for some IIP subtypes.
Patients with IIPs generally present with progressive dyspnea, cough, and other nonspecific respiratory symptoms.
7.6.1 Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF), also known usual pulmonary fibrosis, is a disease characterized by lung fibrosis with unknown cause.
Patient with IPF is typically a 50-year-old patient presenting with progressive dyspnea and nonproductive cough. Clinical examination may show signs of chronic cyanosis, finger clubbing, and basal lung crepitation on auscultation. Diagnosis of IPF by histology is very important because IPF patients usually do not respond to high corticosteroid therapy, with a median survival time ranging from 2 to 4 years after starting symptoms.
History of smoking can be a risk factor for IPF. However, it does not affect the course of the disease.
Signs on Radiographs and HRCT
-
Typically, patients with IPF present with reticular interstitial pattern with reduced lung volume, subpleural reticular opacities, and macrocytic honeycombing (bronchiectatic changes). The distribution of the lung fibrosis characteristically involves the lung bases and decrease toward lung apices (apicobasal gradient) (Figs. 7.6.49 and 7.6.50).
-
Shaggy heart appearance is a term used to describe fibrosis silhouetting the heart borders (Fig. 7.6.50).

An illustration demonstrates the different types of idiopathic interstitial pneumonias (IIPs) and their pathological distribution patterns: (a) idiopathic pulmonary fibrosis, (b) nonspecific interstitial pneumonia (NSIP), (c) cryptogenic organizing pneumonia (COP), (d) respiratory bronchiolitis-associated interstitial lung disease (RB-ILD), (e) lymphoid interstitial pneumonia (LIP), and (f) acute interstitial pneumonia (AIP)

Posteroanterior chest radiograph of a patient with idiopathic pulmonary fibrosis (IPF) shows bilateral reticular interstitial lung pattern located mainly at the base with gradient crawling toward the apices (arrowheads). Notice the shaggy heart appearance
7.6.2 Nonspecific Interstitial Pneumonia
NSIP is a disease with lung fibrosis that is usually difficult to differentiate from IPF. However, differentiating IPF from NSIP is important, since the latter has a better response to high corticosteroid therapy.
Patients with NSIP are typically seen in their 40s with signs and symptoms similar to IPF. NSIP has no obvious relation with cigarette smoking. NSIP may be encountered with other systemic disorders (e.g., connective tissue disorders).
Signs on Radiographs and HRCT
Patients with NSIP show patchy subpleural reticulonodular pattern, bilateral almost homogenous lung involvement, and microcytic honeycombing (Fig. 7.6.49). The main differences between IPF and NSIP are the lack of the apicobasal gradient involvement (seen in IPF) and the macrocytic honeycombing (also seen in IPF).
7.6.3 Cryptogenic Organizing Pneumonia
COP, formerly known as bronchiolitis obliterans with organizing pneumonia (BOOP), is a chronic pulmonary disease characterized by bronchiolar inflammation (bronchiolitis) and obstruction by a polypoid plug of granulation tissue formation (obliterans). The granulation tissue blocks the small airways proximal to the alveoli resulting in patchy parenchymal disease. Pneumonia often develops in bronchiolitis obliterans due to inflammation of the surrounding parenchyma as a consequence to the bronchiolitis (organizing pneumonia).
The bronchioles are classified into terminal bronchioles and respiratory bronchioles. A disease involving the terminal bronchioles will result in a clinical picture that resembles a conductive airways disease. In contrast, when the respiratory bronchioles are affected by a disease, a clinical picture resembles restrictive airway disease that arises because the adjacent alveoli are affected too. COP is a disease of the respiratory bronchioles.
Most cases of COP are unknown and seen in patients between 40 and 60 years of age. COP in adults can arise secondary to a variety of causes such as chronic aspiration pneumonia, radiation therapy, bone marrow transplant, medications (e.g., amiodarone), and connective tissue disorders (e.g., rheumatoid arthritis). Most patients with COP are nonsmokers or ex-smokers.
Patients usually present with persistent nonproductive dry cough that resists antibiotics for duration that can last up to months. Dyspnea, low-grade fever, malaise, and weight loss are other common features. Lab results usually show elevated erythrocyte sedimentation rate (ESR) and C-reactive proteins, with restrictive pattern on pulmonary function tests.
Signs on Radiographs
-
Chest radiographs show peripheral lung field patchy infiltration that can be unilateral or bilateral, often with basilar predominance (Figs. 7.6.49 and 7.6.51).
-
Bilateral interstitial, reticulonodular pattern may be seen.

Anteroposterior chest radiograph of a patient with bone marrow transplant due to leukemia who developed COP shows bilateral patchy infiltrations located at the lung bases with peripheral patchy infiltration
Signs on HRCT
-
The typical HRCT picture of COP is bilateral, patchy, triangular areas of consolidation located in the peripheral subpleural areas (60–90 % of cases) (Fig. 7.6.52). Also, peribronchial patchy consolidations located in the lower lobes are also a common presentation.
-
Bilateral, scattered ground-glass appearance opacities with thickened interlobular septa can be seen in up to 60 % of cases (Fig. 7.6.52). These areas are hyperdense in cases of amiodarone toxicity due to the presence of iodine in the drug.
-
Another uncommon presentation of COP is a focal parenchymal mass often located in the upper lobes in contact with the pleura and fissures (30 % of cases). This presentation cannot be differentiated from cancer by imaging alone; biopsy is needed to confirm the diagnosis.
-
COP also can present as multiple, mass-like parenchymal lesions with speculated margins, another presentation that may mimic metastasis, infections, or lymphoma. Biopsy is needed to confirm the diagnosis. This pattern can be produced by therapy with bleomycin in cancer patients.
-
Bronchocentric COP appears as areas of parenchymal consolidation around the bronchovascular bundle (33 % of cases). This pattern resembles the HRCT picture of patients with vasculitis (e.g., Churg–Strauss syndrome) (Fig. 7.6.52).
-
Atoll sign is seen as an area with ground-glass opacity surrounded by a ring of increased density parenchyma. This sign is typical of COP (Fig. 7.6.52).
-
Band-like opacities are threadlike opacities that run from the bronchi toward the pleura; they may show air-bronchogram sign.

Axial thoracic lung-window HRCT demonstrates the different manifestations of COP: (1) peripheral classical patchy infiltration of COP, (2) bronchogenic COP, (3) bronchocentric COP, (4) thickened interlobar septae, and (5) Atoll sign
7.6.4 Respiratory Bronchiolitis-Associated Interstitial Lung Disease
RB-ILD is a disease that is considered as an exaggerated form of respiratory bronchiolitis, and it is a smoking-related condition. Also, RB-ILD is considered as the early stage of DIP.
Patients with RB-ILD are commonly males in their 30s or 40s with history of chronic smoking. Smoking cessation is an important element in the medical management of RB-ILD.
Signs on Radiographs and HRCT
-
Chest radiograph can be normal.
-
On HRCT, the key findings in RB-ILD are centrilobular nodules in combination with ground-glass opacities and bronchial wall thickening predominantly located in the upper lung zones (Fig. 7.6.49).
7.6.5 Desquamative Interstitial Pneumonia
DIP is a condition that is considered as a severe form of RB-ILD, and it is strongly associated with cigarette smoking. However, it can arise in nonsmokers due to variety of conditions (e.g., exposure to organic dust). Patients with DIP are often between 30 and 40 years old.
Signs on Radiographs and HRCT
-
Radiographic findings are nonspecific.
-
CT finding shows diffuse ground-glass opacity that is predominantly located peripherally and in the lower lobes. However, features overlapped with RB-ILD may be seen.
7.6.6 Lymphoid Interstitial Pneumonia
LIP is a disease characterized by lymphoid tissue proliferation and infiltration of the pulmonary interstitium by lymphocytes.
The normal lymphoid system of the lung is composed of four components:
-
Bronchus-associated lymphoid tissue (BALT): it consists of submucosal lymphoid follicles distributed along distal bronchi and bronchioles, usually at the bifurcation. This complex is analogous to other mucosa-associated lymphoid tissue (MALT) such as Peyer’s patches in the intestine.
-
Hilar lymph nodes: these are seen along the trachea and at the lung helium.
-
Intrapulmonary lymph nodes: they are composed of noncapsulated lymphocytes clusters, usually located in the subpleural parenchyma.
-
Interstitial lymphocytes: they are seen within the lung interstitium with the pulmonary venules.
LIP is a disease characterized by interstitial lymphocyte proliferation, resulting in an interstitial lung disease. It arises commonly secondary to systemic autoimmune disease (e.g., Sjögren’s syndrome) and rarely as idiopathic disease. Most patients are middle aged, who often present with systemic symptoms, dyspnea, and cough. Almost all patients have dysproteinemia, usually polyclonal hyper- or hypogammaglobulinemia.
Signs on Radiographs and HRCT
-
Radiographic signs are nonspecific reticulonodular interstitial pattern that is commonly diffuse.
-
On CT, LIP shows ground-glass opacities plus multiple fine lung cysts located mainly at the center of mid- and lower lung zones (Fig. 7.6.49). The combination of systemic disease, ground-glass opacities, and small lung cysts at the mid- and lower zones are suggestive criteria of LIP.
7.6.7 Acute Interstitial Pneumonia (Hamman–Rich Syndrome)
AIP is a rare fulminant form of lung disease that occurs in previously healthy individuals. Patients present with signs of acute respiratory distress syndrome (ARDS), fever, and cough with rapid deterioration suggesting pneumonia-like illness.
Patients with AIP are usually lung disease-free and are over 40 years old. Most patients develop severe dyspnea that requires mechanical ventilation. The condition is treated with corticosteroid, with a mortality rate that reaches >50 % of cases.
Signs on Radiographs and HRCT
Patients show the radiographic signs of ARDS with lower zone predominance bilaterally and may show spared costophrenic angles (Fig. 7.6.49).
If You Were Given One Investigation to Detect Lung Fibrosis Cause, What Would You Choose?
-
Invasive test: open lung biopsy
-
Noninvasive: HRCT
Further Reading
-
Arakawa H, et al. Bronchiolitis obliterance with organizing pneumonia versus chronic esinophilic pneumonia: high resolution CT findings in 81 patients. AJR. 2001;176:1053–8.
-
Desai SR, et al. Traction bronchiectasis is cryptogenic fibrosing alveolitis: associated computed tomographic features and physiological significance. Eur Radiol. 2003;13:1801–8.
-
Fellrath JM, et al. Idiopathic pulmonary fibrosis/cryptogenic fibrosing alveolitis. Clin Exp Med. 2003;3:65–83.
-
Ghanei M, et al. Bronchiolitis obliterance following exposure to sulfur mustard: chest high resolution computed tomography. Eur J Radiol. 2004;52:164–9.
-
Gibson M, et al. Lymphocytic disorders of the chest: pathology and imaging. Clin Radiol. 1998a;53:469–80.
-
Gibson M, et al. Lymphocytic disorders of the chest: pathology and imaging. Clin Radiol. 1998b;53:469–80.
-
Katzenstein AA, et al. Diagnosis of usual intestitial pneumonia and distinction from other fibrosing interstitial lung diseases. Human Pathol. 2008;39:1275–94.
-
Mueller-Mang C, et al. What every radiologist should know about idiopathic interstitial pneumonias. Radiographics. 2007;27:595–615.
-
Polverosi R, et al. Organizing pneumonia: typical and atypical HRCT patterns. Radiol Med. 2006;111:202–12.
-
Sharief N, et al. Fibrosing alveolitis and desquanative interstitial pneumonitis. Pediatr Pulmonol. 1994;17:359–65.
-
Tempone V, et al. Bronchiolitis obliterance organizing pneumonia secondary to chronic aspiration of pharmaceutical tablets: radiologic-pathologic correlation. Eur J Radiol (Extra). 2008;67:e99–101.
7.7 Histiocytoses
Histiocytoses are a group of diseases characterized by abnormal proliferation and multiorgan infiltration by histiocytes. Different diseases and clinical presentations fall under the umbrella of histiocytoses.
Histiocytes are bone marrow-derived cells, and they fall into two main groups: the mononuclear phagocytes (macrophages) and dendritic cells. Macrophages are part of the immune system, and their main function is to engulf bacteria and damaged tissues (phagocytosis). They are found in all body organs like the liver (Kupffer cells), brain (microglial cells), etc. In contrast, dendritic cells are a cell family that includes Langerhans cells, interdigitating reticulum cells, and follicular dendritic cells. They are found in the reticuloendothelial system, and their main function is to activate the major histocompatibility complex (MHC)-restricted T cells by expressing high levels of MHC class II molecules. The reticuloendothelial system includes the liver, spleen, and lymph nodes.
Histiocytoses are classified into three main classes:
-
Class 1 histiocytoses: Langerhans cell histiocytosis (LCH).
-
Class 2 histiocytoses: Infection-associated hemophagocytic syndrome, Rosai–Dorfman’s syndrome, and Omenn syndrome (OS).
-
Class 3 histiocytoses: True malignant proliferation and include acute monoblastic leukemia and true histiocytic lymphoma.
There are other diseases classified as non-LCH and include Erdheim–Chester disease (ECD) and xanthoma disseminatum (Montgomery syndrome).
7.7.1 Langerhans Cell Histiocytosis
LCH is a disease of unknown origin characterized by proliferation and body infiltration by nonmalignant histiocytes (macrophages and dendrites cells).
The basic lesion in LCH is a granuloma composed of Langerhans cells and lymphocytes (proliferative stage). Next, the granuloma becomes necrotic, with admixture of eosinophils and sometimes multinucleated giant cells (granulomatous stage). Finally, fibrosis of the granuloma occurs with deposition of lipid-laden histiocytes (xanthogranulomatous stage). Electron microscopy reveals characteristic rod-shaped bodies in the cytoplasm of the cells (Birbeck granules).
LCH is a broad spectrum of overlapped syndromes with multiple systemic manifestations. Characteristic bone manifestations include sharply defined, punched-out lytic bony lesions and vertebra plana. Lung involvement is common and shows diffuse reticulonodular interstitial pattern of involvement, cystic bronchiectasis, and pneumothorax. Central nervous system (CNS) involvements mainly affect the hypothalamic–pituitary axis resulting in diabetes insipidus. Other CNS involvements include white matter lesions that are often seen in the cerebellum and the pyramidal tracts, causing ataxia in advanced stage of the disease. Skin involvement is characterized by reddish-brown thoracic and pelvic purperic papules. When these purperic papules are found in a newborn, the condition is called blueberry muffin baby, which is commonly seen in neonates with congenital infection (TORCH) and congenital leukemia (Fig. 7.7.53).

An illustration demonstrates the purpuric papules in a neonate with blueberry muffin baby condition
The disease can be seen in pediatric and adult patients in a localized or diffuse form. In pediatrics, the disease presents in three main forms: eosinophilic granuloma (EG, local form), Hand–Schüller–Christian’s disease (HSCD, chronic form), and Letterer–Siwe disease (LSD, acute form). In adults, the most severe presentation is pulmonary LHC.
Eosinophilic granuloma (EG) is a solitary localized lytic lesion of the bone, which is typically seen in children <15 years of age. Children EG can be asymptomatic or present with pain, swelling, and fracture at the site of the lesion. The bony lesions are classically found in flat bones, such as the skull, mandible, and pelvis. When EG affects long bones, the lytic lesions are typically seen in the diaphyses and maybe the metaphyses. ES affecting the epiphyses is rare.
Hand–Schüller–Christian’s disease (HSCD) is a disease that is seen in children <10 years of age and characterized by a triad of exophthalmos, diabetes insipidus, and hepatosplenomegaly. Cases of HSCD may occur between 20 and 30 years of age. Other manifestations of HSCD include anemia, scaly seborrheic skin rash, restrictive lung diseases (fibrosis), and cerebellar ataxia. Osteolytic bony lesions like EG can be seen.
Letterer–Siwe disease (LSD) is a disease seen in children <2 years old and characterized by hepatosplenomegaly, lymphadenopathy, and sclerosing cholangitis. In LSD, the child grows normally from birth until 2 years of age, where the disease starts to manifest. LSD is the most aggressive form of LCH. Mental retardation, multiple bony fractures, hemorrhagic rash, anemia, and thrombocytopenia may be seen.
Adult pulmonary Langerhans’ cell histiocytosis (PLCH) affects 1–2 cases per million, and it is often seen in young smokers. There is a strong association between PLCH and cigarette smoking. Cigarette smoking was found to increase the number and accumulation of dendritic cells and Langerhans’ cells within the alveolar epithelium in smokers. Most patients with PLCH are asymptomatic. Symptomatic PLCH patients present with dyspnea (35–87 %), pleuritic chest pain (9–18 %), nonproductive cough (50–70 %), pneumothorax (25 %), and fever (15 %).
Signs on Plain Skeletal Radiographs
-
In the clavarium, EG classically presents as osteolytic lesion with geographic, punched-out, sharply defined border measuring 1–4 cm in diameter (Fig. 7.7.54). The lytic lesion may contain bony sequestrum or bony fragments resembling osteomyelitis. Other lesions show osteolytic lesion with bony sequestrum and include bone lymphoma and fibrosarcoma.
-
EG of the long bone is classically present as punched-out lytic lesion in the diaphysis or the metaphysis. Cortical tunneling, permeative cortical destruction, and periostitis may be seen occasionally, giving the lesion a malignant feature. In the vertebrae, a punched-out lytic lesion is commonly observed (Fig. 7.7.55).
-
Vertebra plana (pancake vertebra) is a term that describes a vertebra with severe flattening. In a young patient, the most common causes are Langerhans cell histiocytosis (eosinophilic granuloma) or trauma. In an old patient, the most common causes are multiple myeloma or trauma. Typically, there is vertebral body flattening with normal adjacent vertebral disks and sparing of the posterior elements.
-
Jaw lesions in EG or HSCD (20 %) can present as radicular cysts, periodontal disease, osteomyelitis, and sharply, expansile, punched-out alveolar lesions that spare the roots, making the teeth appear as if they are “floating in air.”

Lateral plain skull radiograph shows two sharply lytic, punched-out lesions with geographic edges (arrows) in a child with eosinophilic granuloma

Anteroposterior thoracic vertebral plain radiograph shows punched-out lytic lesion affecting the vertebral body in a patient with Langerhans cell histiocytosis (LHC) (arrowhead)
Signs on Chest Radiographs and HRCT
-
In early PLCH, lung fields show small nodules (1–10 mm in diameter) with irregular borders. The nodules are predominantly seen in the upper and mid-lung zones, sparing the lung bases (Fig. 7.7.56).
-
Advanced PLCH shows reticulonodular interstitial pattern and cystic bronchiectasis. The cystic interstitial patterns mimic that of bullous emphysema or lymphangiomyomatosis; the latter is typically seen in tuberous sclerosis. The cysts usually measure 2–3 cm in diameter (Fig. 7.7.57).
-
Pneumothorax can be seen in 25 % of cases (Fig. 7.7.57).
-
Hilar lymphadenopathy can be seen in rare cases.

Posteroanterior chest radiograph of a patient with adult pulmonary Langerhans’ cell histiocytosis (PLCH) shows multiple pulmonary nodules with different sizes located at the right upper lung zone (arrowheads)

Axial HRCT illustration of a patient with PLCH shows diffusely bronchiectatic, cystic changes of the lung parenchyma in a bilateral pattern with right pneumothorax
Signs on MRI
-
In the sella, the pituitary stalk is typically thickened and shows contrast enhancement in cases of diabetes mellitus (HSCD).
-
On MRCP, sclerosing cholangitis in LSD shows the absence of the peripheral biliary radicals with intrahepatic biliary tree stenosis. Notice that these findings are seen in a child.
-
Vertebra plana is seen as severely flattened vertebral body (Fig. 7.7.58).

Sagittal thoracic vertebral MRI shows vertebra plana (arrowhead)
7.7.2 Infection-Associated Hemophagocytic Syndrome
Infection-associated hemophagocytic syndrome is a disease characterized by histiocytes hyperplasia (increased numbers), often due to viral infection (e.g., human immunodeficiency virus).
Patients present with high spiking fever, jaundice, lethargy, and generalized lymphadenopathy and hepatomegaly. Laboratory investigations show hemophagocytosis, hypertriglyceridemia, pancytopenia, and consumptive coagulopathy.
7.7.3 Omenn Syndrome
Omenn syndrome (OS) is a rare, autosomal recessive, non-Langerhans cell histiocytosis disorder characterized by severe combined immunodeficiency (SCID), erythroderma, hepatosplenomegaly, lymphadenopathy, and alopecia.
Infants with OS typically present in the first few years of life with generalized cutaneous lesions composed of red, exfoliative dermatitis that involves almost the whole body (erythroderma) (Fig. 7.7.59). Erythroderma in infants combined with splenomegaly or lymphadenopathy is diagnostic of OS.

An illustration demonstrates the features of Omenn syndrome in a neonate
Laboratory investigations may show high serum levels of IgE and hypogammaglobulinemia.
7.7.4 Chédiak–Higashi Disease
Chédiak–Higashi disease (CHD) is an autosomal recessive, rare disorder characterized by oculocutaneous albinism, increased susceptibility to infections due to immunodeficiency, silvery gray hair, photophobia, neurological impairment, and abnormal giant lysosomes in the leukocytes. Most patients with CHD develop lymphoma-like phase characterized by widespread lymphohistiocytic infiltrates in the lymphoreticular organs (85 %).
CHD presents in two main forms: childhood and adult forms. The childhood form is characterized by recurrent pyogenic infections and hepatosplenomegaly. In contrast, the adult form presents in early adulthood with various neurological manifestations that include Parkinsonism, dementia, spinocerebellar degeneration, and peripheral neuropathy.
Skin hyperpigmentation after exposure to sunlight is a characteristic initial feature of CHD. Increased susceptibility to infections in CHD patients is attributed to the defective function of neutrophils. Parental consanguinity is a common feature of CHD.
7.7.5 Differential Diagnoses and Related Diseases
-
Griscelli disease is a rare condition characterized by abnormal transfer of melanin granules resulting in light skin and silver hair (Fig. 7.7.60). Griscelli syndrome may be accompanied by neurological abnormalities (type 1), immunodeficiency and neutropenia (type 2), or no other abnormalities (type 3). Patients with Griscelli disease are typically children presenting with characteristic silver hair in addition to febrile episodes, lymphadenopathy, hepatosplenomegaly, anemia, and pancytopenia. Patients may develop seizures and hemiparesis. Brain CT may show brain atrophy. Griscelli disease can be initially mistaken with CHD.
Fig. 7.7.60 
An illustration demonstrates the features of Griscelli disease
-
Elejalde syndrome (neuroectodermal melanolysosomal syndrome) is a very rare disease characterized by silvery gray hair, bronze-colored skin, profound CNS dysfunction, and a normal immune system. Neurological abnormalities in Elejalde syndrome include severe hypotonia or hyperreflexia, spastic hemi- or quadriplegia, ataxia, seizures, and profound developmental delay. Some investigators believe that Elejalde syndrome and Griscelli syndrome type 1 are the same disease. However, Elejalde syndrome lacks the immunological abnormalities commonly seen in patients with Griscelli and Chédiak–Higashi syndromes.

7.7.6 Rosai–Dorfman’s Disease (Sinus Histiocytosis)
Rosai–Dorfman’s disease (RDD) is a rare benign disease characterized by idiopathic proliferation of the phagocytes within the lymph nodes sinuses.
Patients with RDD are typically male children presenting with bilateral massive cervical lymphadenopathy (Fig. 7.7.61) and low-grade fever, with or without exophthalmos. Extranodal manifestation like any other histiocytosis disease can involve any part of the body.

An illustration demonstrates the features of s’s disease (RDD)
Pathologically, the enlarged lymph nodes show infiltration of the sinuses by large histiocytes. The histiocytes contain engulfed lymphocytes and plasma cells (emperipolesis), which is the hallmark of this disease. Laboratory investigations show leukocytosis, high erythrocyte sedimentation rate, and hypergammaglobulinemia. Burkitt’s lymphoma should be ruled out before establishing the diagnosis of RDD. RDD generally resolves without treatment after several months. However, RDD transformation into true malignant lymphoma may occur.
7.7.7 Xanthoma Disseminatum (Montgomery Syndrome)
Xanthoma disseminatum (XD) is a rare, nonmalignant form of non-Langerhans cells histiocytosis characterized by body xanthomata formation that includes the skin, trachea, and the larynx in a normal lipid profile patient. Other manifestations include hypopituitarism, diabetes insipidus (40 % of patients), multiple osteolytic lesions seen on plain radiographs, and expansile cystic lesions in the bones of the hands. Patient’s brain MRI may show nongliotic brain mass with heterogeneous contrast enhancement. Moreover, multiple spinal cord lesions with heterogeneous high T2 signal intensities and intense contrast enhancement may be found. CT of the neck and the thorax may show laryngeal and tracheal wall thickening.
7.7.8 Erdheim–Chester Disease (Lipogranulomatosis)
ECD is a rare, non-Langerhans cell histiocytosis characterized by almost constant bone lesions and extraosseous manifestations.
ECD affects patients with wide age range (7–78 years). Patients often present with painless bilateral exophthalmos, progressive cerebellar ataxia, diabetes insipidus, and bone pain. Bone involvement is found in 60 % of patients, and it is a diagnostic criteria. Diagnosis of ECD is based on the presence of characteristic bony lesions, visceral involvement (especially the retroperitoneal structures), and pathological findings.
ECD is characterized by the formation of lipid granulomas. The cerebral manifestations of ECD are similar to other histiocytoses. Laboratory findings are usually unremarkable, and cerebrospinal fluid analysis is usually normal.
Signs on Plain Radiographs
-
Bilateral symmetrical osteosclerotic lesions affecting the metaphyses (83 %) and the diaphyses of long bones (98 %), with relative sparing of the epiphyses. The corticomedullary junction is blurred, and the bone marrow is obliterated by dense bone. These lesions classically spare the axial skeleton, hands, and feet.
-
Radiolucent bands separating the sclerotic metaphysis from the sclerotic diaphysis can be found.
-
In <10 % of ECD cases, the bony lesions can be purely lytic with no osteosclerosis.
Signs on MRI
-
The affected long bones diaphyses and metaphyses exhibit low T1 and T2 signal intensities due to osteosclerosis. Periostitis may be seen in some cases. After contrast injection, enhancement of the periosteum is seen as a white line along the bone margin.
-
In CNS, multiple contrast-enhanced lesions on T1W images affecting the meninges, eye, and pituitary are typically found (this combination is very characteristic of ECD).
-
Orbital involvement in ECD is seen in the form of retro-orbital masses or diffuses infiltration of the retro-orbital fat (causing exophthalmos).
7.7.8.1 How Can You Differentiate ECD from HSCD?
-
ECD presents with osteosclerotic lesions, whereas HSCD presents with osteolytic lesions.
-
ECD affects patients in the range of 7–78 years, whereas HSCD affects children <10 years old.
-
Retroperitoneal lesions are almost a characteristic finding in ECD.
-
On pathology, ECD shows lipid-laden histiocytes.
Further Reading
-
Abbott GF, et al. Pulmonary Langerhans cell histocytosis. Radiographics. 2004;24:821–41.
-
Albayram S, et al. Spinal dural involvement in Erdheim-Chester disease: MRI findings. Neuroradiology. 2002;44:1004–7.
-
Alexander AS, et al. Xanthoma disseminatum: a case report and literature review. Br J Radiol. 2005;78:153–7.
-
Baron J, et al. Xanthoma disseminatum: a rare cause of upper airway narrowing. AJR. 2003;180:1180–1.
-
Burihan J, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479–82.
-
Caparros-Lefebvre D, et al. Neuroradiologic aspects of Chester-Erdheim disease. AJNR. 1995;16:735–40.
-
Dion E, et al. Bone involvement in Erdheim-Chester disease: imaging findings, including periostitis and partial epiphyseal involvement. Radiology. 2006;238:632–9.
-
Doyle DJ, et al. Imaging of multisystem Langerhans cell histocytosis in an adult. Eur J Radiol (Extra). 2007;61:109–17.
-
Haraldsson Á, et al. Griscelli disease with cerebral involvement. Eur J Pediatr. 1991;150:419–22.
-
Hauser RA, et al. Adult Chediak-Higashi parkinsonian syndrome with dystonia. Mov Disord. 2000;15:705–8.
-
Hoover KB, et al. Langerhans cell histiocytosis. Skeletal Radiol. 2007;36:95–104.
-
Hurvitz H, et al. A kindred with Griscelli disease: spectrum of neurological involvement. Eur J Pediatr. 1993;152:402–5.
-
Ivanovich J, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313–6.
-
Jurić G, et al. Extranodal sinus histiocytosis (Rosai-Dorfman disease) of the brain parenchyma. Acta Neurochir. 2003;145:145–9.
-
Kaneda T, et al. Langerhans cell histiocytosis in the mandible: computed tomography and magnetic resonance imaging. Oral Radiol. 1997;13:109–14.
-
Kashihara-Sawami M, et al. Letterer-Siwe disease: immunopathologic study with a new monoclonal antibody. J Am Acad Dermatol. 1988;18:646–54.
-
Kumar P, et al. Chediak-Higashi syndrome. Indian J Pediatr. 2000;67:595–7.
-
Leatherwood DL, et al. Pulmonary Langerhans cell histocytosis. Radiographics. 2007;27:265–8.
-
Malhotra AK, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337–40.
-
Meyer JS, et al. Langerhans cell histocytosis: presentation and evolution of radiologic findings with clinical correlation. Radiographics. 1995;15:1135–46.
-
Odell WD, et al. Xanthoma disseminatum: a rare cause of diabetes insipidus. J Clin Endocrinol Metab. 1993;76:777–80.
-
Pupo RA, et al. Omenn’s syndrome and related combined immunodeficiency syndrome: diagnostic considerations in infants with persistent erythroderma and failure to thrive. J Am Acad Dermatol. 1991;25:442–6.
-
Scolozzi P, et al. Multisystem Langerhans’ cell histocytosis (Hand-Schüller-Christian disease) in an adult: a case report and review of the literature. Eur Arch Otorhinolaryngol. 2004;261:326–30.
-
Shaffer MP, et al. Langerhans cell histiocytosis presenting as blueberry muffin baby. J Am Acad Dermatol. 2005;53:S143–6.
-
Simanski C, et al. The Langerhans’ cell histocytosis (eosinophilic granuloma) of the cervical spine: a rare diagnosis of cervical pain. Magn Reson Imaging. 2004;22:589–94.
-
Stéphan JL. Histiocytoses. Eur J Pediatr. 1995;154:600–9.
-
Tamura T, et al. Congenital Letterer-Siwe disease associated with protein losing enteropathy. Eur J Pediatr. 1980;135:77–80.
-
Weidauer S, et al. Cerebral Erdheim-Chester disease: case report and review of the literature. Neuroradiology. 2003;45:241–5.
-
Young PM, et al. Rosai-Dorfman disease presenting as multiple soft tissue masses. Skeletal Radiol. 2005;34:665–9.
7.8 Hemoptysis
Hemoptysis is a term used to describe the pathological condition of coughing blood, typically due to lower respiratory tract disease. Hemoptysis can be a life-threatening condition, and the condition needs urgent evaluation.
The normal lung is supplied by pulmonary arterial system and bronchial arterial system. The pulmonary arteries supply the lungs and take part in gas exchange. In contrast, the bronchial arteries are small arteries (2 mm or less in diameter) that arise from the descending thoracic aorta and supply the trachea, the bronchial tree, the visceral pleura, the esophagus, and part of the mediastinal lymph nodes. Histologically, the two systems are connected by thin-walled capillary anastomoses.
The right bronchial artery arises at the level of the fifth or sixth thoracic vertebra posteriorly and usually forms a common trunk with the intercostals artery. The left bronchial artery arises from the descending thoracic aorta anteriorly, with a second left bronchial artery found in up to 70 % of population. On CT angiography scan, the right bronchial artery is seen as dots or lines of increased density located in the retrotracheal, retrobronchial, and retroesophageal regions (Fig. 7.8.62). The left bronchial artery is identified typically as a linear or nodular hyperdensity in the space below the aortic arch above the pulmonary artery (within the aortopulmonary window) (Fig. 7.8.62). Both bronchial arteries normally have highly tortuous course.

Sequential axial CTA illustration demonstrates the normal anatomy of the right bronchial artery (arrow in a) and the left bronchial artery (arrow in b)
The differential diagnosis of hemoptysis is diverse and can occur due to pulmonary infection (e.g., tuberculosis), malignancy (e.g., bronchogenic carcinoma), vascular event (e.g., pulmonary embolism), and vascular anomaly (e.g., arteriovenous malformation). Cryptogenic hemoptysis is a term used to describe hemoptysis with no identifiable cause, and it is responsible for up to 42 % of hemoptysis episodes, especially in smokers. Cryptogenic hemoptysis is a diagnosis of exclusion, and patients are often recommended to perform another CT several months later to exclude small occult neoplasm formation.
This topic discusses some of the causes of hemoptysis, in which radiology plays an important role in establishing their diagnosis.
7.8.1 Bronchopulmonary Sequestration
Bronchopulmonary sequestration (BPS) is a rare condition characterized by nonfunctioning lung parenchyma mass that is not in continuity with the tracheobronchial tree and is supplied by an anomalous systemic arterial vessel, typically arising from the abdominal aorta and ascending to the mass through the pulmonary ligament. The term sequestration describes disconnected lung tissue with its own anomalous systemic artery.
The primitive lung tissue mass of BPS has its own anomalous systemic vascular supply, usually from the aorta (not bronchial pulmonary circulation). Most cases are diagnosed before the age of 10 years, where the child presents with chronic cough, hemoptysis, and recurrent pneumonias. Hybrid lesion is a term used to describe a lesion, where the sequestered lung mass has a congenital cystic adenomatous malformation lesion within it. The definite diagnosis of hybrid lesion is by histopathology. BPS is divided into intralobar and extralobar forms.
In intralobar BPS, the mass is located inside the lung, surrounded by normal lung parenchyma, and shares the visceral pleura with the normal lung tissue. It accounts for 75 % of the sequestration cases. The mass is located on the left side in the posterior basal segment in 98 % of cases. The mass has its arterial supply from the descending aorta and its venous drainage from the azygos or systemic veins. In extralobar BPS, the mass usually lies within the pleura and has its own pleural layer and has the same radiological features as the interlobar sequestration. It accounts for 1–6 % of the sequestration cases. The mass receives its arterial supply from the thoracic or the abdominal aorta in 80 % of cases and is usually found between the lower lobes and the diaphragm. Most cases present within the first 6 months of life with dyspnea, cyanosis, and feeding difficulties.
Signs on CT
The scan shows a hyperdense pulmonary mass that may contain cystic changes or air–fluid level. The mass may appear as a consolidation or atelectatic mass. After contrast injection, an anomalous vessel arises from the abdominal aorta, or the descending thoracic aorta is seen penetrating the mass, which is a pathognomonic finding (Fig. 7.8.63). The sequestered mass may show homogenous or inhomogeneous contrast enhancement.

Axial (a) and coronal (b) chest CT shows left lower lobe intralobar bronchopulmonary sequestration mass (arrowheads) with its abnormal arterial supply arising from the descending thoracic aorta (arrows)
7.8.2 Anomalous Systemic Artery Supplying Normal Lung Parenchyma
Anomalous systemic artery supplying normal lung parenchyma is a condition characterized by a normal lung parenchyma supplied by anomalous artery in the absence of congenital heart or lung disease. Unlike bronchopulmonary sequestration, there is no abnormal tissue mass, and the vessel is not typically arising from the abdominal aorta. The condition is sometimes referred to as pseudosequestration.
Adult patients with the anomalous systemic artery can be asymptomatic or present with recurrent hemoptysis. In contrast, pediatric patients with this condition often present with cardiac murmur.
Signs on CTA
-
The normal bronchial arteries are <2 mm in diameter and arise directly from the descending thoracic aorta between the levels of T5 and T6 thoracic vertebrae (orthotopic origin). Anomalous bronchial arteries are defined as arteries that originate outside the range of T5 and T6 thoracic vertebrae (ectopic origin). Hypertrophied bronchial arteries are visualized as nodular or linear mediastinal or retrobronchial dilated vessels (>2 mm in size) with early enhancement and density similar to the aorta. The hypertrophied bronchial arteries are classically detected in the retrotracheal area, retroesophageal area, aortopulmonary window, or the posterior wall of the main bronchus.
-
In anomalous systemic artery supplying normal lung parenchyma, a hypertrophied vessel is often identified in an area of normal lung parenchyma in the absence of signs of intra- or extrathoracic abnormal lung mass (differentiate it from bronchopulmonary sequestration) (Fig. 7.8.64). The anomalous systemic arteries usually are very tortuous and are not parallel to the bronchi (differentiate them from normal bronchial arteries).
-
Nonbronchial systemic arteries enter the lung parenchyma through the pulmonary ligament or the adherent pleura. Identification of dilated vessels within extrapleural fat associated with pleural thickening (>3 mm) and lung parenchymal abnormalities may be regarded as nonbronchial systemic arteries responsible for hemoptysis.

Axial CTA illustration demonstrates left pseudosequestration seen as multiple dilated vascular nodules surrounded by normal lung parenchyma (arrowhead)
7.8.3 Pulmonary Vasculitis
Pulmonary vasculitis is a group of disorders characterized by inflammation of the pulmonary vessels with granuloma formation. Patients are typically middle aged presenting with recurrent attacks of fever, dyspnea, and hemoptysis.
The most recognized pulmonary vasculitis diseases are Wegener’s granulomatosis (WG), Churg–Strauss syndrome (CSS) (allergic vasculitis and granulomatosis), and lymphomatoid granulomatosis.
Wegener’s granulomatosis (WG) is a disease characterized by the triad of febrile sinusitis, glomerulonephritis, and pulmonary vasculitis. Patients often present with dyspnea, rhinitis, otitis media, pleuritic chest pain, and hemoptysis. The presence of cytoplasmic pattern of antineutrophil cytoplasmic autoantibody (c-ANCA) in the patient’s serum is a high indicator of WG (96 % sensitive for active WG). The serum level of c-ANCA can be used to monitor the disease activity. Histopathologically, WG is characterized by chronic inflammation of the medium-sized and small pulmonary arteries, veins, and capillaries.
CSS is a disease characterized by the triad of asthma or allergic rhinitis, marked peripheral eosinophilia, and systemic vasculitis involving two or more extrapulmonary organs. Asthma is a disease characterized by reversible airways obstruction. Patients with CSS are presenting with pulmonary distress, hemoptysis, fever, gastrointestinal symptoms, and arthralgia occasionally. The typical laboratory findings include marked eosinophilia in the absence of parasitic disease and high levels of serum rheumatoid factor in 52 % of cases. Extrapulmonary vasculitis may be seen in the form of coronary vasculitis, renal-induced hypertension, glomerulonephritis, cerebral hemorrhage, and purpuric skin lesions. CSS can be differentiated from WG by the characteristic association with asthma (rarely encountered in WG), cardiac involvement (up to 47 % of CSS cases), and less severe paranasal sinus involvement in CSS. Moreover, patients with CSS show serological positivity of perinuclear antineutrophil cytoplasmic autoantibody (p-ANCA), while WG shows serological positivity of c-ANCA.
Lymphomatoid granulomatosis (LG) is a multisystemic disease characterized by lung manifestations (100 % of cases), skin and nervous system manifestations (up to 53 %), and renal disease (up to 40 %). It is characterized pathologically by angiocentric infiltration of the lymphoid tissues and the vessels by atypical lymphocytes, and a mixture of plasma cells and histiocytes, causing tissue destruction and necrosis. Patients typically present with severe generalized symptoms that can be confusing. There is no specific serological marker, and erythrocytes sedimentation rate can be normal in spite of active disease. The main diagnostic method is biopsy of the lesions.
Signs on Radiographs and HRCT
-
In WG, the early stages of the disease show reticular or nodular interstitial lung pattern mainly located at the lung basis. As the disease progresses, diffuse alveolar lung disease may be seen bilaterally due to pulmonary hemorrhage (Fig. 7.8.65). WG may cause areas of consolidation or nodules with a cavity formation in the center.
-
In CSS, the plain radiographs may be normal in 25 % of cases. The pathological signs are similar to WG, with the formation of multiple granulomatous nodules with or without central cavity likely to be seen (Fig. 7.8.66).
-
In LG, there are typically large lung mass-like opacities located within the lung parenchyma (due to pulmonary infarcts). Up to 80 % of cases present as multiple, bilateral nodules located within the middle and the lower lung lobes (Fig. 7.8.67). Unilateral involvement is unusual (21 % of cases). Subpleural nodules may be seen, with pleural effusion likely to be found in 40 % of cases. Mediastinal lymphadenopathy is unusual.

Anteroposterior plain chest radiograph of a Wegener’s granulomatosis (WG) patient in the intensive care unit shows bilateral diffuse alveolar lung disease due to pulmonary hemorrhage

Posteroanterior plain chest radiograph (a) and coronal HRCT (b) of a patient with Churg–Strauss syndrome (CSS) show bilateral nodular patchy lung infiltration due to vasculitis and granulomatosis

Sequential axial HRCT of a patient with lymphomatoid granulomatosis (LG) shows multiple lung masses located at the lung bases in the left lung in (a) and bilaterally in (b) (arrowheads)
7.8.4 Cardiac Bronchus
Cardiac bronchus is a rare congenital anomaly in which there is accessory bronchus that arises from the medial wall of the intermediate bronchus at its proximal third, but occasionally from the right main bronchus. The accessory bronchus runs medially and caudally toward the heart, hence the cardiac appellation.
Cardiac bronchus is not identified on plain chest radiographs and usually incidentally found on CT scans. The anomaly is asymptomatic; however, it may result in hemoptysis when it is infected.
Signs on CT
The cardiac bronchus is typically identified as a small accessory bronchus medial to the intermediate bronchus on the right lung (Fig. 7.8.68).

Axial HRCT illustration demonstrates the typical radiographic sign and location of the cardiac bronchus on CT (arrowhead)
7.8.5 Dieulafoy Disease
Dieulafoy disease is a very rare condition characterized by abnormally dilated submucosal vessels that are prone to bleed and classically described in the colon, small intestine, and the bronchi.
Dieulafoy disease can be seen with cases of chronic bronchitis. On bronchoscopy, the visualization of dilated submucosal blood vessels in the presence of mucosal dilatation should alert the bronchoscopist of the possibility of Dieulafoy disease. Dieulafoy disease can be the case of massive upper gastrointestinal bleeding in 1–2 % of cases.
Further Reading
-
Ahmed M, et al. Multislice CT and CT angiography for noninvasive evaluation of bronchopulmonary sequestration. Eur Radiol. 2004;14:2141–3.
-
Bentala M, et al. Cardiac bronchus: a rare cause of hemoptysis. Eur J Cardiothoracic Surg. 2002;22:643–5.
-
Bolca N, et al. Bronchopulmonary sequestration: radiological findings. Eur J Radiol. 2004;52:185–91.
-
Bruzzi JF, et al. Multi-detector row CT of hemoptysis. Radiographics. 2006;26:3–22.
-
Chung MJ, et al. Bronchial and non-bronchial systemic arteries in patients with hemoptysis: depiction on MDCT angiography. AJR Am J Roentgenol. 2006;186:649–55.
-
Cooper C, et al. CT appearance of the normal inferior pulmonary ligament. AJR Am J Roentgenol. 1983;141:237–40.
-
Do KH, et al. Systemic arterial supply to the lung in adults: spiral CT findings. Radiographics. 2001;21:387–402.
-
Frazier AA, et al. Pulmonary angiitis and granulomatosis: radiologic-pathologic correlation. Radiographics. 1998;18:687–710.
-
Furuse M, et al. Bronchial arteries: CT demonstration with arteriographic correlation. Radiology. 1987;162:393–8.
-
Katayama K, et al. Adult case of accessory cardiac bronchus presenting with bloody sputum. Jpn J Thorac Cardiovasc Surg. 2005;53:641–4.
-
Khalil A, et al. Role of MDCT in identification of the bleeding site and the vessels causing hemoptysis. AJR Am J Roentgenol. 2007;188:W117–25.
-
Löschhorn C, et al. Dieulafoy’s disease of the lung: a potential disaster for the bronchoscopist. Respiration. 2006;73:562–5.
-
Son JS, et al. Anomalous systemic arterial supply to the basal segments of the right lower lobe in neonate. Pediatr Cardiol. 2008;29:1009–10.
-
Temes E, et al. Young patient with recurrent hemoptysis. Resp Med (Extra). 2006;2:64–6.
-
van der Werf TS, et al. Fatal hemorrhage from Dieulafoy’s disease of the bronchus. Thorax. 1999;54:184–5.
-
Wilson SR, et al. CT visualization of mediastinal bronchial artery aneurysm. AJR Am J Roentgenol. 2006;187:W544–5.
-
Yoon YC, et al. Hemoptysis: bronchial and non-bronchial systemic arteries at 16-detector row CT. Radiology. 2005;234:292–8.
7.9 Cystic Fibrosis (Mucoviscidosis)
Cystic fibrosis (CF) is an autosomal recessive, systemic disorder characterized by abnormal function of the exocrine glands due to defect in the permeability of epithelium to chloride ions. The disease is caused by mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene located on the long arm of chromosome 7.
CF affects between 1 in 4500 children of Caucasian origin. Orientals and blacks are rarely affected. Median survival age is 29 years, and up to 95 % of deaths are due to progressive pulmonary disease. Other areas affected by CF include the sweat glands, pancreas, liver, intestine, and Wolffian ducts in males.
7.9.1 Pulmonary Manifestations of Cystic Fibrosis
Patients with cystic CF are prone to progressive pulmonary deterioration due to formation of bronchiectasis. The defective ciliary mechanism within the bronchial tree causes the pulmonary secretions to accumulate within the terminal bronchioles (mucus plugs).
The most frequently encountered pathologies in CF include mucus plugs, sacculations, emphysema, bronchiectasis, atelectasis, and lung fibrosis in terminal lung disease due to chronic inflammation. CF patients are susceptible to airway colonization with specific bacteria such as Staphylococcus aureus, Hemophilus influenza, allergic bronchopulmonary aspergillosis (ABPA), and Pseudomonas species. Hemoptysis is an unusual complication of CF and arises due to enlargement of the bronchial arteries.
Burkholderia cepacia, formerly known as Pseudomonas cepacia, is an anaerobic, catalase-positive, gram-negative rod that was first described by William Burkholder in 1950 as a plant pathogen capable of causing onion rot. B. cepacia emerges as a problematic cystic fibrosis pathogen almost 30 years ago. Unlike other pathogens affecting cystic fibrosis patients, B. cepacia causes rapid, uncontrolled, and fatal clinical disease in 10 % of cystic fibrosis patients, a condition known as cepacia syndrome. The infection is capable of transmission through social contact. B. cepacia uncommonly causes community-acquired infections in immunocompetent patients, causing chronic suppurative otitis media complicated by cerebellopontine abscesses.
Signs on Pulmonary HRCT
-
1.
The most frequent manifestations of CF in chest HRCT include emphysematous air trapping (100 %), peribronchial thickening (97 %), atelectasis, sacculations, bronchiectasis, and fibrosis in long-standing disease (Fig. 7.9.69).
-
2.
Mucus plugs appear as centrilobular densities, typically against isodense background of collapsed pulmonary lobule (Fig. 7.9.70).
-
3.
Sacculations are seen as excessively dilated bronchi (Fig. 7.9.71).
-
4.
On angiography, enlargement of the bronchial arteries may be seen in CF patients presenting with hemoptysis.
-
5.
Pulmonary lobar atelectasis with high-density materials seen within the bronchi can be due to acute hemorrhagic aspiration or more commonly infection with aspergillosis (ABPA). Mucus plugs have typically low attenuation on HRCT; only 30 % of mucus plugs have high attenuation. The high attenuation is due to calcium oxalate crystal deposition within the plugs for unknown reason.

Axial pulmonary HRCT illustration that demonstrates the most common manifestations detected on HRCT in patients with cystic fibrosis

Axial pulmonary CT, postcontrast illustration that demonstrates hyperdense mucus plug sign in bronchial tree in patients with cystic fibrosis (arrowhead)

Axial pulmonary HRCT image of a patient with cystic fibrosis shows bilateral sacculations, seen as excessively dilated bronchi (arrowhead)
7.9.2 Nasal and Sinus Manifestations of Cystic Fibrosis
Patients with CF suffer from sinonasal symptoms in 10 % of cases. Most complaints are due to unilateral or bilateral nasal blockage (81 %), rhinorrhea (50 %), daily headache (51 %), nasal polyposis (48 %), and anosmia (27 %). Up to 33 % of CF patients have broadening of the nasal bridge.
Nasal polyposis is seen in 48 % of CF patients with sinonasal symptoms. Polyps are circumferential, rounded, pedunculated swellings of the nasal and sinus mucosa with an unknown origin. They commonly arise secondary to chronic sinusitis.
Some patients with CF may develop mucoceles. A mucocele is a slow-growing, expansile, cyst-like mass within the paranasal sinus due to blockage of the normal drainage passage. Retention cysts in contrast are due to blockage of a mucous gland duct, not the sinus itself. Mucoceles may take years to grow large enough to cause symptoms. Up to 70 % of mucoceles are located in the frontal and the ethmoid sinuses (maxillary mucocele is rare). Patients with mucocele can present with proptosis and unilateral visual disturbance if the mucocele presses over the globe and the optic pathways. When the mucocele is infected, it is called pyocele and presents clinically with fever and pain. Lastly, hypoplasia or aplasia of the nasal sinuses has been reported in patients with CF.
Signs on Radiograph
-
1.
Polyps are seen as rounded, pedunculated mucosal swelling within the affected sinus (Fig. 7.9.72).
-
2.
Mucoceles are detected as complete sinus opacification with expansion of the affected sinus.

Plain radiograph of the sinuses (water-view) that shows bilateral maxillary sinus polyposis seen as radio-opaque, rounded shadows (arrowheads)
Signs on CT
-
1.
Nasal polyposis on CT is detected as complete or almost-complete opacification of the sinus without destruction of the fine air cell bony septations (differentiate it from mucocele). When polyps occur in the ethmoid sinus, bilateral bowing of the lamina papyracea occurring in the late stage can lead to acquired hypertelorism (lateralization of the total orbit).
-
2.
Mucocele on CT is detected as a mass filling the sinus with thinning and expansion of the sinus walls. Erosion of the sinus wall can occur. The mass does not enhance after contrast injection. Pyocele can show hyperdense mass with air inclusions.
Signs on MRI
-
1.
Nasal polyps on MRI show low T1 and high homogenous T2 signal in MRI (the signal of normal sinus mucosa). After contrast injection, only the mucosa around the mass will enhance, while the center will not enhance as high as the normal mucosa.
-
2.
Mucocele on MRI shows a signal that depends upon the content of the mass. Normal mucocele shows signal similar to normal mucosa (low T1 and high T2 signal intensities). If the mucocele contains hemorrhage, then it will show high signal intensity in all pulses. If the mucocele is filled with dry, inspissated material, then it will show low signal intensity in all pulses.
7.9.3 Gastrointestinal (GI) Manifestations of Cystic Fibrosis
Patients with CF show GI abnormalities that are mainly located within the pancreas and the liver. Pancreatic abnormalities are seen in 85–90 % of CF patients below 30 years of age. Pancreatic insufficiency is common due to the inherent defect in epithelial chloride ion permeability, which is linked to bicarbonate and water secretion. Up to 30–50 % of patients suffer from exocrine dysfunction, fat malabsorption, and glucose intolerance. Pancreatic insufficiency arises due to pancreatic fatty replacement, cystic changes, and pancreatic cystosis. Up to 30 % of patients with CF have hepatic steatosis, biliary cirrhosis (30 %), portal hypertension, and splenomegaly. Other GI complications include cholelithiasis (10 %), appendicitis, and gastroesophageal reflux disease.
The intestines and colon are commonly affected in patients with CF. Complications affecting the intestine and colon include meconium ileus syndrome, distal intestinal obstruction syndrome, intussusception, fibrosing colonopathy, and colonic wall redundancy. Meconium ileus syndrome is seen in 10–15 % of CF infants, causing intestinal obstruction by sticky material that can cause colonic strictures and peritonitis. In older children, intestinal obstruction can occur due to fecal and colonic fecal impaction (distal intestinal obstruction syndrome). Intussusception occurs in 1 % of CF patients and is typically seen between the ages 4 and 10 years. Fibrosing colonopathy is a condition that arises in CF patients receiving high-strength pancreatic enzyme replacement to control intestinal malabsorption. The colon shows marked strictures, longitudinal shortening, and loss of the haustra due to mucosal fibrosis and thickening of the muscularis mucosa. Colonic wall redundancy (CWR) is a frequent condition in adult CF patients (39 % of cases). Patients with CWR can be asymptomatic or present with nonspecific GI symptoms.
Amyloidosis type-AA can develop in older CF patients due to systemic chronic inflammation. Patients with systemic amyloidosis can present with thyroid goiter, proteinuria, and hepatosplenomegaly.
Signs on Plain Radiographs
-
1.
Pancreatic calcifications may be seen due to liposclerosis and calcification of the intraductal secretions.
-
2.
Abdominal punctuated or plaque-like calcifications that may extend along the processus vaginalis to the scrotum may be seen due to meconium peritonitis in meconium ileus syndrome.
Signs on US
-
1.
Hepatic steatosis is detected as highly echogenic liver.
-
2.
Intussusception is seen as a “donut appearance” mass with hypoechoic rim and echogenic center. The Doppler signal is increased due to vascular congestion.
-
3.
Focal cholestasis (intrahepatic biliary ducts >2 mm in diameter), periportal thickening (>2 mm wall thickness), with focal irregular liver contour can be seen in cases of liver cirrhosis due to CF (pathognomonic). This focal cirrhosis and cholestasis are caused by obstruction of small intrahepatic biliary ducts with ductal proliferation and hyperplasia.
-
4.
Multiple scattered areas of fatty infiltration can be seen in the liver in CF patients, and these masses are called pseudomasses. Typically, they are visualized hyperechoic fatty masses with hypoechoic rim and can be easily mistaken for abnormal liver masses.
-
5.
Colonic wall redundancy is detected as hypertrophic colonic wall folds (>4 mm) with overlapping.
Signs on CT
-
1.
In early stages of CF, the pancreas appears heterogeneous due to areas of fat attenuation and normal parenchymal attenuation. In advanced stages of CF with symptoms of pancreatic insufficiency, complete replacement of the pancreas with fat with high-attenuation pancreatic duct within it can be seen (pancreatic lipomatosis) (Fig. 7.9.73). Similar case can be seen in obesity, old age, chronic pancreatitis, and Cushing’s syndrome.
-
2.
Micro- (<3 mm) or macrocysts (>3 mm) can be found within the pancreas due to inspissation of proteins in the acini and ductules, causing their dilatation and atrophy of the acinar tissue (Fig. 7.9.74). Pancreatic cystosis is a term used to describe complete replacement of the pancreas by cysts (Fig. 7.9.75). Similar case can be encountered in polycystic kidney disease, von Hippel–Lindau syndrome, lymphangiomas, and mucinous cystadenoma.
-
3.
In the liver, signs of cholestasis may be seen with pseudomasses. Pseudomasses are seen as multiple hypodense masses with fat attenuation surrounded by normal liver parenchyma.
-
4.
On axial view, intussusception shows “target sign” with crescent hypodense area inside it representing the mesentery. Enhancing mesenteric vessels within the mass is frequently seen (very characteristic; Fig. 7.9.76). On longitudinal axis, the intussusception is seen as an area of “sausage-shaped” mass. The contrast makes a rim around the area of the intussusception due to bowel invagination.
-
5.
Colonic wall redundancy shows overlapping colonic folds, producing a double or triple appearance of the wall mimicking wrinkles (Fig. 7.9.77). Hyperdensity areas within the wall before contrast injection may be seen due to inspissated mucofeculent material. After contrast injection, the inner wall shows marked enhancement due to hypervascularity.

Axial abdomen CT postcontrast image that shows complete fatty replacement of the pancreas in a patient with chronic cystic fibrosis (arrow)

Axial abdomen CT post-contrast image of a patient with cystic fibrosis that shows multiple cysts formation in the pancreas (arrowheads in a and b)

Axial abdomen CT illustration that shows pancreatic cystosis

Axial abdomen CT illustration that shows the classical “target sign” of intussusception

Axial abdomen CT illustration that shows colon wall redundancy CT features
Signs on MRCP
-
1.
Pancreatic cysts or pancreatic cystosis shows multiple pancreatic cysts that has no communication with the pancreatic duct or duct of Wirsung.
-
2.
In the liver, dilatation of the intrahepatic biliary tree with focal areas of stenosis can be seen mimicking primary sclerosing cholangitis.
Signs on MRI
-
1.
In the liver, periportal wall thickening with high signal intensity on T1W images can be seen due to periportal fat deposition instead of cirrhosis.
-
2.
Colonic wall redundancy shows the same overlapping colonic wall folds mimicking wrinkles in CT, with hyperintense signal intensity on T1W and T2W images before contrast injection, and inner wall enhancement after contrast injection due to hypervascularity.
7.9.4 Genitourinary Manifestations of Cystic Fibrosis
About 97 % of males with CF are sterile due to congenital bilateral absence of the vas deference, which can be associated with the absence of the seminal vesicles and absence or atrophy of the distal portion of the epididymis. Testicular calcification or microlithiasis may be seen.
Testicular microlithiasis is a condition characterized by the presence of multiple punctuated 1–3 mm calcifications within the testicular parenchyma. The condition is usually bilateral and can be associated with conditions like Klinefelter syndrome, Down syndrome, Peutz–Jeghers syndrome, alveolar microlithiasis, cryptorchidism, and male infertility. The condition is premalignant, with 10 % of cases turning into germ cell tumors. Annual ultrasound screening is recommended for patients with testicular microlithiasis.
Signs on Radiographs
Rarely, CF patients may show calcified scrotum due to meconium periorchitis (extension of meconium peritonitis into the scrotum).
Signs on US
-
1.
The epididymis may be absent, atrophic, or show internal cysts.
-
2.
Testicular microlithiasis is seen as multiple echogenic foci 1–3 mm in size in a diffuse bilateral fashion.
-
3.
Calcifications within the scrotum can be seen due to meconium periorchitis.
7.9.5 Musculoskeletal Manifestations of Cystic Fibrosis
Cystic fibrosis arthropathy (CFA) is a poorly understood unique form of polyarthritis that occurs in CF patients. CFA arthropathy is characterized by recurrent painful attacks of mono- or polyarthritis associated with erythema nodosum-like rash and purpuric skin lesions (Fig. 7.9.78). The flares of joint inflammation last from day 1 to several weeks (average 1–7 days). Most cases are self-limiting; however, active synovitis persists. Majority of the reported cases are negative for both serum rheumatoid factor and antinuclear antibodies.

An illustration that demonstrates the body’s geographic distribution of cystic fibrosis arthropathy
Signs on Radiographs
In CFA, affected joints show effusion, osteoporosis, and hypertrophic osteoarthropathy.
Selected References
-
Ablin DS, et al. Ulcerative type of colitis associated with the use of high strength pancreatic enzyme supplements in cystic fibrosis. Pediatr Radiol. 1995;25:113–6.
-
Akata D, et al. Hepatobiliary manifestations of cystic fibrosis in children: correlation of CT and US findings. Eur J Radiol. 2002;41:26–33.
-
Akata D, et al. Liver manifestations of cystic fibrosis. Eur J Radiol. 2007;61:11–7.
-
Brihaye P, et al. Pathological changes of the lateral nasal wall in patients with cystic fibrosis (mucoviscidosis). Int J Pediatr Otorhinolaryngol. 1994;28:141–7.
-
Cahill ME, et al. Pancreatic cystosis in cystic fibrosis. Abdom Imaging. 1997;22:313–4.
-
Carucci LR, et al. Focal fatty sparing of the pancreatic head in cystic fibrosis: CT findings. Abdom Imaging. 2003;28:853–5.
-
Chaudry G, et al. Abdominal manifestations of cystic fibrosis in children. Pediatr Radiol. 2006;36:233–40.
-
Chrispin AR, et al. The systematic evaluation of the chest radiograph in cystic fibrosis. Pediatr Radiol. 1974;2:101–6.
-
De Baets F, et al. Achromobacter xylosoxidans in cystic fibrosis: prevalence and clinical relevance. J Cyst Fibros. 2007;6:75–8.
-
De Gruchy S, et al. Pancreatic cystosis in a child with cystic fibrosis. Pediatr Radiol. 2008;38:1142.
-
Demirkazık FB, et al. High resolution CT in children with cystic fibrosis: correlation with pulmonary functions and radiographic scores. Eur J Radiol. 2001;37:54–9.
-
El-Laboudi AH, et al. Acute Burkholderia cepacia pyomyositis in a patient with cystic fibrosis. J Cyst Fibros. 2009;8:273–5.
-
Lang I, et al. Abdominal calcification in cystic fibrosis with meconium ileus: radiologic-pathologic correlation. Pediatr Radiol. 1997;27:523–7.
-
Ledesma-Medina J, et al. Abnormal paranasal sinuses in patients with cystic fibrosis of the pancreas. Radiological findings. Pediatr Radiol. 1980;9:61–4.
-
Lipnick RN, et al. Bone changes associated with cystic fibrosis. Skeletal Radiol. 1992;21:115–6.
-
Lugo-Olivieri CH, et al. Cystic fibrosis: spectrum of thoracis and abdominal CT findings in the adult patient. Clin Imaging. 1998;22:346–54.
-
Mahenthiralingam E, et al. Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. J Appl Microbiol. 2008;104:1539–51.
-
Marioni G, et al. Burkholderia cepacia complex nasal isolation in immunocompetent patients with sinonasal polyposis not associated with cystic fibrosis. Eur J Clin Microbiol Infect Dis. 2007;26:73–5.
-
Mc Laughlin AM, et al. Amyloidosis in cystic fibrosis: a case series. J Cyst Fibros. 2006;5:59–61.
-
Merkel PA. Rheumatic disease and cystic fibrosis. Arthritis Rheum. 1999;42(8):1563–71.
-
Monti L, et al. Pancreatic cystosis in cystic fibrosis: case report. Abdom Imaging. 2001;26:648–50.
-
Morozov A, et al. High-attenuation mucus plugs on MDCT in a child with cystic fibrosis: potential cause and differential diagnosis. Pediatr Radiol. 2007;37:592–5.
-
Paling MR, et al. Scoliosis in cystic fibrosis: an appraisal. Skeletal Radiol. 1982;8:63–6.
-
Quillin SP, et al. Hepatobiliary sonography in cystic fibrosis. Pediatr Radiol. 1993;23:533–5.
-
Rathaus V, et al. Sonographic findings of the genital tract in boys with cystic fibrosis. Pediatr Radiol. 2006;36:162–6.
-
Robertson JM, et al. Nasal and sinus disease in cystic fibrosis. Paediatr Respir Rev. 2008;9:213–9.
-
Ryan S, et al. Pancreatic replacement by cysts in cystic fibrosis. Pediatr Radiol. 2008;38:1141.
-
Sodhi KS, et al. Pancreatic lipomatosis in an infant with cystic fibrosis. Pediatr Radiol. 2005;35:1157–8.
-
Vergesslich KA, et al. Portal venous blood flow in cystic fibrosis: assessment by Duplex Doppler sonography. Pediatr Radiol. 1989;19:371–4.
7.10 Sleep Apnea Syndromes
Sleep apnea syndromes are group of diseases characterized by complete or partial cessation of breathing, lasting at least 10 s, which occurs repeatedly throughout the night. These conditions are defined by a respiratory disturbance index (RDI) >10, and the prevalence is 70 % in elderly men and 56 % in elderly women respectively.
Sleep apnea syndromes can arise due to abnormalities in the pharynx (obstructive), tracheal tree (obstructive), or the respiratory center within the medulla oblongata (central type).
The most common subtypes of sleep apnea disorders:
-
1.
Obstructive sleep apnea/hypopnea syndrome
-
2.
Upper airway resistance syndrome
-
3.
Central alveolar hypoventilation syndrome (Ondine’s curse)
7.10.1 Obstructive Sleep Apnea Syndrome
Obstructive sleep apnea syndrome (OSAS) is a disease characterized by repeated episodes of pharyngeal wall collapse during sleep, causing apnea or hypopnea. The most common symptoms of OSAS patients include chronic loud snoring, excessive daytime sleepiness, personality changes, and deterioration of quality of life.
Patients with OSAS suffer from hypoxemia and hypercapnia with negative consequences in particular on the cardiorespiratory system (pulmonary hypertension, systemic hypertension, cardiac arrhythmias, and myocardial ischemia), central nervous system (cerebral ischemia), and decreased survival. OSAS is classified according to the respiratory distress index (RDI) into:
-
Normal: RDI <5 attacks per hour of sleep
-
Mild: RDI = 5–15 attacks per hour of sleep
-
Moderate: RDI = 16–30 attacks per hour of sleep
-
Severe: RDI >30 attacks per hour of sleep
7.10.1.1 OSAS and Obesity
OSAS often coexists with obesity, with significant OSAS present in approximately 40 % of obese individuals and about 70 % of OSAS patients being obese. Many studies showed that increased levels of leptin in OSAS are due to leptin resistance. Also, the direct relationship between OSAS and leptin is supported by the fact that effective OSAS treatment with continuous positive airway pressure (CPAP) treatment also influences leptin levels.
Adiponectin is an adipocyte-derived cytokine with regulatory functions in glucose and lipid metabolism. It also has profound anti-inflammatory and antiatherogenic effects. Levels of plasma adiponectin are decreased in obesity and metabolic syndrome.
7.10.1.2 OSAS and Endocrine–Metabolic Abnormalities
Men who suffer from OSAS may manifest decreased libido and a decline in morning serum testosterone levels. It is also accepted that obesity in men is associated with reduced androgen secretion, since increased leptin levels is known to cause impairment of testicular Leydig cell function.
Women with the polycystic ovary syndrome (a condition associated with hyperandrogenism and insulin resistance) were found to be much more likely than controls to have sleep-disordered breathing and daytime sleepiness, suggesting a pathogenetic role for insulin resistance in OSAS.
In OSAS, chronic hypoxia, hypercarbia, and respiratory acidosis associated with periodic upper airway obstruction stimulate peripheral and central chemoreceptors in the body that promote a cardiovascular and respiratory sympathetic reflex. This increase in sympathetic activity promotes hyperinsulinemia by stimulating glycogenolysis and gluconeogenesis and produces an increase in circulating free fatty acids via stimulation of lipolysis that promotes insulin resistance.
7.10.1.3 OSAS and Cardiovascular Diseases
There are many evidences which suggest that cyclic intermittent hypoxia in OSAS patient causes hypercoagulability and disturbs nocturnal rennin and aldosterone secretion profiles and increases nighttime urine excretion.
Signs on Plain Radiographs
-
1.
Cephalometry is a lateral plain radiograph of the head and neck that is used to obtain certain cephalometric parameters that describe the relations between the craniofacial structures. The lateral head and neck radiograph should be obtained after complete expiration to ensure standardization. A frontal plain radiograph can be obtained also to complete the measurements. The same measurements can be obtained from CT sinuses in coronal and sagittal planes (Fig. 7.10.79).
-
2.
The most common cephalometric parameters are the same parameters applied to CT:
-
a.
Posterior pharyngeal wall (PPW): the thickness of the posterior pharyngeal wall in front of the tubercle of the atlas (normal MPH >3.7 mm).
-
b.
Posterior airway space (PAS) at C3 level: the measurement of the pharyngeal airway space at C3 (normal >5 mm).
-
c.
Central incisor to the tongue base (TT-TB): OSA <90 mm (Figs. 7.10.80 and 7.10.81).
-
d.
Velum width (Vw): maximal velar width, i.e., thickness of the velum measured on cross section at the widest point (normal Vw >12 mm).
-
e.
Posterior uvular space (PUS): (OSA <4.5 mm).
-
f.
Medial orbit to medial orbit (MOMO) distance: the medio-orbitale (MO) is the point on the medial orbital margin that is the closest to the medial plane (left and right) (Fig. 7.10.79). The distance between the two MO points (MOMO) is normal MOMO <26 mm.
-
g.
Mandibular plane to hyoid (MPH): the distance in mm between the mandibular plane and the most anterior point on the hyoid (normal MPH <20.8 mm).
-
a.

Sagittal CT sinuses images (a–c) and coronal (d) that shows the cephalometric lines used to measure the degree of stenosis in OSA

Sagittal CT sinuses images (b, c) with 3D reconstruction image of a patient with OSA due to prognathism shows reduced central incisor to tongue base (TT-TB) distance (c)

Sagittal (a) and axial (b) CT images of a patient with OSA and narcolepsy show thickened posterior uvula (arrow) and reduced oropharyngeal space (arrowhead)
Signs on CT and MRI
-
1.
The oropharynx provides a common pathway for both swallowing and respiration. There are three main factors that determine the balance between airway patency and collapse:
-
a.
The tone in the dilator muscles of the upper airways: these muscles are activated just before diaphragmatic contraction, and their tone is primarily dependent on the state of consciousness. As a result, prolonged obstruction occurs only during sleep.
-
b.
The magnitude of the negative intra-airway pressure: this negative force is generated by contraction of the diaphragm and is related to the total airway resistance. Narrowing of the nasopharyngeal airway increases total airway resistance and requires additional negative intra-airway pressure to maintain flow.
-
c.
The cross-sectional dimension of the oropharynx: the smaller the oropharyngeal airway, the greater the likelihood of closure at any level of negative intrapharyngeal pressure (Figs. 7.10.81 and 7.10.82).
-
a.
-
2.
Patients with obstructive sleep apnea have abnormally narrow airway (oropharynx) posterior to the tongue, which can be appreciated on dynamic CT imaging of the oropharynx.

Sagittal (a) and axial (b) CT images of a patient with OSA show severe retro-palatal nasopharyngeal stenosis by a hypertrophic soft palate (arrow) and retro-palatal nasopharyngeal space stenosis (arrowhead)
7.10.2 Upper Airway Resistance Syndrome
Upper airways resistance syndrome (UARS) is a disease that lies in between primary snoring and obstructive sleep apnea syndrome in severity (OSAS). UARS is defined as intermittent attacks of sleep apnea associated with snoring and respiratory disturbance index (RDI) less than 5 with daytime sleepiness attacks. Unlike OSAS, UARS is characterized by the absence of frank sleep apneas or oxygen desaturation with daytime hypersomnolence.
UARS patients are typically nonobese (in contrast to OSAS) and have a mean age of 37 years and body mass index <25 kg/m presenting with snoring and daytime hypersomnolence. UARS arises due to retrolingual narrowing, low soft palates, long uvulas, and high, narrow hard palates as determined by cephalometry.
OSAS patients are typically obese (BMI >30) and have RDI >5 and daytime sleepiness (hypersomnolence). In contrast, primary snoring is defined as snoring at night, RDI <5, and no daytime sleepiness.
7.10.3 Central Alveolar Apnea Syndrome (Ondine’s Curse)
Central sleep apnea is defined as an absence of airflow and respiratory effort lasting at least 10 s. Central alveolar apnea syndrome, also known as Ondine’s curse, is a disease characterized by failure of the automatic control of ventilation during sleep due to a lesion affecting the descending anterolateral medullocervical pathway of the reticular formation.
Ondine’s curse is a mythological fairytale about a mermaid who exchanged into a human to marry the man she loves. She made an excellent wife; however, her husband cheated on her with another woman. Ondine was still retaining part of her magic power as a mermaid, so she cursed her husband with a curse that whenever he falls asleep, he suffocates. So he has to be awake forever; otherwise, he will die.
Ondine’s curse can be congenital or acquired. Congenital Ondine’s curse may be associated with Hirschsprung’s disease (known as Haddad syndrome) or neuroblastoma. The disease arises due to mutation in the PHOX2B gene.
Neonates with Ondine’s curse syndrome presents typically with hypotonia and apnea that needs continuous ventilation hours after birth. The pathological process is related to insensitivity to hypercarbia with raised serum pCO2 levels (may be up to 80–90 mmHg), especially during sleep, when the respiration is maximally under chemical control.
Acquired Ondine’s curse is an uncommon condition that arises in adults and has been reported in association with medullary tumors, infection (particularly poliomyelitis), upper cervical trauma with Duret hemorrhage, some mitochondrial diseases, degenerative diseases (e.g., multiple system atrophy), medullary capillary telangiectasias, demyelinating disease (e.g., multiple sclerosis), or nonspecific anoxic–ischemic insults.
Diaphragmatic pacing has been described in series, with reported success rates between 50 and 70 %. Candidates for diaphragmatic pacing must be severely incapacitated by chronic ventilatory insufficiency and are usually receiving ventilatory support before pacing is instituted.
Signs on MRI
-
1.
Congenital Ondine’s curse infants can show molar tooth sign due to cerebellar peduncle atrophy.
-
2.
The brain in congenital Ondine’s curse may show signs of ischemic encephalopathy due to hypoxia and hypercapnia.
7.10.4 Cheyne–Stokes Respiration
Cheyne–Stokes respiration (CSR) is present in up to 50 % of patients with chronic heart failure (CHF) and left ventricular (LV) ejection fraction ≤45 %. CSR is characterized by repetitive apneas and arousal cycles induce sleep fragmentation followed by fatigue, daytime sleepiness, and neurohumoral activation including sympathoadrenergic stimulation.
7.10.5 Uncommon and Rare Causes of Sleep Apnea
-
1.
Arnold–Chiari malformation 1: usually associated with neurological manifestations
-
2.
Atlantoaxial dislocation/subluxation
-
3.
Olivopontocerebellar atrophy (Dejerine–Thomas syndrome)
-
4.
Posterior fossa tumors (e.g., medulloblastoma)
-
5.
Syringobulbia
-
6.
Bulbar poliomyelitis
Selected References
-
Block AJ. Sleep apnea and related disorders. Dis Mon. 1985;31(5):6–56.
-
Calvin JR, et al. Obstructive sleep apnea: diagnosis with ultrafast CT. Radiology. 1989;171:775–8.
-
Cartwright R. Obstructive sleep apnea: a sleep disorder with major effects on health. Dis Mon. 2001;47(4):109–47.
-
D’Souza S, et al. Haddad syndrome: congenital central hypoventilation associated with Hirschsprung’s disease. Indian J Pediatr. 2003;70(7):597–9.
-
Fernbach SK, et al. Radiologic evaluation of adenoids and tonsils in children with obstructive sleep apnea: plain films and fluoroscopy. Pediatr Radiol. 1983;13:258–65.
-
Finkelstein Y, et al. Frontal and lateral cephalometry in patients with sleep-disordered breathing. Laryngoscope. 2001;111:634–41.
-
Gaisie G, et al. Coexistent neuroblastoma and Hirschsprung’s disease: another manifestation of the neurocristopathy? Pediatr Radiol. 1979;8:161–3.
-
Galvin JR, et al. Obstructive sleep apnea: diagnosis with ultrafast CT. Radiology. 1998;171:775–8.
-
Hegstrom T, et al. Obstructive sleep apnea syndrome: preoperative radiologic evaluation. AJR. 1988;150:67–9.
-
Kerbl R, et al. Congenital central hypoventilation syndrome (Ondine’s curse syndrome) in two siblings: delayed diagnosis and successful noninvasive treatment. Eur J Pediatr. 1996;155:977–80.
-
Lam B, et al. Arnold-Chiari malformation presenting as sleep apnea syndrome. Sleep Med. 2000;1:139–44.
-
Macpherson RI, et al. Upper airway obstruction in children: an update. Radiographics. 1985;5(3):339–76.
-
Pracharktam N, et al. Cephalometric assessment in obstructive sleep apnea. Am J Orothod Dentofac Orthop. 1996;109:410–9.
-
Randerath WJ. Treatment options in Cheyne-Stokes respiration. Ther Adv Respir Dis. 2010;4:341–51.
-
Roshkow JE, et al. Hirschsprung’s disease, ondine’s curse, and neuroblastoma-manifestations of neurocristopathy. Pediatr Radiol. 1988;19:45–9.
-
Stein MG, et al. Cine CT in obstructive sleep apnea. AJR. 1987;148:1069–74.
-
Susarla SM, et al. Cephalometric measurement of upper airway length correlates with the presence and severity of obstructive sleep apnea. J Oral Maxillofac Surg. 2010;68:2846–55.
-
Wiegand L, et al. Obstructive sleep apnea. Dis Mon. 1994;40(4):202–52.
-
Zamarron C, et al. Obstructive sleep apnea syndrome is a systemic disease. Current evidence. Eur J Intern Med. 2008;19:390–8.
Author information
Authors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Al-Tubaikh, J.A. (2017). Pulmonology. In: Internal Medicine. Springer, Cham. https://doi.org/10.1007/978-3-319-39747-4_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-39747-4_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-39746-7
Online ISBN: 978-3-319-39747-4
eBook Packages: MedicineMedicine (R0)